 Klaus Ley
Klaus Ley Akula Bala Pramod
Akula Bala Pramod Michael Croft
Michael Croft Kodi S. Ravichandran4
Kodi S. Ravichandran4 Jenny P. Ting
Jenny P. Ting- 1Division of Inflammation Biology, La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA
- 2Department of Bioengineering, University of California San Diego, La Jolla, CA, USA
- 3Division of Immune Regulation, La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA
- 4Department of Microbiology, Immunology, and Cancer Biology, University of Virginia, Charlottesville, VA, USA
- 5Department of Genetics, The Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Macrophages are central to both innate and adaptive immunity. With few exceptions, macrophages are the first cells that sense trouble and respond to disturbances in almost all tissues and organs. They sense their environment, inhibit or kill pathogens, take up apoptotic and necrotic cells, heal tissue damage, and present antigens to T cells. Although the origins (yolk sac versus monocyte-derived) and phenotypes (functions, gene expression profiles, surface markers) of macrophages vary between tissues, they have many receptors in common that are specific to one or a few molecular species. Here, we review the expression and function of almost 200 key macrophage receptors that help the macrophages sense what is going on, including pathogen-derived molecules, the state of the surrounding tissue cells, apoptotic and necrotic cell death, antibodies and immune complexes, altered self molecules, extracellular matrix components, and cytokines, including chemokines.
Introduction
Macrophages are central to proper functioning of the immune system (1–4). Resting tissue macrophages make ornithine (by arginase) and promote repair, called the M2 phenotype (1, 5). Tissue macrophages arise from embryonic precursors and from blood monocytes at varying proportions (6). At the extremes are microglia cells, which are entirely embryonic-derived (7) and intestinal macrophages, which are entirely monocyte-derived (8) with other organs being populated by mixtures of both. When these tissue macrophages sense trouble (1), they express IL12 and iNOS, and become M1 macrophages (5). Macrophages express hundreds of sensor molecules to identify their surroundings (cells, extracellular matrix, and tissue), the state of the tissue (normal and healthy, apoptotic, ischemic, necrotic, altered, or otherwise distressed), metabolites [oxygen, pH, lactic acid, glucose, free fatty acids, sphingosine-1-phosphate (S1P)], lipoproteins (LDL, HDL, and their derivatives), antibodies (IgG, immune complexes), complement (C3a, C3bi, C5a), cytokines (IFN-γ, IL4, IL17), and pathogens (bacteria, viruses, fungi, parasites, and their products). Dendritic cells, another key sensor of pathogens, act in concert with the macrophages in initiating an adaptive immune response. Dendritic cells activate the adaptive immune system by antigen presentation to naive T cells in the context of co-activating receptors. These T cells become licensed to traffic to the infected or inflamed tissue (9, 10), where they see antigen presented by macrophages (11) and differentiate (CD4: Th1, Th2, Th17, Treg, TFH and others, CD8: CTL and others). Many products of the adaptive immune system, such as cytokines, can further activate macrophages and alter their function; for example, IFN-γ enhances M1 polarization (12) and IL4 induces alternative activation (13), thus enhancing M2 polarization (1, 14).
This review is focused on the inputs to the macrophage and dendritic cell system: the cell surface and intracellular receptors by which these cells sense what is going on. For details of the signaling networks and effector systems downstream of these receptors, like TRAFs (15–17), NFkB (18, 19), or inflammasome assembly (20), the reader is referred to other reviews. Although there are human homologs for almost all receptors discussed here, this review is entirely based on mouse data. The innate immune system and macrophages in particular are under enormous evolutionary pressure shaped by the environment and infectious organisms that differ between mice and humans.
Tissue Input at Steady State
At steady state, signals from host tissue cells result in tissue-specific gene expression profiles (21). Langerhans cells of the skin, alveolar macrophages, Kupffer cells of the liver, microglia cells of the CNS, osteoclasts, dendritic cells of the thymus, and other lymphoid organs all have specialized functions and phenotypes. This suggests that tissue-derived signals control the development and polarization of tissue-specific macrophage phenotypes. The first tissue cues were identified in osteoclasts (22) and peritoneal macrophages (23, 24). A key inducer of the peritoneal macrophage phenotype is retinoic acid produced by intestinal cells, which is recognized by the nuclear receptor retinoic acid receptor-β (RAR-β). Retinoic acid induces the transcription factor GATA6, which in turn controls a number of effector molecules (23, 24). Interestingly, RAR-β ligation also induces arginase-1, the defining enzyme of M2 macrophages, underscoring that resting peritoneal macrophages, like macrophages in other tissues, are in the default “healing” M2 state. A second known M2-polarizing stimulus is lactic acid, produced by hypoxic cells in cancer (25). This is consistent with the known M2 phenotype of myeloid-derived suppressor cells (26), a macrophage type found in cancers. Under homeostatic conditions, M2 polarization is maintained by TGF-β, and TGF-β receptor-1 seems to be the major factor in determining microglia phenotype (27). Under inflammatory conditions, TGF-β induces a state of deactivation that is different from M2, and promotes the resolution of inflammation. TGF-β2 is produced by resident peritoneal macrophages (28) and might play a significant role in homeostatic maintenance of surrounding tissues.
To provide a first glimpse at the expression of almost 200 “input” receptors, we compiled heat maps from published data sets on mouse peritoneal macrophages (large, small, and thioglycollate-elicited), microglia (27) (data set GSE62826), and the macrophages from lung, liver, spleen, intestinal, adipose tissue, and bone marrow (23) (data sets GSE56682, 56683, 56684) and (29) (data set GSE47049). The transcriptome data sets were accessed through the Gene Expression Omnibus site1 to examine the gene expression profiles in tissue-specific macrophages for 12 categories of receptors [apoptotic cell receptors, complement receptors, toll-like receptors (TLR), NOD-like receptors (NLR), RNA and DNA receptors, C-type lectins, scavenger receptors, selected cytokine receptors, TNF receptor superfamily members, Fc receptors, selected G-protein-coupled receptors, and integrins]. Cross-platform comparison of gene expression profiles between microarray (GSE47049, GSE56682, GSE56683, and GSE56684) and RNA-Seq (GSE62826) was done on normalized data provided by the authors on the GEO site. The normalized values from different platforms were scaled by log2 transformation for comparison through generation of heat maps (30–32). In brief, MAS5.0 (Affymetrix) and quantile normalized (Illumina GUI) probe data available for the microarray platforms, i.e., GSE47049 and GSE56682/83/84 from Affymetrix Mouse Genome 430 2.0 Array and Illumina mouse expression bead chip, respectively, were annotated by R/Bioconductor packages2 and annotation files downloaded from the GEO site (GPL6105 and GPL6887). The normalized RPKM gene expression levels from the Illumina RNA-sequencing (Illumina HiSeq 2000) dataset (GSE62826) were obtained from the GEO site. Whenever a gene was represented by more than one probe set or entry, the average of the pooled expression values was used. Furthermore, the average value of the genes across the samples for a tissue from different datasets was calculated for log2 transformation. Cluster 3.0 with pair-wise complete linkage was used to perform the hierarchical analysis of log2 transformed expression values of around 200 genes using mean-centered gene expression vectors and visualized with the Java TreeView 10.6 (32, 33). The graphical representation of output as heat maps is presented in color scale from green to red (Z-score transformation), where red and green indicate higher and lower expression levels, respectively (Figure 1). Non-expressed genes are shown in light gray.
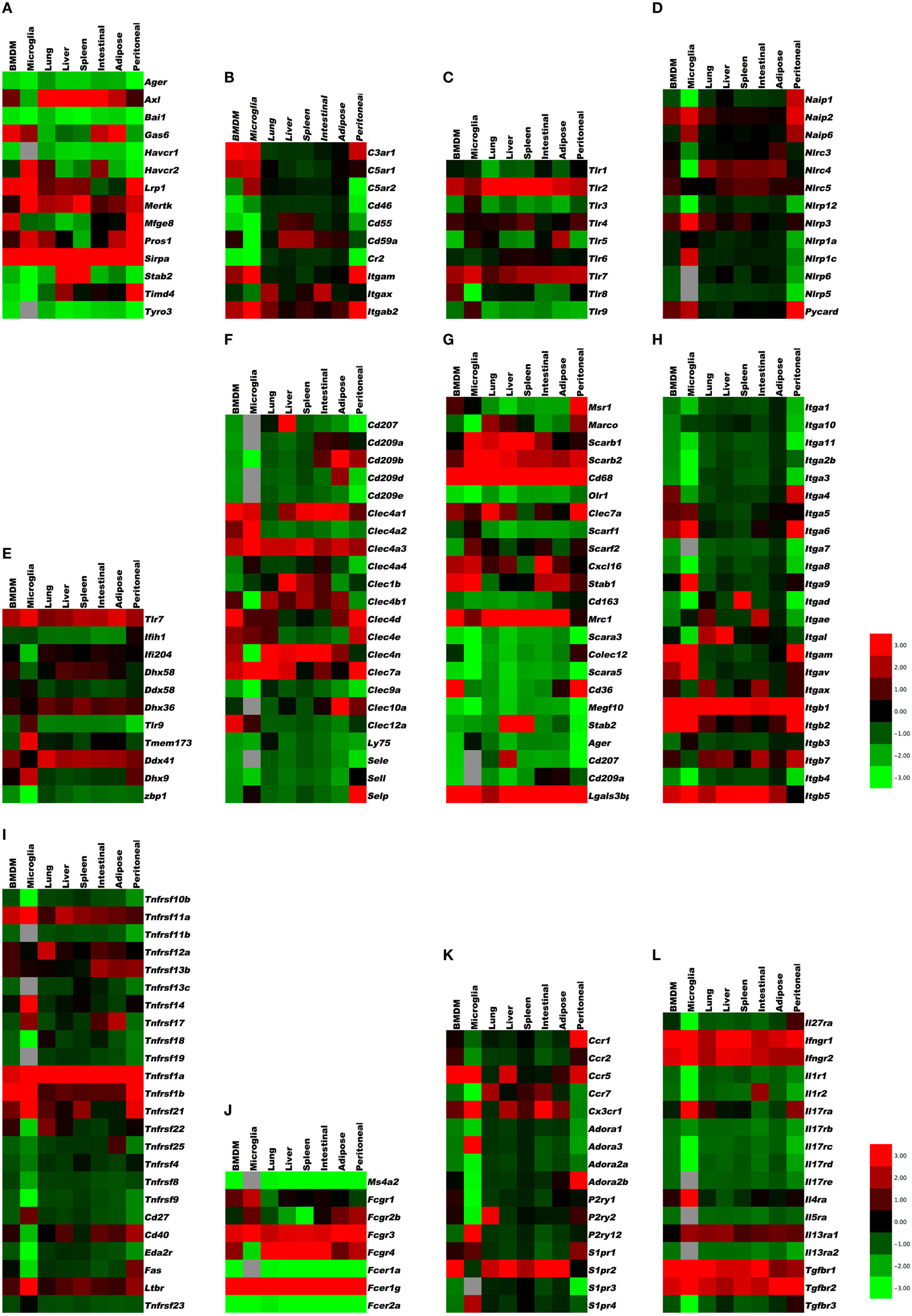
Figure 1. Gene expression in resident tissue-specific macrophages (microglia, lung, liver, spleen, intestinal, adipose) and bone marrow-derived macrophages (BMDM) for comparison. Heat maps based on hierarchical clustering results from microarray and RNA-Seq data for (A) apoptosis receptors, (B) complement receptors, (C) toll-like receptors, (D) NOD-like receptors, (E) RNA and DNA sensors, (F) C-type lectins, (G) scavenger receptors, (H) integrins, (I) tumor necrosis factor receptor superfamily, (J) Fc receptors, (K) G-protein-coupled receptors, and (L) cytokine receptors. Gene expression levels are expressed in log2 values, differences shown in color scale after Z-score transformation. Colors scale bar ranges from red (+3) to green (−3). Light gray indicates undetectable.
Receptors for Apoptotic Cells
In many tissues, there is continuous turnover of cells, where the majority of them die by the process of caspase-dependent apoptosis. When cells undergo apoptosis, their corpses are phagocytosed by macrophages in a process referred to as efferocytosis (34–36). In addition to macrophages, epithelial cells and fibroblasts clear apoptotic cells. Apoptotic cells expose phosphatidylserine (PtdSer) on the outer leaflet of their plasma membrane, which serves as a recognition signal (Eat-me). Apoptotic cells, at the earliest stages of death, also release ATP and other molecules that serve as diffusible “find-me” signals to attract macrophages to their proximity. The find-me signals are sensed by tissue-resident macrophages via some G-protein-coupled receptors, such as P2Y2 sensing ATP and UTP released from apoptotic cells, and CX3CR1 sensing CX3CL1 released from apoptotic cells.
There are several recognition systems for PtdSer (Table 1). Routine, homeostatic uptake of apoptotic cells is inherently anti-inflammatory and helps keep the local tissue inflammation to a very minimal (or below detection) level even in tissues where there is a very high cell turnover (such as the bone marrow or thymus). This is in part achieved through the release of mediators, such as TGF-β, IL10, and prostaglandin E2 (PGE2) from macrophages that engage apoptotic cells. TGF-β and IL10 have anti-inflammatory functions. Depending on the receptor triggered (EP1-EP4), PGE2 can have pro- and anti-inflammatory functions, modulate pain sensation, and can activate mast cells (37). See Ref. (38, 39) for more details on apoptotic cell clearance. αVβ3 integrin (see Integrins) recognizes PtdSer through MFGE8. RAGE and stabilin 2 also bind PtdSer and are listed under scavenger receptors (below). Tyro3, Axl, and Mer bind PtdSer through GAS6 (gene name Gas6) or protein S (gene name Pros1). Mertk, the gene encoding Mer tyrosine kinase, distinguishes macrophages from dendritic cells and has been proposed as a universal mouse macrophage marker (40) when used in combination with other markers like F4/80, CD68, or CD11b. CD91, also known as LDL receptor-related protein LRP, is a receptor for calreticulin, which is exposed in cells undergoing ER stress and apoptosis. SIRPα (gene name Sirpa) can detect CD47 on apoptotic lymphocytes. The results presented in Figure 1A summarize the expression of genes related to apoptotic cell recognition and uptake. The clearance of apoptotic cells by phagocytes is counterbalanced by mechanisms that limit detrimental effects, such as production of reactive oxygen species. Also, efferocytic receptors, such as Mer and CD11b, can be downregulated, a process that limits further signaling and uptake of apoptotic cells.
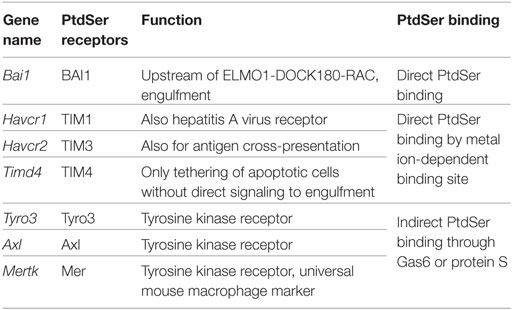
Table 1. PtdSer recognition receptors.
Receptors for Necrotic Cells and Modified Self
Macrophages sense when tissue cells are distressed by events, such as ischemia (pro-angiogenic, pro-inflammatory), reperfusion (pro-inflammatory), tissue necrosis (pro-inflammatory), and altered self induced by enzymatic modification resulting in (lipo) protein citrullination (41), oxidation (42), or nitrosylation (43). Many of the receptors for altered self are among the category of cell surface molecules that are generally referred to as scavenger receptors (see below). Macrophages express many scavenger receptors that are capable of recognizing these necrotic or altered self-molecule on stressed cells within tissues.
Necrotic cells are opsonized by complement factors that are recognized through complement receptors (44, 45) (Table 2; expression in Figure 1B). The scavenger receptor RAGE (see below) also recognizes apoptotic cells and triggers pro-inflammatory mechanisms. The IL33 receptor ST2 detects IL33 released by necrotic cells. CD24 on macrophages binds HMGB1, which in turn binds necrotic cells. Mincle (Clec4e) and CLEC9A are two C-type lectins (listed below) involved in the uptake of necrotic cells. TLRs (see below) can recognize the nuclear protein HMGB1, the Sin3A-Associated Protein SAP130, the heat shock protein HSP90, DNA, and urate crystals, all of which can be exposed on or released from necrotic cells.
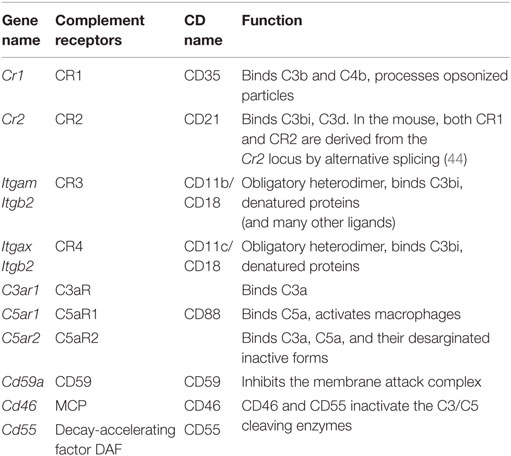
Table 2. Complement receptors.
Pathogen Receptors
Pathogen sensors on macrophages can detect bacterial, viral, fungal, and parasite infections. These receptors bind pathogens directly or recognize their products. They form six main classes: TLR (17, 46), NLR (20, 47), receptors for intracellular RNA, including RIG-I like receptors (RLR) (48), receptors for intracellular DNA, including STING (49, 50), C-type lectins (51, 52), and scavenger receptors (53).
Toll-Like Receptors
Toll-like receptors (Table 3) are a family of homodimeric transmembrane receptors that recognize bacterial, viral, and fungal products. Some also have endogenous ligands. TLRs 1, 2, 4, 5, and 6 are expressed on the plasma membrane, whereas TLRs 3, 7, 8, and 9 are expressed in endosomes (17, 54). Most TLRs signal through the MyD88 pathway, ultimately resulting in NFkB activation, except TLR3, which signals through the intermediary TRIF resulting in type I interferon production. TLR4 signals through both MyD88 and TRIF. (Note: TLR10 is not expressed in mice).

Table 3. Toll-like receptors.
The intracellular vesicles in which TLR3, 7, 8, and 9 are expressed include ER, endosomes, lysosomes, and endolysosomes. An excellent review of TLR expression and function is found in Ref. (17). The results presented in Figure 1C indicate the expression levels of TLRs in various tissue macrophages.
NOD-Like Receptors or NBD–LRR Containing Proteins
This class of pathogen receptors/sensors (20) regulates a number of key inflammatory pathways. NLRs (Table 4) contain a nucleotide-binding domain (NBD) and leucine-rich repeats (LRR). These proteins are conserved in evolution from plants to humans. NLR proteins are subdivided by their N-terminus, which can contain acid transactivation, pyrin, CARD, or BIR domains. A major pathway is the activation of the inflammasome, which is a macromolecular structure that when assembled, causes the cleavage and activation of caspase-1, and thus can produce the active species of IL1β and IL18. There are 22 known human NLRs; here, only the best-studied will be discussed. Some, but not all NLRs assemble an inflammasome. Additionally, some inflammasome-inducing NLRs also exhibit inflammasome-independent functions. The molecular mechanisms of inflammasome activation have been reviewed elsewhere (20). This review will briefly describe the key NLR molecules important for macrophage sensing with a focus on the inflammasomes.
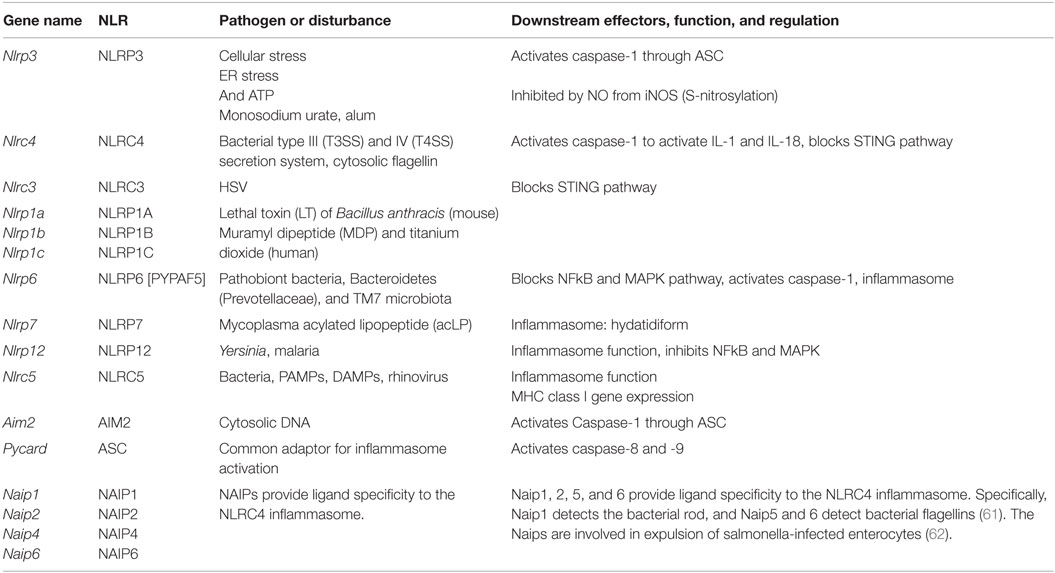
Table 4. NOD-like receptors.
The best-studied inflammasome is anchored by NLRP3, a receptor that does not directly recognize DAMPs or PAMPs but rather responds to cellular stress, including changes in potassium efflux, ROS production, ER stress, and unfolded protein response. The adapter GBP5 promotes NLRP3 inflammasome activation by ATP, nigericin, and bacteria, but not by crystalline agents. Several inhibitory pathways for NLRP3 activation have also been described. For example, nitric oxide can cause the S-nitrosylation of NLRP3 and impair assembly of the inflammasome (55–57). The G Protein signaling modulator-3 protein can associate with the LRR domain of NLRP3 and inhibit inflammasome activation. cAMP binds and inhibits NLRP3, and the neurotransmitter dopamine negatively regulates NLRP3 function via cAMP that promotes NLRP3 ubiquitination and subsequent degradation by the E3 ligase, MARCH7 (58). NLRP3 inflammasome activation depends on ASC (gene name Pycard), a common adaptor required for the activation of caspase-1 by several inflammasomes, and is comprised of a pyrin domain and a CARD domain. Linear ubiquitination via the assembly complex LUBAC is known to enhance ASC function in the activation of NLRP3-dependent inflammasome (59). The NLRP3 inflammasome also senses mitochondrial DNA. Activated caspase-1 or the related caspase-11 can induce pyroptosis (lytic cell death).
The NLRC4 inflammasome activation (60) is enhanced by ASC, and it detects bacterial proteins in the cytosolic compartment, either as markers for the activity of bacterial type III and IV secretion systems or cytosolic invasion by flagellated bacteria. NLRC4 responds to injection of three conserved bacterial proteins: flagellin, rod, and needle. NAIP proteins are also members of the NLR family. NAIP1, 2, 5, and 6 provide ligand specificity to the NLRC4 inflammasome. Specifically, NAIP1 detects the bacterial Needle protein, NAIP2 detects bacterial Rod, and NAIP5 and 6 detect bacterial flagellins (61). The NAIPs are involved in expulsion of salmonella-infected enterocytes (62).
NLRP1 (DEFCAP, NAC, or NALP1) is the NLR that defined the inflammasome (63). Mice express three highly polymorphic paralogs: Nlrp1a, Nlrp1b, and Nlrp1c due to likely gene duplication, while humans only express a single NLRP1. Mouse Nlrp1b and rat Nlrp1, but not human NLRP1 mediates the response to the lethal toxin (LT) of Bacillus anthracis. Reminiscent of the requirement of a cleavage/activation process for caspase-1, IL1β, and IL18 function, a maturation cleavage of the N-terminal of murine NLRP1 is also required for its activation by LT (64). Among pathogens, NLRP1 is also activated by Toxoplasma gondii and is required for host protection (65), although this process does not require the above-mentioned N-terminal cleavage (66). NLRP1 is not the only NLR required for inflammasome activation by T. gondii, as NLRP3 was also shown to play a role in host resistance to this pathogen (67).
NLRP6 was originally found to synergize with ASC to cause NFkB and caspase-1 activation in overexpression systems. Nlrp6−/− mice show spontaneous and induced colitis (68), associated with reduced IL18 and expanded pathobiont bacteria, Bacteroidetes (Prevotellaceae), and TM7 microbiota (69). Other reports indicate alternative roles for the NLRP6 in the control of intestinal cell apoptosis or as a checkpoint regulator of inflammatory mediators and the NFkB/MAPK pathways.
Other NLRs with inflammasome functions also have demonstrated in vitro relevance in other important pathways. For example, NLRP7 mediates inflammasome activation by mycoplasma acylated lipopeptide in a human macrophage cell line, but it has been genetically linked in multiple patient cohorts (70) to the formation of hydatidiform moles, which is an abnormal form of pregnancy. NLRP12 (Monarch-1 or PYPAF7) exhibits at least two functions as an inhibitor of the non-canonical and the canonical NFkB pathway. This was confirmed by in vivo studies of colitis-associated colon carcinoma models (71, 72), a bacterial infection model as well as a study of osteoclasts. Yersinia strains, but not several other bacteria, can cause Nlrp12-dependent inflammasome activation (73), and Plasmodium species induce a NLRP12- and NLRP3-dependent inflammasome activation (74). This dual requirement for two NLR is also observed with NLRC5 and NLRP3, which interact with each other in a human macrophage cell line and PBMCs in response to a host of NLRP3 activators (75). Nlrc5−/− mice show deficiencies in response to NLRP3 activators, such as monosodium urate, alum, and ATP (76). However, the primary function of NLRC5 is the regulation of class I MHC genes, thus demonstrating again the multi-faceted nature of NLRs (77).
In addition to the activation of caspase-1, a non-canonical pathway leading to caspase-11 maturation that requires NLRP3, and can be activated by some bacteria, which appears to be detrimental during endotoxic shock (78). Caspase-11 is activated by LPS from Gram-negative bacteria that invade the cytosol, and protects mice against lethal infection by Burkholderia thailandensis and Burkholderia pseudomallei. By contrast, vacuolar bacteria, such as Salmonella typhimurium are poorly detected by caspase-11. Caspase-11 is non-responsive, unless macrophages are primed by IFN-β or IFN-γ, both of which activate the transcription factor STAT1. Caspase-11 responds to cytosolic lipid A species with five or six acyl groups, but not to species with four acyl groups. This expands the role of LPS as a key microbial pattern detected by the innate immune system: extracellular and vacuolar LPS is detected through TLR4, whereas cytosolic LPS is detected through caspase-11. Interestingly, both Tlr4- and caspase-11-deficient mice are resistant to classical LPS challenge (78). The results presented in Figure 1D reveal the expression levels of NLR and related molecules.
Sensors for Intracellular RNA and DNA
RNA and DNA receptors recognize components of viral genomes. Herpes viruses such as herpes simplex or murine cytomegalovirus (MCMV) are double-stranded (ds)DNA viruses, where the dsDNA is recognized by STING or IFI16 (mouse gene name Ifi204). Single-stranded (ss) viral RNA often forms stem–loop structures that can be detected by TLR7 (see Toll-Like Receptors) and RIG-I (79). ssDNA (not necessarily viral) is recognized by TLR9, especially when it is CpG-rich and not methylated.
These receptors are expressed in macrophages, epithelial cells, and other cells. The immune response starts in infected cells, where the viruses replicate in the cytoplasm. The intracellular viral RNA is detected by the RIG-I family of receptors, including RIG-I (gene name: Ddx58), MDA5 (Ifih1), and LGP2 (Dhx58). RIG-I detects many ssRNA viruses, including influenza virus, hepatitis C, Dengue, yellow fever, west Nile, and Ebola. The RIG-I family receptors require MAVS as an adapter protein and IRF3 or IRF7 as downstream transcription factors, resulting in the elaboration of IFN-α and β.
Single-stranded DNA is recognized by TLR9 (see Toll-Like Receptors). DNA-derived intracellular cyclic dinucleotides are recognized by STING (gene name, Tmem173) (80). Humans have five common STING alleles. Listeria products are processed by cGAS (cyclic GMP AMP synthetase), which produces cGMP that in turn activates STING. STING is also involved in detecting herpes virus. Other sensors for intracellular DNA are DAI (gene name, Zbp1), Aim2 (see under NLRs), DDX41 (Ddx41), DHX9 (Dhx9), IFI16 (gene name, Ifi204), and DHX36 (Dhx36). DAI senses dsDNA. The results in Figure 1E show the mRNA expression of these DNA- and RNA-sensing receptors in various tissue-resident macrophages. Aim2 expression was only detected in bone marrow-derived macrophage (BMDM), peritoneal macrophages, and microglia.
C-Type Lectin Receptors
In general, C-type lectin receptors (Table 5) recognize multivalent carbohydrate ligands (4) that might be present on endothelial cells, epithelial cells, pathogens, and other microorganisms. C-type lectins are also involved in the uptake and clearance of dead cells (6). Macrophages express several C-type lectin receptors, which include collectins, selectins, lymphocyte lectins, and proteoglycans (4). Some are important sensors for fungal infections (81), others such as the selectins recognize host glycans (82). The C-type lectins DCIR and MICL are inhibitory, because their cytoplasmic tails contain ITIM domains. Dectin-2, Mincle, and DCAR are coupled to an ITAM-domain containing adaptor. Dectin-2 is the prototypic receptor of the Dectin-2 subfamily of C-type lectins that also contains CLECSF8, DCIR and Mincle and DCIR2, 3, and 4, DCAR, and DCAR1 in mice only (5). SignR3, Dectin-1, and CLEC9a also contain at least one ITAM domain. DC-Sign, SIGN-R1, Langerin, MGL, DEC-205, and the mannose receptor MR are not coupled to either ITAM or ITIM domains, but signal through adaptor proteins.
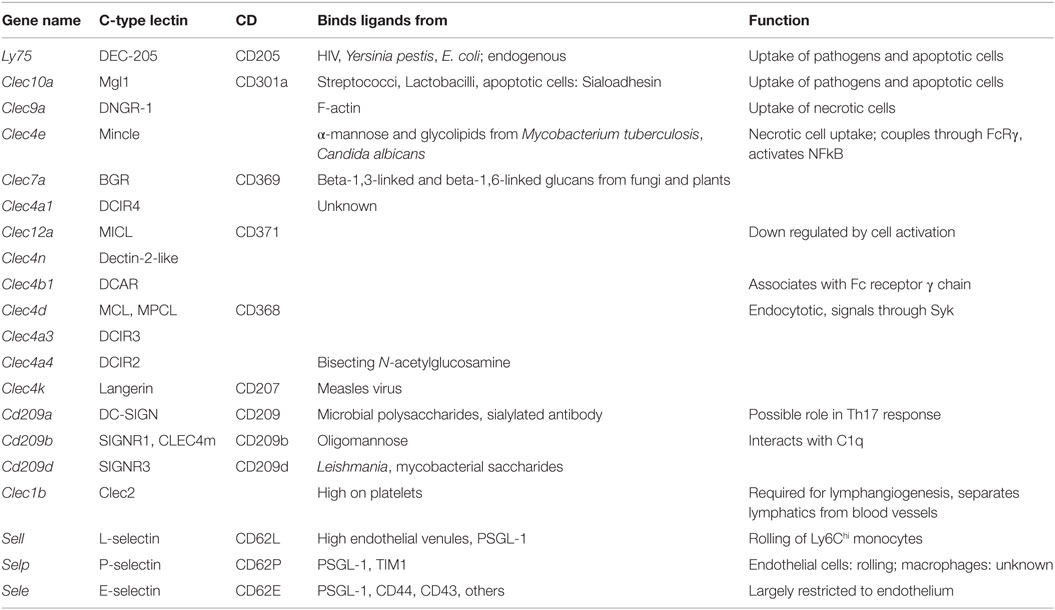
Table 5. C-type lectins.
E-selectin recognizes fucosylated and sialylated ligands of the sialyl-Lewisx type. P-selectin recognizes the glycoproteins PSGL-1 on myeloid cells and TIM1 on activated T cells. L-selectin recognizes 6-sulfated carbohydrates expressed in high endothelial venules and PSGL-1. Many macrophages express P-selectin, but its function in macrophages has not been studied. Monocytes express L-selectin, but its expression is lost upon differentiation to macrophages. L-selectin is an excellent example for an adhesion molecule that is shed upon cell activation (83). Shedding of adhesion molecules, cytokine receptors, and pattern recognition receptors is an important mechanism that limits their signaling (84, 85). In some cases, the shed molecules can have signaling functions in their soluble form. The results in Figure 1F demonstrate the expression levels of C-type lectins.
Scavenger Receptors
Scavenger receptors (Table 6) are cell surface receptors that typically bind multiple ligands and promote the removal of non-self or altered self targets. Both tissue-resident and monocyte-derived macrophages express a number of scavenger receptors. They often function by mechanisms that include endocytosis, phagocytosis, adhesion, and signaling that ultimately lead to the elimination of degraded or harmful substances (7). Scavenger receptors recognize non-self (mostly bacterial products) and altered (oxidized, acetylated) self. They are more involved in uptake and phagocytosis than activation of inflammatory effector systems. Scavenger receptors have recently been classified (53). The systematic names are SR followed by a letter (for the class) and numbers (for the members).
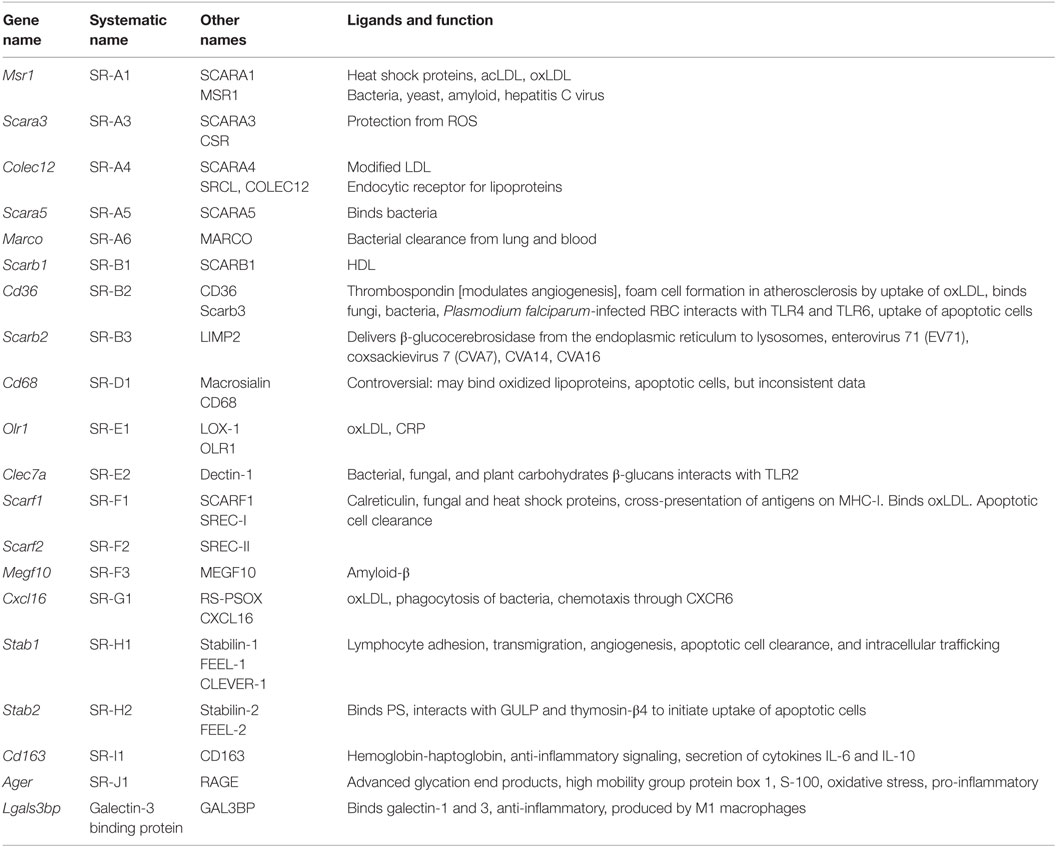
Table 6. Scavenger receptors.
SR-A1, 2, and 3 and MARCO form the SR-A subfamily. Only SR-A1 has an SRCR domain and is the main macrophage receptor for HDL. MARCO is a collagen-like molecule; MARCO defects result in susceptibility to tuberculosis. The SR-B subfamily comprises SR-B1 and SR-B2 (CD36). CD36 is an important receptor for diacyl glycerides, oxidized LDL, and the malaria parasites. CD36 is important for ROS and cytokine production by macrophages. There are no mammalian members of the SR-C subfamily. SR-D is CD68 (macrosialin), an intracellular receptor of unknown function. SR-E1 is Lox1 (Loxin), which detects oxidized LDL. Dectin-1 is SR-E2. The mannose receptor MR is SR-E3 and the asialoglycoprotein receptor is SR-E4. SR-F1 is SCARF1, Ced1 in Caenorhabditis elegans, and SR-F2 is SCARF2. The only known SR-G is CXCL16, a receptor for oxidized LDL and a chemokine that binds CXCR6. SR-H1 is FEEL-1, also known as Stabilin-1 or Clever-1, SR-I is CD163, the macrophage receptor for hemoglobin–haptoglobin complexes. In bovine and ovine species, this subfamily has many more members. SR-J is RAGE, a seven-transmembrane receptor with multiple immunoglobulin domains that sees glycated proteins and advanced glycation end products. SR-L1 is also known as LRP1, and SR-L2 as LRP-2.
Some macrophage scavenger receptors are currently unclassified. Galectin-3-binding protein is a secreted product of M1 macrophages that binds galectin-3 and other cell surface ligands. CD207 (Langerin), MRC1 (CD206), CLEC7A, and CD209 (DC-SIGN) are also unclassified. CD206 is prominently expressed on M2 macrophages. C1q interacts with the Aβ protein relevant in Alzheimer’s disease. The results in Figure 1G show the expression levels of scavenger receptors.
Cytokine Receptors
Another class of macrophage receptors sense products of adaptive immune cells. These receptors can be considered secondary amplifiers, because they do not detect non-self or altered self, but rather amplify the macrophage response. Since most cytokines are produced by activated T cells that require antigen presentation and an ongoing immune response, the cytokine receptors on macrophages sense the nature of that ongoing immune response (Th1, Th2, Th17) (Table 7).

Table 7. Cytokine receptors on macrophages.
IFN-γ receptor is a major activator of macrophages and reinforces the M1 phenotype (12). The Ifngr1 gene encoding this receptor is highly expressed in all macrophages. Its functional antagonist is IL4 receptor. Because of their important role in inflammation, we also discuss IL1 receptors, IL17 receptors, and IL27 receptor. A major anti-inflammatory pathway is initiated by TGF-β, which has three receptors.
IL4, 5, and 13 receptors promote and reinforce the M2 phenotype (3, 86, 87), but their ligation is not required for M2 polarization; in fact, it appears that M2 is the default “healing” phenotype of macrophages (5). IL13 receptor α1 chain is expressed in all macrophages, but the α2 chain is missing from peritoneal macrophages. IL5 receptor is expressed at very low levels on these cells.
TGF-β receptors strongly promote M2 polarization. TGF-β is made by many tissue cells, especially epithelial cells, and seems to signal to the macrophage that the tissue cells are in good health and not infected. Tgfbr1 is highly expressed in all macrophages, with highest levels in microglia. The same is true for Tgfbr2, but with highest levels in adipose tissue macrophages. Tgfbr3 is expressed in all macrophages except microglia and BMDM. Overabundance of TGF-β can lead to excessive collagen formation resulting in fibrosis. Macrophages have receptors for cytokines of the IL17 family. The results in Figure 1L indicate that the IL17 receptor A is expressed in all macrophages, and being highest in microglia. Il17rb, c, d, and e are expressed at low levels. IL1 receptors 1 and 2 are expressed at high levels in all macrophages.
TNF Receptor Superfamily
The TNF receptor superfamily (Tnfrsf) (88–91) bind soluble and cell surface-expressed TNF superfamily members that have diverse biological functions (Table 8). These can range from molecules that induce caspase activation and apoptosis and regulate cell death (Fas, TNFRI, TRAILR) to those that activate NFκB and MAPK pathways and are pro-inflammatory (e.g., TNFRII, RANK, CD40, BAFFR, OX40) inducing a multitude of effects, such as promoting division, survival, cytokine production, chemokine production, and upregulation of other receptors in varied protein families. The summarized expression values in Figure 1I reveal that the Tnfrsf receptors are of specific interest here, because many of them show highly differential expression among tissue macrophages, suggesting that different macrophage subsets have different abilities to see Tnfsf ligands.
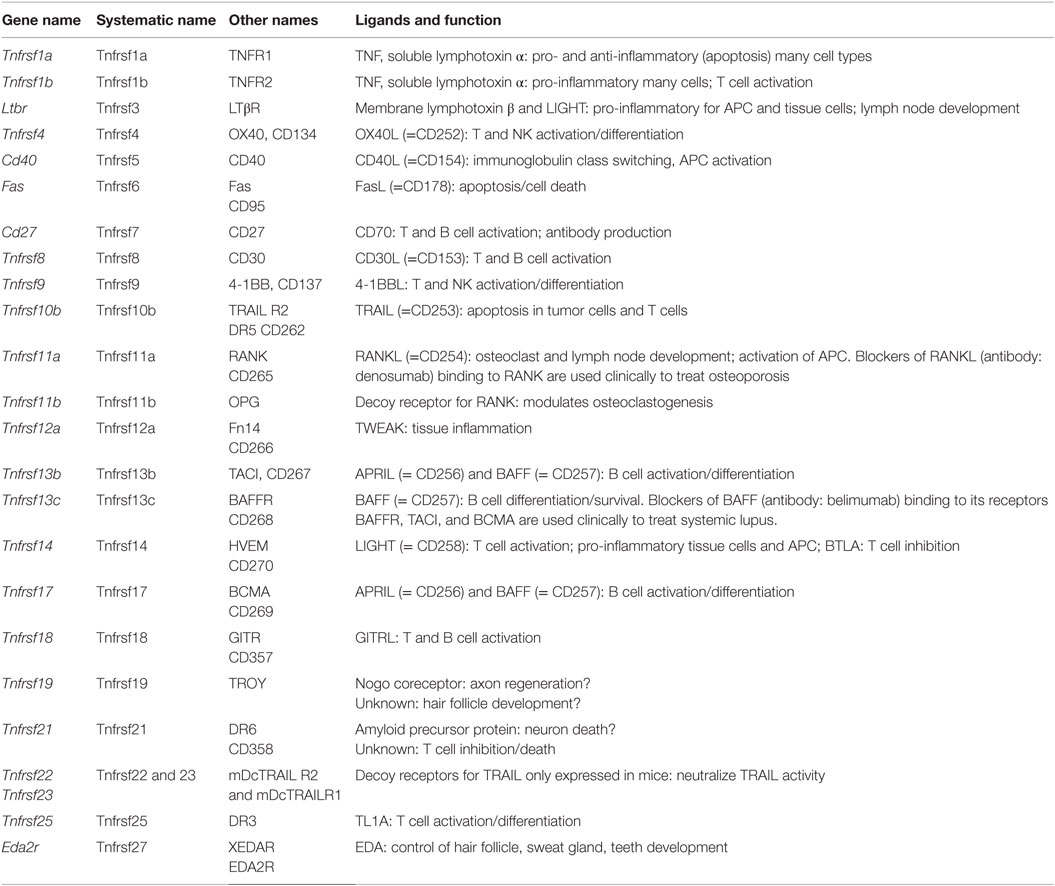
Table 8. TNF receptor superfamily.
Many of the Tnfrsf members are well known for controlling responses of T and B cells, dendritic cells, NK, and NKT cells, as well as inflammatory activity in structural cells, such as fibroblasts, epithelial cells, and osteoclasts. This has resulted in clinical targeting of many of the molecules for autoimmune and inflammatory diseases (89), and approved drugs in blockers of TNF and LTα binding to TNFRI and TNFRII for RA, psoriasis, Crohn’s disease, and others; blockers of RANKL binding to RANK for osteoporosis; and blockers of BAFF binding to its receptors BAFFR, TACI, and BCMA for SLE.
In macrophages, there appears to be strongly divergent expression among tissue macrophages (Figure 1I). Tnfrsf1a (TNFR1), Tnfrsf1b (TNFR2), Tnfrsf11a (RANK), and Tnfrsf3 (LTβR) are highly expressed by most tissue macrophages. Tnfrsf5 (CD40), Tnfrsf12a (Fn14), Tnfrsf14 (HVEM), Tnfrsf21 (DR6) are also highly expressed in some macrophage populations, but variably expressed in others at moderate levels. TNFR2 is largely pro-inflammatory, whereas TNFR1 can display both pro-inflammatory and anti-inflammatory (apoptosis-inducing) activities. Since TNF is a product of macrophages, various reports have suggested both positive and negative effects of TNF on macrophages correlating with these functions of its receptors (92–96). The outcome of TNFR signaling in macrophages may be heavily influenced by the location of the cells and the inflammatory environment in which they are responding. CD40, RANK, and LTβR are stimulatory receptors for antigen-presenting cells, particularly dendritic cells and B cells. CD40 is best known in this regard, but RANK and LTβR can display overlapping and synergistic activities. They have been less studied in macrophages, but parallel activities in promoting survival, differentiation, and inflammatory cytokine production, as well as upregulating co-stimulatory ligands that promote T cell activation, are consequences of engaging these receptors (97, 98). However, some studies of LTβR signaling on macrophages have suggested a regulatory activity in limiting TLR signaling (99). HVEM shares binding to the ligand LIGHT with LTβR. Signaling from either HVEM or LTβR can promote migration of macrophages (98, 100) and can induce inflammatory mediators, such as TNF, IL8, MMPs, and TGF-β (101, 102). Fn14 has been implicated in driving inflammation and remodeling activities in several tissues. TWEAK, the ligand for Fn14, can be made by macrophages (103) and TWEAK can also exert pro-inflammatory activity in macrophages via Fn14, promoting production of molecules, such as IL6, IL8, MCP-1, MMPs, and HMGB1, and driving macrophage migration into inflamed sites (104–107).
The receptors for BAFF and APRIL are best known as regulators of B cell survival or differentiation and also control T cell activity in some cases. Tnfrsf13c (BAFFR) that only binds BAFF was low in all macrophages, but Tnfrsf13b (TACI) and Tnfrsf17 (BCMA) that bind BAFF and APRIL were high in intestinal, adipose, and peritoneal macrophages and adipose macrophages and microglia, respectively. BAFF can be made by macrophages in soluble and membrane form, and membrane BAFF may (reverse) signal to promote inflammatory mediators (108), but the roles of the receptors are not well understood. Recent studies suggest that TACI or BCMA signals can also promote pro-inflammatory cytokines/mediators (109). TACI activity may also suppress differentiation of macrophages into the M2 phenotype, allowing enhanced clearance of intracellular organisms (110).
Tnfrsf9 (4-1BB), Tnfrsf25 (DR3), Tnfrsf4 (OX40), Tnfrsf18 (GITR), Tnfrsf8 (CD30), and Tnfrsf7 (CD27) showed low levels of transcript expression in most tissue macrophages, as did the decoy receptor for RANK (Tnfrsf11b – OPG). 4-1BB, DR3, OX40, GITR, CD30, and CD27 are best known as regulators of conventional T cells as well as Treg cells, NK, or NKT cells. Most have rarely been found on the surface of macrophages, although several studies have seen DR3 and GITR on macrophages/foam cells associated with atherosclerotic plaques, or in joints associated with RA (111–114). Signaling through these receptors, probably via NFkB, may aid production of pro-inflammatory molecules, such as TNF or MMPs. The ligands for 4-1BB, DR3, OX40, GITR, and CD27 can be expressed by many macrophages after they are activated, and can aid the antigen-presenting capacity of macrophages by providing co-stimulatory signals to their receptors on T cells. Certain TNF family ligands, like 4-1BBL, TL1A (ligand for DR3), and GITRL can also signal into macrophages or monocytes (reverse signaling) to modulate their survival, production of cytokines or molecules, such as PGE2, COX-2, and iNOS, and in some cases migration (115–119). In the case of 4-1BBL, it can also associate with TLRs on macrophages independently of binding its receptor and enhance TLR signaling (120, 121).
Tnfrsf27 (EDA2R) was low/absent in most macrophages, not surprising given its role outside the immune system in formation of tissues, such as sweat glands. Tnfrsf19 (TROY) is primarily expressed in the nervous system and has been suggested to regulate axon regeneration, but may also contribute to hair follicle formation. TROY was also low in most tissue macrophages and BMDMs. One prior report had suggested that it could be expressed in microglia in MS patients (122) but this has not been confirmed.
The death receptors Tnfrsf10b (TRAILR2) and Tnfrsf6 (Fas), and the mouse decoy receptors for TRAIL (Tnfrsf22 and 23 – mDcTRAILR1 and R2) showed low levels of expression in most macrophages, except for Fas in peritoneal macrophages and mDcTRAILR1 in BMDM, lung, liver, and peritoneal cells. Fas is likely to promote apoptosis of macrophages in some cases to limit autoimmunity or inflammation (123, 124), but it can be pro-inflammatory by augmenting caspase-dependent production of IL1 and IL18 or other activities (125–127). It is not known when production of the TRAIL decoy receptor by macrophages could be important for limiting TRAILR activity. Tnfrsf21 (DR6) is another death receptor that has been reported to regulate neuronal death and may also limit T and B cell activation, but whether it has other functions is not clear. It was highly expressed in microglia, peritoneal, spleen, and lung macrophages and BMDM (Figure 1I).
Fc Receptors
Fc receptors (Table 9) are receptors for immunoglobulins, which are products of plasma cells, and immune complexes. There are receptors for IgG (FcγRI, II, and III) (128, 129) and IgE (Fce receptor). They vary in their affinity (high to low) and in their downstream signaling effects (activating and inhibitory). Fc receptors are very highly expressed and are of key importance for clearing antibody-opsonized pathogens and antibody-opsonized necrotic cells. In the mouse, most Fc receptors signal through a common adaptor protein called Fc receptor γ chain or CD23.
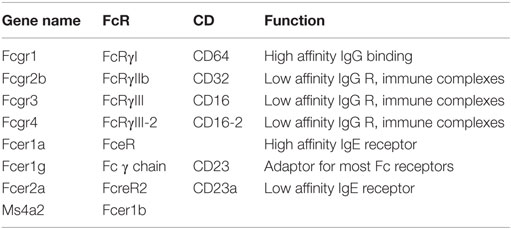
Table 9. Fc receptors.
The results in Figure 1J summarize the expression of Fc receptors in tissue macrophages. Fcgr1, the gene encoding the CD64 Fc γ receptor, is specific for macrophages and has been proposed as a universal mouse macrophage marker (40).
Chemokine Receptors and Other GPCRs
Macrophages and dendritic cells are important sources of many inflammatory chemokines including CCL1, 2, 3, 4, and 5. However, they also express chemokine receptors (Table 10) and, thus, respond to their chemokine environment. The major chemokine receptors on macrophages are CCR2, CCR5, and CX3CR1 (130, 131). CCR2 binds CCL2 and related chemokines of the MCP subfamily and is responsible for release of monocytes from the bone marrow and their trafficking to inflamed tissues. CCR1 and 2 are expressed in all tissue macrophages, but very low in microglia. CCR5 is a receptor for CCL5 and attracts monocytes to the vascular wall. It is expressed in all macrophages, highest in microglia. CX3CR1 is a receptor for CX3CL1 (fractalkine) and promotes macrophage survival. It is expressed in all macrophages, highest in microglia and intestinal macrophages.
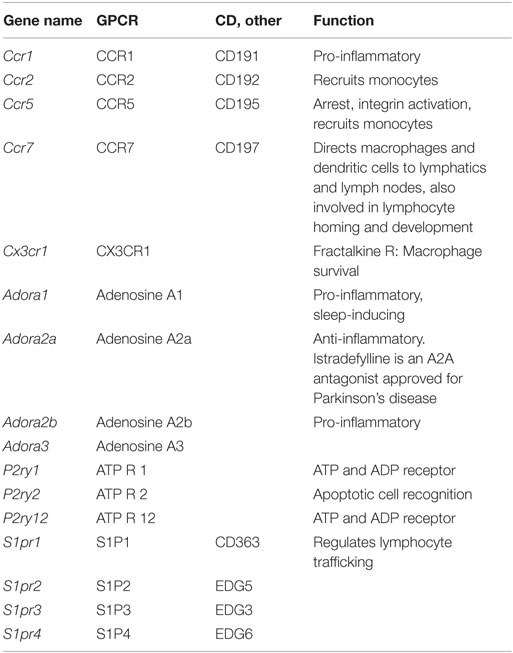
Table 10. GPCRs expressed on macrophages.
Other GPCRs
Other GPCRs (Table 10) include four adenosine receptors, Adora 1, 2a, 2b, and 3. Adenosine is a ubiquitous metabolite found in the extracellular space, and macrophages can respond to adenosine through these receptors. ATP is released by apoptotic cells through a pannexin-dependent mechanism, and ATP and ADP are important metabolites influencing macrophages through P2Y receptors. Sphingosine-1-phosphate is produced by platelets and other blood cells and is sensed by macrophages through S1P receptors S1P1-4 (Figure 1K).
The adenosine receptor A2A (Adora2a) is expressed at low levels in all tissue macrophages except microglia. This receptor downregulates inflammatory responses. A2B (Adora2b) is also expressed at low levels, but much higher in adipose tissue macrophages. This is considered a pro-inflammatory receptor.
P2Y1 (P2ry1) is an ADP receptor with low expression in all macrophages except microglia, where it does not appear to be expressed. By contrast, the ADP receptor P2Y12 (P2ry12) shows low expression in all macrophages but very high expression in microglia. P2Y12 on platelets is a target for anti-thrombotic treatment. P2Y2 is a receptor for UTP and ATP, which is released from apoptotic cells via pannexin (PANX1) (132).
The sphigosine-1-phosphate receptor encoded by S1pr1 is expressed by all tissue macrophages. S1pr2 is also broadly expressed, with highest levels in BMDM and bone marrow macrophages. S1pr3 and S1pr4 are expressed at low levels. Expression of these GPCRs is shown in Figure 1K.
Integrins
Macrophages use integrins (Table 11), αβ heterodimers that bind to extracellular matrix molecules (ECM) and other cells (133, 134). Since integrins, once bound to ligand, promote outside-in signaling, integrin engagement is an important input into macrophage biology. Almost all integrin subunits are expressed at some level in various tissue macrophages.
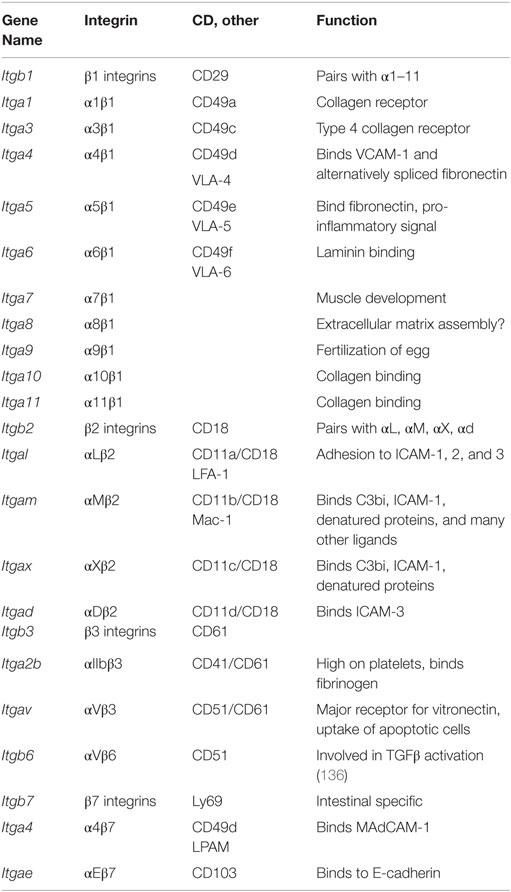
Table 11. Integrins.
Many integrin α subunits pair with β1, which is expressed in all macrophages (Itgb1). Itga1, Itga11, Itga2, Itga2b, Itga3, and Itga7 are expressed at low levels. Itga4 pairs with Itgb1 forming VLA-4, an integrin known to be involved in monocyte recruitment to tissues. Itga4 expression varies widely between tissue macrophages, with highest expression levels seen in large and small peritoneal macrophages. Itga5 pairs with Itgb1 to form a fibronectin receptor that is expressed at moderate levels in all tissue macrophages, highest in thioglycollate-elicited peritoneal macrophages. Itga6/Itgb1 encodes a laminin receptor that is highly expressed in peritoneal macrophages and microglia, but lower in liver, spleen, and adipose tissue macrophages. Itga8 is expressed in all tissue macrophages except peritoneal. Itga9 and 10 are expressed at intermediate to high levels in all macrophages.
The integrin subunits Itgal, Itgam, Itgax, and Itgad all pair with Itgb2 and are leukocyte specific. These β2 integrins are highly expressed in all macrophages, with highest mRNA levels in thioglycollate-elicited peritoneal macrophages. Itgal (LFA-1) is expressed at modest levels in most macrophages but not in microglia. Itgam (Mac-1), also known as complement receptor-3, is expressed at intermediate levels in most macrophages but very high in peritoneal and BMDM. Itgax (CD11c) is widely used as a dendritic cell marker, but this integrin is also expressed in all macrophages, most highly in thioglycollate-elicited peritoneal macrophages. Itgad (CD11d) is expressed at very low levels except for spleen macrophages, where it is very high.
Itgav pairs with Itgb3 to form the vitronectin receptor αVβ3, also known as leukocyte response integrin. Both subunits are expressed across all macrophages. Itgav can also pair with Itgb6 to form αVβ6 integrin, which is involved in TGF-β processing and fibrosis. Itgae and Itga4 pair with Itgb7, a receptor family generally found more in intestinal tissues. Indeed, Itgb7 expression is highest in peritoneal and intestinal macrophages. Itgae is highly specialized and binds to E-cadherin. Its expression is low except in intestinal macrophages, where it is very high (Figure 1H). The collagen receptor α2β1 integrin was not expressed in any of the macrophages studied here.
Concluding Remarks
This review provides a broad overview of receptors used by macrophages to sense their environment, including apoptotic and necrotic cells, pathogens, carbohydrates, modified self molecules, cytokines, chemokines, and other soluble molecules and extracellular matrix components. Some of these macrophage receptors are broadly expressed in all tissue macrophages studied, while others are highly restricted. This suggests that certain macrophages are well equipped for certain functions (listed in the tables) and others are “blind” to certain challenges or inputs. Recent reviews on each of the 12 classes of receptors are cited in the respective subsections for more detailed information. The remarkable heterogeneity in expression of macrophage receptors suggests highly specialized functions of resting tissue macrophages in lung, liver, spleen, intestine, fat tissue, the peritoneal cavity, and the brain (microglia).
This review is limited to mRNA expression data, and it is unknown how these expression patterns correlate with protein levels. In macrophages, the correlation between mRNA and protein levels is modest. Also, we consider the 12 receptor classes individually, but they may cooperate or even compete for ligand recognition, which would affect macrophage function. There could be more than one “danger” that affects a tissue, such as infection with multiple pathogens or viral infection and apoptotic cells. How tissue macrophages respond to such complex inputs remains to be studied. We did not analyze infiltrating monocyte-derived macrophages here, which can be a dominant population in inflammatory diseases, infections, autoimmune diseases, and cancer.
Author Contributions
KL wrote the paper. AP constructed the heat maps and input to the tables. MC wrote and edited the TNFSF part. KR edited the apoptotic cell uptake part. JT wrote and edited the NLR and RLR part.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by the National Institutes of Health.
Footnotes
References
1. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun (2014) 6(6):716–26. doi:10.1159/000364945
2. Mills CD, Ley K, Buchmann K, Canton J. Sequential immune responses: the weapons of immunity. J Innate Immun (2015) 7(5):443–9. doi:10.1159/000380910
3. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi:10.1016/j.immuni.2014.06.008
4. Mantovani A, Locati M. Orchestration of macrophage polarization. Blood (2009) 114(15):3135–6. doi:10.1182/blood-2009-07-231795
5. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi:10.4049/jimmunol.164.12.6166
6. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science (2013) 342(6161):1242974. doi:10.1126/science.1242974
7. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi:10.1126/science.1194637
8. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38(1):79–91. doi:10.1016/j.immuni.2012.12.001
9. Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol (2000) 18:593–620. doi:10.1146/annurev.immunol.18.1.593
10. Ley K, Kansas GS. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat Rev Immunol (2004) 4(5):325–35. doi:10.1038/nri1351
11. Ley K. The second touch hypothesis: T cell activation, homing and polarization. F1000Res (2014) 3:37. doi:10.12688/f1000research.3-37.v2
12. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med (1983) 158(3):670–89. doi:10.1084/jem.158.3.670
13. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med (1992) 176(1):287–92. doi:10.1084/jem.176.1.287
14. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32(5):593–604. doi:10.1016/j.immuni.2010.05.007
15. Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med (2007) 13(11):460–9. doi:10.1016/j.molmed.2007.09.002
16. Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev (2009) 228(1):225–40. doi:10.1111/j.1600-065X.2008.00755.x
17. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34(5):637–50. doi:10.1016/j.immuni.2011.05.006
18. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol (2005) 5(10):749–59. doi:10.1038/nri1703
19. Basak S, Hoffmann A. Crosstalk via the NF-kappaB signaling system. Cytokine Growth Factor Rev (2008) 19(3–4):187–97. doi:10.1016/j.cytogfr.2008.04.005
20. Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity (2013) 39(3):432–41. doi:10.1016/j.immuni.2013.08.037
21. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol (2005) 175(1):342–9. doi:10.4049/jimmunol.175.1.342
22. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature (2003) 423(6937):337–42. doi:10.1038/nature01658
23. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell (2014) 157(4):832–44. doi:10.1016/j.cell.2014.04.016
24. Gautier EL, Ivanov S, Williams JW, Huang SC, Marcelin G, Fairfax K, et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J Exp Med (2014) 211(8):1525–31. doi:10.1084/jem.20140570
25. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi:10.1038/nature13490
26. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475(7355):222–5. doi:10.1038/nature10138
27. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell (2014) 159(6):1327–40. doi:10.1016/j.cell.2014.11.023
28. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159(6):1312–26. doi:10.1016/j.cell.2014.11.018
29. Rosas M, Davies LC, Giles PJ, Liao CT, Kharfan B, Stone TC, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science (2014) 344(6184):645–8. doi:10.1126/science.1251414
30. Raghavachari N, Barb J, Yang Y, Liu P, Woodhouse K, Levy D, et al. A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC Med Genomics (2012) 5:28. doi:10.1186/1755-8794-5-28
31. Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One (2014) 9(1):e78644. doi:10.1371/journal.pone.0078644
32. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A (1998) 95(25):14863–8. doi:10.1073/pnas.95.25.14863
33. Saldanha AJ. Java Treeview – extensible visualization of microarray data. Bioinformatics (2004) 20(17):3246–8. doi:10.1093/bioinformatics/bth349
34. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe (2012) 12(3):289–300. doi:10.1016/j.chom.2012.06.010
35. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol (2011) 13(3):184–90. doi:10.1038/ncb0311-184
36. deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem (2003) 39:105–17. doi:10.1042/bse0390105
37. Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta (2015) 1851(4):414–21. doi:10.1016/j.bbalip.2014.07.008
38. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14(3):166–80. doi:10.1038/nri3607
39. Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol (2007) 7(12):964–74. doi:10.1038/nri2214
40. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13(11):1118–28. doi:10.1038/ni.2419
41. Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis (2014) 73(3):483–91. doi:10.1136/annrheumdis-2013-203844
42. Miller YI, Chang MK, Binder CJ, Shaw PX, Witztum JL. Oxidized low density lipoprotein and innate immune receptors. Curr Opin Lipidol (2003) 14(5):437–45. doi:10.1097/01.mol.0000092631.86399.49
43. Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell (2001) 106(6):675–83. doi:10.1016/S0092-8674(01)00495-0
44. Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity. Immunol Rev (2007) 219:157–66. doi:10.1111/j.1600-065X.2007.00556.x
45. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi:10.1038/ni.1923
46. Cohen HB, Mosser DM. Extrinsic and intrinsic control of macrophage inflammatory responses. J Leukoc Biol (2013) 94(5):913–9. doi:10.1189/jlb.0413236
47. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi:10.1146/annurev-immunol-031210-101405
48. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity (2013) 38(5):855–69. doi:10.1016/j.immuni.2013.05.007
49. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity (2013) 38(5):870–80. doi:10.1016/j.immuni.2013.05.004
50. Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell (2013) 51(2):135–9. doi:10.1016/j.molcel.2013.07.004
51. Sancho D, Reis e Sousa C. Sensing of cell death by myeloid C-type lectin receptors. Curr Opin Immunol (2013) 25(1):46–52. doi:10.1016/j.coi.2012.12.007
52. Sancho D, Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol (2012) 30:491–529. doi:10.1146/annurev-immunol-031210-101352
53. Prabhudas M, Bowdish D, Drickamer K, Febbraio M, Herz J, Kobzik L, et al. Standardizing scavenger receptor nomenclature. J Immunol (2014) 192(5):1997–2006. doi:10.4049/jimmunol.1490003
54. O’Neill LA, Golenbock D, Bowie AG. The history of toll-like receptors – redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi:10.1038/nri3446
55. Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol (2012) 189(11):5113–7. doi:10.4049/jimmunol.1202479
56. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23(2):201–12. doi:10.1038/cr.2013.6
57. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492(7427):123–7. doi:10.1038/nature11588
58. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell (2015) 160(1–2):62–73. doi:10.1016/j.cell.2014.11.047
59. Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med (2014) 211(7):1333–47. doi:10.1084/jem.20132486
60. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi:10.1038/nm.3893
61. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi:10.1038/nature10394
62. Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe (2014) 16(2):237–48. doi:10.1016/j.chom.2014.07.001
63. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi:10.1016/S1097-2765(02)00599-3
64. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog (2012) 8(3):e1002638. doi:10.1371/journal.ppat.1002638
65. Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun (2011) 79(2):756–66. doi:10.1128/IAI.00898-10
66. Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun (2014) 82(1):460–8. doi:10.1128/IAI.01170-13
67. Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio (2014) 5(1):1–12. doi:10.1128/mBio.01117-13
68. Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186(12):7187–94. doi:10.4049/jimmunol.1100412
69. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi:10.1016/j.cell.2011.04.022
70. Slim R, Wallace EP. NLRP7 and the genetics of hydatidiform moles: recent advances and new challenges. Front Immunol (2013) 4:242. doi:10.3389/fimmu.2013.00242
71. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity (2012) 36(5):742–54. doi:10.1016/j.immuni.2012.03.012
72. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell (2011) 20(5):649–60. doi:10.1016/j.ccr.2011.10.022
73. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37(1):96–107. doi:10.1016/j.immuni.2012.07.006
74. Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, et al. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog (2014) 10(1):e1003885. doi:10.1371/journal.ppat.1003885
75. Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol (2011) 186(3):1333–7. doi:10.4049/jimmunol.1003111
76. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res (2012) 22(5):836–47. doi:10.1038/cr.2012.56
77. Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol (2012) 12(12):813–20. doi:10.1038/nri3339
78. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi:10.1038/nature10558
79. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity (2006) 24(1):93–103. doi:10.1016/j.immuni.2005.12.003
81. Pyz E, Marshall AS, Gordon S, Brown GD. C-type lectin-like receptors on myeloid cells. Ann Med (2006) 38(4):242–51. doi:10.1080/07853890600608985
82. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood (2011) 118(26):6743–51. doi:10.1182/blood-2011-07-343566
83. Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K. L-selectin shedding regulates leukocyte recruitment. J Exp Med (2001) 193(7):863–72. doi:10.1084/jem.193.7.863
84. Braley A, Kwak T, Jules J, Harja E, Landgraf R, Hudson BI. Regulation of RAGE ectodomain shedding and its role in cell function. J Biol Chem (2016). doi:10.1074/jbc.M115.702399
85. Chhibber-Goel J, Coleman-Vaughan C, Agrawal V, Sawhney N, Hickey E, Powell JC, et al. gamma-secretase activity is required for regulated intramembrane proteolysis of tumor necrosis factor (TNF) receptor 1 and TNF-mediated pro-apoptotic signaling. J Biol Chem (2016) 291(11):5971–85. doi:10.1074/jbc.M115.679076
86. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science (2011) 332(6035):1284–8. doi:10.1126/science.1204351
87. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med (2013) 210(11):2477–91. doi:10.1084/jem.20121999
88. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9(4):271–85. doi:10.1038/nri2526
89. Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov (2013) 12(2):147–68. doi:10.1038/nrd3930
90. Lee SW, Park Y, Eun SY, Madireddi S, Cheroutre H, Croft M. Cutting edge: 4-1BB controls regulatory activity in dendritic cells through promoting optimal expression of retinal dehydrogenase. J Immunol (2012) 189(6):2697–701. doi:10.4049/jimmunol.1201248
91. Sedy J, Bekiaris V, Ware CF. Tumor necrosis factor superfamily in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2015) 7(4):a016279. doi:10.1101/cshperspect.a016279
92. Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr (2010) 20(2):87–103. doi:10.1615/CritRevEukarGeneExpr.v20.i2.10
93. Flesch IE, Hess JH, Huang S, Aguet M, Rothe J, Bluethmann H, et al. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon gamma and tumor necrosis factor alpha. J Exp Med (1995) 181(5):1615–21. doi:10.1084/jem.181.5.1615
94. Takada Y, Sung B, Sethi G, Chaturvedi MM, Aggarwal BB. Evidence that genetic deletion of the TNF receptor p60 or p80 inhibits Fas mediated apoptosis in macrophages. Biochem Pharmacol (2007) 74(7):1057–64. doi:10.1016/j.bcp.2007.07.005
95. Crisafulli C, Galuppo M, Cuzzocrea S. Effects of genetic and pharmacological inhibition of TNF-alpha in the regulation of inflammation in macrophages. Pharmacol Res (2009) 60(4):332–40. doi:10.1016/j.phrs.2009.05.001
96. Edgel KA, Leboeuf RC, Oram JF. Tumor necrosis factor-alpha and lymphotoxin-alpha increase macrophage ABCA1 by gene expression and protein stabilization via different receptors. Atherosclerosis (2010) 209(2):387–92. doi:10.1016/j.atherosclerosis.2009.10.019
97. Jang SW, Lim SG, Suk K, Lee WH. Activation of lymphotoxin-beta receptor enhances the LPS-induced expression of IL-8 through NF-kappaB and IRF-1. Immunol Lett (2015) 165(2):63–9. doi:10.1016/j.imlet.2015.04.001
98. Grandoch M, Feldmann K, Gothert JR, Dick LS, Homann S, Klatt C, et al. Deficiency in lymphotoxin beta receptor protects from atherosclerosis in apoE-deficient mice. Circ Res (2015) 116(8):e57–68. doi:10.1161/CIRCRESAHA.116.305723
99. Wimmer N, Huber B, Barabas N, Rohrl J, Pfeffer K, Hehlgans T. Lymphotoxin beta receptor activation on macrophages induces cross-tolerance to TLR4 and TLR9 ligands. J Immunol (2012) 188(7):3426–33. doi:10.4049/jimmunol.1103324
100. Wei W, Popov V, Walocha JA, Wen J, Bello-Reuss E. Evidence of angiogenesis and microvascular regression in autosomal-dominant polycystic kidney disease kidneys: a corrosion cast study. Kidney Int (2006) 70(7):1261–8. doi:10.1038/sj.ki.5001725
101. Lee WH, Kim SH, Lee Y, Lee BB, Kwon B, Song H, et al. Tumor necrosis factor receptor superfamily 14 is involved in atherogenesis by inducing proinflammatory cytokines and matrix metalloproteinases. Arterioscler Thromb Vasc Biol (2001) 21(12):2004–10. doi:10.1161/hq1201.098945
102. Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, et al. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med (2011) 17(5):596–603. doi:10.1038/nm.2356
103. Serafini B, Magliozzi R, Rosicarelli B, Reynolds R, Zheng TS, Aloisi F. Expression of TWEAK and its receptor Fn14 in the multiple sclerosis brain: implications for inflammatory tissue injury. J Neuropathol Exp Neurol (2008) 67(12):1137–48. doi:10.1097/NEN.0b013e31818dab90
104. Kim SH, Kang YJ, Kim WJ, Woo DK, Lee Y, Kim DI, et al. TWEAK can induce pro-inflammatory cytokines and matrix metalloproteinase-9 in macrophages. Circ J (2004) 68(4):396–9. doi:10.1253/circj.68.396
105. Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol (2007) 179(11):7949–58. doi:10.4049/jimmunol.179.11.7949
106. Munoz-Garcia B, Moreno JA, Lopez-Franco O, Sanz AB, Martin-Ventura JL, Blanco J, et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler Thromb Vasc Biol (2009) 29(12):2061–8. doi:10.1161/ATVBAHA.109.194852
107. Moreno JA, Sastre C, Madrigal-Matute J, Munoz-Garcia B, Ortega L, Burkly LC, et al. HMGB1 expression and secretion are increased via TWEAK-Fn14 interaction in atherosclerotic plaques and cultured monocytes. Arterioscler Thromb Vasc Biol (2013) 33(3):612–20. doi:10.1161/ATVBAHA.112.300874
108. Jeon HJ, Choi JH, Jung IH, Park JG, Lee MR, Lee MN, et al. CD137 (4-1BB) deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation (2010) 121(9):1124–33. doi:10.1161/CIRCULATIONAHA.109.882704
109. Lee CH, Bose S, Van Vliet KJ, Karp JM, Karnik R. Studying cell rolling trajectories on asymmetric receptor patterns. J Vis Exp (2011) (48):1–2. doi:10.3791/2640
110. Allman WR, Dey R, Liu L, Siddiqui S, Coleman AS, Bhattacharya P, et al. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc Natl Acad Sci U S A (2015) 112(30):E4094–103. doi:10.1073/pnas.1421580112
111. Kang YJ, Kim WJ, Bae HU, Kim DI, Park YB, Park JE, et al. Involvement of TL1A and DR3 in induction of pro-inflammatory cytokines and matrix metalloproteinase-9 in atherogenesis. Cytokine (2005) 29(5):229–35. doi:10.1016/j.cyto.2004.12.001
112. McLaren JE, Calder CJ, McSharry BP, Sexton K, Salter RC, Singh NN, et al. The TNF-like protein 1A-death receptor 3 pathway promotes macrophage foam cell formation in vitro. J Immunol (2010) 184(10):5827–34. doi:10.4049/jimmunol.0903782
113. Kim WJ, Bae EM, Kang YJ, Bae HU, Hong SH, Lee JY, et al. Glucocorticoid-induced tumour necrosis factor receptor family related protein (GITR) mediates inflammatory activation of macrophages that can destabilize atherosclerotic plaques. Immunology (2006) 119(3):421–9. doi:10.1111/j.1365-2567.2006.02453.x
114. Bae E, Kim WJ, Kang YM, Suk K, Koh EM, Cha HS, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein-mediated macrophage stimulation may induce cellular adhesion and cytokine expression in rheumatoid arthritis. Clin Exp Immunol (2007) 148(3):410–8. doi:10.1111/j.1365-2249.2007.03363.x
115. Langstein J, Schwarz H. Identification of CD137 as a potent monocyte survival factor. J Leukoc Biol (1999) 65(6):829–33.
116. Shao Z, Schwarz H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J Leukoc Biol (2011) 89(1):21–9. doi:10.1189/jlb.0510315
117. Shin HH, Lee MH, Kim SG, Lee YH, Kwon BS, Choi HS. Recombinant glucocorticoid induced tumor necrosis factor receptor (rGITR) induces NOS in murine macrophage. FEBS Lett (2002) 514(2–3):275–80. doi:10.1016/S0014-5793(02)02379-7
118. Bae EM, Kim WJ, Suk K, Kang YM, Park JE, Kim WY, et al. Reverse signaling initiated from GITRL induces NF-kappaB activation through ERK in the inflammatory activation of macrophages. Mol Immunol (2008) 45(2):523–33. doi:10.1016/j.molimm.2007.05.013
119. Liao G, van Driel B, Magelky E, O’Keeffe MS, de Waal Malefyt R, Engel P, et al. Glucocorticoid-induced TNF receptor family-related protein ligand regulates the migration of monocytes to the inflamed intestine. FASEB J (2014) 28(1):474–84. doi:10.1096/fj.13-236505
120. Kang YJ, Kim SO, Shimada S, Otsuka M, Seit-Nebi A, Kwon BS, et al. Cell surface 4-1BBL mediates sequential signaling pathways ‘downstream’ of TLR and is required for sustained TNF production in macrophages. Nat Immunol (2007) 8(6):601–9. doi:10.1038/ni1471
121. Ma J, Bang BR, Lu J, Eun SY, Otsuka M, Croft M, et al. The TNF family member 4-1BBL sustains inflammation by interacting with TLR signaling components during late-phase activation. Sci Signal (2013) 6(295):ra87. doi:10.1126/scisignal.2004431
122. Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H. TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol Appl Neurobiol (2007) 33(1):99–107. doi:10.1111/j.1365-2990.2006.00787.x
123. Cuda CM, Agrawal H, Misharin AV, Haines GK III, Hutcheson J, Weber E, et al. Requirement of myeloid cell-specific Fas expression for prevention of systemic autoimmunity in mice. Arthritis Rheum (2012) 64(3):808–20. doi:10.1002/art.34317
124. Jagger AL, Evans HG, Walter GJ, Gullick NJ, Menon B, Ballantine LE, et al. FAS/FAS-L dependent killing of activated human monocytes and macrophages by CD4+CD25- responder T cells, but not CD4+CD25+ regulatory T cells. J Autoimmun (2012) 38(1):29–38. doi:10.1016/j.jaut.2011.11.015
125. Zhao H, Roychoudhury J, Doggett TA, Apte RS, Ferguson TA. Age-dependent changes in FasL (CD95L) modulate macrophage function in a model of age-related macular degeneration. Invest Ophthalmol Vis Sci (2013) 54(8):5321–31. doi:10.1167/iovs.13-12122
126. Uchiyama R, Yonehara S, Tsutsui H. Fas-mediated inflammatory response in Listeria monocytogenes infection. J Immunol (2013) 190(8):4245–54. doi:10.4049/jimmunol.1203059
127. Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol (2012) 189(12):5508–12. doi:10.4049/jimmunol.1202121
128. Daeron M. Fc receptors as adaptive immunoreceptors. Curr Top Microbiol Immunol (2014) 382:131–64. doi:10.1007/978-3-319-07911-0_7
129. Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov (2012) 11(4):311–31. doi:10.1038/nrd2909
130. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med (2015) 212(4):447–56. doi:10.1084/jem.20141539
131. Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol (2011) 31(7):1506–16. doi:10.1161/ATVBAHA.110.221127
132. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature (2010) 467(7317):863–7. doi:10.1038/nature09413
133. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110(6):673–87. doi:10.1016/S0092-8674(02)00971-6
135. Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol (2015) 34:22–7. doi:10.1016/j.coi.2014.12.009
Keywords: macrophages, pathogens, immunity, defense, inflammation
Citation: Ley K, Pramod AB, Croft M, Ravichandran KS and Ting JP (2016) How Mouse Macrophages Sense What Is Going On. Front. Immunol. 7:204. doi: 10.3389/fimmu.2016.00204
Received: 16 March 2016; Accepted: 10 May 2016;
Published: 02 June 2016
Edited by:
Janos G. Filep, University of Montreal, CanadaReviewed by:
Amiram Ariel, University of Haifa, IsraelJoan Clària, Hospital Clinic-Barcelona University School of Medicine, Spain
Ian Dransfield, University of Edinburgh, UK
Marco A. Cassatella, University of Verona, Italy
Copyright: © 2016 Ley, Pramod, Croft, Ravichandran and Ting. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus Ley, a2xhdXNAbGppLm9yZw==