 Jacobo López-Abente
Jacobo López-Abente Rafael Correa-Rocha
Rafael Correa-Rocha Marjorie Pion
Marjorie Pion- Laboratory of Immunoregulation, “Gregorio Marañón” Health Research Institute (IISGM), Madrid, Spain
Regulatory T cells (Tregs) play an important role in infections, by modulating host immune responses and avoiding the overreactive immunity that in the case of human immunodeficiency virus (HIV) infection leads to a marked erosion and deregulation of the entire immune system. Therefore, the suppressive function of Treg in HIV-infected patients is critical because of their implication on preventing the immune hyperactivation, even though it could also have a detrimental effect by suppressing HIV-specific immune responses. In recent years, several studies have shown that HIV-1 can directly infect Treg, disturbing their phenotype and suppressive capacity via different mechanisms. These effects include Foxp3 and CD25 downregulation, and the impairment of suppressive capacity. This review describes the functional mechanisms of Treg to modulate immune activation during HIV infection, and how such control is no longer fine-tune orchestrated once Treg itself get infected. We will review the current knowledge about the HIV effects on the Treg cytokine expression, on pathways implying the participation of different ectoenzymes (i.e., CD39/CD73 axis), transcription factors (ICER), and lastly on cyclic adenosine monophosphate (cAMP), one of the keystones in Treg-suppressive function. To define which are the HIV effects upon these regulatory mechanisms is crucial not only for the comprehension of immune deregulation in HIV-infected patients but also for the correct understanding of the role of Tregs in HIV infection.
Introduction
Human immunodeficiency virus (HIV) infection is hallmarked by a depletion of CD4+ T-cell pool and a persistent immune activation that may lead to a progressive degradation and senescence of the immune system (1, 2). Chronic immune activation is considered at present as a better predictor of HIV-1 disease progression than the extent of viral replication (2, 3). Furthermore, activated CD4+ T cells constitute the main target of HIV infection and its subsequent active replication (4); therefore, the onset of systemic immune activation will be favorable for the HIV spreading and viral persistence. One of the keystones responsible for preventing such immune hyperactivation and controlling immune cell proliferation are regulatory T cells (Tregs). Tregs are a subset of CD4+ T cells characterized by the high surface expression of the high-affinity interleukin 2 receptor alpha (CD25 or IL-2-Rα) and the nuclear transcription factor forkhead box P3 (Foxp3) (5–7). In the majority of infectious episodes, Tregs get activated and, thus, induced to proliferate in order to control overreactive immunity thanks to their suppressive capacity (8). Their suppressive function is achieved by several suppressor mechanisms, including disruption of metabolic pathways (CD39/CD73 axis, cyclic adenosine monophosphate (cAMP), and ICER pathways) (9–13), modulation of APC maturation and function (CTLA-4 and PD1/PD-L1-dependent tolerogenic APC induction) (14–19), production of anti-inflammatory cytokines (IL-10, TGF-β, and IL-35) (20–22), and induction of apoptosis (via Fas/Fas-ligand pathway, granzyme A/B and perforin, TRAIL, the galectin-9/TIM-3 pathway, or galectin-1 production) (23–27). Tregs can suppress immune responses and, therefore, limit collateral tissue injury; however, their function could be also favorable for pathogen persistence by suppressing antigen-specific T-cell responses (28). In fact, it is believed that Treg may contribute to inefficient cell-mediated immunity on chronic HIV infection since some studies showed increased Treg frequencies in such phase (29–31). However, Treg may also have beneficial effects by limiting immune activation and, hence, restricting potential targets for HIV infection, as well as minimizing the pathological effects resulting from HIV-mediated immune hyperactivation (9). Such beneficial versus detrimental effects of Treg in HIV infection have been a subject of controversy in recent years (30, 32–35). In addition, the fact that Tregs are a subset of CD4+ T cells expressing CXCR4 and CCR5 coreceptors makes these cells susceptible of being infected by HIV, complicating even more the comprehension of the real role of Tregs in HIV infection. Recent findings from our group demonstrated that HIV can directly infect Tregs producing a marked deregulation on the phenotype and functionality of the cells (36, 37), and decreased Treg absolute counts described in infected patients (38–40) supports the hypothesis that Treg deregulation might be related to the immune hyperactivation present in HIV-infected patients.
The aim of this review is to provide a detailed scope of those mechanisms and molecules that are crucial for either Treg modulation or functionality but may also play a role in HIV infection (CD25, Foxp3, CD39/CD73, cAMP, ICER, and CTLA-4). The second part of the review will focus on how HIV direct infection of Tregs might jeopardize each of the mechanisms described in the first part by suppressing the Treg cornerstone molecules: CD25 and Foxp3. Finally, we will discuss how HIV-mediated Treg deregulation might contribute to the overall immune-hyperactivation scene characterizing HIV-infected patients and their clinical progression.
Treg Immune-Modulation and Mechanisms of Action
CD25/IL-2 Axis, the Master Regulator in the Maintenance of Balanced Immune Responses
Treg homeostatic role depends on the surface expression of the high-affinity interleukin 2 receptor alpha (CD25 or IL-2-Rα) and IL-2 uptake (41, 42), thus receiving the name of CD25/IL-2 axis. The CD25/IL-2 axis is essential for Treg survival, expansion, homeostasis and their suppressive function. Regarding the CD25/IL-2 axis role in Treg function, it is assumed that activated T cells are the main IL-2 producers; thus, there is a negative feedback control of immune activation through IL-2 availability (37). As soon as the number of IL-2 secreting T cells and IL-2 concentration increase, Treg will react via cellular expansion, uptaking the extracellular IL-2 and, thus, activating their suppressive function. Both Treg production of suppressive cytokines and IL-2 consumption by Treg are pivotal mechanisms to prevent an excessive T-cell expansion and to re-establish the homeostasis of the immune system (43, 44). This mechanism guarantees that the relative Treg:T-effector ratio is continuously maintained even though the number of CD4+ T cells is significantly altered (41, 43). It has been shown that the Treg capacity to sense IL-2 is directly responsible for their function and IL-2 availability is an important mechanism by which Tregs exert their role (44). In humans, IL2RA gene polymorphisms affecting CD25 function have been associated with multiple sclerosis, type 1 diabetes, juvenile idiopathic arthritis, or lymphoproliferative-associated immunodeficiency (43, 45), highlighting the dependency of Treg in this receptor to exert their function. Furthermore, CD25/IL-2 signaling through STAT5 is essential to sustain Forkhead box P3 (Foxp3) expression on Treg (46, 47), which is a critical factor to keep Treg fate and function (6, 48). The CD25/IL-2 axis also plays a critical role in cAMP production, being cAMP a crucial regulator of immune cells. It has been shown that Treg activation by IL-2 leads to a significant upregulation in the adenylyl cyclase (AC) activity and, hence, to the cAMP cytosolic accumulation (11). The high-affinity receptor, CD25, enables the Tregs to uptake extracellular IL-2 in advantage compared to other cells (41). IL-2 removal by Treg will avoid the IL-2-associated downregulation of AC isoform 7 (AC7) in conventional T cell and, therefore, the reduction of intracellular cAMP levels (11). Favoring low cAMP levels in conventional T cells is associated with an increase in T cell proliferation. The role of cAMP in immune response modulation will be extensively studied in following paragraphs.
In the context of HIV infection, CD4+ T cells undergo a marked activation followed by a status of exhaustion and senescence (49). It would be expected to find an increased production of IL-2 due to the extended T-cell activation, which should activate the Treg response to limit an excessive activation/expansion of effector T cells. However, there is evidence that this mechanism is not working properly since it is observed that the CD4+ T cell pool is permanently activated, becoming finally exhausted (50) and the immune activation will persist in HIV-infected patients. Moreover, it was already described a reduction in IL-2-producing cells in moderate and advanced stages of HIV type-1 infection (51). An explanation would be that IL-2 expression is repressed in CD4+ T cells during chronic HIV infection due to the increased methylation of IL-2 promoter observed in infected patients (52). In addition to its role in the Treg/effector balance, IL-2 has proven to inhibit HIV-1 replication in cell lines by the induction of APOBEC3G (53). Moreover, the therapy with recombinant IL-2 has been tested in HIV-infected patients with the goal of both to recover the CD4+ T cell counts and to mobilize the reservoir of latent virus activating the latently infected CD4+ T cells (54–56). However, despite a sustained increase of the CD4+ T cells count, these clinical trials involving recombinant IL-2 plus antiretroviral therapy (ART) did not show any clinical benefit (57). This highlights that there are many factors involved and the modification of IL-2 is not enough to control the fate of the disease.
All that points out the relevance of a deregulation in the CD25/IL-2 axis as one of the mechanisms related to the immune imbalance and subsequent hyperactivation found in HIV-infected patients.
Foxp3, a Determinant Factor of Treg Identity and Functionality
Foxp3 is a crucial transcription factor determining Treg identity, development, and maintenance (6, 48). Expression of Foxp3 can also be induced and converts conventional CD4+ T cells into induced Treg cells (iTreg) (6). This iTreg generation could be observed in periphery or in vitro, and is induced by T-cell receptor (TCR) stimulation in the presence of IL-2 and TGF-β. Even though iTreg may be able to exert suppressive function as natural Treg, their Foxp3 is not stabilized by epigenetic modifications and, therefore, its expression is finally lost in vivo (58). Decreased Foxp3 expression in Treg is related to the switch to a cytokine-secreting profile characteristic from other CD4+ T cell helper lineages (48). Indeed, severe attenuation or ablation of Foxp3 expression resulted in the acquisition of the ability to produce effector cytokines, such as IL-2, IL-4, IL-17, TNF-α, and IFN-γ (48), and correct Foxp3 expression will suppress Th17 differentiation by inhibiting the function of the Th17 lineage-specifying transcription factor retinoid-acid receptor-related orphan receptor (ROR)-γt (59). Moreover, as transcription factor, Foxp3 can regulate gene expression of several genes, between which can be found CD39, adenine cyclase 9, and many others. Within its regulation role, it is interesting to note that Foxp3 forms a cooperative complex with NFAT, which is required to repress the cytokine IL-2 transcription and for upregulation of the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and CD25 expression (60–62), both relevant markers for Treg function. In HIV context, Foxp3 transfection demonstrated inhibitory effects upon HIV transcription in primary human CD4+ T cells (63, 64). HIV transcriptional downregulation could be associated with NF-κB-dependent and -independent mechanisms (63), as well as to the Foxp3-dependent inhibition of NFAT activity. It was shown that Foxp3 decreases the binding of NFAT2 to the HIV promoter in vivo by its capacity to sequestrate it (61, 64).
Several articles describe increased relative frequencies of Tregs (CD4+ T cells expressing Foxp3) in HIV-infected patients (29, 65). This increased Treg proportion could be explained by a preferential depletion of conventional CD4+ T cells or by an increased expansion and/or induction of Tregs (65). Due to the absence of appropriate markers to distinguish natural from induced Treg, it is difficult to know whether the inflammatory or hyperactivated context of HIV infection promotes the differentiation of conventional CD4+ T cells to iTreg. Jenabian et al. recently demonstrated that soluble CD40-ligand (sCD40L) levels are increased in plasma from HIV-infected patients, and this sCD40L induced Treg expansion and favored Treg differentiation from conventional CD4+ T cells (66). Since iTregs are not able to maintain a long-term Foxp3 expression and suppressive activity, and since they can even acquire a pro-inflammatory phenotype, further studies on the Treg dynamics are required to understand the role of iTregs in the context of HIV infection.
Extracellular ATP Metabolism and Signaling in T Cells: CD39/CD73 Axis
From among all the Treg mechanisms related to their suppressive capacity, adenosine triphosphate (ATP) metabolism is one that is well documented. In this context, there are two essential players that constitute the CD39/CD73 axis. CD39 or nucleoside triphosphate diphosphohydrolase 1 (NTDPase 1) is an ectoenzyme that hydrolyzes ATP or ADP to AMP (10). This enzyme is expressed by a subpopulation of Treg and, orchestrated together with another ectonucleotidase named AMPase CD73 present on the Treg surface, they are able to process AMP into adenosine (67). Adenosine exerts immune inhibitory effects as discussed in following paragraphs. It is interesting to note that Foxp3 expression is directly related to adenosine production since retroviral transduction of CD4+ CD25− lymphocytes with Foxp3 induced the expression of CD39 (6, 10), a potent inhibitor of cell proliferation and indirect contributor to the high cAMP levels found in Treg via adenosine generation (9).
In order to understand the formation of adenosine, we will describe the origin and relevance of ATP, which is the CD39/CD73 axis substrate. Extracellular ATP is released under hypoxia, inflammatory responses, metabolic stress, or other types of cell injury. The impact of extracellular ATP on the immune system is critical since its increase induces the activation of the inflammosome and subsequent release of cytokines, such as IL-1β (68, 69), in response to damage-associated molecular patterns (DAMPS) and pathogen-associated molecular patterns (PAMPS) (70). Therefore, extracellular ATP is considered a danger signal liberated by damaged or dying cells that induces pro- and anti-inflammatory signals. In the context of immune chronic activation as in HIV infection, ATP released by activated T cells seems to have an autocrine effect, prolonging activation and IL-2 secretion (71).
In contrast to ATP, adenosine exhibits anti-proliferative and inhibitory effects, hence giving to the CD39/CD73 activity an immune suppressive role (10). In fact, it was shown that induced Treg expressing CD39+ acquired higher suppressive capacity than CD39neg iTreg (72). Adenosine plays an antagonistic role on Treg compared to non-Treg responses by directly binding to the adenosine 2a receptor (A2AR), consequently inducing the adenylyl cyclase activity and, therefore, increasing the intracellular cAMP level. ATP removal and A2AR activation elicits inhibitory functions in dendritic cells and activated T-cell subsets, inducing T-cell anergy (73); whereas in Treg, A2AR induces the generation of Foxp3+ Tregs (73) and enhances Treg immunosuppressive mechanisms (74, 75). Summing up, Treg could dampen immune activation as well as induce activated T-cell dysfunction through CD39/CD73 activity.
It is interesting to note that a study of CD39/CD73 distribution in Treg and conventional CD4+ T cells showed that even though CD39 is largely expressed on human Treg (CD4+ CD25hiFoxp3+ T cells), CD73 is not so widely expressed and <1% of Treg expressed both ectonucleotidases at their surface (76–78). One hypothesis is that only few cells capable of hydrolyzing ATP to adenosine are necessary to induce a local suppression and that this pathway must be finely regulated to avoid any unwanted Treg-suppressive function. Moreover, it was described that CD73 was present in cytoplasmic granules and that its expression at the surface of Treg might be transient, which would be another level of regulation of adenosine production (77, 78).
In the case of HIV infection, there is high expression of CD39 in Treg cells, which remains unaltered even with ART (31, 79). CD39+ Treg frequency and number are elevated in HIV patients, and correlates negatively with CD4+ T-cell recovery and positively with plasma viral load and T-cell activation (80, 81). In addition, through a case-control study, a genetic variant of CD39 associated with lower expression of CD39 enzyme was linked to a slower progression to AIDS (81). Moreover, in vitro suppression assays demonstrated that the suppressive effect of CD39+ Treg upon gag-stimulated CD8+ cytokine production was partially inhibited when adding CD39 blocking monoclonal antibodies (81). All this may highlight a negative role of CD39+ Treg in controlling HIV progression. However, there is still controversy regarding the role of CD39 in HIV infection, Schulze et al. demonstrated that HIV controllers were shown to have similar CD39+ Treg to healthy subjects (79). Additionally, it has been shown that Foxp3 modulates the expression of CD39 at the surface of Treg and also regulates HIV promoter’s transcription activity. Moreover, CD39 might also be contributing to hindering HIV infection as suggested in following paragraphs.
In summary, Treg might have two contrary functions on HIV replication and disease progression. On the one hand, CD39 expression may be involved in Treg-mediated suppression of HIV-specific responses and, thus, on disease progression. On the other hand, Foxp3 induce a negative effect on HIV transcription, which could limit new particles production or induce HIV latency in Foxp3-expressing cells. However, one could claim that CD39+ Tregs may be critical for the inhibition of T-cell immune activation, which may reduce the niche for HIV replication (82, 83). In fact, blocking CD39 and, thus, inducing the decrease of cAMP levels in Treg were shown to abrogate the Treg-mediated suppression of HIV replication (84). Therefore, it is possible that CD39+ Foxp3+ Tregs could control HIV infection, especially during the first days of infection, prior to the HIV dissemination to the secondary lymphoid organs, phase where just a few effector T cells are activated (84).
CD38 and NAD+ as Precursor of Adenosine
Few years ago, an alternate route to producing extracellular adenosine was discovered. This axis had its pivotal role around the NAD+-glycohydrolase CD38, the 5′-ectonucleotidase CD73 and the ecto-nucleotide pyrophosphatase/phosphodiesterase (PDE) CD203A (85). Leaded by CD38, main NAD+ consuming enzyme (86), it has been shown that CD38/CD203A/CD73 will transform NAD+ into adenosine (85), whose function is extensively described in this review. Horenstein et al. and others demonstrated in different in vitro models that this alternate pathway may synergize, flank, or bypass the aforementioned described canonical catabolic pathway orchestrated by CD39 (85–88). Regarding to the Treg population, it has proven to express CD38 (89, 90) and seems that its level of expression positively correlates with their suppressive function (90). Patton et al. demonstrated in a murine model that CD38high Tregs have higher suppressive capacity than CD38low Treg, which failed to upregulate CD73, key molecule for adenosine production, in both canonical and CD38 pathway. However, it is still under evaluation if Tregs are capable of processing NAD+ through this newly discovered pathway (85, 91).
Regarding to the role of CD38 in the context of HIV infection, this enzyme is commonly regarded as a T-cell activation marker, and peripheral blood CD38+ CD8 T cells have been strongly correlated with disease progression in untreated HIV infection (92, 93). It has been shown that rectal Treg frequency is positively related to CD38+ CD4+ and CD8+ rectal T cells in chronic HIV-positive non-controllers (94). However, this may vary depending on the sampling site and time point of infection, since Tregs are inversely correlated to CD38+ CD8+ T cells in blood at primary HIV infection as others have shown (95). Summing up, CD38 is not only a predictive marker of disease progression but also may be another mean for Treg to produce adenosine and exert its suppressive function.
ICER and cAMP, Differential Role on CD4+ T-Cell Subsets
Cyclic adenosine monophosphate is a derivative of ATP. cAMP acts as a secondary messenger involved in many biological processes and in regulation of immune cells, playing an important role on determining the balance between suppression and activation. In fact, cAMP present on conventional T cells or on Treg is implicated in different signaling pathways.
cAMP Is a Major Mediator of Treg-Suppressive Potential
In Treg, the suppressive potential has been demonstrated to depend on cAMP (96). Indeed, Tregs tend to preserve their high levels of intracellular cAMP. To partially explain the maintenance of such high levels, it has been shown in murine nTreg cells that the expression of the cAMP degrading PDE3b is reduced by direct binding of Foxp3 to the PDE3B promoter (97), limiting Treg intracellular cAMP degradation. Treg can induce the intracellular cAMP production by the sequential processing of extracellular ATP to adenosine by the CD39/CD73 axis, inducing adenylyl cyclase activity in the target cell through A2AR (69, 75, 78). The importance of A2AR in mediating the inhibition of inflammation was demonstrated in A2AR null mice, where the transgenic animals suffered of massive tissue destruction and systemic inflammation due to their inability to control inflammation (98). It is interesting to note that A2AR engagement using an agonist not only was able to induce TGF-β and Foxp3 expression on activated T cells (73) but was also able to increase the intracellular cAMP level on purified Treg. Therefore, A2AR engagement increases the number of Tregs, activates their immunoregulatory activity, and allows iTreg generation. Thus, it has already been described how IL-2R, A2AR activation, and Foxp3 expression contribute to the “supraphysiological” intracellular levels of cAMP found in Treg. Treg can mediate cAMP transfer to activated CD4+ T cell and dendritic cells through gap junction intercellular communications (GJICs) (75, 96, 99). The formation of such channels composed by two opposing hemichannels allows the connexion between adjacent cells (100), therefore increasing intracellular cAMP in the target cells (96). In the case of DCs, cAMP induces the rapid downregulation of the co-stimulatory molecules CD80 and CD86, preserving their tolerogenic phenotype and indirectly regulating T-cell function (101). Furthermore, cAMP induction in DCs, following activation of adenosine and prostaglandin receptors, downregulates NF-κB activity not only decreasing the DC production of cytokines, such as TNF-α, IL-6, IL-12, IL-1α, and chemokines MCP-1 and MIP-1α, but also increasing the production of the anti-inflammatory cytokine IL-10 (102).
cAMP and ICER, Key Modulators of Conventional T-Cell Function
On conventional T cells, A2AR expression is induced after TCR stimulation via NF-AT-signaling pathway (73). This upregulation of A2AR expression was followed by an increase in intracellular cAMP level, demonstrated by the use of an A2AR-specific agonist (73). cAMP activation triggers a number of downstream signaling cascades on conventional CD4+ T cells. The main stream pathway is the activation of the cAMP-dependent protein kinase-A (PKA), which is involved in the regulation of several physiological processes as modulation of ion channels and transcription of genes (103). PKA will activate the transcription factor CREB by phosphorylation, consequently triggering the interaction of phospho-CREB and the nuclear protein CBP, which is a coactivator that augments the activity of phosphorylated CREB. Afterwards, CREB binds to a DNA element known as the cAMP-regulated enhancer element (CRE) activating the transcription of cAMP-responsive elements (104). Thus, consequently triggering the formation of the induced cAMP early repressor (ICER), which is generated from the 3′ region of the gene encoding the cAMP response element modulator (CREM) due to alternative promoter binding (105). ICER will bind to CRE and thereby represses the activity of its own promoter, constituting then a negative auto-regulatory loop (105). In fact, ICER was considered a master regulator of the cAMP response, inhibiting T-cell function, most specifically downregulating IL-2 production on effector T cells, critical for T-cell growth (12, 106). When ICER is transgenically over-expressed in mice T cells, these lymphocytes present proliferative defects hallmarked by decreased IL-2 production (13), similar to the effect on IL-2 production resulting from the co-culture of CD4+ T cells and nTreg (12). In parallel, when intracellular cAMP levels rise on effector T cells, NFAT/ICER complexes are formed; this will play an important role on suppressing NFAT activity via its sequestration. Therefore NFAT will not be accessible for the cytokines and chemokines transcriptional induction, such as IL-2, IL-4, TNF-α, IL-13, MIP-1α, MIP-1β, and GM-CSF (107, 108). Taken together, all this information highlights the critical role of ICER induced by cAMP transferred from nTreg and which mediates suppression of several immune mediators, especially IL-2 production on effector T cells (109) and, hence, demonstrates the relevance of the cAMP/PKA pathway in suppressing T-cell responses.
HIV Enhances Intracellular cAMP, a Double-Edged Immune Sword
It has been shown that intracellular cAMP levels of T cells from HIV-infected patients are higher than those from healthy subjects (110, 111). In vitro studies corroborated the presence of higher cAMP levels in HIV-infected primary T cells and T cell lines compared to uninfected cells (111, 112). Interestingly, HIV infection was not required to increase intracellular cAMP, since the T cell treatment with the envelope glycoprotein gp120 was already sufficient to elicit such response and activate cAMP/PKA pathway through Treg function (113, 114). However, the mechanisms underlying gp120-mediated response are still unresolved. The potential benefit or hazard of intracellular cAMP in the context of HIV infection is still a matter of debate.
It has been shown that cAMP/PKA pathway can inhibit HIV-specific T-cell responses and such inhibition will favor viral replication. Besides inhibiting immune responses, HIV induces impairment on proliferation in CD4+ and CD8+ T-cell subsets, through the activation of the cAMP/PKA pathway by viral proteins. In fact, the pre-treatment of the cells with a PKA inhibitor prevented the induction of anergy (112), highlighting the negative role of high intracellular cAMP resulting from HIV infection. In a similar fashion, CD39+ Tregs may be involved in the suppression of HIV-specific T-cell responses through the production of adenosine and consequent rise of cAMP in conventional T cells as shown by Nikolova et al (81). Similar to the PKA inhibitor experiment, it was demonstrated that the use of adenosine deaminase that hydrolyzes adenosine permits to revert the inhibition of immune responses and enhances the HIV-1-specific effector responses in an ex vivo model (115). On the other hand, Tregs could exert a direct inhibition of HIV infection and replication in conventional T cells by cell-to-cell contact. This mechanism is cAMP dependent and involved the PKA activation pathway (84). Therefore, cAMP may also play a positive role by inhibiting viral replication. In fact, there are a number of examples illustrating cAMP benefits; Banas et al. showed in infected monocytes and T-cell lines that the elevation of intracellular cAMP activates CREB protein that will compete for limiting amounts of CBP/p300 thereby mediating a repressive effect on HIV LTR/enhancer transcriptional activity (116). Furthermore, the use of rolipram, a PDE4 inhibitor, prevented the degradation of cAMP consequently inhibiting the transcription of the genes under the control of HIV-LTRs (117, 118). Interestingly, the blockage of PDE4 activity also prevented the nuclear import of HIV DNA in memory T cells (118). cAMP may also limit viral infection by modulating the expression of the T-cell coreceptors needed for viral entry, indeed it was shown a downregulation of CCR5 and CXCR4 in a T-cell line using an agonistic-like anti-A2AR mAb that activated the cAMP/PKA pathway (119). Summing up, cAMP seems to play an important role on preventing HIV entry, DNA nuclear import, viral transcription, and viral replication. All this highlights the beneficial role of Treg on limiting HIV infection in conventional T cells since they remove ATP by generating adenosine and transfer high amounts of cAMP to non-Tregs through GJICs that subsequently limit HIV replication.
CTLA-4
CTLA-4 is a negative immune-modulator receptor of the CD28 superfamily of immune-regulatory molecules (such as CD80 or CD86) expressed in Tregs and with clear involvement in HIV disease. Its suppressive mechanisms have been extensively studied (14–17). Of note are Sakaguchi’s findings, where he demonstrated that nTreg-specific CTLA-4 deficiency or CTLA-4 blockade using anti-CTLA-4, impaired or abrogated nTreg-suppressive function in vivo and in vitro, respectively (14, 16). This is possible since nTreg will interact through CTLA-4 with activated or CD80/CD86 expressing conventional CD4+ T cells and/or APCs, thus mediating suppression. It is claimed that such interaction may act synergistically with expression of ICER in conventional T cells mediated by Treg (12). On the other hand, CTLA-4 has been proved to be selectively upregulated in HIV-specific CD4+ T cells in HIV-infected patients, and its expression correlated positively with disease progression (49). Interestingly, HIV-gp120 protein enhanced Treg-mediated suppression through CTLA-4 upregulation (114). Such enhancement is probably mediated by cAMP/PKA pathway since cAMP itself was shown to induce CTLA-4 upregulation in the absence of full T-cell activation (120). In fact, it could be reasonable to assume that CTLA-4 may synergize with ICER since it will be also upregulated due to cAMP/PKA downstream activation. However, despite the evidence of CTLA-4 involvement in HIV infection, other study showed that blocking CTLA-4 with an anti-CTLA-4 antibody did not demonstrated any differences in the frequency of HIV-p24gag-conventional T cells when these cells were co-cultured with Treg (84). Therefore, CTLA-4 might not play a major role in the Treg-suppressive activity in HIV infection. Taking all this information together, CTLA-4 would not act directly on the HIV replication, but would act through the suppression of HIV-specific CD4+ and CD8+ T cells function. Additionally, the presence of HIV or cAMP that induces CTLA-4 expression at the surface of Treg would increment this capacity.
HIV Infection Impairs Treg Activity
Susceptibility of Treg to Infection
Tregs express HIV-coreceptors CCR5 or CXCR4 at levels comparable to other CD4+ T cells (79), which renders Treg susceptible to HIV infection (36, 37, 121–123). Furthermore, naïve Tregs are capable of upregulating the membrane expression of CXCR4 and CCR5 upon TCR stimulation (122), increasing their susceptibility to infection. The two viral strains CXCR4 and CCR5-tropic HIV showed differential infection dynamics. We described in vitro that CXCR4 virus produce a stronger deregulation than CCR5-HIV in both phenotype and functionality when Tregs are infected (36). Other studies report that HIV-CCR5 infects Tregs at lower levels compared to Teff at both early and late points (121). By contrast, Tregs were more efficiently infected with CXCR4 strain compared to Teff at early time points, but the difference cleared at later time points of the virus life cycle (121). Whether Tregs constitute a major target for HIV or if they are more susceptible than conventional T cells to infection has been a matter of debate in the past years. However, recent in vivo studies have shown that circulating Tregs are not preferentially infected by HIV compared with effector T cells (121), in fact HIV infection induces deep cellular deregulation in CD4+ T cells, including the Treg subset. The HIV-mediated Treg deregulation affects the cell number, their phenotype, and functionality. During chronic HIV infection, the absolute Treg numbers in peripheral blood decline (40) even though the Treg frequency among the total CD4+ T-cell population is increased. In addition, our group has described that direct HIV infection of Tregs modifies the Treg phenotype and induces a strong impairment in their suppressive capacity (36). A similar outcome was observed in a study of HIV-infected patients with immune reconstitution disease after ART, they described that Treg exhibited reduced immunosuppressive capacity that was associated with over-active CD4+ T-cell responses (124). We also demonstrated in vivo an impaired capacity of Tregs to keep the balance between Treg/IL-2-producing cells in viremic HIV-infected patients due to direct Treg viral infection (37), and probably contributing to the immune hyperactivation observed in HIV patients.
These findings provide clarifying information to the debate concerning the beneficial versus the detrimental role of Tregs in HIV infection. Since the Treg number and function is impaired in the presence of HIV, the negative impact on the HIV-specific responses might be limited. However, Treg impairment has probably a higher impact in the absence of an adequate control of immune hyperactivation, which at present is the major factor related to the HIV disease progression in infected patients.
HIV Downregulates Foxp3 Expression and Impairs the Suppressive Activity of Treg
We demonstrated that HIV infection of Treg modifies its phenotype and functionality through a process mediated by intrinsic methylation mechanisms (36), possibly in a Treg attempt to protect its genome against the expression of foreign DNA (125). Foxp3 gene expression is strongly downregulated in HIV-infected Treg, notably with CXCR4-tropic HIV, due to general increase in the CpG methylation pattern of two regulatory sites critical for Foxp3 expression in Tregs (36). It is known that DNA methylation in Treg can be addressed to two known DNA methyl transferases (DNMT1 and DNMT3b), since binding sites for DNMT1 and DNMT3b have been detected in Foxp3 locus (126). Interestingly, we have shown that the expression of DNMT3b was upregulated in HIV-infected Treg compared to non-infected Treg (36). Therefore, the DNMT3b increase might be responsible for the de novo methylation observed in Foxp3 promoter and, thus, be responsible for the subsequent downregulation of Foxp3 expression observed in vitro in HIV-infected Treg (36).
As already described, Foxp3 expression is critical for Treg-suppressive function (127). It was shown that in vitro HIV-infected Treg were less capable of suppressing the proliferation of T-effector cells in co-culture experiments (36, 128). These results support the impaired Treg-suppressive capacity that we observed in untreated HIV patients with high viral load (unpublished data). However, other studies claim that HIV does not impair Treg-suppressive function in patients (35, 129, 130) probably because they studied it in chronic infected patients, and it is known that the number of infected Treg is very low at the late stage of infection (121). Interestingly, the treatment of HIV-infected Treg with antiretroviral drugs, such as ZDV or T20, in vitro reverted the impairment of suppression since the loss of Foxp3 expression was avoided (36). Therefore, it seems that the Foxp3 downregulation may contribute to the Treg loss of suppressive function upon HIV infection. There are many suppressive mechanisms on Tregs susceptible to be affected by Foxp3 reduction as described previously. Foxp3 downregulation may have critical consequences on Foxp3-transcriptional program, being PDE3B gene the most repressed by Foxp3 (60, 127); therefore, its repression might be abrogated if Foxp3 is downregulated and could have detrimental effects upon Treg. In fact, it was shown by Gavin et al. that retroviral gene transfer and expression of PDE3B into Tregs was deleterious to Treg homeostasis (127). Such detrimental effect consisted in a significant reduction of PDE3B-expressing Treg numbers due to cell death or inefficient expansion, and it is most probably related to decreased cAMP intracellular levels due to its degradation by PDE3B (127). Other mechanism related to Foxp3 repression is associated with miR-142-3p transcriptional regulation (131). Foxp3 downregulates the miR-142-3p in order to keep the AC9/cAMP active in Treg since miR-142-3p was described to target AC isoform 9 expression, thus, reducing the intracellular cAMP levels. Therefore, downregulation of Foxp3 expression will lead to miR-142-3p expression and this will have an indirect effect on Treg cAMP levels. Therefore, miR-142-3p might potentially contribute to the decrease in Treg absolute numbers and Treg function impairment shown in HIV, associated with the inability to maintain high cAMP levels. Besides this mechanisms affecting overall cAMP levels, it has been shown that the repression of Foxp3 using siRNA in hepatocellular carcinoma patients induces a downregulation of CTLA-4 in those Treg with lower Foxp3 levels, compromising their function (132). Thus, similar outcomes may be expected from HIV-infected Treg whose Foxp3 gene is downregulated.
Besides transcriptional regulation, it is known that Foxp3 can also be regulated by various post-translational modifications, such as acetylation, ubiquitination, and phosphorylation, thus affecting its activity (133, 134). A decade ago, Li et al. and others showed that the transcriptional repression by Foxp3 involves a histone acetyltransferase (HAT) complex that includes HAT TIP60 and class II histone deacetylase 7 (HDAC7) (135, 136). Within this complex, TIP60 is the principal responsible for Foxp3 acetylation and Foxp3-mediated transcriptional regulation via STAT3 in vivo (136). Therefore, the association of HDAC7 and TIP60 to the Foxp3 N-terminal domain resulted to be a requirement for Foxp3-mediated transcriptional repression in terms of suppression of IL-2 production in T cells after TCR stimulation (136). In the same line as the previous comment, there are a number of animal models such as the TIP60 conditional knock out mice supporting the relevance of Foxp3 complex formation (133). Indeed, several studies showed that the failure of Foxp3 to associate with TIP60 and HDAC7 leads to altered Foxp3-dependent transcription (higher promoter activity and production of IL-2) and epigenetic modifications, limiting nTreg function at inflammatory sites and decreasing the Treg population in peripheral immune organs, consequently resulting in accelerated fatal autoimmune disease (133, 136, 137).
In terms of HIV infection, it has been shown that HIV-1 transactivator Tat protein represses TIP60 activity (138). Indeed, Tat induces the poly-ubiquitination and, thus, the degradation of TIP60 by the p300/CBP-associated E4-type ubiquitin-ligase activity (139), consequently affecting the Foxp3–TIP60–HDAC7 complex formation. Therefore, it could be expected a limited Treg function or decreased Treg levels as shown in the animal models (133, 137) (See Figure 1). In fact, the HIV-dependent disruption of Foxp3 complex may be contributing to the low Treg absolute numbers detected in HIV patients (140, 141).
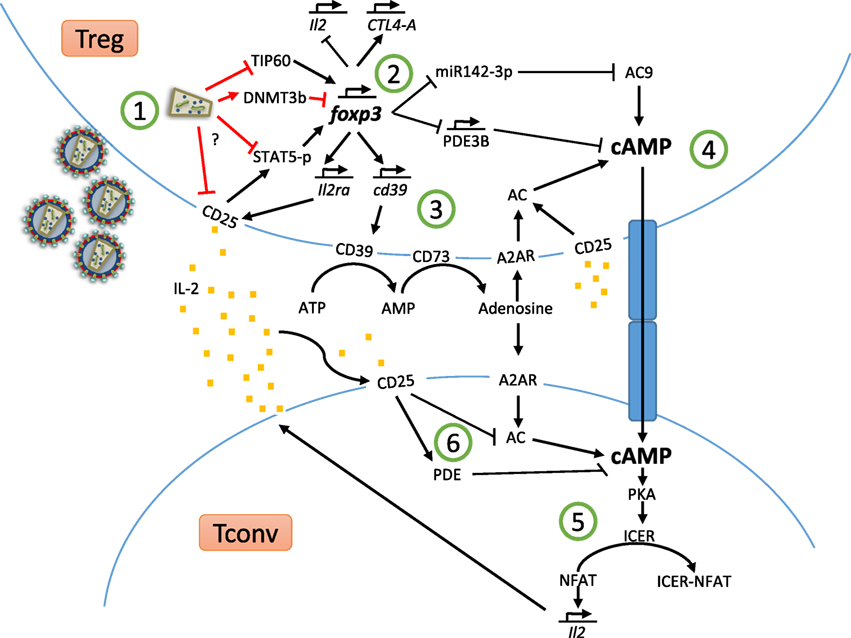
Figure 1. Treg suppressor mechanisms and HIV-dependent Treg dysregulation.
Finally, HIV infection also leads to a disturbance on interleukin secretion profile (36) probably associated with Foxp3 downregulation (48, 142). Indeed, HIV-infected Tregs have shown a significant increase in IL-4 production and a decrease in TGF-β production (36). Since Foxp3 has a deep influence on Treg-associated genes expression (62) and that Foxp3 expression is highly modified during HIV infection (36), we can assume that such dysregulation in the production of cytokines may affect Treg function, since this dysregulation correlates with Treg switch to a T helper type 2 cytokine profile that may contribute to the hyperactivation shown in HIV patients.
Impaired Balance between Regulatory T Cells and IL-2 Producing CD4+ T Cells in HIV Infection
The preserved and constant ratio between Treg and the number of activated T cells constitutes a homeostatic mechanism ensuring that T-cell activation and expansion remain under control (41). However, the persistent immune hyperactivation that drives HIV disease suggest that there is a dysfunction on such mechanism, since the Treg values are decreased despite high levels of activated T cells (34, 123, 140). In fact, a recent study indicates that the balance between Treg and IL-2 producing T-cell numbers is deeply disturbed in viremic HIV-infected patients (37). Such balance impairment is motivated by a HIV-mediated downregulation of IL-2 receptor alpha (CD25) at the surface of Treg from viremic patients, as confirmed by in vitro experiments of Treg infection with HIV (37). Therefore, the lower expression of CD25 observed in Treg from infected patients could affect their capacity of expansion due to the impossibility of responding to extracellular IL-2 produced by effector CD4+ T cells. Furthermore, decreased CD25 expression may also reduce Treg-suppressive function. Indeed, it has been reported in healthy donors that a lower CD25 expression correlates with impaired suppressive capacity and lower capability to maintain Foxp3 expression (143). CD25 downregulation correlates with the reduction of STAT5 activation in HIV-infected Treg (37), being STAT5 a key player on CD25 activation downstream cascade. Loss of phospho-STAT5 has been shown to abrogate Treg-suppressive activity and to induce the downregulation of Foxp3 expression (144). STAT5 and CD25 genes contain differentially methylated regions, and many of these regions present DNA methylation-dependent enhancer activity (145). Therefore, STAT5 and CD25 expression could be controlled by DNA methylation as occurs with Foxp3, thus explaining their downregulation upon infection. Therefore, the downregulation of Foxp3 expression, the functional impairment of Treg (36), and the impairment of CD25 pathway (37) observed in Treg from infected patients could be critical in the immune hyperactivation that hallmarks HIV disease.
HIV Modifies the Treg Cellular Transcriptome Following Infection
It was shown by using an ultrasensitive multiplexed NanoString gene expression assay that HIV-1 modifies the Treg transcriptome after infection (128). Infected and non-infected Treg formed two distinct clusters showing differential expression in a total of 23 genes (128). HIV-infected Tregs show downregulation of genes linked to TCR signaling (PTPRC, TCR, IKBKE, CD3E), which may affect Treg antigen recognition and activation, and CD27 and PPAR-gamma that are related to suppressor function and enhancement of Treg function, respectively (146, 147). Furthermore, many other genes involved in function and Treg lineage were downregulated (TRAF6, STAT5, ETS-1, LEF-1) (148–151) but also Treg function inhibitor genes were upregulated after infection (LTA; IL-18RAP, BCL6, IL-7) (152–155), probably contributing to the HIV-dependent Treg impairment. Moreover, LEF-1, a gene that contributes together with Foxp3 in controlling transcription in Treg (151), and STAT5a, essential for Foxp3 expression, were downregulated (36, 46). However, in the Angin et al. study, Foxp3 downregulation was not detected and suggested a possible destabilization of the master transcription factor by downregulation of ETS-1 (148). Despite of these results, in recent years, we have shown that indeed HIV infection downregulates Foxp3 expression (36). Summing up, HIV-1 infection seems to exert Treg deregulation through a number of pathways simultaneously. The transcription pattern modification of critical genes involved in Treg function will probably play a detrimental role allowing HIV-1 hyperactivation and pathogenesis.
Mechanism Summary
In Figure 1, we have gathered most of the Treg suppressor mechanisms covered in the review and how HIV-dependent dysregulation of Foxp3, CD25, and STAT5 may hinder Treg function.
Upon viral entry and replication (Figure 1 ①), HIV-1 Tat protein induces the poly-ubiquitination and degradation of TIP60, consequently disrupting the Foxp3–TIP60–HDAC7 complex formation, critical for Foxp3 function. Furthermore, Tregs suffer a downregulation of CD25 expression and, hence, a reduction in phosphorylated STAT5 levels. Additionally, HIV infection leads to DNMT3b upregulation, this methylase will methylate two regulatory sites critical for Foxp3 expression, inhibiting Foxp3. Moreover, STAT5 is essential to sustain Foxp3 expression, its downregulation further compromises Foxp3 level. With a reduced expression of Foxp3 (Figure 1 ②), miR142-3p and PDE3B expression will not be downregulated; therefore, PDE3B will start to degrade intracellular cAMP and miR142-3p will block AC9 mRNA, inhibiting cAMP production. Furthermore, Foxp3 induces CD39 gene expression; therefore, the lack of Foxp3 could result in decreased levels of CD39 and, hence, lower conversion of extracellular ATP to adenosine (Figure 1 ③). Low levels of extracellular adenosine will result in decreased signaling through A2AR and, hence, less induction of AC activity and lower cAMP production in either Tregs or conventional T cells. After viral infection, Treg may not be able to maintain their cAMP supraphysiological levels (Figure 1 ④), hence transferring less cAMP to conventional T cells through GJICs compared to optimal conditions. Insufficient cAMP levels in conventional T cells upon cellular contact, together with the decreased endogenous production of cAMP (Figure 1 ⑤), will result in lower ICER production via PKA activation and, therefore, NFAT will maintain the induction of pro-inflammatory cytokines, such as IL-2. NFAT will also bind to HIV-promoter enhancing HIV genes transcription. HIV-infected Treg may not only be unable to suppress the activity and IL-2 production by conventional T cells, but also will not be able to sense an increase in extracellular IL-2 levels as a result of T-cell hyperactivation (Treg CD25 is downregulated), thus impairing Treg proliferation and suppressive response. In addition, CD25 downregulation renders Treg less capable of consuming IL-2, this means higher IL-2 availability for conventional T cells. IL-2 captured by conventional T cells induces activation of PDE and AC7 downregulation, thus leading to intracellular cAMP degradation. Low intracellular cAMP is related to increased T-cells proliferation and activation. As a result of these HIV-mediated alterations of regulatory mechanisms, the balance between activated T cells and Treg will be broken, compromising the homeostatic fine-tuned balance of the immune system.
Concluding Remarks
Retroviruses, such as HIV, have coexisted with the human race for millions of years. By that, host species have evolved to protect themselves by different mechanisms, such as restriction factors, but also by modulating the immune system to reach a fine-tuned response against the virus and other pathogens. As a consequence, virus counter-evolution leads to an evolutionary arms race. This phenomenon is well illustrated in Leigh Van Valens’s Red Queen Hypothesis, where two evolutionary systems, such as host/pathogen are constantly developing in order to maintain its fitness relative to the system they are co-evolving with. In this review it has been described how Treg exert a number of immune-modulatory mechanisms capable of hindering viral entry, replication and transcription but also guaranteeing the immune system homeostasis. However, HIV is capable of infecting and abrogating Treg function by inducing the downregulation of key molecules essential to enable Treg-dependent suppression. Impairment of Treg immune-modulatory function and induction of hyper-immune activation, provide an immunological condition favorable for viral infection and replication. The fact that HIV might exert a suppressive effect upon Treg function help us to understand that Treg role might be detrimental for HIV fitness and disease progression. This hypothesis means that the beneficial role of Treg on preventing immune hyperactivation outweighs the detrimental effect of Treg upon HIV CD4+ and CD8+-specific responses, which has been a matter of controversy in recent years. Up to date, it has been extensively described by others the pros and cons of Treg immunomodulatory mechanisms in HIV-infected patients and its effect upon disease progression. However, the increasing evidence of HIV’s direct impact upon the Treg population adds an additional layer of complexity considering the dysregulation of HIV-infected Treg and the contribution of such alterations to the disease progression. Understanding the immune-regulatory mechanisms that are impaired or deregulated will facilitate to unveil the rationale behind the immune hyperactivation that hallmarks HIV disease. This will open new avenues to find potential targets that could restore the immune homeostasis and, hence, decrease the systemic immune activation and exhaustion that determine the disease progression.
Author Contributions
JL-A, RC-R, and MP have all participated in the conception, discussion, and elaboration of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
All the authors declare that they have no competing financial interests. This work was supported by grants from Instituto de Salud Carlos III (ISCIII) co-financed by FEDER funds (PI12/00934; PI12/016763; ICI14/00282; PI15/00011; PI15/00923). RC-R is supported by the ISCIII through the “Miguel Servet II” program (CPII13/00033) and M P is supported by the Spanish MICINN through the Ramón y Cajal I3 Program. JL-A is supported by an IISGM pre-doctoral grant.
References
1. Massanella M, Negredo E, Perez-Alvarez N, Puig J, Ruiz-Hernandez R, Bofill M, et al. CD4 T-cell hyperactivation and susceptibility to cell death determine poor CD4 T-cell recovery during suppressive HAART. AIDS (2010) 24(7):959–68. doi:10.1097/QAD.0b013e328337b957
2. Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS (2003) 17(13):1881–8. doi:10.1097/00002030-200309050-00006
3. Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol (2002) 169(6):3400–6. doi:10.4049/jimmunol.169.6.3400
4. Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol (2003) 21:265–304. doi:10.1146/annurev.immunol.21.120601.141053
5. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi:10.1146/annurev.immunol.21.120601.141122
6. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
7. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
8. Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, et al. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol (2005) 79(12):7860–7. doi:10.1128/JVI.79.12.7860-7867.2005
9. Moreno-Fernandez ME, Rueda CM, Velilla PA, Rugeles MT, Chougnet CA. cAMP during HIV infection: friend or foe? AIDS Res Hum Retroviruses (2012) 28(1):49–53. doi:10.1089/AID.2011.0265
10. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110(4):1225–32. doi:10.1182/blood-2006-12-064527
11. Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+ T cells. Mol Immunol (2010) 47(4):678–84. doi:10.1016/j.molimm.2009.10.032
12. Bodor J, Fehervari Z, Diamond B, Sakaguchi S. ICER/CREM-mediated transcriptional attenuation of IL-2 and its role in suppression by regulatory T cells. Eur J Immunol (2007) 37(4):884–95. doi:10.1002/eji.200636510
13. Bodor J, Feigenbaum L, Bodorova J, Bare C, Reitz MS Jr, Gress RE. Suppression of T-cell responsiveness by inducible cAMP early repressor (ICER). J Leukoc Biol (2001) 69(6):1053–9.
14. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
15. Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med (2000) 192(2):295–302. doi:10.1084/jem.192.2.295
16. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192(2):303–10. doi:10.1084/jem.192.2.303
17. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206(8):1717–25. doi:10.1084/jem.20082492
18. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood (2009) 113(23):5811–8. doi:10.1182/blood-2009-02-203141
19. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi:10.1016/j.immuni.2004.08.010
20. Kindlund B, Sjoling A, Yakkala C, Adamsson J, Janzon A, Hansson LE, et al. CD4 regulatory T cells in gastric cancer mucosa are proliferating and express high levels of IL-10 but little TGF-beta. Gastric Cancer (2016). doi:10.1007/s10120-015-0591-z
21. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 limits anti-tumor immunity. Immunity (2016) 44(2):316–29. doi:10.1016/j.immuni.2016.01.013
22. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119(19):4430–40. doi:10.1182/blood-2011-11-392324
23. Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol (2009) 182(3):1469–80. doi:10.4049/jimmunol.182.3.1469
24. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27(4):635–46. doi:10.1016/j.immuni.2007.08.014
25. Pillai MR, Collison LW, Wang X, Finkelstein D, Rehg JE, Boyd K, et al. The plasticity of regulatory T cell function. J Immunol (2011) 187(10):4987–97. doi:10.4049/jimmunol.1102173
26. Ju Y, Shang X, Liu Z, Zhang J, Li Y, Shen Y, et al. The Tim-3/galectin-9 pathway involves in the homeostasis of hepatic Tregs in a mouse model of concanavalin A-induced hepatitis. Mol Immunol (2014) 58(1):85–91. doi:10.1016/j.molimm.2013.11.001
27. Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood (2007) 109(5):2058–65. doi:10.1182/blood-2006-04-016451
28. Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*). Annu Rev Immunol (2009) 27:551–89. doi:10.1146/annurev.immunol.021908.132723
29. Bi X, Suzuki Y, Gatanaga H, Oka S. High frequency and proliferation of CD4+ FOXP3+ Treg in HIV-1-infected patients with low CD4 counts. Eur J Immunol (2009) 39(1):301–9. doi:10.1002/eji.200838667
30. Nilsson J, Boasso A, Velilla PA, Zhang R, Vaccari M, Franchini G, et al. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood (2006) 108(12):3808–17. doi:10.1182/blood-2006-05-021576
31. Presicce P, Orsborn K, King E, Pratt J, Fichtenbaum CJ, Chougnet CA. Frequency of circulating regulatory T cells increases during chronic HIV infection and is largely controlled by highly active antiretroviral therapy. PLoS One (2011) 6(12):e28118. doi:10.1371/journal.pone.0028118
32. Chevalier MF, Weiss L. The split personality of regulatory T cells in HIV infection. Blood (2013) 121(1):29–37. doi:10.1182/blood-2012-07-409755
33. Moreno-Fernandez ME, Presicce P, Chougnet CA. Homeostasis and function of regulatory T cells in HIV/SIV infection. J Virol (2012) 86(19):10262–9. doi:10.1128/JVI.00993-12
34. Hunt PW, Landay AL, Sinclair E, Martinson JA, Hatano H, Emu B, et al. A low T regulatory cell response may contribute to both viral control and generalized immune activation in HIV controllers. PLoS One (2011) 6(1):e15924. doi:10.1371/journal.pone.0015924
35. Eggena MP, Barugahare B, Jones N, Okello M, Mutalya S, Kityo C, et al. Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol (2005) 174(7):4407–14. doi:10.4049/jimmunol.174.7.4407
36. Pion M, Jaramillo-Ruiz D, Martinez A, Munoz-Fernandez MA, Correa-Rocha R. HIV infection of human regulatory T cells downregulates Foxp3 expression by increasing DNMT3b levels and DNA methylation in the FOXP3 gene. AIDS (2013) 27(13):2019–29. doi:10.1097/QAD.0b013e32836253fd
37. Mendez-Lagares G, Jaramillo-Ruiz D, Pion M, Leal M, Munoz-Fernandez MA, Pacheco YM, et al. HIV infection deregulates the balance between regulatory T cells and IL-2-producing CD4 T cells by decreasing the expression of the IL-2 receptor in Treg. J Acquir Immune Defic Syndr (2014) 65(3):278–82. doi:10.1097/QAI.0000000000000092
38. Card CM, Keynan Y, Lajoie J, Bell CP, Dawood M, Becker M, et al. HIV controllers are distinguished by chemokine expression profile and HIV-specific T-cell proliferative potential. J Acquir Immune Defic Syndr (2012) 59(5):427–37. doi:10.1097/QAI.0b013e3182454fcd
39. Simonetta F, Lecuroux C, Girault I, Goujard C, Sinet M, Lambotte O, et al. Early and long-lasting alteration of effector CD45RA(-)Foxp3(high) regulatory T-cell homeostasis during HIV infection. J Infect Dis (2012) 205(10):1510–9. doi:10.1093/infdis/jis235
40. Thorborn G, Pomeroy L, Isohanni H, Perry M, Peters B, Vyakarnam A. Increased sensitivity of CD4+ T-effector cells to CD4+CD25+ Treg suppression compensates for reduced Treg number in asymptomatic HIV-1 infection. PLoS One (2010) 5(2):e9254. doi:10.1371/journal.pone.0009254
41. Almeida AR, Zaragoza B, Freitas AA. Indexation as a novel mechanism of lymphocyte homeostasis: the number of CD4+CD25+ regulatory T cells is indexed to the number of IL-2-producing cells. J Immunol (2006) 177(1):192–200. doi:10.4049/jimmunol.177.1.192
42. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi:10.1016/j.cell.2008.05.009
43. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol (2013) 146(3):248–61. doi:10.1016/j.clim.2013.01.004
44. James CR, Buckle I, Muscate F, Otsuka M, Nakao M, Oon JS, et al. Reduced interleukin-2 responsiveness impairs the ability of T cells to compete for IL-2 in nonobese diabetic mice. Immunol Cell Biol (2016). doi:10.1038/icb.2016.7
45. Hinks A, Ke X, Barton A, Eyre S, Bowes J, Worthington J, et al. Association of the IL2RA/CD25 gene with juvenile idiopathic arthritis. Arthritis Rheum (2009) 60(1):251–7. doi:10.1002/art.24187
46. Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A. Upregulation of Foxp3 expression in mouse and human Treg is IL-2/STAT5 dependent: implications for the NOD STAT5B mutation in diabetes pathogenesis. Ann N Y Acad Sci (2006) 1079:198–204. doi:10.1196/annals.1375.031
47. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi:10.1182/blood-2006-02-004747
48. Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol (2007) 8(3):277–84. doi:10.1038/ni1437
49. Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol (2007) 8(11):1246–54. doi:10.1038/ni1515
50. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi:10.1038/nature05115
51. Tanaka M, Hirabayashi Y, Gatanaga H, Aizawa S, Hachiya A, Takahashi Y, et al. Reduction in interleukin-2-producing cells but not Th1 to Th2 shift in moderate and advanced stages of human immunodeficiency virus type-1-infection: direct analysis of intracellular cytokine concentrations in CD4+ CD8- T cells. Scand J Immunol (1999) 50(5):550–4. doi:10.1046/j.1365-3083.1999.00627.x
52. Nakayama-Hosoya K, Ishida T, Youngblood B, Nakamura H, Hosoya N, Koga M, et al. Epigenetic repression of interleukin 2 expression in senescent CD4+ T cells during chronic HIV type 1 infection. J Infect Dis (2015) 211(1):28–39. doi:10.1093/infdis/jiu376
53. Oguariri RM, Dai L, Adelsberger JW, Rupert A, Stevens R, Yang J, et al. Interleukin-2 inhibits HIV-1 replication in some human T cell lymphotrophic virus-1-infected cell lines via the induction and incorporation of APOBEC3G into the virion. J Biol Chem (2013) 288(24):17812–22. doi:10.1074/jbc.M113.468975
54. Pett SL, Kelleher AD, Emery S. Role of interleukin-2 in patients with HIV infection. Drugs (2010) 70(9):1115–30. doi:10.2165/10898620-000000000-00000
55. Sued O, Ambrosioni J, Nicolas D, Manzardo C, Aguero F, Claramonte X, et al. Structured treatment interruptions and low doses of IL-2 in patients with primary HIV infection. Inflammatory, virological and immunological outcomes. PLoS One (2015) 10(7):e0131651. doi:10.1371/journal.pone.0131651
56. Levy Y, Thiebaut R, Gougeon ML, Molina JM, Weiss L, Girard PM, et al. Effect of intermittent interleukin-2 therapy on CD4+ T-cell counts following antiretroviral cessation in patients with HIV. AIDS (2012) 26(6):711–20. doi:10.1097/QAD.0b013e3283519214
57. Abrams D, Levy Y, Losso MH, Babiker A, Collins G, Cooper DA, et al. Interleukin-2 therapy in patients with HIV infection. N Engl J Med (2009) 361(16):1548–59. doi:10.1056/NEJMoa0903175
58. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5(2):e38. doi:10.1371/journal.pbio.0050038
59. Zhou L, Littman DR. Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol (2009) 21(2):146–52. doi:10.1016/j.coi.2009.03.001
60. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126(2):375–87. doi:10.1016/j.cell.2006.05.042
61. Lozano T, Casares N, Lasarte JJ. Searching for the Achilles heel of FOXP3. Front Oncol (2013) 3:294. doi:10.3389/fonc.2013.00294
62. Xie X, Stubbington MJ, Nissen JK, Andersen KG, Hebenstreit D, Teichmann SA, et al. The regulatory T cell lineage factor Foxp3 regulates gene expression through several distinct mechanisms mostly independent of direct DNA binding. PLoS Genet (2015) 11(6):e1005251. doi:10.1371/journal.pgen.1005251
63. Grant C, Oh U, Fugo K, Takenouchi N, Griffith C, Yao K, et al. Foxp3 represses retroviral transcription by targeting both NF-kappaB and CREB pathways. PLoS Pathog (2006) 2(4):e33. doi:10.1371/journal.ppat.0020033
64. Selliah N, Zhang M, White S, Zoltick P, Sawaya BE, Finkel TH, et al. FOXP3 inhibits HIV-1 infection of CD4 T-cells via inhibition of LTR transcriptional activity. Virology (2008) 381(2):161–7. doi:10.1016/j.virol.2008.08.033
65. Suchard MS, Mayne E, Green VA, Shalekoff S, Donninger SL, Stevens WS, et al. FOXP3 expression is upregulated in CD4T cells in progressive HIV-1 infection and is a marker of disease severity. PLoS One (2010) 5(7):e11762. doi:10.1371/journal.pone.0011762
66. Jenabian MA, Patel M, Kema I, Vyboh K, Kanagaratham C, Radzioch D, et al. Soluble CD40-ligand (sCD40L, sCD154) plays an immunosuppressive role via regulatory T cell expansion in HIV infection. Clin Exp Immunol (2014) 178(1):102–11. doi:10.1111/cei.12396
67. Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, et al. CD73 is involved in lymphocyte binding to the endothelium: characterization of lymphocyte-vascular adhesion protein 2 identifies it as CD73. J Exp Med (1995) 182(5):1603–8. doi:10.1084/jem.182.5.1603
68. MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity (2001) 15(5):825–35. doi:10.1016/S1074-7613(01)00229-1
69. Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112(2):358–404. doi:10.1016/j.pharmthera.2005.04.013
70. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi:10.1038/ni.2919
71. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol (2011) 11(3):201–12. doi:10.1038/nri2938
72. Lu Y, Wang X, Gu J, Lu H, Zhang F, Li X, et al. iTreg induced from CD39(+) naive T cells demonstrate enhanced proliferate and suppressive ability. Int Immunopharmacol (2015) 28(2):925–30. doi:10.1016/j.intimp.2015.03.039
73. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111(1):251–9. doi:10.1182/blood-2007-03-081646
74. Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, McRae JL, et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant (2010) 10(11):2410–20. doi:10.1111/j.1600-6143.2010.03291.x
75. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi:10.3389/fimmu.2012.00190
76. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem (2010) 285(10):7176–86. doi:10.1074/jbc.M109.047423
77. Saze Z, Schuler PJ, Hong CS, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood (2013) 122(1):9–18. doi:10.1182/blood-2013-02-482406
78. Schuler PJ, Schilling B, Harasymczuk M, Hoffmann TK, Johnson J, Lang S, et al. Phenotypic and functional characteristics of CD4+ CD39+ FOXP3+ and CD4+ CD39+ FOXP3neg T-cell subsets in cancer patients. Eur J Immunol (2012) 42(7):1876–85. doi:10.1002/eji.201142347
79. Schulze Zur Wiesch J, Thomssen A, Hartjen P, Toth I, Lehmann C, Meyer-Olson D, et al. Comprehensive analysis of frequency and phenotype of T regulatory cells in HIV infection: CD39 expression of FoxP3+ T regulatory cells correlates with progressive disease. J Virol (2011) 85(3):1287–97. doi:10.1128/JVI.01758-10
80. Burton CT, Westrop SJ, Eccles-James I, Boasso A, Nelson MR, Bower M, et al. Altered phenotype of regulatory T cells associated with lack of human immunodeficiency virus (HIV)-1-specific suppressive function. Clin Exp Immunol (2011) 166(2):191–200. doi:10.1111/j.1365-2249.2011.04451.x
81. Nikolova M, Carriere M, Jenabian MA, Limou S, Younas M, Kok A, et al. CD39/adenosine pathway is involved in AIDS progression. PLoS Pathog (2011) 7(7):e1002110. doi:10.1371/journal.ppat.1002110
82. Seddiki N, Cook L, Hsu DC, Phetsouphanh C, Brown K, Xu Y, et al. Human antigen-specific CD4(+) CD25(+) CD134(+) CD39(+) T cells are enriched for regulatory T cells and comprise a substantial proportion of recall responses. Eur J Immunol (2014) 44(6):1644–61. doi:10.1002/eji.201344102
83. Jenabian MA, Ancuta P, Gilmore N, Routy JP. Regulatory T cells in HIV infection: can immunotherapy regulate the regulator? Clin Dev Immunol (2012) 2012:908314. doi:10.1155/2012/908314
84. Moreno-Fernandez ME, Rueda CM, Rusie LK, Chougnet CA. Regulatory T cells control HIV replication in activated T cells through a cAMP-dependent mechanism. Blood (2011) 117(20):5372–80. doi:10.1182/blood-2010-12-323162
85. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology (2013) 2(9):e26246. doi:10.4161/onci.26246
86. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev (2008) 88(3):841–86. doi:10.1152/physrev.00035.2007
87. Quarona V, Ferri V, Chillemi A, Bolzoni M, Mancini C, Zaccarello G, et al. Unraveling the contribution of ectoenzymes to myeloma life and survival in the bone marrow niche. Ann N Y Acad Sci (2015) 1335:10–22. doi:10.1111/nyas.12485
88. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16- NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J Immunol (2015) 195(3):965–72. doi:10.4049/jimmunol.1500591
89. Park A, Govindaraj C, Xiang SD, Halo J, Quinn M, Scalzo-Inguanti K, et al. Substantially modified ratios of effector to regulatory T cells during chemotherapy in ovarian cancer patients return to pre-treatment levels at completion: implications for immunotherapy. Cancers (Basel) (2012) 4(2):581–600. doi:10.3390/cancers4020581
90. Patton DT, Wilson MD, Rowan WC, Soond DR, Okkenhaug K. The PI3K p110delta regulates expression of CD38 on regulatory T cells. PLoS One (2011) 6(3):e17359. doi:10.1371/journal.pone.0017359
91. Horenstein AL, Chillemi A, Quarona V, Zito A, Roato I, Morandi F, et al. NAD(+)-metabolizing ectoenzymes in remodeling tumor-host interactions: the human myeloma model. Cells (2015) 4(3):520–37. doi:10.3390/cells4030520
92. Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood (2004) 104(4):942–7. doi:10.1182/blood-2003-09-3333
93. Giorgi JV, Lyles RH, Matud JL, Yamashita TE, Mellors JW, Hultin LE, et al. Predictive value of immunologic and virologic markers after long or short duration of HIV-1 infection. J Acquir Immune Defic Syndr (2002) 29(4):346–55. doi:10.1097/00126334-200204010-00004
94. Shaw JM, Hunt PW, Critchfield JW, McConnell DH, Garcia JC, Pollard RB, et al. Increased frequency of regulatory T cells accompanies increased immune activation in rectal mucosae of HIV-positive noncontrollers. J Virol (2011) 85(21):11422–34. doi:10.1128/JVI.05608-11
95. Ndhlovu LC, Loo CP, Spotts G, Nixon DF, Hecht FM. FOXP3 expressing CD127lo CD4+ T cells inversely correlate with CD38+ CD8+ T cell activation levels in primary HIV-1 infection. J Leukoc Biol (2008) 83(2):254–62. doi:10.1189/jlb.0507281
96. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204(6):1303–10. doi:10.1084/jem.20062129
97. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463(7282):808–12. doi:10.1038/nature08750
98. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414(6866):916–20. doi:10.1038/414916a
99. Ring S, Karakhanova S, Johnson T, Enk AH, Mahnke K. Gap junctions between regulatory T cells and dendritic cells prevent sensitization of CD8(+) T cells. J Allergy Clin Immunol (2010) 125(1):.e1–7. doi:10.1016/j.jaci.2009.10.025
100. Mendoza-Naranjo A, Bouma G, Pereda C, Ramirez M, Webb KF, Tittarelli A, et al. Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J Immunol (2011) 187(6):3121–32. doi:10.4049/jimmunol.1100378
101. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol (2010) 265(2):91–6. doi:10.1016/j.cellimm.2010.07.007
102. Zhou W, Hashimoto K, Goleniewska K, O’Neal JF, Ji S, Blackwell TS, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol (2007) 178(2):702–10. doi:10.4049/jimmunol.178.2.702
103. Gray PC, Scott JD, Catterall WA. Regulation of ion channels by cAMP-dependent protein kinase and A-kinase anchoring proteins. Curr Opin Neurobiol (1998) 8(3):330–4. doi:10.1016/S0959-4388(98)80057-3
104. Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, et al. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature (1994) 370(6486):223–6. doi:10.1038/370223a0
105. Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell (1993) 75(5):875–86. doi:10.1016/0092-8674(93)90532-U
106. Bodor J, Spetz AL, Strominger JL, Habener JF. cAMP inducibility of transcriptional repressor ICER in developing and mature human T lymphocytes. Proc Natl Acad Sci U S A (1996) 93(8):3536–41. doi:10.1073/pnas.93.8.3536
107. Bodor J, Habener JF. Role of transcriptional repressor ICER in cyclic AMP-mediated attenuation of cytokine gene expression in human thymocytes. J Biol Chem (1998) 273(16):9544–51. doi:10.1074/jbc.273.16.9544
108. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev (2003) 17(18):2205–32. doi:10.1101/gad.1102703
109. Bodor J, Bopp T, Vaeth M, Klein M, Serfling E, Hunig T, et al. Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol (2012) 42(6):1375–84. doi:10.1002/eji.201141578
110. Hofmann B, Nishanian P, Nguyen T, Liu M, Fahey JL. Restoration of T-cell function in HIV infection by reduction of intracellular cAMP levels with adenosine analogues. AIDS (1993) 7(5):659–64. doi:10.1097/00002030-199305000-00008
111. Aandahl EM, Aukrust P, Skalhegg BS, Muller F, Froland SS, Hansson V, et al. Protein kinase A type I antagonist restores immune responses of T cells from HIV-infected patients. FASEB J (1998) 12(10):855–62.
112. Hofmann B, Nishanian P, Nguyen T, Insixiengmay P, Fahey JL. Human immunodeficiency virus proteins induce the inhibitory cAMP/protein kinase A pathway in normal lymphocytes. Proc Natl Acad Sci U S A (1993) 90(14):6676–80. doi:10.1073/pnas.90.14.6676
113. Masci AM, Galgani M, Cassano S, De Simone S, Gallo A, De Rosa V, et al. HIV-1 gp120 induces anergy in naive T lymphocytes through CD4-independent protein kinase-A-mediated signaling. J Leukoc Biol (2003) 74(6):1117–24. doi:10.1189/jlb.0503239
114. Becker C, Taube C, Bopp T, Michel K, Kubach J, Reuter S, et al. Protection from graft-versus-host disease by HIV-1 envelope protein gp120-mediated activation of human CD4+CD25+ regulatory T cells. Blood (2009) 114(6):1263–9. doi:10.1182/blood-2009-02-206730
115. Naval-Macabuhay I, Casanova V, Navarro G, Garcia F, Leon A, Miralles L, et al. Adenosine deaminase regulates Treg expression in autologous T cell-dendritic cell cocultures from patients infected with HIV-1. J Leukoc Biol (2016) 99(2):349–59. doi:10.1189/jlb.3A1214-580RR
116. Banas B, Eberle J, Schlondorff D, Luckow B. Modulation of HIV-1 enhancer activity and virus production by cAMP. FEBS Lett (2001) 509(2):207–12. doi:10.1016/S0014-5793(01)03182-9
117. Navarro J, Punzon C, Jimenez JL, Fernandez-Cruz E, Pizarro A, Fresno M, et al. Inhibition of phosphodiesterase type IV suppresses human immunodeficiency virus type 1 replication and cytokine production in primary T cells: involvement of NF-kappaB and NFAT. J Virol (1998) 72(6):4712–20.
118. Sun Y, Li L, Lau F, Beavo JA, Clark EA. Infection of CD4+ memory T cells by HIV-1 requires expression of phosphodiesterase 4. J Immunol (2000) 165(4):1755–61. doi:10.4049/jimmunol.165.4.1755
119. By Y, Durand-Gorde JM, Condo J, Lejeune PJ, Fenouillet E, Guieu R, et al. Monoclonal antibody-assisted stimulation of adenosine A2A receptors induces simultaneous downregulation of CXCR4 and CCR5 on CD4+ T-cells. Hum Immunol (2010) 71(11):1073–6. doi:10.1016/j.humimm.2010.08.010
120. Vendetti S, Riccomi A, Sacchi A, Gatta L, Pioli C, De Magistris MT. Cyclic adenosine 5′-monophosphate and calcium induce CD152 (CTLA-4) up-regulation in resting CD4+ T lymphocytes. J Immunol (2002) 169(11):6231–5. doi:10.4049/jimmunol.169.11.6231
121. Moreno-Fernandez ME, Zapata W, Blackard JT, Franchini G, Chougnet CA. Human regulatory T cells are targets for human immunodeficiency Virus (HIV) infection, and their susceptibility differs depending on the HIV type 1 strain. J Virol (2009) 83(24):12925–33. doi:10.1128/JVI.01352-09
122. Antons AK, Wang R, Oswald-Richter K, Tseng M, Arendt CW, Kalams SA, et al. Naive precursors of human regulatory T cells require FoxP3 for suppression and are susceptible to HIV infection. J Immunol (2008) 180(2):764–73. doi:10.4049/jimmunol.180.2.764
123. Oswald-Richter K, Grill SM, Shariat N, Leelawong M, Sundrud MS, Haas DW, et al. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol (2004) 2(7):E198. doi:10.1371/journal.pbio.0020198
124. Seddiki N, Sasson SC, Santner-Nanan B, Munier M, van Bockel D, Ip S, et al. Proliferation of weakly suppressive regulatory CD4+ T cells is associated with over-active CD4+ T-cell responses in HIV-positive patients with mycobacterial immune restoration disease. Eur J Immunol (2009) 39(2):391–403. doi:10.1002/eji.200838630
125. Waterhouse PM, Wang MB, Lough T. Gene silencing as an adaptive defence against viruses. Nature (2001) 411(6839):834–42. doi:10.1038/35081168
126. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol (2009) 182(1):259–73. doi:10.4049/jimmunol.182.1.259
127. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445(7129):771–5. doi:10.1038/nature05543
128. Angin M, Sharma S, King M, Murooka TT, Ghebremichael M, Mempel TR, et al. HIV-1 infection impairs regulatory T-cell suppressive capacity on a per-cell basis. J Infect Dis (2014) 210(6):899–903. doi:10.1093/infdis/jiu188
129. Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, Daucher M, et al. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med (2004) 200(3):331–43. doi:10.1084/jem.20032069
130. Kinter AL, Horak R, Sion M, Riggin L, McNally J, Lin Y, et al. CD25+ regulatory T cells isolated from HIV-infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV-specific CD8+ T cells in vitro. AIDS Res Hum Retroviruses (2007) 23(3):438–50. doi:10.1089/aid.2006.0162
131. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep (2009) 10(2):180–5. doi:10.1038/embor.2008.224
132. Zhang HH, Fei R, Xie XW, Wang L, Luo H, Wang XY, et al. [Specific suppression in regulatory T cells by Foxp3 siRNA contributes to enhance the in vitro anti-tumor immune response in hepatocellular carcinoma patients]. Beijing Da Xue Xue Bao (2009) 41(3):313–8.
133. Xiao Y, Nagai Y, Deng G, Ohtani T, Zhu Z, Zhou Z, et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep (2014) 7(5):1471–80. doi:10.1016/j.celrep.2014.04.021
134. van Loosdregt J, Coffer PJ. Post-translational modification networks regulating FOXP3 function. Trends Immunol (2014) 35(8):368–78. doi:10.1016/j.it.2014.06.005
135. Xiao H, Chung J, Kao HY, Yang YC. Tip60 is a co-repressor for STAT3. J Biol Chem (2003) 278(13):11197–204. doi:10.1074/jbc.M210816200
136. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A (2007) 104(11):4571–6. doi:10.1073/pnas.0700298104
137. Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity (2012) 36(5):717–30. doi:10.1016/j.immuni.2012.03.020
138. Creaven M, Hans F, Mutskov V, Col E, Caron C, Dimitrov S, et al. Control of the histone-acetyltransferase activity of Tip60 by the HIV-1 transactivator protein, Tat. Biochemistry (1999) 38(27):8826–30. doi:10.1021/bi9907274
139. Col E, Caron C, Chable-Bessia C, Legube G, Gazzeri S, Komatsu Y, et al. HIV-1 Tat targets Tip60 to impair the apoptotic cell response to genotoxic stresses. EMBO J (2005) 24(14):2634–45. doi:10.1038/sj.emboj.7600734
140. Weiss L, Piketty C, Assoumou L, Didier C, Caccavelli L, Donkova-Petrini V, et al. Relationship between regulatory T cells and immune activation in human immunodeficiency virus-infected patients interrupting antiretroviral therapy. PLoS One (2010) 5(7):e11659. doi:10.1371/journal.pone.0011659
141. Jiao Y, Fu J, Xing S, Fu B, Zhang Z, Shi M, et al. The decrease of regulatory T cells correlates with excessive activation and apoptosis of CD8+ T cells in HIV-1-infected typical progressors, but not in long-term non-progressors. Immunology (2009) 128(1 Suppl):e366–75. doi:10.1111/j.1365-2567.2008.02978.x
142. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445(7129):766–70. doi:10.1038/nature05479
143. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi:10.1016/j.immuni.2009.03.019
144. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol (2007) 178(1):280–90. doi:10.4049/jimmunol.178.1.280
145. Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res (2009) 19(7):1165–74. doi:10.1101/gr.091470.109
146. Duggleby RC, Shaw TN, Jarvis LB, Kaur G, Gaston JS. CD27 expression discriminates between regulatory and non-regulatory cells after expansion of human peripheral blood CD4+ CD25+ cells. Immunology (2007) 121(1):129–39. doi:10.1111/j.1365-2567.2006.02550.x
147. Wohlfert EA, Nichols FC, Nevius E, Clark RB. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol (2007) 178(7):4129–35. doi:10.4049/jimmunol.178.7.4129
148. Mouly E, Chemin K, Nguyen HV, Chopin M, Mesnard L, Leite-de-Moraes M, et al. The Ets-1 transcription factor controls the development and function of natural regulatory T cells. J Exp Med (2010) 207(10):2113–25. doi:10.1084/jem.20092153
149. Shimo Y, Yanai H, Ohshima D, Qin J, Motegi H, Maruyama Y, et al. TRAF6 directs commitment to regulatory T cells in thymocytes. Genes Cells (2011) 16(4):437–47. doi:10.1111/j.1365-2443.2011.01500.x
150. Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood (2007) 109(10):4368–75. doi:10.1182/blood-2006-11-055756
151. Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol (2012) 13(10):972–80. doi:10.1038/ni.2420
152. Carroll RG, Carpenito C, Shan X, Danet-Desnoyers G, Liu R, Jiang S, et al. Distinct effects of IL-18 on the engraftment and function of human effector CD8 T cells and regulatory T cells. PLoS One (2008) 3(9):e3289. doi:10.1371/journal.pone.0003289
153. Dasgupta G, Chentoufi AA, You S, Falatoonzadeh P, Urbano LA, Akhtarmalik A, et al. Engagement of TLR2 reverses the suppressor function of conjunctiva CD4+CD25+ regulatory T cells and promotes herpes simplex virus epitope-specific CD4+CD25- effector T cell responses. Invest Ophthalmol Vis Sci (2011) 52(6):3321–33. doi:10.1167/iovs.10-6522
154. Heninger AK, Theil A, Wilhelm C, Petzold C, Huebel N, Kretschmer K, et al. IL-7 abrogates suppressive activity of human CD4+CD25+FOXP3+ regulatory T cells and allows expansion of alloreactive and autoreactive T cells. J Immunol (2012) 189(12):5649–58. doi:10.4049/jimmunol.1201286
Keywords: Treg cells, HIV Infections, regulatory mechanisms, IL-2, immune hyperactivation
Citation: López-Abente J, Correa-Rocha R and Pion M (2016) Functional Mechanisms of Treg in the Context of HIV Infection and the Janus Face of Immune Suppression. Front. Immunol. 7:192. doi: 10.3389/fimmu.2016.00192
Received: 14 March 2016; Accepted: 02 May 2016;
Published: 19 May 2016
Edited by:
Josef Bodor, Institute of Experimental Medicine, Czech RepublicReviewed by:
Bin Li, Institut Pasteur of Shanghai, Chinese Academy of Sciences, ChinaFabio Malavasi, University of Torino Medical School, Italy
Copyright: © 2016 López-Abente, Correa-Rocha and Pion. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Correa-Rocha, cmFmYWVsLmNvcnJlYUBpaXNnbS5jb20=;
Marjorie Pion, bWFyam9yaWUucGlvbkBpaXNnbS5jb20=