 Sebastian Carotta
Sebastian Carotta- 1Immune Modulation Department, Boehringer Ingelheim RCV, Vienna, Austria
- 2The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
The recent success of checkpoint blockade has highlighted the potential of immunotherapy approaches for cancer treatment. Although the majority of approved immunotherapy drugs target T cell subsets, it is appreciated that other components of the immune system have important roles in tumor immune surveillance as well and thus represent promising additional targets for immunotherapy. Natural killer (NK) cells are the body’s first line of defense against infected or transformed cells, as they kill target cells in an antigen-independent manner. Although several studies have clearly demonstrated the active role of NK cells in cancer immune surveillance, only few clinically approved therapies currently exist that harness their potential. Our increased understanding of NK cell biology over the past few years has renewed the interest in NK cell-based anticancer therapies, which has lead to a steady increase of NK cell-based clinical and preclinical trials. Here, the role of NK cells in cancer immune surveillance is summarized, and several novel approaches to enhance NK cell cytotoxicity against cancer are discussed.
Overview
Natural killer (NK) cells were identified in 1975 as a unique lymphocyte population that is clearly distinct from the other lymphoid lineages, such as T- and B-cells. NK cells were shown to differ from adaptive lymphocytes in respect to their morphology as well as in their capability to kill tumor cells without prior sensitization (1, 2). Since their discovery, research over the past 40 years significantly improved our understanding of the regulation of NK cells and has established several essential roles of NK cells during development in healthy individuals and during disease that can be therapeutically utilized.
Natural killer cells are the founding member of the innate lymphoid cell (ILC) family and are generally grouped based on their organ of development and tissue localization: we distinguish bone marrow-derived or adult conventional NK (cNK) cells, thymic-derived, fetal liver-derived, liver-resident, uterine-resident, and intestinal-resident NK cells. Adult cNK cells develop from the common lymphoid progenitor in the bone marrow in mice and humans and are considered the major NK cell subset responsible for tumor immune surveillance, albeit a role of the other subsets cannot completely be ruled out. During murine adult hematopoietic development, NK cell precursors are thought to be derived from a common innate lymphoid progenitor (CILP) and then mature through several progenitor stages into mature NK cells and migrate to several lymphoid and non-lymphoid tissues (3, 4). Peripheral NK cell maturation is then defined by the differential expression of CD11b, CD27, and KLRG1. Immature NK cells are defined as CD11b−CD27+KLRG1− and mature NK cells as CD11b+CD27+KLRG1− (M1) or CD11b+CD27−KLRG1+ (M2) (4–6). These different subsets differ in their ability to lyse target cells and their ability to secrete cytokines (5, 6). The mature CD11bhighCD27−KLRG1+ NK cells are the dominant population in non-lymphoid organs except for the liver, where a distinct TNF-related apoptosis-inducing ligand (TRAIL)+CD49b−CD11blow expressing population exists (7–9).
In contrast to murine NK cell development, the NK cell precursor populations in humans are currently not as well defined (10). Mature NK cells make up around 5–20% of peripheral blood lymphocytes. They are usually defined as CD3−CD56+ lymphoid cells and are subdivided into two major subpopulations, CD56dimCD16+ and CD56brightCD16− cells. CD56dim NK cells are the dominant subset in peripheral blood and spleen, express perforin, and are the most potent one in killing cancer cells (11–13). CD56bright NK cells represent the main NK cell subset in lymph nodes and tonsils, lack perforin expression but are efficient producers of cytokines, such as IFN-γ, in response to the interleukins (IL)-12, IL-15, and IL-18. Thus, this subset is considered to be one of the key regulators of immune responses.
A major difference between NK and T cells is that NK cells can kill target cells instantly without needing prior sensitization, giving the adaptive immune reaction enough time to mount an antigen-specific immune response. Although the speed in which NK cells can kill infected or malignant cells is a big advantage when fast immune reactions are required, the ready-to-kill status of NK cells could be potentially dangerous for the body. Thus, NK cell activation is tightly regulated by activating the inhibitory receptors and the balance of the signaling through these receptors dictates if NK cells kill their target cells or remain inactive (14, 15). To prevent autoreactivity, NK cells express MHC class-I-specific receptors such as the killer cell immunoglobulin-like receptors (KIRs) in human, the lectin-like Ly49 dimers in the mouse, and the CD94–NKG2A heterodimers which exist on both, mice and humans. Binding of MHC-I molecules to these inhibitory receptors prevents cytolytic activity against healthy cells. During cancer progression, cancerous cells often decrease or even loose the expression of MHC-I on the surface, which allows them to evade T cell recognition and killing. However, loss of the MHC-I-mediated inhibitory signal on NK cells results in NK cell activation and cancer cell killing if no other inhibitory signals are active. In addition to the loss of inhibitory receptor signaling, NK cells can be directly activated by activating receptors, such as NKG2D, NKp30, NKp44, NKp46, 2B4, DNAM-1 (CD226), or CD16 (4, 6, 14, 16, 17). Although the ligands for some activating receptors have not yet been identified, it is currently believed that activating ligands are not expressed on healthy cells but are upregulated on diseased cells and that signaling through the activating receptors will dominate over the MHC-I-mediated inhibitory signaling. Besides the direct ligand–receptor interaction, NK cell functions are as well modulated by several cytokines. NK cells can be activated through type I interferons, IL-2, IL-12, IL-15, IL-18, and IL-21, whereas suppressive cytokines, such as transforming growth factor (TGF)-β or IL-10, can render NK cells inactive (18).
Several different pathways exist through which NK cells kill their target cells. On the one hand, NK cells induce apoptosis in their target cells by releasing lytic granules, such as granzyme B and perforin, via the formation of a lytic immunological synapse between the NK and target cells (19). Released perforin induces membrane perforation allowing the secretion of granzymes into the intracellular space inducing either caspase-dependent or -independent apoptosis. Another mechanism to kill is the induction of the death receptor-mediated apoptosis pathway. Here, FasL and TRAIL expressed on NK cells bind to Fas and TRAIL receptor triggering target cell apoptosis. In addition, NK cell-derived TNF-α can as well induce target cell apoptosis.
Despite the majority of current NK cell-mediated anticancer therapies focus on the lytic capability of NK cells, the indirect antitumor immunity capacity of NK cells should not be disregarded. NK cells are known to regulate the innate and adaptive immune response through the secretion of various cytokines, chemokines, adenosine, and growth factors (20, 21). NK cell-derived IFN-γ induces dendritic cell (DC) maturation leading to increased IL-12 production. IFN-γ as well induces the differentiation of CD8+ T cells into cytotoxic T cells (CTLs) and promotes the differentiation of CD4+ cells into Th1 T cells, which in turn promote the CTL response. NK cells not only enhance immune responses but also dampen T cell responses by either killing DC or inhibiting CD8+ T cell responses directly through IL-10 secretion. Our current understanding of the immune modulatory role of NK cells is, however, still limited and a better understanding will certainly open the door to novel NK cell-based immunotherapy approaches.
Evidence for the Importance of NK Cells in Anticancer Immunosurveillance
An essential role for NK cells in human immune surveillance has been clearly established. Defects in human NK cell development or effector functions result in recurrent viral infections and in an increased risk of cancer development (22). Probably, the best evidence for the role of NK cells in anticancer immune surveillance comes from an epidemiological 11-year follow-up cohort study among a Japanese general population: the study demonstrated that high cytotoxic activity in peripheral blood lymphocytes is associated with reduced cancer risk, whereas low activity is associated with increased risk to develop various types of cancer (23). Subsequently, several other studies found that high levels of tumor infiltrating NK cells are associated with favorable outcome in patients with colorectal carcinoma, gastric cancer, and squamous cell lung cancer (24). Indicative of an important role of NK cells in tumor control, cancer cells have developed several strategies to escape from NK cell recognition. Tumor cells can upregulate ligands for inhibitory receptors or secrete immune suppressive factors, including TGF-β, IL-10, prostaglandin E2, indoleamine 2,3-dioxygenase (Ido), and adenosine (25–29). Shedding of ligands for activating receptors represents another potential strategy by tumor cells to reduce the amount of activating ligands on the surface of tumor cells and/or induce NK cell desensitization (30–33). However, a recent report questioned the shedding mechanism as a way to invade the immune surveillance. In the mouse model, Deng et al. demonstrated that a shed form of the mouse NKG2D ligand MULT1 can lead to boosting of NK cell activity (34).
Despite ample evidence that NK cells participate in the fight against cancerous cells, very few therapeutical approaches currently exist that are targeting NK cells. However, support for the potential of NK cells as therapeutic targets is coming from approved cancer cell-targeting therapies as several drugs have been recently demonstrated to additionally modulate NK cell activity. In the next section, I will review the effect of a few of such therapies.
Cancer Cell-Targeting Drugs with NK Cell-Modulating Activity
Noteworthy, many targets of current cancer therapies are expressed in cancer cells and immune cells. It is therefore not surprising that few cancer therapies not only impact on cancer cell survival and proliferation but also influence the immune system. But because the majority of cancer-targeting drugs is generally tested preclinically for their efficacy and safety in xenograft models that lack a functional immune system, this effect is often not apparent. Indeed, recent studies have shown that radiotherapy or chemotherapies, such as Ara-C, cisplantin, or 5-FU, can lead to increased expression of NK cell activating ligands and thus enhance NK cell recognition and killing (35). More recently, several precision medicine drugs have additionally been demonstrated to increase NK cell-mediated tumor killing (36, 37). The proteasome inhibitor bortezomib, currently successfully used in the treatment of multiple myeloma, can induce the expression of ligands of NK cell activating receptors. Another example is the immunomodulatory (IMiD) drug lenalidomide, which is approved for the treatment of multiple myeloma and myelodysplastic syndromes (MDS). Besides having a direct effect on cancer cells and angiogenesis, lenalidomide modulates the immune response by increasing the NK cell number in the periphery. The exact mode of action of lenalidomide on NK cells is currently not clear. Several modes of actions have been proposed. Lenalidomide might increase NK cell activation indirectly by upregulating ligands on tumor cells and induce the expression of NK cell stimulatory cytokines such as T cell-derived IL-2 or directly by lowering the threshold for NK cell activation (38, 39). A better understanding of the mode of actions of lenalidomide on NK cells will be certainly crucial to design rational combination therapies. This is highlighted by the fact that lenalidomide in combination with the anti-CD20 antibody rituximab can lead to increased efficacy in B cell malignancies by enhancing the antibody-dependent cell-mediated cytotoxicity (ADCC) effect, but the combination with dexamethasone inhibits the immune-stimulatory effect of lenalidomide on NK cells, potentially via suppressing IL-2 production in CD4+ T cells (40–42).
However, cancer-targeting drugs not always enhance the activity of immune cells, but in some cases, have been reported to exert detrimental effects on the immune system. Ibrutinib is a novel irreversible inhibitor of Bruton’s tyrosine kinase that shows promising effects in the treatment of mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL). Rituximab in combination with chemotherapy is currently standard of care in CD20+ B-cell malignancies and thus a potential combination of ibrutinib with rituximab is attractive. However, recent studies demonstrated that ibrutinib actually antagonizes the ADCC effect of rituximab in CD20+ B-cell lymphoma due to Ibrutinib irreversible binding to IL-2 inducible tyrosine kinase (ITK), which is required for FcR-stimulated NK cell function (43, 44).
Another example is ruxolitinib, a small molecule inhibitor of the JAK 1/2/3 signaling pathway. Ruxolitinib is currently approved for the treatment of myelofibrosis (MPN). As several cytokines regulate NK cell development and function via the JAK/STAT signaling pathway, patients who were treated with ruxolitinib had drastically reduced circulating NK cell numbers. In vitro studies further demonstrated that ruxolitinib potently inhibited the cytokine-induced cytolytic activity of NK cells (45). However, importantly, NK cell depletion by ruxolitinib was reversible as the NK cell levels rose back to normal values in patients who stopped ruxolitinib treatment. Thus, when combined with NK cell-based immunotherapies, proper scheduling of therapeutic drugs will be crucial.
Clinical or Preclinical Therapies Augmenting NK Cell Function
Checkpoint Inhibitors
PD-1
Checkpoint inhibitors are currently the most promising approaches among immunotherapies. Treatment with anti-CTLA4 or anti-PD-1 antibodies restores T cell activity in cancer patients and has resulted in durable tumor regression in some patients. And the combination of both checkpoint inhibitors was able to further enhance the therapeutic benefit significantly (46, 47). The expression of PD-1, however, is not restricted to activated and exhausted T cells but can be detected on subsets of other immune cells and even on melanoma cells (48–51). A recent report demonstrated that NK cells from multiple myeloma and renal carcinoma patients expressed PD-1 on their surface and engagement of PD-1 signaling reduced their cytolytic potential (Figure 1) (50, 51). Treatment of patient-derived PD-1+ NK cells with an anti-PD-1 antibody (pidilizumab, CT-011) was able to increase NK cell-mediated killing of autologous cancer cells in vitro (50). A recent phase II trial tested the efficacy of pidilizumab with rituximab in patients with relapsed follicular lymphoma and found that the combination is well tolerated and indicated favorable therapeutic effects when compared to rituximab single treatment (52). The therapeutic benefit of re-invigorating PD-1+ NK cells in cancer patients is currently not well understood, and the major therapeutical effect is certainly due to re-activation of exhausted T cells. However, a contribution of NK cells to the observed therapeutic benefit cannot be excluded, especially in hematological malignancies, and thus warrants further investigation.
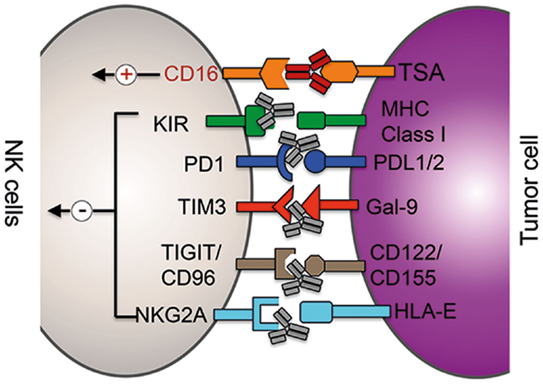
Figure 1. Clinical and preclinical therapies augmenting NK cells function. mAb (gray)-mediated blockade of the interaction between inhibitor receptors (in black) expressed on NK cells with the respective ligands on tumor cells (or suppressor cells) results in increased cytolytic potential of NK cells. ADCC therapy: binding of mAbs (red) to tumor-specific antigens (TSA) results in the activation of NK cells via the activation of the activating receptor CD16 (red).
TIM-3
TIM-3, also known as HAVCR2, is another immune checkpoint receptor that is currently being tested in preclinical models for its potential to re-invigorate exhausted T cells in cancer patients (53). Resting T cells express low levels of TIM-3, and its expression is strongly upregulated in activated and exhausted T cells. Antibody-mediated blockade of TIM-3 signaling was able to reverse the exhausted phenotype of CD4+ and CD8+ T cells in melanoma patients proving the inhibitory function of TIM-3 in T cells (54). Human tumor-derived CD8+ T cells often coexpress TIM-3 and PD-1, and preclinical studies in several murine tumor models demonstrated that the combination of TIM-3 and PD1 blocking antibodies can significantly increase the reversal of T-cell exhaustion. Like PD-1, TIM-3 expression is not restricted to T cells but can also be detected on murine and human NK cells (55, 56). In contrast to T cells, where TIM-3 surface expression marks dysfunctional T cells, TIM-3 is expressed on virtually all human NK cells and is further upregulated on cytokine-activated NK cells (Figure 1). Thus, TIM-3 expression is regarded as a marker for mature NK cells. Currently, the functional role of TIM-3 on NK cells is highly controversial. Ndhlovu et al. recently demonstrated that crosslinking of TIM-3 via anti-TIM-3 antibodies on the human NK cell line NKL or on human PBMC-derived NK cells significantly decreased their cytolytic ability (57). In stark contrast to these findings, Gleason et al. showed that activation of TIM-3 through the ligand Gal-9 actually increased the production of IFN-γ in NK cells (58). A more recent report suggested that the discrepancy between these two studies might origin from the different experimental layout as well as by the fact that NK cell lines and NK cells from healthy donors have been analyzed (56). Therefore, they tested the effect of TIM-3 blockade on NK cells derived from advanced melanoma patients. da Silva et al. found that TIM-3 surface expression increases with the progression of the cancer, TIM-3+ NK cells display an exhausted phenotype and that high expression levels correlated with poor prognosis. More importantly, when TIM-3+ NK cells derived from melanoma patients were incubated with anti-TIM-3-coated beads, TIM-3 activation resulted in modest, but statistically significant decrease in IFN-γ secretion and degranulation. In summary, the function of TIM-3 on NK cells is currently controversial, and more detailed studies on the role of TIM-3 on NK cells derived from cancer patients are required to fully understand the role and therapeutic potential of TIM-3 blockade in NK cell therapy.
NKG2A
The heterodimer CD94/NKG2A is another checkpoint inhibitor complex whose expression is shared between T and NK cells (Figure 1). Human CD94–NKG2A/C/E heterodimers recognize the non-classical MHC class-I molecule HLA-E in humans and Qa-1 in mice, which is expressed on many lymphoid cells (59). The NKG2A chain of the CD94/NKG2A receptor contains two immunoreceptor Tyr-based inhibitory motifs (ITIMs) in its cytoplasmic tail and HLA-E/NKG2A interaction results in a dominant inhibitory signaling event that causes a strong decrease in NK cell effector functions. Several solid cancer and hematological malignancies use the upregulation of HLA-E expression as an immune escape mechanism in order to evade killing by NK cells and T cells (60, 61). Therefore, the use of a blocking NKG2A antibody could be another useful addition to the steadily growing list of T/NK cell-targeting immunotherapy approaches. Monalizumab (previously IPH2201) represents such an anti-NKG2A checkpoint inhibitor and is currently under clinical investigation. In a phase I/II trial, monalizumab is currently being evaluated in head and neck cancer and ovarian cancer. Furthermore, the effects of the combination of monalizumab with ibrutinib (CLL, phase I/II), cetuximab (head and neck, phase I/II), and duvalumab (solid tumors, phase I/II) are currently investigated.
TIGIT and CD96
TIGIT, CD96, and CD226 (DNAM-1) belong to the same immunoglobulin family of receptors that interact with nectin and nectin-like proteins (16). Although all three receptors are expressed on NK cells and can bind CD155 and CD112, ligand binding is triggering different responses (Figure 1). CD226 is an activating receptor that is important for NK cell-mediated tumor surveillance and ligand binding increases the cytotoxic potential of NK cells against target cells (16). On the other hand, TIGIT and CD96 contain ITIM motifs in their cytoplasmic domains and are inhibitory receptors. Although CD155 present on tumor cells can induce CD226-dependent immunosurveillance, the expression of CD96 and TIGIT on the same cell can counterbalance CD226 activity. While activation of TIGIT on human NK cells inhibited in vitro cell killing of target cells, antibody-mediated blocking of TIGIT significantly increased the cytolytic activity (62). Using CD96−/− mice, Chan et al. recently demonstrated that loss of CD96 expression resulted in improved tumor control of methylcholanthrene (MCA)-induced fibrosarcoma and lung metastasis (63). Although blockade of TIGIT in vitro increased the cytolytic activity of NK cells, the improved antitumor response in CD96-deficient mice was dependent on IFN-γ production by NK cells. It is currently not clear why NK cells express simultaneously two inhibitory receptors on the same cell to counteract CD226 activation, but data from the Smyth group indicate that the two receptors control different NK cell effector functions: TIGIT may predominantly inhibit the cytolytic potential of NK cells, whereas CD96 regulates the production of IFN-γ (16, 63). Future research will unravel which of these two receptors should be inhibited to increase tumor surveillance in human patients or if inhibition of both receptors simultaneously will result in improved NK/T cell-mediated cancer cell control.
Killer Cell Immunoglobulin-Like Receptors
Natural killer cells express inhibitory KIRs that recognize self-MHC class-I molecules to prevent cytotoxicity against host cells (Figure 1). As tumor cells express the same MHC class-I molecules than healthy tissue, the interaction between self-HLA on cancer cells with KIRs on NK cells reduces the cytolytic activity of NK cells against tumor cells (64–68). Therefore, KIRs represent an interesting class of targets for NK cell-specific checkpoint inhibition. Following this reasoning, a humanized KIR-blocking monoclonal antibody (mAb), IPH2101, has been generated and is currently tested in clinical trials. IPH2101 is specific against three inhibitory KIRs, namely, KIR2DL-1, -2, and -3, that are specific for all HLA-C molecules. Preclinical in vitro and in vivo studies demonstrated that IPH2101-mediated blockade of KIRs on human NK cells significantly increased cytolytic activity against tumor cells (69–71). Importantly, no sign of autoimmunity was observed in treated mice. Confirming the results of the preclinical studies, no severe side effects were observed in clinical phase I and phase II trials in patients with acute lymphoblastic leukemia or multiple myeloma (72–74). Although the current clinical trials using IPH2101 as a monotherapy did not demonstrate significant antitumor efficacy, based on the encouraging preclinical data of IPH2101 and the recent success of combining checkpoint inhibitors, there is still hope that the rational combination with other drugs can lead to improved clinical antitumor responses. Potential combination partners could be the above described checkpoint inhibitors and other IMiD drugs, such as lenalidomide or NK cell activating cytokines.
Redirection of NK Cell Cytotoxicity via Biologics
Antibodies recognizing tumor-specific epitopes represent a highly efficient strategy to direct the cytolytic activity of NK cells against malignant cells. One approach that is currently successfully used in the clinics is ADCC-based therapies. NK cells express the activating surface receptor CD16 (FcγRIIIA), which specifically binds the constant region (Fc) of immunoglobulin G (IgG) antibodies. The interaction between CD16 on NK cells and the Fc portion of a tumor-specific IgG antibody bound on cancer cells results in the activation of NK cells and subsequently killing of respective tumor cells (Figure 1). Currently, several ADCC therapies are tested in clinical trials or are already successfully used in the clinics, such as α-CD20, α-GD2, α-Her2, and α-EGFR mAbs. The current status of ADCC therapies were summarized in recent review (75). However, it is important to mention that CD16 is expressed not only on NK cells but also on activated myeloid subsets. Therefore, several hematopoietic lineages are likely to contribute to the observed therapeutic effects of ADCC (75). Besides mAbs, bispecific or trispecific killer engagers (BiKEs and TriKEs) are currently developed. These antibodies are able to target either one (BiKE) or two (TriKE) different antigens on the tumor cell and bind to another epitope of the CD16 receptor leading to improved NK cell-mediated ADCC effect [for a review on BiKEs and TriKEs, please see Wang et al. (75) and Kontermann and Brinkmann (76)].
Targeting Immune Suppressive Signaling
Transforming Growth Factor-β
Secretion of TGF-β by tumor cells or the tumor microenvironment has copious effects on tumor progression and on the immune system (77). During cancer progression, TGF-β can play a key role in tumor immune escape. TGF-β levels are often increased in the serum of cancer patients and elevated levels correlate with systemic inhibition of the immune system and poor prognosis (78, 79). Like CD8+ T cells, NK cells from patients with elevated TGF-β levels displayed reduced cytotoxicity and had reduced expression levels of the activation markers, NKG2D, NKp46, or increased expression of NKG2A (Figure 2) (28, 80). Ex vivo treatment of patient-derived NK cells with neutralizing anti-TGF-β mAbs was able to restore activating receptor expression, proliferation, and cytokine secretion (29). Coculture of healthy human NK cells with human ALL blasts reduced their cytolytic activity and IFN-γ production. This effect was mediated by ALL-derived TGF-β as an anti-TGF-β blocking antibody was able to rescue NK cell functions (28). In line with a direct effect of TGF-β signaling on NK cell receptor expression and NK cell function, in vitro incubation of human NK cells with TGF-β resulted in downregulation of NKp30 and NKG2D, inhibition of IL-15 induced NK cell proliferation and IFN-γ secretion (81). Therefore, targeting TGF-β signaling in NK cells represents an attractive immunotherapy approach in cancer patients with elevated TGF-β levels. However, due to the many functions of TGF-β in normal tissue, cancer cells, tumor microenvironment, and immune cells, developing potent inhibitors with a low toxicity profile is challenging (82, 83). Currently, several approaches to inhibit TGF-β signaling are pursued to increase efficacy and limit toxicity in preclinical and in clinical trials with various successes. Approaches include ligand traps, antisense oligonucleotides, receptor kinase inhibitors, and peptide aptamers (84). In summary, while it is currently to early to judge if anti-TGF-β immunotherapies will become reality, preclinical studies yielded enough convincing results that interference with the TGF-β pathway is able to increase NK cell (and T cell) effector functions to warrant further new drug development.
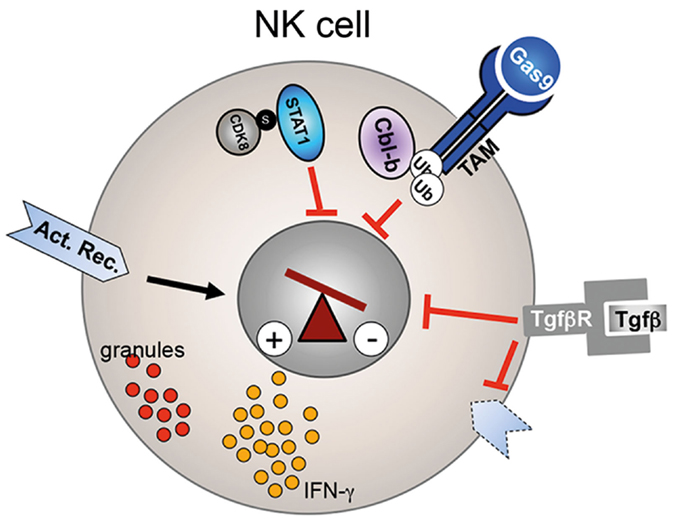
Figure 2. Regulation of NK cell activity. CDK8-mediated STAT1–Ser727 phosphorylation inhibits the cytolytic activity of NK cells. Similarly, Cbl-b-mediated ubiquitinylation of the TAM receptor results in reduced perforin, granzyme B (red granules), and IFN-γ secretion. TGF-β signaling reduces as well NK cell effector function and leads to downregulation of activating receptors (dotted receptor).
Exploratory Targets – The Next Wave?
Intracellular Targets
The majority of approved immunotherapies target surface receptors on immune cells via mAbs that either inhibit protein–protein interactions between immune cells and other cell types (antagonistic antibodies) or activate target receptor on certain immune cells (agonistic antibodies). Whereas in cancer-targeting therapeutic approaches small molecule drugs dominate, this class of therapies is conspicuously missing or at least under-represented in current anticancer immunotherapy approaches. Targeting intracellular proteins via small molecules significantly extends the pool of potential novel immunotherapy targets. Different inhibitory receptors often use the same intracellular pathways to relay their inhibitory signal into the nucleus. Thus, by inhibiting such pathways, it might be possible to therapeutically affect several inhibitory receptors at the same time via inhibiting one molecule. The phosphatases Src homology region 2 domain-containing phosphatase (SHP)-1 (PTPN6) and SHP-2 (PTPN11) are two good examples as several inhibitory receptors on T cells and NK cells have been shown to recruit SHP-1 and/or SHP-2 after activation (85). Of course, targeting intracellular pathways with such broad activity can come with a cost, in this case, the potential increase of toxicity. Other advantages of small molecules over biologicals are excellently summarized in a recent review (86). In this next section, I will discuss a few recently published molecules that play a role in the regulation of NK cell function and might represent potential future targets for NK cell-mediated immunotherapy.
Casitas B-Lineage Lymphoma Proto-Oncogene-b
Post-transcriptional modification of proteins, such as ubiquitination, is an important regulatory mechanism for the fine-tuning of several pathways. Recent studies demonstrated that the E3 ligase Casitas B-lineage lymphoma proto-oncogene-b (Cbl-b) is a key regulator of the immune response against cancer (87). Cbl-b is highly expressed in most murine and human immune cells, including T and NK cells. The importance of Cbl-b in antitumor immune response was discovered when Cbl-b-deficient mice spontaneously rejected several different tumors (88, 89). Tumor rejection was first considered to be mainly mediated by CD8+ T cells. However, a recent study elegantly demonstrated that Cbl-b also plays a key role in NK cell-mediated tumor control. Deletion or pharmacological inhibition of Cbl-b increased the cytolytic potential, proliferative capacity, and IFN-γ secretion of NK cells in vitro (Figure 2) (90). More importantly, tumor growth and metastasis were significantly decreased in Rag2−/−Cbl-b−/− mice when compared to Rag2−/−Cbl-bwt mice. Antibody-mediated NK cell depletion and inactivation via anti-NK1.1 and anti-NKG2D antibodies, respectively, abrogated this antitumor response, identifying the NK cell lineage as the main mediator of the observed antitumor effect in these mice. Furthermore, NK cell activity was completely dependent on the catalytic domain of the E3 ligase of Cbl-b. Through an in vitro ubiquitinylation screen of 9000 human proteins, the authors then identified the TAM receptor tyrosine kinases AXL, TYRO3, and MER as targets of Cbl-b. Indeed, activation of TAM receptors on wild-type NK cells via the natural ligand Gas6 suppressed IFN-γ secretion in vitro, whereas Cbl-b-deficient NK cells were resistant to this inhibition. These date therefore indicate that TAM or Cbl-b is potentially suitable targets for NK cell-mediated immunotherapy. Indeed, Paolino et al. developed a highly selective TAM kinase inhibitor, LDC1267, which increased the lytic activity of NK cell against B16F10 melanoma cells in vitro and in vivo in an adoptive transfer mouse model. Furthermore, intra-peritoneal injection of LDC1267 resulted in a decrease of micro-metastases in mice that were injected with the syngenic tumor cell line 4T1. In summary, interference of TAM receptor activity represents an interesting novel immunotherapy approach. Alternatively, a small molecule inhibitor of Cbl-b potentially could increase NK cell and T cell effector function against cancer cells (87). It is currently unclear how toxic such a small molecule would be as Cbl-b is expressed in many hematopoietic cell types. However, Cbl-b-deficient mice are viable and do not show signs of severe autoimmunity, thus a therapeutic window might exist.
CDK8
Natural killer cell development and functions are tightly regulated by several cytokines, such as IL-2, IL-12, IL-15, or type I interferons. The JAK/STAT pathway is playing a central role in relaying the effect of these different cytokines into the nucleus. IL-2 and IL-15 promote NK cell development and homeostasis mainly via activation of the transcription factor signal transducer and activator of transcription protein 5 (STAT5) (91, 92). On the other hand, STAT1 plays an essential role on the regulation of NK cell effector function (93). Type I interferon and IL12 induce STAT1 activation, resulting in increased cytotoxicity and IFN-γ secretion. Not surprisingly, therefore, deletion of STAT1 results in impaired NK cytolytic activity in vitro and reduced tumor rejection in vivo, despite normal numbers of NK cells, whereas STAT5-deficient mice lack NK cells completely (94–96). The activity of STAT1 is mainly regulated post-transcriptionally. For activation and translocation of STAT1 into the nucleus, STAT1 has to be phosphorylated at tyrosine 701 (Y701) by the Janus kinase JAK. A recent report demonstrated a role of STAT1–Ser727 phosphorylation in the regulation of the lytic potential of NK cells (96). Resting NK cells showed a basal level of STAT1–Ser727 phosphorylation, which increased after in vitro stimulation with either IFN-β or IL-12. Interestingly, in contrast to Y701 phosphorylation, Ser727 phosphorylation resulted in an inhibitory effect on NK cell activity, indicating that phosphorylation of STAT1–Ser727 represents a negative feedback in activated NK cells loop to prevent over-stimulation. Ex vivo isolated STAT1–Ser727A mutant NK cells had increased lytic potential against a range of tumor cell lines in vitro and secreted increased levels of granzyme B and perforin. In vivo, STAT1–Ser727A mutant mice showed increased anticancer immunosurveillance against the murine tumor lines B16F10, 4T1, and a v-abl transformed leukemic cell line. Through the generation of Rag1−/− STAT1–Ser727A mice that lack B and T cells but have NK cells, Putz et al. demonstrated that the antitumor effect was strictly dependent on NK cell activity. Interestingly, By contrast, the molecules mentioned above which limited both, the production of lytic granules and IFN-γ secretion, STAT–1Ser727 repressed perforin and granzyme B but induced IFN-γ secretion slightly. The authors then identified the cyclin-dependent kinase 8 (CDK8) as the kinase responsible for the phosphorylation of STAT1 as Ser727 (Figure 2). Knock-down of CDK8 reduced STAT-1–Ser727 phosphorylation and slightly increased target cell lysis in an in vitro killing assay. Thus, pharmacological inhibition of CDK8 kinase activity might represent an attractive approach to augment NK cell-mediated anticancer immunosurveillance. Currently, the open questions are if the same effect will be seen in human NK cells and how toxic a CDK8 inhibitor will be given the broad expression of CDK8. However, CDK8 therapy could potentially have another positive anticancer effect: CDK8 has been previously shown to be an oncogenic driver in colorectal cancer, breast cancer, and melanoma (97–99). Therefore, one could envision that CDK8 inhibitors, on the one hand, induce tumor cell death and, on the other hand, stimulate NK cell activity.
EZH2
The H3K27 methyltransferase enhancer of zeste homolog 2 (EZH2) is essential for many biological processes, including the regulation of immune responses, and is overexpressed in several cancers. Therefore, the pharmacological targeting of EZH2 is an interesting approach for future immunotherapies (100, 101). A recent study demonstrated a role of EZH2 in NK cell development (102). Absence of EZH2 in human and murine hematopoietic progenitors resulted in an increased commitment to the NK cell lineage. In addition, EZH2−/− NK cells expressed higher levels of NKG2D, IL2Rα, IL7Rα, and the lytic proteases granzyme A and B. The negative regulation of NK cell development and function by EZH2 was dependent on its methyltransferase activity as pharmacological inhibition of EZH2 resulted in a similar phenotype when compared to EZH2−/− NK cells. These data suggest that EZH2 inhibitors may not only have an effect on cancer cell growth and survival but potentially can augment NK cell number and function in patients. However, this remains to be tested as the above-mentioned study mainly focused on the effect of EZH2 on in vitro NK cell differentiation, and little data are currently available on the effect on mature NK cells. Nevertheless, several studies are testing the efficacy of adoptive transfer of ex vivo expanded NK cells as an immunotherapy approach. Thus, it will be of interest if the inhibition of EZH2 during the NK cell differentiation/expansion phase can lead to an increase in cell number and augment the activity of NK cells.
Conclusion
Although the ability of NK cells to kill malignant cells efficiently has been demonstrated several decades ago, the potential of NK cell-based immunotherapy is often questioned due to modest clinical responses of current therapies. Recent advances in our understanding of NK cell biology yielded already in promising new therapeutic approaches and continuous investigation of the mechanisms that regulate NK cell function will result in improved and more efficacious therapies in the future.
Many of the above described targets are not specific to NK cells, but often also function in other therapeutically interesting immune cells, such as T cells. Although the close relationship between NK and T cells makes if often difficult to identify how much of the therapeutic effect is due to NK cell activity, therapies that activate both effector cells are highly interesting as they are able to combine the therapeutic effects of both cell types (103). Until recently, only few experimental approaches existed to test the potential of NK cells in antitumor therapy. The most common and most feasible approach represented the antibody-mediated depletion of NK cells to investigate tumor growth in the presence or absence of NK cells. However, due to the lack of specific NK cell markers that can be targeted for depletion, it is often unclear if other cell types, such as T cells, have been affected as well. Recently, a novel NK cell-less mouse model has been established via the conditionally deletion of Mcl1 in NK cells (Mcl1fl/fl NCR1Cre) (104). As MCL1 expression is essential for NK cell survival, virtually no residual NK cell subsets in all anatomical locations tested have been detected. As NCR1 is expressed as well on a subset of ILC3 cells in the gut, this mouse models lacked all NK cells and NCR1+ ILC3 cells. Nevertheless, this genetically engineered mouse represents an attractive model to test the specific role of NK cells in various disease settings.
Natural killer cell therapy as a monotherapy is unlikely to be curative for most, if not all, cancer types and a critical parameter for successful NK cell therapies will be the choice of combination partners. Therefore, future studies that investigate the interaction of NK cells with the other components of the immune system will be crucial for the optimal design of combination therapies.
In summary, while the potential of NK cell therapy is currently still not entirely clear, the recent advances in our understanding of NK cells certainly have resulted in novel, promising approaches, and it is very likely that future discoveries will continue to improve the efficacy of NK cell-based therapies.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author is an employee of Boehringer Ingelheim.
Acknowledgments
The author would like to thank Martina Minnich and Alina Neunkirchner for critical feed back on the manuscript.
References
1. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16:216–29. doi:10.1002/ijc.2910160204
2. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–7. doi:10.1002/eji.1830050208
3. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity (2014) 41:354–65. doi:10.1016/j.immuni.2014.09.005
4. Huntington ND, Nutt SL, Carotta S. Regulation of murine natural killer cell commitment. Front Immunol (2013) 4:14. doi:10.3389/fimmu.2013.00014
5. Hayakawa Y, Huntington ND, Nutt SL, Smyth MJ. Functional subsets of mouse natural killer cells. Immunol Rev (2006) 214:47–55. doi:10.1111/j.1600-065X.2006.00454.x
6. Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol (2007) 7:703–14. doi:10.1038/nri2154
7. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med (2014) 211:563–77. doi:10.1084/jem.20131560
8. Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife (2014) 3:e01659. doi:10.7554/eLife.01659
9. Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, et al. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood (2005) 105:2082–9. doi:10.1182/blood-2004-08-3262
10. Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol (2013) 34:573–82. doi:10.1016/j.it.2013.07.005
11. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity (2006) 25:331–42. doi:10.1016/j.immuni.2006.06.013
12. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22:633–40. doi:10.1016/S1471-4906(01)02060-9
13. Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev (2006) 214:56–72. doi:10.1111/j.1600-065X.2006.00451.x
14. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi:10.1038/ni1582
15. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol (2011) 89:216–24. doi:10.1038/icb.2010.78
16. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol (2015) 15:243–54. doi:10.1038/nri3799
17. Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol (2003) 3:304–16. doi:10.1038/nri1055
18. Marcais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:450. doi:10.3389/fimmu.2013.00450
19. Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol (2008) 8:713–25. doi:10.1038/nri2381
20. Crouse J, Xu HC, Lang PA, Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol (2015) 36:49–58. doi:10.1016/j.it.2014.11.001
21. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16- NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J Immunol (2015) 195:965–72. doi:10.4049/jimmunol.1500591
22. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132:515–25. doi:10.1016/j.jaci.2013.07.020
23. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356:1795–9. doi:10.1016/S0140-6736(00)03231-1
24. Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov (2014) 4:522–6. doi:10.1158/2159-8290.CD-13-0985
25. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol Immunother (2011) 60:1577–86. doi:10.1007/s00262-011-1064-9
26. Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (review). Int J Oncol (2008) 32:527–35.
27. Pietra G, Vitale M, Moretta L, Mingari MC. How melanoma cells inactivate NK cells. Oncoimmunology (2012) 1:974–5. doi:10.4161/onci.20405
28. Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, et al. The TGF-beta/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia (2015) 30(4):800–11. doi:10.1038/leu.2015.327
29. Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human tumour immune evasion via TGF-beta blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS One (2011) 6:e22842. doi:10.1371/journal.pone.0022842
30. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419:734–8. doi:10.1038/nature01112
31. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature (2007) 447:482–6. doi:10.1038/nature05768
32. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169:4098–102. doi:10.4049/jimmunol.169.8.4098
33. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2015) 16:7–19. doi:10.1038/nrc.2015.5
34. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science (2015) 348:136–9. doi:10.1126/science.1258867
35. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ (2014) 21:15–25. doi:10.1038/cdd.2013.67
36. Romagne F, Vivier E. Natural killer cell-based therapies. F1000 Med Rep (2011) 3:9. doi:10.3410/M3-9
37. Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov (2015) 14:487–98. doi:10.1038/nrd4506
38. Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood (2015) 126:50–60. doi:10.1182/blood-2015-01-625004
39. Fionda C, Abruzzese MP, Zingoni A, Cecere F, Vulpis E, Peruzzi G, et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget (2015) 6:23609–30. doi:10.18632/oncotarget.4603
40. Wang M, Fayad L, Wagner-Bartak N, Zhang L, Hagemeister F, Neelapu SS, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol (2012) 13:716–23. doi:10.1016/S1470-2045(12)70200-0
41. Badoux XC, Keating MJ, Wen S, Wierda WG, O’Brien SM, Faderl S, et al. Phase II study of lenalidomide and rituximab as salvage therapy for patients with relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol (2013) 31:584–91. doi:10.1200/JCO.2012.42.8623
42. Hsu AK, Quach H, Tai T, Prince HM, Harrison SJ, Trapani JA, et al. The immunostimulatory effect of lenalidomide on NK-cell function is profoundly inhibited by concurrent dexamethasone therapy. Blood (2011) 117:1605–13. doi:10.1182/blood-2010-04-278432
43. Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood (2014) 123:1957–60. doi:10.1182/blood-2014-01-547869
44. Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica (2015) 100:77–86. doi:10.3324/haematol.2014.107011
45. Schönberg K, Rudolph J, Vonnahme M, Parampalli Yajnanarayana S, Cornez I, Hejazi M, et al. JAK inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res (2015) 75:2187–99. doi:10.1158/0008-5472.CAN-14-3198
46. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol (2014) 4:385. doi:10.3389/fonc.2014.00385
47. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373:1270–1. doi:10.1056/NEJMoa1504030
48. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell (2015) 162:1242–56. doi:10.1016/j.cell.2015.08.052
49. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi:10.1111/j.1600-065X.2010.00923.x
50. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116:2286–94. doi:10.1182/blood-2010-02-271874
51. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol (2013) 132:536–44. doi:10.1016/j.jaci.2013.07.006
52. Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol (2014) 15:69–77. doi:10.1016/S1470-2045(13)70551-5
53. Anderson AC. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res (2014) 2:393–8. doi:10.1158/2326-6066.CIR-14-0039
54. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi:10.1084/jem.20100637
55. Gallois A, Silva I, Osman I, Bhardwaj N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Oncoimmunology (2014) 3:e946365. doi:10.4161/21624011.2014.946365
56. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res (2014) 2:410–22. doi:10.1158/2326-6066.CIR-13-0171
57. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood (2012) 119:3734–43. doi:10.1182/blood-2011-11-392951
58. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood (2012) 119:3064–72. doi:10.1182/blood-2011-06-360321
59. Vivier E, Anfossi N. Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat Rev Immunol (2004) 4:190–8. doi:10.1038/nri1306
60. Fruci D, Lo Monaco E, Cifaldi L, Locatelli F, Tremante E, Benevolo M, et al. T and NK cells: two sides of tumor immunoevasion. J Transl Med (2013) 11:30. doi:10.1186/1479-5876-11-30
61. Lo Monaco E, Tremante E, Cerboni C, Melucci E, Sibilio L, Zingoni A, et al. Human leukocyte antigen E contributes to protect tumor cells from lysis by natural killer cells. Neoplasia (2011) 13:822–30. doi:10.1593/neo.101684
62. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A (2009) 106:17858–63. doi:10.1073/pnas.0903474106
63. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol (2014) 15:431–8. doi:10.1038/ni.2850
64. Jaeger BN, Vivier E. When NK cells overcome their lack of education. J Clin Invest (2012) 122:3053–6. doi:10.1172/JCI63524
65. Jaeger BN, Vivier E. Natural killer cell tolerance: control by self or self-control? Cold Spring Harb Perspect Biol (2012) 4:1–10.
66. Thielens A, Vivier E, Romagne F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol (2012) 24:239–45. doi:10.1016/j.coi.2012.01.001
67. Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol (2009) 101:27–79. doi:10.1016/S0065-2776(08)01002-X
68. Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol (2006) 6:520–31. doi:10.1038/nri1863
69. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114:2667–77. doi:10.1182/blood-2009-02-206532
70. Vahlne G, Lindholm K, Meier A, Wickstrom S, Lakshmikanth T, Brennan F, et al. In vivo tumor cell rejection induced by NK cell inhibitory receptor blockade: maintained tolerance to normal cells even in the presence of IL-2. Eur J Immunol (2010) 40:813–23. doi:10.1002/eji.200939755
71. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123:678–86. doi:10.1182/blood-2013-08-519199
72. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120:4317–23. doi:10.1182/blood-2012-06-437558
73. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120:4324–33. doi:10.1182/blood-2012-06-438028
74. Benson DM Jr, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res (2015) 21:4055–61. doi:10.1158/1078-0432.CCR-15-0304
75. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol (2015) 6:368. doi:10.3389/fimmu.2015.00368
76. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today (2015) 20:838–47. doi:10.1016/j.drudis.2015.02.008
77. Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer (2013) 13:788–99. doi:10.1038/nrc3603
78. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10:554–67. doi:10.1038/nri2808
79. Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity (2000) 12:171–81. doi:10.1016/S1074-7613(00)80170-3
80. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol (2010) 12:7–13. doi:10.1093/neuonc/nop009
81. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100:4120–5. doi:10.1073/pnas.0730640100
82. Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-beta targeted cancer therapy. Int J Biol Sci (2012) 8:964–78. doi:10.7150/ijbs.4564
83. Sheen YY, Kim MJ, Park SA, Park SY, Nam JS. Targeting the transforming growth factor-beta signaling in cancer therapy. Biomol Ther (Seoul) (2013) 21:323–31. doi:10.4062/biomolther.2013.072
84. Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther (2015) 147:22–31. doi:10.1016/j.pharmthera.2014.11.001
85. Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev (2009) 228:342–59. doi:10.1111/j.1600-065X.2008.00760.x
86. Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov (2015) 14:603–22. doi:10.1038/nrd4596
87. Lutz-Nicoladoni C, Wolf D, Sopper S. Modulation of immune cell functions by the E3 ligase Cbl-b. Front Oncol (2015) 5:58. doi:10.3389/fonc.2015.00058
88. Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med (2007) 204:879–91. doi:10.1084/jem.20061699
89. Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest (2007) 117:1029–36. doi:10.1172/JCI29472
90. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature (2014) 507:508–12. doi:10.1038/nature12998
91. Delconte RB, Shi W, Sathe P, Ushiki T, Seillet C, Minnich M, et al. The helix-loop-helix protein ID2 governs NK cell fate by tuning their sensitivity to interleukin-15. Immunity (2016) 44:103–15. doi:10.1016/j.immuni.2015.12.007
92. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity (1998) 9:669–76. doi:10.1016/S1074-7613(00)80664-0
93. Lee CK, Smith E, Gimeno R, Gertner R, Levy DE. STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-gamma. J Immunol (2000) 164:1286–92. doi:10.4049/jimmunol.164.3.1286
94. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med (1998) 188:2067–74. doi:10.1084/jem.188.11.2067
95. Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science (1995) 270:800–2. doi:10.1126/science.270.5237.800
96. Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep (2013) 4:437–44. doi:10.1016/j.celrep.2013.07.012
97. Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature (2008) 455:547–51. doi:10.1038/nature07179
98. Li XY, Luo QF, Wei CK, Li DF, Fang L. siRNA-mediated silencing of CDK8 inhibits proliferation and growth in breast cancer cells. Int J Clin Exp Pathol (2014) 7:92–100.
99. Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature (2010) 468:1105–9. doi:10.1038/nature09590
100. Yamaguchi H, Hung MC. Regulation and role of EZH2 in cancer. Cancer Res Treat (2014) 46:209–22. doi:10.4143/crt.2014.46.3.209
101. Prinjha R, Tarakhovsky A. Chromatin targeting drugs in cancer and immunity. Genes Dev (2013) 27:1731–8. doi:10.1101/gad.221895.113
102. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci U S A (2015) 112:15988–93. doi:10.1073/pnas.1521740112
103. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol (2011) 11:645–57. doi:10.1038/nri3044
Keywords: natural killer cells, checkpoint inhibitors, immunotherapy, cancer, immune therapy
Citation: Carotta S (2016) Targeting NK Cells for Anticancer Immunotherapy: Clinical and Preclinical Approaches. Front. Immunol. 7:152. doi: 10.3389/fimmu.2016.00152
Received: 06 February 2016; Accepted: 07 April 2016;
Published: 21 April 2016
Edited by:
Chiara Romagnani, Deutsches Rheuma Forschungszentrum, GermanyReviewed by:
Stephan Gasser, National University of Singapore, SingaporeJacques Zimmer, Luxembourg Institute of Health (LIH), Luxembourg
Copyright: © 2016 Carotta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastian Carotta, c2ViYXN0aWFuLmNhcm90dGFAYm9laHJpbmdlci1pbmdlbGhlaW0uY29t