 Peter Isaac Lobo
Peter Isaac Lobo- Department of Internal Medicine, Division of Nephrology, Center of Immunology, Inflammation and Regenerative Medicine, University of Virginia Health Center, Charlottesville, VA, USA
We review how polyreactive natural IgM autoantibodies (IgM-NAA) protect the host from invading micro-organisms and host neo-antigens that are constantly being produced by oxidation mechanisms and cell apoptosis. Second, we discuss how IgM-NAA and IgM anti-leukocyte antibodies (IgM-ALA) inhibits autoimmune inflammation by anti-idiotypic mechanisms, enhancing removal of apoptotic cells, masking neo-antigens, and regulating the function of dendritic cells (DC) and effector cells. Third, we review how natural IgM prevents autoimmune disorders arising from pathogenic IgG autoantibodies, triggered by genetic mechanisms (e.g., SLE) or micro-organisms, as well as by autoreactive B and T cells that have escaped tolerance mechanisms. Studies in IgM knockout mice have clearly demonstrated that regulatory B and T cells require IgM to effectively regulate inflammation mediated by innate, adaptive, and autoimmune mechanisms. It is, therefore, not surprising why the host positively selects such autoreactive B1 cells that generate IgM-NAA, which are also evolutionarily conserved. Fourth, we show that IgM-ALA levels and their repertoire can vary in normal humans and disease states and this variation may partly explain the observed differences in the inflammatory response after infection, ischemic injury, or after a transplant. We also show how protective IgM-NAA can be rendered pathogenic under non-physiological conditions. We also review IgG-NAA that are more abundant than IgM-NAA in plasma. However, we need to understand if the (Fab)2 region of IgG-NAA has physiological relevance in non-disease states, as in plasma, their functional activity is blocked by IgM-NAA having anti-idiotypic activity. Some IgG-NAA are produced by B2 cells that have escaped tolerance mechanisms and we show how such pathogenic IgG-NAA are regulated to prevent autoimmune disease. The Fc region of IgG-NAA can influence inflammation and B cell function in vivo by binding to activating and inhibitory FcγR. IgM-NAA has therapeutic potential. Polyclonal IgM infusions can be used to abrogate on-going inflammation. Additionally, inflammation arising after ischemic kidney injury, e.g., during high-risk elective cardiac surgery or after allograft transplantation, can be prevented by pre-emptively infusing polyclonal IgM or DC pretreated ex vivo with IgM or by increasing in vivo IgM with a vaccine approach. Cell therapy is appealing as less IgM will be required.
Introduction
The study of natural IgM autoantibodies (IgM-NAA) has given us a good insight on how nature, by creating polyreactive pentavalent antibodies, has accomplished a difficult task of trying to protect the host from both diverse foreign pathogens and from diverse self neo-antigens that are constantly produced within the host. Poly-reactivity with low binding affinity, but with high avidity, has enabled these IgM-NAA-producing B cell clones to be rapidly activated by a foreign or an auto neo-antigen for deploying protective mechanisms to the host. Such a response provides time for the adaptive immune system to mount a highly specific immune response to foreign antigens and, in addition, lessens the burden on the host to maintain diverse B cell clones producing highly specific IgG auto-antibodies, which has the potential of causing autoimmune disease owing to their high-affinity binding. Second, since both foreign and self neo-antigens can induce an inflammatory response, these IgM-NAA have taken over another task of subduing an excessive inflammatory response that can injure the host. This regulation of inflammatory cells without damage is made possible by the low binding affinity of IgM-ALA to live leukocytes, combined with its inability to effectively activate the lytic component of complement at body temperature (37°C). Hence, it is not surprising why these IgM-NAA antibodies, which first arose in cartilaginous fish, have been conserved during evolution [reviewed in Ref. (1)] and why IgM-NAA makes up about 70–80% of circulating IgM (2, 3). Additionally, some studies have shown that the binding of natural IgM to autologous receptors is also evolutionarily conserved among mammalian species as human IgM has the same functional effect as murine IgM on murine cells in vitro or when used in vivo in mice (4–6).
Natural autoantibodies of different isotypes have been intensively studied during the last 40 years (7–17). These autoantibodies have been termed “natural antibodies” as they are produced at birth in the absence of exposure to foreign antigens. The full repertoire of NAA develops by early childhood. In mice, NAA are predominantly produced by the CD5+ B1 cells, while marginal-zone splenic B (MZB) cells contribute the remainder. These B1 cells produce predominantly IgM, IgA, and IgG3 autoantibodies (18, 19), independently of T cell help, and exhibit an enhanced response to innate immune signals, such as TLR agonist. Hence, B1 and MZB cells differ from B2 cells in that the response of these cells in vivo is rapid and can be driven by TLR agonists independently of antigen binding to their BCR (20–23). Additionally, there are data to indicate that autoantibody-producing B1 cells, unlike self-reactive T cells, are positively selected for their self-reactivity, thus implying that NAA are conserved by design (24–27). Further support for their importance comes from studies in chimeric mice demonstrating that IgM-NAA comprise the majority of circulating IgM (2, 3). Most cross-sectional studies in humans and rodents would indicate that IgM-NAA decrease with age (28–31) or lose their effectiveness with age (32) except for one report where follow-up of five healthy individuals for 25 years revealed no change in IgM-NAA levels (33). However, IgG-NAA can increase (34) but do not decrease with age (35, 36).
Innately produced natural IgM-NAA should not be confused with immune IgM and IgG that are produced several days later after exposure to foreign antigens or pathogens. Such immune IgM and IgG are not natural autoantibodies and in general are antigen specific and are produced by B2 cells that require antigen binding to BcR and additional T cell help to generate anti-protein antibodies. However, production of immune IgM is limited as these IgM secreting B2 cells migrate to B cell follicles, where with the help of T cells, these B2 cells undergo isotype switching and somatic hyper-mutation, thus generating long-lived memory B cells and differentiating into plasma cells that produce IgG antibodies with high-affinity binding.
The human equivalent of the murine CD5+ B1 subset has been recently identified and characterized. This CD20+ CD43+ CD27+ human B1 subset that can spontaneously secrete antibody represents about 50% of umbilical cord B cells and 15–20% of circulating adult B cells, and is the predominant source of human IgM-NAA (29). In humans, CD5 is not a specific marker of B1 as this marker is expressed by both B1 and B2 cells. Similarly, CD43 and CD27 are not specific markers for human B1 as about 20% of CD43+ CD 27+ B cells have characteristics of pre-plasmablasts that are derived from T-dependent B cells present in germinal centers (37, 38). Human IgM-NAA are also polyreactive and bind similar autoantigens as in mice, including oxidized neo-determinants and leukocyte receptors (4, 39–41).
IgM-NAA, IgG-NAA, and Pathogenic IgG Autoantibodies
One physiological role of NAA is to protect the host from pathogenic IgG autoantibodies. We will, therefore, briefly describe the biology of natural IgM and IgG-NAA and pathogenic IgG autoantibodies in health and disease and then discuss the different mechanisms used by NAA to counter pathogenic IgG. B1 cells have been shown to secrete IgM, IgA, and IgG3-NAA that are encoded by minimally or non-mutated germ line genes that are enriched for heavy chain variable region rearrangements and with H-L pairings that allow for poly-reactivity (18, 19). NAA generated by B1 cells rarely undergo isotype switching or somatic hyper-mutation to acquire antigen specificity. IgA in the gut is predominantly produced by B1 cells present in the gut lymphoid tissue but there is increasing evidence to show that B2 cells can also participate in gut mucosal immunity and this could account for the observed somatic hyper-mutation and antigen specificity of gut mucosal IgA (42–46).
Natural IgM Autoantibodies
In mice, the majority of IgM-NAA are produced by splenic B1 and MZB cells (47–49). B1 and MZB cells, such as memory B cells, express CD27 and spontaneously produce IgM, but IgM production can also be increased by TLR activation [e.g., lipopolysaccharide (LPS)] or via the BCR in response to pathogens. Normal levels of IgM-NAA are present at birth even under germ-free conditions and in nude mice, indicating that IgM-NAA production is not dependent on exposure to foreign antigens, TLR or T cell activation (50). IgM-NAA repertoire is shaped by T-independent antigen activation, especially of MZB cells (51, 52). An important characteristic of these antibodies is their low binding affinity (53). It is possible that the membrane expressed IgM or BCR of B1 and MZB cells producing NAA also exhibit low binding affinity and perhaps this latter characteristic may be involved in preventing autoreactive B1 cells from being deleted or undergoing negative selection. There are data to show that autoreactive B1 cells are positively selected and this process requires both the autoantigen and the relevant BCR (24–27). The need to positively select B1 cells secreting IgM-NAA would indicate that these antibodies have an important physiological role that will be reviewed later.
Natural IgM autoantibodies have been shown to be polyclonal with clones having specificity for some, but not all self-antigens, i.e., certain common epitopes present on phylogenetically conserved self-antigens. Some of these IgM clones with reactivity to self-antigens have been identified, e.g., IgM clones with specificity for leukocyte receptors [IgM anti-leukocyte antibodies (IgM-ALA)] (4, 39), Fc domain of IgG (rheumatoid factor) (14, 15), complement components (17), collagen, thyroglobulin, intracellular constituents, such as cytoskeletal proteins, cytosolic enzymes, dsDNA, or nucleosomes, neutrophil cytoplasmic enzymes (ANCA) (50, 54) and oxidized neo-determinants [e.g., phosphorylcholine (PC)] that are exposed when lipids are oxidized or cells undergo apoptosis (55, 56). While some IgM-ALA have mono-reactivity, e.g., to some cytokines, most are polyreactive with each polyreactive IgM-NAA clone having a selective binding profile (50). For example, IgM anti-PC NAA will bind to ABO blood type antigens, endotoxins, and oxidized neo-determinants on apoptotic cells but this autoantibody has no binding reactivity to nuclear antigens or to IgG (57). Additionally, these IgM-NAA, by virtue of being polyreactive, also cross-react with pathogen-expressed molecules, for example, PC on Streptococcus pneumoniae and other antigens expressed by various viruses and parasites (55–57) Hence, it has been suggested that these natural IgM antibodies are protective, serving as a first line of defense against infections and protecting the host from pathogen-mediated apoptotic cells and oxidized neo-determinants that can induce pathogenic IgG autoantibodies (55, 56). Additionally, polyreactive IgM-NAA have been shown to bind to idiotypic determinants on self-reactive IgG, thus providing another mechanism to protect the host from high-affinity binding IgG autoantibodies that are potentially pathogenic (31, 50).
In mice, B1 cells are rare in the bone-marrow, lymph nodes, and splenic B cell follicles (white pulp) while significant numbers of B1 cells are located in the splenic marginal zone as well as in the peritoneal and pleural cavities. However, under normal conditions, these peritoneal B1 cells do not contribute significantly to circulating IgM-NAA, but during sepsis, peritoneal B1 cells rapidly migrate to the splenic marginal zone (23, 58) where most of the circulating IgM-NAA are produced (59). B1 cells are distinct from B2 cells in many respects and they are derived from different progenitors (18, 19, 60). Importantly B1cells express CD27, a memory B cell marker, and such as memory B cells, on encountering antigen, spontaneously secrete IgM without requiring signaling via co-stimulatory molecules (60). B1 cells do not require to traffic or reside in the splenic B2/T cell follicles as they are T independent and secrete IgM without isotype switching or somatic mutation (25, 59, 61). In fact, isotype switching and somatic hyper-mutation of immunoglobulins in B1 cells is kept in check during an immune response to prevent the development of high affinity, anti-self IgG antibodies (61, 62). In this regard, SPA-1 in B-1 cells inhibits Rap-1 GTP, which enhances somatic hyper-mutation and hence SPA-1 deficient mice generate high-affinity IgG anti-dsDNA and anti-red blood cell autoantibodies and develop lupus nephritis as they age (62). Recent data would indicate that levels of peritoneal B1 and splenic B1 and MZB cells is regulated by serum levels of polyclonal (but not monoclonal) IgM (63–65) that interacts with FcμR expressed by these cells (66) as well as by IgG binding to FcγRIIB (67).
IgG-NAA
One can demonstrate the existence of IgG-NAA in normal adult murine or human serum under conditions where serum IgM is either removed or diluted out (31, 68–70), indicating therefore that in serum the functional activity of IgG-NAA is blocked by polyreactive IgM with anti-idiotypic activity (31, 50, 54). There is, however, a large amount of IgG-NAA as significant levels of IgG-NAA can be detected even after diluting serum at 1:500 (34). Both IgG-NAA and IgM-NAA bind to the same phylogenetically conserved self-antigens and 15–20% of normal mouse IgG has been found to be polyreactive (50, 68). IgG produced by B1 cells are characteristically of the IgG3 isotype that have been shown to be polyreactive (18, 19). Clones of B cells secreting IgG4κ with high-affinity binding to dsDNA has been shown to exist in human umbilical cord blood and in the same umbilical cord there were other B cell clones producing IgM antibodies with binding reactivity to idiotypic determinants on IgG4 anti-dsDNA but not to idiotypic determinants on other IgG4 antibodies (34). The above observations would suggest that B cells generating IgG-NAA of different isotypes and affinities are present at birth and the IgG-NAA are rendered functionally inactive by polyreactive IgM with anti-idiotypic activity.
Importantly, IgG-NAA-producing B cells, unlike IgM-NAA-producing B cells, are in an inactive state at birth and in mice these B cells start producing IgG-NAA after exposure to bowel bacteria or foreign antigens (71–73). In humans, it may take more than 2 years before significant levels of IgG-NAA can be detected in the serum (74). It is also possible that autoreactive T cells, which have been shown to exist in normal individuals (75) or infectious agents could activate B cells to produce IgG-NAA later on in life. For example, levels of polyreactive IgG anti-dsDNA have been shown to increase after various infections and these polyreactive IgG anti-dsDNA have been found to cross-react with antigens on micro-organisms, including bacteria (76). It is, therefore, not surprising that low levels of IgG-NAA at birth increase after exposure to infections (71–73).
Studies on IgG-NAA isotypes in normal sera or on the B cell subsets that produce these antibodies are lacking. There are, however, extensive data on an IgG-NAA, i.e., IgG anti-DNA, that is both mono-reactive and polyreactive and present in both normal sera and SLE sera and some of the findings with IgG anti-DNA may apply to other IgG-NAA. Several lines of evidence would favor that B2 cells, with the help of T cells, can generate IgG-NAA. Wardemann et al. clearly demonstrated that 55–75% of early B cell precursors, present in human adult bone-marrow, display auto-reactivity to conserved intracellular cytoplasmic and nuclear constituents. About 80% of these autoreactive B cells are removed at two check points, i.e., centrally in the bone-marrow and in the periphery (77). Second, in human umbilical cord, B cells secreting high-affinity binding IgG4 anti-dsDNA have been isolated and IgG1 anti-myeloperoxidase (ANCA) antibodies have been found in normal human sera, indicating that these high-affinity isotype-switched antibodies are produced by the T cell-dependent B2 cells (34, 78). Furthermore, in the MLR/lpr murine model of SLE, IgG anti-dsDNA has been shown to be produced by B2 cells under the influence of excess T helper cells as a result of Fas deficiency (79). Hence, removing T cells ameliorates SLE and reduces production of IgG anti-DNA in this model of SLE (80–82).
Under normal conditions, it is unclear whether the (Fab)2 region of IgG-NAA is physiologically active especially since their activity is blocked by poly reactive IgM with anti-idiotypic activity (31, 50). Hence, most of the physiological functions of IgG-NAA have been derived from purified IgG or pooled purified IgG (IVIG). In vivo infusion of IVIG has been shown to ameliorate cell-mediated inflammatory processes, e.g., in Kawasaki’s disease or autoantibody-mediated disorders, e.g., in idiopathic thrombocytopenic purpura, anti-factor VIII autoimmune disease, or myasthenia gravis [reviewed in Ref. (83)]. One can show, in in vitro studies, that the beneficial effects of purified IgG-NAA can be mediated by both the (Fab)2 region and the Fc region of IgG-NAA. In these in vitro studies, the (Fab)2 region has been shown to (i) block function of activating FcγR receptors expressed by monocytes/macrophages. Such activating FcγR are involved in phagocytosis of IgG autoantibody complexed to platelets, (ii) block idiotypic determinants on IgG autoantibodies, thus neutralizing these antibodies, e.g., their binding to Factor VIII or platelets, (iii) inhibit function of certain pro-inflammatory cytokines by binding to these cytokines or their receptors, and (iv) provide protection against bacterial infection by binding of IgG-NAA to lectins on bacterial surfaces thus enhancing opsonization (84). Similarly, in in vitro studies, the Fc region of IgG-NAA has been shown to inhibit the function of B cells, plasma cells, dendritic cells (DC), macrophages, and neutrophils by binding to the inhibitory FcγRIIB receptor expressed by these cells. Activation of FcγRIIB by Fc region of IgG-NAA inhibits production of autoantibodies by B cells and plasma cells. Additionally IVIG ameliorates cell-mediated inflammation, e.g., in Kawasaki’s disease, by activating FcγRIIB and inhibiting the function of dendritic cells, macrophages, and neutrophils.
IgG with anti-idiotypic activity have been found in pathological conditions especially when there is minimal or no IgM with anti-idiotypic activity (85) and increased levels of in vivo anti-idiotypic IgG has been associated with amelioration of disease activity mediated by pathogenic IgG autoantibodies, thus indicating that under pathological conditions, in vivo IgG with anti-idiotypic activity is functionally active (85, 86). There are data to show that normal levels of IgG autoantibodies, e.g., to myeloperoxidase (MPO) increase >10-fold during disease states (87) and it is possible that under pathological conditions when IgM-NAA with anti-idiotypic activity is low, there is initially a compensatory increase in IgG antibodies with anti-idiotypic activity and the disease does not manifest clinically (85). However, with time, the data would also indicate that there is a decrease in the protective IgM/IgG anti-idiotypic antibodies and the disease manifests clinically (85). Hence, administering IVIG, which is known to have anti-idiotypic activity, could be beneficial in this situation especially if levels of both IgG and IgM-NAA are relatively low.
Pathogenic IgG Autoantibodies
Pathogenic autoantibodies that cause disease differ from IgM and IgG3-NAA in that they are predominantly of the IgG isotype, undergo somatic mutation with addition of N regions and these IgG autoantibodies can be either polyreactive or exhibit mono-reactivity. IgG autoantibodies, unlike IgM-NAA, have high-binding affinity (53). However, not all IgG autoantibodies with high-binding affinity are pathogenic in vivo and this is best exemplified by IgG anti-dsDNA autoantibodies (88–91). There are conflicting data on the B cell precursors that generate these pathogenic IgG autoantibodies. Data would indicate that both B1 and B2 cells can generate pathogenic IgG autoantibodies depending on the animal model used. For example, studies in the Fas-deficient MRL-lpr murine SLE model would indicate that pathogenic self-reactive IgG antibodies are generated by B2 cells (79). However, in the NZB/W disease model of SLE and in a murine transgenic model of hemolytic anemia, the pathogenic murine IgG anti-dsDNA and anti-erythrocyte transgenic autoantibodies were shown to be produced by B1 lymphocytes (21, 92–94). Hence, removing T cells did not affect production of IgG anti-dsDNA or disease activity in the NZB/W model of SLE (80).
There are several mechanisms that increase pathogenic IgG antibody production and IgG anti-dsDNA has been most studied in this regard. In adolescent females with SLE and rheumatoid arthritis (RA) patients, there are data to show that production of IgG polyreactive autoantibodies results from breakdown of B cell tolerance mechanisms where during B cell development, B cells producing high-affinity IgG autoantibodies have not been removed or silenced either centrally in the bone-marrow or in the periphery before they mature into naïve immuno-competent lymphocytes (95, 96). Studies in healthy individuals indicate that 55–70% of newly generated bone-marrow B cells express self-reactive antibodies and most of these B cells are removed or silenced such that in healthy humans there remains 5–20% of circulating naïve B cells that continue to generate self-reactive IgG autoantibodies (77). However, in SLE and RA patients, owing to breakdown of B cell tolerance mechanisms, 25–50% of mature naïve B cells were found to generate self-reactive IgG autoantibodies with diverse specificities (95, 96). Second, excess BAFF could also contribute to the breakdown of tolerance in the periphery as BAFF has been shown to inhibit deletion or apoptosis of self-reactive B cells (92, 97). Elevated BAFF levels have been shown to be present in serum of RA and Sjogren’s syndrome (98). Third, there are murine studies to show that IgM-NAA-producing B1 cells can be induced, especially with repeated immunization, to generate polyreactive IgG anti-self that can also react to the immunizing antigen. For example, B1 cells can be induced to generate pathogenic polyreactive IgG anti-DNA and anti-myosin antibodies by immunization with a hapten or an antigen derived from a micro-organism (99–101). Other mechanisms may, however, operate in certain other autoimmune disorders where highly specific IgG autoantibodies are produced, e.g., in autoimmune hemolytic anemia and in Factor VIII deficiency. It is possible that such autoimmune disorders could arise from a breakdown in peripheral mechanisms such as specific deficiency of anti-idiotypic IgM-NAA that blocks pathogenic autoantibody or specific deficiency of IgM-NAA that binds and masks autologous neo-determinants such as dsDNA.
Based on observations in the preceding paragraph, it appears that different mechanisms could operate in generating pathogenic IgG autoantibodies depending on the murine disease model. Additionally, the mechanisms may differ in humans even though the disease has similar disease manifestations. A summary of the existing evidence would indicate that IgG autoantibodies can be induced by the following mechanisms:
(i) An excessive increase in production of pathogenic IgG autoantibodies relative to IgM-NAA-producing B1 cells. Potential mechanisms that increase self-reactive IgG antibodies include lack of helper T cell apoptosis [e.g., in Fas-deficient MRL-lpr mice (79)], excess neoantigen-driven B cells [e.g., in human C1q deficiency with decreased removal of apoptotic cells (102)], excess BAFF production [e.g., in patients with Sjogren’s syndrome (98)], a breakdown in B cell tolerance mechanisms resulting in excess naïve self-reactive B cells [e.g., in SLE affecting young female patients (95)] and a deficiency in the inhibitory FcγRIIB receptor that normally inhibits B cells and plasma cells from producing IgG antibodies (103, 104).
(ii) Genetic predisposition that permits some antigen-driven B1 cell clones to acquire the ability for isotype switching and somatic mutation as, for example, in the NZB/W murine lupus model and in SPA-1 deficient mice (105, 106, 62). Hence, diseases induced by such defective B1 cells are ameliorated with administration of either polyclonal IgM or the specific IgM-NAA (107) or depleting B1 cells (94).
(iii) Repeated immunization that induces isotype switching in B1 cells that normally generate IgM autoantibodies with germline genes. For example, repeated immunization with hapten or a streptococcal antigen can induce B1 cells to generate IgG antibodies of different isotypes that are polyreactive and bind to the immunizing antigen as well as to self-antigens, such as DNA or myosin (99–101).
(iv) Excess production of a highly specific pathogenic IgG autoantibody by autoreactive B2 cell clones as, for example, in myasthenia gravis (108). Expansion of such B2 cell clones could have been driven by antigen, e.g., with cross-reactive micro-organisms in patients without thymoma (109) or be driven by acetylcholine receptor specific autoreactive helper CD4+ T cells in thymoma-associated myasthenia gravis where such autoreactive T cells have escaped tolerance mechanisms (110, 111).
(v) Deficiency of IgM-NAA, especially IgM-NAA with anti-idiotypic activity to a specific IgG autoantibody, thus incompletely neutralizing the increase in pathogenic IgG autoantibodies, such as antibodies to neutrophil cytoplasmic enzymes (myeloperoxidase, proteinase 3), which are commonly referred to as ANCA, dsDNA, and glomerular basement membrane (GBM) that are present in normal individuals (31, 50, 87, 112). Additionally, certain autoimmune disorders, such as ANCA vasculitis and anti-GBM nephritis, frequently occur in elderly patients who, with aging, can develop lower levels of IgM-NAA (28–32) but not IgG-NAA (33, 35, 36), which can increase with aging (34).
Figure 1 summarizes the role of natural and immune antibodies in normal and pathological states.
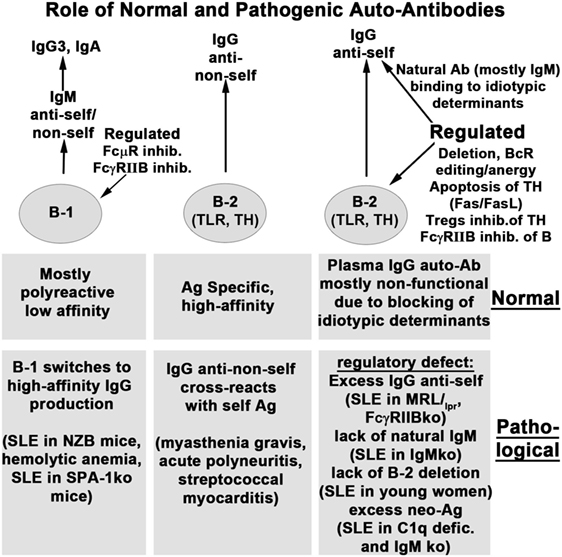
Figure 1. Role of normal and pathogenic autoantibodies.
Role of IgM-NAA in Protecting Against Pathogenic IgG Autoantibodies
SLE is the most studied autoimmune disorder where high levels of IgM and IgG autoantibodies, directed against nuclear antigens, especially dsDNA, are produced in the majority of patients. Before we discuss the role of IgM-NAA in protecting against pathogenic IgG anti-dsDNA, it would be important to indicate that a subset of lupus nephritis (approximately 25%) can occur in the absence of detectable circulating IgG anti-DNA and this is commonly referred to as C1q nephropathy (113, 114). The latter observations have led some investigators to question whether IgG anti-dsDNA is indeed pathogenic (115). In some patients with C1q nephropathy, circulating IgG anti-dsDNA can be detected several years later but this subset of patients with C1q nephropathy have in general a better prognosis even though histologically there is similar glomerular immunoglobulin staining and immune deposits as in classical lupus nephritis (114). It is tempting to speculate that patients with C1q nephropathy may have high IgM-NAA and IgM anti-dsDNA that may be preventing detection of circulating IgG anti-dsDNA. A murine model with lupus nephritis but with no IgG anti-dsDNA in both the circulation and in kidney eluates has also been described (116). It is possible that in this murine model, the immune deposits detected in the kidney may be caused by IgG complexes containing another antigen, other than dsDNA, and a similar mechanism could also operate in some patients with C1q nephropathy without circulating IgG anti-dsDNA.
However, there is evidence indicating that IgG anti-dsDNA, together with C1q, is an active participant in inducing or initiating glomerular damage in classical lupus nephritis with circulating IgG anti-dsDNA. First, SLE in the MRL/lpr lupus-prone mice is ameliorated when rendered IgG deficient by crossing these mice with either activation-induced deaminase (AID) or CD40L-deficient mice (117, 118). Second, one can induce lupus nephritis in normal mice by either infusing purified IgG anti-dsDNA or by increasing in vivo IgG anti-dsDNA with use of transgenic cells or hybridoma cells (88–91). Importantly, not all IgG anti-dsDNA are pathogenic as defined by induction of lupus nephritis in vivo and this also may explain why certain patients with high levels of circulating IgG anti-DNA are spared from lupus nephritis (88–91). In SLE, a potential protective mechanism involves binding of IgM anti-dsDNA to DNA with masking of the antigenic determinants that bind to IgG anti-dsDNA. Hence, lupus nephritis can occur if there is excess IgG anti-dsDNA or if there is a relative deficiency of IgM anti-dsDNA. Such a concept will explain the accelerated development of lupus nephritis in MRL/lpr mice lacking IgM and will also explain the lack of lupus nephritis in patients having high IgG anti-dsDNA and high IgM anti-dsDNA or IgM-NAA (119, 120). This latter possibility may also explain why FcγRIIB-deficient Balb/c mice are more resistant to developing lupus nephritis when compared to FcγRIIB-deficient C57BL/6 mice (121). In both mice, there is excess IgG anti-dsDNA production due to lack of the inhibitory FcγRIIB receptor but Balb/c mice have significantly higher levels of plasma IgM when compared to C57BL/6 mice (122–124).
Several observations would suggest a role of IgM-NAA in protecting normal individuals and patients with autoimmune disorders from such self-reactive IgG antibodies. The protective role of IgM-NAA is best exemplified in murine models of SLE, where enhancing IgM anti-dsDNA levels or infusing IgM anti-dsDNA, ameliorates disease activity (107, 125, 126). Well studied mechanisms by which IgM-NAA can induce protection and prevent inflammation include (i) removing neo-antigens by binding of IgM-NAA to oxidized neo-determinants and removal of IgM/C1q bound antigens by phagocytosis, e.g., IgM anti-PC (127), (ii) inhibition of the pathogenic IgG autoantibody induced inflammatory response by IgM-NAA, e.g., IgM-ALA (128), (iii) neutralization of pathogenic IgG autoantibodies by IgM-NAA having anti-idiotypic activity (129), (iv) preventing binding of IgG to antigenic determinants by competitive inhibition even though these IgM-NAA have lower binding affinity (129), (v) inhibiting complement activity by binding of IgM to complement components (17), and (vi) inhibiting generation of IgG autoantibodies possibly by binding of IgM-NAA to endogenous autoantigens/neo-antigens or to the B cell FcμR (64, 65, 119, 130–132).
The above mechanisms may explain why mice without secretory IgM have increased IgG autoantibodies, including anti-dsDNA, with aging (119, 120). This would also explain why B1-deficient SJL mice are more susceptible to induction of experimental autoimmune thyroiditis or allergic encephalitis cells (133). Conversely, such a concept would also explain why disease activity in SLE patients is less severe with high levels of IgM anti-dsDNA and other IgM-NAA (125, 134, 135). The above mechanisms may also explain why several autoimmune disorders are helped by either increasing production of in vivo IgM-NAA (126) or administering either pooled polyclonal IgM or a specific IgM-NAA (110, 129, 136). The latter is best exemplified by IgM anti-dsDNA where infusion of a specific IgM-NAA ameliorated SLE-induced nephritis in NZB mice (107).
Presence of pathogenic IgG autoantibodies leads to a compensatory increase in IgM–NAA, such as IgM anti-PC, IgM-ALA, and IgM/IgG anti-idiotypic antibodies, to counter these pathogenic IgG antibodies as well as inflammation initiated by the excess IgG self-reactive antibody. It is also possible, that over time, the production of IgM and IgG anti-idiotypic antibodies may not be able to keep up with the relatively high production of pathogenic IgG autoantibody and the disease becomes symptomatic. Hence, the initial increase in IgM-NAA and IgG anti-idiotypic antibody levels could explain the asymptomatic nature of the disease despite increased levels of IgG anti-dsDNA and other anti-nuclear antibodies several years before clinical onset of disease (85, 137). Additionally, there could be a heightened compensatory response of other immuno-regulatory mechanisms, e.g., increase in T-regs or IL-10 in B1 cells, which could inhibit the clinical onset of disease or ameliorate disease activity in these patients and other animal models. The latter is best exemplified in mice with secretory IgM deficiency, where despite total lack of circulating IgM and increased IgG3 anti-dsDNA (120), these cellular compensatory mechanisms inhibit development of autoimmune disease including lupus nephritis (63, 119). It is also possible that IgG3 anti-DNA is not pathogenic and their increased production by B1 cells, in secretory IgM deficiency (120), may be protective. However, in situations where inflammation has been experimentally induced or there is an excessive increase in production of pathogenic IgG autoantibodies or increased apoptosis, then these cellular regulatory mechanisms cannot effectively compensate for lack of IgM and this leads to a more severe and rapid worsening in disease activity. For example, SLE disease is more severe in the MRL-lpr mice when these mice are crossed with secretory IgM deficient mice (120). Similarly, secretory IgM-deficient mice are prone to develop a more rapid and severe inflammatory response after renal ischemia or allogeneic cardiac transplantation (138).
Physiological Role of IgM-NAA
IgM–NAA, which are spontaneously secreted by B1 and MZB cells, are polyclonal and exhibit a diverse repertoire of antigen specificities, many of which are polyreactive and, hence, cross-react with foreign antigens or pathogens. Additionally, B1 and MZB cell clones can be activated either by their TLR or BCR by both endogenous and foreign antigens. These special characteristics of IgM-NAA, together with the low-binding affinity (but with high-binding avidity owing to its pentameric structure), have allowed these antibodies to acquire functions that are not characteristic of the highly specific immune IgM and IgG antibodies. Physiological functions that have been attributed to IgM-NAA have included the following:
(i) Countering the pathogenic effects of the normally present IgG autoantibodies that escape B cell tolerance mechanisms. Potential strategies employed by IgM-NAA for this purpose include (a) neutralization of these IgG autoantibodies by anti-idiotypic mechanisms (129), (b) masking endogenous antigens (129), and (c) inhibiting the expansion of B2 cell clones that produce these IgG autoantibodies (79, 126). We have discussed these mechanisms in an earlier section of this review. However, the most compelling data demonstrating that IgM-NAA can counter pathogenic IgG autoantibodies are studies in murine models of SLE where administration of IgM anti-DNA or increasing expression of IgM anti-DNA in MRL-lpr mice (by crossing MRL-lpr mice with transgenic mice expressing IgM anti-DNA) ameliorated disease activity (107, 126).
(ii) Providing the first line of defense against pathogens while the adaptive immune system, i.e., B2 and T cells, are being deployed to mediate a more specific and effective immune response that is long lasting and has memory. The role of NAA in mediating protection against pathogens was first brought to light by the observation that NAA recognize bacterial toxins and several viral antigens (139, 140). Second, the creation of mice deficient in secretory IgM clearly aided in analyzing the protective role of both natural and immune IgM (63, 141). This topic will be briefly discussed as there are several excellent and detailed reviews on this subject (55, 141). Briefly, IgM-NAA mediates this protection through several mechanisms. First, each natural IgM clone, by having a pentavalent structure and by being polyreactive, can simultaneously bind to different conserved structures, such as nucleic acids, phospholipids, and carbohydrates, on the same pathogen and inhibit the pathogen from invading cells and disseminating into different organs. Second, the early components of complement, such as C1q, bind to the IgM complexed to the invading organism. Complement binding to IgM not only enhances neutralization of pathogens, e.g., certain viruses and bacteria, but also enhances phagocytosis of pathogens by macrophages and dendritic cells. The importance of either natural or immune IgM in limiting infection varies with different pathogens and this subject has been well reviewed (55, 141). Third, normally present IgM-NAA has significant levels of IgM anti–PC, which can readily bind to PC that is attached to sugar residues on the cell membranes and cell walls of many invading organisms (142). PC was first detected in 1967 on S. pneumoniae (143) and subsequently found to be present on many bacteria, protozoa, fungi, and nematodes (142). Infection with many of these organisms increases levels of IgM anti-PC, which in the presence of C1q enhances phagocytosis of these organisms and inhibits an excess inflammatory response by mechanisms discussed below. Finally, we have shown that IgM-NAA can limit HIV infection, both in vitro and in humanized SCID mice through another mechanism, that is by inhibiting T cell activation (39) as well as by binding of natural IgM to CD4 and chemokine receptors and inhibiting HIV entry into cells (Figure 2) (39, 144).
(iii) Inhibiting IgG autoantibody production and inflammatory responses by clearing apoptotic cells and binding to oxidized neo-determinants. Apoptotic cells that are normally produced in large numbers, express oxidized neo-determinants. Several homeostatic mechanisms, including binding of C1q and mannose binding lectin (MBL) to apoptotic cells, are involved in inducing macrophages and DC to phagocytose apoptotic cells and nuclear antigens [reviewed in Ref. (145)]. IgM-NAA are also involved in enhancing this phagocytic process, especially after tissue injury of nerves (69), and in inducing an anti-inflammatory effect (56). Several IgM-NAA antibodies have been identified, each with binding specificity to certain oxidized neo-determinants, including PC, malondialdehyde (MDA), cardiolipin, phosphatidylserine, and Annexin IV present on apoptotic cells and not live cells (146). The best-studied IgM-NAA reactive to apoptotic cells is the IgM anti-PC antibody (also referred to as T-15), which binds to a dominant oxidized neo-determinant on apoptotic cells. Both in vitro and in vivo studies, using monoclonal IgM anti-PC, have clearly shown that phagocytosis by DC and macrophage is enhanced when C1q or MBL binds to the IgM/apoptotic cell complex. In these in vivo studies, either endogenous production of IgM anti-PC was increased by administering large numbers of apoptotic thymocytes (2.5 × 107) or mice were given 1.5–2 mg of a monoclonal IgM anti-PC preparation that bound to endogenous apoptotic cells. Hence, natural IgM anti-PC inhibits production of pathogenic IgG autoantibodies by masking oxidized neo-determinants and enhancing removal of apoptotic cells. However, enhancing apoptotic cell phagocytosis by DC also inhibits DC maturation/activation and induces an anti-inflammatory effect with decreased production of pro-inflammatory cytokines and an increase in levels of splenic regulatory B cells (127, 147). Additionally, repeated weekly infusions of monoclonal IgM anti-PC or apoptotic thymocytes inhibited development of arthritis and induced regulatory B cells in mice (127, 147).
Natural IgM autoantibodies has also been shown to bind to various oxidation-specific neo-determinants, specifically PC and MDA present on oxidized LDL lipids, but not native LDL (146, 148, 149). High levels of IgM anti-PC and IgM anti-MDA were also found in ApoE-deficient mice that have high circulating cholesterol and severe atherosclerosis. It has been suggested that oxidized lipids, in this murine model, induces these high IgM anti-PC/MDA antibodies as a protective mechanism. Supporting such a concept are other observations showing an association of high in vivo levels of IgM anti-PC and reduced atherosclerotic plaques in both patients and different animal models [reviewed in Ref. (56)]. Similarly, there are studies showing that immunization of LDL receptor knockout mice with the S. pneumoniae vaccine induces IgM anti-PC and this is associated with less atherosclerotic lesions (150). Several mechanisms have been postulated to explain the amelioration of atherosclerotic lesions by IgM anti-PC in these murine models of atherosclerosis. First, IgM anti-PC masks the PC containing neo-determinants on oxidized lipids, thus inhibiting uptake of these lipids by macrophages and this leads to a decrease in foamy macrophages that are part of the atherosclerotic plaque (151). Second, IgM anti-PC, together with C1q, enhances phagocytosis of apoptotic foamy macrophages containing high levels of lipids. Enhanced phagocytosis of apoptotic macrophages, especially by DC, inhibits DC maturation/activation and induces an anti-inflammatory milieu (127, 147). Data demonstrating increased atherosclerosis in secretory IgM-deficient mice or in C1q ko mice would lend support to the role of IgM anti-PC and C1q in inhibiting atherosclerosis (102, 141, 152–154).
The finding that both IgM-deficient mice and C1q knockout mice have increased apoptotic cells as well as develop SLE would indicate that both IgM anti-PC and C1q are required to effectively remove or clear apoptotic cells (155, 156). The latter studies would also indicate that inefficient clearance of apoptotic cells, including nuclear antigens, induces development of pathogenic IgG autoantibodies and SLE. However, ineffectual clearance of apoptotic cells cannot entirely explain the increased propensity of IgM-deficient mice to develop autoimmunity and more severe atherosclerosis when compared to C1q ko mice as both mice have similar number of apoptotic cells in the atherosclerotic plaques and the C1q ko mice has in addition, circulating apoptotic cells that should have worsened atherosclerosis (102, 141, 155). Such observations would indicate that IgM-NAA regulates inflammation by other additional mechanisms (to be presented below) besides enhancing phagocytosis of apoptotic cells.
(iv) Inhibiting inflammation by binding of IgM-NAA to receptors on live leukocytes, i.e., via IgM-ALA. The existence of this IgM-NAA subset was known since 1970 (157). Their role in inflammation was recognized when several investigators showed that the level of these antibodies, like IgM anti-PC, increased with diverse infections and inflammatory states [reviewed in Ref. (39)]. However, it was unclear whether IgM-ALA were pathogenic or not. The idea that IgM-ALA may have anti-inflammatory function and may be protective came from observations in allograft recipients where patients with high levels of IgM-ALA were found to have significantly less rejections and developed less alloantibodies after allo-immunization (Figure 3) (39, 131). We hypothesized that these natural IgM-ALA increase during inflammatory states to regulate leukocyte function and prevent excess inflammation that may be detrimental to the host. Two characteristics inherent in IgM-ALA allowed us to make this hypothesis. First, we showed that these antibodies bind with low affinity to different leukocyte receptors, including co-stimulatory molecules and chemokine receptors (4, 39). Second, we and other investigators observed that these complement fixing antibodies did not lyse leukocytes at body temperature but readily lysed cells at room temperature or colder temperatures in the presence of complement (131, 157, 159). This non-lytic nature of these antibodies at body temperature led us to hypothesize that IgM-ALA, by binding to receptors on live leukocytes at body temperature, could alter or regulate their function. Initially, with in vitro studies using human IgM, we showed that the repertoire of IgM-ALA varies among individuals during health and in disease and, second, that IgM-ALA regulates the function of both human and murine T cells and dendritic cells (4, 39, 138). Additionally, in murine models, we showed that increasing in vivo levels of IgM-ALA with physiological doses of purified polyclonal IgM, i.e., 150 μg/mouse, every 3 days, protected mice from (a) renal ischemia reperfusion injury (IRI), (b) ameliorated rejection in a fully mis-matched cardiac allograft model, and (c) protected NOD mice from developing autoimmune insulitis and diabetes. In further experiments, we show that the protective effect in the model of innate inflammation (i.e., renal IRI) is mediated in part by IgM-ALA binding to activated/mature DC and regulating their function (4). In a later section, we will review in more detail the above data on IgM-ALA. We plan to show that the observed anti-inflammatory effects of polyclonal IgM was predominantly from IgM-ALA and not from IgM anti-PC that bound to apoptotic cells.
(v) Inhibiting expansion of B1 cells and enhancing antigen presentation to B2 and helper T cells in splenic lymphoid follicles. This physiological function of IgM became obvious in mice with secretory IgM deficiency as well as in mice with FcμR deficiency. B1 cells in the splenic marginal zone increased several fold in mice deficient in IgM while levels of IgM increased in FcμR-deficient mice indicating that IgM maintains B1 cell homeostasis and levels of natural IgM by binding to FcμR (63, 64, 66, 160). Second, both these mice have impaired immune IgG response to protein antigens, especially with low dose antigen, indicating that IgM facilitates antigen trafficking from the splenic marginal zone to the B2 cell rich lymphoid follicles via FcμR bound IgM/antigen complexes (63, 160).
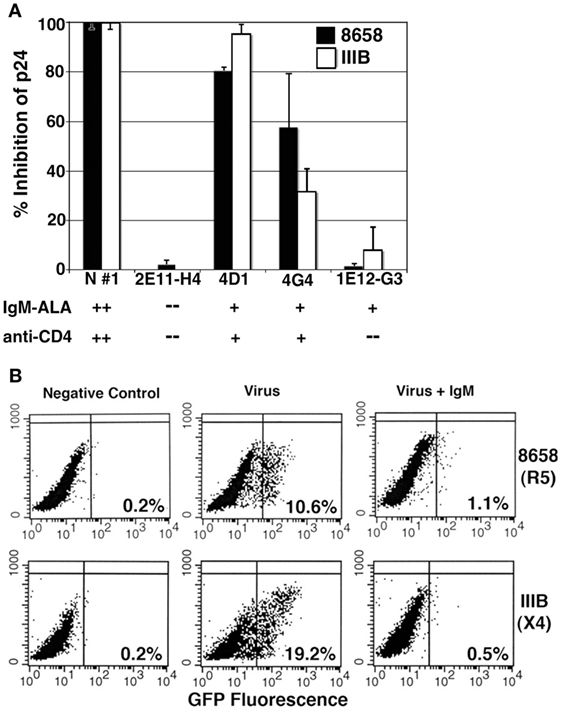
Figure 2. Evaluating the inhibitory effect of human monoclonal IgM and polyclonal IgM on R5-8658 and X4-IIIB HIV viruses. (A) Specificity of umbilical cord B cell clone IgM was characterized by binding of IgM, to recombinant CD4 and to leukocytes, which is depicted below each clone. IgM from B cell clones was used at concentration of 3 μg/ml, while purified IgM from normal serum (positive control) was used at 10 μg/ml. B cell clone 2E11-H4 with no IgM-ALA and no anti-CD4 activity was used as a negative control. Without IgM, p-24 levels for R5-8658 and X4-IIIB were 87,500 and 126,000 pg/ml, respectively. These data are representative of three different experiments. (B) The inhibitory effect of normal polyclonal IgM (5 μg/ml) on HIV-1 infection (single cycle replication) of GHOST cells that express GFP upon integration on viral HIV-1 DNA. IgM was present throughout the culture period and added to cells 15 min before adding virus. These are representative examples of five separate experiments. Figure and legend reproduced with permission from Ref. (144). Copyright 2008. The American Association of Immunologists, Inc.
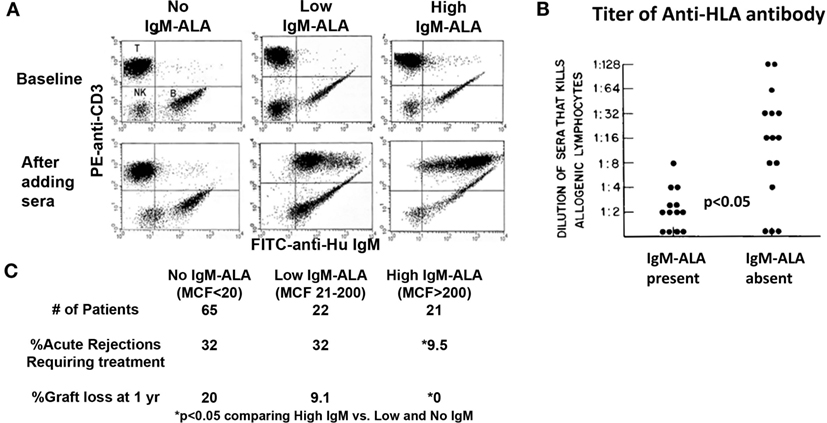
Figure 3. High levels of serum IgM-ALA in transplant recipients are associated with better kidney allograft survival and decreased alloantibody levels. (A) Dot plots labeled “baseline” depict IgM staining on B lymphocytes, but not on T cells, in the absence of sera. The lower panels (after adding sera) depict differences in the level of IgM bound to donor T lymphocytes (IgM-ALA) after addition of pre-transplant serum from different ESRD patients. (B) Alloantibody titer in sera measured by cytotoxicity is correlated to presence of IgM autoantibody to leukocytes. (C) The difference in percentage of acute rejections and graft loss comparing high IgM-ALA vs. the no and low IgM-ALA groups. MCF indicates increase in mean channel fluorescence of anti-IgM staining after addition of serum. Figure and legend reproduced with permission from Ref. (131) copyright 1981 Wolters Kluwer Health Inc. (B) Figure and legend reproduced with permission from Ref. (158). Copyright 2010 Springer Science + Business Media LLC (A,C).
The above observations indicate that IgM-NAA protect the host from invading organisms and more importantly maintain several homeostatic mechanisms primarily aimed at preventing autoimmunity and over exuberant inflammation, which can have detrimental effects on the host. Table 1 summarizes some of the physiological and pathological concepts outlined above. Several observations indicate that infective and other inflammatory states increase all IgM-NAA subsets, especially IgM anti-PC to clear the increased production of apoptotic cells that could trigger autoimmunity and, second, increase IgM-ALA to subdue excess inflammation that can be detrimental to the host [reviewed in Ref. (56) and next section]. Based on the preceding observations, one could predict that a decrease in IgM-NAA, as can occur in aging (28–30), could predispose to increased autoimmunity and increased morbidity and mortality from an excess inflammatory response, for example with infections.
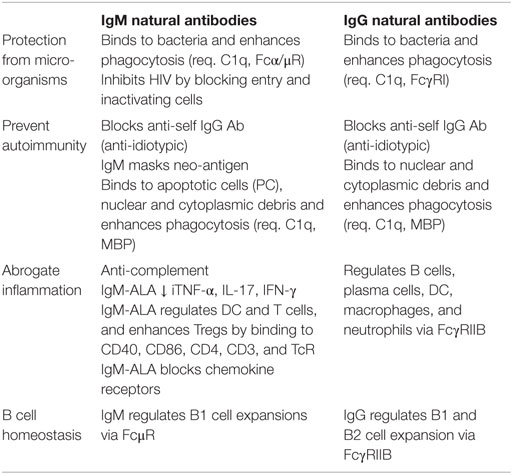
Table 1. Physiologic function of non-pathogenic autoantibodies.
Pathogenic Effects of IgM-NAA Under Non-Physiological Conditions
Certain IgM-NAA which under physiological conditions are protective and anti-inflammatory can under non-physiological conditions become pathogenic and induce inflammation. In this section, we will present some of the conditions that can induce certain protective IgM-NAA to become pathogenic.
(i) Induction of pathogenesis by binding of IgM-NAA at cold temperatures: This is best exemplified in human renal allograft transplant recipients having high IgM-ALA and IgM-anti endothelial cell antibody (IgM-AEA) levels at the time of the kidney transplant. These recipients have a high incidence of delayed kidney graft function (DGF) (161, 162) resulting from high levels of IgM-AEA which cause glomerular endothelial cell injury when, after successful vascular anastomosis, warm blood is allowed to flow into a cold kidney. Fortunately, this self-limiting injury is prevented by warming the kidney prior to re-instituting blood flow. Such observations highlight the nature of IgM-NAA, i.e., their potential for complement mediated cytotoxicity under non-physiological cold conditions (157, 159).
(ii) Induction of pathogenesis by binding of natural IgM to unmasked neo-antigens. This is best exemplified by the ubiquitous neo-antigen “non-muscle myosin heavy chain type IIA and C (NMM)” which is unmasked in murine models of acute ischemia to the small bowel, skeletal muscle (hind limb) and heart. About 1–2% of IgM-NAA B1 cell clones in mice secrete IgM anti-NMM probably to protect against NMM derived from infectious organisms (163, 164). In murine models of acute ischemia to the bowel or hind limb, injury is predominantly mediated during reperfusion by innate inflammation triggered by IgM binding to the unmasked NMM and activation of complement (6, 163, 165, 166). RAG-1 ko mice are normally protected from this ischemic injury to the small bowel or hind limb (163, 166). However, RAG-1 ko mice succumb to this ischemic injury after infusion of polyclonal IgM or monoclonal IgM anti-NMM. Additionally, one can clearly demonstrate binding of IgM and complement to NMM in bowel epithelial cells or to hind limb striated muscle cells (163, 165).
Interestingly, one does not observe innate inflammation mediated by IgM anti-NMM after murine renal ischemia even though endothelial cells in glomeruli and peri-tubular capillaries express NMM (167, 168). One possibility is that NMM in the peri-tubular capillaries or in the tubules is not unmasked after ischemia. After renal IRI, most of the ischemia induced kidney injury occurs in the outer medullary renal tubules. However, after renal IRI, one can detect increased IgM binding to glomeruli but not to the extensive network of NMM containing capillaries that surround the tubules or the renal tubules (168). Additionally, depleting B1 cells, decreased binding of IgM to glomeruli, and decreased glomerular injury, but did not protect tubules from inflammation-mediated renal injury, thus indicating that the innate inflammation seen after renal IRI is not mediated by natural IgM and complement (168). Other studies would indicate that the inflammatory response after renal IRI is mediated by ischemia-induced renal tubular injury that activates innate immune cellular mechanisms involving NK and NKT cells (169). This latter study would also explain why Rag-1 ko mice and IgM ko mice without secretory IgM are not protected from renal IRI (4, 138, 170–172).
(iii) Induction of pathogenesis by non-physiologic expansion of specific IgM-NAA clones. This is best exemplified by expansion of certain B1 cell clones that specifically secrete rheumatoid factor (RhF), an IgM-NAA that binds to self IgG. Excess production of RhF predisposes to generation of large circulating IgM/IgG complexes (referred to as cryoglobulins as these complexes precipitate ex vivo in the cold) that cause thrombosis of small blood vessels especially in the kidney glomeruli and skin (173, 174). This serious clinical problem is treated by plasmapheresis (to remove cryoglobulins) and agents to deplete B cells. The most common etiology for monoclonal or polyclonal expansion of RhF secreting B1 cell clones is chronic hepatitis C infection. Currently, it is unclear why this viral infection is associated with clonal expansion of RhF secreting B1 clones. More importantly, even though RhF was the first IgM-NAA to be discovered, we do not understand their normal physiological role.
Nature of IgM-ALA and Understanding Their Anti-Inflammatory Function
In this section, we review some of our pertinent observations on IgM-ALA that initially were discovered because of their lymphocytotoxic activity at room temperature in the presence of complement [reviewed in Ref. (39)]. Here, we show (a) that a substantial subset (8–10%) of IgM secreting human B cell clones, obtained from the umbilical cord, has IgM-ALA activity, (b) that polyclonal IgM-ALA clones have binding specificity to certain leukocyte subsets and that polyclonal IgM-ALA will bind to only some leukocyte receptors and not others. More importantly, we show that the repertoires of IgM-ALA vary in disease states among different individuals, and (c) that IgM-ALA exhibits anti-inflammatory effects, both in vitro and in vivo. We show that the anti-inflammatory effects are in part explained by IgM-ALA binding to co-stimulatory receptors and regulating the function of T effector cells and DC without decreasing Tregs. These in vitro studies involve human and murine cells, while in vivo studies were performed in mice.
Human Umbilical Cord B Cell Clones Produce IgM-ALA that Exhibit Leukocyte Receptor Specificity – Binding of IgM to Leukocytes was Not Mediated by FcμR
We initially wanted to determine if IgM-ALA exhibited leukocyte receptor specificity as natural antibodies are polyreactive and, hence, each monoclonal IgM could non-specifically bind to carbohydrate or other moieties on several leukocyte receptors. We isolated B cell clones from human umbilical cord blood and observed that >90% of B cells were IgM secreting with about 10% of the IgM clones having IgM-ALA binding activity when examined by flowcytometry on a cell mixture comprising of B (Daudi), T (Jurkat, Sup T1), and macrophage (U937) human cell lines. The lack of demonstrable leukocyte reactivity by the majority of IgM clones would indicate that IgM did not bind to FcμR on human B cells and macrophages (175). Second, we observed that these IgM-ALA clones exhibited leukocyte subset specificity in that some of the IgM-ALA monoclonal antibodies only bound to receptors expressed by all leukocytes or either T cells (SupT-1, Jurkat) or macrophages (U937) or B cells (Daudi) (Figure 4) (39). Further studies revealed that only human monoclonal IgM, having T cell reactivity, immunoprecipitated CD4 from cell lysates and bound to recombinant soluble CD4, thus indicating that IgM-ALA can exhibit both receptor and cell specificity.
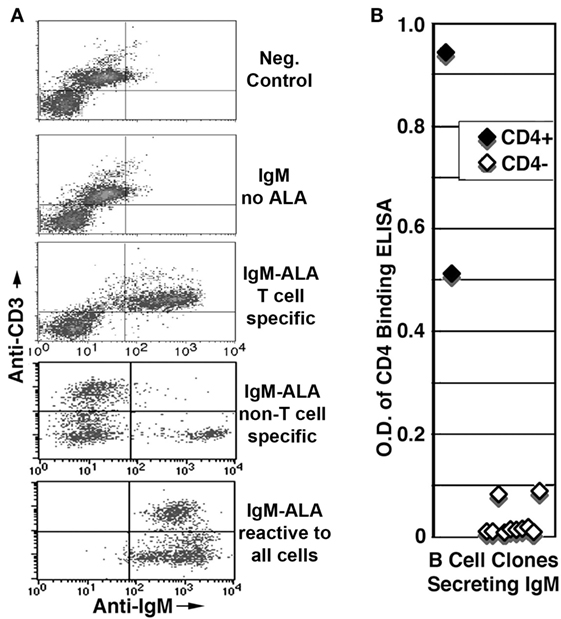
Figure 4. Presence of IgM-ALA in supernatants from umbilical cord B cells. (A,B) IgM-ALA reactivity in IgM containing supernatants from B cell clones activated with the EBV virus. IgM-ALA reactivity was detectable in 8 of 79 supernatants. (A) Supernatants were interacted with cells containing a mixture of cell lines, i.e., Jurkat, SupT-1, Daudi, and U937. Note that the supernatants have IgM-ALA that is specific for either T or non-T cells or all four cell lines. Subsequent studies revealed that IgM anti-non-T cell had binding reactivity to only U937 cells and not to Daudi cells. (B) Data on IgM anti-CD4 reactivity using an ELISA technique. Note that only 2 of 79 IgM-containing supernatants had IgM anti-CD4 reactivity and both these supernatants also had IgM with binding to leukocytes. Figure and legend reproduced with permission from Ref. (39). Copyright 2008. The American Association of Immunologists, Inc.
Polyclonal IgM from Different Human Sera Differ in Their Repertoire for Receptor Binding. IgM, in Addition, Regulates Human T Effector Cells and DC Without Affecting Tregs or Chemokine Production
We used polyclonal IgM, purified by size exclusion chromatography, from sera of normals, HIV patients and renal dialysis (ESRD) patients (39). Ammonium chloride precipitation was not used as this method affected IgM-ALA binding [see method details in Ref. (39)]. We showed that IgM could immunoprecipitate CD3, CD4, CCR5, and CXCR4 from lysates of cell lines (Figure 5). However, the repertoire of IgM-ALA was found to be different among individuals, especially patients as exemplified with HIV patients (see Figure 5). Such differences in the repertoires of IgM-ALA may explain why clinical manifestations of inflammation are different among different individuals. It is also possible that prior exposure to different infective agents or foreign antigens may explain the observed differences in the repertoire of IgM-ALA among different individuals (31).
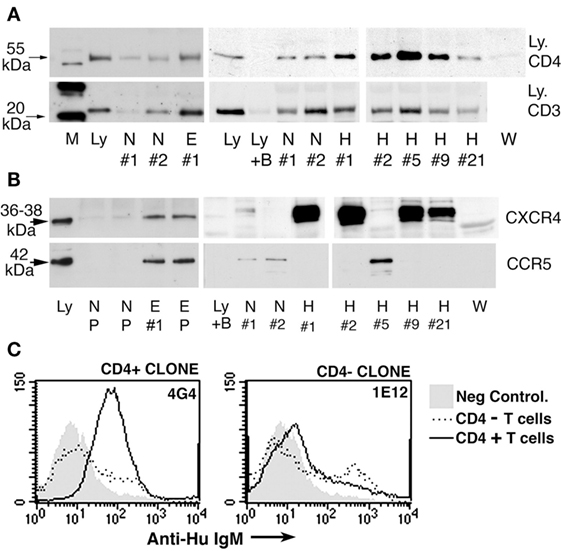
Figure 5. Immunoprecipitation experiments to show binding of human polyclonal IgM to CD3, CD4, CCR5, and CXCR4. (A,B) Identical quantities of individual (labeled no. 1, 2, etc.) or pooled (labeled P) IgM from normal (labeled N), HIV (labeled H), ESRD (labeled E), or Waldenstrom (labeled W) were used to immunoprecipitate leukocyte receptors from equal amounts of whole cell lysates or recombinant soluble CD4. As controls, Western blots were performed with cell lysates in the absence of agarose beads [to control for binding of primary Ab to leukocyte receptor and to determine receptor size (labeled Ly)]. In another control, agarose beads without IgM were added to lysate to determine whether the leukocyte receptor non-specifically bound to the bead (B plus Ly). Note that several fold more receptors were immunoprecipitated by ESRD and HIV IgM when compared with normal IgM. (C) Significantly increased binding of IgM from B cell clone 4G4 to CD4+ T cells when compared with CD4− T cells (MCF 71.6 vs. 16.6). Clone 4G4 secreted IgM with anti-CD4 reactivity. Note that no increased binding was observed on CD4+ T cells using a B cell clone (IE12) secreting IgM without anti-CD4 reactivity (MCF 22.5 vs. 22.3). Figure and legend reproduced with permission from Ref. (39). Copyright 2008. The American Association of Immunologists, Inc.
In in vitro studies with human peripheral blood mononuclear cells (PBMC), addition of polyclonal human IgM, but not human IgG or Waldenstrom’s IgM lacking IgM-ALA, differentially inhibited co-stimulatory receptor upregulation, cytokine production, and proliferation of T cells (39). Both normal and patient IgM downregulated expression of CD4, CD2, and CD86 but not CD8 and CD28 on blood PBMC activated with alloantigens (MLR) (39). Additionally, both normal and patient IgM inhibited production of the same set of cytokines, i.e., TNF-α, IL-13, and IL-2 but not IL-6 and chemokines when human PBMC were activated by alloantigens [Figure 6E; Ref. (39)]. Other investigators working with a monoclonal IgM-ALA with reactivity to TcR have shown that IgM-ALA can inhibit IL-2 production and T cell proliferation by binding to the TcR (5, 176). Similarly, we showed that IgM inhibited T cell proliferation induced by alloantigens or CD3 ligation and also inhibited Zap-70 phosphorylation induced by anti-CD3 [Figure 6F; Ref. (39)]. However, IgM did not alter chemokine production by activated PBMC but inhibited chemokine-induced chemotaxis by binding to the receptor and blocking chemokine binding (39). Importantly, IgM did not alter Treg levels in an MLR, despite inhibiting T cell proliferation. In general, identical quantities of patient IgM had a more inhibitory effect in the above studies when compared to normal IgM (39). These functional differences between normal and patient IgM may be explained by differences in their quantity and IgM-NAA repertoire (Figure 5). Other investigators have also shown that polyclonal human IgM can inhibit proliferation of human T cells (5, 177).
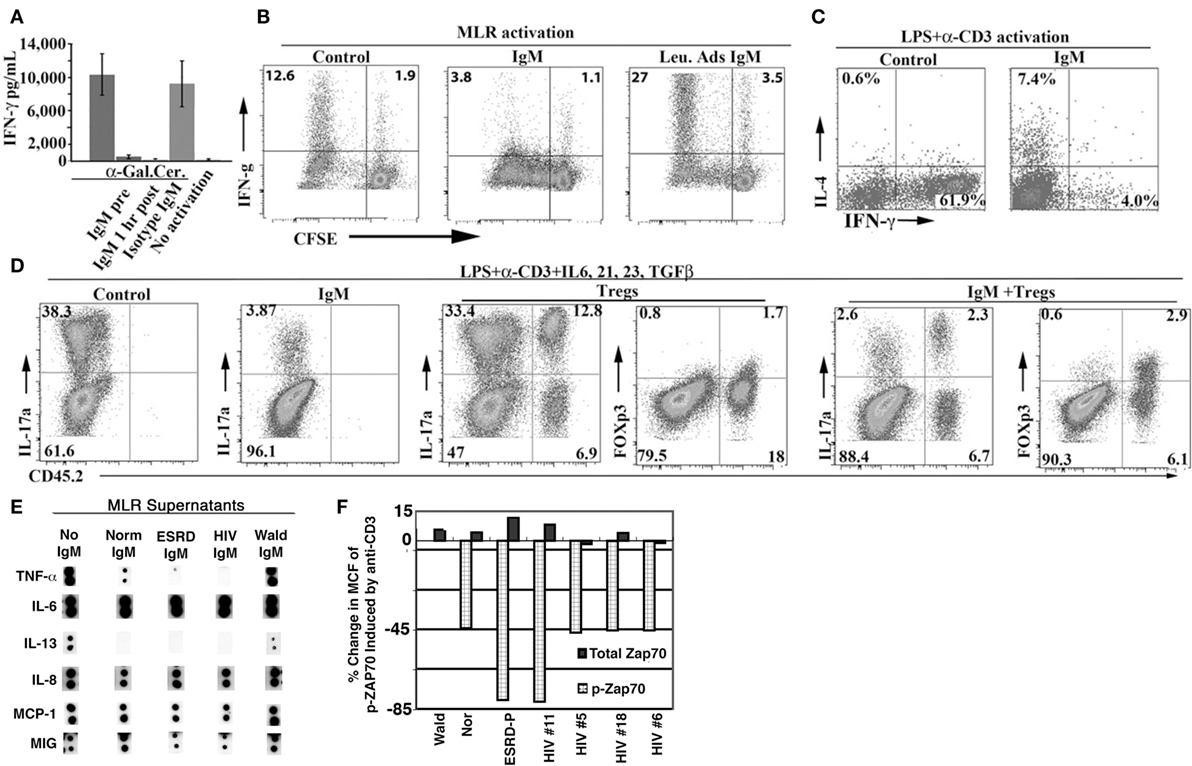
Figure 6. Polyclonal IgM inhibits IFN-γ production and T cell proliferation and differentiation into TH-1 and TH-17 cells of murine splenic cells and specific pro-inflammatory cytokines from human leukocytes activated with alloantigens. (A) Supernatant IFN-γ in 48 h culture media of splenic cells activated with a-gal-ceramide that specifically activates NKT-1 cells. IgM was added either 0.5 h before activation (IgM pre) or 1 h post activation. (B–D) CFSE labeled WT-B6 splenocytes (2.5 × 105 in 0.5 ml media) were activated either in a one-way MLR (using 7.5 × 05 BALB/c irradiated splenocytes) or LPS (350 ng) and soluble anti-CD3. Cells were cultured for 4 to 5 days. IgM (10–15 μg) was added at the initiation of culture unless otherwise indicated. In (D), the effect of Tregs was evaluated by co-culturing 2.5 × 105 CD45.1 WT-B6 splenic leukocytes, containing 1.8% CD4+ Foxp3+ cells, with 0.5 × 105 CD45.2 WT-B6 Tregs (76% Foxp3+) under cytokine conditions favoring TH-17 differentiation. (E) Pooled human normal, ESRD, and HIV IgM but not Waldenstrom IgM significantly inhibit the increase in TNF-α and IL-13 but not that of IL-6, IL-8, MIG, and MCP-1 produced in response to alloantigen activation of T cells. Supernatants were obtained from day 5 MLR cultures stimulated in the presence or absence of pooled IgM (15 μg/ml), added on day 0. (F) Pooled human IgM inhibits anti-CD3-mediated Zap-70 phosphorylation of human T cells. IgM was added 30 min before anti-CD3/28 and cells were cultured overnight before quantitation with flowcytometry. Figures and legend reproduced with permission from Ref. (138) (A–D) and (39) (E,F). Copyright 2008 and 2012. The American Association of Immunologists, Inc.
In summary, the data indicate that polyclonal IgM, in physiological doses, inhibit human T effector cell activation and proliferation as well as regulates production of certain cytokines by binding to certain co-stimulatory molecules (CD4, CD3, TcR). IgM does not inhibit T regs. We also show that the quantity and repertoire of IgM-ALA varies in different individuals especially in disease. Interestingly, IgM-ALA does not appear to affect the production of chemokines by leukocytes, but interferes with their action by binding to chemokine receptors.
Polyclonal Murine IgM Binds to Specific Co-Stimulatory Receptors and Regulates the Function of Murine T Effector Cells, DC, and NKT Cells but Not Tregs
In murine studies, we initially observed that IgM-ALA bound to pronased splenic leukocytes (Figures 7B,C) and that IgM-ALA had several fold increased binding to live splenic granulocytes, DC, and B cells when compared to T cells and IgM binding to all leukocytes was enhanced when cells were activated (see Figure 7A). Furthermore, we showed that IgM-ALA bound to splenic leukocytes independently of FcμR and, second, showed that IgM, after binding to leukocytes, could immunoprecipitate several different leukocyte receptors (Figure 7D) (138). These observations, together with the above findings with human leukocytes, led us to investigate if IgM had an inhibitory effect on the function of T cells, DC, and NKT cells by binding to receptors, e.g., antigen-presenting receptors and co-stimulatory receptors, that get upregulated during activation. Both T cells and DC have an important role in adaptive immunity, e.g., in transplantation and NKT cells, together with DC, have an important role in innate immunity.
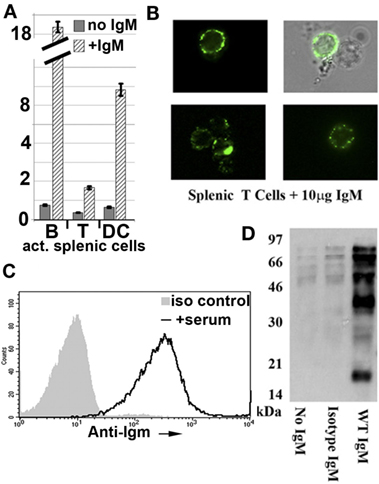
Figure 7. Polyclonal murine WT IgM bind to membrane receptors on leukocytes. (A) Polyclonal IgM has several fold increased binding to activated murine splenic B cells and dendritic cells (DC). Splenic leukocytes, activated for 48 h were interacted with purified mouse IgM at 4°C and evaluated for IgM binding using IgG anti-IgM (clone 11/41). Isotype monoclonal IgM with reactivity to KLH did not bind to activated leukocytes (data not shown). IgM binding to B cells was evaluated by blocking intrinsically expressed IgM with unlabeled IgG anti-IgM (clone 11/41). (B) It depicts immunofluorescence microscopy images of IgM binding to cell membranes of splenic T lymphocytes. (C) It compares binding of IgM and isotype IgM on CD3+WT-B6 pronase pretreated splenic leukocytes. Spleen cells were pronase digested to remove FcμR and to show that IgM-ALA can bind to other receptors on cell membranes. (D) It depicts a representative example of a Western blot from two separate experiments demonstrating immunoprecipitation by WT-polyclonal IgM of biotinylated membrane proteins from the murine macrophage cell line J77. In this experiment, WT-polyclonal IgM is compared with an equal amount of isotype IgM that has no binding activity to leukocytes using flow cytometry. Figure and legend reproduced with permission from Ref. (4) (A) and Ref. (138) (B–D). Copyright 2012 and 2015. The American Association of Immunologists, Inc.
With murine leukocytes, we showed in in vitro studies, that physiological doses of murine polyclonal IgM inhibited naïve T cells from differentiating into TH-1 and Th-17 cells even when IgM was added 48 h after activation (Figures 6B–D) (138). This inhibitory effect of IgM-ALA on T cells did not depend on presence of DC as the same inhibitory effect was noted when T cells were activated with insoluble anti-CD3/28. IgM inhibition of anti-CD3-mediated Zap-70 phosphorylation would indicate that the inhibitory effect of IgM on T cell function is mediated by IgM binding to CD3 [Figure 6F; Ref. (39)]. IgM did not, however, inhibit differentiation of murine T cells into Foxp3+ cells and, furthermore, under Th-17 differentiating cytokine conditions, IgM inhibited sorted Foxp3+ cells from differentiating into TH-17 cells (Figure 6D) (138).
Since there are <1.5% of DC in murine splenic leukocytes, we used 7–8 day cultured murine bone-marrow DC (BMDC) to investigate the functional effects of IgM on DC (4). We showed that polyclonal murine IgM, but not IgM pre-adsorbed with activated splenic leukocytes, bound to recombinant soluble CD40 and PD1 but not PDL-1, CD40L, and CD80, indicating, therefore, that IgM-ALA has binding specificity to certain DC receptors, just as we observed with human T cell receptors where IgM bound to CD4, CD3, and CD2 but not to CD8 (39). The functional effect of IgM binding to certain specific co-stimulatory receptors was tested on LPS-activated BMDC. We show that IgM, even when added 2 h after LPS, inhibited LPS-induced CD40 upregulation, but not upregulation of CD86, PDL-1, and MHC-II of BMDC and downregulated basal expression of PD1 on BMDC. IgM, in addition, downregulated p65NF-κB activation induced by LPS (Figure 8D) but not by LPS + anti-CD40 (agonistic Ab), thus indicating that IgM can inhibit p65NF-κB upregulation mediated by TLR4 activation, but not when both TLR4 and CD40 are activated (4). Interestingly, IgM inhibited TLR4 activation by a mechanism that did not involve inhibition of LPS binding to cell receptors (4). However, despite downregulation by IgM of TLR4-induced p65NF-κB, there was no decrease in IL-12 production or increase in IL-10 production indicating that LPS activates other transcription factors, besides p65NF-κB, to upregulate these cytokines and certain other co-stimulatory receptors that were not downregulated with IgM (4). In in vivo studies (to be presented in the next section), we show that IgM pretreatment of LPS-activated BMDC switches these activated BMDC to a regulatory phenotype possibly by a mechanism involving downregulation of CD40 and NF-κB.
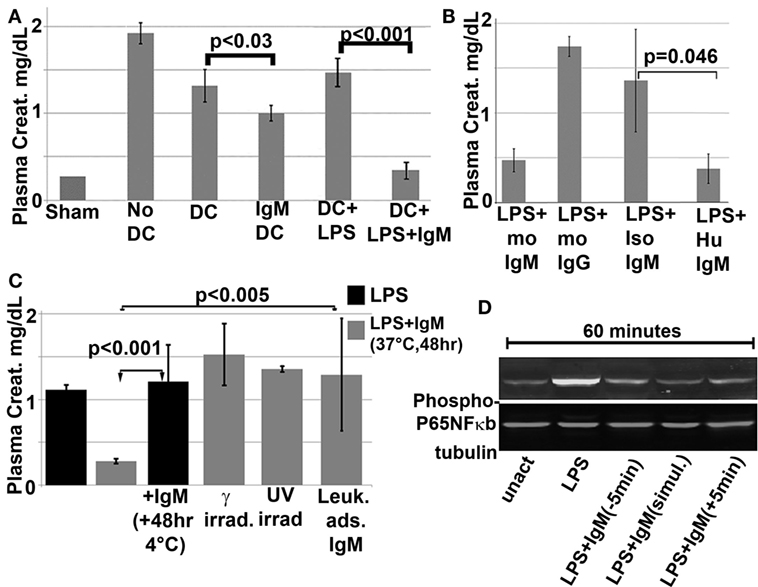
Figure 8. In vivo infusion of polyclonal human or murine IgM pretreated WT-BMDC with downregulated NF-κB protects mice from renal IRI. (A) There is more protection when using WT-BMDC pretreated with both IgM and LPS as evaluated by SCr. WT-BMDC were pretreated with IgM for 1 h before adding LPS and culturing BMDC at 37°C for 48 h. (B) LPS-activated BMDC pretreated with either mouse or human IgM are protective in renal IRI. (C) Pretreating 48 h LPS-activated WT-BMDC with IgM for 1 h at 4°C is not protective in IRI. Fourty-eight hour IgM/LPS-pretreated WT-BMDC are non-protective in IRI after being irradiated (3000 Rad) or rendered apoptotic by exposure to UV light. Additionally, polyclonal IgM adsorbed with splenic leukocytes to deplete IgM-ALA clones was not protective in IRI. (D) Western blot depicting IgM-mediated downregulation of LPS-induced phosphorylation of p65 NF-κB in BMDC. IgM was added either before or after LPS activation. Figure and legend reproduced with permission from Ref. (4) Copyright 2015. The American Association of Immunologists, Inc.
We next did in vitro studies to test the effect of polyclonal IgM on Type1 NKT cells, which together with DC, have an important role in inducing innate inflammation, e.g., after renal reperfusion injury (IRI). In these studies, we used α-gal-ceramide, a glycolipid that is taken up by DC and presented via the CD1d receptor to Type 1 NKT cells, which get activated and secrete IFN-γ. This assay is specific for determining Type 1 NKT function as only Type 1 NKT, but not T effector cells, will secrete IFN-γ after exposure to α-gal-ceramide, which is recognized, in context of CD1d presentation, by the invariant TcR on Type 1 NKT cells. We show that physiological doses of IgM inhibits α-gal-ceramide induced IFN-γ production of splenic leukocytes even when IgM is introduced 1 h after α-gal-ceramide (Figure 6A) (138). We have not defined the mechanism for the inhibitory effect of IgM on Type 1 NKT function.
In summary (see Table 2), the in vitro data with both human and murine cells would indicate that IgM-ALA regulates leukocyte function by binding and downregulating certain leukocyte receptors (e.g., CD4 and CD2 on T cells, CD40, and CD86 on DC) and inducing regulatory function in DC. Importantly, IgM does not decrease Foxp3+ Tregs. IgM regulates leukocyte activation, proliferation, and chemotaxis to attenuate excess inflammation [Figure 6; Ref. (39)]. The marked individual variation in the repertoire of IgM-ALA, with specificity to the different leukocyte receptors, observed in both normal and disease states could potentially explain the differences in the vigor and character of inflammatory responses in different individuals exposed to the same inciting agent. Additionally, differences in inflammatory response may also be influenced by total levels of IgM-NAA or IgM-ALA as we observed in transplant recipients (Figure 3). Finally, IgM-ALA, by binding to leukocyte receptors and inhibiting cell activation, can provide another mechanism to limit viral entry into cells and replication as we have shown with the HIV-1 virus (Figure 2) (144).
Table 2. In vitro effects of IqM-ALA on human and murine leukocytes.
IgM-ALA Inhibits the Innate Immune Inflammatory Response in Renal Ischemia Reperfusion Injury
To test the in vitro inhibitory effects of IgM-ALA on DC and NKT cells, we used an in vivo murine model of renal IRI (169). In this model, renal vessels to both kidneys are completely occluded with clamps for 26 or 32 min to induce either mild or severe ischemic renal tubular injury. The kidneys are then allowed to re-perfuse by unclamping the blood vessels and the extent of renal injury or decrease in renal function is quantitated at 24 h of reperfusion by measuring for accumulation in the plasma of waste products (e.g., creatinine) that are normally only removed by the kidneys. In this model, the initial ischemic injury is not sufficient to impair renal function as quantitated by measuring plasma creatinine, but it is the innate inflammatory response to products released by ischemic renal cells (e.g., DAMPS and glycolipids) that significantly worsen kidney injury that leads to loss of function. In this model, DAMPS and glycolipids released by ischemic renal cells are taken up by DC and in the splenic marginal zone, DC present glycolipids in the context of CD1d to activate NKT cells, which rapidly release IFN-γ to activate innate effector cells especially granulocytes, macrophages, and NK cells (169). Activated innate effectors migrate to the kidney, where chemokines, released by ischemic cells, enhance extravasation of inflammatory cells into the kidney interstitium. The inflammatory effector cells in the kidney interstitium cause further renal tubular injury with loss of kidney function that leads to an increase in plasma creatinine. This acute loss in kidney function is referred to as acute kidney injury (AKI).
We used two approaches to test the protective role of IgM in the suppression of this ischemia-induced innate inflammatory response. First, we performed renal IRI in B6/S4-IgMko mice (referred to as IgM ko) that lack circulating IgM but have normal levels of other immunoglobulins. These mice have normal or increased levels of Tregs, B regs, and IL-10 and their normal functioning B cells express membrane IgM or BcR but are unable to secrete IgM. Unlike their WT counterpart, we demonstrated that these mice are very sensitive to renal ischemia, developing AKI with mild ischemia (26 min clamp time) that is insufficient to cause AKI in their WT counterparts (Figures 9A,B). Replenishing IgM in the IgM ko mice with a single 240 μg dose of polyclonal IgM, to achieve plasma levels similar to that in their WT counterparts, protected these IgM ko mice from developing AKI with mild ischemia, thus indicating that sensitivity to ischemia in the IgM ko mice resulted from a lack of circulating IgM (Figures 9A,B).
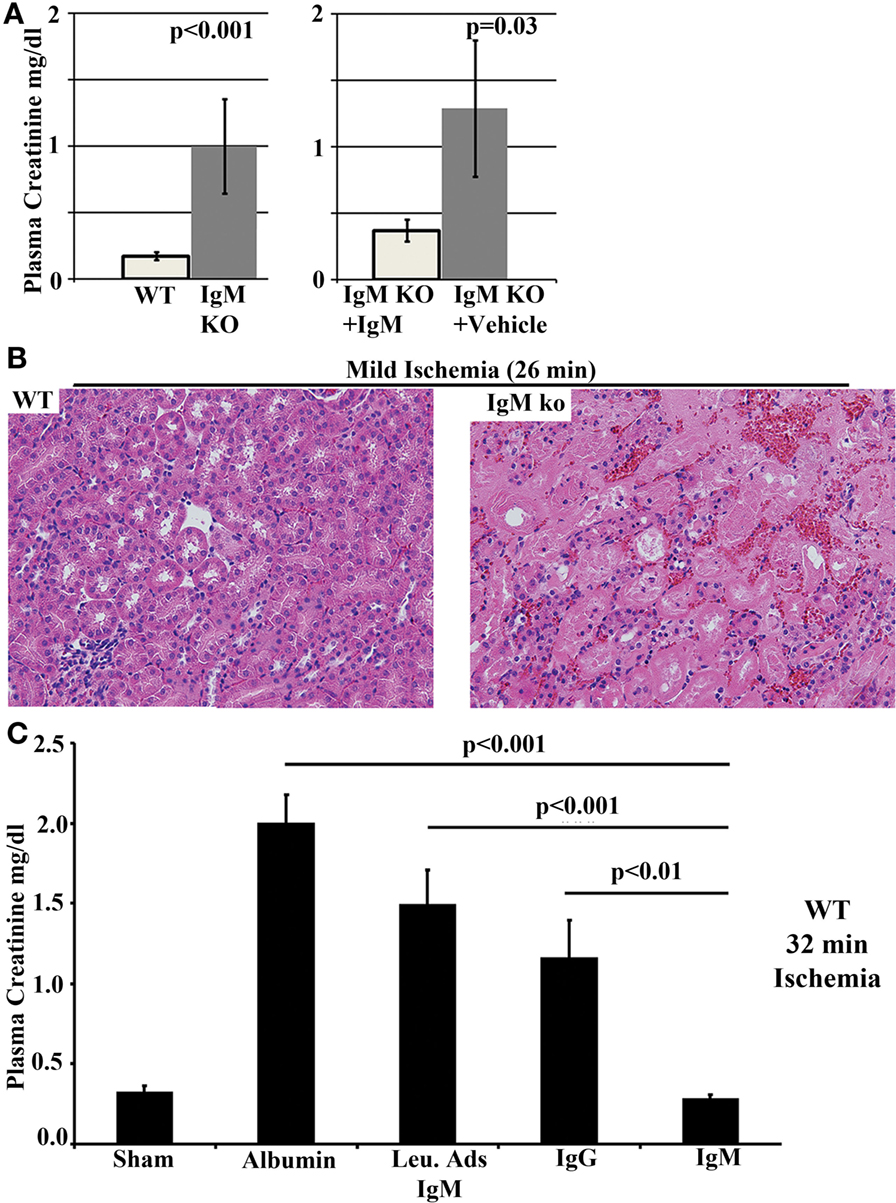
Figure 9. B6/S4-IgMko mice are more sensitive to renal IRI when compared to their WT counterparts (WT-B6/S4). (A, B) Kidneys from B6/S4-IgMko mice and their WT counterparts (WT-B6/S4) were subjected to mild ischemia (26 min) and then reperfused. Data depict 24 h plasma creatinine comparing (WT-B6/S4) WT mice with B6/S4-IgMko mice, and B6/S4-IgMko pretreated with 240 μg IgM, 24 h before ischemic injury. Histology depicts H&E staining of renal outer medulla after 24 h of reperfusion. (C) Polyclonal IgM, but not leukcocyte adsorbed IgM (Leu-Ads IgM), protects against renal IRI in WT-B6 mice. In these studies, WT-B6 mice were pretreated with equal quantities (150 μg in 0.75 ml) of IgM or Leu-Ads IgM or IgG, 24 h before subjecting the kidneys to severe ischemia (32 min). Kidneys were reperfused for 24 h prior to determining plasma creatinine (C). Control mice were pretreated with 0.75 ml RPMI containing 150 μg bovine albumin to exclude variables, such as volume/colloid, that can protect against ischemic injury. Figure and legend reproduced with permission from Ref. (138). Copyright 2012. The American Association of Immunologists, Inc.
In the second approach, a single dose (150 μg) of normal purified polyclonal IgM was administered intravenously to wild-type C57BL6 (WT-B6) mice to increase baseline circulating IgM by about 30–50% and to determine if increasing IgM would protect these WT-B6 from severe renal ischemia (32 min clamp time). These studies clearly indicated that increasing circulating IgM levels protected mice from severe renal IRI (Figure 9C). We next determined that this protection was mediated by IgM-ALA as administering similar quantity of polyclonal IgM pre-adsorbed with activated splenic leukocytes to remove IgM-ALA failed to protect these WT-B6 mice from severe renal IRI (Figure 9C).
In both approaches, physiological doses of intravenous polyclonal IgM mediated protection by decreasing the innate ischemia-induced inflammatory response. Protected kidneys had a very minimal inflammatory response with no or minimal tubular injury that could be detected on histology. Based on our in vitro data, in vivo IgM-ALA could mediate protection through several mechanisms, including regulation of NKT and DC and maintaining or enhancing Tregs, which also mediates protection in this model of innate inflammation (178). Our prior observations showing that IgM-ALA had several fold increased binding to splenic DC, when compared to T cells, prompted us to investigate the role of IgM-ALA in regulating DC in this model. These studies are presented in the next section.
Protection from Renal Ischemia is Mediated by IgM Induced Regulatory DC. Regulatory DC Require Tregs, B cells, Circulating IgM, and IL-10 to Mediate In Vivo Protection
To examine the in vivo role of IgM on DC, we used 7 to 8-day-old cultured BMDC. In these studies, BMDC were activated ex vivo for 48 h with LPS with or without polyclonal IgM. Activated BMDC were washed and then 0.5 × 106 BMDC were infused intravenously into mice 24 h before performing renal ischemia. In these studies, IgM + LPS-pretreated BMDC protected mice from ischemia induced AKI (Figures 8A) by inhibiting the increased generation of circulating granulocytes and inhibiting innate activated leukocytes from infiltrating the ischemic kidney [Figure 10, Ref. (4)]. Importantly, protection with LPS-activated BMDC was only observed when IgM was present during the 48-h culture and not when IgM was added at the end of the 48 h LPS activation (Figure 8C), indicating therefore that regulation of BMDC by IgM is an active process requiring both NF-κB and CD40 downregulation induced by IgM (Figure 8D). Preventing downregulation of NF-κB and CD40 by adding the agonistic anti-CD40 antibody to LPS + IgM during activation, negated the protective effect, thus supporting that NF-κB and CD40 downregulation are required to switch activated BMDC to a regulatory phenotype (4). It is possible that IgM by binding to CD40 induces this regulatory phenotype. Ex-vivo pretreatment of murine BMDC with human IgM was also protective in renal IRI (Figure 8B), indicating that the function of IgM-NAA is evolutionarily conserved among species.
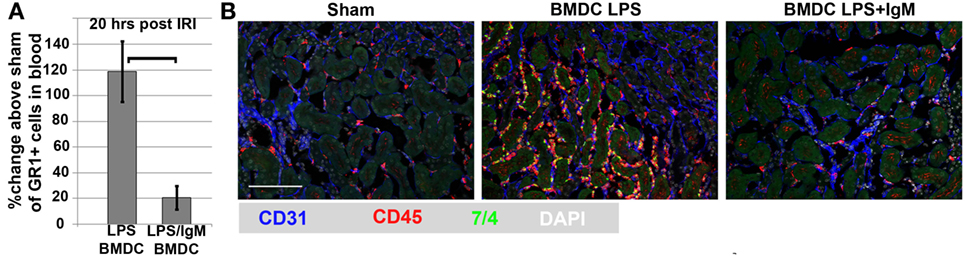
Figure 10. Murine IgM/LPS-pretreated BMDC inhibit the inflammatory response after renal IRI. There is significantly less inflammatory cell response 20 h post ischemia in mice administered IgM/LPS-pretreated BMDC as determined by circulating granulocytes (CD45+, GR1+) in blood (A) and infiltrating CD45+ and GR1+ leukocytes in kidneys (B). CD31 is a marker for endothelial cells. 7/4 is a marker for GR1+ granulocytes. Figure and legend reproduced with permission from Ref. (4). Copyright 2015. The American Association of Immunologists, Inc.
However, since LPS activation of BMDC generates apoptotic cells, it is possible that protection from ischemia could be mediated by complexes of IgM anti-PC and apoptotic cells, which when injected, regulate endogenous DC (127). Such a possibility seemed unlikely, as in the in vivo murine studies to demonstrate the anti-inflammatory role of apoptotic cells, large quantities of apoptotic cells (2.5 × 107 thymocytes) were used (127), while in our studies, we only used 0.5 × 106 BMDC (4). However, to exclude the possibility of apoptotic cell-mediated protection, we used apoptotic BMDC by subjecting activated LPS + IgM pretreated BMDC (0.5 × 106 cells) to UV irradiation. Such apoptotic LPS + IgM pretreated BMDC failed to protect mice from ischemia induced AKI, thus excluding the role of apoptotic cell/IgM complexes in inducing protection (Figure 8C). The latter experiments indicated that IgM-ALA mediated protection by switching LPS-activated BMDC to a regulatory phenotype. BMDC required IL-10 but not IDO (indoleamine 2, 3-dioxygenase) to switch to a regulatory phenotype as IgM + LPS pretreatment of IL-10 ko BMDC, but not IDO ko BMDC, failed to protect mice from developing AKI after renal ischemia (4).
We hypothesize that intravenously injected regulatory BMDC inhibit the innate inflammatory response by entering the splenic marginal zone where they inhibit NKT function. However, in further studies, we show that injected regulatory BMDC require the presence of other in vivo suppressive mechanisms, such as circulating IgM, IL-10, Tregs, and B cells, to mediate protection (4).
In summary, IgM-ALA inhibits the ischemia-induced innate inflammatory response by several mechanisms, including switching activated DC to a regulatory phenotype, inhibiting NKT cell IFN-γ production, and inhibiting chemotaxis of leukocytes by binding to chemokine receptors. However, IgM-ALA is in-effective on its own in inhibiting the ischemia-induced innate inflammatory response and requires the presence of other in vivo suppressive mechanisms, such as IL-10, Tregs and B cells. Conversely, these other in vivo suppressive mechanisms, such as Tregs and Bregs, cannot effectively protect against ischemia induced innate inflammation without IgM-NAA as evidenced in our renal ischemia experiments using IgM ko mice that lack secretory IgM (Figures 9A,B).
Polyclonal IgM Inhibits Inflammation Mediated by Adaptive Immune Mechanisms in Allograft Transplantation
Because of our clinical observations (Figure 3) and the in vitro studies demonstrating that IgM (a) inhibited alloantigen-activated T cell proliferation and differentiation into Th-1 and Th-17 independently of DC (Figure 6) and (b) could induce regulatory function in DC, we performed experiments aimed at determining whether IgM could also inhibit allograft rejection, which is an in vivo model of inflammation mediated by alloantigen-activated DC and T cells (138). Two approaches were used to test the role of polyclonal IgM. First, cardiac transplants were performed intra-abdominally in B6/S4-IgM ko mice (referred to as IgM ko) or their WT littermates using B6-bm12 donor hearts, which are minimally incompatible at the MHC class II locus (Ia) with the recipient. In this transplant model, there is a mild chronic form of cellular rejection and a vasculopathy that is initiated by a T cell-mediated inflammatory process and cardiac graft loss (defined as loss of intra-abdominal cardiac pulsation) occurs at >2 months in WT recipients. However, in IgM ko recipients, there is a more severe acute cellular rejection and graft loss occurs in 2–3 weeks, which is significantly earlier compared with their WT-B6/S4 counterparts, where cardiac graft loss occurs after >2 months (Figure 11). Histologically, there are considerably more TH-17 cells infiltrating the cardiac allograft in the IgM ko recipient despite no significant difference in infiltrating Tregs between the groups (138). The T cell findings on histology mirror the in vitro studies where IgM inhibited naïve T cells and Foxp3+ T cells from differentiating into TH-17 cells without affecting levels of Tregs (Figure 6D).
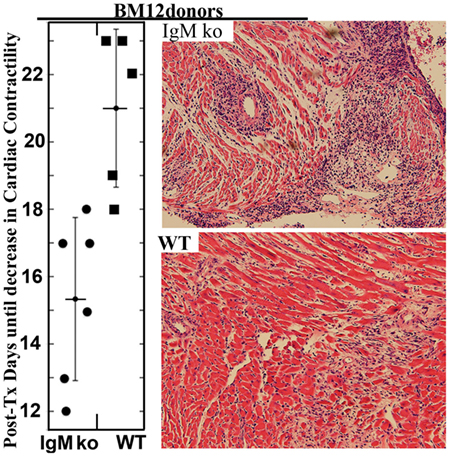
Figure 11. Allograft rejection is more rapid and severe in B6/S4-IgMko mice. B6/S4-IgMko mice and their WT counterparts (WT-B6/S4) received cardiac allografts from B6-bm-12 donors that are only incompatible at the MHC-Ia locus. Graph depicts the post-transplant day when cardiac contractility was found to be decreased by finger palpation. Histology depicts Day 10 post-transplant B6-bm12 cardiac allograft histology. Figure and legend reproduced with permission from Ref. (138). Copyright 2012. The American Association of Immunologists, Inc.
In the second approach, we wanted to determine whether increasing circulating levels of IgM in WT-B6 mice inhibited the severe and rapid rejection that occurs in the setting of fully MHC-incompatible donor hearts (i.e., from BALB/c donors). In this model, rejection in WT-B6 recipients is detectable by day 5 and graft loss occurs by 7–9 days (138). In these studies, 175 μg IgM was intravenously administered 24 h after ascertaining that cardiac surgery was successful, and the dose of IgM was repeated on days 3 and 5. Mice were euthanized on day 6. The data clearly show that IgM inhibited the severe inflammation in the cardiac allograft induced by rejection on day 6, as detected by H&E staining and immunofluorescence staining for neutrophils (7/4) and T cells (CD3). Importantly, with immunohistochemistry, this lack of leukocyte infiltration in the cardiac parenchyma of IgM-treated recipients was also associated with no or minimal CXCL1+ leukocytes and with no or minimal fragmentation of capillaries, as identified by the endothelial cell marker CD31 (138).
In summary, we show that physiological doses (175 μg) of polyclonal IgM can subdue inflammatory responses mediated by an adaptive immune mechanism. Potential mechanisms include the following (a) a direct inhibitory effect of IgM-ALA on T effector cells, but not Tregs, possibly by binding and down-modulating CD3/TcR and certain specific co-stimulatory receptors, such as CD4 and CD2 but not CD8. As a result, T effector cells are inhibited from proliferating or producing certain specific cytokines (e.g., TNF, IFN-γ, IL-17 but not IL-6, and chemokines) or from differentiating into TH-1 and TH-17 pro-inflammatory cells. Importantly IgM-ALA does not affect levels of Foxp3+ Tregs, but prevents Foxp3 + cells from differentiating into TH-17 cells under pro-inflammatory conditions, (b) by binding to CD40 and switching activated DC to a regulatory phenotype with downregulation of CD40 and p65NF-κB, and (c) by inhibiting chemotaxis. It is highly unlikely that IgM anti-PC could have a significant role in inhibiting allograft rejection in our studies as we used small doses of polyclonal IgM (175 μg) while 1.5–2.0 mg of a monoclonal IgM anti-PC (T15 idiotype) was used to inhibit an arthritis model of inflammation mediated by adaptive immune mechanisms (127).
Polyclonal IgM Antibodies Inhibit Autoimmune-Mediated Insulitis in NOD Mice
Insulitis in the NOD mouse is primarily mediated by autoimmune T cells but there are data to indicate that B cells are also involved. Depleting B cells or only B1 cells ameliorates insulitis indicating that some of the IgG autoantibodies detected in NOD mice may also be pathogenic (179–181). The role of B1 cells in generating pathogenic IgG autoantibodies has also been described in other autoimmune murine models (20, 94). Because our in vitro studies demonstrated that polyclonal IgM inhibited T cell proliferation and differentiation into Th-1 and Th-17 cells (Figures 6B–D) and can also counter pathogenic IgG autoantibodies (as previously discussed), we performed studies to determine whether IgM could inhibit autoimmune insulitis that results in islet cell destruction and diabetes mellitus (DM) in NOD mice (182). In these mice, the autoimmune inflammatory process begins spontaneously around 4–5 weeks after birth and the initial phase is characterized by a silent and non-destructive leukocyte infiltration of the perivascular and periductal regions in the pancreas as well as the peripheral islet regions by a heterogeneous mixture of CD4 and CD8 T cells, B cells, macrophages, and DC (peri-insulitis). In the invasive phase that begins at 8–12 weeks of age, the immune infiltrate enters the islet inducing beta cell destruction (insulitis). Significant destruction first becomes evident around 12–13 weeks of age with mice exhibiting overt diabetes (DM).
We wanted to determine the effect of increasing IgM levels on development of DM (182). NOD mice were administered bi-weekly intra-peritoneal polyclonal IgM (50 μg/dose) beginning either at 5 or 11 weeks of age and ending when mice were 18 weeks old. At 25 weeks of age, 80% of control mice (n = 30) became diabetic, while 0% of mice (n = 30) treated with IgM beginning at 5 weeks developed DM. Importantly, only 20% of pre-diabetic mice (n = 20) treated with IgM beginning at 11 weeks of age developed DM at 25 weeks of age. This latter observation is particularly notable as prior studies using co-stimulatory blockade failed to prevent DM in this murine model. At 18–25 weeks of age the pancreas revealed no or minimal insulitis in NOD mice treated with IgM beginning at 5 weeks of age. Other investigators using monoclonal polyreactive natural IgM in the neonatal period have also obtained similar results (183, 184).
In summary, these studies indicate that polyclonal IgM inhibits insulitis via several potential mechanisms, including inhibition of autoimmune T effectors and possibly countering IgG autoantibodies via anti-idiotypic mechanisms and by inhibiting the B cells that produce them. Additionally, IgM by switching activated DC to a regulatory phenotype and maintaining Tregs could enhance this protective effect.
Conclusion
Figures 12 and 13 summarize our concepts of the inter-relationship between pathogens and natural antibodies. In both murine and human models, the evidence shows that these polyreactive and low-affinity binding IgM-NAA function under physiological conditions to (i) provide a first line of defense against invading organisms, (ii) protect the host from autoimmune inflammation mediated by autoimmune B and T cells that have escaped tolerance mechanisms, (iii) protect the host from endogenous oxidized neo-determinants and other neo-antigens that are unmasked during tissue damage, and (iv) regulate excess inflammation, mediated by both innate and adaptive immune mechanisms. Even though the full repertoire of IgM-NAA develop during the first few years of life, both their levels and repertoire differ in healthy individuals as well as in disease and could contribute to the varying inflammatory response, for example, after an infection or alloantigen exposure (see Figures 2 and 5). We hypothesize that high protective levels of IgM-NAA are maintained by infections that have been shown to increase IgM-NAA, especially IgM-ALA and anti-PC [reviewed in Ref. (31, 39, 148)]. Such a hypothesis could explain the significantly low incidence of autoimmune disorders, such as SLE or sarcoidosis in rural parts of Africa where malaria and other infections are endemic (185–187). We have shown that IgM-ALA increases in active sarcoidosis (188). There are other suppressive mechanisms (e.g., Tregs, B regs, IL-10, TGF-β) that regulate inflammation and based on the different animal models of inflammation, it would appear that IgM-NAA have a more prominent role in regulating inflammation that involves pathogenic IgG autoantibodies, macrophages, and NKT cells. However, we show that IgM-NAA require Tregs, B cells, and IL-10 to be fully effective in controlling inflammation (4). Conversely, our studies and that of others, using mice deficient in IgM secretion, would also indicate that Tregs and Bregs also require IgM-NAA to effectively control inflammation (39, 119, 120). Finally, it may be easier to develop a vaccine to increase IgM-NAA especially if we understand how diverse infectious agents increase IgM-ALA. Studies are also needed to determine if prolonged high IgM-NAA levels can induce excess immunosuppression. One could also use enriched IgM intravenous preparations to acutely treat patients with uncontrolled inflammation. Cell therapy, especially with IgM pretreated DC, could provide an alternative approach requiring minimal quantities of IgM to prevent ischemic acute renal failure (e.g., in high-risk patients undergoing cardiac surgery) or delayed graft function after renal transplantation (4).
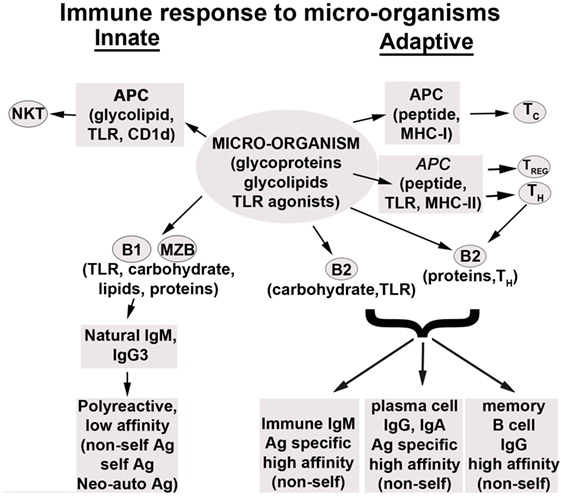
Figure 12. Immune response to micro-organisms.
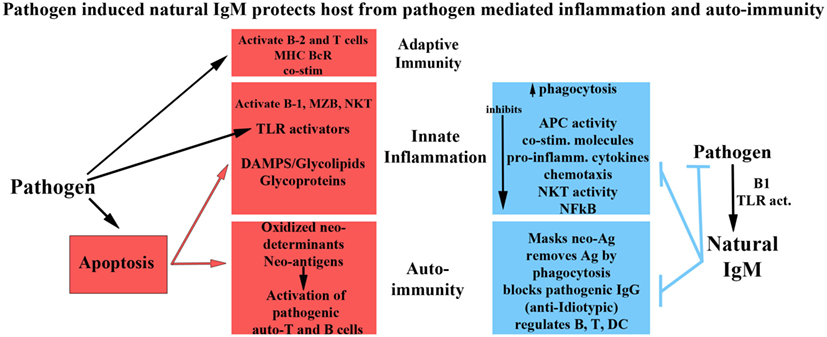
Figure 13. Pathogen-induced natural IgM protects host from pathogen-mediated inflammation and autoimmunity.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dooley H, Flajnik MF. Antibody repertoire development in cartilaginous fish. Dev Comp Immunol (2006) 30(1–2):43–56. doi:10.1016/j.dci.2005.06.022
2. Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, Herzenberg LA. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc Natl Acad Sci U S A (1999) 96(5):2250–5. doi:10.1073/pnas.96.5.2250
3. Thurnheer MC, Zuercher AW, Cebra JJ, Bos NA. B1 cells contribute to serum IgM, but not to intestinal IgA, production in gnotobiotic Ig allotype chimeric mice. J Immunol (2003) 170(9):4564–71. doi:10.4049/jimmunol.170.9.4564
4. Lobo PI, Schlegel KH, Bajwa A, Huang L, Kurmaeva E, Wang B, et al. Natural IgM switches the function of lipopolysaccharide-activated murine bone marrow-derived dendritic cells to a regulatory dendritic cell that suppresses innate inflammation. J Immunol (2015) 195(11):5215–26. doi:10.4049/jimmunol.1500052
5. Robey IF, Schluter SF, Akporiaye E, Yocum DE, Marchalonis JJ. Human monoclonal natural autoantibodies against the T-cell receptor inhibit interleukin-2 production in murine T cells. Immunology (2002) 105(4):419–29. doi:10.1046/j.1365-2567.2002.01389.x
6. Zhang M, Alicot EM, Carroll MC. Human natural IgM can induce ischemia/reperfusion injury in a murine intestinal model. Mol Immunol (2008) 45(15):4036–9. doi:10.1016/j.molimm.2008.06.013
7. Steele E, Cunningham A. High proportion of Ig-producing cells making autoantibody in normal mice. Nature (1978) 274:483–4. doi:10.1038/274483a0
8. Dighiero G, Guilbert B, Fermand J, Lymberi P, Danon F, Avrameas S. Thirty-six human monoclonal immunoglobulins with antibody activity. Blood (1983) 62(2):264–70.
9. Hardy RR, Hayakawa K. Development and physiology of Ly-1 B and its human homolog, Leu-1 B. Immunol Rev (1986) 93(1):53–80. doi:10.1111/j.1600-065X.1986.tb01502.x
10. Kantor AB, Herzenberg LA. Origin of murine B cell lineages. Annu Rev Immunol (1993) 11(1):501–38. doi:10.1146/annurev.iy.11.040193.002441
11. Nakamura M, Burastero SE, Ueki Y, Larrick JW, Notkins AL, Casali P. Probing the normal and autoimmune B cell repertoire with Epstein-Barr virus. Frequency of B cells producing monoreactive high affinity autoantibodies in patients with Hashimoto’s disease and systemic lupus erythematosus. J Immunol (1988) 141(12):4165–72.
12. Mouthon L, Lacroix-Desmazes S, Nobrega A, Barreau C, Coutinho A, Kazatchkine M. The self-reactive antibody repertoire of normal human serum IgM is acquired in early childhood and remains conserved throughout life. Scand J Immunol (1996) 44(3):243–51. doi:10.1046/j.1365-3083.1996.d01-306.x
13. Clarke SH, Arnold LW. B-1 cell development: evidence for an uncommitted immunoglobulin (Ig)M+ B cell precursor in B-1 cell differentiation. J Exp Med (1998) 187(8):1325–34. doi:10.1084/jem.187.8.1325
14. Casali P, Burastero SE, Nakamura M, Inghirami G, Notkins AL. Human lymphocytes making rheumatoid factor and antibody to ssDNA belong to Leu-1+ B-cell subset. Science (1987) 236(4797):77–81. doi:10.1126/science.3105056
15. Hardy RR, Hayakawa K, Shimizu M, Yamasaki K, Kishimoto T. Rheumatoid factor secretion from human Leu-1+ B cells. Science (1987) 236(4797):81–3. doi:10.1126/science.3105057
16. Kasaian MT, Casali P. Autoimmunity-prone Bl (CD5 B) cells, natural antibodies and self recognition. Autoimmunity (1993) 15(4):315–29. doi:10.3109/08916939309115755
17. Rieben R, Roos A, Muizert Y, Tinguely C, Gerritsen AF, Daha MR. Immunoglobulin M-enriched human intravenous immunoglobulin prevents complement activation in vitro and in vivo in a rat model of acute inflammation. Blood (1999) 93(3):942–51.
18. Sidman CL, Shultz LD, Hardy RR, Hayakawa K, Herzenberg LA. Production of immunoglobulin isotypes by ly-1+ B cells in viable motheaten and normal mice. Science (1986) 232(4756):1423–5. doi:10.1126/science.3487115
19. Solvason N, Lehuen A, Kearney JF. An embryonic source of Ly1 but not conventional B cells. Int Immunol (1991) 3(6):543–50. doi:10.1093/intimm/3.6.543
20. Murakami M, Tsubata T, Shinkura R, Nisitani S, Okamoto M, Yoshioka H, et al. Oral administration of lipopolysaccharides activates B-1 cells in the peritoneal cavity and lamina propria of the gut and induces autoimmune symptoms in an autoantibody transgenic mouse. J Exp Med (1994) 180(1):111–21. doi:10.1084/jem.180.1.111
21. Nisitani S, Tsubata T, Murakami M, Honjo T. Administration of interleukin -5 or -10 activates peritoneal B-1 cells and induces autoimmune hemolytic anemia in anti-erythrocyte autoantibody-transgenic mice. Eur J Immunol (1995) 25(11):3047–52. doi:10.1002/eji.1830251110
22. Yang Y, Tung JW, Ghosn EEB, Herzenberg LA, Herzenberg LA. Division and differentiation of natural antibody-producing cells in mouse spleen. Proc Natl Acad Sci U S A (2007) 104(11):4542–6. doi:10.1073/pnas.0700001104
23. Ha S, Tsuji M, Suzuki K, Meek B, Yasuda N, Kaisho T, et al. Regulation of B1 cell migration by signals through Toll-like receptors. J Exp Med (2006) 203(11):2541–50. doi:10.1084/jem.20061041
24. Hayakawa K, Asano M, Shinton SA, Gui M, Allman D, Stewart CL, et al. Positive selection of natural autoreactive B cells. Science (1999) 285(5424):113–6. doi:10.1126/science.285.5424.113
25. Martin F, Kearney JF. Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity (2000) 12(1):39–49. doi:10.1016/S1074-7613(00)80157-0
26. Cancro MP, Kearney JF. B cell positive selection: road map to the primary repertoire? J Immunol (2004) 173(1):15–9. doi:10.4049/jimmunol.173.1.15
27. Tian Q, Beardall M, Xu Y, Li J, Parker DC, Casanova N, et al. B cells expressing a natural polyreactive autoantibody have a distinct phenotype and are overrepresented in immunoglobulin heavy chain transgenic mice. J Immunol (2006) 177(4):2412–22. doi:10.4049/jimmunol.177.4.2412
28. Love SD, Lee W, Nakamura YC, Platt JL, Bollinger RR, Parker W. Natural anti-carbohydrate IgM in mice: dependence on age and strain. J Immunol Methods (2000) 246(1):61–8. doi:10.1016/S0022-1759(00)00296-9
29. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-. J Exp Med (2011) 208(1):67–80. doi:10.1084/jem.20101499
30. Simell B, Lahdenkari M, Reunanen A, Kayhty H, Vakevainen M. Effects of ageing and gender on naturally acquired antibodies to pneumococcal capsular polysaccharides and virulence-associated proteins. Clin Vaccine Immunol (2008) 15(9):1391–7. doi:10.1128/CVI.00110-08
31. Adib M, Ragimbeau J, Avrameas S, Ternynck T. IgG autoantibody activity in normal mouse serum is controlled by IgM. J Immunol (1990) 145(11):3807–13.
32. Nicoletti C, Yang X, Cerny J. Repertoire diversity of antibody response to bacterial antigens in aged mice. III. Phosphorylcholine antibody from young and aged mice differ in structure and protective activity against infection with Streptococcus pneumoniae. J Immunol (1993) 150(2):543–9.
33. Lacroix-Desmazes S, Mouthon L, Kaveri SV, Kazatchkine MD, Weksler ME. Stability of natural self-reactive antibody repertoires during aging. J Clin Immunol (1999) 19(1):26–34. doi:10.1023/A:1020510401233
34. Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, Nagele RG. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One (2013) 8(4):e60726. doi:10.1371/journal.pone.0060726
35. Lacroix-Desmazes S, Mouthon L, Coutinho A, Kazatchkine MD. Analysis of the natural human IgG antibody repertoire: life-long stability of reactivities towards self antigens contrasts with age-dependent diversification of reactivities against bacterial antigens. Eur J Immunol (1995) 25(9):2598–604. doi:10.1002/eji.1830250929
36. Bachi AL, Suguri VM, Ramos LR, Mariano M, Vaisberg M, Lopes JD. Increased production of autoantibodies and specific antibodies in response to influenza virus vaccination in physically active older individuals. Results Immunol (2013) 3:10–6. doi:10.1016/j.rinim.2013.01.001
37. Covens K, Verbinnen B, Geukens N, Meyts I, Schuit F, Van Lommel L, et al. Characterization of proposed human B-1 cells reveals pre-plasmablast phenotype. Blood (2013) 121(26):5176–83. doi:10.1182/blood-2012-12-471953
38. Tangye SG. To B1 or not to B1: that really is still the question! Blood (2013) 121(26):5109–10. doi:10.1182/blood-2013-05-500074
39. Lobo PI, Schlegel KH, Spencer CE, Okusa MD, Chisholm C, McHedlishvili N, et al. Naturally occurring IgM anti-leukocyte autoantibodies (IgM-ALA) inhibit T cell activation and chemotaxis. J Immunol (2008) 180(3):1780–91. doi:10.4049/jimmunol.180.3.1769
40. Chen ZJ, Wheeler CJ, Shi W, Wu AJ, Yarboro CH, Gallagher M, et al. Polyreactive antigen-binding B cells are the predominant cell type in the newborn B cell repertoire. Eur J Immunol (1998) 28(3):989–94. doi:10.1002/(SICI)1521-4141(199803)28:03<989::AID-IMMU989>3.0.CO;2-1
41. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest (2009) 119(5):1335–49. doi:10.1172/JCI36800
42. Kroese FG, Butcher EC, Stall AM, Herzenberg LA. A major peritoneal reservoir of precursors for intestinal IgA plasma cells. Immunol Invest (1989) 18(1–4):47–58. doi:10.3109/08820138909112226
43. Murakami M, Honjo T. Involvement of B-1 cells in mucosal immunity and autoimmunity. Immunol Today (1995) 16(11):534–9. doi:10.1016/0167-5699(95)80047-6
44. Fagarasan S, Kinoshita K, Muramatsu M, Ikuta K, Honjo T. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature (2001) 413(6856):639–43. doi:10.1038/35098100
45. Bowman EP, Kuklin NA, Youngman KR, Lazarus NH, Kunkel EJ, Pan J, et al. The intestinal chemokine thymus-expressed chemokine (CCL25) attracts IgA antibody-secreting cells. J Exp Med (2002) 195(2):269–75. doi:10.1084/jem.20010670
46. Golby SJ, Spencer J. Where do IgA plasma cells in the gut come from? Gut (2002) 51(2):150–1. doi:10.1136/gut.51.2.150
47. Herzenberg LA. B-1 cells: the lineage question revisited. Immunol Rev (2000) 175(1):9–22. doi:10.1111/j.1600-065X.2000.imr017520.x
48. Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Annu Rev Immunol (2002) 20(1):253–300. doi:10.1146/annurev.immunol.20.100301.064833
49. Rothstein TL. Cutting edge commentary: two B-1 or not to be one. J Immunol (2002) 168(9):4257–61. doi:10.4049/jimmunol.168.9.4257
50. Avrameas S. Natural autoantibodies: from ‘horror autotoxicus’ to ‘gnothi seauton’. Immunol Today (1991) 12(5):154–9. doi:10.1016/S0167-5699(05)80045-3
51. Kretschmer K, Jungebloud A, Stopkowicz J, Kleinke T, Hoffmann R, Weiss S. The selection of marginal zone B cells differs from that of B-1a cells. J Immunol (2003) 171(12):6495–501. doi:10.4049/jimmunol.171.12.6495
52. Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol (2002) 2(5):323–35. doi:10.1038/nri799
53. Zhou Z, Tzioufas AG, Notkins AL. Properties and function of polyreactive antibodies and polyreactive antigen-binding B cells. J Autoimmun (2007) 29(4):219–28. doi:10.1016/j.jaut.2007.07.015
54. Vittecoq O, Brard F, Jovelin F, Le Loet X, Tron F, Gilbert D. IgM anti-myeloperoxidase antibody-secreting lymphocytes are present in the peripheral repertoire of lupus mice but rarely differentiate into IgG-producing cells. Clin Exp Immunol (1999) 118(1):122–30. doi:10.1046/j.1365-2249.1999.00995.x
55. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol (2011) 11(1):34–46. doi:10.1038/nri2901
56. Gronwall C, Vas J, Silverman GJ. Protective roles of natural IgM antibodies. Front Immunol (2012) 3:66. doi:10.3389/fimmu.2012.00066
57. Baxendale HE, Johnson M, Stephens RC, Yuste J, Klein N, Brown JS, et al. Natural human antibodies to pneumococcus have distinctive molecular characteristics and protect against pneumococcal disease. Clin Exp Immunol (2008) 151(1):51–60. doi:10.1111/j.1365-2249.2007.03535.x
58. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science (2012) 335(6068):597–601. doi:10.1126/science.1215173
59. Holodick NE, Tumang JR, Rothstein TL. Immunoglobulin secretion by B1 cells: differential intensity and IRF4-dependence of spontaneous IgM secretion by peritoneal and splenic B1 cells. Eur J Immunol (2010) 40(11):3007–16. doi:10.1002/eji.201040545
60. Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol (2006) 7(3):293–301. doi:10.1038/ni1301
61. Matejuk A, Beardall M, Xu Y, Tian Q, Phillips D, Alabyev B, et al. Exclusion of natural autoantibody-producing B cells from IgG memory B cell compartment during T cell-dependent immune responses. J Immunol (2009) 182(12):7634–43. doi:10.4049/jimmunol.0801562
62. Ishida D, Su L, Tamura A, Katayama Y, Kawai Y, Wang SF, et al. Rap1 signal controls B cell receptor repertoire and generation of self-reactive B1a cells. Immunity (2006) 24(4):417–27. doi:10.1016/j.immuni.2006.02.007
63. Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol (1998) 160(10):4776–87.
64. Lino AC, Mohr E, Demengeot J. Naturally secreted immunoglobulins limit B1 and MZ B-cell numbers through a microbiota-independent mechanism. Blood (2013) 122(2):209–18. doi:10.1182/blood-2012-08-447136
65. Baker N, Ehrenstein MR. Cutting edge: selection of B lymphocyte subsets is regulated by natural IgM. J Immunol (2002) 169(12):6686–90. doi:10.4049/jimmunol.169.12.6686
66. Choi SC, Wang H, Tian L, Murakami Y, Shin DM, Borrego F, et al. Mouse IgM Fc receptor, FCMR, promotes B cell development and modulates antigen-driven immune responses. J Immunol (2013) 190(3):987–96. doi:10.4049/jimmunol.1202227
67. Amezcua Vesely MC, Schwartz M, Bermejo DA, Montes CL, Cautivo KM, Kalergis AM, et al. FcγRIIb and BAFF differentially regulate peritoneal B1 cell survival. J Immunol (2012) 188(10):4792–800. doi:10.4049/jimmunol.1102070
68. Berneman A, Ternynck T, Avrameas S. Natural mouse IgG reacts with self antigens including molecules involved in the immune response. Eur J Immunol (1992) 22(3):625–33. doi:10.1002/eji.1830220303
69. Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A (2010) 107(26):11993–8. doi:10.1073/pnas.1001948107
70. Hurez V, Kaveri S, Kazatchkine MD. Expression and control of the natural autoreactive IgG repertoire in normal human serum. Eur J Immunol (1993) 23(4):783–9. doi:10.1002/eji.1830230402
71. Pereira P, Forni L, Larsson E, Cooper M, Heusser C, Coutinho A. Autonomous activation of B and T cells in antigen-free mice. Eur J Immunol (1986) 16(6):685–8. doi:10.1002/eji.1830160616
72. Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol (1995) 7(6):812–8. doi:10.1016/0952-7915(95)80053-0
73. Bos NA, Kimura H, Meeuwsen CG, Visser HD, Hazenberg MP, Wostmann BS, et al. Serum immunoglobulin levels and naturally occurring antibodies against carbohydrate antigens in germ-free BALB/c mice fed chemically defined ultrafiltered diet. Eur J Immunol (1989) 19(12):2335–9. doi:10.1002/eji.1830191223
74. Hamanova M, Chmelikova M, Nentwich I, Thon V, Lokaj J. Anti-gal IgM, IgA and IgG natural antibodies in childhood. Immunol Lett (2015) 164(1):40–3. doi:10.1016/j.imlet.2015.02.001
75. Cohen IR, Young DB. Autoimmunity, microbial immunity and the immunological homunculus. Immunol Today (1991) 12(4):105–10. doi:10.1016/0167-5699(91)90093-9
76. Watts RA, Isenberg DA. Autoantibodies and antibacterial antibodies: from both sides now. Ann Rheum Dis (1990) 49(12):961–5. doi:10.1136/ard.49.12.961
77. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science (2003) 301(5638):1374–7. doi:10.1126/science.1086907
78. Xu PC, Cui Z, Chen M, Hellmark T, Zhao MH. Comparison of characteristics of natural autoantibodies against myeloperoxidase and anti-myeloperoxidase autoantibodies from patients with microscopic polyangiitis. Rheumatology (Oxford) (2011) 50(7):1236–43. doi:10.1093/rheumatology/ker085
79. Reap EA, Sobel ES, Jennette JC, Cohen PL, Eisenberg RA. Conventional B cells, not B1 cells, are the source of autoantibodies in chronic graft-versus-host disease. J Immunol (1993) 151(12):7316–23.
80. Wofsy D, Ledbetter JA, Hendler PL, Seaman WE. Treatment of murine lupus with monoclonal anti-T cell antibody. J Immunol (1985) 134(2):852–7.
81. Jevnikar AM, Grusby MJ, Glimcher LH. Prevention of nephritis in major histocompatibility complex class II-deficient MRL-lpr mice. J Exp Med (1994) 179(4):1137–43. doi:10.1084/jem.179.4.1137
82. Steinberg AD, Roths JB, Murphy ED, Steinberg RT, Raveche ES. Effects of thymectomy or androgen administration upon the autoimmune disease of MRL/Mp-lpr/lpr mice. J Immunol (1980) 125(2):871–3.
83. Kaveri S, Dietrich G, Hurez V, Kazatchkine M. Intravenous immunoglobulins (IVIg) in the treatment of autoimmune diseases. Clin Exp Immunol (1991) 86(2):192–8. doi:10.1111/j.1365-2249.1991.tb05794.x
84. Panda S, Zhang J, Tan NS, Ho B, Ding JL. Natural IgG antibodies provide innate protection against ficolin-opsonized bacteria. EMBO J (2013) 32(22):2905–19. doi:10.1038/emboj.2013.199
85. Abdou NI, Wall H, Lindsley HB, Halsey JF, Suzuki T. Network theory in autoimmunity. in vitro suppression of serum anti-DNA antibody binding to DNA by anti-idiotypic antibody in systemic lupus erythematosus. J Clin Invest (1981) 67(5):1297–304. doi:10.1172/JCI110158
86. Dwyer DS, Bradley RJ, Urquhart CK, Kearney JF. Naturally occurring anti-idiotypic antibodies in myasthenia gravis patients. Nature (1983) 301(5901):611–4.
87. Cui Z, Zhao M, Segelmark M, Hellmark T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int (2010) 78(6):590–7. doi:10.1038/ki.2010.198
88. Ohnishi K, Ebling FM, Mitchell B, Singh RR, Hahn BH, Tsao BP. Comparison of pathogenic and non-pathogenic murine antibodies to DNA: antigen binding and structural characteristics. Int Immunol (1994) 6(6):817–30. doi:10.1093/intimm/6.6.817
89. Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int (1995) 48(3):705–11. doi:10.1038/ki.1995.341
90. Tsao BP, Ohnishi K, Cheroutre H, Mitchell B, Teitell M, Mixter P, et al. Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J Immunol (1992) 149(1):350–8.
91. Radic MZ, Ibrahim SM, Rauch J, Camper SA, Weigert M. Constitutive secretion of transgene-encoded IgG2b autoantibodies leads to symptoms of autoimmune disease. J Immunol (1995) 155(6):3213–22.
92. Murakami M, Tsubata T, Okamoto M, Shimizu A, Kumagai S, Imura H, et al. Antigen-induced apoptotic death of Ly-1 B cells responsible for autoimmune disease in transgenic mice. Nature (1992) 357:77–80. doi:10.1038/357077a0
93. Takahashi T, Strober S. Natural killer T cells and innate immune B cells from lupus-prone NZB/W mice interact to generate IgM and IgG autoantibodies. Eur J Immunol (2008) 38(1):156–65. doi:10.1002/eji.200737656
94. Murakami M, Yoshioka H, Shirai T, Tsubata T, Honjo T. Prevention of autoimmune symptoms in autoimmune-prone mice by elimination of B-1 cells. Int Immunol (1995) 7(5):877–82. doi:10.1093/intimm/7.5.877
95. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med (2005) 201(5):703–11. doi:10.1084/jem.20042251
96. Samuels J, Ng Y, Paget D, Meffre E. Impaired early B-cell tolerance in patients with rheumatoid arthritis. Arthritis Res Ther (2005) 7(Suppl 1):78. doi:10.1186/ar1599
97. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity (2004) 20(4):441–53. doi:10.1016/S1074-7613(04)00079-2
98. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren’s syndrome. J Clin Invest (2002) 109(1):59–68. doi:10.1172/JCI0214121
99. Guilbert B, Mahana W, Gilbert M, Mazie JC, Avrameas S. Presence of natural autoantibodies in hyperimmunized mice. Immunology (1985) 56(3):401–8.
100. Naparstek Y, Andre-Schwartz J, Manser T, Wysocki LJ, Breitman L, Stollar BD, et al. A single germline VH gene segment of normal A/J mice encodes autoantibodies characteristic of systemic lupus erythematosus. J Exp Med (1986) 164(2):614–26. doi:10.1084/jem.164.2.614
101. Cunningham MW, Hall NK, Krisher KK, Spanier AM. A study of anti-group A streptococcal monoclonal antibodies cross-reactive with myosin. J Immunol (1986) 136(1):293–8.
102. Lewis M, Botto M. Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity (2006) 39(5):367–78. doi:10.1080/08916930600739233
103. Li F, Smith P, Ravetch JV. Inhibitory Fcγ receptor is required for the maintenance of tolerance through distinct mechanisms. J Immunol (2014) 192(7):3021–8. doi:10.4049/jimmunol.1302934
104. Baerenwaldt A, Lux A, Danzer H, Spriewald BM, Ullrich E, Heidkamp G, et al. Fcγ receptor IIB (FcγRIIB) maintains humoral tolerance in the human immune system in vivo. Proc Natl Acad Sci U S A (2011) 108(46):18772–7. doi:10.1073/pnas.1111810108
105. Tillman DM, Jou NT, Hill RJ, Marion TN. Both IgM and IgG anti-DNA antibodies are the products of clonally selective B cell stimulation in (NZB x NZW)F1 mice. J Exp Med (1992) 176(3):761–79. doi:10.1084/jem.176.3.761
106. Ikematsu H, Kasaian MT, Schettino EW, Casali P. Structural analysis of the VH-D-JH segments of human polyreactive IgG mAb. Evidence for somatic selection. J Immunol (1993) 151(7):3604–16.
107. Werwitzke S, Trick D, Kamino K, Matthias T, Kniesch K, Schlegelberger B, et al. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB x NZW)F1 mouse. Arthritis Rheum (2005) 52(11):3629–38. doi:10.1002/art.21379
108. Heidenreich F, Jovin T. Synthesis of anti-acetylcholine receptor antibodies by CD5-B cells from peripheral blood of myasthenia gravis patients. J Neurol (1996) 243(1):57–62. doi:10.1007/BF00878532
109. Lubetzki Korn I, Abramsky O. Myasthenia gravis following viral infection. Eur Neurol (1981) 20(6):435–9. doi:10.1159/000115275
110. Hohlfeld R, Kalies I, Kohleisen B, Heininger K, Conti-Tronconi B, Toyka KV. Myasthenia gravis: stimulation of antireceptor autoantibodies by autoreactive T cell lines. Neurology (1986) 36(5):618–21. doi:10.1212/WNL.36.5.618
111. Wang ZY, Okita DK, Howard J Jr, Conti-Fine BM. T-cell recognition of muscle acetylcholine receptor subunits in generalized and ocular myasthenia gravis. Neurology (1998) 50(4):1045–54. doi:10.1212/WNL.50.4.1045
112. Jennette JC, Falk RJ. The rise and fall of horror autotoxicus and forbidden clones. Kidney Int (2010) 78(6):533–5. doi:10.1038/ki.2010.237
113. Jennette JC, Hipp CG. Clq nephropathy: a distinct pathologic entity usually causing nephrotic syndrome. Am J Kidney Dis (1985) 6(2):103–10. doi:10.1016/S0272-6386(85)80150-5
114. Sharman A, Furness P, Feehally J. Distinguishing C1q nephropathy from lupus nephritis. Nephrol Dial Transplant (2004) 19(6):1420–6. doi:10.1093/ndt/gfh139
115. Jacob N, Stohl W. Autoantibody-dependent and autoantibody-independent roles for B cells in systemic lupus erythematosus: past, present, and future. Autoimmunity (2010) 43(1):84–97. doi:10.3109/08916930903374600
116. Waters ST, McDuffie M, Bagavant H, Deshmukh US, Gaskin F, Jiang C, et al. Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. J Exp Med (2004) 199(2):255–64. doi:10.1084/jem.20031519
117. Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol (2007) 178(11):7422–31. doi:10.4049/jimmunol.178.11.7422
118. Ma J, Xu J, Madaio MP, Peng Q, Zhang J, Grewal IS, et al. Autoimmune lpr/lpr mice deficient in CD40 ligand: spontaneous ig class switching with dichotomy of autoantibody responses. J Immunol (1996) 157(1):417–26.
119. Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J Exp Med (2000) 191(7):1253–8. doi:10.1084/jem.191.7.1253
120. Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci U S A (2000) 97(3):1184–9. doi:10.1073/pnas.97.3.1184
121. Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity (2000) 13(2):277–85. doi:10.1016/S1074-7613(00)00027-3
122. Côrte-Real J, Rodo J, Almeida P, Garcia J, Coutinho A, Demengeot J, et al. Irf4 is a positional and functional candidate gene for the control of serum IgM levels in the mouse. Genes Immun (2009) 10(1):93–9. doi:10.1038/gene.2008.73
123. Klein-Schneegans A, Kuntz L, Fonteneau P, Loor F. Serum concentrations of IgM, IgG1, IgG2b, IgG3 and IgA in C57BL6 mice and their congenics at the lpr (lymphoproliferation) locus. J Autoimmun (1989) 2(6):869–75. doi:10.1016/0896-8411(89)90013-9
124. Kalpaktsoglou PK, Hong R, Good RA. The five classes of immunoglobulins in normal C3H and BALB/c mice. Immunology (1973) 24(2):303–14.
125. Witte T. IgM antibodies against dsDNA in SLE. Clin Rev Allergy Immunol (2008) 34(3):345–7. doi:10.1007/s12016-007-8046-x
126. Mannoor K, Matejuk A, Xu Y, Beardall M, Chen C. Expression of natural autoantibodies in MRL-lpr mice protects from lupus nephritis and improves survival. J Immunol (2012) 188(8):3628–38. doi:10.4049/jimmunol.1102859
127. Chen Y, Khanna S, Goodyear CS, Park YB, Raz E, Thiel S, et al. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J Immunol (2009) 183(2):1346–59. doi:10.4049/jimmunol.0900948
128. Lobo PI, Brayman KL, Okusa MD. Natural IgM anti-leucocyte autoantibodies (IgM-ALA) regulate inflammation induced by innate and adaptive immune mechanisms. J Clin Immunol (2014) 34(Suppl 1):S22–9. doi:10.1007/s10875-014-0027-2
129. Hurez V, Kazatchkine MD, Vassilev T, Ramanathan S, Pashov A, Basuyaux B, et al. Pooled normal human polyspecific IgM contains neutralizing anti-idiotypes to IgG autoantibodies of autoimmune patients and protects from experimental autoimmune disease. Blood (1997) 90(10):4004–13.
130. Chen Y, Park YB, Patel E, Silverman GJ. IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J Immunol (2009) 182(10):6031–43. doi:10.4049/jimmunol.0804191
131. Lobo PI. Nature of autolymphocytotoxins present in renal hemodialysis patients: their possible role in controlling alloantibody formation. Transplantation (1981) 32(3):233–7. doi:10.1097/00007890-198109000-00010
132. Honjo K, Kubagawa Y, Jones DM, Dizon B, Zhu Z, Ohno H, et al. Altered Ig levels and antibody responses in mice deficient for the Fc receptor for IgM (FcmuR). Proc Natl Acad Sci U S A (2012) 109(39):15882–7. doi:10.1073/pnas.1206567109
133. Hutchings PR, Varey AM, Cooke A. Immunological defects in SJL mice. Immunology (1986) 59(3):445–50.
134. Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest (2005) 115(12):3428–39. doi:10.1172/JCI23587
135. Jost SA, Tseng LC, Matthews LA, Vasquez R, Zhang S, Yancey KB, et al. IgG, IgM, and IgA antinuclear antibodies in discoid and systemic lupus erythematosus patients. ScientificWorldJournal (2014) 2014:171028. doi:10.1155/2014/171028
136. Vassilev T, Yamamoto M, Aissaoui A, Bonnin E, Berrih-Aknin S, Kazatchkine MD, et al. Normal human immunoglobulin suppresses experimental myasthenia gravis in SCID mice. Eur J Immunol (1999) 29(8):2436–42. doi:10.1002/(SICI)1521-4141(199908)29:08<2436::AID-IMMU2436>3.0.CO;2-9
137. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med (2003) 349(16):1526–33. doi:10.1056/NEJMoa021933
138. Lobo PI, Bajwa A, Schlegel KH, Vengal J, Lee SJ, Huang L, et al. Natural IgM anti-leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune mechanisms involving Th-17. J Immunol (2012) 188(4):1675–85. doi:10.4049/jimmunol.1101762
139. Gobet R, Cerny A, Ruedi E, Hengartner H, Zinkernagel RM. The role of antibodies in natural and acquired resistance of mice to vesicular stomatitis virus. Exp Cell Biol (1988) 56(4):175–80.
140. Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science (1999) 286(5447):2156–9. doi:10.1126/science.286.5447.2156
141. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol (2010) 10(11):778–86. doi:10.1038/nri2849
142. Harnett W, Harnett MM. Phosphorylcholine: friend or foe of the immune system? Immunol Today (1999) 20(3):125–9. doi:10.1016/S0167-5699(98)01419-4
143. Tomasz A. Choline in the cell wall of a bacterium: novel type of polymer-linked choline in pneumococcus. Science (1967) 157(3789):694–7. doi:10.1126/science.157.3789.694
144. Lobo PI, Schlegel KH, Yuan W, Townsend GC, White JA. Inhibition of HIV-1 infectivity through an innate mechanism involving naturally occurring IgM anti-leukocyte autoantibodies. J Immunol (2008) 180(3):1769–79. doi:10.4049/jimmunol.180.3.1769
145. Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol (2007) 7(12):964–74. doi:10.1038/nri2214
146. Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest (2000) 105(12):1731–40. doi:10.1172/JCI8472
147. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci U S A (2007) 104(35):14080–5. doi:10.1073/pnas.0700326104
148. Kearney JF. Immune recognition of OxLDL in atherosclerosis. J Clin Invest (2000) 105(12):1683–5. doi:10.1172/JCI10426
149. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med (2002) 196(5):655–65. doi:10.1084/jem.20020542
150. Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med (2003) 9(6):736–43. doi:10.1038/nm876
151. Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest (1999) 103(1):117–28. doi:10.1172/JCI4533
152. Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol (2007) 170(1):416–26. doi:10.2353/ajpath.2007.060406
153. Fraser DA, Tenner AJ. Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J Immunol (2010) 185(7):3932–9. doi:10.4049/jimmunol.1002080
154. Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation (2009) 120(5):417–26. doi:10.1161/CIRCULATIONAHA.109.868158
155. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology (1998) 199(2):265–85. doi:10.1016/S0171-2985(98)80032-6
156. Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity (2005) 38(4):259–64. doi:10.1080/08916930500124452
157. Terasaki PI, Mottironi VD, Barnett EV. Cytotoxins in disease. N Engl J Med (1970) 283(14):724–8. doi:10.1056/NEJM197010012831403
158. Lobo PI, Schlegel KH, Vengal J, Okusa MD, Pei H. Naturally occurring IgM anti-leukocyte autoantibodies inhibit T-cell activation and chemotaxis. J Clin Immunol (2010) 30(1):S31–6. doi:10.1007/s10875-010-9412-7
159. Winfield JB, Winchester RJ, Wernet P, Fu SM, Kunkel HG. Nature of cold-reactive antibodies to lymphocyte surface determinants in systemic lupus erythematosus. Arthritis Rheum (1975) 18(1):1–8. doi:10.1002/art.1780180101
160. Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, et al. Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc Natl Acad Sci U S A (2012) 109(40):E2699–706. doi:10.1073/pnas.1210706109
161. Lobo PI, Sturgill BC, Bolton WK. Cold-reactive alloantibodies and allograft malfunction occurring immediately posttransplant. Transplantation (1984) 37(1):76–80. doi:10.1097/00007890-198401000-00021
162. Sturgill B, Lobo P, Bolton W. Cold-reacting IgM antibody-induced renal allograft failure. Nephron (1984) 36(2):125–7. doi:10.1159/000183131
163. Zhang M, Austen WG Jr, Chiu I, Alicot EM, Hung R, Ma M, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A (2004) 101(11):3886–91. doi:10.1073/pnas.0400347101
164. Betapudi V. Life without double-headed non-muscle myosin II motor proteins. Front Chem (2014) 2:45. doi:10.3389/fchem.2014.00045
165. Austen WG, Zhang M, Chan R, Friend D, Hechtman HB, Carroll MC, et al. Murine hindlimb reperfusion injury can be initiated by a self-reactive monoclonal IgM. Surgery (2004) 136(2):401–6. doi:10.1016/j.surg.2004.05.016
166. Zhang M, Michael LH, Grosjean SA, Kelly RA, Carroll MC, Entman ML. The role of natural IgM in myocardial ischemia-reperfusion injury. J Mol Cell Cardiol (2006) 41(1):62–7. doi:10.1016/j.yjmcc.2006.02.006
167. Arrondel C, Vodovar N, Knebelmann B, Grunfeld JP, Gubler MC, Antignac C, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol (2002) 13(1):65–74.
168. Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol (2010) 185(7):4393–400. doi:10.4049/jimmunol.0903239
169. Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol (2007) 178(9):5899–911. doi:10.4049/jimmunol.178.9.5899
170. Burne-Taney MJ, Yokota-Ikeda N, Rabb H. Effects of combined T- and B-cell deficiency on murine ischemia reperfusion injury. Am J Transplant (2005) 5(6):1186–93. doi:10.1111/j.1600-6143.2005.00815.x
171. Gigliotti JC, Huang L, Ye H, Bajwa A, Chattrabhuti K, Lee S, et al. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol (2013) 24(9):1451–60. doi:10.1681/ASN.2013010084
172. Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol (2002) 282(2):F352–7. doi:10.1152/ajprenal.00160.2001
173. Gorevic PD. Rheumatoid factor, complement, and mixed cryoglobulinemia. Clin Dev Immunol (2012) 2012:6. doi:10.1155/2012/439018
174. Charles ED, Dustin LB. Hepatitis C virus-induced cryoglobulinemia. Kidney Int (2009) 76(8):818–24. doi:10.1038/ki.2009.247
175. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang D, et al. Identity of the elusive IgM Fc receptor (FcμR) in humans. J Exp Med (2009) 206(12):2779–93. doi:10.1084/jem.20091107
176. Marchalonis JJ, Schluter SF, Wang E, Dehghanpisheh K, Lake D, Yocum DE, et al. Synthetic autoantigens of immunoglobulins and T-cell receptors: their recognition in aging, infection, and autoimmunity. Proc Soc Exp Biol Med (1994) 207(2):129–47. doi:10.3181/00379727-207-43801
177. Vassilev T, Mihaylova N, Voynova E, Nikolova M, Kazatchkine M, Kaveri S. IgM-enriched human intravenous immunoglobulin suppresses T lymphocyte functions in vitro and delays the activation of T lymphocytes in hu-SCID mice. Clin Exp Immunol (2006) 145(1):108–15. doi:10.1111/j.1365-2249.2006.03098.x
178. Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol (2009) 20(8):1744–53. doi:10.1681/ASN.2008111160
179. Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Peritoneal B cells govern the outcome of diabetes in non-obese diabetic mice. Eur J Immunol (2004) 34(9):2387–95. doi:10.1002/eji.200324744
180. Ryan GA, Wang CJ, Chamberlain JL, Attridge K, Schmidt EM, Kenefeck R, et al. B1 cells promote pancreas infiltration by autoreactive T cells. J Immunol (2010) 185(5):2800–7. doi:10.4049/jimmunol.1000856
181. Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol (2008) 180(5):2863–75. doi:10.4049/jimmunol.180.5.2863
182. Chhabra P, Schlegel K, Okusa MD, Lobo PI, Brayman KL. Naturally occurring immunoglobulin M (nIgM) autoantibodies prevent autoimmune diabetes and mitigate inflammation after transplantation. Ann Surg (2012) 256(4):634–41. doi:10.1097/SLA.0b013e31826b4ba9
183. Andersson Å, Forsgren S, Söderström Å, Holmberg D. Monoclonal, natural antibodies prevent development of diabetes in the non-obese diabetic (NOD) mouse. J Autoimmun (1991) 4(5):733–42. doi:10.1016/0896-8411(91)90169-D
184. Andersson A, Ekstrand-Hammarstrom B, Eriksson B, Overmo C, Holmberg D. Neonatal treatment with monoclonal natural antibodies restores a normal pattern of VH gene utilization in the non-obese diabetic mouse. Int Immunol (1994) 6(4):623–30. doi:10.1093/intimm/6.4.623
185. Symmons DP. Frequency of lupus in people of African origin. Lupus (1995) 4(3):176–8. doi:10.1177/096120339500400303
186. Lobo PI. The first reported case of sarcoidosis in an East African. East Afr Med J (1972) 49(1):37–49.
187. Jacyk WK. Sarcoidosis in the West African. A report of eight Nigerian patients with cutaneous lesions. Trop Geogr Med (1984) 36(3):231–6.
Keywords: natural IgG autoantibodies, natural IgM autoantibodies, natural autoantibodies, natural IgM anti-leukocyte antibodies, allograft rejection, TH-17, renal IRI, autoimmune insulitis, regulation of co-stimulatory receptors, regulatory dendritic cells
Citation: Lobo PI (2016) Role of Natural Autoantibodies and Natural IgM Anti-Leucocyte Autoantibodies in Health and Disease. Front. Immunol. 7:198. doi: 10.3389/fimmu.2016.00198
Received: 16 December 2015; Accepted: 06 May 2016;
Published: 06 June 2016
Edited by:
Nichol E. Holodick, The Feinstein Institute for Medical Research, USAReviewed by:
Ziaur S. M. Rahman, Pennsylvania State University, USAChristoph J. Binder, Medical University of Vienna, Austria
Copyright: © 2016 Lobo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Isaac Lobo, cGlsQHZpcmdpbmlhLmVkdQ==