 Cesar M. Rueda
Cesar M. Rueda Courtney M. Jackson
Courtney M. Jackson Claire A. Chougnet
Claire A. Chougnet- Division of Immunobiology, Department of Pediatrics, Cincinnati Children’s Hospital Research Foundation, University of Cincinnati College of Medicine, Cincinnati, OH, USA
Regulatory T-cells (Tregs) mediate their suppressive action by acting directly on conventional T-cells (Tcons) or dendritic cells (DCs). One mechanism of Treg suppression is the increase of cyclic adenosine 3′,5′-monophosphate (cAMP) levels in target cells. Tregs utilize cAMP to control Tcon responses, such as proliferation and cytokine production. Tregs also exert their suppression on DCs, diminishing DC immunogenicity by downmodulating the expression of costimulatory molecules and actin polymerization at the immunological synapse. The Treg-mediated usage of cAMP occurs through two major mechanisms. The first involves the Treg-mediated influx of cAMP in target cells through gap junctions. The second is the conversion of adenosine triphosphate into adenosine by the ectonucleases CD39 and CD73 present on the surface of Tregs. Adenosine then binds to receptors on the surface of target cells, leading to increased intracellular cAMP levels in these targets. Downstream, cAMP can activate the canonical protein kinase A (PKA) pathway and the exchange protein activated by cyclic AMP (EPAC) non-canonical pathway. In this review, we discuss the most recent findings related to cAMP activation of PKA and EPAC, which are implicated in Treg homeostasis as well as the functional alterations induced by cAMP in cellular targets of Treg suppression.
Introduction
Regulatory T-cells (Tregs), first described by Sakaguchi et al. in 1995 (1), are essential to maintain immune homeostasis and protection against autoimmunity. This CD4+ T-cell subset highly expresses IL-2 receptor alpha chain (CD25) and Forkhead box P3 (FOXP3), the central transcription factor for Treg development and function. Defects in the FOXP3 gene in both mice and humans lead to a fatal lymphoproliferative and autoimmune disease (2, 3).
Regulatory T-cells control immune activation by acting directly on conventional CD4+ and CD8+ conventional T cells (Tcons) and antigen-presenting cells, such as dendritic cells (DCs). Tregs preferentially localize to DC aggregates, subsequently preventing Tcon activation in vivo and in vitro (4, 5), suggesting DCs are the primary targets of Treg suppression (6, 7). Cyclic adenosine 3′,5′-monophosphate (cAMP) was recognized in 2007 as being essential to Treg suppression (8). cAMP is a common intracellular second messenger found in various cell types, which was discovered in the year 1957 (9). It is generated after the initial binding of hormones, neurotransmitters, and other ligands to cell-surface receptors (10). cAMP activates the canonical protein kinase A (PKA) pathway and the exchange protein activated by cyclic AMP (EPAC) non-canonical pathway (11, 12). In this review, we will discuss how cAMP regulates Tcon and DC function, as well as describing downstream PKA and EPAC intracellular pathways within Tregs, Tcons, and DCs.
Elevated cAMP Concentration in Tregs is Determined by Adenylyl Cyclase and Phosphodiesterase Expression
Intracellular cAMP levels are regulated by adenylyl cyclases (ACs) that catalyze the formation of cAMP and phosphodiesterases (PDEs), which hydrolyze cAMP to 5′-AMP. Overall, there are 11 PDEs and 10 AC families. ACs 3, 6, 7, and 9 are expressed in murine T cells (13, 14). PDEs 3, 4, 7, and 8 are expressed in human T-cells, with PDE4 being the most abundant (15–17). Importantly, the differential expression and activation of ACs and PDEs in Tregs and Tcons explain the high level of intracellular cAMP in murine and human Tregs compared to Tcons (8, 18, 19).
Similar to its expression in murine Tregs, AC7 is expressed in resting and activated human Tregs (20). Activation of AC7 downstream of IL-2 signaling plays an important role in promoting high cAMP levels in resting Tregs (18). However, since CD25 expression is upregulated in Tcons following activation, preferential IL-2-mediated AC7 activation is not sufficient to explain the increased cAMP levels present in activated Tregs compared to activated Tcons. Elevated expression of AC9 has also been shown to be important for cAMP accumulation in murine Tregs (13) (Figure 1A), which is regulated in part by microRNA miR-142-3p targeting of AC9 mRNA expression. Although FOXP3 downregulates miR-142-3p to keep the AC9/cAMP pathway active in Tregs (13), miR-142-3p is elevated in other CD4+ subsets, keeping AC9 inactive and thus cAMP levels low. Additionally, an isoform of PDE (PDE3b) is one of the most FOXP3-repressed genes in murine Treg (21), resulting in low cAMP degradation and subsequent elevation of cAMP levels in Tregs (Figure 1A). Further demonstrating the involvement of FOXP3 in cAMP regulation, T cells programed to be Tregs, but that did not express functional FOXP3 protein due to a frame-shift mutation, had substantially lower intracellular cAMP levels than FOXP3-expressing Tregs (22). However, we recently reported that neonatal human Tregs have lower expression of FOXP3, but higher intracellular cAMP levels compared to adult Tregs, suggesting that cAMP levels may also be regulated in a FOXP3-independent manner (23). Several mechanisms may explain this profile exhibited by human neonatal Tregs, and neonatal plasma contains high adenosine concentrations due to a low degradation rate (24, 25). In addition, the adenosine receptors in neonatal mononuclear cells seem to be more sensitive than those in adults, leading to higher intracellular cAMP (24, 25).
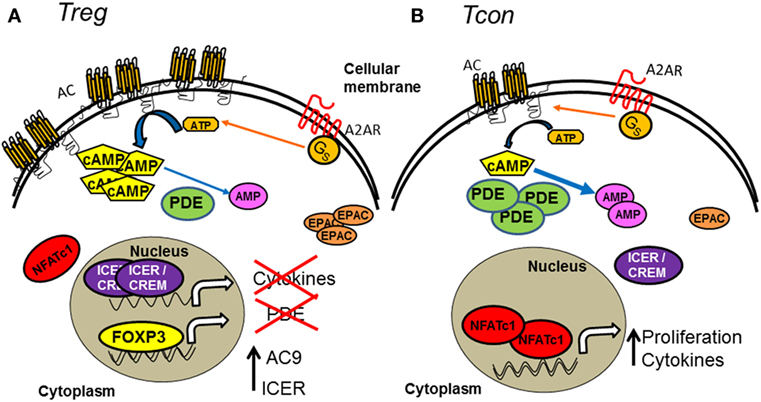
Figure 1. Comparison of cAMP metabolism and intracellular signaling pathway in Treg and Tcon subsets. (A) Tregs contain a high concentration of cAMP compared to Tcons as a consequence of their high cAMP anabolism. Tregs express mainly AC compared to Tcons, and AC catalyzes the conversion of ATP into cyclic adenosine monophosphate (cAMP). In addition, G protein-coupled receptors, such as A2A, are able to activate AC. In contrast to Tcons, Tregs exhibit low cAMP catabolism due to a low expression of PDEs, which decompose cAMP into AMP. The presence of FOXP3 in Tregs, but not in Tcons, suppresses PDE transcription, while it favors ICER and AC expression. The localization and expression of molecules, such as ICER/CREM (high expression and nuclear), NFAT (low expression and cytoplasmic) and EPAC (high expression), are associated with the maintenance of Treg phenotype and function. (B) Tcons contain low levels of cAMP due to their reduced AC but high PDE expression. In contrast to Treg, NFAT is active in the nucleus of Tcons. The low and cytoplasmic expression of ICER/CREM and EPAC are also associated with cell cycle progression and active cytokine secretion in Tcons.
Tregs Increase cAMP Levels in Target Cells through cAMP Influx and Adenosine Production
The ability of Tregs to generate and accumulate high levels of cAMP gives them the capacity to transfer it through gap junctions (GJ) into target cells (8, 19, 26, 27). These channels allow intercellular exchange of ions, metabolites, and other molecules between adjacent cells (28). GJ are formed by two opposing hemichannels from each cell, called connexons, which are made of six proteins called connexins (Cx) (28). Tregs and Tcons both express Cx31.1, Cx32, Cx43, Cx45, and Cx46, and their expression increases after activation (8).
An additional mechanism to increase cAMP in the target cell involves the ecto-5′-nucleotidases CD39 and CD73 expressed on the surface of Tregs, which cleave extracellular adenosine triphosphate (ATP) into adenosine (29, 30). The binding of adenosine to its G protein-coupled receptors (GPCRs) on target cells leads to the stimulatory G protein alpha subunit (Gsα) directly activating ACs and generating cAMP (31–33).
Extracellular ATP, which is a hallmark of inflammation (34, 35), is first degraded to AMP by CD39, which CD73 converts into adenosine (36). CD39 and CD73 are coexpressed on the surface of murine Tregs (29, 37). In contrast to murine Tregs, very few (<5%) human CD39+ Tregs appear to express CD73 on their surface (38–41). However, there is high intracellular expression of CD73 in human Tregs, which seems to be readily shed from their surface, potentially explaining the low surface expression of CD73 on human Tregs (39). Furthermore, human Tregs produce exosomes that carry CD39 and CD73 and are able to hydrolyze ATP (42–44).
Adenosine binds to several receptors (A1, A2A, A2B, and A3) that are expressed on various cells, including T cells and APCs. High affinity A2A appears to be the main receptor involved in mediating adenosine-dependent Treg suppressive function (39). Binding of adenosine to A2A on target cells, such as T cells and APCs, activates AC, leading to cAMP accumulation (33, 45). The A2A receptor is also expressed by resting and activated Tregs (30) and treatment for Tregs by adenosine analogs increased their cAMP levels (46) (Figure 1A). A2A stimulation not only expanded FOXP3+ Treg cells but also increased their suppressive function (46, 47). The fact that Tregs produce and respond to adenosine thus suggests that adenosine might act as an autocrine factor to optimize Treg anti-inflammatory function, as proposed by Ernst et al. (48).
The Role of cAMP in Treg Control of Tcon Proliferation and Cytokine Production
Pharmacological inhibition of PDE3 and PDE4 increases cAMP levels in Tregs and leads to their enhanced suppression of Tcons, both in vivo and in vitro (49, 50). Conversely, treatment for human Tregs with interferon-α before activation decreased intracellular cAMP through PDE4 activation, which led to a loss of Treg suppression (51). Tregs were shown to inhibit in vitro activation of Tcons by using the mechanisms, as described previously, e.g., by transferring cAMP through GJ (8, 30) and via CD39-mediated generation of adenosine. We have shown that Tregs limit in vitro HIV infection in Tcons using these same two mechanisms (40).
The relevance of CD39 expression as a mechanism of suppression was also shown in Tregs from CD39-null mice, which failed to suppress CD4+CD25− cell proliferation (30). In addition, in a murine melanoma model, the adenosine generated by the increased frequency of CD39+ Tregs was associated with the suppression of T-cell effector functions (52). Similarly, human CD39+ Tregs suppressed IL-2 and IL-17 expression and proliferation of activated Tcons more efficiently than their CD39− counterparts (53–55).
The Role of cAMP in Treg Control of DC Function
Regulatory T-cells also use cAMP to downregulate the expression of several costimulatory molecules on DCs, such as CD40, CD80, CD86, and CD83 (23, 26, 27, 29, 56–58), while upregulating the expression of several inhibitory molecules (B7-H, B7-H3, and B7-DC) (26, 29, 56). In contrast, cAMP did not modify cytokine production by DCs (26). Again, both mechanisms (cAMP influx and adenosine) appear to be active in this suppression (29, 56, 58).
Tregs Control DC–Tcon Interactions through cAMP-Dependent Mechanisms
The duration of contact between Tcons and antigen-loaded DCs are shortened in the presence of Tregs (7), indicating an early effect of Tregs on the induction of immune responses. Additionally, Tregs are more mobile than Tcons in vitro and out-compete the latter in aggregating around DCs (5). Tregs were also found to form long-lasting conjugates with islet antigen-bearing DCs, which lost the capacity to effectively present antigens (4). Confirming the idea that DCs are the primary targets of Treg suppression, the amount of cAMP transferred from Tregs to DCs was significantly higher than that transferred from Tregs to Tcons in in vitro cocultures with DC–Tcon–Treg (56). In concordance with this, we have recently shown that cAMP, together with CTLA-4 and TGF-β, are essential for adult Tregs to suppress the formation of DC–Tcon aggregates (23). We have also shown that the influx of cAMP by adult Tregs suppresses actin polymerization at the interface of DCs and Tcons (23, 57). Importantly, due to the role of the immunological synapse in the transmission of HIV particles from DCs to Tcons, we showed that Tregs could blunt this transmission by cAMP-dependent mechanisms (57).
Interestingly, Ring et al. showed that Tregs specifically directed the migration of DCs toward them (58) and that adenosine played a major role in this phenomenon because CD39− Tregs were unable to attract DCs (58). These data are consistent with the fact that cAMP could stimulate DC chemotaxis by increasing the expression of the lymph node-homing chemokines CCL19 and CCL21 (59). Taken together, these data suggest that Tregs use cAMP at multiple levels to prevent Tcon activation by DCs, they not only keep the DCs in an immature state but they also attract them away from Tcons.
Treg-Mediated Suppression of Tcons and DCs by Different Intracellular Pathways Downstream of cAMP
PKA and EPAC Intracellular Signaling Pathways
Once inside the cell, cAMP triggers various downstream pathways, mainly the canonical PKA and the non-canonical EPAC pathway. PKA contains an evolutionarily conserved cAMP-binding domain (CBD) that acts as a sensor of intracellular cAMP levels (11). cAMP binding to CBD on the PKA regulatory subunit induces its activation and releases the catalytic subunit. Downstream of PKA dissociation, several signaling pathways are regulated (activated or inactivated) by the catalytic subunit of PKA through phosphorylation. For example, cAMP response element binding protein (CREB) is phosphorylated at serine133 by PKA, leading to the complex formation of CREB with CSK (CBP) (60, 61). This complex binds to cAMP responsive elements in the promoter regions of genes such as cAMP response element modulator (CREM). CREM regulates multiple transcriptional activators or inhibitors in T cells. Increased cellular cAMP levels enhance the expression of an isoform of CREM, named the inducible cAMP early repressor (ICER). cAMP promotes ICER translocation from the cytoplasm to the nucleus (62). This translocation is crucial for ICER action because ICER binds activator protein-1 (AP-1) and nuclear factor of activated T-cells (NFAT) in the nucleus. These nuclear interactions between ICER and NFAT/AP-1 mediate ICER transcriptional regulation of many genes involved either in cell cycle control or cytokine secretion (63, 64).
Cyclic adenosine 3′,5′-monophosphate can also activate the non-canonical EPAC pathway. EPAC proteins (EPAC1 and EPAC2) contain a CBD that is homologous to the one contained within PKA. cAMP binding to EPAC proteins activates the Ras superfamily small GTPases RAP-1 and RAP-2 by promoting the exchange of GDP for GTP (11). Depending on the cell type, EPAC and PKA may act independently or synergistically or oppose each other in regulating specific cellular functions (11).
Activation of cAMP–PKA and cAMP–EPAC Control the Phenotype and Suppressive Activity of Tregs
Several authors have shown that the cAMP–PKA pathway in Tregs differs from that in Tcons. Although the expression of PKA is similar in Tregs and Tcons (12), other molecules, such as ICER/CREM, are markedly more expressed in Tregs than in Tcons (65). In addition, ICER/CREM is mainly localized in the nucleus in Tregs (Figure 1A) (65). This particular phenotype in Tregs may be explained by the presence of FOXP3, as forced expression of FOXP3 in murine Tcons induced constitutive expression of ICER/CREM (66). cAMP signaling and the presence of ICER in the nucleus seems to be important to blunt Treg cytokine production (19). However, deletion of ICER did not alter the number or function of murine Tregs (12). In contrast to ICER/CREM, NFATc1 is primarily localized in the cytoplasm of Tregs (65, 67, 68) (Figure 1A). Decreasing cAMP increased induction and nuclear translocation of NFATc1 in human Tregs, leading to increased Treg proliferation and blunted suppression (19). cAMP/PKA signaling in Tregs also modulates the expression of other functionally important molecules, such as CTLA-4 (69). Furthermore, PKA/CREB activation promotes the TGFβ-mediated generation of Tregs from naive Tcons and contributes to the maintenance of FOXP3 expression in these induced Tregs both in vitro and in vivo (69–71).
Murine Tregs express 10-fold more EPAC1 than naive and activated Tcons (12) (Figure 1A). In contrast to ICER, EPAC1 is critical for Treg-mediated suppression as genetic deletion of EPAC1 attenuated their suppression of Tcons (72). Furthermore, RAP-1 seems to be more activated in human Tregs compared to Tcons (73). Transgenic mice with a constitutively active RAP-1 have increased Treg frequency, and their Tregs are more suppressive (74, 75). These data suggest a feedback cycle in Tregs (particularly in peripheral Tregs), whereby sustained and elevated levels of cAMP and FOXP3 expression intensify each other, helping Tregs maintain their phenotype and functionality.
Tregs Induce PKA and EPAC Activation in Tcons
Interestingly, the intracellular signaling pathways downstream of cAMP appear to differ between target cells. Tregs redundantly induce the activation of PKA and EPAC in Tcons. The role of PKA in blunting Tcon proliferation was demonstrated through the use of cAMP analogs that bind PKA (76), long before Tregs were discovered to use this pathway to blunt Tcon activation. Downstream of PKA, ICER/CREM is normally localized in the cytoplasm of Tcons (Figure 1B). These molecules translocate and accumulate within the nucleus of Tcons in presence of Tregs, blunting IL-2 synthesis (65) (Figure 2). However, ICER−/− Tcons were still significantly suppressed by wild-type Tregs, suggesting that EPAC may also contribute to Treg suppression (12). Confirming this hypothesis, EPAC1−/− Tcons were poorly suppressed by Tregs (72). Taken together, these data suggest that Tregs induce both PKA and EPAC activation in Tcons, and these mechanisms could work cooperatively to suppress Tcon activation.
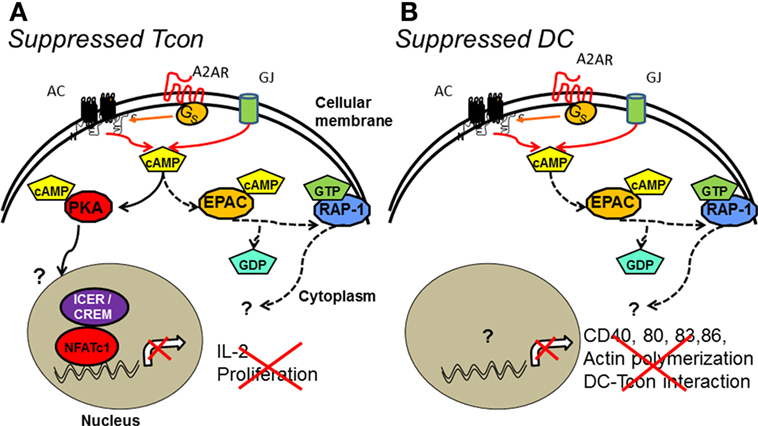
Figure 2. cAMP intracellular signaling pathways in target cells of Treg suppression. Activation of AC induced by adenosine interaction with its receptor (A2A) and/or influx of cAMP through gap junctions (GJ) increases cAMP concentration in Treg target cells. (A) cAMP-mediated suppression by Treg decreases effector responses (cytokine production and proliferation) in Tcon by PKA and EPAC. PKA activation in Tcon results in the translocation of ICER/CREM into the nucleus inhibiting NFATc1-driven transcription. Downstream signaling in the EPAC pathway includes the activation of the small GTPase RAP-1, promoting the exchange of GDP for GTP. (B) In DCs, the suppression of costimulatory molecules expression, actin polymerization, and DC–Tcon interaction downstream of cAMP seems to be due to EPAC activation. Intermediate and final molecules downstream of PKA and RAP-1, respectively, are still unknown.
Tregs Induce EPAC Activation in DCs
Exchange protein activated by cyclic AMP proteins regulate many mechanisms in APCs, such as integrin-dependent cell adhesion, polarization, chemotaxis, and phagocytosis (77, 78). Therefore, not surprisingly, triggering the cAMP–EPAC1 axis through Treg-derived adenosine activated RAP-1–GTP and increased DC migration (58). Tregs were also able to induce a re-localization of RAP-1 to the membrane of DCs, from its cytoplasmic localization when DCs are not in contact with Tregs (58) (Figure 2B). By contrast, Tregs do not prevent CREB activation in DCs, suggesting that Treg-mediated DC suppression is not PKA dependent (58). Similarly, cAMP does not inhibit the PKA/CREB-dependent production of inflammatory chemokines CCL3 and CCL4 by DCs (79). In addition, PKA activation by cAMP analogs induces maturation of human DCs, as evidenced by the increased surface expression of MHC class II, costimulatory molecules, and CD83 by treated DCs (80). These studies thus suggest that increased cAMP levels in DCs due to contact with Tregs is suppressive; however, PKA and EPAC appear to mediate opposing effects in DC.
The mechanisms that explain these cell-specific differential effects are still unclear. Interestingly, although pharmacological cAMP analogs that directly bind PKA are active in DCs, EPAC activity in DC appears to suppress PKA activation (80), which could thus explain why cAMP is globally suppressive in DCs. Second, the cellular effect of cAMP may also vary depending on the relative cellular abundance/distribution of EPAC and PKA and/or the amount of cAMP injected by Tregs in the target cells (56, 81). Finally, EPAC and PKA have opposite regulatory effects on downstream targets such as PKB (81). Activation of EPAC leads to a phosphatidylinositol 3-kinase-dependent PKB activation, whereas stimulation of PKA inhibits PKB activity (81). Future experiments are required to evaluate what controls cAMP downstream pathways in a cell-specific context.
Conclusion
It is now accepted that the cAMP-dependent intracellular signaling induced by Tregs in target cells is much more complex than initially assumed, and the classic PKA pathway is only part of the story. Although both human and murine Tregs mediate suppression indistinctly by cAMP influx and/or the CD39/adenosine pathway, the downstream pathways differ in Tcons and DCs. On the one hand, Treg suppression of Tcon cytokine production and proliferation requires the integration of EPAC and PKA in a cooperative manner. On the other hand, the suppression of DC function seems to be mainly mediated by EPAC, with a paradoxical opposite effect of PKA. Although our knowledge of the EPAC pathway in DCs has greatly progressed in the past years, much remains to be discovered. In particular, we still lack a full understanding of the physiological role of EPAC and RAP isoforms, of the mechanisms of PKA inactivation, and of the effector molecules downstream of RAP-1. A cautious dissection of the individual role and comparative contribution of EPAC and PKA within the overall cAMP signaling in various models will continue to be an important goal for upcoming investigations. This could lead to the development of targeted approaches fine-tuning Treg suppression for therapeutic applications.
Author Contributions
CR and CJ wrote the manuscript, and CC discussed and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Eunice Kennedy Shriver National Institute of Child Health and Human Development diversity supplement (ROIHD078127) supported CJ.
References
1. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
2. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi:10.1038/83713
3. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
4. Tang Q, Krummel MF. Imaging the function of regulatory T cells in vivo. Curr Opin Immunol (2006) 18:496–502. doi:10.1016/j.coi.2006.05.007
5. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A (2008) 105:10113–8. doi:10.1073/pnas.0711106105
6. Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol (2004) 34:2996–3005. doi:10.1002/eji.200425143
7. Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, et al. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med (2006) 203:505–11. doi:10.1084/jem.20050783
8. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
9. Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem (1958) 232:1077–92.
10. Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol (2002) 3:710–8. doi:10.1038/nrm911
11. Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) (2008) 40:651–62. doi:10.1111/j.1745-7270.2008.00438.x
12. Vang AG, Housley W, Dong H, Basole C, Ben-Sasson SZ, Kream BE, et al. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: a potential role for Epac. Biochem J (2013) 456:463–73. doi:10.1042/BJ20130064
13. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen G-X, et al. miR-142-3p restricts cAMP production in CD4(+)CD25(−) T cells and CD4(+)CD25(+) T(REG) cells by targeting AC9 mRNA. EMBO Rep (2009) 10:180–5. doi:10.1038/embor.2008.224
14. Duan B, Davis R, Sadat EL, Collins J, Sternweis PC, Yuan D, et al. Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J Immunol (2010) 185:335–44. doi:10.4049/jimmunol.0903474
15. Giembycz MA, Corrigan CJ, Seybold J, Newton R, Barnes PJ. Identification of cyclic AMP phosphodiesterases 3, 4 and 7 in human CD4+ and CD8+ T-lymphocytes: role in regulating proliferation and the biosynthesis of interleukin-2. Br J Pharmacol (1996) 118:1945–58. doi:10.1111/j.1476-5381.1996.tb15629.x
16. Glavas NA, Ostenson C, Schaefer JB, Vasta V, Beavo JA. T cell activation up-regulates cyclic nucleotide phosphodiesterases 8A1 and 7A3. Proc Natl Acad Sci USA (2001) 98:6319–24. doi:10.1073/pnas.101131098
17. Peter D, Jin SLC, Conti M, Hatzelmann A, Zitt C. Differential expression and function of phosphodiesterase 4 (PDE4) subtypes in human primary CD4+ T cells: predominant role of PDE4D. J Immunol (2007) 178:4820–31. doi:10.4049/jimmunol.178.8.4820
18. Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+ T cells. Mol Immunol (2010) 47:678–84. doi:10.1016/j.molimm.2009.10.032
19. Klein M, Vaeth M, Scheel T, Grabbe S, Baumgrass R, Berberich-Siebelt F, et al. Repression of cyclic adenosine monophosphate upregulation disarms and expands human regulatory T cells. J Immunol (2012) 188:1091–7. doi:10.4049/jimmunol.1102045
20. Birzele F, Fauti T, Stahl H, Lenter MC, Simon E, Knebel D, et al. Next-generation insights into regulatory T cells: expression profiling and FoxP3 occupancy in Human. Nucleic Acids Res (2011) 39:7946–60. doi:10.1093/nar/gkr444
21. Zheng Y, Josefowicz SZ, Kas A, Chu T-T, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature (2007) 445:936–40. doi:10.1038/nature05563
22. Lahl K, Mayer CT, Bopp T, Huehn J, Loddenkemper C, Eberl G, et al. Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J Immunol (2009) 183:5662–72. doi:10.4049/jimmunol.0803762
23. Rueda CM, Moreno-Fernandez ME, Jackson CM, Kallapur SG, Jobe AH, Chougnet CA. Neonatal regulatory T cells have reduced capacity to suppress dendritic cell function. Eur J Immunol (2015) 45:2582–92. doi:10.1002/eji.201445371
24. Levy O, Coughlin M, Cronstein BN, Roy RM, Desai A, Wessels MR. The adenosine system selectively inhibits TLR-mediated TNF-alpha production in the human newborn. J Immunol (2006) 177:1956–66. doi:10.4049/jimmunol.177.3.1956
25. Pettengill M, Robson S, Tresenriter M, Millan JL, Usheva A, Bingham T, et al. Soluble ecto-5’-nucleotidase (5’-NT), alkaline phosphatase, and adenosine deaminase (ADA1) activities in neonatal blood favor elevated extracellular adenosine. J Biol Chem (2013) 288:27315–26. doi:10.1074/jbc.M113.484212
26. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol (2010) 265:91–6. doi:10.1016/j.cellimm.2010.07.007
27. Ring S, Karakhanova S, Johnson T, Enk AH, Mahnke K. Gap junctions between regulatory T cells and dendritic cells prevent sensitization of CD8(+) T cells. J Allergy Clin Immunol (2010) 125(237–246):e231–7. doi:10.1016/j.jaci.2009.10.025
28. Fonseca PC, Nihei OK, Savino W, Spray DC, Alves LA. Flow cytometry analysis of gap junction-mediated cell-cell communication: advantages and pitfalls. Cytometry A (2006) 69:487–93. doi:10.1002/cyto.a.20255
29. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi:10.1182/blood-2006-12-064527
30. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
31. Van Calker D, Muller M, Hamprecht B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem (1979) 33:999–1005. doi:10.1111/j.1471-4159.1979.tb05236.x
32. Londos C, Cooper DM, Wolff J. Subclasses of external adenosine receptors. Proc Natl Acad Sci U S A (1980) 77:2551–4. doi:10.1073/pnas.77.5.2551
33. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2A extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood (1997) 90:1600–10.
34. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. doi:10.1038/nri2873
35. Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis (2014) 5:e1102. doi:10.1038/cddis.2014.70
36. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi:10.3389/fimmu.2014.00304
37. Alam MS, Kurtz CC, Rowlett RM, Reuter BK, Wiznerowicz E, Das S, et al. CD73 is expressed by human regulatory T helper cells and suppresses proinflammatory cytokine production and Helicobacter felis-induced gastritis in mice. J Infect Dis (2009) 199:494–504. doi:10.1086/596205
38. Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, Mcrae JL, et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant (2010) 10:2410–20. doi:10.1111/j.1600-6143.2010.03291.x
39. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4(+)CD25(high)FOXP3(+) regulatory T cells. J Biol Chem (2010) 285:7176–86. doi:10.1074/jbc.M109.047423
40. Moreno-Fernandez ME, Rueda CM, Rusie LK, Chougnet CA. Regulatory T cells control HIV replication in activated T cells through a cAMP-dependent mechanism. Blood (2011) 117:5372–80. doi:10.1182/blood-2010-12-323162
41. Schuler PJ, Schilling B, Harasymczuk M, Hoffmann TK, Johnson J, Lang S, et al. Phenotypic and functional characteristics of CD4+ CD39+ FOXP3+ and CD4+ CD39+ FOXP3neg T-cell subsets in cancer patients. Eur J Immunol (2012) 42:1876–85. doi:10.1002/eji.201142347
42. Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol (2011) 187:676–83. doi:10.4049/jimmunol.1003884
43. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, et al. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol (2013) 43:2430–40. doi:10.1002/eji.201242909
44. Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol (2014) 177:531–43. doi:10.1111/cei.12354
45. Panther E, Idzko M, Herouy Y, Rheinen H, Gebicke-Haerter PJ, Mrowietz U, et al. Expression and function of adenosine receptors in human dendritic cells. FASEB J (2001) 15:1963–70. doi:10.1096/fj.01-0169com
46. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi:10.3389/fimmu.2012.00190
47. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111:251–9. doi:10.1182/blood-2007-03-081646
48. Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol (2010) 185:1993–8. doi:10.4049/jimmunol.1000108
49. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol (2009) 182:4017–24. doi:10.4049/jimmunol.0803310
50. Feng G, Nadig SN, Bäckdahl L, Beck S, Francis RS, Schiopu A, et al. Functional regulatory T cells produced by inhibiting cyclic nucleotide phosphodiesterase type 3 prevent allograft rejection. Sci Transl Med (2011) 3:83ra40. doi:10.1126/scitranslmed.3002099
51. Bacher N, Raker V, Hofmann C, Graulich E, Schwenk M, Baumgrass R, et al. Interferon-α suppresses cAMP to disarm human regulatory T cells. Cancer Res (2013) 73:5647–56. doi:10.1158/0008-5472.CAN-12-3788
52. Umansky V, Shevchenko I, Bazhin AV, Utikal J. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol Immunother (2014) 63:1073–80. doi:10.1007/s00262-014-1553-8
53. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol (2009) 183:7602–10. doi:10.4049/jimmunol.0901881
54. Jenabian MA, Seddiki N, Yatim A, Carriere M, Hulin A, Younas M, et al. Regulatory T cells negatively affect IL-2 production of effector T cells through CD39/adenosine pathway in HIV infection. PLoS Pathog (2013) 9:e1003319. doi:10.1371/journal.ppat.1003319
55. Herrath J, Chemin K, Albrecht I, Catrina AI, Malmstrom V. Surface expression of CD39 identifies an enriched Treg-cell subset in the rheumatic joint, which does not suppress IL-17A secretion. Eur J Immunol (2014) 44:2979–89. doi:10.1002/eji.201344140
56. Weber M, Lupp C, Stein P, Kreft A, Bopp T, Wehler TC, et al. Mechanisms of cyclic nucleotide phosphodiesterases in modulating T cell responses in murine graft-versus-host disease. PLoS One (2013) 8:e58110. doi:10.1371/journal.pone.0058110
57. Moreno-Fernandez ME, Joedicke JJ, Chougnet CA. Regulatory T cells diminish HIV infection in dendritic cells – conventional CD4(+) T cell clusters. Front Immunol (2014) 5:199. doi:10.3389/fimmu.2014.00199
58. Ring S, Pushkarevskaya A, Schild H, Probst HC, Jendrossek V, Wirsdorfer F, et al. Regulatory T cell-derived adenosine induces dendritic cell migration through the Epac-Rap1 pathway. J Immunol (2015) 194:3735–44. doi:10.4049/jimmunol.1401434
59. Adkins I, Kamanova J, Kocourkova A, Svedova M, Tomala J, Janova H, et al. Bordetella adenylate cyclase toxin differentially modulates toll-like receptor-stimulated activation, migration and T cell stimulatory capacity of dendritic cells. PLoS One (2014) 9:e104064. doi:10.1371/journal.pone.0104064
60. Brindle P, Linke S, Montminy M. Protein-kinase-A-dependent activator in transcription factor CREB reveals new role for CREM repressors. Nature (1993) 364:821–4. doi:10.1038/364821a0
61. Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature (1993) 365:855–9. doi:10.1038/365855a0
62. Yehia G, Schlotter F, Razavi R, Alessandrini A, Molina CA. Mitogen-activated protein kinase phosphorylates and targets inducible cAMP early repressor to ubiquitin-mediated destruction. J Biol Chem (2001) 276:35272–9. doi:10.1074/jbc.M105404200
63. Bodor J, Bodorova J, Bare C, Hodge DL, Young HA, Gress RE. Differential inducibility of the transcriptional repressor ICER and its role in modulation of Fas ligand expression in T and NK lymphocytes. Eur J Immunol (2002) 32:203–12. doi:10.1002/1521-4141(200201)32:1<203::AID-IMMU203>3.0.CO;2-C
64. Barabitskaja O, Foulke JS Jr, Pati S, Bodor J, Reitz MS Jr. Suppression of MIP-1beta transcription in human T cells is regulated by inducible cAMP early repressor (ICER). J Leukoc Biol (2006) 79:378–87. doi:10.1189/jlb.0505255
65. Vaeth M, Gogishvili T, Bopp T, Klein M, Berberich-Siebelt F, Gattenloehner S, et al. Regulatory T cells facilitate the nuclear accumulation of inducible cAMP early repressor (ICER) and suppress nuclear factor of activated T cell c1 (NFATc1). Proc Natl Acad Sci U S A (2011) 108:2480–5. doi:10.1073/pnas.1009463108
66. Bodor J, Fehervari Z, Diamond B, Sakaguchi S. ICER/CREM-mediated transcriptional attenuation of IL-2 and its role in suppression by regulatory T cells. Eur J Immunol (2007) 37:884–95. doi:10.1002/eji.200636510
67. Nayak A, Glockner-Pagel J, Vaeth M, Schumann JE, Buttmann M, Bopp T, et al. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J Biol Chem (2009) 284:10935–46. doi:10.1074/jbc.M900465200
68. Bodor J, Bopp T, Vaeth M, Klein M, Serfling E, Hunig T, et al. Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol (2012) 42:1375–84. doi:10.1002/eji.201141578
69. Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, et al. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol (2013) 43:1001–12. doi:10.1002/eji.201243005
70. Hansen W, Loser K, Westendorf AM, Bruder D, Pfoertner S, Siewert C, et al. G protein-coupled receptor 83 overexpression in naive CD4+CD25- T cells leads to the induction of Foxp3+ regulatory T cells in vivo. J Immunol (2006) 177:209–15. doi:10.4049/jimmunol.177.1.209
71. Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, et al. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol (2006) 18:1197–209. doi:10.1093/intimm/dxl060
72. Almahariq M, Mei FC, Wang H, Cao AT, Yao S, Soong L, et al. Exchange protein directly activated by cAMP modulates regulatory T-cell-mediated immunosuppression. Biochem J (2015) 465:295–303. doi:10.1042/BJ20140952
73. Li L, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, et al. CD4+CD25+ regulatory T-cell lines from human cord blood have functional and molecular properties of T-cell anergy. Blood (2005) 106:3068–73. doi:10.1182/blood-2005-04-1531
74. Li L, Greenwald RJ, Lafuente EM, Tzachanis D, Berezovskaya A, Freeman GJ, et al. Rap1-GTP is a negative regulator of Th cell function and promotes the generation of CD4+CD103+ regulatory T cells in vivo. J Immunol (2005) 175:3133–9. doi:10.4049/jimmunol.175.5.3133
75. Li L, Kim J, Boussiotis VA. Rap1A regulates generation of T regulatory cells via LFA-1-dependent and LFA-1-independent mechanisms. Cell Immunol (2010) 266:7–13. doi:10.1016/j.cellimm.2010.08.014
76. Skålhegg BS, Landmark BF, Døskeland SO, Hansson V, Lea T, Jahnsen T. Cyclic AMP-dependent protein kinase type I mediates the inhibitory effects of 3’,5’-cyclic adenosine monophosphate on cell replication in human T lymphocytes. J Biol Chem (1992) 267:15707–14.
77. Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol (2005) 174:595–9. doi:10.4049/jimmunol.174.2.595
78. Lorenowicz MJ, Van Gils J, De Boer M, Hordijk PL, Fernandez-Borja M. Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J Leukoc Biol (2006) 80:1542–52. doi:10.1189/jlb.0506357
79. Jing H, Yen JH, Ganea D. A novel signaling pathway mediates the inhibition of CCL3/4 expression by prostaglandin E2. J Biol Chem (2004) 279:55176–86. doi:10.1074/jbc.M409816200
80. Garay J, D’Angelo JA, Park Y, Summa CM, Aiken ML, Morales E, et al. Crosstalk between PKA and Epac regulates the phenotypic maturation and function of human dendritic cells. J Immunol (2010) 185:3227–38. doi:10.4049/jimmunol.0903066
81. Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem (2002) 277:11497–504. doi:10.1074/jbc.M110856200
Keywords: cyclic adenosine monophosphate, T regulatory cells, suppression, dendritic cells, T cells
Citation: Rueda CM, Jackson CM and Chougnet CA (2016) Regulatory T-Cell-Mediated Suppression of Conventional T-Cells and Dendritic Cells by Different cAMP Intracellular Pathways. Front. Immunol. 7:216. doi: 10.3389/fimmu.2016.00216
Received: 01 March 2016; Accepted: 19 May 2016;
Published: 02 June 2016
Edited by:
Josef Bodor, Institute of Experimental Medicine, Czech RepublicReviewed by:
Janis K. Burkhardt, Children’s Hospital of Philadelphia, USAAvery August, Cornell University, USA
Copyright: © 2016 Rueda, Jackson and Chougnet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire A. Chougnet, Y2xhaXJlLmNob3VnbmV0QGNjaG1jLm9yZw==
†Cesar M. Rueda and Courtney M. Jackson contributed equally.