 Kamalakannan Rajasekaran1Matthew J. Riese2,3,4*Sridhar Rao5,6Li Wang3Monica S. Thakar1,6Charles L. Sentman7*
Kamalakannan Rajasekaran1Matthew J. Riese2,3,4*Sridhar Rao5,6Li Wang3Monica S. Thakar1,6Charles L. Sentman7* Subramaniam Malarkannan1,3,4,6*
Subramaniam Malarkannan1,3,4,6*
- 1Laboratory of Molecular Immunology and Immunotherapy, Blood Research Institute, Milwaukee, WI, USA
- 2Laboratory of Lymphocyte Biology, Blood Research Institute, Milwaukee, WI, USA
- 3Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, USA
- 4Department of Microbiology and Molecular Genetics, Medical College of Wisconsin, Milwaukee, WI, USA
- 5Laboratory of Stem Cell Transcriptional Regulation, Blood Research Institute, Milwaukee, WI, USA
- 6Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, USA
- 7Department of Microbiology and Immunology, Center for Synthetic Immunity at the Geisel School of Medicine at Dartmouth, Lebanon, NH, USA
Receptors on T and NK cells systematically propagate highly complex signaling cascades that direct immune effector functions, leading to protective immunity. While extensive studies have delineated hundreds of signaling events that take place upon receptor engagement, the precise molecular mechanism that differentially regulates the induction or repression of a unique effector function is yet to be fully defined. Such knowledge can potentiate the tailoring of signal transductions and transform cancer immunotherapies. Targeted manipulations of signaling cascades can augment one effector function such as antitumor cytotoxicity while contain the overt generation of pro-inflammatory cytokines that contribute to treatment-related toxicity such as “cytokine storm” and “cytokine-release syndrome” or lead to autoimmune diseases. Here, we summarize how individual signaling molecules or nodes may be optimally targeted to permit selective ablation of toxic immune side effects.
Introduction
Immune responses by NK and T cells control infection and nascent malignancy. Generation of optimal immune responses requires efficient lymphocyte differentiation, proliferation, trafficking, recognition of target antigens, production of inflammatory cytokines, and lysis of infected and tumor cells. Functions of effector lymphocytes must be tightly regulated to prevent generation of uncontrolled immune responses. When they become chronic, uncontrolled immune responses can lead to autoimmune diseases such as rheumatoid arthritis (1), autoimmune vasculitis (2), or encephalomyelitis (3). Recent advances in utilizing synthetic receptors such as chimeric antigen receptors (CARs) have significantly augmented antitumor responses. CAR consist of an antigen-binding domain, linked to a primary signaling domain, such as CD3zeta, and often contain a co-stimulation domain from CD28 or 4-1BB to enhance NK or T cell activation (4). This enhancement also results in a heightened level of inflammatory cytokine and chemokine production. When this inflammatory response is acute, it can lead to “tumor lysis syndrome” (TLS), “cytokine storm” (CS), or “cytokine release syndrome” (CRS), which result in multi-organ failure leading to life-threatening situations.
Tumor lysis syndrome represents rapid death of malignant cells in patients with bulky tumors and release of cellular contents into circulation. This occurs following responsive treatments in patients with tumors that are highly proliferative. In addition, TLS can also occur spontaneously (5, 6). In most cases, kidney functions of the patients are compromised due to the large volume of intracellular contents released into the blood circulation (6, 7).
Cytokine storm also known as hypercytokinemia is the uncontrolled production of proinflammatory cytokines largely due to viral infections (8). Acute lung injury is the most common pathological outcome of CS (9). The cause of CS and the resultant elevated cytokine production is primarily has been associated with the activation of T cells. The central cause of CS is yet to be determined; however, a loss of feedback controls assumed to be the basis. CS can result in tissue and organ damage and at chronic conditions, can result in death.
Cytokine release syndrome is a complex clinical phenomenon characterized by the high activation of immune cells and production of proinflammatory cytokines. CRS can occur during severe infections, graft-versus-host disease, and after treatment with a variety of immunomodulating therapies, such as monoclonal antibodies, bispecific antibodies, or CAR T cells (10–12). Clinical manifestations of CRS include fever, nausea, diarrhea, headaches, confusion, seizure, hypotension, tachycardia, tachypenea, rash, liver alterations, and renal failure. Hours to days after treatment, patients experience symptoms and have elevated amounts of proinflammatory cytokines in their serum, which may include IFN-γ, IL-2, TNF-α, MIP-1, GM-CSF, IL-6, IL-8, or IL-10 (13, 14). In some cases, the CRS appeared in patients 1–4 weeks after infusion of CD19-specific CAR T cells at a time when there was a great CAR T cell expansion (15, 16). The outcome of CRS can be life-threatening, and there have been a number of patient deaths after treatment with novel immunotherapy agents (17). When identified early, CRS can be clinically managed, and treatment for CRS is focused on anti-cytokine therapies, such as corticosteroids and anti-IL-6R mAbs (18, 19). Davila and colleagues have defined five criteria for severe CRS, and Lee et al. describes a grading system and treatment algorithm for CRS (20, 21). Currently, numerous NK cell-based clinical trials are underway to treat hematological and solid tumors (22–36). Pioneering clinical applications have been developed using CAR-transduced primary T cells (20, 37). However, development of CRS following CAR T cell treatment is a major concern (12). Life-threatening CRS is an impediment to the clinical utilization and curative efficacy of CAR therapies (18). The identification of a unique signaling pathway that regulates inflammation may help to reduce toxicity and is of high clinical relevance. Therefore, understanding the molecular basis of signal transduction and subsequent regulation of effector functions within lymphocytes has the potential to limit CRS mediated by T cells or NK cells transduced with CARs or after treatment with other immunotherapeutic agents.
NK cells and cytotoxic T cells utilize multiple components of similar signal transduction machineries, but differ in the mode they utilize to activate these signaling components. Unique clonotypic T cell receptors (TCR) equip T cells with exquisite antigen specificity and functions as the most important driver of their activation (Figure 1). In contrast, NK cells integrate signals from a variety of activating and inhibitory receptors in a hierarchical manner to establish an activation state (Figure 1). Conserved non-variant receptors expressed on NK cells recognize “induced-self” (NKG2D), pathogen-derived ligands (NCR1), or the yet to be defined mechanism that governs “missing-self” (38). Additional classes of receptors (CD16, CD244, Ly49s, and KIRs) are also important in mediating NK cell effector functions. NKG2D and CD137 function as independent activation receptor in NK cells while they play an important co-stimulatory role in T cells (39).
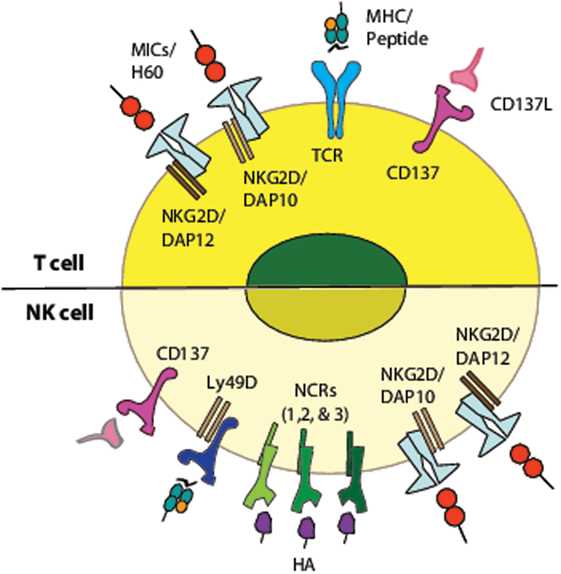
Figure 1. Activating and co-stimulatory receptors in T and NK cells and their corresponding ligands. Schematic representation of activating receptors on T cells and NK cells. While T cell receptor (TCR) functions as the primary receptor on T cells, NKG2D and CD137 function as co-stimulatory receptors along with CD28. Whereas, in NK cells, NKG2D, CD137, LY49D, NCR1, and 2B4 function as independent activating receptors and a cumulative effect of their engagement determines the final outcome of NK cell effector functions. The differences in the cytoplasmic domain of these receptors contribute to the interaction of various adapter molecules. These differences govern the signaling cascade that is engaged downstream of these receptors.
Two of the most important functions of cytotoxic T cells and NK cells are target cell killing and production of pro-inflammatory cytokines. The latter forms a major basis for TLS, CS, and in particular CRS. The qualitative differences in the temporal kinetics and duration of cytotoxicity and pro-inflammatory cytokine production suggest how these might be differentially regulated at the molecular level (40). Here, we summarize membrane proximal and intermediate signal transductions that govern NK cell or T cell activation and how shared or unique signaling molecules elicit specific effector functions. Defining these molecular regulations will help tailor effector functions of T and NK cells in immunotherapeutic strategies.
Membrane Proximal Signaling
Src Family Kinases: Lck, Fyn, and Lyn
Tyrosine phosphorylation of substrates by Src-family tyrosine kinases represents the first step in NK or T cell activation. Src family members are expressed in numerous tissues and play a vital role in all hematopoietic cell types. Lck, one of the Src members, is central to signal transduction downstream of the TCR and NK cell-activating receptors. Following receptor–ligand interaction, conformational changes take place in the intracellular domains of adapters, such as CD3ζ, that expose ITAMs (Immune Tyrosine Activation Motifs) for Lck, which localizes to the site of receptor activation through constitutive/induced interaction with co-stimulatory receptors, such as CD4 or CD8 in T cells; CD137 (4-1BB) or NK1.1 (NKR-P1) in NK cells, via its Cys–X–Cys–Pro domain; or an inducible interaction with NKG2D in NK cells (41–43). Best-characterized downstream targets of Lck are Syk and Zap-70 (Figure 2). Other imputed interactions of Lck include possible binding to DAP10 and DAP12 adapter proteins (44), and binding to the inhibitory cell surface phosphatase CD45, an interaction that may physically sequester CD45 from TCR and its downstream signaling events (45, 46).
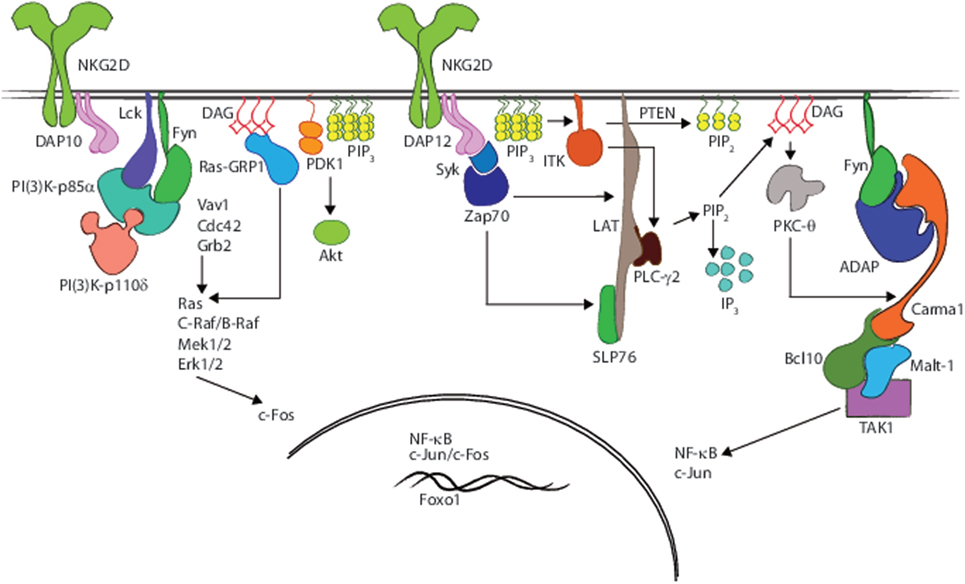
Figure 2. Receptor interacting and nucleating signaling molecules that regulate the effector functions. A graphical rendering of membrane proximal signaling events and resultant involvement of scaffold proteins, adapter molecules, and second messengers that are critical for eliciting effector functions such as cytotoxicity and proinflammatory cytokine production following NKG2D-mediated activation in NK cells.
Lck plays a complex role in NK cell signal transduction. Germ-line deletion of Lck results in NK cells with normal development and capacity for activation after stimulation with poly (I:C) or Interleukin (IL)-2 (47). In contrast, either inhibition or knockdown of Lck resulted in significant reductions in NKG2D- and CD137-mediated cytotoxicity and cytokine production in NK cells (Table 1), but no change in the cytokine production mediated by IL-12 and IL-18 stimulation (48). This suggests selective utilization of Lck playing a dominant role downstream of some, but not all activating receptors.
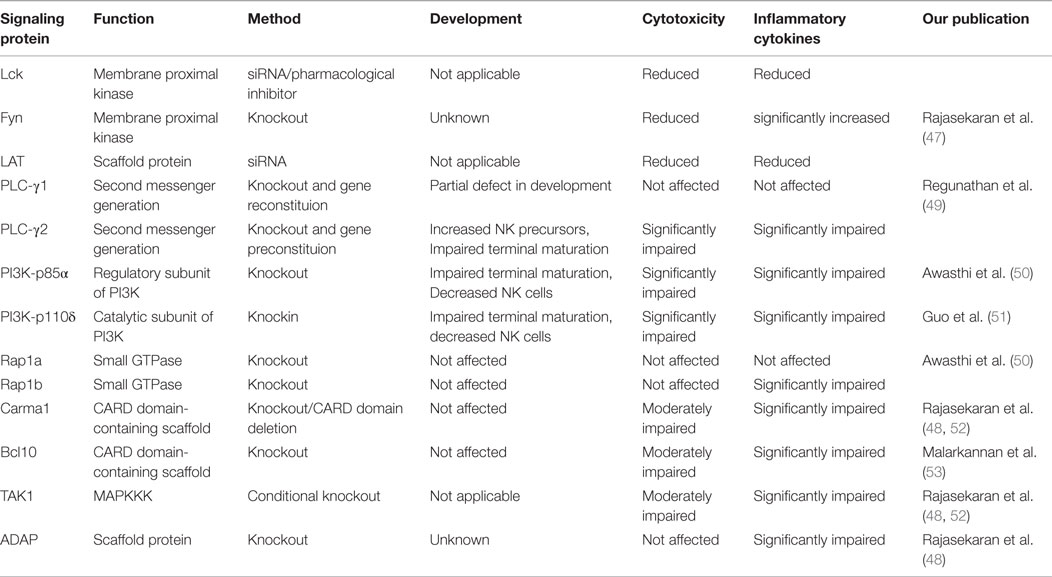
Table 1. Critical signaling molecules that regulate the development, cytotoxicity, or cytokine production from NK cells.
Fyn is another well-characterized Src family tyrosine kinase with a molecular weight of 59 kDa (54). Although the target substrates of Lck and Fyn appear redundant, it is evident that they play non-overlapping roles (55, 56). For instance, mice deficient of Fyn demonstrate minor impairments in T cell development, while lack of Lck results in a significant block in their development (55). T cells deficient in both Lck and Fyn demonstrate a complete block in T cell development (57). NK cells deficient in Fyn demonstrate a proliferative defect with only a modest enhancement observed with concurrent deficiency of Lck (58). Additionally, NK cells also utilize other Src family kinases such as Src itself, Lyn, and Fgr, although the relative importance of these kinases is uncertain (59–61).
In T cells, Lck has been shown to phosphorylate Fyn (62, 63) following ligand-induced TCR–CD4 co-aggregation. Fyn phosphorylation by Lck does not require other components of the TCR signaling apparatus, since ectopic expression of Fyn and Lck in NIH 3T3 fibroblast results in Fyn phosphorylation in a manner dependent on Lck kinase activity (63). Like Lck, Fyn subsequently phosphorylates Syk family members such as Zap-70 (64). While deficiency of Fyn is insufficient to significantly affect downstream TCR signaling events such as activation of Zap-70, LAT, and PLC-γ1, concurrent loss of Fyn and abrogation of Lck-CD4-TCR complex formation results in impaired downstream signals (65). This suggests that function of Fyn is largely redundant with that of Lck, but may play a more specialized role in facilitating TCR signaling.
Apart from its role in activation, Fyn may also play a suppressive role in T cells (66) and NK cells (48). For instance, activation of Fyn−/− NK cells via NKG2D or CD137 results in significantly elevated levels of proinflammatory cytokine and chemokine production (Table 1) compared to that of wild type (WT) (48), or following Ly49D cross linking (67). Additionally, co-culture of WT or Fyn−/− NK cells with target cells such as H60- or CD137L-expressing EL4 or RMA/S and YAC1 resulted in significantly reduced production of proinflammatory cytokines and chemokines in Fyn−/− NK cells (48, 58). These findings indicate a high level of complexity in Src family kinase activation that reflects components of redundancy and, perhaps, antagonism in lymphocyte activation.
Distinct from Lck and Fyn, the third Src-kinase family member Lyn acts predominantly as a negative regulator of lymphocyte functions (68). Hyperactivity associated with increased basal and inducible PI(3)K activity in Lyn−/− B cells indicates that it may impose inhibitory regulation of other Src-family kinases (69). Consistent with this observation, Lyn−/− mice develop autoimmunity resulting, in part, from hyperactive and increased absolute numbers of B and T cells (70). The mechanism underlying these changes results from hypophosphorylation of PAG/Cbp (phosphoprotein associated with glycosphingolipid-enriched microdomains/Csk-binding protein) and increased activity of Fyn (69); however, the direct substrates of Lyn responsible for these changes are yet to be determined. Further studies are needed to delineate the exact mechanism whereby Lyn regulates other signaling molecules.
PI(3)-Kinase
Activation of Src family kinases in NK cells is crucial for the stimulation of multiple downstream signaling events. This includes PI(3)-Kinase [PI(3)K] that are comprised of regulatory (p85α, p55α, p50α, p85β, and p55γ) and catalytic (p110α, p110β, p110γ, and p110δ) subunits that function to generate the second messenger phosphoinositol (3,4,5) trisphosphate [PI(3,4,5)P3] (71). Absence of regulatory or catalytic subunits of PI(3)K significantly impairs the development and functions of lymphocytes (50, 51, 72, 73). PI(3)K-p85α requires membrane localization for optimal activation, and can be recruited to the membrane through multiple mechanisms including binding to a YXXM motif present in the cytoplasmic tail of co-stimulatory receptors (CD19 and CD28) (74, 75), inhibitory receptors (CTLA4), signaling protein (Grb2), or adapter protein (DAP10) (76, 77). In addition, CD137 (4-1BB) recruits PI(3)K-p85α via an association with Lck and Fyn (48). In T cells, activation of PI(3)K and generation of PIP3 is largely driven by ligation of co-stimulatory receptors, such as CD28 (78). Once localized to the inner leaflet of plasma membrane, using their SH3 domains, Lck and Fyn can bind to the N-terminal proline-rich region (PRR) of the PI(3)K-p85α subunit (79), leading to the phosphorylation of the p85 and recruitment of catalytic p110 isoforms (76). Thus, Src family kinases, through high-affinity interaction with PI(3)K-p85α, function as a critical link between an activation receptor and generation of PIP3 (56, 76, 79).
Once generated, PIP3 binds and anchors multiple signaling molecules to the plasma membrane including Akt permitting its subsequent phosphorylation by phosphoinositide-dependent kinase-1 (PDK-1) at Thr308 (Figure 2). PIP3-dependent signaling is terminated through the dephosphorylation of PIP3 by lipid phosphatase PTEN that produces PI(4,5)P2 (80, 81) or SHIP-1 that generates PI(3,4)P2 (82) (Figure 2). Though the phosphoinositides generated by PTEN and SHIP-1 are incapable of binding to Akt, the reduction in the Ser473 phosphorylation of Akt in the bone marrow-derived mast cells obtained from SHIP-1 knockout mice and a concomitant reduction in its kinase activity demonstrate the relevance of these signaling intermediates (83). In general, PI(3)K has pro-growth effects in T and NK cells, and PTEN has growth suppressive effects such that deletion of PTEN from TH1 cells results in increased proliferation and Akt phosphorylation when compared with WT cells (84).
In NK cells, activation of PI(3)K is crucial for effector functions (50, 72, 73, 85–87). NK cells that lack p85α− (50) or expressing a mutant form of p110δ (p110δD910A) that lack kinase activity (51) are impaired in both cytotoxicity and cytokine production (Table 1). Moreover, perforin-dependent cryptococcal microbicidal activity of NK cells requires PI(3)K-mediated activation of ERK1/2 (88), and cytotoxic granule mobilization in NK cell is dependent on the PI(3)K–Rac–PAK–ERK1/2 pathway (89). These findings indicate that PI(3)K is a critical proximal signaling module responsible for regulating multiple effector functions.
Deletion of PI(3)K subunits leads to hypo-responsive effector T cells, whereas deletion of E3 ubiquitin ligases that induce degradation of p85 subunits are functionally hyper-responsive (90, 91). Interestingly, modulation of various subunits of PI(3)K has variable impacts on different T cell subsets. For instance, deletion of p110δ in T cells results in enhanced tumor clearance and relative increases in cytotoxic T cell activity, largely because any changes in cytotoxic T cells are countered by profound inactivation of immunosuppressive regulatory T cells (92).
The Signalosome: Contribution of LAT and SLP-76
Syk family kinases phosphorylate adapter proteins that nucleate signal transduction complexes. In T cells, Zap70 phosphorylates LAT (Linker of activated T cells) and SLP-76 (SH2 domain-containing leukocyte protein of 76 kDa) (Figure 2). LAT and SLP-76 subsequently serve as a scaffold to bind proteins that mediate downstream signals including GADS and Grb2 to LAT, and Itk, adhesion and degranulation promoting adapter protein (ADAP), Vav1, and PLC-γ1 to SLP-76 (93–97). Despite the lack of enzymatic activity, the presence of LAT and SLP-76 are obligatory during T cell development, since LAT−/− or SLP-76−/− lack mature T cells (98, 99).
Role of LAT in signaling in NK cells is not clearly defined. NK cells deficient in LAT alone or LAT and NTAL displayed efficient cytotoxicity against all target cell lines (100), but decreased capacity to generate IFN-γ following co-culture with some (e.g., Rae1β+ B16 cells) but no other (e.g., YAC-1 or RMA/S cells) target cells (100). These results suggest that LAT is required (Table 1) for signal transduction in NK cells downstream of “induced-self” receptors, but not receptors that facilitate activation from “missing-self” or “non-self.” Similarly, NK cells from LAT−/− mice demonstrated normal development and cytotoxicity (98), likely because of compensation from other scaffolds including SLP-76 (Table 1) (101). Therefore, the requirement of LAT is limited to the generation of pro-inflammatory cytokines and is not required for cytotoxicity. Interestingly, recent data suggest that SLP-76 is required for efficient IFN-γ production and cytotoxicity in NK cells in a manner independent of LAT (102), indicating that T and NK cells exploit these adapter molecules in unique ways to coordinate effector functions (Figure 2).
Intermediate Signaling Events in Cytotoxic T and NK Cell Lymphocytes
Second Messengers: DAG and IP3
One event crucial to NK and T cell activation is the localization and activation of PLC-γ molecules into the adapter scaffold complexes (103). LAT, anchored to the plasma membrane, serves as a docking site for PLC-γ. While PI(3,4,5)P3 is required for the activation of PLC-γ, action of PTEN on the former leads to the generation of PI(4,5)P2 permitting the cleavage of membrane-bound PI(4,5)P2 by PLC-γ into the second messengers diacylglycerol (DAG) and IP3. Whereas DAG, a membrane signaling lipid, binds and activates RasGRP1 (a Ras-activating molecule) and PKC-θ, soluble IP3 binds to proteins that facilitate calcium flux from the endoplasmic reticulum (Figure 2). NK cells and T cells utilize different isoforms of PLC-γ in hydrolysis of PIP2; T cells primarily utilize PLC-γ1, whereas NK cells predominantly utilize PLC-γ2 (49, 104–108). Although these cell-specific differences partially result from alterations in protein expression levels of individual isoforms (109), overexpression of PLC-γ1 in NK cells can only partially compensate for PLC-γ2 deficiency in NK cells, for instance, restoring the terminal maturation of NK cells, but failing to fully restore impaired cytotoxicity and cytokine production (Table 1) (49). Further dissection of the defects in NK cell-mediated cytotoxicity in the Plc-γ2−/− NK cells revealed that NK cells were capable of forming conjugates with target cells and developing Microtubule organizing center (MTOC) polarization, but that calcium mobilization and resulting signaling events were compromised (108). In T cells, in addition to LAT, a unique complex containing SLP-76 and the Tec family kinase Itk (IL-2 inducible kinase) are required for PLC-γ activation (110). While phosphorylation of at least three tyrosines at the N-terminus of SLP-76 by Zap70 appear essential for efficient signal transduction, phosphorylation at Y145 appears to be most important for recruitment and activation of Itk, and the subsequent phosphorylation and activation of PLC-γ1 (111) (Figure 2). Mutation of SLP-76 at Y145, in general, phenocopies the loss of Itk (112), likely because other Tec family kinases, such as Rlk, can only partially compensate for the lack of Itk-mediated PLC-γ1 activation (113). Thus, apart from binding LAT, PLC-γ1 requires interaction with multiple components of the signalosome in order to optimally coordinate enzymatic activity (94).
Once activated, PLC-γ1 generates IP3 and DAG, allowing DAG to bind RasGRP1 to facilitate activation of Ras in T cells (114), along with at least three isoforms of PKC, including PKC-ϵ, PKC-ŋ, and PKC-θ (115). Whereas PKC-ϵ and PKC-ŋ serve in a redundant manner to recruit PKC-θ to the immune synapse (115, 116), the role of PKC-θ is obligatory for initiation of downstream signaling events such as NF-κB activation. The localization of DAG is crucial to its ability to mediate signaling and cytoskeletal changes such as dynein-mediated MTOC orientation and T cell polarization (116).
Second Messenger to PKC-θ
Apart from binding to DAG, PKC-θ is also regulated by multiple other factors. PKC-θ exists as an inactive high-molecular disulfide-linked complex in naive T cells. During T cell activation, PKC-θ is gradually reduced from a 85 kDa inactive form to a 82 kDa active form by two major redox regulators, glutathione and thioredoxin (117), thus implicating the intracellular redox state as another node for PKC-θ regulation. Further, a C2 phosphotyrosine-binding domain also appears important for optimizing activity of PKC-θ (118). In T cells, efficient PKC-θ activity also requires phospohorylation downstream of PDK1, a PIP3-regulated kinase, either directly through PDK1 or through an intermediary serine/threonine kinase (119), indicating the role of PI(3)K and thereby a requirement for co-stimulatory receptor ligation for optimal PKC-θ activation (Figure 2).
Activated PKC-θ is required for efficient in vivo function of NK cells, since PKC-θ−/− mice are unable to eliminate tumor cells lacking expression of MHC-I, secondary to reduced NK cell intra-tumoral migration and decreased NK cell activation (120). Similarly, PKC-θ−/− NK cells demonstrate reduced cytotoxicity after treatment with Toll receptor agonists, such as poly I:C (121, 122) and reduced cytokine production after stimulation through NK1.1 or Ly49D (123). Though the signaling events responsible for the phenotype of PKC-θ−/− NK cells has undergone limited analysis, it appears that sustained Erk1/2 and Jnk1/2 activation is abrogated, resulting in decreased nuclear translocation of AP-1 and NFAT (123). Intriguingly, the activation of NF-κB and degradation of IκBα does not appear to be impacted by loss of PKC-θ in NK cells (123), in contrast to T cells in which loss of PKC-θ results in major defects in NFκB signaling (124), leading to abrogation of TCR-induced IL-2 production and proliferation (125).
Unique Signaling That Regulate Selective Effector Functions
Our recent work has shown that “ADAP” serves as a node of signaling divergence and it could be manipulated to differentially limit unique effector responses. ADAP [Fyn-binding protein (Fyb)] was first identified in T cells as a 120/130 kDa component of the TCR–CD3ζ–p59FynT activation (126). ADAP binds to the SH2 domain of Fyn in a specific manner, such that other SH2-domain-containing Src family kinases, such as Lck, do not (127). Fyb was renamed ADAP in consideration of the specific functional defects identified in lymphocytes from mice deficient of this protein (128). ADAP utilizes several domains to facilitate binding with partner proteins (Figure 3) (129–131) and is known to undergo phosphorylation following TCR ligation, an event that likely contributes to its protein–protein interactions with SLP-76, Nck1, and Nck2 (132). ADAP also binds to the monomeric GTPase Rap1, contributing to its localization at the plasma membrane (133). Interaction of the SH2 domain of SLP-76 with ADAP takes place at three specific sites within ADAP, permitting oligomerization and the formation of SLP-76-based microclusters (134) (Figure 3). ADAP also contains SH3 (hSH3) domains that bind to phosphoinositides that likely contribute to its role in cellular migration, cytokine production, and integrin activation (135).
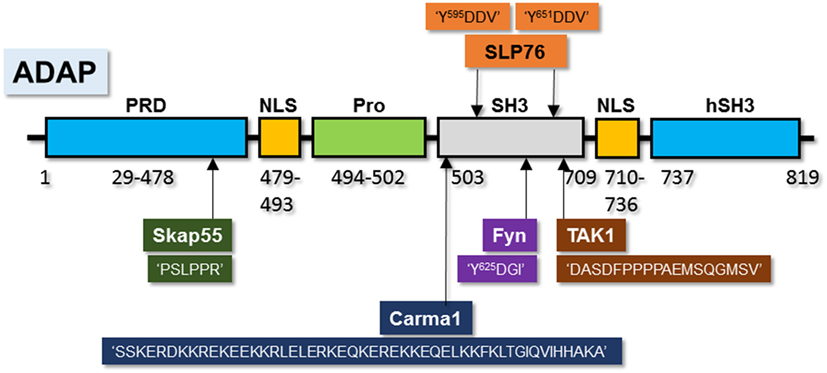
Figure 3. Unique amino acid motifs in ADAP scaffold facilitate its interactions with multiple-binding partners. Pictorial depiction of ADAP protein and its amino acid sequences (or motifs) that are required to interact with Fyn, Carma1, TAK1, SLP76, and SKAP55. Amino acid sequences within ‘’ are from ADAP with the name of the interacting partner listed above or below this sequence.
In NK and effector T cells, ADAP has been shown to play important roles in both ITAM-dependent signaling pathways, and activation of β1 and β2 integrins such as LFA1 (134 and 147). In T cells, loss of ADAP results in a maturation block of double-positive thymocytes resulting from alterations in both positive and negative selection of developing T cells (136). Peripheral T cells that manage to emigrate from the thymus are also impaired as evidenced by reduced tissue rejection in heart (137) and intestinal allografts in ADAP−/− mice (138), and significant amelioration of pathology in Experimental Autoimmune Encephalitis (EAE) (139). Loss of ADAP in T cells further decreases their proliferation and cytokine production efficiency in response to limiting antigen doses (140). Crucially, ADAP also plays a role in integrin activation of T and NK cells (141). Integrins are components of cell–cell interactions that are required for extravasation and tissue localization of lymphocytes to sites of infection. In resting cells, integrins bind weakly to ligands, such as ICAM1, but in activated cells, integrins are conformationally altered to bind ligands with high affinity. These conformational changes in integrins are due to “inside-out” signaling in contrast to “outside-in” signaling transduced by integrins themselves after ligand binding (142–144). In T cells, the chemokine receptor CCR7 induces activation of the LFA-1 through a mechanism that requires binding of ADAP, and an additional adapter molecule, SKAP55, to LFA-1 (145). Studies using ADAP/SKAP55 chimeric proteins in Adap−/− T cells identified distinct roles for ADAP in facilitating NF-κB signaling and LFA-1 activation. Expression of a SKAP55/ADAP chimeric molecule in Adap−/− T cells was sufficient and necessary for integrin function and NF-κB activation (146–148), whereas expression of a chimeric molecule with a point mutation in the PH-domain of SKAP55 permitted restoration of NF-κB activation in NK cells but not integrin function (148). It is currently thought that recruitment of ADAP to LFA1 complexes through the PH-domain of SKAP55 restricts the ability of ADAP to interact with the CBM signalosome and to activate NF-κB signal transduction (148).
Stimulation through TCR and CD28 utilizes ADAP to facilitate signaling downstream of the Carma1–Bcl10–Malt1 (CBM) complex, which leads to phosphorylation and degradation of IκBα and nuclear translocation of NF-κB (149). While the molecular mechanism whereby ADAP regulates the formation of the CBM have not been fully elucidated, the essential function of ADAP in linking CBM via Carma1 to PKC-θ is well documented (136). NF-κB, which is sequestered in the cytosol through binding to IκBα, translocates into the nucleus (136). Carma1 and Bcl10 play an obligatory role in the nuclear translocation of NF-kB following activation of NK cells through NK1.1, Ly49D, NKG2D, and CD137 (52, 150). In addition to interactions with Carma1, ADAP also recruit TAK1, which facilitates phosphorylation of IKK alpha and beta (149), components of NF-κB signaling pathway. In NK cells, ADAP contributes to CBM complex formation in response to ITAM-containing receptors such as NK1.1, Ly49D, Ly49H, and response to activation through NKG2D, NCR1, and CD137 (150).
In contrast to T cells, analyses of NK cell development in ADAP−/− mice revealed no significant alterations (151). Recently, we (48), and others (102), have extended these initial findings to provide a more detailed evaluation of NK cell functions in response to a variety of NK cell receptors. While ADAP−/− and WT NK cells demonstrated comparable cytotoxicity, we and others have observed attenuated cytokine production in ADAP−/− NK cells following activation via Ly49D (48, 102), NKG2D, NCR1, 2B4, and CD137 (4-1BB) (48). Thus, ADAP could act as a divergent point to selectively attenuate cytokine production without affecting cytotoxicity in NK cells.
Future Perspectives
Identifying unique signaling myriads of signaling cascades in NK and T cells is complex, but improved understanding can serve to enhance existing immune-based therapies. For instance, new treatments that use genetically engineered lymphocytes to target malignancy have had successes in elimination of tumors such as refractory B cell malignancies, but have been limited by CRS. Targeting ADAP could help to regulate inflammatory cytokine production from effector lymphocytes while preserving cytotoxicity. Further, studies are also ongoing to test the efficacy of several pharmacological PI(3)K inhibitors in regulating tumor growth and survival without affecting the function of the immune cells. Continued detailed, rigorous study of lymphocyte signaling has the potential to unlock additional targets that could permit tailored responses for clinical therapy related to chronic autoimmunity and cancer.
Author Contributions
KR, MR, CS, and SM wrote the major sections of the manuscript. SR, LW, and MT provided critical intellectual input and helped in writing. KR and SM compiled and revised the text; prepared the figures and the table.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
We thank Lucia Sammarco and her Lulu’s Lemonade Stand for inspiration, motivation, and support. This work was supported in part by NIH R01 AI064826, NIH R01 AI102893 and NCI R01 CA179363 (SM); NHLBI-HL087951 (SR); NIH-CA151893-K08 (MR); NCI 1R01CA164225 (LW); Alex Lemonade Stand Foundation (SM); HRHM Program of MACC Fund (SM, MT, and SR), Nicholas Family Foundation (SM); Gardetto Family (SM); Hyundai Scholars Program (MT); Hyundai Hope on Wheels (SR); Pavlove Foundation (MT); Rebecca Jean Slye Endowment (MT); MCW-Cancer Center-Large Seed Grant (SM and MT; SR); MACC Fund (MT and SM); American Cancer Society Grants (SM and MT, SR, MR); and by the Clinical & Translational Science Institute of Southeastern Wisconsin (NIH UL1RR031973) (MT and SM). Children’s Research Institute, MCW (SR); Kathy Duffey Fogerty Award (MR). NIH R01 CA164178 (CS).
References
1. Finnegan A, Ashaye S, Hamel KM. B effector cells in rheumatoid arthritis and experimental arthritis. Autoimmunity (2012) 45:353–63. doi: 10.3109/08916934.2012.665526
2. Lintermans LL, Stegeman CA, Heeringa P, Abdulahad WH. T cells in vascular inflammatory diseases. Front Immunol (2014) 5:504. doi:10.3389/fimmu.2014.00504
3. Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci (2013) 16:1618–26. doi:10.1038/nn.3531
4. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov (2013) 3:388–98. doi:10.1158/2159-8290.CD-12-0548
5. Okamoto K, Kinoshita T, Shimizu M, Okura I, Kawada A, Mizobuchi K, et al. A case of spontaneous tumor lysis syndrome in a patient with ovarian cancer. Case Rep Obstet Gynecol (2015) 2015:461870. doi:10.1155/2015/461870
6. Mavani G, Wolf V, Orin G. Spontaneous tumor lysis syndrome in a case of B-cell non-Hodgkin’s lymphoma. Clin Kidney J (2014) 7:422–3. doi:10.1093/ckj/sfu061
7. Saleh RR, Rodrigues J, Lee TC. A tumour lysis syndrome in a chemotherapy naive patient with metastatic pancreatic adenocarcinoma. BMJ Case Rep (2015) 2015. doi:10.1136/bcr-2014-207748
8. Rattanaburee T, Junking M, Panya A, Sawasdee N, Songprakhon P, Suttitheptumrong A, et al. Inhibition of dengue virus production and cytokine/chemokine expression by ribavirin and compound A. Antiviral Res (2015) 124:83–92. doi:10.1016/j.antiviral.2015.10.005
9. Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med (2008) 26:711–5. doi:10.1016/j.ajem.2007.10.031
10. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355:1018–28. doi:10.1056/NEJMoa063842
11. Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol (2015) 16:57–66. doi:10.1016/S1470-2045(14)71170-2
12. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18:843–51. doi:10.1038/mt.2010.24
13. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5:177ra38. doi:10.1126/scitranslmed.3005930
14. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Trans Med (2011) 3:95ra73. doi:10.1126/scitranslmed.3002842
15. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi:10.1056/NEJMoa1103849
16. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368:1509–18. doi:10.1056/NEJMoa1215134
17. Xu XJ, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett (2013) 343:172–8. doi:10.1016/j.canlet.2013.10.004
18. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J (2014) 20:119–22. doi:10.1097/PPO.0000000000000035
19. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi:10.1016/S0140-6736(14)61403-3
20. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Trans Med (2014) 6:224ra25. doi:10.1126/scitranslmed.3008226
21. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124:188–95. doi:10.1182/blood-2014-05-552729
22. Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol (2008) 9:486–94. doi:10.1038/ni1580
23. Locatelli F, Pende D, Maccario R, Mingari MC, Moretta A, Moretta L. Haploidentical hemopoietic stem cell transplantation for the treatment of high-risk leukemias: how NK cells make the difference. Clin Immunol (2009) 133:171–8. doi:10.1016/j.clim.2009.04.009
24. Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood (2009) 113:3119–29. doi:10.1182/blood-2008-06-164103
25. Moretta L, Locatelli F, Pende D, Mingari MC, Moretta A. Natural killer alloeffector responses in haploidentical hemopoietic stem cell transplantation to treat high-risk leukemias. Tissue Antigens (2010) 75:103–9. doi:10.1111/j.1399-0039.2009.01404.x
26. Moretta L, Locatelli F, Pende D, Marcenaro E, Mingari MC, Moretta A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood (2011) 117:764–71. doi:10.1182/blood-2010-08-264085
27. Willemze R, Rodrigues CA, Labopin M, Sanz G, Michel G, Socié G, et al. KIR-ligand incompatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia (2009) 23:492–500. doi:10.1038/leu.2008.365
28. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood (2010) 116:2411–9. doi:10.1182/blood-2010-05-283051
29. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol (2008) 143:641–53. doi:10.1111/j.1365-2141.2008.07340.x
30. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi:10.1182/blood-2004-07-2974
31. Barkholt L, Alici E, Conrad R, Sutlu T, Gilljam M, Stellan B, et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: a phase I clinical study. Immunotherapy (2009) 1:753–64. doi:10.2217/imt.09.47
32. Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy (2009) 11:341–55. doi:10.1080/14653240902807034
33. Yong AS, Keyvanfar K, Hensel N, Eniafe R, Savani BN, Berg M, et al. Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood (2009) 113:875–82. doi:10.1182/blood-2008-05-158253
34. Berg M, Childs R. Ex-vivo expansion of NK cells: what is the priority – high yield or high purity? Cytotherapy (2010) 12:969–70. doi:10.3109/14653249.2010.536216
35. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J Cancer (2011) 2:383–5. doi:10.7150/jca.2.383
36. Boyiadzis M, Memon S, Carson J, Allen K, Szczepanski MJ, Vance BA, et al. Up-regulation of NK cell activating receptors following allogeneic hematopoietic stem cell transplantation under a lymphodepleting reduced intensity regimen is associated with elevated IL-15 levels. Biol Blood Marrow Transplant (2008) 14:290–300. doi:10.1016/j.bbmt.2007.12.490
37. Sadelain M, Brentjens R, Riviere I, Park J. CD19 CAR therapy for acute lymphoblastic leukemia. Am Soc Clin Oncol Educ Book (2015):e360–3. doi:10.14694/EdBook_AM.2015.35.e360
38. Malarkannan S. The balancing act: inhibitory Ly49 regulate NKG2D-mediated NK cell functions. Semin Immunol (2006) 18:186–92. doi:10.1016/j.smim.2006.04.002
39. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med (1997) 186:47–55. doi:10.1084/jem.186.1.47
40. Vivier E, Ugolini S, Nunes JA. ADAPted secretion of cytokines in NK cells. Nat Immunol (2013) 14:1108–10. doi:10.1038/ni.2737
41. Salcedo TW, Kurosaki T, Kanakaraj P, Ravetch JV, Perussia B. Physical and functional association of p56lck with Fc gamma RIIIA (CD16) in natural killer cells. J Exp Med (1993) 177:1475–80. doi:10.1084/jem.177.5.1475
42. Pignata C, Prasad KV, Robertson MJ, Levine H, Rudd CE, Ritz J. Fc gamma RIIIA-mediated signaling involves src-family lck in human natural killer cells. J Immunol (1993) 151:6794–800.
43. Campbell KS, Giorda R. Thy cytoplamic domain of rat NKR-P1 receptor interacts with the N-terminal domain of p56(lck) via cysteine residues. Eur J Immunol (1997) 27:802. doi:10.1002/eji.1830270111
44. Wu J, Cherwinski H, Spies T, Phillips JH, Lanier LL. DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J Exp Med (2000) 192:1059–68. doi:10.1084/jem.192.7.1059
45. Rossy J, Williamson DJ, Gaus K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front Immunol (2012) 3:167. doi:10.3389/fimmu.2012.00167
46. Rossy J, Owen DM, Williamson DJ, Yang Z, Gaus K. Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat Immunol (2013) 14:82–9. doi:10.1038/ni.2488
47. Wen T, Zhang L, Kung SK, Molina TJ, Miller RG, Mak TW. Allo-skin graft rejection, tumor rejection and natural killer activity in mice lacking p56lck. Eur J Immunol (1995) 25:3155–9. doi:10.1002/eji.1830251125
48. Rajasekaran K, Kumar P, Schuldt KM, Peterson EJ, Vanhaesebroeck B, Dixit V, et al. Signaling by Fyn-ADAP via the Carma1-Bcl-10-MAP3K7 signalosome exclusively regulates inflammatory cytokine production in NK cells. Nat Immunol (2013) 14:1127–36. doi:10.1038/ni.2708
49. Regunathan J, Chen Y, Kutlesa S, Dai X, Bai L, Wen R, et al. Differential and nonredundant roles of phospholipase Cgamma2 and phospholipase Cgamma1 in the terminal maturation of NK cells. J Immunol (2006) 177:5365–76. doi:10.4049/jimmunol.177.8.5365
50. Awasthi A, Samarakoon A, Dai X, Wen R, Wang D, Malarkannan S. Deletion of PI3K-p85alpha gene impairs lineage commitment, terminal maturation, cytokine generation and cytotoxicity of NK cells. Genes Immun (2008) 9:522–35. doi:10.1038/gene.2008.45
51. Guo H, Samarakoon A, Vanhaesebroeck B, Malarkannan S. The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Med (2008) 205:2419–35. doi:10.1084/jem.20072327
52. Gross O, Grupp C, Steinberg C, Zimmermann S, Strasser D, Hannesschläger N, et al. Multiple ITAM-coupled NK-cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-kappaB and MAPK activation to selectively control cytokine production. Blood (2008) 112:2421–8. doi:10.1182/blood-2007-11-123513
53. Malarkannan S, Regunathan J, Chu H, Kutlesa S, Chen Y, Zeng H, et al. Bcl10 plays a divergent role in NK cell-mediated cytotoxicity and cytokine generation. J Immunol (2007) 179:3752–62. doi:10.4049/jimmunol.179.6.3752
54. Rudd CE, Janssen O, Prasad KV, Raab M, da Silva A, Telfer JC, et al. src-related protein tyrosine kinases and their surface receptors. Biochim Biophys Acta (1993) 1155:239–66.
55. Olszowy MW, Leuchtmann PL, Veillette A, Shaw AS. Comparison of p56lck and p59fyn protein expression in thymocyte subsets, peripheral T cells, NK cells, and lymphoid cell lines. J Immunol (1995) 155:4236–40.
56. Susa M, Rohner D, Bichsel S. Differences in binding of PI 3-kinase to the src-homology domains 2 and 3 of p56 lck and p59 fyn tyrosine kinases. Biochem Biophys Res Commun (1996) 220:729–34. doi:10.1006/bbrc.1996.0472
57. Zamoyska R, Basson A, Filby A, Legname G, Lovatt M, Seddon B. The influence of the src-family kinases, Lck and Fyn, on T cell differentiation, survival and activation. Immunol Rev (2003) 191:107–18. doi:10.1034/j.1600-065X.2003.00015.x
58. Dong Z, Davidson D, Pérez-Quintero LA, Kurosaki T, Swat W, Veillette A. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity (2012) 36:974–85. doi:10.1016/j.immuni.2012.03.023
59. Cerný J, Fiserová A, Horváth O, Bezouska K, Pospísil M, Horejsí V. Association of human NK cell surface receptors NKR-P1 and CD94 with Src-family protein kinases. Immunogenetics (1997) 46:231–6. doi:10.1007/s002510050267
60. Sasanuma H, Tatsuno A, Hidano S, Ohshima K, Matsuzaki Y, Hayashi K, et al. Dual function for the adaptor MIST in IFN-gamma production by NK and CD4+NKT cells regulated by the Src kinase Fgr. Blood (2006) 107:3647–55. doi:10.1182/blood-2005-10-4102
61. Oykhman P, Timm-McCann M, Xiang RF, Islam A, Li SS, Stack D, et al. Requirement and redundancy of the Src family kinases Fyn and Lyn in perforin-dependent killing of Cryptococcus neoformans by NK cells. Infect Immun (2013) 81:3912–22. doi:10.1128/IAI.00533-13
62. Filipp D, Zhang J, Leung BL, Shaw A, Levin SD, Veillette A, et al. Regulation of Fyn through translocation of activated Lck into lipid rafts. J Exp Med (2003) 197:1221–7. doi:10.1084/jem.20022112
63. Filipp D, Moemeni B, Ferzoco A, Kathirkamathamby K, Zhang J, Ballek O, et al. Lck-dependent Fyn activation requires C terminus-dependent targeting of kinase-active Lck to lipid rafts. J Biol Chem (2008) 283:26409–22. doi:10.1074/jbc.M710372200
64. Michel F, Grimaud L, Tuosto L, Acuto O. Fyn and ZAP-70 are required for Vav phosphorylation in T cells stimulated by antigen-presenting cells. J Biol Chem (1998) 273:31932–8. doi:10.1074/jbc.273.48.31932
65. Sugie K, Jeon MS, Grey HM. Activation of naive CD4 T cells by anti-CD3 reveals an important role for Fyn in Lck-mediated signaling. Proc Natl Acad Sci U S A (2004) 101:14859–64. doi:10.1073/pnas.0406168101
66. Filby A, Seddon B, Kleczkowska J, Salmond R, Tomlinson P, Smida M, et al. Fyn regulates the duration of TCR engagement needed for commitment to effector function. J Immunol (2007) 179:4635–44. doi:10.4049/jimmunol.179.7.4635
67. Mason LH, Willette-Brown J, Taylor LS, McVicar DW. Regulation of Ly49D/DAP12 signal transduction by Src-family kinases and CD45. J Immunol (2006) 176:6615–23. doi:10.4049/jimmunol.176.11.6615
68. Krebs DL, Chehal MK, Sio A, Huntington ND, Da ML, Ziltener P, et al. Lyn-dependent signaling regulates the innate immune response by controlling dendritic cell activation of NK cells. J Immunol (2012) 188:5094–105. doi:10.4049/jimmunol.1103395
69. Xu Y, Huntington ND, Harder KW, Nandurkar H, Hibbs ML, Tarlinton DM. Phosphatidylinositol-3 kinase activity in B cells is negatively regulated by Lyn tyrosine kinase. Immunol Cell Biol (2012) 90:903–11. doi:10.1038/icb.2012.31
70. Tsantikos E, Quilici C, Harder KW, Wang B, Zhu HJ, Anderson GP, et al. Perturbation of the CD4 T cell compartment and expansion of regulatory T cells in autoimmune-prone Lyn-deficient mice. J Immunol (2009) 183:2484–94. doi:10.4049/jimmunol.0804346
71. Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem (2001) 70:535–602. doi:10.1146/annurev.biochem.70.1.535
72. Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science (1999) 283:393–7. doi:10.1126/science.283.5400.393
73. Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, et al. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science (1999) 283:390–2. doi:10.1126/science.283.5400.390
74. Carter RH, Tuveson DA, Park DJ, Rhee SG, Fearon DT. The CD19 complex of B lymphocytes. Activation of phospholipase C by a protein tyrosine kinase-dependent pathway that can be enhanced by the membrane IgM complex. J Immunol (1991) 147:3663–71.
75. Beitz LO, Fruman DA, Kurosaki T, Cantley LC, Scharenberg AM. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J Biol Chem (1999) 274:32662–6. doi:10.1074/jbc.274.46.32662
76. Raab M, Cai YC, Bunnell SC, Heyeck SD, Berg LJ, Rudd CE. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: implications for T-cell costimulation. Proc Natl Acad Sci U S A (1995) 92:8891–5. doi:10.1073/pnas.92.19.8891
77. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science (1999) 285:730–2. doi:10.1126/science.285.5428.730
78. Pagès F, Ragueneau M, Rottapel R, Truneh A, Nunes J, Imbert J, et al. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature (1994) 369:327–9. doi:10.1038/369327a0
79. Kapeller R, Prasad KV, Janssen O, Hou W, Schaffhausen BS, Rudd CE, et al. Identification of two SH3-binding motifs in the regulatory subunit of phosphatidylinositol 3-kinase. J Biol Chem (1994) 269:1927–33.
80. Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol (1999) 11:219–25. doi:10.1016/S0955-0674(99)80029-5
81. Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity (2001) 14:523–34. doi:10.1016/S1074-7613(01)00134-0
82. Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev (1996) 10:1084–95. doi:10.1101/gad.10.9.1084
83. Scheid MP, Huber M, Damen JE, Hughes M, Kang V, Neilsen P, et al. Phosphatidylinositol (3,4,5)P3 is essential but not sufficient for protein kinase B (PKB) activation; phosphatidylinositol (3,4)P2 is required for PKB phosphorylation at Ser-473: studies using cells from SH2-containing inositol-5-phosphatase knockout mice. J Biol Chem (2002) 277:9027–35. doi:10.1074/jbc.M106755200
84. Locke FL, Zha YY, Zheng Y, Driessens G, Gajewski TF. Conditional deletion of PTEN in peripheral T cells augments TCR-mediated activation but does not abrogate CD28 dependency or prevent anergy induction. J Immunol (2013) 191:1677–85. doi:10.4049/jimmunol.1202018
85. Fruman DA, Snapper SB, Yballe CM, Alt FW, Cantley LC. Phosphoinositide 3-kinase knockout mice: role of p85alpha in B cell development and proliferation. Biochem Soc Trans (1999) 27:624–9. doi:10.1042/bst0270624
86. Okkenhaug K, Vanhaesebroeck B. New responsibilities for the PI3K regulatory subunit p85 alpha. Sci STKE (2001) 2001:65. doi:10.1126/stke2001.65.pe1
87. Glassford J, Soeiro I, Skarell SM, Banerji L, Holman M, Klaus GG, et al. BCR targets cyclin D2 via Btk and the p85alpha subunit of PI3-K to induce cell cycle progression in primary mouse B cells. Oncogene (2003) 22:2248–59. doi:10.1038/sj.onc.1206425
88. Wiseman JC, Ma LL, Marr KJ, Jones GJ, Mody CH. Perforin-dependent cryptococcal microbicidal activity in NK cells requires PI3K-dependent ERK1/2 signaling. J Immunol (2007) 178:6456–64. doi:10.4049/jimmunol.178.10.6456
89. Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, et al. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol (2000) 1:419–25. doi:10.1038/80859
90. Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol (2001) 2:870–5. doi:10.1038/ni0901-870
91. Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem (2001) 276:4872–8. doi:10.1074/jbc.M008901200
92. Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan AM, Tkaczyk C, et al. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature (2004) 431:1007–11. doi:10.1038/nature02991
93. Wang H, McCann FE, Gordan JD, Wu X, Raab M, Malik TH, et al. ADAP-SLP-76 binding differentially regulates supramolecular activation cluster (SMAC) formation relative to T cell-APC conjugation. J Exp Med (2004) 200:1063–74. doi:10.1084/jem.20040780
94. Beach D, Gonen R, Bogin Y, Reischl IG, Yablonski D. Dual role of SLP-76 in mediating T cell receptor-induced activation of phospholipase C-gamma1. J Biol Chem (2007) 282:2937–46. doi:10.1074/jbc.M606697200
95. Wang H, Wei B, Bismuth G, Rudd CE. SLP-76-ADAP adaptor module regulates LFA-1 mediated costimulation and T cell motility. Proc Natl Acad Sci U S A (2009) 106:12436–41. doi:10.1073/pnas.0900510106
96. Block H, Herter JM, Rossaint J, Stadtmann A, Kliche S, Lowell CA, et al. Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J Exp Med (2012) 209:407–21. doi:10.1084/jem.20111493
97. Coussens NP, Hayashi R, Brown PH, Balagopalan L, Balbo A, Akpan I, et al. Multipoint binding of the SLP-76 SH2 domain to ADAP is critical for oligomerization of SLP-76 signaling complexes in stimulated T cells. Mol Cell Biol (2013) 33:4140–51. doi:10.1128/MCB.00410-13
98. Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, et al. Essential role of LAT in T cell development. Immunity (1999) 10:323–32. doi:10.1016/S1074-7613(00)80032-1
99. Kumar L, Pivniouk V, de la Fuente MA, Laouini D, Geha RS. Differential role of SLP-76 domains in T cell development and function. Proc Natl Acad Sci U S A (2002) 99:884–9. doi:10.1073/pnas.022619199
100. Chiesa S, Mingueneau M, Fuseri N, Malissen B, Raulet DH, Malissen M, et al. Multiplicity and plasticity of natural killer cell signaling pathways. Blood (2006) 107:2364–72. doi:10.1182/blood-2005-08-3504
101. Hidano S, Sasanuma H, Ohshima K, Seino K, Kumar L, Hayashi K, et al. Distinct regulatory functions of SLP-76 and MIST in NK cell cytotoxicity and IFN-gamma production. Int Immunol (2008) 20:345–52. doi:10.1093/intimm/dxm150
102. May RM, Okumura M, Hsu CJ, Bassiri H, Yang E, Rak G, et al. Murine natural killer immunoreceptors use distinct proximal signaling complexes to direct cell function. Blood (2013) 121:3135–46. doi:10.1182/blood-2012-12-474361
103. Inoue H, Miyaji M, Kosugi A, Nagafuku M, Okazaki T, Mimori T, et al. Lipid rafts as the signaling scaffold for NK cell activation: tyrosine phosphorylation and association of LAT with phosphatidylinositol 3-kinase and phospholipase C-gamma following CD2 stimulation. Eur J Immunol (2002) 32:2188–98. doi:10.1002/1521-4141(200208)32:8<2188::AID-IMMU2188>3.0.CO;2-T
104. Tassi I, Cella M, Castro I, Gilfillan S, Khan WN, Colonna M. Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur J Immunol (2009) 39:1369–78. doi:10.1002/eji.200839313
105. Cambier JC, Pleiman CM, Clark MR. Signal transduction by the B cell antigen receptor and its coreceptors. Annu Rev Immunol (1994) 12:457–86. doi:10.1146/annurev.iy.12.040194.002325
106. Irvin BJ, Williams BL, Nilson AE, Maynor HO, Abraham RT. Pleiotropic contributions of phospholipase C-gamma1 (PLC-gamma1) to T-cell antigen receptor-mediated signaling: reconstitution studies of a PLC-gamma1-deficient Jurkat T-cell line. Mol Cell Biol (2000) 20:9149–61. doi:10.1128/MCB.20.24.9149-9161.2000
107. Weiss A, Irving BA, Tan LK, Koretzky GA. Signal transduction by the T cell antigen receptor. Semin Immunol (1991) 3:313–24.
108. Caraux A, Kim N, Bell SE, Zompi S, Ranson T, Lesjean-Pottier S, et al. Phospholipase C-gamma2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood (2006) 107:994–1002. doi:10.1182/blood-2005-06-2428
109. Tassi I, Presti R, Kim S, Yokoyama WM, Gilfillan S, Colonna M. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. J Immunol (2005) 175:749–54. doi:10.4049/jimmunol.175.2.749
110. Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell (1998) 92:83–92. doi:10.1016/S0092-8674(00)80901-0
111. Jordan MS, Sadler J, Austin JE, Finkelstein LD, Singer AL, Schwartzberg PL, et al. Functional hierarchy of the N-terminal tyrosines of SLP-76. J Immunol (2006) 176:2430–8. doi:10.4049/jimmunol.176.4.2430
112. Prince AL, Kraus Z, Carty SA, Ng C, Yin CC, Jordan MS, et al. Development of innate CD4+ and CD8+ T cells in Itk-deficient mice is regulated by distinct pathways. J Immunol (2014) 193:688–99. doi:10.4049/jimmunol.1302059
113. Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol (2005) 23:549–600. doi:10.1146/annurev.immunol.22.012703.104743
114. Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, et al. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol (2000) 1:317–21. doi:10.1038/79766
115. Quann EJ, Liu X, tan-Bonnet G, Huse M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat Immunol (2011) 12:647–54. doi:10.1038/ni.2033
116. Quann EJ, Merino E, Furuta T, Huse M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat Immunol (2009) 10:627–35. doi:10.1038/ni.1734
117. von Essen MR, Kongsbak M, Levring TB, Hansen AK, Boding L, Lauritsen JP, et al. PKC-θ exists in an oxidized inactive form in naive human T cells. Eur J Immunol (2013) 43:1659–66. doi:10.1002/eji.201243140
118. Stahelin RV, Kong KF, Raha S, Tian W, Melowic HR, Ward KE, et al. Protein kinase C-θ C2 domain is a phosphotyrosine binding module that plays a key role in its activation. J Biol Chem (2012) 287:30518–28. doi:10.1074/jbc.M112.391557
119. Villalba M, Bi K, Hu J, Altman Y, Bushway P, Reits E, et al. Translocation of PKC[theta] in T cells is mediated by a nonconventional, PI3-K- and Vav-dependent pathway, but does not absolutely require phospholipase C. J Cell Biol (2002) 157:253–63. doi:10.1083/jcb.200201097
120. Anel A, Aguiló JI, Catalán E, Garaude J, Rathore MG, Pardo J, et al. Protein Kinase C-θ (PKC-θ) in Natural Killer Cell Function and Anti-Tumor Immunity. Front Immunol (2012) 3:187. doi:10.3389/fimmu.2012.00187
121. Garaude J, Kaminski S, Charni S, Aguilò JI, Jacquet C, Plays M, et al. Impaired anti-leukemic immune response in PKCtheta-deficient mice. Mol Immunol (2008) 45:3463–9. doi:10.1016/j.molimm.2008.03.016
122. Aguilo JI, Garaude J, Pardo J, Villalba M, Anel A. Protein kinase C-theta is required for NK cell activation and in vivo control of tumor progression. J Immunol (2009) 182:1972–81. doi:10.4049/jimmunol.0801820
123. Tassi I, Cella M, Presti R, Colucci A, Gilfillan S, Littman DR, et al. NK cell-activating receptors require PKC-theta for sustained signaling, transcriptional activation, and IFN-gamma secretion. Blood (2008) 112:4109–16. doi:10.1182/blood-2008-02-139527
124. Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor kappa B. Immunity (2006) 25:701–15. doi:10.1016/j.immuni.2006.10.010
125. Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature (2000) 404:402–7. doi:10.1038/35006090
126. da Silva AJ, Yamamoto M, Zalvan CH, Rudd CE. Engagement of the TcR/CD3 complex stimulates p59fyn(T) activity: detection of associated proteins at 72 and 120-130 kD. Mol Immunol (1992) 29:1417–25. doi:10.1016/0161-5890(92)90215-J
127. da Silva AJ, Janssen O, Rudd CE. T cell receptor zeta/CD3-p59fyn(T)-associated p120/130 binds to the SH2 domain of p59fyn(T). J Exp Med (1993) 178:2107–13. doi:10.1084/jem.178.6.2107
128. Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, et al. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science (2001) 293:2263–5. doi:10.1126/science.1063486
129. da Silva AJ, Raab M, Li Z, Rudd CE. TcR zeta/CD3 signal transduction in T-cells: downstream signalling via ZAP-70, SLP-76 and FYB. Biochem Soc Trans (1997) 25:361–6. doi:10.1042/bst0250361
130. Liu J, Kang H, Raab M, da Silva AJ, Kraeft SK, Rudd CE. FYB (FYN binding protein) serves as a binding partner for lymphoid protein and FYN kinase substrate SKAP55 and a SKAP55-related protein in T cells. Proc Natl Acad Sci U S A (1998) 95:8779–84. doi:10.1073/pnas.95.15.8779
131. Geng L, Raab M, Rudd CE. Cutting edge: SLP-76 cooperativity with FYB/FYN-T in the Up-regulation of TCR-driven IL-2 transcription requires SLP-76 binding to FYB at Tyr595 and Tyr651. J Immunol (1999) 163:5753–7.
132. Sylvester M, Kliche S, Lange S, Geithner S, Klemm C, Schlosser A, et al. Adhesion and degranulation promoting adapter protein (ADAP) is a central hub for phosphotyrosine-mediated interactions in T cells. PLoS One (2010) 5:e11708. doi:10.1371/journal.pone.0011708
133. Wu JN, Gheith S, Bezman NA, Liu QH, Fostel LV, Swanson AM, et al. Adhesion- and degranulation-promoting adapter protein is required for efficient thymocyte development and selection. J Immunol (2006) 176:6681–9. doi:10.4049/jimmunol.176.11.6681
134. Kliche S, Worbs T, Wang X, Degen J, Patzak I, Meineke B, et al. CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood (2012) 119:777–85. doi:10.1182/blood-2011-06-362269
135. Heuer K, Arbuzova A, Strauss H, Kofler M, Freund C. The helically extended SH3 domain of the T cell adaptor protein ADAP is a novel lipid interaction domain. J Mol Biol (2005) 348:1025–35. doi:10.1016/j.jmb.2005.02.069
136. Medeiros RB, Burbach BJ, Mueller KL, Srivastava R, Moon JJ, Highfill S, et al. Regulation of NF-kappaB activation in T cells via association of the adapter proteins ADAP and CARMA1. Science (2007) 316:754–8. doi:10.1126/science.1137895
137. Tian J, Pabst O, Römermann D, Skubich S, Förster R, Beckmann J, et al. Inactivation of T-cell receptor-mediated integrin activation prolongs allograft survival in ADAP-deficient mice. Transplantation (2007) 84:400–6. doi:10.1097/01.tp.0000269724.06142.92
138. Tian J, Rodriguez-Barbosa JI, Pabst O, Roemermann D, Foerster R, Beckmann J, et al. ADAP deficiency combined with costimulation blockade synergistically protects intestinal allografts. Transpl Int (2010) 23:71–9. doi:10.1111/j.1432-2277.2009.00924.x
139. Engelmann S, Togni M, Thielitz A, Reichardt P, Kliche S, Reinhold D, et al. T cell-independent modulation of experimental autoimmune encephalomyelitis in ADAP-deficient mice. J Immunol (2013) 191:4950–9. doi:10.4049/jimmunol.1203340
140. Mueller KL, Thomas MS, Burbach BJ, Peterson EJ, Shimizu Y. Adhesion and degranulation-promoting adapter protein (ADAP) positively regulates T cell sensitivity to antigen and T cell survival. J Immunol (2007) 179:3559–69. doi:10.4049/jimmunol.179.6.3559
141. Geng L, Pfister S, Kraeft SK, Rudd CE. Adaptor FYB (Fyn-binding protein) regulates integrin-mediated adhesion and mediator release: differential involvement of the FYB SH3 domain. Proc Natl Acad Sci U S A (2001) 98:11527–32. doi:10.1073/pnas.191378198
142. Menasche G, Kliche S, Bezman N, Schraven B. Regulation of T-cell antigen receptor-mediated inside-out signaling by cytosolic adapter proteins and Rap1 effector molecules. Immunol Rev (2007) 218:82–91. doi:10.1111/j.1600-065X.2007.00543.x
143. Horn J, Wang X, Reichardt P, Stradal TE, Warnecke N, Simeoni L, et al. Src homology 2-domain containing leukocyte-specific phosphoprotein of 76 kDa is mandatory for TCR-mediated inside-out signaling, but dispensable for CXCR4-mediated LFA-1 activation, adhesion, and migration of T cells. J Immunol (2009) 183:5756–67. doi:10.4049/jimmunol.0900649
144. Raab M, Wang H, Lu Y, Smith X, Wu Z, Strebhardt K, et al. T cell receptor ‘‘inside-out’’ pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity (2010) 32:541–56. doi:10.1016/j.immuni.2010.03.007
145. Kliche S, Breitling D, Togni M, Pusch R, Heuer K, Wang X, et al. The ADAP/SKAP55 signaling module regulates T-cell receptor-mediated integrin activation through plasma membrane targeting of Rap1. Mol Cell Biol (2006) 26:7130–44. doi:10.1128/MCB.00331-06
146. Burbach BJ, Medeiros RB, Mueller KL, Shimizu Y. T-cell receptor signaling to integrins. Immunol Rev (2007) 218:65–81. doi:10.1111/j.1600-065X.2007.00527.x
147. Burbach BJ, Srivastava R, Medeiros RB, O’Gorman WE, Peterson EJ, Shimizu Y. Distinct regulation of integrin-dependent T cell conjugate formation and NF-kappa B activation by the adapter protein ADAP. J Immunol (2008) 181:4840–51. doi:10.4049/jimmunol.181.7.4840
148. Burbach BJ, Srivastava R, Ingram MA, Mitchell JS, Shimizu Y. The pleckstrin homology domain in the SKAP55 adapter protein defines the ability of the adapter protein ADAP to regulate integrin function and NF-kappaB activation. J Immunol (2011) 186:6227–37. doi:10.4049/jimmunol.1002950
149. Srivastava R, Burbach BJ, Shimizu Y. NF-kappaB activation in T cells requires discrete control of IkappaB kinase alpha/beta (IKKalpha/beta) phosphorylation and IKKgamma ubiquitination by the ADAP adapter protein. J Biol Chem (2010) 285:11100–5. doi:10.1074/jbc.M109.068999
150. Rajasekaran K, Chu H, Kumar P, Xiao Y, Tinguely M, Samarakoon A, et al. Transforming growth factor-beta-activated kinase 1 regulates natural killer cell-mediated cytotoxicity and cytokine production. J Biol Chem (2011) 286:31213–24. doi:10.1074/jbc.M111.261917
Keywords: signaling, NK and T cells, immunotherapy, target molecules, NKG2D
Citation: Rajasekaran K, Riese MJ, Rao S, Wang L, Thakar MS, Sentman CL and Malarkannan S (2016) Signaling in Effector Lymphocytes: Insights toward Safer Immunotherapy. Front. Immunol. 7:176. doi: 10.3389/fimmu.2016.00176
Received: 07 February 2016; Accepted: 20 April 2016;
Published: 12 May 2016
Edited by:
Anahid Jewett, UCLA School of Dentistry and Medicine, USAReviewed by:
Stephan Gasser, National University of Singapore, SingaporeEliana Ruggiero, Ospedale San Raffaele, Italy
Copyright: © 2016 Rajasekaran, Riese, Rao, Wang, Thakar, Sentman and Malarkannan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew J. Riese, bWF0dGhldy5yaWVzZUBiY3cuZWR1;
Charles L. Sentman, Y2hhcmxlcy5sLnNlbnRtYW5AZGFydG1vdXRoLmVkdQ==;
Subramaniam Malarkannan, c3VicmEubWFsYXJAYmN3LmVkdQ==