 Andrew S. Kimball
Andrew S. Kimball Andrea T. Obi
Andrea T. Obi- Section of Vascular Surgery, Conrad Jobst Vascular Research Laboratories, Department of Surgery, University of Michigan, Ann Arbor, MI, USA
Venous thrombosis (VT), a leading cause of morbidity and mortality worldwide, has recently been linked to neutrophil activation and release of neutrophil extracellular traps (NETs) via a process called NETosis. The use of various in vivo thrombosis models and genetically modified mice has more precisely defined the exact role of NETosis in the pathogenesis of VT. Translational large animal VT models and human studies have confirmed the presence of NETs in pathologic VT. Activation of neutrophils, with subsequent NETosis, has also been linked to acute infection. This innate immune response, while effective for bacterial clearance from the host by formation of an intravascular bactericidal “net,” also triggers thrombosis. Intravascular thrombosis related to such innate immune mechanisms has been coined immunothrombosis. Dysregulated immunothrombosis has been proposed as a mechanism of pathologic micro- and macrovascular thrombosis in sepsis and autoimmune disease. In this focused review, we will address the dual role of NETs in the pathogenesis of VT and immunothrombosis.
Introduction
Neutrophils (PMNs) have frequently been touted as the pawns of the immune system. As our knowledge of immune system grows and as our techniques for evaluating dynamic cell populations improve – we are learning that this could not be further from the truth. While a PMNs principle function remains as a key player in the front line of innate immunity and host defense against bacteria, they are proving to have a multifaceted role in coagulation and have also been implicated as major contributors in the pathophysiology of many systemic illnesses.
Until the early 2000s, the associations between PMN activation and systemic disease had not been well understood; but in March of 2004, Brinkmann et al. published a landmark study in Science (1), where they described a fragile fibrillar material extruded from PMNs in the presence of lipopolysaccharide (LPS) by transmission electron microscopy (TEM). In actuality, these fragile fibers were decondensed chromatin and DNA, as they stained strongly for DNA and histones, they were resistant to proteases, and they disappeared upon instillation of DNase. Bacteria were found to colocalize with the extruded DNA in vivo in a rabbit model of shigellosis and in human specimens of acute appendicitis. In summation, they demonstrated that these large webs of DNA trap bacteria and allow adjacent or connected PMNs to drive bactericidal activity with proteases and reactive oxygen species. Brinkmann et al. coined these nuclear extrusions “neutrophil extracellular traps” or NETs.
Since that time, there has been a flurry of exciting new work in the field of NET formation (NETosis). NETosis has been demonstrated to be a distinct form of cell death outside of necrosis and apoptosis (2). Also, more interestingly, NETs have been indicted in the pathophysiology of many systemic diseases, including venous thrombosis (VT) (3), sepsis (4, 5), trauma (6), cancer-related thrombosis (7), and autoimmune diseases (8–12). Despite the apparent widespread influence of NETs on disease, there remains a common theme throughout that NETs drive micro- or macrovascular thrombosis leading to ischemia and further injury (13, 14).
In this article, we will review the role of NETs in pathologic thrombosis. Specifically, we will review the findings of NET pathophysiology in murine models of VT, NETs in primate models and human studies of VT, and NETs in immunothrombosis.
NETs in Murine VT Models
Murine models have been essential to our understanding of the role of NETosis in the pathophysiology of thrombosis. PMNs were first shown to be essential for immune-mediated microvascular thrombosis in a murine model of glomerulonephritis, in which CD11b−/− or PMN-depleted mice were resistant to glomeruli thrombosis and renal failure (15). At that time, it was not widely recognized that NETs contributed to thrombosis; however, this changed in 2010, when Fuchs et al. showed that NETs caused platelet adhesion, activation, and aggregation (3). Stimulation of platelets with purified histones was sufficient for aggregation, and interestingly, DNase and heparin dismantled the NET scaffold and prevented thrombus formation. Brill et al. later demonstrated that NETs are principle effectors in an IVC stenosis model (16). In mice with uninterrupted IVC side-branches, levels of extracellular DNA increased in plasma 6 h after thrombus initiation. Citrullinated histone H3 (CitH3), an element of NETs’ structure, was present in thrombi and was frequently associated with the Gr-1 antigen. Furthermore, immunofluorescent staining of thrombi showed proximity of extracellular CitH3 and von Willebrand factor (vWF), a platelet adhesion molecule crucial for thrombus development in this particular model.
Neutrophils, monocytes, and NETs have also been found to affect the clotting cascade in murine models of thrombosis (17–20). For example, myeloid cells roll along the venous endothelium in a P-selectin-dependent manner and produce thrombogenic tissue factor (TF) in the IVC stenosis model (17). TF, then contributes to thrombin generation and extensive fibrin deposition along the vein wall. Despite this finding, TF alone was inadequate for thrombus propagation. Neutropenia, genetic ablation of Factor XII, and disintegration of NETs were all protective against thrombus propagation. Later, activated PMNs within the fibrin matrix were found to produce NETs that associate with secreted Factor XII, activate the intrinsic pathway, and lead to thrombus extension (17, 21). However, these conclusions were questioned, as during the same year, TF was also found to be secreted by PMNs in an inflammatory signaling and autophagy-dependent manner, with adherence to extruded NETs, activation of the extrinsic pathway, and eventual propagation of the thrombus (18, 22). Regardless, the presence of PMNs and/or monocytes at the endothelial interface has long been assumed to have a major role in VT and now appears unquestionable (17, 18, 23, 24).
Many proteins have been implicated in contributing to NETosis and thrombosis. In 2013, Martinod et al. demonstrated that the enzyme peptidyl arginine deiminase 4 (PAD4) – an enzyme essential for the citrullination and decondensation of chromatin was – not only essential for NET formation but also important for thrombus formation in the IVC stenosis model of murine VT (25). PAD4−/− mice formed IVC thrombus in less than 10% of cases of IVC stenosis at 48 h compared to 90% in C57BL/6 controls (25). This antithrombotic tendency was rescued in the PAD4−/− mice with adoptive transfer of wild-type PMNs. Conversely, in a stasis IVC ligation model of VT, either preemptive administration of DNase to wild-type mice or PAD4−/− mice did not have an effect on thrombogenesis, suggesting a model-dependent effect (19).
Other proteins that have been implicated in NET formation and NET-associated thrombosis include cathepsin G, serine proteases, PMN elastase, P-selectin, high-mobility group box protein 1 (HMGB-1), platelet glycoprotein Ib, integrin beta-2, vWF, and platelet factor 4. Pharmacological inhibition or genetic deletion of cathepsin G increases mouse bleeding time, decreases platelet activation, and decreases circulating neutrophil–platelet conjugates (26). While the serine proteases and PMN elastase have been implicated in pathologic thrombosis and have been found to colocalize with NETs (27), it was recently demonstrated that PMN elastase is neither necessary for NET formation in mice nor essential for IVC thrombosis in murine IVC stenosis model (28). Indeed, it is likely that the prothrombotic effect of the serine proteases is better explained by their inhibition of tissue factor pathway inhibitor (TFPI) (29). Signaling through P-selectin by P-selectin glycoprotein ligand 1 (PSGL-1) is known to assist in PMN migration to sites of injury or inflammation (30), but beyond that, the P-selectin axis has now been postulated to promote NETosis in the setting of activated platelets (24). However, this conclusion was recently challenged in a study that demonstrated that HMGB-1 expressed by activated platelets was individually capable of activating PMNs via the receptor for advanced glycosylation end-products (RAGE) to induce NETosis (31). Similarly, in 2016, an in vitro study demonstrated activated platelets binding to vWF through glycoprotein Ib, in turn, linked to PMNs via integrin beta-2 and stimulated by platelet factor 4 led to NETosis independent of P-selectin (32). While our knowledge of NET formation continues to grow, the specific proteomic cascade that leads to NETosis remains contested. These findings also underline the heterogeneity and limitations of model systems as well.
Given that thrombi consist of significant quantities of DNA combined with proteins, thrombosis resolution and associated inflammation becomes less straight forward. We have explored this topic in our laboratory by examining toll-like receptor 9 (TLR9) function in VT resolution. TLR9 is a conserved pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP) receptor that recognizes CpG DNA repeats and alerts the immune system to invading pathogens or local damage. In an IVC ligation model of VT (complete stasis), TLR9−/− mice had significantly increased thrombus size at 2 and 8 days despite increased numbers of PMNs and monocyte/macrophages (33). Further, TLR9−/− mice had increased apoptosis, citrullinated histones, PAD4, and neutrophil elastase; and reduced TFPI (19), suggesting that TLR9 is important for normal thrombogenesis and thrombosis resolution. Lastly, M1-like (CCR2+) monocyte/macrophages were decreased in TLR9−/− thrombi, consistent with impaired inflammatory cell influx, and this divergence was corrected with adoptive transfer of TLR9+/+ bone marrow-derived monocytes with normalization of thrombus size (20). The new found composition of DNA-rich thrombi will likely have long-standing implications for future research in VT resolution.
NETs in Primate and Human VT Studies
Although first thought to only occur in pathologic states, NETosis with release of extracellular DNA has been shown to occur in healthy individuals (34). Following exhaustive treadmill or cycling exercise, circulating levels of cell-free DNA (cfDNA) and myeloperoxidase (MPO) rise, and isolated PMNs from the circulation develop swollen nuclei and emanating DNA. A concomitant rise in circulating DNase occurs, suggesting that similar to the ongoing processes of fibrin formation and fibrinolysis, NETosis may be a tightly regulated and constantly ongoing homeostatic process. Dysregulation of NETosis and its relationship to thrombosis has been recognized in a variety of clinical scenarios: NETs are present in fresh thrombi from individuals with acute myocardial infarction (31, 35), they are found in high circulating levels in patients with severe trauma and microvascular thrombosis with acute lung injury (6); and in patients with thrombotic microangiopathies (TMAs) (8). Circulating cfDNA rises 24 h following chemotherapy in breast cancer patients, corresponding to peak in thrombin–antithrombin levels (36). This has been proposed as a potential causative mechanism for the high rate of thrombotic events experienced by individuals undergoing chemotherapy.
Ex vivo platelet activation studies with recombinant human histones demonstrate that not all histones are created equal in NETosis: only histone 3 (H3) and histone 4 (H4) induce functional platelet response (37). Recent advances in techniques of PMN isolation (38) and recognition of NETs in human pathologic thrombus specimens have improved the ability to study NETosis in humans (39).
The link between human venous thromboembolism (VTE) events and NETosis has been established in a handful of studies to date. Analysis of balloon occlusion-induced iliac thrombosis in a baboon model of VT demonstrated increased circulating NETs at 48 h post-thrombosis and persisting through 6 days (3). Consistently, extracellular DNA markers were present in the experimental thrombus. In 2013, a human study composed of healthy controls and symptomatic patients (swelling and leg pain) with and without VT examined circulating NET markers (40). Extracellular DNA and MPO were significantly elevated in symptomatic patients with VT compared to both groups of non-thrombosed patients. A direct correlation was also seen between common predictors of thrombosis including D-dimer, Wells score, and plasma DNA, suggesting that NETs may be useful in diagnostic evaluation. Development of more accurate methods to diagnose VT is of particular interest as the current “gold standard,” duplex ultrasound, is often limited in availability and in evaluation of central veins (41). In the same year, a similar case–control study of 195 individuals with and without VT examined nucleosomes and α-1 antitrypsin elastase (as a PMN activation marker) in relation to thrombosis. Levels of nucleosomes and PMN activation above the 80th percentile were associated with a threefold risk of VT (42).
The precise role of NETs in VT initiation, formation, and propagation, as well as optimal area to intervene in human VT remains relatively unknown. In a study evaluating unorganized, organizing, and organized thrombi from patients with either VT or PE, CD11b, and MPO positive cells were seen in organizing thrombi along with intra- and extracellular CitH3 and PAD4 (43). Unorganized and organized thrombi failed to demonstrate the similar histopathology, suggesting the predominant role of NETs during the inflammatory response and thrombus organization (43). Another study of 29 VT patients and controls demonstrated increased neutrophil adhesion and inflammatory cytokine profile among patients with residual vein obstruction and elevated D-dimer (44). Ex vivo studies of NETs related hypercoagulability in inflammatory bowel disease (IBD), and TMA patients have demonstrated efficacy of DNAse I in decreasing the thrombotic response, although this has yet to be shown in human VT (11, 12). To date, no clinical trial has targeted NETs in humans as a mechanism to prevent or treat VT.
NETs in Immunothrombosis
While the ability to form thrombus in the face of vessel injury has long been known to be essential for the maintenance of hemostasis, until recently, it had not been recognized as an intravascular mechanism of immune defense. Engelmann and Massberg recently reviewed the mechanism of NET-mediated microvascular thrombosis and proposed it as a potential biological adjunct for containing uncontrolled infection, coining this process “Immunothrombosis” (45). In their review of the topic, they proposed four mechanisms by which immunothrombosis prevents the spread of infection (1) it captures and ensnares pathogens in the microvasculature and prevents dissemination, (2) it prevents distant tissue invasion by forming microthrombi in microvessels, (3) it concentrates pathogens in one area for bactericidal killing by innate immune cells, and (4) it recruits other immune cells to the site of inflammation for further bacterial killing. Indeed, coagulation has been previously postulated to play a role in host defense against bacteria (46, 47), but if left unchecked, immunothrombosis may contribute to significant systemic pathology.
Severe sepsis accounts for 10–40% of ICU admissions in the United States and carries a mortality rate of 20–50% (48, 49). NET-related immunothrombosis, cfDNA, and histones have been implicated in the morbidity and mortality of sepsis. In in vitro and in vivo models of sepsis, LPS has been shown to activate platelets and PMNs via toll-like receptor 4 (TLR4) to induce NETosis (31, 50, 51). Intravascular NETs can be digested by DNase to release cfDNA and histones, and further, cfDNA obtained from human specimens has been shown to be at ~150–300 bp sizes consistent with nucleosome units (52). In the same study, cfDNA levels in peripheral blood of patients with severe sepsis were found to be highly predictive of ICU mortality. In 2009, Xu et al. demonstrated that extracellular histones led to endothelial dysfunction, organ failure, and death in animal models of sepsis (4). Specifically, histone administration to mice resulted in neutrophil activation, endothelial toxicity, acute lung injury, and microvascular thrombosis. Interestingly, antihistone antibodies and activated protein C (APC) improved mortality rates in sepsis models in these mice. In addition, NETosis has also been linked to prothrombotic activity because NET-associated enzymes break down TFPI (29), while also enhancing antifibrinolytic activity because cfDNA inhibits plasmin-mediated fibrin degradation (13, 14, 28). Further, H3 levels were found to correlate with ICU mortality and were inversely correlated with antithrombin and platelet levels (53). Structurally, NET-induced immunothrombosis leads to more sturdy thrombi with less permeability and decreased susceptibility to lysis, although this may be overcome with DNase (54). In a murine cecal ligation and puncture model of sepsis (CLP), DNase given at 6 h after injury reduced organ damage and mortality (55). Lastly, bacterial resistance to NET trapping has been demonstrated in vitro and has been postulated to contribute to widespread immunothrombosis in the septic host leading to disseminated intravascular coagulation (DIC) (56, 57).
In addition to its inflammatory role in sepsis, NETs and immunothrombosis have also been implicated in autoimmune disease. Phosphatidylserine (PS) and TF-bearing microparticles (MP) were found to be markedly increased in the sera of IBD patients (11). Increased TF-bearing MPs correlated with increased NETosis, markedly shortened coagulation times, and increased levels of fibrin, thrombin, and factor Xa. Similarly, in an in vitro study of antineutrophil cytoplasmic antibody-associated vasculitis (ANCA-AV or AAV), C5a-primed PMNs treated with ANCA IgG-released NETs and TF-bearing MPs contributing to a prothrombotic state, and interestingly, this was remedied partially by DNase treatment (58). TMAs, more commonly known as thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), and DIC, are characterized by microvascular thrombosis and coagulopathy. In vitro studies of TMAs demonstrate that the sera of TMA patients are incapable of degrading NETs due to a deficiency of DNase I and this may contribute to widespread microvascular thrombosis and organ dysfunction (12). Intriguingly, supplementation of TMA sera with recombinant human DNase I reestablished NET degradation capability. Lastly, PMNs from patients with antiphospholipid antibody syndrome (APS) were inclined for spontaneous NETosis and thrombin generation, and further, APS sera and APS-isolated IgG stimulated NETosis in control PMNs (59). These findings highlight the prothrombotic role of NETs in autoimmune-related immunothrombosis.
Finally, NETs have also been implicated in multiple organ failure related to severe trauma. In 2013, Abrams et al. examined a cohort of 52 patients with severe non-thoracic blunt trauma and correlated circulating histone levels with acute lung injury and sequential organ failure assessment scores (SOFA) (6). Circulating histone levels increased dramatically immediately following trauma and high levels correlated with acute lung injury, SOFA scores, endothelial injury, and coagulation activation. In the same study, with translational animal models of blunt trauma, circulating histone levels corresponded with organ edema, hemorrhage, microvascular thrombosis, and neutrophil congestion. Multiple organ dysfunction syndrome (MODS) is a leading cause of delayed mortality following major trauma and has been correlated with circulating cfDNA levels (52). Inherently, future research should look to address NETosis and immunothrombosis as potential therapeutic targets for preventing immune-mediated MODS after severe injury.
Summary, Integration, and Potential Translation
Over the past 12 years, since its discovery, NETosis has been catapulted to the forefront of innate immunity research. Nowhere is its effect more relevant in human disease than in its implication in immune-mediated micro- and macrovascular thrombosis (Figure 1). In this review and others, NETs have been shown to have a significant role in pathogenic thrombosis through platelet and PMN recruitment to the endothelial wall, subsequent activation and NETosis, and proteomic activation of the intrinsic and extrinsic coagulation cascades, ultimately leading to thrombosis (3, 13, 14, 16, 17, 31, 32, 45, 50, 60, 61).
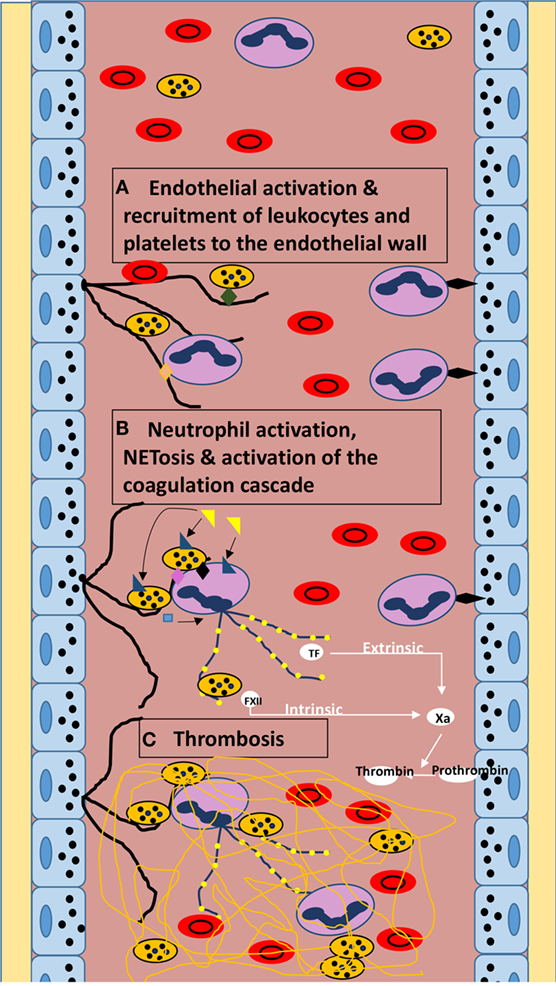
Figure 1. Diagram of theoretical mechanism of NET-mediated microvascular thrombosis in sepsis. (A) Sepsis-induced endothelial activation leading to the release of vWF (black lines) from endothelial cell Weibel–Palade bodies provoking the recruitment of leukocytes and platelets to the endothelial wall via vWF A1 domain–beta-2 integrin interaction (vWF–PMN interaction; orange diamond) and vWF A1 domain–glycoprotein Ib interaction (vWF–platelet interaction; green diamond). PMNs are also recruited to the endothelium via P-selectin-to-PSGL-1 interaction (black diamond) inducing neutrophil rolling. (B) Neutrophil and platelet activation by LPS (yellow triangle) via TLR4 (blue triangle). Platelet activation leading to the release of PF4 (blue square) and expression of P-selectin (black diamond) and HMGB-1 (pink diamond) further stimulate NETosis [purple lines of decondensed chromatin with attached histones (yellow circles)]. NETosis also leads to autophagy-induced release of TF, and NETs further stimulate FXII activation, inciting both the intrinsic and extrinsic coagulation cascades. (C) Thrombin activation by factor Xa leads to the conversion of fibrinogen to fibrin (yellow lines) culminating in microvascular thrombosis.
This unique development in the etiology of immune-mediated thrombosis affords novel targets for the prevention of pathologic thrombosis in a susceptible patient (Table 1) (4, 25, 55, 62–70). One obvious target for the prevention of NET-mediated VTE is PAD4 inhibition (25, 60). Enzymatic inhibition of PAD4 would prevent NETosis, and hopefully, its prothrombotic effects as well; however, this may also leave the host susceptible to bacterial infection (71). DAMP-mediated NETosis by HMGB-1 is also a potential therapeutic target. HMGB-1 is released from damaged cells and expressed on the surface of activated platelets and leads to immune system activation via RAGE, TLR2, and TLR4 (5, 72, 73). HMGB-1-mediated PMN activation subsequently contributes to microvascular thrombosis and NETosis (31, 74). HMGB-1 is cleaved by thrombomodulin–thrombin complexes in vivo, and recombinant thrombomodulin is presently approved for the treatment of DIC in Japan and is in phase III trials here in the United States (5, 70).
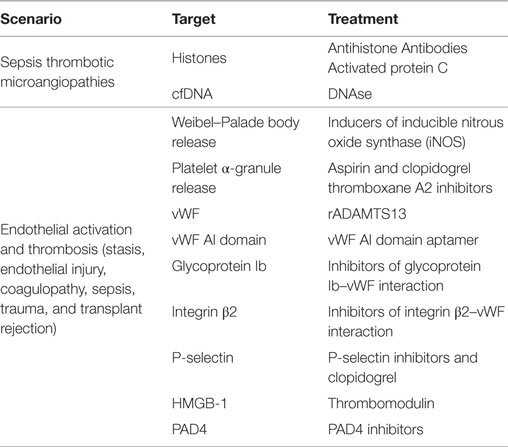
Other targets include the components of PMN and platelet recruitment to the endothelium. Specifically, blockade of platelet alpha-granule release or endothelial Weibel–Palade body release would decrease P-selectin and vWF-mediated platelet and PMN recruitment to the endothelium decreasing NETosis (24, 32, 75, 76). Similarly, blockade of P-selectin (24), vWF A1 domain (16), glycoprotein Ib, or integrin beta-2 may reach a similar end, however, more specifically (32). Lastly, the vWF degradation enzyme, ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif – member 13), may be given in a recombinant form (rADAMTS13) to degrade vWF and prevent PMN recruitment with subsequent NETosis (77). Although, each of the above-listed countermeasures may result in mild immunodeficiency, if given in a dose-dependent manner, they may abrogate the pathologic immune-mediated thrombosis without sacrificing immune competence.
Despite the relatively recent discovery of NETs as a contributor to VTE, some long-standing traditional VTE therapies already affect NETs. Polyanionic heparin, long considered the gold standard therapy for prevention and treatment of VTE, has a secondary effect of displacing histones from chromatin (78, 79). This allows increased accessibility of nucleases to the exposed chromatin, further permitting degradation of NETs (78, 79). Similarly, aspirin, recently shown to decrease risk of recurrent VTE (80), inhibits NETosis in vitro by inhibition of thromboxane A2 synthesis (63, 81). Finally, clopidogrel has also been shown to decrease inflammation and platelet-mediated expression of soluble P-selectin, further decreasing PMN–platelet interactions and hence NETosis (68). It is currently unknown as to the effects of the new oral anticoagulants on NETs, although this may represent an important area for future investigation.
Thrombolysis has become a key weapon in the arsenal against pathologic thrombosis; however, not all thrombotic events are susceptible to thrombolysis with tissue plasminogen activator (tPA) (82). Indeed, the addition of DNA and histones to a fibrin matrix has been shown to make artificial thrombus more stable, more rigid, and more resistant to tPA, and this is partially remedied by DNase (83). Preliminary data from murine models of VT demonstrate inhibition of thrombus formation with DNase instillation prior to and during IVC stenosis (16, 17). In another study of limb ischemia-reperfusion injury, DNase instillation decreased tissue NETs but did not effect end tissue damage or inflammatory infiltrate (84). In spite of this, more recent studies have shown decreased inflammation, increased tissue perfusion, and improved survival with (1) late DNase treatment in cecal ligation puncture (CLP) model of murine sepsis (55), (2) concurrent DNase and tPA treatment in a rat model of myocardial ischemia (85), and (3) preemptive DNase treatment in a rat model of renal ischemia-reperfusion injury (86). Combination tPA and DNase therapy for thrombolysis of acute ischemic events has yet to be studied in human patients but will likely constitute a major area of research in the near future.
There are still many unanswered questions in the field of NETosis. What are the exact proteomic mechanisms that lead to neutrophil activation and subsequent NET formation? What is the role or RAGE, and does this have implications for diabetics? What is the intracellular cascade that leads to PAD4 induction? After NETosis and thrombosis, how are NETs naturally removed from the resolving thrombus? Do NETs play a role in the post-thrombotic syndrome? Will the addition of DNase to tPA broaden the spectrum of patients that can be treated with thrombolysis as opposed to surgery? These questions and more will be answered with time and continued diligent, meticulous, and conscientious research. We look forward to the future with great expectations for upcoming discoveries in the field of NETosis.
Author Contributions
Dr. Andrew S. Kimball did the literature review, wrote the paper, and approved of the submission. Drs. Andrea T. Obi and Jose A. Diaz did a literature review, wrote sections of the paper, and approved of the submission. Dr. Peter K. Henke wrote and edited the paper, approved of the submission, and is responsible for the content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Whitney Townsend, Librarian at the University of Michigan Taubman Health Sciences Library, for her assistance with the comprehensive literature search.
Funding
Andrew S. Kimball is funded by an NIH T32 Research Fellowship (NIH-T32 HL076123).
References
1. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi:10.1126/science.1092385
2. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176(2):231–41. doi:10.1083/jcb.200606027
3. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107(36):15880–5. doi:10.1073/pnas.1005743107
4. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15(11):1318–21. doi:10.1038/nm.2053
5. Liaw PC, Ito T, Iba T, Thachil J, Zeerleder S. DAMP and DIC: the role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev (2016). doi:10.1016/j.blre.2015.12.004
6. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187(2):160–9. doi:10.1164/rccm.201206-1037OC
7. Demers M, Wagner DD. Neutrophil extracellular traps: a new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology (2013) 2(2):e22946. doi:10.4161/onci.22946
8. Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood (2012) 120(6):1157–64. doi:10.1182/blood-2012-02-412197
9. Nakazawa D, Tomaru U, Yamamoto C, Jodo S, Ishizu A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front Immunol (2012) 3:333. doi:10.3389/fimmu.2012.00333
10. Simon D, Simon HU, Yousefi S. Extracellular DNA traps in allergic, infectious, and autoimmune diseases. Allergy (2013) 68(4):409–16. doi:10.1111/all.12111
11. He Z, Si Y, Jiang T, Ma R, Zhang Y, Cao M, et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb Haemost (2016) 115(4):738–51. doi:10.1160/TH15-09-0710
12. Jiménez-Alcázar M, Napirei M, Panda R, Köhler EC, Kremer Hovinga JA, Mannherz HG, et al. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J Thromb Haemost (2015) 13(5):732–42. doi:10.1111/jth.12796
13. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol (2015) 35(12):2544–53. doi:10.1161/ATVBAHA.115.306035
14. Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost (2015) 13(S1):S82–91. doi:10.1111/jth.12977
15. Hirahashi J, Hishikawa K, Kaname S, Tsuboi N, Wang Y, Simon DI, et al. Mac-1 (CD11b/CD18) links inflammation and thrombosis after glomerular injury. Circulation (2009) 120(13):1255–65. doi:10.1161/CIRCULATIONAHA.109.873695
16. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost (2012) 10(1):136–44. doi:10.1111/j.1538-7836.2011.04544.x
17. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi:10.1084/jem.20112322
18. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One (2012) 7(9):e45427. doi:10.1371/journal.pone.0045427
19. El-Sayed OM, Dewyer NA, Luke CE, Elfline M, Laser A, Hogaboam C, et al. Intact toll-like receptor 9 signaling in neutrophils modulates normal thrombogenesis in mice. J Vasc Surg (2015). doi:10.1016/j.jvs.2015.08.070
20. Dewyer NA, El-Sayed OM, Luke CE, Elfline M, Kittan N, Allen R, et al. Divergent effects of Tlr9 deletion in experimental late venous thrombosis resolution and vein wall injury. Thromb Haemost (2015) 114(5):1028–37. doi:10.1160/TH14-12-1031
21. Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol (2014) 34(9):1977–84. doi:10.1161/ATVBAHA.114.304114
22. Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front Immunol (2012) 3:385. doi:10.3389/fimmu.2012.00385
23. Darbousset R, Thomas GM, Mezouar S, Frère C, Bonier R, Mackman N, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood (2012) 120(10):2133–43. doi:10.1182/blood-2012-06-437772
24. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126(2):242–6. doi:10.1182/blood-2015-01-624023
25. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A (2013) 110(21):8674–9. doi:10.1073/pnas.1301059110
26. Faraday N, Schunke K, Saleem S, Fu J, Wang B, Zhang J, et al. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS One (2013) 8(8):e71447. doi:10.1371/journal.pone.0071447
27. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16(8):887–96. doi:10.1038/nm.2184
28. Martinod K, Witsch T, Farley K, Gallant M, Remold-O’Donnell E, Wagner DD. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost (2016) 14(3):551–8. doi:10.1111/jth.13239
29. Petersen LC, Bjorn SE, Nordfang O. Effect of leukocyte proteinases on tissue factor pathway inhibitor. Thromb Haemost (1992) 67(5):537–41.
30. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science (2014) 346(6214):1234–8. doi:10.1126/science.1256478
31. Maugeri N, Campana L, Gavina M, Covino C, de Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12(12):2074–88. doi:10.1111/jth.12710
32. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol (2016) 99(1):153–62. doi:10.1189/jlb.3A0415-161R
33. Henke PK, Mitsuya M, Luke CE, Elfline MA, Baldwin JF, Deatrick KB, et al. Toll-like receptor 9 signaling is critical for early experimental deep vein thrombosis resolution. Arterioscler Thromb Vasc Biol (2011) 31(1):43–9. doi:10.1161/ATVBAHA.110.216317
34. Beiter T, Fragasso A, Hudemann J, Schild M, Steinacker J, Mooren FC, et al. Neutrophils release extracellular DNA traps in response to exercise. J Appl Physiol (2014) 117(3):325–33. doi:10.1152/japplphysiol.00173.2014
35. de Boer OJ, Li X, Teeling P, Mackaay C, Ploegmakers HJ, van der Loos CM, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost (2013) 109(2):290–7. doi:10.1160/TH12-06-0425
36. Swystun LL, Mukherjee S, Liaw PC. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J Thromb Haemost (2011) 9(11):2313–21. doi:10.1111/j.1538-7836.2011.04465.x
37. Carestia A, Rivadeneyra L, Romaniuk MA, Fondevila C, Negrotto S, Schattner M. Functional responses and molecular mechanisms involved in histone-mediated platelet activation. Thromb Haemost (2013) 110(5):1035–45. doi:10.1160/TH13-02-0174
38. Barrientos L, Marin-Esteban V, de Chaisemartin L, Le-Moal VL, Sandré C, Bianchini E, et al. An improved strategy to recover large fragments of functional human neutrophil extracellular traps. Front Immunol (2013) 4:166. doi:10.3389/fimmu.2013.00166
39. de Boer OJ, Li X, Goebel H, van der Wal AC. Nuclear smears observed in H&E-stained thrombus sections are neutrophil extracellular traps. J Clin Pathol (2016) 69(2):181–2. doi:10.1136/jclinpath-2015-203019
40. Diaz JA, Fuchs TA, Jackson TO, Kremer Hovinga JA, Lämmle B, Henke PK, et al. Plasma DNA is elevated in patients with deep vein thrombosis. J Vasc Surg Venous Lymphat Disord (2013) 1(4). doi:10.1016/j.jvsv.2012.12.002
41. Coleman DM, Wakefield TW. Biomarkers for the diagnosis of deep vein thrombosis. Expert Opin Med Diagn (2012) 6(4):253–7. doi:10.1517/17530059.2012.692674
42. van Montfoort ML, Stephan F, Lauw MN, Hutten BA, Van Mierlo GJ, Solati S, et al. Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arterioscler Thromb Vasc Biol (2013) 33(1):147–51. doi:10.1161/ATVBAHA.112.300498
43. Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost (2014) 12(6):860–70. doi:10.1111/jth.12571
44. Zapponi KC, Mazetto BM, Bittar LF, Barnabé A, Santiago-Bassora FD, De Paula EV, et al. Increased adhesive properties of neutrophils and inflammatory markers in venous thromboembolism patients with residual vein occlusion and high D-dimer levels. Thromb Res (2014) 133(5):736–42. doi:10.1016/j.thromres.2014.01.035
45. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi:10.1038/nri3345
46. Zinsser HH, Pryde AW. Experimental study of physical factors, including fibrin formation, influencing the spread of fluids and small particles within and from the peritoneal cavity of the dog. Ann Surg (1952) 136(5):818–27. doi:10.1097/00000658-195211000-00006
47. Flick MJ, Du X, Witte DP, Jirousková M, Soloviev DA, Busuttil SJ, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest (2004) 113(11):1596–606. doi:10.1172/JCI20741
48. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med (2003) 348(16):1546–54. doi:10.1056/NEJMoa022139
49. Dreiher J, Almog Y, Sprung CL, Codish S, Klein M, Einav S, et al. Temporal trends in patient characteristics and survival of intensive care admissions with sepsis: a multicenter analysis. Crit Care Med (2012) 40(3):855–60. doi:10.1097/CCM.0b013e318236f7b8
50. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi:10.1038/nm1565
51. Ma AC, Kubes P. Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J Thromb Haemost (2008) 6(3):415–20. doi:10.1111/j.1538-7836.2007.02865.x
52. Dwivedi DJ, Toltl LJ, Swystun LL, Pogue J, Liaw KL, Weitz JI, et al. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Critical Care (2012) 16(4):R151. doi:10.1186/cc11466
53. Wildhagen KC, Wiewel MA, Schultz MJ, Horn J, Schrijver R, Reutelingsperger CP, et al. Extracellular histone H3 levels are inversely correlated with antithrombin levels and platelet counts and are associated with mortality in sepsis patients. Thromb Res (2015) 136(3):542–7. doi:10.1016/j.thromres.2015.06.035
54. Varjú I, Longstaff C, Szabó L, Farkas ÁZ, Varga-Szabó VJ, Tanka-Salamon A, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost (2015) 113(6):1289–98. doi:10.1160/TH14-08-0669
55. Mai SH, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed but not early treatment with DNase reduces organ damage and improves outcome in a murine model of sepsis. Shock (2015) 44(2):166–72. doi:10.1097/SHK.0000000000000396
56. Lappann M, Danhof S, Guenther F, Olivares-Florez S, Mordhorst IL, Vogel U. In vitro resistance mechanisms of Neisseria meningitidis against neutrophil extracellular traps. Mol Microbiol (2013) 89(3):433–49. doi:10.1111/mmi.12288
57. Ito T. PAMPs and DAMPs as triggers for DIC. J Intensive Care (2014) 2(1):67. doi:10.1186/s40560-014-0065-0
58. Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol (2015) 67(10):2780–90. doi:10.1002/art.39239
59. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol (2015) 67(11):2990–3003. doi:10.1002/art.39247
60. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood (2014) 123(18):2768–76. doi:10.1182/blood-2013-10-463646
61. Xu Z, Huang Y, Mao P, Zhang J, Li Y. Sepsis and ARDS: the dark side of histones. Mediators Inflamm (2015) 2015:205054. doi:10.1155/2015/205054
62. Qian Z, Gelzer-Bell R, Yang Sx SX, Cao W, Ohnishi T, Wasowska BA, et al. Inducible nitric oxide synthase inhibition of weibel-palade body release in cardiac transplant rejection. Circulation (2001) 104(19):2369–75. doi:10.1161/hc4401.098471
63. Tarantino E, Amadio P, Squellerio I, Porro B, Sandrini L, Turnu L, et al. Role of thromboxane-dependent platelet activation in venous thrombosis: aspirin effects in mouse model. Pharmacol Res (2016) 107:415–25. doi:10.1016/j.phrs.2016.04.001
64. Coppo P, Froissart A. Treatment of thrombotic thrombocytopenic purpura beyond therapeutic plasma exchange. Hematology Am Soc Hematol Educ Program (2015) 2015:637–43. doi:10.1182/asheducation-2015.1.637
65. Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, et al. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost (2009) 7(7):1155–62. doi:10.1111/j.1538-7836.2009.03459.x
66. Vanhoorelbeke K, Ulrichts H, Van de Walle G, Fontayne A, Deckmyn H, et al. Inhibition of platelet glycoprotein Ib and its antithrombotic potential. Curr Pharm Des (2007) 13(26):2684–97. doi:10.2174/138161207781662867
67. Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood (1996) 88(1):146–57.
68. Thomas MR, Storey RF. Effect of P2Y12 inhibitors on inflammation and immunity. Thromb Haemost (2015) 114(3):490–7. doi:10.1160/TH14-12-1068
69. Krishnamurthy VR, Sardar MY, Ying Y, Song X, Haller C, Dai E, et al. Glycopeptide analogues of PSGL-1 inhibit P-selectin in vitro and in vivo. Nat Commun (2015) 6:6387. doi:10.1038/ncomms7387
70. Ito T, Kawahara K, Okamoto K, Yamada S, Yasuda M, Imaizumi H, et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler Thromb Vasc Biol (2008) 28(10):1825–30. doi:10.1161/ATVBAHA.107.150631
71. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207(9):1853–62. doi:10.1084/jem.20100239
72. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi:10.1038/nature00858
73. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol (2005) 5(4):331–42. doi:10.1038/nri1594
74. Ito T, Kawahara K, Nakamura T, Yamada S, Nakamura T, Abeyama K, et al. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost (2007) 5(1):109–16. doi:10.1111/j.1538-7836.2006.02255.x
75. Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell (2003) 115(2):139–50. doi:10.1016/S0092-8674(03)00803-1
76. Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med (2013) 19(10):1281–7. doi:10.1038/nm.3288
77. Schettert IT, Pereira AC, Lopes NH, Hueb WA, Krieger JE. Association between ADAMTS13 polymorphisms and risk of cardiovascular events in chronic coronary disease. Thromb Res (2010) 125(1):61–6. doi:10.1016/j.thromres.2009.03.008
78. Villeponteau B. Heparin increases chromatin accessibility by binding the trypsin-sensitive basic residues in histones. Biochem J (1992) 288(Pt 3):953–8. doi:10.1042/bj2880953
79. Napirei M, Ludwig S, Mezrhab J, Klöckl T, Mannherz HG. Murine serum nucleases – contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J (2009) 276(4):1059–73. doi:10.1111/j.1742-4658.2008.06849.x
80. Simes J, Becattini C, Agnelli G, Eikelboom JW, Kirby AC, Mister R, et al. Aspirin for the prevention of recurrent venous thromboembolism: the INSPIRE collaboration. Circulation (2014) 130(13):1062–71. doi:10.1161/CIRCULATIONAHA.114.008828
81. Lapponi MJ, Carestia A, Landoni VI, Rivadeneyra L, Etulain J, Negrotto S, et al. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J Pharmacol Exp Ther (2013) 345(3):430–7. doi:10.1124/jpet.112.202879
82. Oklu R, Albadawi H, Watkins MT, Monestier M, Sillesen M, Wicky S. Detection of extracellular genomic DNA scaffold in human thrombus: implications for the use of deoxyribonuclease enzymes in thrombolysis. J Vasc Interv Radiol (2012) 23(5):712–8. doi:10.1016/j.jvir.2012.01.072
83. Longstaff C, Varjú I, Sótonyi P, Szabó L, Krumrey M, Hoell A, et al. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J Biol Chem (2013) 288(10):6946–56. doi:10.1074/jbc.M112.404301
84. Albadawi H, Oklu R, Raacke Malley RE, O’Keefe RM, Uong TP, Cormier NR, et al. Effect of DNase I treatment and neutrophil depletion on acute limb ischemia-reperfusion injury in mice. J Vasc Surg (2015). doi:10.1016/j.jvs.2015.01.031
85. Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol (2015) 308(5):H500–9. doi:10.1152/ajpheart.00381.2014
Keywords: NETs, extracellular DNA, venous thromboembolism, venous thrombosis, sepsis, immunothrombosis
Citation: Kimball AS, Obi AT, Diaz JA and Henke PK (2016) The Emerging Role of NETs in Venous Thrombosis and Immunothrombosis. Front. Immunol. 7:236. doi: 10.3389/fimmu.2016.00236
Received: 02 May 2016; Accepted: 02 June 2016;
Published: 27 June 2016
Edited by:
Mariana Julieta Kaplan, National Institutes of Health, USACopyright: © 2016 Kimball, Obi, Diaz and Henke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter K. Henke, aGVua2VAbWVkLnVtaWNoLmVkdQ==