 Michelle A. Sugimoto
Michelle A. Sugimoto Lirlândia P. Sousa1,2
Lirlândia P. Sousa1,2 Mauro Perretti
Mauro Perretti Mauro M. Teixeira
Mauro M. Teixeira- 1Laboratório de Sinalização Inflamação, Departamento de Análises Clínicas e Toxicológicas, Faculdade de Farmácia, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 2Laboratório de Imunofarmacologia, Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 3Laboratório de Resolução da Resposta Inflamatória, Departamento de Morfologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 4William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, London, UK
An effective resolution program may be able to prevent the progression from non-resolving acute inflammation to persistent chronic inflammation. It has now become evident that coordinated resolution programs initiate shortly after inflammatory responses begin. In this context, several mechanisms provide the fine-tuning of inflammation and create a favorable environment for the resolution phase to take place and for homeostasis to return. In this review, we focus on the events required for an effective transition from the proinflammatory phase to the onset and establishment of resolution. We suggest that several mediators that promote the inflammatory phase of inflammation can simultaneously initiate a program for active resolution. Indeed, several events enact a decrease in the local chemokine concentration, a reduction which is essential to inhibit further infiltration of neutrophils into the tissue. Interestingly, although neutrophils are cells that characteristically participate in the active phase of inflammation, they also contribute to the onset of resolution. Further understanding of the molecular mechanisms that initiate resolution may be instrumental to develop pro-resolution strategies to treat complex chronic inflammatory diseases, in humans. The efforts to develop strategies based on resolution of inflammation have shaped a new area of pharmacology referred to as “resolution pharmacology.”
Introduction
Inflammation is a reaction of the host to infectious or sterile tissue damage and has the physiological purpose of restoring tissue homeostasis (1). However, uncontrolled or unresolved inflammation can lead to tissue damage, giving rise to a plethora of chronic inflammatory diseases, including metabolic syndromes and autoimmunity pathologies with eventual loss of organ function (2). In fact, signs of persistent unresolved inflammation are not only typical of classical inflammatory diseases but also an underlying feature of a variety of human conditions not previously thought to have an inflammatory component (3), including Alzheimer’s disease (4), atherosclerosis (5), cardiovascular disease (6), and cancer (7). This justifies the increasing interest in studying inflammatory processes. In this context, an important milestone has been reached with the awareness that engagement of resolution of acute inflammation is crucial to avoid persistent chronic inflammation and ensure proper return to homeostasis (8).
Historically, the first acknowledged report on resolution of inflammation was published in 1907 (9). This report shows that, in experimental irritant-induced pleurisy, a fluid containing fibrin and leukocytes was formed, disappearing after 5 days, with the clearance of “polynuclear leukocytes” and the persistence of mononuclear cells in the pleural cavity (9). For many years, resolution of inflammation was considered a passive phenomenon, merely associated with the removal of inflammatory stimuli, end of chemoattractant production, dilution of chemokine gradients over time, and prevention of further leukocyte recruitment. Some years later, the existence of endogenous inhibitors of leukocyte trafficking was reported, acting as a counteractive mechanism against promoters of cell recruitment, such as chemoattractants and adhesion molecules [reviewed in Ref. (10)]. Since then, several studies, especially those from Serhan’s lab at Harvard, showed that the resolution of inflammation is an active process brought about by the biosynthesis of active mediators, which act on key events of inflammation to promote the return to homeostasis (11–14). In this context, homeostasis is recovered after the production of pro-resolving mediators that act on specific receptor targets to (i) shutdown polymorphonuclear leukocyte recruitment, (ii) counteract signaling pathways associated with leukocyte survival to promote apoptosis (or programmed cell death), and (iii) activate the clearance of apoptotic cells (especially by macrophages through a non-phlogistic process), yielding (iv) macrophage reprogramming from a proinflammatory to a pro-resolving phenotype (15, 16).
Inadequate or insufficient resolution can lead to chronic inflammation, excessive tissue damage, and dysregulation of tissue healing, leading to fibrosis. Additionally, it has been implicated in multiple disease states, including the development of autoimmunity (2, 8, 17). Thus, understanding the mechanisms required for the resolution of inflammation may not only unveil new mechanisms of pathogenesis but also support the development of drugs that are able to manage inflammatory processes in directed and controlled ways. Resolution of inflammation requires pro-resolving molecular pathways that are triggered as part of the host response, during the inflammatory phase. This concept challenges a linear model of induction and resolution of inflammation, suggesting a more complex balance between proinflammatory and anti-inflammatory events that are initiated, at least partly, in parallel (18). The inflammatory cells involved in the active phase of inflammation undergo a functional repolarization and contribute to the onset of resolution. Additionally, an accumulating body of evidence suggests that many proinflammatory mediators that promote the inflammatory phase can simultaneously initiate a program for active resolution. For this reason, it is important to understand that adequate resolution of inflammation follows on a coordinated and florid proinflammatory phase with marked leukocyte accumulation. In this context, Sehran, who uncovered the most important pro-resolving lipid mediators, and Savill elegantly stated that “the beginning programs the end” meaning that the events occurring early in acute inflammation engage an active and coordinated “resolution program” (18). In this review, we reason on the events required for an effective transition from the proinflammatory phase to the onset and establishment of resolution (Figure 1).
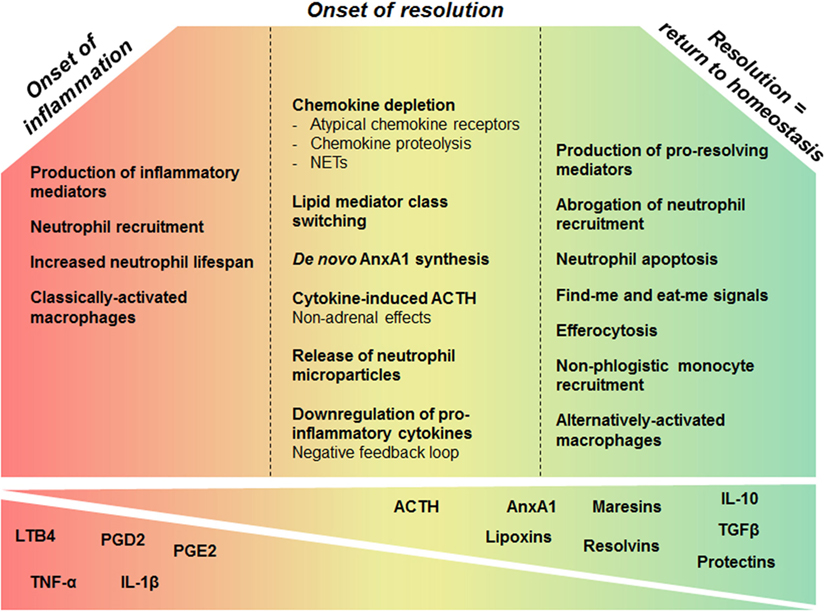
Figure 1. Overview of cellular and molecular processes that govern inflammation and its resolution. During early phase of inflammation, production of inflammatory mediators promotes leukocyte accumulation and survival in the inflammatory site. While the inflammatory response evolves, several mechanisms enable the fine-tuning of these phenomena creating a favorable environment for the resolution phase leading to return to tissue homeostasis. Chemokine proteolysis, sequestration by atypical receptors, and degradation by neutrophil extracellular traps (NETs) are important mechanisms to shape chemokine gradients restricting the influx of neutrophils, once sufficient numbers of cells have been recruited. In addition, inflammatory mediators may induce a negative-feedback loop downregulating the production of inflammatory cytokines. Prostaglandins generated in the active phase of inflammation are involved in the switch from proinflammatory lipid production to the synthesis of lipoxins and other pro-resolving lipids, within inflammatory exudates. Mediators released early in inflammation, like ACTH, can also enable the induction of the pro-resolving phase. Upon activation, neutrophils release microparticles containing pro-resolution mediators that control further granulocyte ingress and turn on a resolution and tissue reparative programs. AnxA1 is a major component of the pro-resolving properties of neutrophil-derived microvesicles. Many resolution mediators downregulate survival pathways and activate apoptosis of granulocytes. Apoptotic neutrophils release pro-resolving mediators that contribute to inhibition of continued neutrophil infiltration and to recruitment of monocytes in a non-phlogistic manner. Upon apoptosis, neutrophils also promote their own clearance by expressing find me and eat me signals that attract scavengers and allow the identification of the dying cell, respectively. In response to local mediators and upon efferocytosis, proinflammatory macrophages switch to resolution-phase macrophages. These events will reestablish tissue homeostasis.
Cellular Events in the Resolution of Acute Inflammation
The molecular and cellular events of the inflammatory response are well known and typically characterized by increased blood flow, capillary dilatation, leukocyte infiltration, and production of chemical mediators. Acute inflammation is mainly characterized by the presence of neutrophils, which are highly motile leukocytes, able to rapidly migrate to the site of injury or infection. Although neutrophils are essential for proper elimination of the inflammogen, exaggerated influx of leukocytes can be more deleterious than the infection or injury itself and has been considered a bad marker of tissue homeostasis (19). Therefore, the key histological feature in the resolution of acute inflammation is the depletion of neutrophils from the local inflamed sites. This is achieved through programmed processes that occur in an overlapping fashion and are actively regulated at multiple levels (20, 21). The cardinal signs of resolution entail the limitation or cessation of blood-borne cell extravasation, the counter regulation of chemokines and cytokines, the switching off of signaling pathways associated with leukocyte survival, the induction of leukocyte apoptosis and their subsequent removal through efferocytosis by macrophages, the reprogramming of macrophages from classically activated to alternatively activated cells, the return of non-apoptotic cells to the vasculature or lymph, and finally the initiation of healing processes. Altogether, these events avoid excessive tissue damage and culminate in the return to tissue homeostasis, giving little opportunity for the development of chronic, non-resolving inflammation. On the other hand, failure of one or more steps in the resolution of inflammation may be involved in the pathogenesis of several human chronic inflammatory diseases (8).
Pro-Resolving Mediators
Similar to the onset phase of inflammation, resolution of inflammation is coordinated and regulated by a large panel of mediators. The pioneer authors in the field of resolution and other investigators worldwide have focused on defining the endogenous mediators of resolution and the mechanisms through which the body regulates effector cells (PMNs, monocytes, and macrophages). It is worth noting that anti-inflammatory effects and pro-resolving effects are not totally overlapping: anti-inflammation mainly refers to an inhibitory/blocking action (e.g., stopping immune cell extravasation, which is a hallmark of acute inflammation), whereas pro-resolving actions indicate an inherent stimulation and activation of specific processes, such as apoptosis or efferocytosis. In both cases, the end point is the inhibition of inflammation, but pro-resolving mediators are those that genuinely enable resolution to take place (12, 22, 23). In the same vein, there is a mechanistic difference between an anti-inflammatory drug that blocks some specific pathways and a pro-resolving drug that is expected to activate a plethora of actions. Hence, the distinction is between blocking/inhibiting particular mediators, which can cause tissue damage, and agonism/activating cellular processes that participate in limiting or preventing damage, the latter enabling an amplifying effect. It is reasoned that pro-resolving-based therapies will promote both anti-inflammatory and pro-resolution actions, differing from traditional anti-inflammatory agents that solely inhibit key proinflammatory mediators (20). In addition, we have recently pointed out that pro-resolving molecules are characterized by “mild-to-moderate actions,” since they balance pro- and anti-inflammatory responses to reach an equilibrium (22).
According to the first consensus report from leading authorities on definitions and mechanisms in resolution (3) and subsequent reviews (16, 21), pro-resolving mediators should ideally fulfill some fundamental criteria that include:
• Stop: the limitation or cessation of neutrophil tissue infiltration;
• Sink: the counter regulation of chemokines and cytokines;
• Kill: the induction of apoptosis in spent neutrophils and their subsequent efferocytosis by macrophages;
• Skew: the reprogramming of macrophages from classically activated to alternatively activated cells;
• Leave: the return of non-apoptotic cells to the blood or lymphatic vasculature and egress of immune cells – following efferocytosis, the macrophages and dendritic cells leave the site of inflammation;
• Inform: the instruction of suppressive immune cells and adaptive immune response to help dealing with subsequent encounters;
• Heal: the induction of tissue repair – return to homeostasis without fibrosis or scar formation marks the final step of resolution.
Molecules that fulfill the criteria above, which qualify a pro-resolving mediator, are very diverse in nature (21) and include specialized lipid mediators [lipoxins (e.g., LXA4), resolvins (e.g., RvD1), protectins, and maresins] (14), proteins and peptides [e.g., annexin A1 (AnxA1), adrenocorticotropic hormone, chemerin peptides, and galectin-1] (24), gaseous mediators (e.g., H2S and CO) (25), a purine (adenosine) (26–28), as well as neuromodulators (acetylcholine and other neuropeptides) released under the control of the vagus nerve (29, 30).
Failure to produce adequate amounts of these anti-inflammatory and pro-resolving mediators or yet a failure to bind to their receptor could lead to the persistence of inflammation, playing a significant etiopathogenic role in chronic inflammatory and autoimmune diseases. This is highly plausible for inflammatory bowel diseases (IBDs), such as Crohn’s disease (CD) and ulcerative colitis (UC), chronic relapsing inflammatory conditions of the gastrointestinal tract that are characterized by intestinal inflammation and epithelial injury (31, 32). Resolution mediators (e.g., AnxA1, lipoxins, and resolvins) regulate intestinal mucosal injury, inflammation, and repair, supporting the resolution of inflammation in the gut. Therefore, defective expression of pro-resolution mediators may contribute to the chronic inflammatory response associated with IBD. Notably, colonic mucosa from UC patients demonstrates defective LXA4 biosynthesis, which may contribute to the inability of these patients to resolve persistent colonic inflammation (33). Complete loss of AnxA1 protein was detected in colonic tissues from chronic CD patients, which correlated with the clinical status, response to therapy, TNF-α expression, and lymphocyte activation (34). Vong and coworkers (35) documented an increase in mucosal synthesis of AnxA1 and LXA4, in individuals in medically induced remission from UC. Besides, during anti-TNF-α therapy, AnxA1 expression was upregulated in patients with a successful intervention, whereas non-responsive patients did not show the same expression profile (34). The contribution of AnxA1 to the remission of IBD was validated with a model of dextran sulfate sodium (DSS)-induced colitis in TNFR knockout (KO) mice, mimicking the anti-TNF-α therapy. Mucosal levels of AnxA1 increased in the absence of TNF-α signaling, allowing early recovery of colitis as compared to wild-type (WT) mice (36). According to these findings, changes in pro-resolving mediator levels may predict therapeutic efficacy. Moreover, inflammation-resolution agonists prevent immune-mediated tissue damage and restore tissue homeostasis. Interestingly, pharmacological treatment with LXA4 or Resolvin E1 (RvE1) effectively promoted the resolution of trinitrobenzenesulphonate (TNBS)-induced colitis (37, 38). The beneficial effect of lipid mediators in colitis was accompanied by decreased leukocyte infiltration and proinflammatory cytokines. In addition, TNBS-specific IgG serum levels decreased after treatment with RvE1, suggesting diminished antigen presentation and antibody production (38). Moreover, AnxA1 peptides encapsulated in nanoparticles accelerated the recovery of experimentally induced colitis and the healing of colonic biopsy-induced wounds (39).
Persistent airway inflammation in lung diseases, including asthma, may also be due to a defect in counter regulatory signaling (40, 41). Clinical findings suggest that severe asthma is associated with diminished expression of LXA4, its receptor FPR2, and 15-lipoxygenase, the major enzyme involved in LXs generation (42–46). Thus, LXA4-deficient production and/or signaling might have a role in the progression of the disease. In a recent study, AnxA1 and LXA4 plasma levels were lower in wheezy infants than in control group (47). Once persistent wheezing in children may progress to asthma, this reduced level of pro-resolving molecules could be an early event in asthma progression (48).
In some cases, failure in the activity of specific mediators may contribute to the inflammatory process even when the expression is normal or higher, when compared to healthy controls. For example, CD-related inflammation is characterized by reduced activity of the immunosuppressive cytokine transforming growth factor (TGF)-β1. TGF-β is a crucial cytokine in inflammation resolution due to its immunoregulatory activities, essential to tolerance and homeostasis, and its role in epithelial restitution and fibrosis (49). Indeed, in vitro and in vivo studies have demonstrated that TGF-β1 acts as a potent negative regulator of mucosal inflammation (50). Although TGF-β is found in high levels in human IBD tissue, it has reduced activity due to the overexpression of an inhibitor of TGF-β1 signaling, SMAD7 (51). As a result, TGF-β is unable to reduce the chronic production of proinflammatory cytokines that drives the inflammatory process in IBD and, consequently, inflammation is maintained (51). Notably, therapeutic strategies that restore TGF-β signaling pathway may downregulate the inflammatory response and induce remission in patients with CD (51, 52).
Positive Networks in Resolution
Evidence is accumulating that a pro-resolving cascade becomes operative during resolution, whereby one pro-resolving mediator would induce another one. We reported one of the first evidence that fundamental pro-resolving mediators, such as AnxA1 and LXA4, induce the production of further anti-inflammatory molecules in vivo, such as IL-10 (53). Later, Brancaleone and colleagues (54) provided strong evidence that the engagement of FPR2/ALX by LXA4 induces AnxA1 phosphorylation and mobilization in human PMN. Similarly, the pro-resolving mediator RvE1 stimulates endogenous LXA4 production (55).
Other examples and modus operandi of this cross talk in resolution are emerging, as the cross talk between AnxA1 and glucocorticoid (GC)-induced leucine zipper (GILZ) during certain inflammatory events (56). GILZ mediate and mimic several anti-inflammatory actions of GCs (57). Besides demonstrating that GILZ expression depends on AnxA1, we identified that the lack of endogenous GILZ during the resolution of inflammation is compensated by AnxA1 overexpression. In the model of lipopolysaccharide (LPS)-induced pleurisy, GILZ deficiency was associated with an early increase of AnxA1 and equal neutrophil influx and resolution as compared to WT mice. Likewise, we demonstrated that dexamethasone-induced resolution was not altered in GILZ KO mice due to compensatory expression and action of AnxA1 (56). These studies indicate that pro-resolution mediators not only communicate in positive loops but also enact compensatory actions to guarantee the effective engagement of resolution pathways.
We predict that a further definition of the positive loops of resolution is crucial for the discovery of new pharmacological targets that could resolve inflammation, especially in the context of chronic inflammatory diseases. A better understanding of the key controlling points of resolution networks may allow us to design specific strategies to promote resolution.
How Does Resolution Start?
Briefly, the acute inflammatory response can be divided in two stages: initiation (productive and transition phases) and resolution (Figure 1) (58). Interestingly, molecular and cellular mechanisms involved in the first phase of inflammation contribute to the initiation of the pro-resolving response. It has now become evident that coordinated programs of resolution initiate shortly after the beginning of the inflammatory response (18). In this context, several anti-inflammatory and pro-resolving mediators are endogenously produced to temper the inflammatory events. However, here we intend to highlight the existence of events and pathways that do not fulfill all criteria to be classified as pro-resolving, but do contribute to the initiation of resolution. These mechanisms provide the fine-tuning of inflammation, creating a favorable environment for the resolution phase to take place, and for homeostasis to return. As “contributors of resolution” these events, pathways, and mediators deserve special attention since they may be key targets for the pharmacological input or enacting of resolution, especially when it has not turned on, such as in chronic inflammatory settings.
Aside its well-known proinflammatory functions, nuclear factor kappa B (NF-κB) also has a crucial role in the initiation of resolution of inflammation. NF-κB proteins are a family of transcription factors of central importance in inflammation and immunity (59, 60). NF-κB and its activating IκB kinase (IKK)β play important roles in driving the inflammatory response by activating the expression of proinflammatory and anti-apoptotic genes (61). However, several reports have shown that NF-κB and IKKβ also influence anti-inflammatory response, pointing to their involvement in both onset and resolution of acute inflammation (62–64). The functional transcription factors consist in homo- or hetero-dimers comprising five subunits (p50, p52, p65, cRel, and RelB), which utilize Rel homology domain (RHD) for DNA binding and dimerization (65). Dimers containing at least one subunit with transactivating domains (TAD) in their C-terminus (p65, RelB, or cRel) are required to induce gene transcription. In contrast, dimers that contain only subunits without TAD (p50 and p52) are transcriptionally inactive and may prevent transcriptionally active NF-κB dimers from binding to κB sites (66). In resting cells, NF-κB dimers are sequestered to the cytoplasm and maintained inactivated by reversible association with its inhibitor IκB or unprocessed forms of cytoplasmic p50/p105 (NF-κB1) and p52/p100 (NF-κB2) (60, 65, 67). NF-κB activation in response to proinflammatory stimuli is regulated by IKK, which phosphorylates IκB and promotes its proteasome degradation and the release of NF-κB for nuclear translocation and gene transcription activation (61).
Nuclear factor kappa B activates many promoters containing highly divergent κB-site sequences. The fact that the regulation of gene expression is dimer-specific explains, in part, how NF-κB pathways can modulate both inflammation and resolution (65, 68). Differential expression of NF-κB subunits and the differential effects of NF-κB dimers may be intimately associated with the temporal regulation of inflammatory responses (69). p65/p50 heterodimer is the predominant form of functionally active NF-κB with proinflammatory activity, since this dimer enhances the transcription of genes related to the proinflammatory phase. On the other hand, p50/cRel, p65/cRel, or p50/p50 seems to be involved in the transcription of genes related to the recovery phase (70). Accordingly, the genes regulated by p50/cRel and p65/cRel are activated in later points after inflammatory stimulation, providing the necessary period between the burst of the proinflammatory response and the recovery phase (69, 71). p50/p50 homodimer exerts important anti-inflammatory and pro-resolving effects and competes with p65/50 heterodimer for DNA binding (72, 73). Unlike p65/p50, p50/p50 lacks the transactivation domain and may repress proinflammatory genes (74–76). Bohuslav and colleagues demonstrated that increased expression of p50 subunit of NF-κB directly results in the downregulation of LPS-induced TNF production (72). Recently, the enhancement of efferocytosis mediated by RvD1 was associated with p50/p50-mediated suppression of TNF-α expression (77). In this context, RvD1 modulates at least two different NF-κB pathways leading to enhanced localization of p50 in the nucleus, while it suppresses dissociation from IκBα and concurrent nuclear translocation of p65 (77). Moreover, upon LPS stimulation, macrophages express p65/p50 heterodimer in predominance over p50/50 homodimer, thereby provoking the proinflammatory state. However, in later time points, these macrophages show p105 degradation, nuclear translocation of p50, and formation of p50/p50 homodimer, presumably as an adaptive cellular response to proinflammatory insult.
During the proinflammatory phase, besides inducing proinflammatory genes, p65/p50 also induces the transcription of genes that will provide the control of the recovery phase, such as Rel, the gene that codifies cRel (71). For example, Muxel and colleagues showed that the expression of p65/cRel, crucial for inflammation resolution, is induced by p65/p50, which is earlier expressed in LPS-stimulated macrophages (69). The authors identified that temporal regulation of cRel promoted the synthesis of melatonin (via p65/cRel) by macrophages, a modulator of phagocyte function preventing over-activation of this cell type (78, 79). In addition, NF-κB negatively regulates NLRP3-inflammasome activation and IL-1β production (63). In macrophages, NF-κB prevents premature and excessive NLRP3-inflammasome activation, acting as a negative regulator of IL-1β secretion (63). Although the precise molecular mechanism underlying NF-κB-mediated inhibition of NLRP3-inflammasome activation remains unclear, NF-κB has been suggested to promote autophagy (80), a cellular process that negatively regulates NLRP3 inflammasome activity (81–83). Reinforcing this observation, a recent study revealed that NF-κB restricts inflammasome activation in macrophages via elimination of damaged mitochondria (84). This allows NF-κB to restrain its own inflammation-promoting activity in macrophages (84).
Clearly, NF-κB may have dual function in inflammation, which is likely the result of the central role of this molecule in the convergence of several inflammatory signals (62). This results in divergent effects of NF-κB pharmacological inhibition in inflammatory models. On the one hand, NF-κB inhibitors may attenuate inflammation and promote resolution in different experimental models of inflammation (62). For example, NF-κB inhibitors possess anti-inflammatory effects in models of LPS-induced lung injury (85), traumatic brain injury (86), colitis (87), and pulmonary arterial hypertension (88). Our research group showed that inhibition of NF-κB promotes resolution in established murine models of neutrophilic and eosinophilic inflammation associated with enhanced apoptosis of inflammatory cells (89, 90). On the other hand, inhibition of NF-κB during the resolution of inflammation prolonged the inflammatory response and prevented apoptosis (62). In addition, IKKβ has also been shown to have an anti-inflammatory role, such as the suppression of M1 macrophage activation during infection through the inhibition of signal transducer and activator of transcription (STAT)1 pathway (91). In accordance with this observation, IKKβ ablation results in severe neutrophilia and inflammation mediated by IL-1β (92). Notably, mice lacking IKKβ had hyperproliferative granulocyte–macrophage progenitors and pregranulocytes and a prolonged lifespan of mature neutrophils that correlated with the induction of genes encoding pro-survival molecules (92). Of clinical relevance, enhanced inflammation and neutrophilia were observed in human subjects that were treated with IKKβ inhibitors.
Notably, proinflammatory and resolution phases of inflammation are under the control of both transcriptional and post-transcriptional mechanisms, which regulate the expression of proteins that initiate and resolve inflammation. Reviewing this topic in 2010, Anderson (93) pointed out that post-transcriptional controlling mechanisms link the initiation/productive phase to the resolution phase of inflammation. mRNA translation is a highly regulated process governed by post-transcriptional mechanisms. Transcription is the first step in the regulation of gene expression, but since mRNA can be long-lived, turning off its synthesis does not rapidly redirect or stop the progress of inflammation. On the other hand, the second step, i.e., post-transcriptional regulation, can rapidly suppress protein expression by promoting mRNA degradation or by inhibiting its translation (93). Post-transcriptional control mechanisms may rapidly limit the expression of potentially toxic inflammatory mediators and help protecting the host against the pathological overexpression of potentially injurious proteins. For instance, a number of cytokine mRNAs can be regulated at the level of mRNA stability (94). mRNA decay and translational repression of target transcripts are promoted by RNA-induced silencing complex (RISC) that is composed by argonaute proteins bound to small non-coding RNAs, microRNAs (miRNAs). Importantly, the mechanisms used to ensure limited production of the proteins involved in the inflammatory response are highly variable, and in some cases, interact with each other to define protein expression levels. It remains not fully understood whether post-transcriptional controlling mechanisms play a role in the resolution of inflammation, but exciting possibilities for pharmacological intervention against the overproduction of many inflammatory proteins are likely to emerge from this elucidation (95).
Importantly, miRNAs triggered by immune mediators have a central role in modulating NF-κB signaling pathways and might be involved in controlling the switch from a strong early-inflammatory response to the resolution phase of the inflammatory process, in a timely and orchestrated manner (96, 97). The endotoxin-responsive gene miR-146a was the first one to be discovered to suppress the activation of the NF-κB pathway (98). miR-146a has been described as a negative regulator of the canonical NF-κB inflammatory cascade by targeting IL-1 receptor-associated kinase (IRAK) 1 and TNF receptor-associated factor (TRAF) 6 (98, 99). Moreover, miR-146a targets RelB, which is mostly implicated in the non-canonical NF-κB pathway, and controls monocyte responses during inflammatory challenge (100, 101). Some studies indicate that miR-146a can regulate proinflammatory gene expression by controlling RelB-dependent reversible chromatin remodeling (102, 103). Notably, deletion of miR-146a gene results in the production of higher levels of inflammatory cytokines by macrophages (104). Remarkably, the expression of many miRNAs is induced in an NF-κB-dependent manner after inflammatory stimulus or pathogen infection, promoting the control of the strength and longevity of an inflammatory response (97, 98, 104–109). miR-146a was the first reported miRNA whose expression can be induced through the NF-κB-dependent pathway in response to various immune mediators, such as LPS, IL-1β, and TNF-α (98, 105, 110–113). Since then, many studies have further identified subsets of miRNAs related to the TLR-induced NF-κB-dependent pathway. Another example, miR-9 expression is directly induced by LPS via the TLR4-MyD88-NF-κB-dependent pathway in human monocytes and neutrophils. In turn, miR-9 operates a feedback control of the NF-κB-dependent responses by fine-tuning NF-κB1 expression. Bazzoni and colleagues suggest that miR-9 induction probably acts as a tuning mechanism to prevent negative regulation by p50 homodimers, as occurs in monocytes in systemic anti-inflammatory response syndrome (SIRS) (109).
Because miRNA-mediated post-transcriptional control is important to fine-tune the expression of genes involved in inflammation, dysregulation of expression levels of miRNAs can lead to chronic infections, autoimmunity, allergic inflammation, or immune deficiency. Recent studies have identified dysregulated miRNAs in tissue samples of IBD patients and have demonstrated similar differences in circulating miRNAs in the serum of these patients [reviewed in Ref. (114)]. In fact, dysregulated expression of tissue and blood miRNAs in IBD already numbers >100 (114) and may be involved in the reduced apoptosis of T-cells, which is an important mechanism in T-cell homeostasis, and cell activation (115).
Also important for resolution initiation, the pituitary hormone adrenocorticotrophin (ACTH) is released quite early during inflammation, in response to proinflammatory cytokines, including IL-1β (116). For a long time, ACTH has only been thought to modulate host response through the rapid generation of adrenal-derived GCs, which are de novo synthesized from cholesterol. However, recent works have revealed important immune-modulatory properties of ACTH, through the activation of specific receptors in the periphery (117), expressed on macrophages and other stromal cells such as chondrocytes [reviewed by Montero-Melendez (118)]. Molecules that activate these receptors on macrophages are able to promote resolution of inflammation with a downstream impact on experimental arthritis (119, 120).
Chemokine Depletion Decreases Infiltration of Neutrophils into Tissue
As discussed above, successful inflammation depends on the regulation of neutrophil recruitment, allowing the proper elimination of the inflammogen but avoiding the tissue damage induced by excessive neutrophil influx and toxic content release. According to Headland and Norling – who recently reviewed this subject (21) – restricting the influx of neutrophils, once sufficient number of cells has been recruited, is a process through which chemokine and cytokine gradients are reduced, proinflammatory lipid mediators are switched to pro-resolving mediators, and circulating neutrophils are no longer activated and recruited to the inflammatory site. Chemokines are low molecular weight cytokines that orchestrate the migration of target cells to the site of inflammation. Chemokine depletion through mechanisms, such as chemokine cleavage by proteolysis and chemokine sequestration, is necessary to achieve a resolving environment and to abrogate neutrophil influx (16). Chemokines directly induce cell migration through a set of conventional chemokine G protein-coupled receptors. However, chemokines are also recognized by a small subfamily of atypical chemokine receptors (ACKR), previously called decoys, interceptors, scavengers, or chemokine-binding proteins (121). The binding of chemokines to their respective atypical receptors does not promote leukocyte migration due to the inability of ACKR to initiate classic G protein-dependent signaling pathways. Instead, ACKR sequestrate chemokines from the environment, an important mechanism to shape chemokine gradients. Therefore, ACKR are now emerging as crucial regulatory components of chemokine networks in a wide range of physiologic and pathologic contexts (122).
Chemokine proteolysis is another important mechanism for chemokine depletion and consequently the decrease of neutrophil recruitment and activation. Matrix metalloproteinases (MMPs) are traditionally associated with extracellular matrix protein degradation in several physiological and pathological processes. However, it is now clear that MMPs mediate homeostasis of the extracellular environment by modulating the biological activity of many bioactive molecules involved in cell function (123, 124) and innate immunity (125), including chemokines (123, 126–130), TNF-α (124, 131), α-defensin (132), and mannose-binding lectin (133). In this context, Dean and colleagues (134) proposed that macrophages aid the regulation of acute inflammatory responses by precise proteolysis of chemokines through MMP-12. Macrophage-specific MMP-12 cleaves CXC chemokines in the ELR motif, which is fundamental for receptor binding, thus rendering the mediators unable to recruit neutrophils (134). In some cases, cleaved chemokines continue to bind to their corresponding receptors, but fail to induce downstream signaling and chemotaxis, thus acting as antagonists dampening inflammation (126, 127).
Pro- and Anti-Inflammatory Networks Help to Turn on the Resolution Program
A great number of evidence indicates that proinflammatory molecules can be involved in the initiation of the resolution program. In order to limit the undesirable consequences of an excessive inflammatory process, many mediators involved in the onset of the inflammatory response simultaneously trigger a program that actively resolves inflammation. In this context, our group has observed, in two complementary studies, the intricate balance and cross talk between pro- and anti-inflammatory cytokines during a systemic inflammatory response. In 2003, we described a network of TNF-α, IL-1β, and IL-10 during severe intestinal ischemia and reperfusion injury (135). Both, IL-1β and TNF-α triggered an anti-inflammatory cascade resulting in the production of IL-10. We identified that IL-1β plays a major role in driving endogenous IL-10 production and protecting against TNF-α-dependent systemic and local acute inflammatory response. IL-1β has been implicated in inflammatory events, such as the expression of adhesion molecules and neutrophil influx following reperfusion of ischemic tissues. However, some studies have failed to show a protective effect of IL-1β inhibition during ischemia/reperfusion (I/R) injury (136–138). In our investigations, we associated neutralizing strategies or selective receptor antagonism to prevent the actions of IL-1β with an overall enhancement of tissue injury, proinflammatory cytokine expression (TNF-α), and lethality (135). Members of the IL-1 family of cytokines (e.g., IL-1β, IL-18, and IL-36γ) display a dual role in regulating IBD, reinforcing the concept that proinflammatory cytokines may contribute to both proinflammatory responses and resolution of inflammation. These cytokines are upregulated in the inflamed mucosa during experimental colitis as well as in human IBD. Remarkably, they not only contribute to intestinal inflammation (139) but also to resolution of inflammation, as demonstrated by the increased susceptibility to DSS-induced colitis by mice lacking IL-1β, IL-18, and IL-36 receptors or components of their processing (140–144). In humans, polymorphisms leading to decreased Nlrp3 expression, and consequent hypoproduction of IL-1β, are associated with increased risk of developing CD (145).
Moreover, we and others have observed that TNF-α is central to the pathogenesis of reperfusion-associated injury and lethality (135, 146, 147). However, this proinflammatory cytokine also contributes to the production of IL-10 during intestinal ischemia and reperfusion (147). Furthermore, we reported that TNF-α modulates IL-1β production: first, inhibition of TNF-α was accompanied by enhanced reperfusion-induced production of IL-1β (147); second, administration of exogenous IL-10 was linked to decreased TNF-α concentration and enhanced IL-1β. Based on these results, we hypothesized that TNF-α could be inducing an intermediate molecule that controls IL-1β production (147). It is interesting to note that recent investigations have identified a central role for TNF-α in upregulating a pro-resolving master receptor that transduces the actions of AnxA1, LXA4, and RvD1 (148).
Several studies have identified a mechanism feedback for IL-10 as a potent repressor of proinflammatory cytokine production by macrophages, acting therefore as a key anti-inflammatory mediator (149, 150). In murine bone marrow-derived macrophages (BMDM) activated by LPS, IL-10 attenuated proinflammatory cytokine production via reduction of mRNA stability. IL-10 initiates a STAT3-dependent increase of the expression of the RNA destabilizing factor tristetraprolin (TTP) accompanied by the release from p38 MAPK-mediated inhibition. As a result, IL-10 diminishes mRNA and protein levels of TNF-α and IL-1β (151).
Resolution of Inflammation Is Accompanied by an Active Switch in the Mediators That Predominate in Exudates
In a classical acute inflammatory response, proinflammatory lipid mediators, such as the classical eicosanoids [prostaglandins (PGs) and leukotrienes (LTs)], are generated during the initial phase of the inflammatory response through enzymatic modification of arachidonic acid (AA) by cyclooxygenases (COX) and lipoxygenases (LO) (152). These proinflammatory molecules have important roles in initiating leukocyte trafficking and stimulating blood flow changes, increasing vasopermeability to yield edema formation, all leading to neutrophil influx to the site of inflammation (14). In addition, PGs and LTB4 are involved in the initiating steps that permit leukocytes to leave postcapillary venules via diapedesis (153). Thereby, a switch in lipid mediators from proinflammatory PGs to lipoxins, which are anti-inflammatory/pro-resolving mediators, is crucial for the transition from inflammation to resolution (154). As Serhan pointed out in a scholar review (20), during inflammation, neutrophils undergo a phenotype switch to produce different profiles of lipid mediators depending on the cells and substrates present in the local environment. Neutrophils in the peripheral blood generate and release LTB4 on activation, as one of their main bioactive products. During spontaneous resolution of acute inflammation, there is a switch in PMN-LO pathway products expression, from LTs to lipoxins and resolvins. Evidence indicates that first-phase proinflammatory eicosanoids “reprogram” the exudate PMN to produce pro-resolving lipid mediators and hence promote resolution. For instance, Levy and colleagues suggested that when circulating PMNs begin diapedesis, they are exposed to autacoid gradients (e.g., PGE2) that initiate phenotypic changes via gene expression regulation (12). In this context, local PGE2 and PGD2 stimulate the processing of 15-LO mRNA in leukocytes to produce functional enzymes for the synthesis of lipoxin. AA is then converted to anti-inflammatory lipid mediators, such as LXs (e.g., lipoxin A4 and lipoxin B2), which harness dual anti-inflammatory and pro-resolving actions, in vitro and in vivo (20). Lipoxins are generated by transcellular biosynthesis, involving two or more cell types, since the required enzymes are differentially expressed in the cells. Thus, at the sites of injury or inflammation, LXs are generated via biosynthetic routes engaged during cell–cell interactions. Mobilization of LX biosynthetic circuit occurs, for example, when infiltrating PMNs (which express 5-LO) interact with tissue resident cells (which express 15-LO) in inflamed target organs. In an autocrine, paracrine, or juxtacrine manner, newly formed LXs can interact with specific receptors on leukocytes to regulate their function (12).
Cyclooxygenase-2 apparently has a dual role in the inflammatory process, initially contributing to the onset of inflammation and later helping to resolve the process. Gilroy and colleagues reported that COX-2 expression and PGE2 levels transiently increased in the early stage of carrageenan-induced pleurisy in rats (155). Later in the response, COX-2 was induced again to even greater levels and generated anti-inflammatory PGs, such as PGD2 and 15-deoxy-Delta(12,14)-PGJ2 (15d-PGJ2), but only low levels of proinflammatory PGE2. Anti-inflammatory actions mediated by 15d-PGJ2, a terminal product of COX-2 pathway, represent another negative feedback that explains how once-initiated immunologic and inflammatory responses are switched off and terminated. 15d-PGJ2, a terminal product of COX-2 pathway, is abundantly produced in inflamed sites, suggesting its potential role in facilitating the resolution of inflammation (156). 15d-PGJ2 exerts potent anti-inflammatory actions, in part by antagonizing the activities of NF-κB, STAT3, and activator protein 1 (AP1), while stimulating the anti-inflammatory nuclear factor E2-related factor 2 (Nrf2). Besides targeting the transcriptional machinery, 15d-PGJ2 is a potent inhibitor of protein translation. Interestingly, 15d-PGJ2-mediated translational repression triggers a stress response program that results in the assembly of stress granules containing untranslated mRNAs. Stress granules have an important role in reprogramming gene expression to allow stressed cells to survive to noxious stimuli (157, 158). Altogether, these mechanisms might combine to effectively dampen inflammation (93). Thus, 15d-PGJ2, especially formed during the late phase of inflammation, might inhibit cytokine secretion and other events by antigen-presenting cells such as dendritic cells or macrophages. Production of the 15d-PGJ2 is a consequence of a series of dehydration (oxidation) of PGD2 (159). The latter is a major COX-2 product formed in various cells (e.g., mast cells) and tissues during inflammatory processes by the action of PGD2 synthase, which catalyzes the isomeric conversion of PGH2 to PGD2. The pathogenic relevance of PGJ2 is suggested by clinical findings of reduced levels of PGD2 in some human diseases, such as the cerebrospinal fluid of patients suffering from multiple sclerosis and schizophrenia (160). Other evidence of clinical relevance comes from atherosclerosis, where PGE2 is over-expressed in symptomatic plaques of patients who underwent carotid endoarterectomy, while in asymptomatic ones, the PGD2 pathway prevails, known to be associated with NF-κB inactivation and MMP-9 suppression. These clinical findings suggest that PGE2-dominated eicosanoid profile is associated with cerebral ischemic syndromes, possibly through MMP-induced plaque rupture (161).
Although therapeutic inhibition of COX-2 by non-steroidal anti-inflammatory drugs (NSAIDs) may have beneficial effects in the early phase of inflammation by preventing prostanoid production, it may also be “resolution-toxic,” by disrupting the production of anti-inflammatory PGs and LXs (3, 155, 162, 163). Disturbance of physiologic lipid mediator class switching by COX-2 inhibitors has deleterious consequences in humans (164) as well as in murine peritonitis (163), arthritis (165), and lung acute injury (ALI) models (166). In the study from Fukunaga and colleagues, COX-2 inhibition resulted in an exacerbation of ALI with longer recovery times. Reinforcing the dual role of COX-2 during inflammation, inhibition of COX-2 activity by pharmacologic treatment or gene targeting decreased early PMN trafficking to the lung but paradoxically led to dramatic increases in inflammation at later time points, mainly due to the disruption of LXA4 production (166). Furthermore, COX-2 inhibition decreased macrophage phagocytosis of apoptotic PMNs in vitro and reduced prostaglandin E2 and LXA4 expression (163). During peritonitis, treatment with specialized pro-resolving lipid mediators [aspirin (ASA)-triggered lipoxins, RvE1, and protectin D1] rescued the resolution deficit promoted by COX-2 inhibition (163).
Aspirin is unique among other NSAIDs because it irreversibly inhibits COX-2 by acetylation of an amino acid serine residue preventing prostanoid generation (167) yet enabling the biosynthesis of endogenous anti-inflammatory mediators. Therefore, the generation of ASA-triggered specialized lipid mediators (AT-SLM) (11, 168–170) may enhance resolution and counteract the loss in prostaglandin production by ASA (18). Low-dose ASA triggers the resolution phase by activating endogenous epimers of specialized pro-resolving lipid mediators in humans and several animal models (3). Low-dose ASA triggers 15-epi-LXA4 in skin blisters in humans to reduce PMN infiltration by inducing antiadhesive nitric oxide, thereby dampening leukocyte/endothelial cell interaction and subsequent extravascular leukocyte migration (171). In addition, low-dose ASA administration to mice triggered the formation of 15-epi-LXA4, which in turn attenuated I/R-mediated vascular inflammation (172). In a randomized controlled study, low-dose ASA administration to volunteers augmented plasma ATL levels while inhibiting thromboxane (173). These observations support the idea that low-dose ASA may be considered “resolution friendly” (18), since it mimics endogenous biosynthetic mechanisms to trigger new mediators, leading to a favorable net change (173) for pro-resolution (174, 175).
Neutrophils: Important Cells to Turn on Resolution
Aborted neutrophil recruitment is one of the steps required to reconstitute tissue homeostasis, followed by apoptosis and clearance by macrophages. Interestingly, neutrophils have pivotal roles in attenuating inflammatory diseases and seem to orchestrate both elimination of microorganisms and resolution of inflammation (21). In view of that, wound healing is delayed in neutrophil depletion models, indicating a critical role of these cells in the resolution of inflammation (176). Moreover, depletion of neutrophils aggravates different types of experimental UC (177, 178) and extends joint inflammation in a murine model of gout (179). Among the anti-inflammatory functions of neutrophils, it is worth mentioning its capacity to disrupt chemokine gradients via several mechanisms. For instance, neutrophils release proteases that not only degrade extracellular matrices and cells surrounding the inflammatory milieu but also deactivate inflammatory cytokines (180). Additionally, neutrophils modulate the cytokine production stimulated by bacterial peptidoglycans and LPS (181). In vitro studies have shown that PMN lysates and neutrophil elastase can degrade recombinant human IL-1β and TNF-α but not IL-10, and alpha1-antitrypsin can inhibit this process (180). Neutrophil-derived proteases are also involved in the downregulation of IL-1β and TNF-α produced by mononuclear cells, an effect that is independent on ROS production or phagocytosis (180). Serine proteases released by activated neutrophils may also be associated with NETs, which are web-like structures composed of nuclear material in complex with neutrophil proteins that display exquisite antibacterial properties (182). A recent article by Schauer and colleagues revealed that at the very high neutrophil densities that occur at the site of inflammation, NETs build aggregates that trap and degrade proinflammatory mediators via the proteolytic action of inherent neutrophil serine proteases (179). However, it remains to be investigated if this anti-inflammatory effect can be reproduced in physiological conditions where concentrations of NET may be lower. Conversely, NETs are also related to proinflammatory effects that in part induce further neutrophil recruitment (183). Recent observations suggest that NETs are effective activators of the inflammasome machinery in both human and murine macrophages, resulting in the release of active IL-1β and IL-18 (184). Indeed, pharmacological and genetic strategies that prevent NETosis have been shown to be protective in murine models of lupus (185), cardiac infarction (186), deep vein thrombosis (187), atherosclerosis (183), and diabetes (188). In addition, a recent work suggests that damage-associated molecular patterns (DAMPs) released during liver I/R result in formation of NETs which subsequently exacerbate organ damage and initiate inflammatory responses (189). Moreover, the presence of DNAse-sensitive NETs in skin wounds impairs wound healing in diabetes (188). Timely degradation/removal of NETs is critical since its components may serve as autoantigen or DAMPs leading to inflammatory and chronic autoimmune diseases, including systemic lupus erythematosus (SLE) [reviewed in Ref. (189–194)]. Furthermore, mitochondrial ROS-dependent NETosis may promotes externalization of proinflammatory oxidized mtDNA and subsequent activation of type I interferon (IFN) synthesis, what may contribute to lupus-like disease (195). Finally, serum of SLE patients show an increase in various NET proteins [e.g., defensins, high-mobility group box protein 1 (HMGB1), and bactericidal proteins] compared to healthy-donor blood, indicating that NETosis may be implicated in the genesis and/or amplification of the disease (196, 197). Therefore, like uncleaned apoptotic and necrotic cell remnants, uncleaned NETs may contribute to inflammation and autoimmunity.
Another neutrophil-related mechanism that is worth mentioning here is the release of S100A8 and S100A9 proteins and their calprotectin heterocomplex, upon stimulation. These proteins have been shown to have dual biological functions on inflammation (198–201). Abundant in neutrophils, calprotectin is released at sites of infection where it exerts antimicrobial activity, which is attributed to its ability to chelate manganese and zinc (200–205). In addition, calprotectin activates the innate immune system through activation of the receptor of advanced glycation end products (RAGE) and TLR4, resulting in downstream NF-κB activation and secretion of proinflammatory cytokines, such as TNF-α and IL-17 (206–208). Diverging properties of calprotectin related to PMN recruitment and functions have been described. Calprotectin was shown to activate the recruitment of PMNs and stimulate their adhesion by activating MAC-1 β2 integrin (209). Moreover, the functional blockage of calprotectin reduced PMN recruitment stimulated by LPS in vivo (210). Conversely, studies have pointed to the ability of S100A8 and S100A9 to repel PMNs (fugetaxis) and inhibit their chemotaxis toward chemokines in vitro. Additionally, calprotectin inhibited LPS-induced recruitment of PMNs in the rat air-pouch model of inflammation in vivo (211, 212). S100A9 differentially modified the responsiveness of neutrophils and dendritic cells to LPS, suggesting that the effects of calprotectin may be cell specific. While S100A9-deficient neutrophils exhibited a reduced secretion of cytokines (e.g., TNF-α and MCP-1) in response to LPS stimulation, inflammatory cytokine production in dendritic cells was exacerbated by S100A9 deficiency (213). Circulating concentrations of calprotectin increase with acute inflammation and during sepsis (214, 215), which has led some authors to suggest a proinflammatory role for this protein (216). Supporting this notion, Pepper and coworkers (217) showed that calprotectin plays a critical role during glomerulonephritis, amplifying autocrine and paracrine proinflammatory effects on BMDMs, renal endothelial cells, and mesangial cells. Indeed, calprotectin have an established clinical role as a biomarker in IBD (218).
In contradiction to these findings, anti-inflammatory, antinociceptive, and protective properties of calprotectin have also been described. In addition, regulation of S100A8 by GCs reinforces the idea of an anti-inflammatory role for this protein (219). For instance, calprotectin was suggested to be involved in the regulation of inflammatory processes in joints, since it produced marked anti-inflammatory and protective effects in models of adjuvant-induced arthritis in rats (220). Indeed, calprotectin deficiency was found in wound fluid from patients with non-healing venous leg ulcers, when compared with that from patients with healing open-granulating acute wounds (221). Sun and colleagues proposed protective and anti-inflammatory functions for calprotectin in sepsis. The authors showed that mice treated with S100A8 increased their survival rates and reduced tissue damage, inflammation, and oxidative injuries to major organ systems in a model of LPS-induced endotoxemia (222). Calprotectin was also shown to inhibit the oxidative metabolism of LPS-activated PMNs in vitro, which could contribute to reduce the oxidative organ injury seen in sepsis (223–225). Calprotectin suppressed NF-κB expression, proinflammatory cytokines, and inflammation in experimental autoimmune myocarditis (226), while the loss of calprotectin exacerbated T-cell activation and cardiac allograft rejection (227). In opposition, calprotectin aggravated post-ischemic heart failure through activation of RAGE-dependent NF-κB signaling (228). The diverging biological functions reported for calprotectin and its subunits suggest that their effects might be concentration dependent and influenced by the cellular and biochemical composition of the local milieu (229).
A novel and intriguing pro-resolving mechanism centered on neutrophils involves the generation of membrane borne microvesicles, also called microparticles or ectosomes (21). In 2004, Gasser and Schifferli (230) found that these microvesicles blocked the inflammatory response of macrophages exposed to zymosan and LPS. Further studies on neutrophil microparticles revealed that these microstructures could carry a variety of anti-inflammatory and pro-revolving mediators, enabling important modulatory functions in inflammation. Dalli and colleagues (231) defined the proteomic content of neutrophil microparticles. These authors observed that neutrophils have the ability to respond to a specific stimulus by producing microparticles loaded with a distinct proteomic profile, supporting the notion that microparticles production is a regulated process and might be endowed with very discrete functions (231). Some proteins, such as alpha-2-microglobulins, were identified to be selectively confined in vesicles generated from neutrophils adhered to an endothelial monolayer, whereas AnxA1 was more enriched in vesicles from exudate neutrophils. AnxA1 +ve vesicles possess anti-inflammatory properties (232) and allow the proper externalization of this pro-resolving mediator to gain access to extracellular surface receptors (i.e., FPR2) and to exert anti-inflammatory effects (39). AnxA1 acts as an exquisite brake for neutrophil adhesion to the microvascular wall, preventing over-exuberant cell transmigration to the inflammatory site (21, 233–235). We recently identified new properties for AnxA1 +ve vesicles, specifically those abundant in human synovial fluids collected from patients suffering from rheumatoid arthritis: these vesicles ensure the delivery of AnxA1 (and presumably other factors) to the chondrocyte in deep cartilage, enabling the activation of reparative circuits (236). In a recent review, we discussed the newly discovered modulatory roles of AnxA1 on neutrophil recruitment and other features of the resolution of inflammation (237).
Distinct Macrophage Populations Mediate Acute Inflammation and Resolution Phases of Inflammation
Macrophages are one of the first cells to sense injury, infection, and other types of noxious conditions, triggering the immune response through the production of proinflammatory mediators (1). During resolution, macrophages play an anti-inflammatory role and are required for the clearance of apoptotic cells. Following efferocytosis, macrophages undergo a functional repolarization, switching from a pro- to an anti-inflammatory phenotype (238). Accordingly, efferocytosis is coupled with increased release of TGF-β and IL-10 and lower levels of proinflammatory cytokines, such as IL-6 (238–240). This change in the phenotype of macrophages also activates pro-resolving mechanisms, because they generate LXA4, which stimulates phagocytic activities without releasing proinflammatory mediators. This is an important non-phlogistic process typical of resolution, and shared, for instance, by GCs (241).
In addition to participating in the lipid mediator class switching discussed above, PGE2 is also important in macrophage reprogramming, mediating the transition from the acute to the resolution phase of inflammation. Early data from Kunkel’s group showed a suppressive effect of PGE2 on macrophage TNF-α and IL-1β production (242, 243), and this has been confirmed by other investigators (238). This inhibitory feature allows proinflammatory cytokines to regulate their own production using PGE2 as a self-induced modulator (242). Recently, MacKenzie and colleagues (244) reported that the addition of PGE2 to LPS-stimulated macrophages represses proinflammatory cytokine production but induces IL-10. In particular, PGE2 displayed a biphasic effect on IL-6 transcription: at early time points, this eicosanoid promoted IL-6 transcription but at later time points, it repressed the induction of IL-6 mRNA (244). Another study showed that PGE2 from activated bone marrow stromal cells promotes IL-10 in LPS-stimulated macrophages, an effect mediated by prostaglandin EP2 and EP4 receptors (245). PGE2 in combination with LPS was able to induce the mRNA for Arginase 1, LIGHT (TNFSF14), and SPHK1, potential markers of alternatively activated and regulatory macrophages (245), again suggesting long-lasting roles for this prostaglandin in macrophage reprogramming.
PGE2 has also been implicated in tissue maintenance and regeneration. This is supported by reports that indicate that increased levels of PGE2 were associated with increased regenerative capacity. In this regard, Zhang and colleagues showed that the inhibition of 15-hydroxyprostaglandin dehydrogenase (15-PGDH), a prostaglandin-degrading enzyme, potentiates tissue regeneration in multiple organs in mice. In a model of DSS-induced colitis, PGE2 elevation diminished colon ulcers, suppressed mucosal inflammation, and reduced colitis symptoms, in conjunction with increased cell proliferation in the DSS-damaged mucosa. Interestingly, the pharmacological induction of higher levels of PGE2 was associated with markedly increased rate and extent of liver regeneration in mice after partial hepatectomy as compared to control groups (246). In the lung, PGE2 is the major eicosanoid produced by fibroblasts, alveolar macrophages, and other lung cells, playing important roles in tissue repair processes and in immune-inflammatory response limitation (247). PGE2 directly inhibits several major pathobiologic functions of lung fibroblasts and myofibroblasts, including proliferation, migration, collagen secretion, and myofibroblast differentiation [reviewed in Ref. (248)]. Of note, diminished PGE2 production and/or signaling can be observed in human and animal lung fibrosis, reinforcing its relevance for proper resolution (249, 250).
What the Future Reserves for Resolution
Undoubtedly, the inflammatory system is greatly complex. The history of the discovery of proinflammatory mediators reminds us that several decades of research were required to define the biology and pharmacology of the currently known mediators of inflammation. Since Sir Henry Dale and Patrick Laidlaw described some physiological effects of histamine in vivo, in 1910, immunological research has tremendously advanced (251). Pharmacological research has accompanied this progress, as historically represented by the discovery of antihistamines by Daniel Bovet and the identification of anti-H2R antagonists by Sir James Black, both awarded with the Nobel Prize in Physiology and Medicine (251). Subsequently, we made progress in the immunological and pharmacological fields of research, appreciating and shaping the concept of resolution of inflammation, and the mechanisms underpinning it. Fundamental concept here is the acceptance that resolution of inflammation is an active process evoked by specific classes of pro-resolving mediators, which differ from classical “anti-inflammatories” due to their ability to stimulate selective molecular and cellular programs of resolution. In the last decade, it has become evident that the enormous complexity of the proinflammatory system is mirrored at the level of pro-resolution pathways. Despite these remarkable advancements in the field, it seems that we have just started to scratch the surface of resolution mediators and other new cellular players are likely to be identified and defined in the near future. Likewise, we need to identify the major triggering pathway of these pro-resolving events, a phenomenon likely to be tissue- and/or disease-specific, as well as appreciate the complex networks among pro-resolving mediators. Such knowledge would be instrumental in developing pro-resolution based strategies to treat complex chronic inflammatory diseases in man, thus establishing a new area of pharmacology to be referred to as “resolution pharmacology” (22).
Author Contributions
MS, LS, VP, MP, and MT conceived and wrote the manuscript, and realized the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer VP and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors would like to acknowledge the funding agencies, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Comissão de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES, Brazil), Fundação do Amparo a Pesquisa de Minas Gerais (FAPEMIG, Brazil), Instituto Nacional de Ciência e Tecnologia (INCT in Dengue), and the European Community’s Seventh Framework Programme (FP7-2007-2013, Timer consortium) under grant agreement HEALTH-F4-2011-281608. MP acknowledges funding from the Wellcome Trust (program 086867/Z/08), the Medical Research Council UK (MR/K013068/1), and the William Harvey Research Foundation. The authors apologize to their colleagues whose original contributions are not included in the list of references due to space limitations.
References
1. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140(6):771–6. doi: 10.1016/j.cell.2010.03.006
2. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140(6):871–82. doi:10.1016/j.cell.2010.02.029
3. Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J (2007) 21(2):325–32. doi:10.1096/fj.06-7227rev
4. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci (2015) 16(6):358–72. doi:10.1038/nrn3880
5. Viola J, Soehnlein O. Atherosclerosis – a matter of unresolved inflammation. Semin Immunol (2015) 27(3):184–93. doi:10.1016/j.smim.2015.03.013
6. Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet (2004) 36(3):233–9. doi:10.1038/ng1311
7. Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. C-reactive protein and the risk of incident colorectal cancer. JAMA (2004) 291(5):585–90. doi:10.1001/jama.291.5.585
8. Lawrence T, Gilroy DW. Chronic inflammation: a failure of resolution? Int J Exp Pathol (2007) 88(2):85–94. doi:10.1111/j.1365-2613.2006.00507.x
9. Opie EL. Experimental pleurisy-resolution of a fibrinous exudate. J Exp Med (1907) 9(4):391–413. doi:10.1084/jem.9.4.391
10. Perretti M. Endogenous mediators that inhibit the leukocyte-endothelium interaction. Trends Pharmacol Sci (1997) 18(11):418–25. doi:10.1016/S0165-6147(97)01116-4
11. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med (2000) 192(8):1197–204. doi:10.1084/jem.192.8.1197
12. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol (2001) 2(7):612–9. doi:10.1038/89759
13. Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem (2003) 278(17):14677–87. doi:10.1074/jbc.M300218200
14. Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol (2015) 7(2):a016311. doi:10.1101/cshperspect.a016311
15. Stables MJ, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, et al. Transcriptomic analyses of murine resolution-phase macrophages. Blood (2011) 118(26):e192–208. doi:10.1182/blood-2011-04-345330
16. Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med (2013) 5(5):661–74. doi:10.1002/emmm.201202382
17. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40(3):315–27. doi:10.1016/j.immuni.2014.02.009
18. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6(12):1191–7. doi:10.1038/ni1276
19. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6(3):173–82. doi:10.1038/nri1785
20. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8(5):349–61. doi:10.1038/nri2294
21. Headland SE, Norling LV. The resolution of inflammation: principles and challenges. Semin Immunol (2015) 27:149–60. doi:10.1016/j.smim.2015.03.014
22. Perretti M, Leroy X, Bland EJ, Montero-Melendez T. Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol Sci (2015) 36(11):737–55. doi:10.1016/j.tips.2015.07.007
23. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510(7503):92–101. doi:10.1038/nature13479
24. Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol (2009) 9(1):62–70. doi:10.1038/nri2470
25. Wallace JL, Ianaro A, Flannigan KL, Cirino G. Gaseous mediators in resolution of inflammation. Semin Immunol (2015) 27(3):227–33. doi:10.1016/j.smim.2015.05.004
26. Hasko G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol (2013) 4:85. doi:10.3389/fimmu.2013.00085
27. Koroskenyi K, Duro E, Pallai A, Sarang Z, Kloor D, Ucker DS, et al. Involvement of adenosine A2A receptors in engulfment-dependent apoptotic cell suppression of inflammation. J Immunol (2011) 186(12):7144–55. doi:10.4049/jimmunol.1002284
28. Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26(1):376–86. doi:10.1096/fj.11-190934
29. Mirakaj V, Dalli J, Granja T, Rosenberger P, Serhan CN. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J Exp Med (2014) 211(6):1037–48. doi:10.1084/jem.20132103
30. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex – linking immunity and metabolism. Nat Rev Endocrinol (2012) 8(12):743–54. doi:10.1038/nrendo.2012.189
31. Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med (2011) 365(18):1713–25. doi:10.1056/NEJMra1102942
32. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet (2012) 380(9853):1590–605. doi:10.1016/s0140-6736(12)60026-9
33. Mangino MJ, Brounts L, Harms B, Heise C. Lipoxin biosynthesis in inflammatory bowel disease. Prostaglandins Other Lipid Mediat (2006) 79(1–2):84–92. doi:10.1016/j.prostaglandins.2005.10.004
34. Sena A, Grishina I, Thai A, Goulart L, Macal M, Fenton A, et al. Dysregulation of anti-inflammatory annexin A1 expression in progressive Crohns disease. PLoS One (2013) 8(10):e76969. doi:10.1371/journal.pone.0076969
35. Vong L, Ferraz JG, Dufton N, Panaccione R, Beck PL, Sherman PM, et al. Up-regulation of Annexin-A1 and lipoxin A(4) in individuals with ulcerative colitis may promote mucosal homeostasis. PLoS One (2012) 7(6):e39244. doi:10.1371/journal.pone.0039244
36. Sena AA, Pedrotti LP, Barrios BE, Cejas H, Balderramo D, Diller A, et al. Lack of TNFRI signaling enhances annexin A1 biological activity in intestinal inflammation. Biochem Pharmacol (2015) 98(3):422–31. doi:10.1016/j.bcp.2015.09.009
37. Fiorucci S, Wallace JL, Mencarelli A, Distrutti E, Rizzo G, Farneti S, et al. A beta-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proc Natl Acad Sci U S A (2004) 101(44):15736–41. doi:10.1073/pnas.0404722101
38. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U S A (2005) 102(21):7671–6. doi:10.1073/pnas.0409271102
39. Leoni G, Neumann PA, Kamaly N, Quiros M, Nishio H, Jones HR, et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest (2015) 125(3):1215–27. doi:10.1172/JCI76693
40. Haworth O, Levy BD. Lipoxins, resolvins and protectins: new leads for the treatment of asthma. Expert Opin Drug Discov (2008) 3(10):1209–22. doi:10.1517/17460441.3.10.1209
41. Planaguma A, Levy BD. Uncontrolled airway inflammation in lung disease represents a defect in counter-regulatory signaling. Future Lipidol (2008) 3(6):697–704. doi:10.2217/17460875.3.6.697
42. Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E, et al. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med (2005) 172(7):824–30. doi:10.1164/rccm.200410-1413OC
43. Celik GE, Erkekol FO, Misirligil Z, Melli M. Lipoxin A4 levels in asthma: relation with disease severity and aspirin sensitivity. Clin Exp Allergy (2007) 37(10):1494–501. doi:10.1111/j.1365-2222.2007.02806.x
44. Planagumà A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med (2008) 178(6):574–82. doi:10.1164/rccm.200801-061OC
45. Vachier I, Bonnans C, Chavis C, Farce M, Godard P, Bousquet J, et al. Severe asthma is associated with a loss of LX4, an endogenous anti-inflammatory compound. J Allergy Clin Immunol (2005) 115(1):55–60. doi:10.1016/j.jaci.2004.09.038
46. Wu SH, Yin PL, Zhang YM, Tao HX. Reversed changes of lipoxin A4 and leukotrienes in children with asthma in different severity degree. Pediatr Pulmonol (2010) 45(4):333–40. doi:10.1002/ppul.21186
47. Eke Gungor H, Tahan F, Gokahmetoglu S, Saraymen B. Decreased levels of lipoxin A4 and annexin A1 in wheezy infants. Int Arch Allergy Immunol (2014) 163(3):193–7. doi:10.1159/000358490
48. Bhatt JM, Smyth AR. The management of pre-school wheeze. Paediatr Respir Rev (2011) 12(1):70–7. doi:10.1016/j.prrv.2010.09.001
49. Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell (2008) 134(3):392–404. doi:10.1016/j.cell.2008.07.025
50. Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol (1998) 16:137–61. doi:10.1146/annurev.immunol.16.1.137
51. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest (2001) 108(4):601–9. doi:10.1172/JCI12821
52. Monteleone G, Neurath MF, Ardizzone S, Di Sabatino A, Fantini MC, Castiglione F, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N Engl J Med (2015) 372(12):1104–13. doi:10.1056/NEJMoa1407250
53. Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, et al. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol (2007) 179(12):8533–43. doi:10.4049/jimmunol.179.12.8533
54. Brancaleone V, Dalli J, Bena S, Flower RJ, Cirino G, Perretti M. Evidence for an anti-inflammatory loop centered on polymorphonuclear leukocyte formyl peptide receptor 2/lipoxin A4 receptor and operative in the inflamed microvasculature. J Immunol (2011) 186(8):4905–14. doi:10.4049/jimmunol.1003145
55. Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol (2008) 9(8):873–9. doi:10.1038/ni.1627
56. Vago JP, Tavares LP, Garcia CC, Lima KM, Perucci LO, Vieira EL, et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J Immunol (2015) 194(10):4940–50. doi:10.4049/jimmunol.1401722
57. Ronchetti S, Migliorati G, Riccardi C. GILZ as a mediator of the anti-inflammatory effects of glucocorticoids. Front Endocrinol (2015) 6:170. doi:10.3389/fendo.2015.00170
58. Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, Teixeira MM. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther (2013) 139(2):189–212. doi:10.1016/j.pharmthera.2013.04.006
59. Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol (1994) 12:141–79. doi:10.1146/annurev.iy.12.040194.001041
60. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol (2004) 25(6):280–8. doi:10.1016/j.it.2004.03.008
61. Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov (2004) 3(1):17–26. doi:10.1038/nrd1279
62. Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med (2001) 7(12):1291–7. doi:10.1038/nm1201-1291
63. Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell (2007) 130(5):918–31. doi:10.1016/j.cell.2007.07.009
64. Lawrence T, Fong C. The resolution of inflammation: anti-inflammatory roles for NF-kappaB. Int J Biochem Cell Biol (2010) 42(4):519–23. doi:10.1016/j.biocel.2009.12.016
65. Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J (2003) 22(20):5530–9. doi:10.1093/emboj/cdg534
66. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol (2009) 1(4):a000034. doi:10.1101/cshperspect.a000034
67. Huxford T, Malek S, Ghosh G. Structure and mechanism in NF-kappa B/I kappa B signaling. Cold Spring Harb Symp Quant Biol (1999) 64:533–40. doi:10.1101/sqb.1999.64.533
68. Wang VY, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, et al. The transcriptional specificity of NF-kappaB dimers is coded within the kappaB DNA response elements. Cell Rep (2012) 2(4):824–39. doi:10.1016/j.celrep.2012.08.042
69. Muxel SM, Laranjeira-Silva MF, de Carvalho Sousa CE, Floeter-Winter LM, Markus RP. The RelA/cRel nuclear factor-kappaB (NF-kappaB) dimer, crucial for inflammation resolution, mediates the transcription of the key enzyme in melatonin synthesis in RAW 264.7 macrophages. J Pineal Res (2016). 60(4):394–404. doi:10.1111/jpi.12321
70. Woo JI, Kil SH, Pan H, Lee YJ, Lim DJ, Moon SK. Distal NF-kB binding motif functions as an enhancer for nontypeable H. influenzae-induced DEFB4 regulation in epithelial cells. Biochem Biophys Res Commun (2014) 443(3):1035–40. doi:10.1016/j.bbrc.2013.12.091
71. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi:10.1038/ni.1937
72. Bohuslav J, Kravchenko VV, Parry GC, Erlich JH, Gerondakis S, Mackman N, et al. Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J Clin Invest (1998) 102(9):1645–52. doi:10.1172/JCI3877
73. Ma XY, Wang H, Ding B, Zhong H, Ghosh S, Lengyel P. The interferon-inducible p202a protein modulates NF-kappaB activity by inhibiting the binding to DNA of p50/p65 heterodimers and p65 homodimers while enhancing the binding of p50 homodimers. J Biol Chem (2003) 278(25):23008–19. doi:10.1074/jbc.M302105200
74. Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res (2006) 66(23):11432–40. doi:10.1158/0008-5472.CAN-06-1867
75. Ziegler-Heitbrock L. The p50-homodimer mechanism in tolerance to LPS. J Endotoxin Res (2001) 7(3):219–22. doi:10.1179/096805101101532701
76. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A (2009) 106(35):14978–83. doi:10.1073/pnas.0809784106
77. Lee HN, Kundu JK, Cha YN, Surh YJ. Resolvin D1 stimulates efferocytosis through p50/p50-mediated suppression of tumor necrosis factor-alpha expression. J Cell Sci (2013) 126(Pt 17):4037–47. doi:10.1242/jcs.131003
78. Aparicio-Soto M, Alarcon-de-la-Lastra C, Cardeno A, Sanchez-Fidalgo S, Sanchez-Hidalgo M. Melatonin modulates microsomal PGE synthase 1 and NF-E2-related factor-2-regulated antioxidant enzyme expression in LPS-induced murine peritoneal macrophages. Br J Pharmacol (2014) 171(1):134–44. doi:10.1111/bph.12428
79. Lardone PJ, Carrillo-Vico A, Naranjo MC, De Felipe B, Vallejo A, Karasek M, et al. Melatonin synthesized by Jurkat human leukemic T cell line is implicated in IL-2 production. J Cell Physiol (2006) 206(1):273–9. doi:10.1002/jcp.20461
80. Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, et al. The IKK complex contributes to the induction of autophagy. EMBO J (2010) 29(3):619–31. doi:10.1038/emboj.2009.364
81. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi:10.1038/nature09663
82. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi:10.1038/ni.1980
83. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456(7219):264–8. doi:10.1038/nature07383
84. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164(5):896–910. doi:10.1016/j.cell.2015.12.057
85. Chian CF, Chiang CH, Chuang CH, Liu SL. Inhibitor of nuclear factor-kappaB, SN50, attenuates lipopolysaccharide-induced lung injury in an isolated and perfused rat lung model. Transl Res (2014) 163(3):211–20. doi:10.1016/j.trsl.2013.10.002
86. Sun YX, Dai DK, Liu R, Wang T, Luo CL, Bao HJ, et al. Therapeutic effect of SN50, an inhibitor of nuclear factor-kappaB, in treatment of TBI in mice. Neurol Sci (2013) 34(3):345–55. doi:10.1007/s10072-012-1007-z
87. El-Salhy M, Umezawa K, Gilja OH, Hatlebakk JG, Gundersen D, Hausken T. Amelioration of severe TNBS induced colitis by novel AP-1 and NF-kappa B inhibitors in rats. ScientificWorldJournal (2014) 2014:813804. doi:10.1155/2014/813804
88. Hosokawa S, Haraguchi G, Sasaki A, Arai H, Muto S, Itai A, et al. Pathophysiological roles of nuclear factor kappaB (NF-kB) in pulmonary arterial hypertension: effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovasc Res (2013) 99(1):35–43. doi:10.1093/cvr/cvt105
89. Sousa LP, Carmo AF, Rezende BM, Lopes F, Silva DM, Alessandri AL, et al. Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-kappaB. Biochem Pharmacol (2009) 78(4):396–405. doi:10.1016/j.bcp.2009.04.030
90. Lopes F, Coelho FM, Costa VV, Vieira EL, Sousa LP, Silva TA, et al. Resolution of neutrophilic inflammation by H2O2 in antigen-induced arthritis. Arthritis Rheum (2011) 63(9):2651–60. doi:10.1002/art.30448
91. Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, et al. An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J Exp Med (2008) 205(6):1269–76. doi:10.1084/jem.20080124
92. Hsu LC, Enzler T, Seita J, Timmer AM, Lee CY, Lai TY, et al. IL-1beta-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKbeta. Nat Immunol (2011) 12(2):144–50. doi:10.1038/ni.1976
93. Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol (2010) 10(1):24–35. doi:10.1038/nri2685
94. Anderson P. Post-transcriptional control of cytokine production. Nat Immunol (2008) 9(4):353–9. doi:10.1038/ni1584
95. Mazumder B, Li X, Barik S. Translation control: a multifaceted regulator of inflammatory response. J Immunol (2010) 184(7):3311–9. doi:10.4049/jimmunol.0903778
96. Boldin MP, Baltimore D. MicroRNAs, new effectors and regulators of NF-kappaB. Immunol Rev (2012) 246(1):205–20. doi:10.1111/j.1600-065X.2011.01089.x
97. Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol (2011) 3(3):159–66. doi:10.1093/jmcb/mjr007
98. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103(33):12481–6. doi:10.1073/pnas.0605298103
99. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol (2010) 10(2):111–22. doi:10.1038/nri2708
100. Etzrodt M, Cortez-Retamozo V, Newton A, Zhao J, Ng A, Wildgruber M, et al. Regulation of monocyte functional heterogeneity by miR-146a and Relb. Cell Rep (2012) 1(4):317–24. doi:10.1016/j.celrep.2012.02.009
101. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
102. El Gazzar M, Church A, Liu T, McCall CE. MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J Leukoc Biol (2011) 90(3):509–19. doi:10.1189/jlb.0211074
103. Brudecki L, Ferguson DA, McCall CE, El Gazzar M. MicroRNA-146a and RBM4 form a negative feed-forward loop that disrupts cytokine mRNA translation following TLR4 responses in human THP-1 monocytes. Immunol Cell Biol (2013) 91(8):532–40. doi:10.1038/icb.2013.37
104. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med (2011) 208(6):1189–201. doi:10.1084/jem.20101823
105. Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol (2008) 180(8):5689–98. doi:10.4049/jimmunol.180.8.5689
106. Jurkin J, Schichl YM, Koeffel R, Bauer T, Richter S, Konradi S, et al. miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation. J Immunol (2010) 184(9):4955–65. doi:10.4049/jimmunol.0903021
107. Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, et al. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J (2011) 30(1):57–67. doi:10.1038/emboj.2010.296
108. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature (2008) 451(7182):1125–9. doi:10.1038/nature06607
109. Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A (2009) 106(13):5282–7. doi:10.1073/pnas.0810909106
110. Cheng Y, Kuang W, Hao Y, Zhang D, Lei M, Du L, et al. Downregulation of miR-27a* and miR-532-5p and upregulation of miR-146a and miR-155 in LPS-induced RAW264.7 macrophage cells. Inflammation (2012) 35(4):1308–13. doi:10.1007/s10753-012-9443-8
111. Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A (2009) 106(37):15819–24. doi:10.1073/pnas.0901216106
112. Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity (2009) 31(6):965–73. doi:10.1016/j.immuni.2009.09.019
113. Lagos D, Pollara G, Henderson S, Gratrix F, Fabani M, Milne RS, et al. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat Cell Biol (2010) 12(5):513–9. doi:10.1038/ncb2054
114. Coskun M, Bjerrum JT, Seidelin JB, Nielsen OH. MicroRNAs in inflammatory bowel disease – pathogenesis, diagnostics and therapeutics. World J Gastroenterol (2012) 18(34):4629–34. doi:10.3748/wjg.v18.i34.4629
115. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol (2008) 9(4):405–14. doi:10.1038/ni1575
116. Besedovsky H, del Rey A, Sorkin E, Dinarello C. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science (1986) 233(4764):652–4. doi:10.1126/science.3014662
117. Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum (2002) 46(10):2765–75. doi:10.1002/art.10526
118. Montero-Melendez T. ACTH: the forgotten therapy. Semin Immunol (2015) 27(3):216–26. doi:10.1016/j.smim.2015.02.003
119. Montero-Melendez T, Patel HB, Seed M, Nielsen S, Jonassen TEN, Perretti M. The melanocortin agonist AP214 exerts anti-inflammatory and proresolving properties. Am J Pathol (2011) 179(1):259–69. doi:10.1016/j.ajpath.2011.03.042
120. Montero-Melendez T, Gobbetti T, Cooray SN, Jonassen TEN, Perretti M. Biased agonism as a novel strategy to harness the proresolving properties of melanocortin receptors without eliciting melanogenic effects. J Immunol (2015) 194(7):3381–8. doi:10.4049/jimmunol.1402645
121. Vacchini A, Locati M, Borroni EM. Overview and potential unifying themes of the atypical chemokine receptor family. J Leukoc Biol (2016). doi:10.1189/jlb.2MR1015-477R
122. Nibbs RJ, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol (2013) 13(11):815–29. doi:10.1038/nri3544
123. Dean RA, Overall CM. Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol Cell Proteomics (2007) 6(4):611–23. doi:10.1074/mcp.M600341-MCP200
124. Tam EM, Morrison CJ, Wu YI, Stack MS, Overall CM. Membrane protease proteomics: isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc Natl Acad Sci U S A (2004) 101(18):6917–22. doi:10.1073/pnas.0305862101
125. Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol (2004) 4(8):617–29. doi:10.1038/nri1418
126. McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase a cleavage of monocyte chemoattractant protein-3. Science (2000) 289(5482):1202–6. doi:10.1126/science.289.5482.1202
127. McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood (2002) 100(4):1160–7.
128. McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem (2001) 276(47):43503–8. doi:10.1074/jbc.M107736200
129. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood (2000) 96(8):2673–81.
130. Moelants EA, Mortier A, Van Damme J, Proost P. In vivo regulation of chemokine activity by post-translational modification. Immunol Cell Biol (2013) 91(6):402–7. doi:10.1038/icb.2013.16
131. Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature (1994) 370(6490):555–7. doi:10.1038/370555a0
132. Wilson CL. Regulation of intestinal – defensin activation by the metalloproteinase matrilysin in innate host defense. Science (1999) 286(5437):113–7. doi:10.1126/science.286.5437.113
133. Butler GS, Sim D, Tam E, Devine D, Overall CM. Mannose-binding lectin (MBL) mutants are susceptible to matrix metalloproteinase proteolysis: potential role in human MBL deficiency. J Biol Chem (2002) 277(20):17511–9. doi:10.1074/jbc.M201461200
134. Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood (2008) 112(8):3455–64. doi:10.1182/blood-2007-12-129080
135. Souza DG, Guabiraba R, Pinho V, Bristow A, Poole S, Teixeira MM. IL-1-Driven endogenous IL-10 production protects against the systemic and local acute inflammatory response following intestinal reperfusion injury. J Immunol (2003) 170(9):4759–66. doi:10.4049/jimmunol.170.9.4759
136. Burne MJ, Elghandour A, Haq M, Saba SR, Norman J, Condon T, et al. IL-1 and TNF independent pathways mediate ICAM-1/VCAM-1 up-regulation in ischemia reperfusion injury. J Leukoc Biol (2001) 70(2):192–8.
137. Haq M, Norman J, Saba SR, Ramirez G, Rabb H. Role of IL-1 in renal ischemic reperfusion injury. J Am Soc Nephrol (1998) 9(4):614–9.
138. Fu LW, Longhurst JC. Interleukin-1beta sensitizes abdominal visceral afferents of cats to ischaemia and histamine. J Physiol (1999) 521(Pt 1):249–60. doi:10.1111/j.1469-7793.1999.00249.x
139. Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine (2012) 59(3):451–9. doi:10.1016/j.cyto.2012.06.014
140. Takagi H, Kanai T, Okazawa A, Kishi Y, Sato T, Takaishi H, et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand J Gastroenterol (2003) 38(8):837–44. doi:10.1080/00365520310004047
141. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32(3):379–91. doi:10.1016/j.immuni.2010.03.003
142. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity (2010) 32(3):367–78. doi:10.1016/j.immuni.2010.02.012
143. Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, et al. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut (2014) 63(4):598–609. doi:10.1136/gutjnl-2012-303329
144. Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, et al. Cutting edge: IL-36 receptor promotes resolution of intestinal damage. J Immunol (2016) 196(1):34–8. doi:10.4049/jimmunol.1501312
145. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet (2009) 41(1):71–6. doi:10.1038/ng.285
146. Souza DG, Soares AC, Pinho V, Torloni H, Reis LF, Teixeira MM, et al. Increased mortality and inflammation in tumor necrosis factor-stimulated gene-14 transgenic mice after ischemia and reperfusion injury. Am J Pathol (2002) 160(5):1755–65. doi:10.1016/S0002-9440(10)61122-4
147. Souza DG, Cassali GD, Poole S, Teixeira MM. Effects of inhibition of PDE4 and TNF-alpha on local and remote injuries following ischaemia and reperfusion injury. Br J Pharmacol (2001) 134(5):985–94. doi:10.1038/sj.bjp.0704336
148. Gobbetti T, Coldewey SM, Chen J, McArthur S, le Faouder P, Cenac N, et al. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc Natl Acad Sci U S A (2014) 111(52):18685–90. doi:10.1073/pnas.1410938111
149. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi:10.1146/annurev-immunol-031210-101312
150. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10(3):170–81. doi:10.1038/nri2711
151. Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, et al. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol (2009) 183(2):1197–206. doi:10.4049/jimmunol.0803883
152. Samuelsson B. Role of basic science in the development of new medicines: examples from the eicosanoid field. J Biol Chem (2012) 287(13):10070–80. doi:10.1074/jbc.X112.351437
153. Malawista SE, de Boisfleury Chevance A, van Damme J, Serhan CN. Tonic inhibition of chemotaxis in human plasma. Proc Natl Acad Sci U S A (2008) 105(46):17949–54. doi:10.1073/pnas.0802572105
154. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(7203):428–35. doi:10.1038/nature07201
155. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med (1999) 5(6):698–701. doi:10.1038/9550
156. Surh YJ, Na HK, Park JM, Lee HN, Kim W, Yoon IS, et al. 15-Deoxy-delta(1)(2),(1)(4)-prostaglandin J(2), an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem Pharmacol (2011) 82(10):1335–51. doi:10.1016/j.bcp.2011.07.100
157. Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci (2008) 33(3):141–50. doi:10.1016/j.tibs.2007.12.003
158. Kim WJ, Kim JH, Jang SK. Anti-inflammatory lipid mediator 15d-PGJ2 inhibits translation through inactivation of eIF4A. EMBO J (2007) 26(24):5020–32. doi:10.1038/sj.emboj.7601920
159. Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem (2002) 277(12):10459–66. doi:10.1074/jbc.M110314200
160. Harrington MG, Aebersold R, Martin BM, Merril CR, Hood L. Identification of a brain-specific human cerebrospinal fluid glycoprotein, beta-trace protein. Appl Theor Electrophor (1993) 3(5):229–34.
161. Cipollone F, Fazia M, Iezzi A, Ciabattoni G, Pini B, Cuccurullo C, et al. Balance between PGD synthase and PGE synthase is a major determinant of atherosclerotic plaque instability in humans. Arterioscler Thromb Vasc Biol (2004) 24(7):1259–65. doi:10.1161/01.ATV.0000133192.39901.be
162. Bandeira-Melo C, Serra MF, Diaz BL, Cordeiro RS, Silva PM, Lenzi HL, et al. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: relationship with concurrent eosinophilia. J Immunol (2000) 164(2):1029–36. doi:10.4049/jimmunol.164.2.1029
163. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature (2007) 447(7146):869–74. doi:10.1038/nature05877
164. Markworth JF, Vella L, Lingard BS, Tull DL, Rupasinghe TW, Sinclair AJ, et al. Human inflammatory and resolving lipid mediator responses to resistance exercise and ibuprofen treatment. Am J Physiol Regul Integr Comp Physiol (2013) 305(11):R1281–96. doi:10.1152/ajpregu.00128.2013
165. Chan MM, Moore AR. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J Immunol (2010) 184(11):6418–26. doi:10.4049/jimmunol.0903816
166. Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol (2005) 174(8):5033–9. doi:10.4049/jimmunol.174.8.5033
167. Vane JR. Adventures and excursions in bioassay: the stepping stones to prostacyclin. Br J Pharmacol (1983) 79(3):821–38. doi:10.1111/j.1476-5381.1983.tb10020.x
168. Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A (1995) 92(21):9475–9. doi:10.1073/pnas.92.21.9475
169. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins. J Exp Med (2002) 196(8):1025–37. doi:10.1084/jem.20020760
170. Serhan CN, Fredman G, Yang R, Karamnov S, Belayev LS, Bazan NG, et al. Novel proresolving aspirin-triggered DHA pathway. Chem Biol (2011) 18(8):976–87. doi:10.1016/j.chembiol.2011.06.008
171. Morris T, Stables M, Hobbs A, de Souza P, Colville-Nash P, Warner T, et al. Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol (2009) 183(3):2089–96. doi:10.4049/jimmunol.0900477
172. Brancaleone V, Gobbetti T, Cenac N, le Faouder P, Colom B, Flower RJ, et al. A vasculo-protective circuit centered on lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 operative in murine microcirculation. Blood (2013) 122(4):608–17. doi:10.1182/blood-2013-04-496661
173. Chiang N, Bermudez EA, Ridker PM, Hurwitz S, Serhan CN. Aspirin triggers antiinflammatory 15-epi-lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci U S A (2004) 101(42):15178–83. doi:10.1073/pnas.0405445101
174. Morris T, Stables M, Colville-Nash P, Newson J, Bellingan G, de Souza PM, et al. Dichotomy in duration and severity of acute inflammatory responses in humans arising from differentially expressed proresolution pathways. Proc Natl Acad Sci U S A (2010) 107(19):8842–7. doi:10.1073/pnas.1000373107
175. Pillai PS, Leeson S, Porter TF, Owens CD, Kim JM, Conte MS, et al. Chemical mediators of inflammation and resolution in post-operative abdominal aortic aneurysm patients. Inflammation (2012) 35(1):98–113. doi:10.1007/s10753-011-9294-8
176. Harty MW, Muratore CS, Papa EF, Gart MS, Ramm GA, Gregory SH, et al. Neutrophil depletion blocks early collagen degradation in repairing cholestatic rat livers. Am J Pathol (2010) 176(3):1271–81. doi:10.2353/ajpath.2010.090527
177. Kuhl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology (2007) 133(6):1882–92. doi:10.1053/j.gastro.2007.08.073
178. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40(1):66–77. doi:10.1016/j.immuni.2013.11.020
179. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med (2014) 20(5):511–7. doi:10.1038/nm.3547
180. Gresnigt MS, Joosten LA, Verschueren I, van der Meer JW, Netea MG, Dinarello CA, et al. Neutrophil-mediated inhibition of proinflammatory cytokine responses. J Immunol (2012) 189(10):4806–15. doi:10.4049/jimmunol.1103551
181. van der Meer JH, Netea MG, Dinarello CA. Modulation of muramyl dipeptide stimulation of cytokine production by blood components. Clin Exp Immunol (2009) 156(3):428–33. doi:10.1111/j.1365-2249.2009.03926.x
182. Branzk N, Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol (2013) 35(4):513–30. doi:10.1007/s00281-013-0384-6
183. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349(6245):316–20. doi:10.1126/science.aaa8064
184. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol (2013) 190(3):1217–26. doi:10.4049/jimmunol.1202388
185. Knight JS, Zhao W, Luo W, Subramanian V, O’Dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest (2013) 123(7):2981–93. doi:10.1172/JCI67390
186. Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood (2014) 123(1):141–8. doi:10.1182/blood-2013-07-514992
187. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A (2013) 110(21):8674–9. doi:10.1073/pnas.1301059110
188. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med (2015) 21(7):815–9. doi:10.1038/nm.3887
189. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62(2):600–14. doi:10.1002/hep.27841
190. Podolska MJ, Biermann MH, Maueroder C, Hahn J, Herrmann M. Inflammatory etiopathogenesis of systemic lupus erythematosus: an update. J Inflamm Res (2015) 8:161–71. doi:10.2147/JIR.S70325
191. Knight JS, Kaplan MJ. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol (2012) 24(5):441–50. doi:10.1097/BOR.0b013e3283546703
192. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107(21):9813–8. doi:10.1073/pnas.0909927107
193. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi:10.1126/scitranslmed.3001201
194. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi:10.1126/scitranslmed.3001180
195. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi:10.1038/nm.4027
196. Ma CY, Jiao YL, Zhang J, Yang QR, Zhang ZF, Shen YJ, et al. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatol Int (2012) 32(2):395–402. doi:10.1007/s00296-010-1636-6
197. Vordenbaumen S, Fischer-Betz R, Timm D, Sander O, Chehab G, Richter J, et al. Elevated levels of human beta-defensin 2 and human neutrophil peptides in systemic lupus erythematosus. Lupus (2010) 19(14):1648–53. doi:10.1177/0961203310377089
198. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol (2012) 10(8):525–37. doi:10.1038/nrmicro2836
199. Diaz-Ochoa VE, Jellbauer S, Klaus S, Raffatellu M. Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front Cell Infect Microbiol (2014) 4:2. doi:10.3389/fcimb.2014.00002
200. Sohnle PG, Collins-Lech C, Wiessner JH. The zinc-reversible antimicrobial activity of neutrophil lysates and abscess fluid supernatants. J Infect Dis (1991) 164(1):137–42. doi:10.1093/infdis/164.1.137
201. Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science (2008) 319(5865):962–5. doi:10.1126/science.1152449
202. Kehl-Fie TE, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, et al. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe (2011) 10(2):158–64. doi:10.1016/j.chom.2011.07.004
203. Hayden JA, Brophy MB, Cunden LS, Nolan EM. High-affinity manganese coordination by human calprotectin is calcium-dependent and requires the histidine-rich site formed at the dimer interface. J Am Chem Soc (2013) 135(2):775–87. doi:10.1021/ja3096416
204. Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc Natl Acad Sci U S A (2013) 110(10):3841–6. doi:10.1073/pnas.1220341110
205. Brophy MB, Nakashige TG, Gaillard A, Nolan EM. Contributions of the S100A9 C-terminal tail to high-affinity Mn(II) chelation by the host-defense protein human calprotectin. J Am Chem Soc (2013) 135(47):17804–17. doi:10.1021/ja407147d
206. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13(9):1042–9. doi:10.1038/nm1638
207. Riva M, Kallberg E, Bjork P, Hancz D, Vogl T, Roth J, et al. Induction of nuclear factor-kappaB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology (2012) 137(2):172–82. doi:10.1111/j.1365-2567.2012.03619.x
208. Loser K, Vogl T, Voskort M, Lueken A, Kupas V, Nacken W, et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med (2010) 16(6):713–7. doi:10.1038/nm.2150
209. Newton RA, Hogg N. The human S100 protein MRP-14 is a novel activator of the beta 2 integrin Mac-1 on neutrophils. J Immunol (1998) 160(3):1427–35.
210. Ryckman C, McColl SR, Vandal K, de Medicis R, Lussier A, Poubelle PE, et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum (2003) 48(8):2310–20. doi:10.1002/art.11079
211. Sroussi HY, Berline J, Dazin P, Green P, Palefsky JM. S100A8 triggers oxidation-sensitive repulsion of neutrophils. J Dent Res (2006) 85(9):829–33. doi:10.1177/154405910608500910
212. Sroussi HY, Berline J, Palefsky JM. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J Leukoc Biol (2007) 81(3):818–24. doi:10.1189/jlb.0706433
213. Averill MM, Barnhart S, Becker L, Li X, Heinecke JW, Leboeuf RC, et al. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: implications for atherosclerosis and adipose tissue inflammation. Circulation (2011) 123(11):1216–26. doi:10.1161/CIRCULATIONAHA.110.985523
214. Rau M, Schiller M, Krienke S, Heyder P, Lorenz H, Blank N. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J Rheumatol (2010) 37(11):2369–76. doi:10.3899/jrheum.100247
215. Terrin G, Passariello A, Manguso F, Salvia G, Rapacciuolo L, Messina F, et al. Serum calprotectin: an antimicrobial peptide as a new marker for the diagnosis of sepsis in very low birth weight newborns. Clin Dev Immunol (2011) 2011:291085. doi:10.1155/2011/291085
216. Youssef P, Roth J, Frosch M, Costello P, Fitzgerald O, Sorg C, et al. Expression of myeloid related proteins (MRP) 8 and 14 and the MRP8/14 heterodimer in rheumatoid arthritis synovial membrane. J Rheumatol (1999) 26(12):2523–8.
217. Pepper RJ, Wang HH, Rajakaruna GK, Papakrivopoulou E, Vogl T, Pusey CD, et al. S100A8/A9 (calprotectin) is critical for development of glomerulonephritis and promotes inflammatory leukocyte-renal cell interactions. Am J Pathol (2015) 185(5):1264–74. doi:10.1016/j.ajpath.2015.01.015
218. van den Bergh FA, Kolkman JJ, Russel MG, Vlaskamp RT, Vermes I. [Calprotectin: a fecal marker for diagnosis and follow-up in patients with chronic inflammatory bowel disease]. Ned Tijdschr Geneeskd (2003) 147(48):2360–5.
219. Hsu K, Passey RJ, Endoh Y, Rahimi F, Youssef P, Yen T, et al. Regulation of S100A8 by glucocorticoids. J Immunol (2005) 174(4):2318–26. doi:10.4049/jimmunol.174.4.2318
220. Brun JG, Haland G, Haga HJ, Fagerhol MK, Jonsson R. Effects of calprotectin in avridine-induced arthritis. APMIS (1995) 103(3):233–40. doi:10.1111/j.1699-0463.1995.tb01100.x
221. Trostrup H, Lundquist R, Christensen LH, Jorgensen LN, Karlsmark T, Haab BB, et al. S100A8/A9 deficiency in nonhealing venous leg ulcers uncovered by multiplexed antibody microarray profiling. Br J Dermatol (2011) 165(2):292–301. doi:10.1111/j.1365-2133.2011.10384.x
222. Sun Y, Lu Y, Engeland CG, Gordon SC, Sroussi HY. The anti-oxidative, anti-inflammatory, and protective effect of S100A8 in endotoxemic mice. Mol Immunol (2013) 53(4):443–9. doi:10.1016/j.molimm.2012.10.002
223. Schwartz R, Lu Y, Villines D, Sroussi HY. Effect of human immunodeficiency virus infection on S100A8/A9 inhibition of peripheral neutrophils oxidative metabolism. Biomed Pharmacother (2010) 64(8):572–5. doi:10.1016/j.biopha.2010.03.005
224. Sroussi HY, Lu Y, Zhang QL, Villines D, Marucha PT. S100A8 and S100A9 inhibit neutrophil oxidative metabolism in-vitro: involvement of adenosine metabolites. Free Radic Res (2010) 44(4):389–96. doi:10.3109/10715760903431434
225. Sroussi HY, Lu Y, Villines D, Sun Y. The down regulation of neutrophil oxidative metabolism by S100A8 and S100A9: implication of the protease-activated receptor-2. Mol Immunol (2012) 50(1–2):42–8. doi:10.1016/j.molimm.2011.12.001
226. Otsuka K, Terasaki F, Ikemoto M, Fujita S, Tsukada B, Katashima T, et al. Suppression of inflammation in rat autoimmune myocarditis by S100A8/A9 through modulation of the proinflammatory cytokine network. Eur J Heart Fail (2009) 11(3):229–37. doi:10.1093/eurjhf/hfn049
227. Shimizu K, Libby P, Rocha VZ, Folco EJ, Shubiki R, Grabie N, et al. Loss of myeloid related protein-8/14 exacerbates cardiac allograft rejection. Circulation (2011) 124(25):2920–32. doi:10.1161/CIRCULATIONAHA.110.009910
228. Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, Wienbrandt AR, et al. S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol (2012) 107(2):250. doi:10.1007/s00395-012-0250-z
229. Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm (2013) 2013:828354. doi:10.1155/2013/828354
230. Gasser O, Schifferli JA. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood (2004) 104(8):2543–8. doi:10.1182/blood-2004-01-0361
231. Dalli J, Montero-Melendez T, Norling LV, Yin X, Hinds C, Haskard D, et al. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteomics (2013) 12(8):2205–19. doi:10.1074/mcp.M113.028589
232. Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, Perretti M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood (2008) 112(6):2512–9. doi:10.1182/blood-2008-02-140533
233. Perretti M, Croxtall J, Wheller S, Goulding N, Hannon R, Flower R. Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med (1996) 2(11):1259–62. doi:10.1038/nm1196-1259
234. Gastardelo TS, Damazo AS, Dalli J, Flower RJ, Perretti M, Oliani SM. Functional and ultrastructural analysis of annexin A1 and its receptor in extravasating neutrophils during acute inflammation. Am J Pathol (2009) 174(1):177–83. doi:10.2353/ajpath.2009.080342
235. Pederzoli-Ribeil M, Maione F, Cooper D, Al-Kashi A, Dalli J, Perretti M, et al. Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood (2010) 116(20):4288–96. doi:10.1182/blood-2010-02-270520
236. Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci Transl Med (2015) 7(315):315ra190. doi:10.1126/scitranslmed.aac5608
237. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res (2016) 2016:1–13. doi:10.1155/2016/8239258
238. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. doi:10.1172/JCI1112
239. Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J Immunol (2005) 174(6):3727–33. doi:10.4049/jimmunol.174.6.3727
240. Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol (2007) 178(7):4595–605. doi:10.4049/jimmunol.178.7.4595
241. Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol (1999) 162(6):3639–46.
242. Kunkel SL, Chensue SW, Phan SH. Prostaglandins as endogenous mediators of interleukin 1 production. J Immunol (1986) 136(1):186–92.
243. Katakami Y, Nakao Y, Koizumi T, Katakami N, Ogawa R, Fujita T. Regulation of tumour necrosis factor production by mouse peritoneal macrophages: the role of cellular cyclic AMP. Immunology (1988) 64(4):719–24.
244. MacKenzie KF, Clark K, Naqvi S, McGuire VA, Noehren G, Kristariyanto Y, et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol (2013) 190(2):565–77. doi:10.4049/jimmunol.1202462
245. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med (2009) 15(1):42–9. doi:10.1038/nm.1905
246. Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, et al. Tissue regeneration. Inhibition of the prostaglandin-degrading enzyme 15-pgdh potentiates tissue regeneration. Science (2015) 348(6240):aaa2340. doi:10.1126/science.aaa2340
247. Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol (2004) 25(1):40–6. doi:10.1016/j.it.2003.11.001
248. Bozyk PD, Moore BB. Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol (2011) 45(3):445–52. doi:10.1165/rcmb.2011-0025RT
249. Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest (1995) 95(4):1861–8. doi:10.1172/JCI117866
250. Hodges RJ, Jenkins RG, Wheeler-Jones CPD, Copeman DM, Bottoms SE, Bellingan GJ, et al. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E2 production. Am J Pathol (2004) 165(5):1663–76. doi:10.1016/s0002-9440(10)63423-2
Keywords: resolution, chemokine depletion, eicosanoids, pro-resolving mediators, tissue homeostasis
Citation: Sugimoto MA, Sousa LP, Pinho V, Perretti M and Teixeira MM (2016) Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 7:160. doi: 10.3389/fimmu.2016.00160
Received: 04 February 2016; Accepted: 12 April 2016;
Published: 26 April 2016
Edited by:
Brigitta Stockinger, MRC National Institute for Medical Research, UKReviewed by:
Yongsheng Li, Harvard Medical School, USAGabor Csanyi, Medical College of Georgia, USA
Venizelos Papayannopoulos, Crick, UK
Copyright: © 2016 Sugimoto, Sousa, Pinho, Perretti and Teixeira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Perretti, bS5wZXJyZXR0aUBxbXVsLmFjLnVr;
Mauro M. Teixeira, bW10ZXhAaWNiLnVmbWcuYnI=
†Mauro Perretti and Mauro M. Texeira are co-senior authors.