 Verena Katharina Raker
Verena Katharina Raker Christian Becker
Christian Becker Kerstin Steinbrink
Kerstin Steinbrink- Department of Dermatology, University Medical Center Mainz, Johannes Gutenberg-University Mainz, Mainz, Germany
Nucleotide signaling molecules contribute to the regulation of cellular pathways. In the immune system, cyclic adenosine monophosphate (cAMP) is well established as a potent regulator of innate and adaptive immune cell functions. Therapeutic strategies to interrupt or enhance cAMP generation or effects have immunoregulatory potential in autoimmune and inflammatory disorders. Here, we provide an overview of the cyclic AMP axis and its role as a regulator of immune functions and discuss the clinical and translational relevance of interventions with these processes.
Introduction
Cells must be able to sense and integrate countless extracellular and intracellular signals and adapt their cellular functions. Second messengers serve as initiating components of intracellular signal transduction cascades that transmit signals by cellular messengers that depend on extracellular signaling molecules (1). Thereby second messengers serve to greatly amplify the strength of the original first signal. Cyclic adenosine monophosphate (cAMP) was the first discovered intracellular second messenger of extracellular ligand action (2). Within the immune system, cAMP regulates pro- and anti-inflammatory activities: drugs that elevate intracellular cAMP levels reduce the production of pro-inflammatory mediators and increase the production of anti-inflammatory factors in numerous immune cells. This review aims to shed light on the variety of processes influenced by cAMP in the immune system with regard to treatment options in diseases.
The cAMP Pathway
Cyclic adenosine monophosphate, identified in 1957 (2) as the first intracellular second messenger of extracellular ligand action, is now established as a universal regulator of metabolism and gene expression in all life forms (3). A family of enzymes called adenylate cyclases (AC) catalyzes cAMP formation from ATP. In vertebrates, AC comprise nine membrane-bound isoforms and one soluble isoform (4). AC vary in distribution and developmental expression and their regulation is complex and isozyme specific. In addition to AC expression and activity cAMP homeostasis is regulated by a superfamily of phosphodiesterases (PDE) that degrade intracellular cyclic nucleotides. PDE comprise more than 100 enzyme variants divided into 11 families (5) based on their structure, specificity for, and modulation by, cyclic nucleotides. Certain PDE increase their activities in response to cAMP and cAMP stimulates the synthesis of new PDE mRNA (6, 7), resulting in a feedback loop between cAMP levels and PDE activity. Contributing to the complexity of the pathway, some PDE families are strictly cAMP-specific (PDE 4, 7, and 8), whereas others are cyclic guanosine monophosphate (cGMP)-specific (PDE 5, 6, and 9) (8, 9). Additional families hydrolyze both cAMP and cGMP (PDE 1, 2, 3, 10, and 11), establishing cross-regulation of both pathways with important implications in the utility of pharmacotherapeutic agents targeting cyclic nucleotide metabolism (10, 11).
As a second messenger, cyclic AMP serves in multiple downstream pathways. Most prominent, it activates the cAMP-dependent protein kinase A I (PKA) (12) (see Figure 1). Upon binding of cAMP to the regulatory subunits, PKA dissociates into its regulatory and catalytic subunits and the catalytic subunits phosphorylate specific Ser and Thr residues on numerous target proteins initiating successive signaling cascades, particularly in nutrient metabolism (13). In addition, cAMP-activated PKA binds and phosphorylates cAMP-responsive transcription factors, including cAMP-response element binding protein (CREB), members of the cAMP-responsive element modulator/inducible cAMP early repressor (CREM/ICER) protein family (14), activating transcription factor-1 (ATF-1), NFκB, and nuclear receptors (see Figure 1). Phosphorylated CREB, CREM, and ATF-1 interact with the transcriptional coactivators CREB-binding protein (CBP) and p300 when bound to cAMP-response elements (CREs) in target genes (15). In addition to PKA activation, cAMP also directly modulates the activity of guanine-nucleotide-exchange factor (GEF) exchange proteins (Epacs) and cyclic nucleotide-gated channels (CNGs) (16) all with important roles in cellular functions (17, 18). In addition to PKA, CREB, CREM, and ATF-1 can all be phosphorylated by many other kinases, and the action of PKA is counterbalanced by specific protein phosphatases.
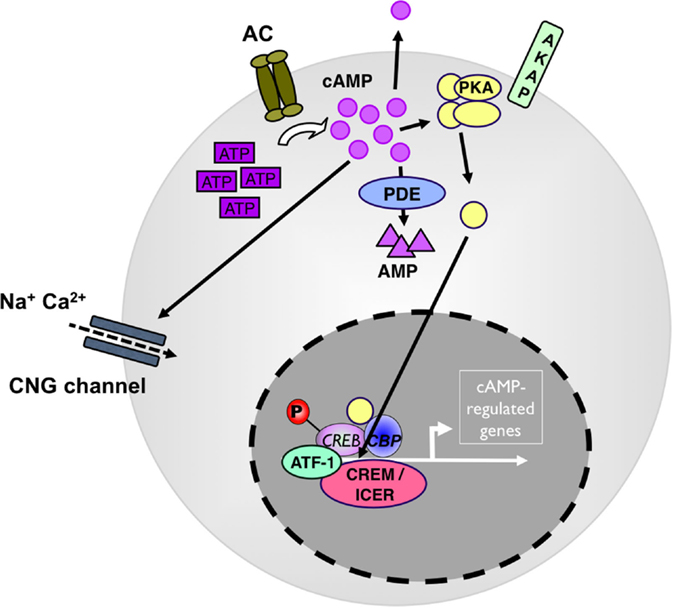
Figure 1. cAMP as a regulator of immunity. Adenylate cyclases (AC) produce cAMP from adenosin-tri-phosphate (ATP). High levels of cytosolic cAMP lead to activation of protein kinase A (PKA). PKA stimulation induces the phosphorylation of transcription factors, such as CREB, ICER/CREM, ATF-1, and CBP to drive camp-driven genes. Phosphodiesterase 4 (PDE4) decreases intracellular cAMP levels and counterbalances the intracellular cAMP effect. ATF, cAMP-dependent transcription factor; CBP, cAMP-binding protein; CNG, cyclic nucleotide-gated ion channel; CREB, cAMP response element-binding protein; ICER, inducible cAMP early repressor; P, phosphorylation.
Basal cytosolic cAMP levels are in the low micrometer range (19). In the cytosol, cAMP is not evenly distributed but rather forms submembranous spatially discrete pools generated in microdomains containing AC, PDE next to PKA localized by A-kinase-anchoring proteins (AKAPs) (20). Specificity in cAMP signaling and fine and selective tuning of its different tasks is ensured by the differential expression of distinct isoforms and splice variants of anabolic, katabolic, and signaling molecules in various tissues and cell types and by differential composition of cAMP microdomains (21). Although various cAMP activities can have redundant, independent, or opposing effects within the same cell (22), some individual AC and PDE knockout and transgenic mice (23, 24) show specific phenotypes. In particular, individual PDE control select cyclic nucleotide-regulated events by being integrated into non-overlapping multi-molecular regulatory signaling complexes, suggesting cell or tissue-specific interference points (25, 26).
Eventually, an important, often overlooked aspect of the pathway consists in the secretion of cAMP into extracellular space and its transmission via gap junctions between cells (27). Whereas transmitted cAMP directly contributes to intracellular cAMP levels, excreted cAMP is converted into AMP and adenosine by cell surface bound PDE and ecto-5′-nucleotidases CD39 and CD73. By signaling through A2A and A2B adenosine receptors, extracellular adenosine stimulates AC and increases intracellular cAMP generation (28). Knockout mice with disrupted CD39 and CD73 have underscored the importance of the extracellular cAMP–adenosine feedback mechanism in physiological processes (29, 30). In the immune system extracellular cAMP may contribute to regulatory T cells (Treg) function (31, 32) and has been shown to promote monocyte differentiation into dendritic cells (DCs) (33).
Cyclic AMP in Immune Homeostasis and Pathophysiology
Due to its multiple roles in cell physiology cAMP exerts broad modulatory effects on a variety of cells (see Figure 2). In the immune system, cyclic AMP regulates both innate and adaptive immune cell activities (34).
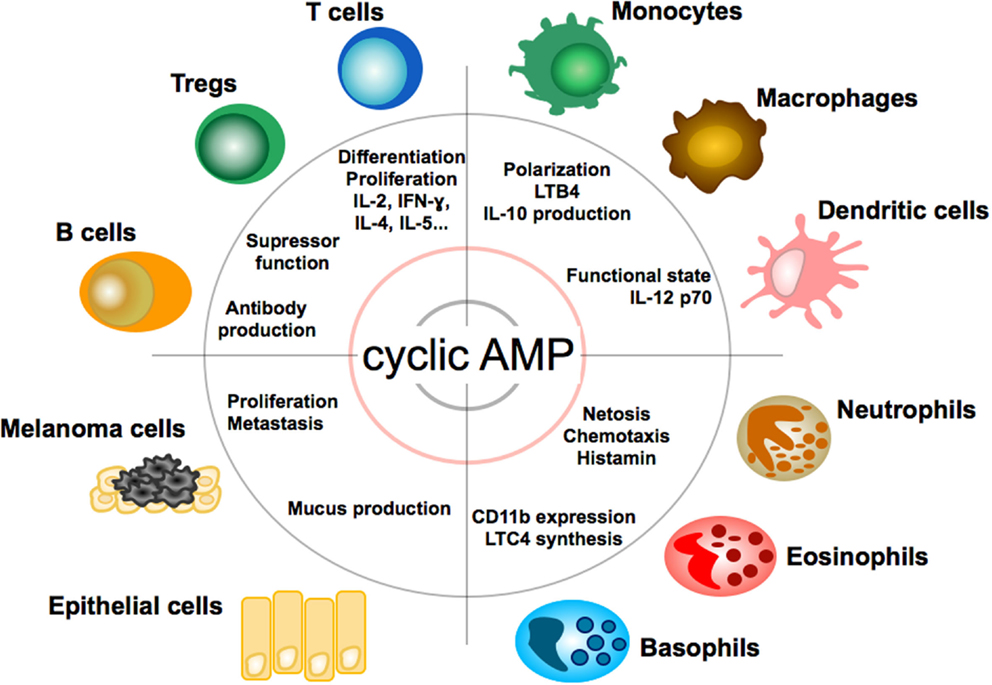
Figure 2. Effect of cAMP on immune, tumor, and epithelial cells. Impact and function of cyclic adenosin monophopshate (cAMP) on T and B lymphocytes, granulocytes, monocytes, macrophages, dendritic cells, epithelial cells, and melanoma cells. LTB4, leukotriene B4; LTC4, leukotriene C4.
Monocytes and Granulocytes
The functional state of monocytes orchestrates inflammatory and reparative phases in inflammatory responses and appears to be accompanied by changes in their intracellular cAMP levels. In the mouse, two major types of monocytes, Ly6Chigh and Ly6Clow, circulate in blood. Ly6Chigh monocytes display pro-inflammatory activity, whereas Ly6Clow monocytes are patrolling cells, monitor tissue integrity, and exert anti-inflammatory and tissue repair activities (35). The orphan nuclear receptor Nr4a1 (Nur77) regulates the expression of genes linked to inflammation. Inflammatory stimuli inhibit its expression and induce an inflammatory Ly6Chigh phenotype (36, 37). In turn, Nur77 is upregulated and represses numerous inflammatory genes in the transition from an inflammatory Ly6Chigh to anti-inflammatory Ly6Clow/neg state (38–40). Elevated cAMP levels induce Nur77 expression (41) and, thus, favor a reparatory monocyte phenotype (42). Through these effects on phagocytes increased cAMP levels affect myeloid cell immunity against pathogen and parasites (43–45) and may also affect the differentiation of tumor-infiltrating myeloid-derived suppressor cells (MDSCs) by repression of TNF-α production. In regard of the latter CREB activation has been shown to upregulate miR-9 expression that promotes the differentiation of the so-called MDSCs with significantly increased immunosuppressive function (46).
In sum, increased cAMP levels appear to generally weaken monocyte inflammatory functions (47–50). Interestingly, bacteria and fungi have taken advantage of this effect in the course of evolution. Pathogen capture and programed destruction are among the most important activities of innate immune cells to prevent tissue invasion and pathogen dissemination. Certain microbacteria and fungi have evolved to hijack the host cAMP axis by introducing microbial adenylyl and guanylyl cyclases (51) and by intoxicating the host cell with preformed cAMP or adenylate cyclase toxins (52–54). Bordetella pertussis, for example, suppresses neutrophil extracellular trap (NET) formation by overwhelming leukocytes with supraphysiologic intracellular cAMP levels (55). Likewise, bacterial-derived or -induced cAMP facilitates intracellular bacterial survival by multiple actions, including CREB-dependent anti-apoptotic signaling and repression of intracellular bacterial killing in invaded monocytes and macrophages.
NK Cells
Natural killer (NK) cells are capable of destroying tumor cells and virally infected cells (cytolysis) without prior sensitization. In NK cells, cAMP levels regulate target cell adherence and cytotoxic function. Both pharmacological repression and induction of cAMP inhibit perforin-mediated and CD95 ligand-mediated target cell lysis (56–60).
Dendritic Cells
As professional antigen-presenting cells of the immune system, DCs are equipped with a unique capability to induce and regulate adaptive immune responses. In DC, cyclic AMP suppresses the release of pro-inflammatory mediators (TNF-α, IL-17, IFN-γ) (61) and promotes the release of anti-inflammatory mediators, such as IL-10 (62). As a functional consequence, cAMP concentrations in DC regulate T cell immunity (63). Pharmacological inhibition of cyclic nucleotide PDE4, which is highly expressed in DC, for example, suppresses the DC Th1-polarizing capacity (64, 65) and commands secretion of IL-6 and TGF-beta and subsequent induction of Th17 differentiation (66). It, thus, appears that cAMP levels differentially regulate cytokine production by DC as a response to changes in the microenvironment. Apart from spatio-temporal fine-tuning of DC activities, cAMP activities in DC depend on the stage of DC maturation: prostaglandin E2 (PGE2), a key inducer of cAMP, exerts a stimulatory function for immature DCs in peripheral tissues (67) but inhibitory function for mature DCs in lymph nodes (68).
B and T Cells
In addition to innate cell function, cAMP also controls numerous adaptive immune cell activities. In adaptive immune cells, cAMP is essentially required in the induction of antigen-stimulated activation (69–72) but subsequently limits activation by negatively regulating signaling through B cell and T cell receptors (TCR). In B cells, it provides an essential signal in the induction of antigen-stimulated proliferation and antibody production (69, 70, 72). Elevation of intracellular cAMP enhances IgE production by promoting recombination of the Ig heavy chain loci and by favoring Th2 differentiation. In T cells, cAMP participates in the regulation of nearly all functional activities ranging from peripheral maintenance of naïve T cells (73) to their activation via the TCR (74), acquisition of effector function (75, 76), and memory (77). In cognate activation, cAMP acts as a temporary inhibitory feedback signal that limits T cell activation through the cAMP–PKA–Csk signaling pathway (74). Unlike temporary increases, continuously elevated cAMP levels induce an anergy-like state (78, 79). Likewise, anergizing TCR signals result in increased intracellular cAMP concentrations that upregulate the cyclin-dependent kinase (CDK) inhibitor p27kip1, sequester cyclin D2–cdk4, and cyclin E/cdk2 complexes and prevent progression through the G1 restriction point of the cell cycle (80). Furthermore, cAMP levels regulate the acquisition of effector function. Pharmacological upregulation of cAMP by inhibition of PDE activity, for example, prevents the development and function of cytotoxic T lymphocyte (CTL) (81). The significance of cAMP in acquisition of effector functions in T cells is also reflected by the observation that CREB mutant mice have normal T cell numbers in the thymus but exhibit a marked defect in peripheral T cell proliferation and IL-2 production, resulting from G1 cell-cycle arrest and apoptotic cell death (82). Most prominent, cAMP forms an essential component of the suppressive mechanism in Treg (83–92). Treg contain increased levels of cytosolic cAMP, further upregulate their cAMP level upon activation and consign cAMP to target cells via gap junctions (83, 85). In the target cell, cAMP inhibits the proliferation and differentiation of effector functions, in part by interfering with gene expression via ICER (90). Repression of cAMP accumulation in Treg by either adenylyl cyclase inhibition, application of a cAMP-specific antagonist, or PDE overexpression abrogates murine and human Treg suppression (83, 84, 86, 91, 93). Inversely, blockade of cAMP degradation by PDE inhibition improves Treg-mediated suppression in a murine asthma model (85). In line, non-functional Treg in Foxp3-mutant scurfy mice harbor significantly reduced levels of cytosolic cAMP (94).
Increased cAMP formation in Treg is a prerequisite for their suppressive activity (95) (see Figures 3 and 4). Constitutively high cAMP levels in Treg appear to be caused by Foxp3-induced decreased PDE3B expression (96) and increased AC9 activity (87) driven by their constitutive active state (95). During Treg-mediated suppression, cAMP is transferred via gap junctions to conventional T cells (Tcon), where it represses IL-2 production and inhibits the proliferative response (83). Pharmacological inhibition of cAMP formation abrogates the suppressive function of Treg (see Figure 3) (91).
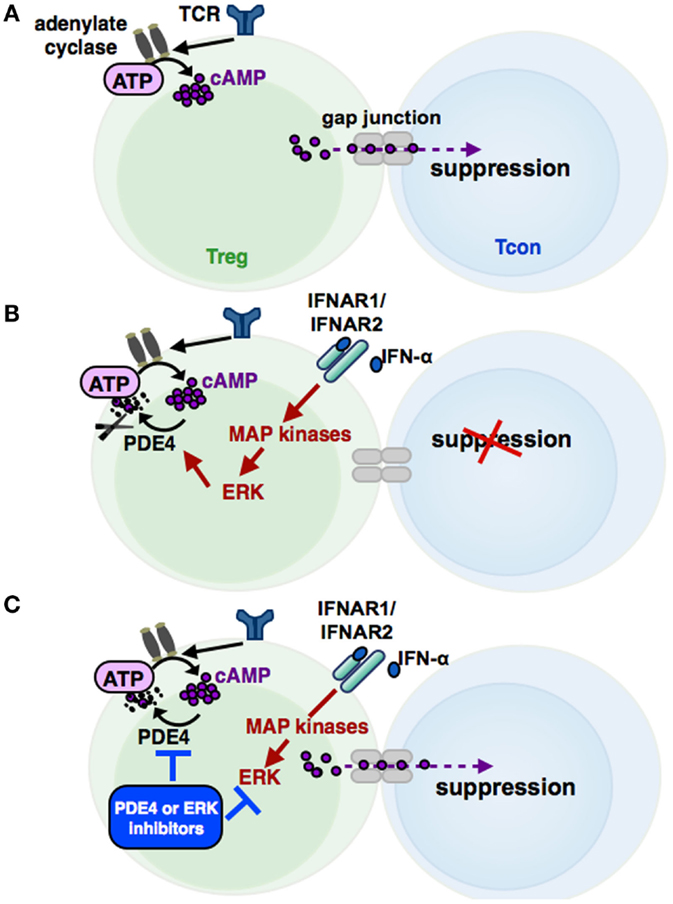
Figure 3. The cAMP pathway in Treg and its regulation by IFN-α. Signaling via the T cell receptor (TCR) leads to an activation of adenylate cyclases, resulting in high cAMP levels in regulatory T cells (Treg). cAMP can be transferred via gap junctions into conventional T cells (Tcon), thereby mediating the suppressive activity of Treg (A). Phosphodiesterase 4 (PDE4), which can be activated by MAP kinase ERK-related pathways, reduces cAMP amounts in Treg by enzymatic cleavage, impairing the regulatory activity of Treg (B). IFN-α abolishes the suppressive function of Treg by cAMP reduction, restoring the Tcon activation. Inhibition of the ERK or PDE4 pathway, respectively, results in a renewed suppressive capacity of IFN-α treated Treg (C).
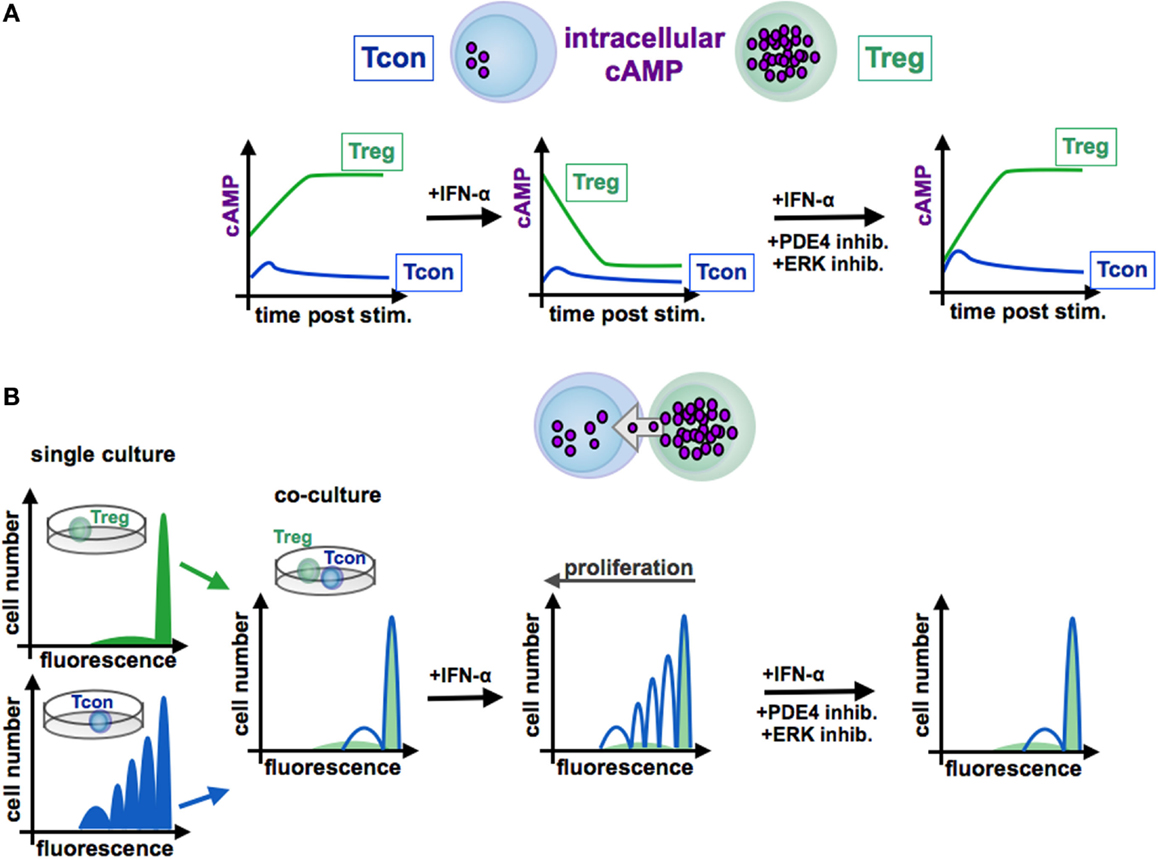
Figure 4. Function of cAMP in the interaction of conventional and regulatory T cells. In contrast to Tcon (blue line), Treg (green line) exhibit high levels of cAMP (A). Stimulated Tcon display a high proliferation whereas Treg are characterized by a low proliferative capacity [(B), left panels, single culture]. Treg efficiently inhibit Tcon proliferation in co-culture experiments by cAMP transfer via gap junctions to Tcon [(B), co-culture]. By contrast, IFN-α abrogates the suppressive function of Treg through reduction of cAMP levels [(A), centered panel], resulting in a restored Tcon activation [(B), centered panel]. Blockade of the ERK or PDE4 pathway, respectively, increases intracellular cAMP amounts [(A), right panel], renews the suppressive activity of Treg [(B), right panel]. Tcon, conventional T cells; Treg, regulatory T cells.
In this context, Bacher et al. showed that IFN-α, an antineoplastic agent with well-known autoimmune side effects, disturbs the immunosuppressive activity of human CD4+CD25+Foxp3+ Treg by disabling cAMP upregulation upon activation (92, 97) (see Figure 3 and 4). IFN-α-mediated inhibition of Treg suppression can be partially restored by pharmacological inhibitors blocking ERK and PDE/PDE4 activity through specific inhibitors (92, 97) (see Figures 3 and 4). These results are in line with the observation that human T cells predominantly express the short PDE4B and PDE4D isoforms, functionally regulated by the ERK2 MAP kinase (98, 99). As PDE have an essential role in the IFN-α-mediated inhibition of Treg, PDE4 interference by specific inhibitors may represent a therapeutic option to restore immune regulation in autoimmune diseases, such as psoriasis or lupus erythematosus, accompanied by reduced Treg function (64, 100).
Next to its role in the Treg-suppressive mechanism cAMP is required for the generation and maintenance of Treg: the cAMP-responsive transcription factor CREB stabilizes FoxP3 expression and promotes and maintains the Treg phenotype (101, 102). Treg essentially depend on IL-2 for their peripheral maintenance and suppressive activity (103, 104) and their number and activity can be therapeutically manipulated by low-dose IL-2 and particular IL-2/anti-IL-2 complexes (105, 106) to control autoimmune diseases and inflammation (107). Interestingly, IL-2 may contribute to increased cAMP production in Treg by increasing adenylate cyclase AC7 activity (88). In conjunction with its role in control of the Treg phenotype, its transmission via gap junctions to and from Treg also appears to play a role in the Treg lifecycle as evidence by the observation that Treg numbers are significantly reduced in connexin 43 knockout mice (108).
Some viruses prevent their rejection by the immune system by interfering with the cAMP pathway in T cells. HIV-1 surface glycoprotein gp120 induces anergy in naive T lymphocytes (109, 110) and increases cAMP levels and suppressive activity in Treg (86, 111, 112). In turn, cAMP repression restores antiviral T cell function in HIV patients (113).
Beyond their role in immune regulation, Treg take on homeostatic functions by regulating metabolic activity in visceral fat and participating in tissue repair. Functionally distinct Treg accumulate in injured skeletal muscle and contribute to repair processes. Muscle Treg distinctly express the growth factor amphiregulin, which improves muscle repair by directly acting on muscle satellite cells (114). In line with outlined role of cAMP in Treg function, amphiregulin synthesis is inhibited by PKA inhibitors and enhanced by ligands that increased cAMP or directly activate the PKA (115).
Together these findings classify cAMP as a key component of immune cell function and disclose cAMP-regulating enzymes as molecular targets for therapeutic intervention with immune activities in pathological processes like allergy and autoimmunity.
Modulation of cAMP in Autoimmune and Inflammatory Diseases
Cyclic AMP is a central player in the network of signaling pathways underlying pathogenesis of several diseases and several interference points are used therapeutically in a variety of conditions. Although the clinical impact of changes in cAMP remains incompletely defined, one fundamental conclusion can nevertheless be drawn: interventions that enhance cAMP generation or actions have immune dampening potential; conversely, repression of cAMP or cAMP signaling has immunostimulatory capability.
Formation of cAMP by AC and degradation by PDE identifies AC and PDE as major targets for therapeutic intervention with cAMP levels. To date, the AC activity has been mostly pharmacologically targeted through agonists or antagonists affecting upstream G-protein-coupled receptors (GPCR) (23, 116). However, AC knockout and transgenic mice revealed individual and clearly distinct physiological functions for AC isoforms (23). The observation that individual isoforms play a dominant role in specific tissues has led to AC being considered as main drug targets (117). In order to achieve selective interference, isoform-selective compounds are required. Such compounds are currently being sought and tested. Here, the idea is pursued, that selective inhibitors intervene in a tissue-specific manner, but remain ineffective in tissues that express various AC isoforms (118).
AC-specific compounds already reached preclinical stages and others have been approved for particular diseases, such as colforsin daropate hydrochloride (NKH447), a AC5 selective forskolin (FSK) derivate, for the treatment of advanced congestive heart failure (119, 120). Thus, even though AC isoform-targeted drugs are still in early stages of the development, the finding that AC have clearly separated physiological functions at least suggests AC as pharmacologic targets in a broad spectrum of diseases ranging from neurodegenerative disorders to congestive heart failure and lung diseases as asthma and chronic obstructive pulmonary disease (COPD).
Since their identification in 1958 (2), continuing efforts have been undertaken to advance the understanding of PDE biology and function, and PDE have been considered pharmacological targets in various diseases, such as pulmonary diseases like COPD and asthma, depression, schizophrenia, erectile dysfunction, and autoimmune disease like psoriasis/psoriasis arthritis and rheumatoid arthritis (8, 100, 121–125). Although numerous PDE inhibitors have been developed, their introduction into the clinic has been hampered by their narrow therapeutic window and side effects, such as nausea and emesis, occurring even at sub-therapeutic levels.
In the immune system, PDE family 3, 4, and 7 members represent the predominant cAMP-degrading enzymes (126). PDE4 are encoded by four separate genes (PDE4A–D) and each PDE4 controls non-redundant cellular function (127). In addition, more than 20 PDE4 variants arise from alternative mRNA splicing or the use of different transcriptional units (5). While PDE4A, PDE4B, and PDE4D are expressed in immune cells (T and B cells, neutrophils, eosinophils, DCs, monocytes, macrophages), PD4C is minimally active or absent (128, 129). PDE3 and PDE7 are detected in most inflammatory cells, including T and B cells, NK, and myeloid cells (6, 59, 127, 130–132). However, PDE4s are the predominant cAMP-degrading isoenzymes (126, 127). In addition, the expression levels of the PDE isoenzymes are differentially regulated by a variety of inflammatory stimuli (126, 127). Apart from immune cells, PDE4 members are also expressed in chondrocytes, smooth muscle cells, epithelial cells, and vascular endothelium (127). By increasing levels of intracellular cAMP, PDE4 inhibitors show anti-inflammatory effects in almost all inflammatory and immune cells and are known to suppress a multitude of inflammatory responses, including proliferation, chemotaxis, phagocytosis, and release of pro-inflammatory mediators, such as cytokine and chemokines, reactive oxygen species, lipid mediators, and hydrolytic enzymes (34, 126, 129). Numerous selective PDE4 inhibitors have been patented and some of them have been evaluated in clinical trials, including diseases, such as asthma, COPD, atopic dermatitis, rheumatoid arthritis, and psoriasis/psoriasis arthritis. However, most of these compounds had to be discontinued because of narrow therapeutic windows. Doses needed for an efficient treatment could not be reached due to side effects, such as nausea, emesis, diarrhea, and abdominal pain being the most common. It has been hypothesized that adverse side effects of the PDE4 inhibitors are a result of their non-selectivity to all four PDE4 subtypes and PDE4 inhibition in non-target tissues at doses similar (or lower) than needed for therapeutic efficacy. It is postulated that blocking of PDE4D in non-target organs promotes emesis (133). In view of side effect profile of second-generation PDE4 inhibitors, new strategies for the design of active and non-emetic compounds have been employed to overcome the adverse effects and to improve therapeutic effects. In this context, despite highly conserved catalytic domains of PDE4 isoenzymes, PDE4 subtype-specific inhibitors have been generated. For example, potent PDE4B inhibitors with more than 100-fold selectivity over PDE4D have been synthesized (134, 135). Compared with the non-selective PDE4 inhibitor cilomilast (134), selective PDE4B inhibitors demonstrated a potent anti-inflammatory activity and significantly less gastrointestinal side effects. In order to circumvent side effects observed upon oral administration, inhalation (136) and topical application (137) of PDE4 inhibitors have been explored in the treatment of airway inflammation and inflammatory cutaneous diseases. Two phase studies conducted with a PDE4 inhibitor (AN2728) in psoriasis and atopic dermatitis patients showed promising results (138, 139). The interest for PDE4 anti-inflammatory activity arose from early studies with the prototypic PDE4 inhibitor, rolipram (140). However, although PDE4 inhibitors have been mostly developed to treat lung diseases, such as asthma or COPD, no compound has yet reached the market for asthma treatment. By contrast, the orally active PDE4 inhibitor roflumilast (Daliresp®, Forest Pharmaceuticals) has been approved for COPD by the European Medicines Agency in 2010 and the U.S. Food and Drug Administration in 2011 based on four clinical trials. These studies have shown that roflumilast improves lung function and reduces the frequency of COPD exacerbations in patients with chronic bronchitis symptoms (141–144). Although side effects were generally mild to moderate, nausea, diarrhea, weight loss, and headache were still reported (145). Despite these side effects, roflumilast received approval for COPD with severe air flow limitations, symptoms of chronic bronchitis, and a history of exacerbations in several countries (146, 147).
Another currently marketed oral PDE4 inhibitor is apremilast (Otezla®, Celgene Corporation) that has been approved by the EMA and FDA for psoriasis and psoriasis arthritis, two autoimmune diseases, characterized by chronic inflammation, tissue and organ involvement, and accelerated growth cycle of skin cells. Apremilast was developed based on the rolipram and roflumilast pharmacophore by coupling a series of phthalimide analogs in order to optimize its activity and to decrease side effects (148). The safety and efficacy of apremilast for the treatment of patients with plaque psoriasis and psoriasis arthritis were evaluated in numerous multicenter, randomized, double-blind, placebo-controlled clinical trials (ESTEEM-1 and -2 for psoriasis, PALACE-1, -2, and -3 for psoriasis arthritis) (149–152). In the two ESTEEM trials, apremilast reduced the severity and extent of moderate-to-severe plaque psoriasis (including nail, scalp, and palmoplantar manifestations) versus placebo in adults. Similarly, in three PALACE trials (PALACE 1–3), apremilast improved the signs and symptoms of psoriasis arthritis relative to placebo in adults with active disease despite treatment with conventional synthetic and/or biologic disease-modifying anti-rheumatic drugs. According to the published clinical trials, apremilast was well tolerated in all study groups analyzed. Throughout phase II and III trials, the most frequently reported side effects consisted of headache, nausea, diarrhea, emesis, and nasopharyngitis and upper respiratory tract infection under continued treatment. However, the studies showed that the gastrointestinal adverse effects usually subside within a month of therapy.
It is an interesting result of the clinical studies that improved inhibitor specificity does not prevent side effects. This result suggests that the same or overlapping cell populations caused both wanted and unwanted effects. In view of recent research results regarding the expression and activities of anabolic and catabolic cAMP enzymes in immune cells, the question arises whether particular PDE4 inhibitor effects are caused by alteration of immune cell functions. This question is underlined by the similarity of side effects in PDE4 inhibitor studies and some immunotherapeutic approaches. Unfortunately, effects in individual immune cell populations have not been considered in clinical studies with PDE inhibitors so far. For a better understanding of the underlying causes of wanted and unwanted effects, such studies appear urgently needed. Alongside their specificity, effective interference with the cAMP pathway through inhibitors depends on their mechanism of action. Basically, inhibitors may act reversibly or irreversibly. Irreversible inhibitors bind to enzymes through covalent bonds. Covalent inhibitors have many desirable features, including increased biochemical efficiency of target disruption, reduced sensitivity toward pharmacokinetic parameters and increased duration of action that outlasts the pharmacokinetics of the compound. Only few inhibitors of this type, however, exist for anabolic and catabolic cAMP enzymes with the common ADCY inhibitor MDL-12,330A, a cyclo-alkyllactamide derivative supposedly representing an exception (153). Most inhibitors are reversible, bind to enzyme through non-covalent bonds, and typically address the ATP-binding site or the catalytic portion. With non-covalent inhibitors, cells can quickly become insensitive by recovering enzyme activity. To increase their activity, however, inhibitors can be coupled to proteins that regulate protein expression. A favorable example exists in proteolytic targeting, such as the ubiquitin proteasome system (UPS) (154). Proteolytic targeting chimeric molecules, or PROTACS comprise a UPS recognition motif coupled to an inhibitor via a linker. While a first generation of PROTACs suffered from limited cell-permeability, the second generation has been improved by using a HIF1α peptide fragment as an E3 ubiquitin ligase recognition motif to increase permeability (155). Thus, in addition to the development of more specific inhibitors to achieve selective interference, their inhibitory activity may be improved through proteolytic targeting, particularly by preventing target cell resistance.
Conclusion and Perspective
Because of its central importance as a universal regulator of metabolism and gene expression, systemic intervention of the cAMP metabolism is associated with numerous, sometimes considerable, side effects. Additionally or alternatively to the development of isoform-specific AC and PDE inhibitors, new methods need to be found by which these inhibitors may be delivered to tissues and cells specifically. Novel strategies may encompass the development of highly specific agents, new routes of delivery (cutaneous, inhalation) or the use of nanoparticles for tissue or even cell-specific drug delivery. Since cAMP signaling controls very different processes in different cells, a better understanding of the cAMP-mediated activities in particular cell types could help to pave the way to more specific interventions in cell function. Unlike anabolic and catabolic cAMP metabolism, very few drugs engage in signal transduction yet and, thus, the potential use of such actions remains unclear. Although known for over 60 years, the cAMP signaling still reveals new functional details. Therapeutic intervention of its activities, thus, requires further elucidation of its role in individual cell types and its entanglements with other signaling and metabolic pathways.
Author Contributions
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the German Research Foundation (DFG): STE791/6-1, STE791/9-1, CRC 1066/B6, TR156/A4/C5; by the German Cancer Aid (110631), and by grants from the University Medical Center Mainz (all to KS).
References
1. Taskén K, Skålhegg BS, Taskén KA, Solberg R, Knutsen HK, Levy FO, et al. Structure, function, and regulation of human cAMP-dependent protein kinases. Adv Second Messenger Phosphoprotein Res (1997) 31:191–204. doi: 10.1016/S1040-7952(97)80019-5
2. Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem (1958) 232:1077–91.
3. Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol (2002) 3:710–8. doi:10.1038/nrm911
4. Dessauer CW. Adenylyl cyclase – A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol (2009) 76:935–41. doi:10.1124/mol.109.059345
5. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem (2007) 76:481–511. doi:10.1146/annurev.biochem.76.060305.150444
6. Erdogan S, Houslay MD. Challenge of human Jurkat T-cells with the adenylate cyclase activator forskolin elicits major changes in cAMP phosphodiesterase (PDE) expression by up-regulating PDE3 and inducing PDE4D1 and PDE4D2 splice variants as well as down-regulating a novel PDE4A splice variant. Biochem J (1997) 321(Pt 1):165–75.
7. Loriaux MM, Rehfuss RP, Brennan RG, Goodman RH. Engineered leucine zippers show that hemiphosphorylated CREB complexes are transcriptionally active. Proc Natl Acad Sci U S A (1993) 90:9046–50. doi:10.1073/pnas.90.19.9046
8. Page CP, Spina D. Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handb Exp Pharmacol (2011) 204:391–414. doi:10.1007/978-3-642-17969-3_17
9. Cote RH. Characteristics of photoreceptor PDE (PDE6): similarities and differences to PDE5. Int J Impot Res (2004) 16(Suppl 1):S28–33. doi:10.1038/sj.ijir.3901212
10. Begum N, Shen W, Manganiello V. Role of PDE3A in regulation of cell cycle progression in mouse vascular smooth muscle cells and oocytes: implications in cardiovascular diseases and infertility. Curr Opin Pharmacol (2011) 11:725–9. doi:10.1016/j.coph.2011.10.006
11. Wang Z-Z, Zhang Y, Zhang H-T, Li Y-F. Phosphodiesterase: an interface connecting cognitive deficits to neuropsychiatric and neurodegenerative diseases. Curr Pharm Des (2015) 21:303–16. doi:10.2174/1381612820666140826115559
12. Walsh DA, Perkins JP, Krebs EG. An adenosine 3',5'-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem (1968) 243:3763–5.
13. Shabb JB. Physiological substrates of cAMP-dependent protein kinase. Chem Rev (2001) 101:2381–411. doi:10.1021/cr000236l
14. Sassone-Corsi P. Transcription factors responsive to cAMP. Annu Rev Cell Dev Biol (1995) 11:355–77. doi:10.1146/annurev.cb.11.110195.002035
15. Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol (2001) 2:599–609. doi:10.1038/35085068
16. Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol (2003) 19:23–44. doi:10.1146/annurev.cellbio.19.110701.154854
17. de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature (1998) 396:474–7. doi:10.1038/24884
18. Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science (1998) 282:2275–9. doi:10.1126/science.282.5397.2275
19. Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, et al. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J Biol Chem (2006) 281:21500–11. doi:10.1074/jbc.M603116200
20. Edwards AS, Scott JD. A-kinase anchoring proteins: protein kinase A and beyond. Curr Opin Cell Biol (2000) 12:217–21. doi:10.1016/S0955-0674(99)00085-X
21. Filadi R, Pozzan T. Generation and functions of second messengers microdomains. Cell Calcium (2015) 58:405–14. doi:10.1016/j.ceca.2015.03.007
22. Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol (2005) 174:595–9. doi:10.4049/jimmunol.174.2.595
23. Pierre S, Eschenhagen T, Geisslinger G, Scholich K. Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov (2009) 8:321–35. doi:10.1038/nrd2827
24. Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov (2014) 13:290–314. doi:10.1038/nrd4228
25. Houslay MD, Baillie GS, Maurice DH. cAMP-specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res (2007) 100:950–66. doi:10.1161/01.RES.0000261934.56938.38
26. Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS. AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol (2012) 52:351–8. doi:10.1016/j.yjmcc.2011.05.002
27. Kumar NM, Gilula NB. The gap junction communication channel. Cell (1996) 84:381–8. doi:10.1016/S0092-8674(00)81282-9
28. Gödecke A. cAMP: fuel for extracellular adenosine formation? Br J Pharmacol (2008) 153:1087–9. doi:10.1038/bjp.2008.7
29. Enjyoji K, Sévigny J, Lin Y, Frenette PS, Christie PD, Esch JS, et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med (1999) 5:1010–7. doi:10.1038/12447
30. Koszalka P, Ozüyaman B, Huo Y, Zernecke A, Flögel U, Braun N, et al. Targeted disruption of cd73/ecto-5’-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res (2004) 95:814–21. doi:10.1161/01.RES.0000144796.82787.6f
31. Kurtz CC, Alam MS, Ernst PB. Extracellular adenosine production is essential for Treg-mediated Th cell suppression. FASEB J (2008) 22:848.26. doi:10.1096/fj.1530-6860
32. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
33. Sciaraffia E, Riccomi A, Lindstedt R, Gesa V, Cirelli E, Patrizio M, et al. Human monocytes respond to extracellular cAMP through A2A and A2B adenosine receptors. J Leukoc Biol (2014) 96:113–22. doi:10.1189/jlb.3A0513-302RR
34. Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol (2008) 39:127–32. doi:10.1165/rcmb.2008-0091TR
35. Xu XJ, Reichner JS, Mastrofrancesco B, Henry WL, Albina JE. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol (2008) 180:2125–31. doi:10.4049/jimmunol.180.4.2125
36. Papac-Milicevic N, Breuss JM, Zaujec J, Ryban L, Plyushch T, Wagner GA, et al. The interferon stimulated gene 12 inactivates vasculoprotective functions of NR4A nuclear receptors. Circ Res (2012) 110:e50–63. doi:10.1161/CIRCRESAHA.111.258814
37. Jutila MA, Kroese FG, Jutila KL, Stall AM, Fiering S, Herzenberg LA, et al. Ly-6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon-gamma. Eur J Immunol (1988) 18(11):1819–26. doi:10.1002/eji.1830181125
38. Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol (2011) 12:778–85. doi:10.1038/ni.2063
39. Carry J-C, Clerc F, Minoux H, Schio L, Mauger J, Nair A, et al. SAR156497, an exquisitely selective inhibitor of aurora kinases. J Med Chem (2015) 58:362–75. doi:10.1021/jm501326k
40. Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J Biol Chem (2005) 280:29256–62. doi:10.1074/jbc.M502606200
41. Maxwell MA, Muscat GEO. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal (2006) 4:e002. doi:10.1621/nrs.04002
42. Bystrom J, Evans I, Newson J, Stables M, Toor I, van Rooijen N, et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood (2008) 112:4117–27. doi:10.1182/blood-2007-12-129767
43. Nokta MA, Pollard RB. Human immunodeficiency virus replication: modulation by cellular levels of cAMP. AIDS Res Hum Retroviruses (1992) 8:1255–61. doi:10.1089/aid.1992.8.1255
44. Fülöp T, Fóris G, Wórum I, Leövey A. Age-dependent alterations of Fc gamma receptor-mediated effector functions of human polymorphonuclear leucocytes. Clin Exp Immunol (1985) 61:425–32.
45. Wirth JJ, Kierszenbaum F. Macrophage mediation of the inhibitory effects of elevated intracellular levels of adenosine-3’:5’ cyclic monophosphate (cAMP) on macrophage-Trypanosoma cruzi association. Int J Parasitol (1984) 14:401–4. doi:10.1016/0020-7519(84)90096-1
46. Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, et al. MicroRNA-9 regulates the differentiation and function of myeloid-derived suppressor cells via targeting Runx1. J Immunol (2015) 195:1301–11. doi:10.4049/jimmunol.1500209
47. Dent G, Giembycz MA, Rabe KF, Wolf B, Barnes PJ, Magnussen H. Theophylline suppresses human alveolar macrophage respiratory burst through phosphodiesterase inhibition. Am J Respir Cell Mol Biol (1994) 10:565–72. doi:10.1165/ajrcmb.10.5.8179921
48. Rowe J, Finlay-Jones JJ, Nicholas TE, Bowden J, Morton S, Hart PH. Inability of histamine to regulate TNF-alpha production by human alveolar macrophages. Am J Respir Cell Mol Biol (1997) 17:218–26. doi:10.1165/ajrcmb.17.2.2722
49. Abrahamsen H, Baillie G, Ngai J, Vang T, Nika K, Ruppelt A, et al. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J Immunol (2004) 173:4847–58. doi:10.4049/jimmunol.173.8.4847
50. Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, et al. Suppression of LPS-induced TNF-α production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal (2009) 2:ra28–28. doi:10.1126/scisignal.2000202
51. Baker DA, Kelly JM. Structure, function and evolution of microbial adenylyl and guanylyl cyclases. Mol Microbiol (2004) 52:1229–42. doi:10.1111/j.1365-2958.2004.04067.x
52. Pezard C, Weber M, Sirard JC, Berche P, Mock M. Protective immunity induced by Bacillus anthracis toxin-deficient strains. Infect Immun (1995) 63:1369–72.
53. Coote JG. Structural and functional relationships among the RTX toxin determinants of Gram-negative bacteria. FEMS Microbiol Rev (1992) 8:137–61. doi:10.1111/j.1574-6968.1992.tb04961.x
54. Agarwal N, Lamichhane G, Gupta R, Nolan S, Bishai WR. Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature (2009) 460:98–102. doi:10.1038/nature08123
55. Eby JC, Gray MC, Hewlett EL. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect Immun (2014) 82:5256–69. doi:10.1128/IAI.02487-14
56. Whalen MM, Bankhurst AD. Effects of beta-adrenergic receptor activation, cholera toxin and forskolin on human natural killer cell function. Biochem J (1990) 272:327–31. doi:10.1042/bj2720327
57. Bariagaber AK, Whalen MM. Decreased adenylyl cyclase and cAMP-dependent protein kinase activities inhibit the cytotoxic function of human natural killer cells. Hum Immunol (2003) 64:866–73. doi:10.1016/S0198-8859(03)00154-X
58. Zhao W, Huang Y, Liu Z, Cao B-B, Peng Y-P, Qiu Y-H. Dopamine receptors modulate cytotoxicity of natural killer cells via cAMP-PKA-CREB signaling pathway. PLoS One (2013) 8:e65860. doi:10.1371/journal.pone.0065860
59. Goto M, Murakawa M, Kadoshima-Yamaoka K, Tanaka Y, Inoue H, Murafuji H, et al. Phosphodiesterase 7A inhibitor ASB16165 suppresses proliferation and cytokine production of NKT cells. Cell Immunol (2009) 258:147–51. doi:10.1016/j.cellimm.2009.04.005
60. Torgersen KM, Vaage JT, Levy FO, Hansson V, Rolstad B, Taskén K. Selective activation of cAMP-dependent protein kinase type I inhibits rat natural killer cell cytotoxicity. J Biol Chem (1997) 272:5495–500. doi:10.1074/jbc.272.9.5495
61. Bäumer W, Hoppmann J, Rundfeldt C, Kietzmann M. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets (2007) 6:17–26. doi:10.2174/187152807780077318
62. Oger S, Méhats C, Dallot E, Cabrol D, Leroy M-J. Evidence for a role of phosphodiesterase 4 in lipopolysaccharide-stimulated prostaglandin E2 production and matrix metalloproteinase-9 activity in human amniochorionic membranes. J Immunol (2005) 174:8082–9. doi:10.4049/jimmunol.174.12.8082
63. Lee J, Kim TH, Murray F, Li X, Choi SS, Broide DH, et al. Cyclic AMP concentrations in dendritic cells induce and regulate Th2 immunity and allergic asthma. Proc Natl Acad Sci U S A (2015) 112:1529–34. doi:10.1073/pnas.1417972112
64. Schett G, Sloan VS, Stevens RM, Schafer P. Apremilast: a novel PDE4 inhibitor in the treatment of autoimmune and inflammatory diseases. Ther Adv Musculoskelet Dis (2010) 2:271–8. doi:10.1177/1759720X10381432
65. Heystek HC, Thierry A-C, Soulard P, Moulon C. Phosphodiesterase 4 inhibitors reduce human dendritic cell inflammatory cytokine production and Th1-polarizing capacity. Int Immunol (2003) 15:827–35. doi:10.1093/intimm/dxg079
66. Datta SK, Sabet M, Nguyen KPL, Valdez PA, Gonzalez-Navajas JM, Islam S, et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc Natl Acad Sci U S A (2010) 107:10638–43. doi:10.1073/pnas.1002348107
67. Legler DF, Krause P, Scandella E, Singer E, Groettrup M. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J Immunol (2006) 176:966–73. doi:10.4049/jimmunol.176.2.966
68. Harizi H, Juzan M, Grosset C, Rashedi M, Gualde N. Dendritic cells issued in vitro from bone marrow produce PGE(2) that contributes to the immunomodulation induced by antigen-presenting cells. Cell Immunol (2001) 209:19–28. doi:10.1006/cimm.2001.1785
69. Gilbert KM, Hoffmann MK. cAMP is an essential signal in the induction of antibody production by B cells but inhibits helper function of T cells. J Immunol (1985) 135:2084–9.
70. Levy FO, Rasmussen AM, Taskén K, Skålhegg BS, Huitfeldt HS, Funderud S, et al. Cyclic AMP-dependent protein kinase (cAK) in human B cells: co-localization of type I cAK (RI alpha 2 C2) with the antigen receptor during anti-immunoglobulin-induced B cell activation. Eur J Immunol (1996) 26:1290–6. doi:10.1002/eji.1830260617
71. Taskén K, Stokka AJ. The molecular machinery for cAMP-dependent immunomodulation in T-cells. Biochem Soc Trans (2006) 34:476–9. doi:10.1042/BST0340476
72. Wortis HH, Teutsch M, Higer M, Zheng J, Parker DC. B-cell activation by crosslinking of surface IgM or ligation of CD40 involves alternative signal pathways and results in different B-cell phenotypes. Proc Natl Acad Sci U S A (1995) 92:3348–52. doi:10.1073/pnas.92.8.3348
73. Cekic C, Sag D, Day Y-J, Linden J. Extracellular adenosine regulates naive T cell development and peripheral maintenance. J Exp Med (2013) 210:2693–706. doi:10.1084/jem.20130249
74. Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skålhegg BS, et al. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med (2001) 193:497–507. doi:10.1084/jem.193.4.497
75. Liopeta K, Boubali S, Virgilio L, Thyphronitis G, Mavrothalassitis G, Dimitracopoulos G, et al. cAMP regulates IL-10 production by normal human T lymphocytes at multiple levels: a potential role for MEF2. Mol Immunol (2009) 46:345–54. doi:10.1016/j.molimm.2008.10.025
76. Hedrich CM, Crispin JC, Rauen T, Ioannidis C, Apostolidis SA, Lo MS, et al. cAMP response element modulator α controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc Natl Acad Sci U S A (2012) 109:16606–11. doi:10.1073/pnas.1210129109
77. Vig M, George A, Sen R, Durdik J, Rath S, Bal V. Commitment of activated T cells to secondary responsiveness is enhanced by signals mediated by cAMP-dependent protein kinase A-I. Mol Pharmacol (2002) 62:1471–81. doi:10.1124/mol.62.6.1471
78. Cone RE, Cochrane R, Lingenheld EG, Clark RB. Elevation of intracellular cyclic AMP induces an anergic-like state in Th1 clones. Cell Immunol (1996) 173:246–51. doi:10.1006/cimm.1996.0274
79. Powell JD, Lerner CG, Ewoldt GR, Schwartz RH. The -180 site of the IL-2 promoter is the target of CREB/CREM binding in T cell anergy. J Immunol (1999) 163:6631–9.
80. Appleman LJ, Tzachanis D, Grader-Beck T, van Puijenbroek AA, Boussiotis VA. Helper T cell anergy: from biochemistry to cancer pathophysiology and therapeutics. J Mol Med (Berl) (2001) 78:673–83. doi:10.1007/s001090000180
81. Kadoshima-Yamaoka K, Murakawa M, Goto M, Tanaka Y, Inoue H, Murafuji H, et al. Effect of phosphodiesterase 7 inhibitor ASB16165 on development and function of cytotoxic T lymphocyte. Int Immunopharmacol (2009) 9:97–102. doi:10.1016/j.intimp.2008.10.005
82. Bodor J, Spetz AL, Strominger JL, Habener JF. cAMP inducibility of transcriptional repressor ICER in developing and mature human T lymphocytes. Proc Natl Acad Sci U S A (1996) 93:3536–41. doi:10.1073/pnas.93.8.3536
83. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
84. Oberle N, Eberhardt N, Falk CS, Krammer PH, Suri-Payer E. Rapid suppression of cytokine transcription in human CD4+CD25 T cells by CD4+Foxp3+ regulatory T cells: independence of IL-2 consumption, TGF-beta, and various inhibitors of TCR signaling. J Immunol (2007) 179:3578–87. doi:10.4049/jimmunol.179.6.3578
85. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol (2009) 182:4017–24. doi:10.4049/jimmunol.0803310
86. Becker C, Taube C, Bopp T, Becker C, Michel K, Kubach J, et al. Protection from graft-versus-host disease by HIV-1 envelope protein gp120-mediated activation of human CD4+CD25+ regulatory T cells. Blood (2009) 114:1263–9. doi:10.1182/blood-2009-02-206730
87. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen G-X, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep (2009) 10:180–5. doi:10.1038/embor.2008.224
88. Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+ T cells. Mol Immunol (2010) 47:678–84. doi:10.1016/j.molimm.2009.10.032
89. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol (2010) 265:91–6. doi:10.1016/j.cellimm.2010.07.007
90. Vaeth M, Gogishvili T, Bopp T, Klein M, Berberich-Siebelt F, Gattenloehner S, et al. Regulatory T cells facilitate the nuclear accumulation of inducible cAMP early repressor (ICER) and suppress nuclear factor of activated T cell c1 (NFATc1). Proc Natl Acad Sci U S A (2011) 108:2480–5. doi:10.1073/pnas.1009463108
91. Klein M, Vaeth M, Scheel T, Grabbe S, Baumgrass R, Berberich-Siebelt F, et al. Repression of cyclic adenosine monophosphate upregulation disarms and expands human regulatory T cells. J Immunol (2012) 188:1091–7. doi:10.4049/jimmunol.1102045
92. Bacher N, Raker V, Hofmann C, Graulich E, Schwenk M, Baumgrass R, et al. Interferon-α suppresses cAMP to disarm human regulatory T cells. Cancer Res (2013) 73:5647–56. doi:10.1158/0008-5472.CAN-12-3788
93. Martin H, Reuter S, Dehzad N, Heinz A, Bellinghausen I, Saloga J, et al. CD4-mediated regulatory T-cell activation inhibits the development of disease in a humanized mouse model of allergic airway disease. J Allergy Clin Immunol (2012) 129:e1–7. doi:10.1016/j.jaci.2011.09.038
94. Lahl K, Mayer CT, Bopp T, Huehn J, Loddenkemper C, Eberl G, et al. Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J Immunol (2009) 183:5662–72. doi:10.4049/jimmunol.0803762
95. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity (2014) 41:722–36. doi:10.1016/j.immuni.2014.10.012
96. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A (2006) 103:6659–64. doi:10.1073/pnas.0509484103
97. Becker C, Bopp T, Steinbrink K. Interferon α interferes with immunological tolerance. Oncoimmunology (2013) 2:e27528. doi:10.4161/onci.27528
98. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi:10.1038/nri1604
99. Peter D, Jin SLC, Conti M, Hatzelmann A, Zitt C. Differential expression and function of phosphodiesterase 4 (PDE4) subtypes in human primary CD4+ T cells: predominant role of PDE4D. J Immunol (2007) 178:4820–31. doi:10.4049/jimmunol.178.8.4820
100. Chiricozzi A, Caposiena D, Garofalo V, Cannizzaro MV, Chimenti S, Saraceno R. A new therapeutic for the treatment of moderate to severe plaque psoriasis: apremilast. Expert Rev Clin Immunol (2015) 12:237–49. doi:10.1586/1744666X.2016.1134319
101. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi:10.1038/nri2785
102. Kim H-P, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med (2007) 204:1543–51. doi:10.1084/jem.20070109
103. de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol (2004) 34:2480–8. doi:10.1002/eji.200425274
104. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol (2010) 185:6426–30. doi:10.4049/jimmunol.0903940
105. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311:1924–7. doi:10.1126/science.1122927
106. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med (2009) 206:751–60. doi:10.1084/jem.20082824
107. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol (2015) 15:283–94. doi:10.1038/nri3823
108. Kuczma M, Lee JR, Kraj P. Connexin 43 signaling enhances the generation of Foxp3+ regulatory T cells. J Immunol (2011) 187:248–57. doi:10.4049/jimmunol.1003785
109. Rahmouni S, Aandahl EM, Nayjib B, Zeddou M, Giannini S, Verlaet M, et al. Cyclo-oxygenase type 2-dependent prostaglandin E2 secretion is involved in retrovirus-induced T-cell dysfunction in mice. Biochem J (2004) 384:469–76. doi:10.1042/BJ20031859
110. Masci AM, Galgani M, Cassano S, De Simone S, Gallo A, De Rosa V, et al. HIV-1 gp120 induces anergy in naive T lymphocytes through CD4-independent protein kinase-A-mediated signaling. J Leukoc Biol (2003) 74:1117–24. doi:10.1189/jlb.0503239
111. Moreno-Fernandez ME, Rueda CM, Rusie LK, Chougnet CA. Regulatory T cells control HIV replication in activated T cells through a cAMP-dependent mechanism. Blood (2011) 117:5372–80. doi:10.1182/blood-2010-12-323162
112. Hofmann B, Nishanian P, Nguyen T, Liu M, Fahey JL. Restoration of T-cell function in HIV infection by reduction of intracellular cAMP levels with adenosine analogues. AIDS (1993) 7:659–64. doi:10.1097/00002030-199305000-00008
113. Hofmann B, Nishanian P, Nguyen T, Insixiengmay P, Fahey JL. Human immunodeficiency virus proteins induce the inhibitory cAMP/protein kinase A pathway in normal lymphocytes. Proc Natl Acad Sci U S A (1993) 90:6676–80. doi:10.1073/pnas.90.14.6676
114. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155:1282–95. doi:10.1016/j.cell.2013.10.054
115. Braeuning A. The connection of β-catenin and phenobarbital in murine hepatocarcinogenesis: a critical discussion of Awuah et al., PLoS ONE 7(6):e39771, 2012. Arch Toxicol (2013) 87:401–2. doi:10.1007/s00204-012-1002-4
116. Pavan B, Biondi C, Dalpiaz A. Adenylyl cyclases as innovative therapeutic goals. Drug Discov Today (2009) 14:982–91. doi:10.1016/j.drudis.2009.07.007
117. Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, Tomlinson JE, et al. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem (2004) 279:40938–45. doi:10.1074/jbc.M314238200
118. Okumura S, Kawabe J, Yatani A, Takagi G, Lee M-C, Hong C, et al. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ Res (2003) 93:364–71. doi:10.1161/01.RES.0000086986.35568.63
119. Alasbahi RH, Melzig MF. Plectranthus barbatus: a review of phytochemistry, ethnobotanical uses and pharmacology – part 2. Planta Med (2010) 76:753–65. doi:10.1055/s-0029-1240919
120. Toya Y, Schwencke C, Ishikawa Y. Forskolin derivatives with increased selectivity for cardiac adenylyl cyclase. J Mol Cell Cardiol (1998) 30:97–108. doi:10.1006/jmcc.1997.0575
121. Diamant Z, Spina D. PDE4-inhibitors: a novel, targeted therapy for obstructive airways disease. Pulm Pharmacol Ther (2011) 24:353–60. doi:10.1016/j.pupt.2010.12.011
122. Hatzelmann A, Morcillo EJ, Lungarella G, Adnot S, Sanjar S, Beume R, et al. The preclinical pharmacology of roflumilast – a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther (2010) 23:235–56. doi:10.1016/j.pupt.2010.03.011
123. Zhang H-T. Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr Pharm Des (2009) 15:1688–98. doi:10.2174/138161209788168092
124. Siuciak JA. The role of phosphodiesterases in schizophrenia: therapeutic implications. CNS Drugs (2008) 22:983–93. doi:10.2165/0023210-200822120-00002
125. Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science (2005) 310:1187–91. doi:10.1126/science.1112915
126. Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med (1998) 157:351–70. doi:10.1164/ajrccm.157.2.9708012
127. Jin S-LC, Ding S-L, Lin S-C. Phosphodiesterase 4 and its inhibitors in inflammatory diseases. Chang Gung Med J (2012) 35:197–210.
128. Spina D, McFadzean I, Bertram FKR, Page CP. Peripheral mechanisms II: the pharmacology of peripherally active antitussive drugs. Handb Exp Pharmacol (2009) 187:155–86. doi:10.1007/978-3-540-79842-2_8
129. Press NJ, Banner KH. PDE4 inhibitors – a review of the current field. Prog Med Chem (2009) 47:37–74. doi:10.1016/S0079-6468(08)00202-6
130. Giembycz MA, Smith SJ. Phosphodiesterase 7A: a new therapeutic target for alleviating chronic inflammation? Curr Pharm Des (2006) 12:3207–20. doi:10.2174/138161206778194123
131. Tenor H, Hatzelmann A, Kupferschmidt R, Stanciu L, Djukanović R, Schudt C, et al. Cyclic nucleotide phosphodiesterase isoenzyme activities in human alveolar macrophages. Clin Exp Allergy (1995) 25:625–33. doi:10.1111/j.1365-2222.1995.tb01110.x
132. Tenor H, Staniciu L, Schudt C, Hatzelmann A, Wendel A, Djukanović R, et al. Cyclic nucleotide phosphodiesterases from purified human CD4+ and CD8+ T lymphocytes. Clin Exp Allergy (1995) 25:616–24. doi:10.1111/j.1365-2222.1995.tb01109.x
133. Robichaud A, Stamatiou PB, Jin S-LC, Lachance N, MacDonald D, Laliberté F, et al. Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest (2002) 110:1045–52. doi:10.1172/JCI15506
134. Naganuma K, Omura A, Maekawara N, Saitoh M, Ohkawa N, Kubota T, et al. Discovery of selective PDE4B inhibitors. Bioorg Med Chem Lett (2009) 19:3174–6. doi:10.1016/j.bmcl.2009.04.121
135. Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol (2010) 28:63–70. doi:10.1038/nbt.1598
136. Page CP, Spina D. Selective PDE inhibitors as novel treatments for respiratory diseases. Curr Opin Pharmacol (2012) 12:275–86. doi:10.1016/j.coph.2012.02.016
137. Kagayama K, Morimoto T, Nagata S, Katoh F, Zhang X, Inoue N, et al. Synthesis and biological evaluation of novel phthalazinone derivatives as topically active phosphodiesterase 4 inhibitors. Bioorg Med Chem (2009) 17:6959–70. doi:10.1016/j.bmc.2009.08.014
138. Nazarian R, Weinberg JM. AN-2728, a PDE4 inhibitor for the potential topical treatment of psoriasis and atopic dermatitis. Curr Opin Investig Drugs (2009) 10:1236–42.
139. Akama T, Baker SJ, Zhang Y-K, Hernandez V, Zhou H, Sanders V, et al. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg Med Chem Lett (2009) 19:2129–32. doi:10.1016/j.bmcl.2009.03.007
140. Conti M, Richter W, Mehats C, Livera G, Park J-Y, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem (2003) 278:5493–6. doi:10.1074/jbc.R200029200
141. Rennard SI, Calverley PMA, Goehring UM, Bredenbröker D, Martinez FJ. Reduction of exacerbations by the PDE4 inhibitor roflumilast – the importance of defining different subsets of patients with COPD. Respir Res (2011) 12:18. doi:10.1186/1465-9921-12-18
142. Rabe KF, Bateman ED, O’Donnell D, Witte S, Bredenbröker D, Bethke TD. Roflumilast – an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trial. Lancet (2005) 366:563–71. doi:10.1016/S0140-6736(05)67100-0
143. Calverley PMA, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2007) 176:154–61. doi:10.1164/rccm.200610-1563OC
144. Calverley PMA, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ. M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet (2009) 374:685–94. doi:10.1016/S0140-6736(09)61255-1
145. Giembycz MA, Field SK. Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des Devel Ther (2010) 4:147–58.
146. Beghè B, Rabe KF, Fabbri LM. Phosphodiesterase-4 inhibitor therapy for lung diseases. Am J Respir Crit Care Med (2013) 188:271–8. doi:10.1164/rccm.201301-0021PP
147. Mulhall AM, Droege CA, Ernst NE, Panos RJ, Zafar MA. Phosphodiesterase 4 inhibitors for the treatment of chronic obstructive pulmonary disease: a review of current and developing drugs. Expert Opin Investig Drugs (2015) 24:1597–611. doi:10.1517/13543784.2015.1094054
148. Man H-W, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, et al. Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor. J Med Chem (2009) 52:1522–4. doi:10.1021/jm900210d
149. Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, et al. Longterm (52-week) results of a phase III randomized, controlled trial of apremilast in patients with psoriatic arthritis. J Rheumatol (2015) 42:479–88. doi:10.3899/jrheum.140647
150. Zerilli T, Ocheretyaner E. Apremilast (Otezla): a new oral treatment for adults with psoriasis and psoriatic arthritis. P T (2015) 40:495–500.
151. Papp K, Reich K, Leonardi CL, Kircik L, Chimenti S, Langley RGB, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (efficacy and safety trial evaluating the effects of apremilast in psoriasis [ESTEEM] 1). J Am Acad Dermatol (2015) 73:37–49. doi:10.1016/j.jaad.2015.03.049
152. Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis (2014) 73:1020–6. doi:10.1136/annrheumdis-2013-205056
153. Delpiano MA, Acker H. Hypoxia increases the cyclic AMP content of the cat carotid body in vitro. J Neurochem (1991) 57:291–7. doi:10.1111/j.1471-4159.1991.tb02127.x
154. Carmony KC, Kim K-B. PROTAC-induced proteolytic targeting. Methods Mol Biol (2012) 832:627–38. doi:10.1007/978-1-61779-474-2_44
Keywords: cyclic AMP, autoimmunity, targeted therapies, inflammation, T cells, Tregs, T regulatory cells
Citation: Raker VK, Becker C and Steinbrink K (2016) The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 7:123. doi: 10.3389/fimmu.2016.00123
Received: 27 January 2016; Accepted: 18 March 2016;
Published: 31 March 2016
Edited by:
Josef Bodor, Institute of Experimental Medicine, Czech RepublicReviewed by:
Gottfried Baier, Medical University of Innsbruck, AustriaJacques A. Nunes, Centre de Recherche en Cancerologie de Marseille, France
Copyright: © 2016 Raker, Becker and Steinbrink. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Verena Katharina Raker, cmFrZXJ2QHVuaS1tYWluei5kZQ==