 Daniel Söderberg
Daniel Söderberg Mårten Segelmark
Mårten Segelmark- 1Department of Medical and Health Sciences, Linköping University, Linköping, Sweden
- 2Department of Nephrology, Linköping University, Linköping, Sweden
A group of pauci-immune vasculitides, characterized by neutrophil-rich necrotizing inflammation of small vessels and the presence of antineutrophil cytoplasmic antibodies (ANCAs), is referred to as ANCA-associated vasculitis (AAV). ANCAs against proteinase 3 (PR3) (PR3-ANCA) or myeloperoxidase (MPO) (MPO-ANCA) are found in over 90% of patients with active disease, and these ANCAs are implicated in the pathogenesis of AAV. Dying neutrophils surrounding the walls of small vessels are a histological hallmark of AAV. Traditionally, it has been assumed that these neutrophils die by necrosis, but neutrophil extracellular traps (NETs) have recently been visualized at the sites of vasculitic lesions. AAV patients also possess elevated levels of NETs in the circulation. ANCAs are capable of inducing NETosis in neutrophils, and their potential to do so has been shown to be affinity dependent and to correlate with disease activity. Neutrophils from AAV patients are also more prone to release NETs spontaneously than neutrophils from healthy blood donors. NETs contain proinflammatory proteins and are thought to contribute to vessel inflammation directly by damaging endothelial cells and by activating the complement system and indirectly by acting as a link between the innate and adaptive immune system through the generation of PR3- and MPO-ANCA. Injection of NET-loaded myeloid dendritic cells into mice results in circulating PR3- and MPO-ANCA and the development of AAV-like disease. NETs have also been shown to be essential in a rodent model of drug-induced vasculitis. NETs induced by propylthiouracil could not be degraded by DNaseI, implying that disordered NETs might be important for the generation of ANCAs. NET degradation was also highlighted in another study showing that AAV patients have reduced DNaseI activity resulting in less NET degradation. With this in mind, it might be that prolonged exposure to proteins in the NETs due to the overproduction of NETs and/or reduced clearance of NETs is important in AAV. However, not all ANCAs are pathogenic and some might possibly also aid in the clearance of NETs. A dual role for ANCAs in relation to circulating NET levels has been proposed because a negative correlation was observed between PR3-ANCA and NET remnants in patients in remission.
Antineutrophil Cytoplasmic Antibody-Associated Vasculitis
Vasculitides are inflammations in the walls of blood vessels, and they can affect any organ system in the body. They are divided into broad groups based on the size of the vessels predominantly being affected. A subgroup of small-vessel vasculitides is characterized by a scarcity of immune depositions (pauci-immune) and the presence of antineutrophil cytoplasmic antibodies (ANCAs) and is referred to as ANCA-associated vasculitis (AAV) (1). AAV comprise three diseases, including granulomatosis with polyangiitis [GPA, previously known as Wegener’s granulomatosis (2)], microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA, previously known as Churg–Strauss syndrome) (3). GPA and EGPA share the feature of necrotizing granulomatous inflammation of the lower respiratory tract, whereas MPA is characterized by the absence of this component. Also, GPA often affects the upper respiratory tract and can result in rhinitis, otitis, and cartilage destruction, while eosinophilia and asthma are defining features of EGPA. Renal involvement is observed in as many as 90% of the patients with MPA, compared to 80% of the patients with GPA and 45% in EGPA. All three diseases affect the skin, joints, eyes, and nerves to various extents (1, 4). There is also an increased incidence of venous thromboembolism in AAV patients, especially during active disease (5, 6). AAVs are relapsing–remitting diseases, and 50% of the patients have a relapse within 5 years of successful treatment. The mortality rate is around 80% at 1 year when left untreated, but with current treatments, the mortality rate is reduced to 25% within 5 years (7).
Autoantibodies specific for proteinase 3 (PR3) (PR3-ANCA) or myeloperoxidase (MPO) (MPO-ANCA) are found in over 90% of patients with active disease (8), and these are important as diagnostic tools. The association between PR3- and MPO-ANCAs and active disease in AAV suggests a pathogenic role for the autoantibodies, and such a role is supported by results from animal models (9, 10) and in vitro studies showing that PR3- and MPO-ANCAs can activate neutrophils to produce reactive oxygen species (ROS) and proteolytic enzymes (11). ANCA-induced neutrophil activation also leads to increased adhesion of the neutrophils (12) and the activation of the alternative complement pathway (13) with the generation of C5a. C5a in turn potentiates the inflammatory response by priming neutrophils and acting as a chemoattractant to recruit more neutrophils to the inflammatory site (14). However, ANCA levels do not conclusively predict relapses (15, 16), and there is an unmet need for biomarkers for this purpose.
Neutrophil Extracellular Traps
Neutrophil extracellular traps (NETs) were first described in 2004 as a means for neutrophils to trap and kill bacteria (17) and are released as a result of a programed cell death mechanism referred to as NETosis (18, 19). NETs consist of a DNA backbone and various proteins with proinflammatory characteristics, such as histones, high-mobility group box 1 (HMGB1), LL37, neutrophil elastase (NE), calprotectin (S100A8/S100A9, MRP8/14), and, interestingly, MPO (Figure 1) and PR3 (20, 21). All described ANCA antigens are components of NETs. NETosis depends on a cascade of events that lead to the mixing of nuclear, cytoplasmic, and granular components before the NETs are released into the surrounding matrix (18). NETosis has been shown to depend on NADPH oxidase and ROS production as well as on autophagy and histone citrullination. Peptidyl arginine deiminase 4 (PAD4), NE, and MPO have been shown to play important roles in this signaling pathway (18, 22, 23). More recently, other forms of NETosis have also been described, including NETosis with the release of mitochondrial DNA (mtDNA ETs) (24) instead of nuclear DNA and ROS-independent NETosis (25–27). Interestingly, when releasing mtDNA ETs, the neutrophils can also remain viable (24). In addition to their role as antimicrobial agents, NETs of both nuclear and mitochondrial origin have also been connected to various autoinflammatory and autoimmune diseases (28–33).
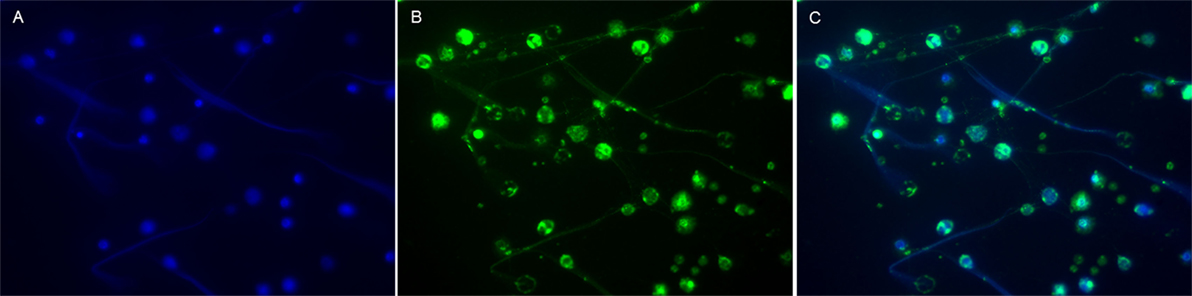
Figure 1. Visualization of MPO in NETs from human neutrophils. Neutrophils isolated from human peripheral whole blood were cultured for 4 h at 37°C with 25 nM PMA. NETs were then visualized by immunofluorescence microscopy using a 40× objective. (A) DNA, the backbone of NETs, was labeled with DAPI (blue). (B) MPO (clone 2B11), a granulae protein within the NETs (17), was labeled with a Dylight 488-conjugated antibody (green). (C) DNA and MPO (merged) co-localized in the NETs. NETs, neutrophil extracellular traps; PMA, phorbol-12-myristate-13-acetate. Blood samples were collected after obtaining informed consent in accordance with the declaration of Helsinki, and the study was approved by the Regional Ethical Review Board in Linköping. This figure is not intended to be quantitative, but only to serve as a representative image of common prior knowledge regarding the presence of MPO in NETs (17).
NETs are Present in Glomeruli, Skin Lesions, and Thrombi of AAV Patients
Dying neutrophils surrounding the walls of small vessels are a histological hallmark of AAV. Traditionally, it has been assumed that these neutrophils die by necrosis, but in 2009, Kessenbrock et al. showed that NETs were present in the glomeruli in kidney biopsies from AAV patients (34). They reported the presence of NETs as co-localizations of DNA, histones, and the granule proteins PR3, LL37, NE, and MPO in various combinations (34). This phenomenon was later confirmed by others (35–38). Although the method for detecting NETs in glomeruli was rather similar in these studies, i.e., visualization of DNA and histones (although some looked at citrullinated histones) in combination with the granule proteins already described – one study also reported the presence of PAD4 in the NETs, which is necessary for histone citrullination (35), and another study detected LAMP2, which is also an ANCA antigen (36, 39).
Neutrophil extracellular traps have also been shown to be present in skin lesions (40, 41) and thrombi from AAV patients (38, 42). In the studies investigating NETs in skin lesions, the presence of NETs was based on extracellular MPO (40, 41) or on DNA in combination with MPO (41). The presence of NETs in thrombi was defined not only as co-localizations of DNA and MPO but also as citrullinated histones alone (38). Another study also defined NETs based only on the presence of citrullinated histones (42).
Increased Levels of NETs and NET-Associated Proteins in the Circulation of AAV Patients
In addition to the presence of NETs in various lesions from AAV patients, it has been shown that these patients also have elevated levels of NETs in the circulation (34, 43–46) (Table 1). In these studies, NETs were defined as nucleosome/MPO complexes (34, 43, 46), total DNA or DNA/MPO or citrullinated histone 3 (H3) complexes (45), DNA/histone complexes (46), or as nuclear DNA or mtDNA (44). There are also several observations regarding circulating neutrophil components that are the main constituents of NETs. Important examples are HMGB1, calprotectin (S100A8/S100A9, MRP8/14), PR3, MPO, and NE (46–55) (Table 1). The study measuring calprotectin used longitudinally collected samples from the NORAM trial and found that calprotectin levels correlated with disease activity (47), and the studies measuring NE observed a correlation between NE and Birmingham Vasculitis Activity Score (i.e., disease activity) (51). However, the presence of these proteins in the circulation does not reveal whether they are released as a result of NETosis or by other mechanisms, but it was recently shown that the levels of MPO and NE correlate with the levels of DNA/MPO complexes in the circulation (46). The capability of using NETs as a biomarker to monitor disease activity in AAV has not been evident in previous studies. No study has so far measured the levels of NETs longitudinally in patients at multiple time points. In some cross-sectional studies, the levels of NETs have been measured in patients during both remission and active disease, but with inconclusive results regarding their correlation with disease activity (43, 45).
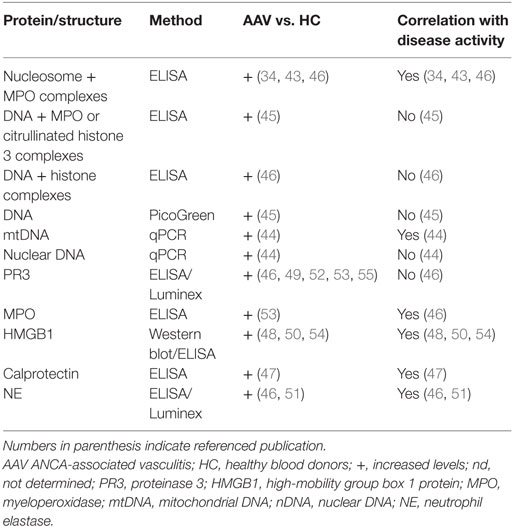
Table 1. NET-associated proteins and structures present in the circulation of AAV patients.
Proinflammatory Aspects of NETs in AAV
Neutrophil extracellular traps have previously been described as double-edged swords of innate immunity (56), considering that they are involved in both fighting pathogens and in contributing to autoinflammatory and autoimmune conditions. Various proinflammatory aspects of NETs in general might also be important in the pathogenesis of AAV. For example, NETs can cause endothelial damage (57–59) and can activate the alternative complement pathway (60), which, as already mentioned, plays an important role in amplifying the inflammatory process in AAV. Further, anti-histone antibodies have been shown to ameliorate experimental glomerulonephritis, emphasizing the proinflammatory aspect of histones in the NETs (61). It has also been shown that the presence of histones in NETs can contribute to thrombus formation (62) and that the presence of tissue factor (63, 64) in NETs can contribute to the generation of thrombin. In turn, it has been demonstrated that activated platelets can stimulate neutrophils to release NETs and that platelet-induced NETs propagate deep vein thrombosis in mice (65). Others have shown that HMGB1 expressed on platelets mediate the formation of platelet-induced NETs and that this process is dependent on autophagy (66), and in mice, it has been shown that platelet-derived p-selectin can induce NETosis (67). Increased levels of platelet-neutrophil aggregates and soluble P-selectin have been observed in the circulation of AAV patients during active disease and to correlate with disease activity (46). Additionally, HMGB1 has also been shown to potentiate the effect of ANCAs on NET formation (68). Oxidized mtDNA ETs released from neutrophils in systemic lupus erythematosus (SLE) have been shown to possess proinflammatory characteristics (33), and the role of mtDNA in general as a danger-associated molecular pattern has been extensively described (69).
Spontaneous NET Formation In Vitro
Earlier studies have shown that neutrophils from AAV patients are less prone to undergo apoptosis (70), suggesting that these neutrophils are more prone to other forms of cell death. Indeed, in vitro studies have shown that neutrophils from AAV patients are more prone to release NETs spontaneously than neutrophils from healthy blood donors (36, 43, 71). A subpopulation of neutrophils, referred to as low-density granulocytes (LDGs), have been shown to spontaneously release NETs significantly more than normal-density neutrophils, and these LDGs haves been proposed to be the major source of NETs in AAV (71). However, the same study also showed that normal-density neutrophils from AAV patients spontaneously released more NETs than normal-density neutrophils from healthy blood donors (71). More detailed studies of LDGs in SLE have revealed that LDGs express increased levels of mRNA of various immunostimulatory bactericidal proteins and alarmins compared to normal-density neutrophils (59). It is important to note that during the various isolation procedures normally used to obtain neutrophils from peripheral whole blood, LDGs will not be included because they will be found in the fraction of peripheral blood mononuclear cells. This is important to consider in future in vitro studies of neutrophils and NET formation.
ANCAs as Mediators of NETosis
In addition to the effects already ascribed to PR3- and MPO-ANCA in terms of neutrophil activation, they are also capable of inducing NETosis (Figure 2) (34). Although the exact mechanism for neutrophil activation by ANCAs is not clear, full activation requires binding of autoantibodies to both Fc-receptors and to PR3/MPO on the surface of neutrophils (72). It has been suggested that neutrophil activation, in this case evaluated as ROS production by MPO-ANCA, is epitope-specific, that epitope specificity varies with disease activity and that ANCAs activate neutrophils more robustly during active disease (73). Furthermore, in vitro studies have shown that neutrophils from patients are more easily activated (they produce more ROS) by ANCAs (in this case PR3-ANCA) than neutrophils from healthy blood donors (74). It has previously been shown that neutrophils from AAV patients possess increased membrane expression of PR3 (75, 76), which could possibly be explained by disrupted epigenetic silencing of the PR3 and MPO gene in these patients (77). However, in the study by Ohlsson et al. the results could not be explained by increased PR3 expression on the cell surface of neutrophils from patients or the ANCA levels (74). Rather, epitope specificity and affinity seemed to be of importance for the antibodies’ ability to activate neutrophils (74). It has also been shown that MPO-ANCA has higher affinity for MPO during active disease and that MPO-ANCA induces more NETs during active disease (78), and the observation that the affinity for MPO-ANCA is important for the ability to induce NETs was recently confirmed by another group (79). In summary, it seems that both epitope specificity and affinity are important for neutrophil activation by ANCAs and that at least the affinity is important for their ability to induce NETs.
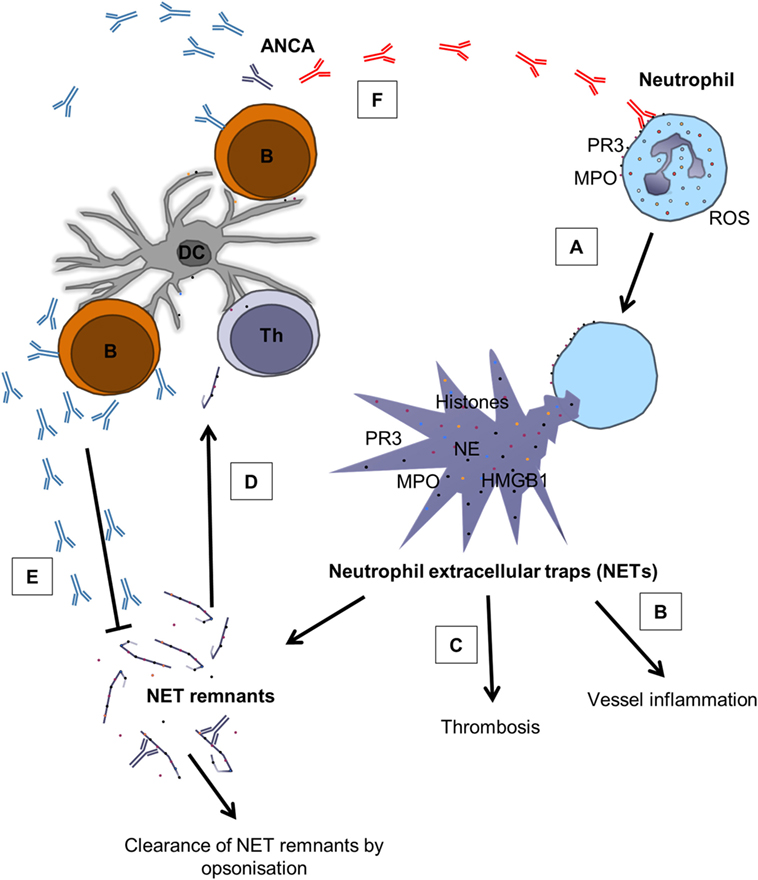
Figure 2. The role of NETs in AAV and the complex relation between ANCAs and NETs. (A) Pathogenic ANCAs (red) reacting with PR3 and MPO on the surface of neutrophils cause ROS production and the release of NETs through NETosis (34, 78, 79). (B) NETs contain various proinflammatory mediators, such as histones, HMGB1, PR3, MPO, and NE (17, 20, 21), and contribute to vessel inflammation by damaging endothelial cells (57–59) and by activating the complement system (60). (C) NETs do also promote thrombosis through the expression of histones (62) and tissue factor (63, 64). (D) NETs can also act as a link between the innate and adaptive immune system through the generation of ANCAs (41, 80). (E) ANCAs seem to belong to repertoire of “natural” antibodies (81), indicating that not all ANCAs are pathogenic, and it has been proposed that ANCAs can aid in clearance of circulating NET remnants (43). (F) However, under unfavorable circumstances, pathogenic ANCAs (red) are produced, creating a vicious circle that promotes inflammation. B, B cell; Th, T helper cell; DC, dendritic cell. Modified from Ref. (43) with permission from Oxford University Press.
NETs: Bridging Innate and Adaptive Immunity
It has been shown using NETotic neutrophils from mice that MPO and PR3 can be taken up from the NETs by myeloid dendritic cells (mDCs) and that injection of NET-loaded mDCs into mice results in circulating MPO- and PR3-ANCA and development of AAV-like disease (41). The addition of DNaseI to the in vitro cultures prevented PR3 and MPO uptake by the mDCs from the NETs, and when mice were injected with those mDCs, the mice did not develop disease (41). In the same study, injection of mDCs cocultured with apoptotic neutrophils into mice also caused autoantibody production, but those mice did not develop AAV-like disease. These experiments indicate that NETs show higher immunogenicity than apoptotic cells and that the structural integrity of the NETs is important for transferring NET-antigens to mDCs and the subsequent production of pathogenic autoantibodies. This is in line with a previous study showing that rats immunized with apoptotic neutrophils do develop ANCAs, but not disease (82). In another study, rats were immunized with NETs induced by phorbol-12-myristate-13-acetate (PMA) and propylthiouracil (PTU) (which together induced abnormal NETs that could not be degraded by DNaseI) or were given PTU orally in combination with PMA (intraperitoneal injection), and these rats developed MPO-ANCA and pulmonary capillaritis or glomerulonephritis and pulmonary capillaritis, respectively (80). This resembles the situation in humans, where over 20% of patients with Graves’ disease treated with PTU develop MPO-ANCA and some also AAV-like disease (83, 84).
NET Formation vs. Clearance: The Importance of Balance
The studies described earlier imply that NETs can act as a link between the innate and adaptive immune system with the production of pathogenic ANCAs. With this in mind, it might be that prolonged exposure to the proteins in the NETs due to the overproduction of NETs and/or reduced clearance of NETs is important in AAV. In line with this, it has been shown that AAV patients have reduced capacity to degrade NETs in vitro (78). This could possibly be due to the reduced DNaseI activity observed in these patients compared to healthy blood donors, although DNaseI activity did not correlate with disease activity. Thus, the elevated levels of NETs in the circulation of AAV patients might also be explained by the reduced capacity to clear the NETs from the circulation. Interestingly, low levels of both PR3-ANCA and MPO-ANCA can be found in the circulation of healthy individuals (81), indicating that the presence of ANCAs does not necessarily lead to disease development. Rather, ANCAs might be part of the repertoire of natural antibodies that are important for maintaining homeostasis (85). In line with this, a dual role for ANCAs was recently suggested, where the autoantibodies in addition to inducing NET formation can also aid in the clearance of NETs (Figure 2) (43), possibly through opsonization and the formation of immune complexes. This hypothesis was proposed because a negative correlation was observed between PR3-ANCA and circulating NET remnants in AAV patients in remission (43). As others have shown that the pathogenicity of ANCAs seems to vary with both epitope specificity (73) and affinity (78) and that these parameters change with disease activity, it appears that ANCAs might play different roles at different stages of AAV. Together, these studies might suggest how and why all individuals can possess ANCAs but only some develop AAV.
Infections and ANCAs
Antineutrophil cytoplasmic antibodies are common in chronic infections, such as Pseudomonas aeruginosa infections, in patients with cystic fibrosis, tuberculosis, HIV, and infective endocarditis (86–89). Infections are also implicated in the pathogenesis of AAV and as a trigger of relapses. Molecular mimicry, either directly (90) or indirectly through autoantigen complementarity (91), is the traditional way to explain the relationship between AAV and infection. However, infections lead to neutrophil activation, which triggers NETosis. Lipopolysaccharide-activated platelets can also activate neutrophils to release NETs (92), and this suggests an indirect way in which bacteria can contribute to NETosis as well as to the coagulation cascade and thrombosis formation discussed earlier. In sepsis, the liver sinusoids are filled with neutrophils undergoing NETosis (93), and in infective endocarditis, a role for NETs has also been described (94). ANCAs are found in up to 20% of patients with endocarditis (95), and many of these patients have symptoms resembling vasculitis, such as fever, increased CRP, weight loss, malaise, multiform skin lesions, and renal involvement (1, 96–98).
Concluding Remarks/Discussion
This review has outlined the role of NETs in the pathogenesis of AAV. There is compelling evidence that NETs contribute to vessel inflammation directly by damaging endothelial cells and by activating the complement system and indirectly by acting as a link between the innate and adaptive immune system through the generation of PR3-ANCA and MPO-ANCA. This can lead to a vicious circle because ANCAs can activate neutrophils. However, ANCA pathogenicity is dependent on both affinity and epitope specificity, and there also seem to be ANCAs that are non-pathogenic and even beneficial. NETs are most probably formed at a constant rate in healthy individuals, but NET formation can become highly elevated by infections, certain drugs, and possibly by epigenetic changes as one age. Increased NET formation must be balanced by clearance mechanisms, which seem to include DNaseI and possibly autoantibodies with ANCA specificity. We hypothesize that under unfavorable circumstances some individuals (partly depending on genetics) develop pathogenic autoantibodies that can activate neutrophils thus creating a vicious circle resulting in widespread vessel wall inflammation.
Author Contributions
DS has written most of the text, but in close collaboration with MS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This review has been funded by Ingrid Asps Foundation, Swedish Society of Nephrology, and Swedish Renal Foundation.
References
1. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med (1997) 337(21):1512–23. doi:10.1056/NEJM199711203372106
2. Falk RJ, Gross WL, Guillevin L, Hoffman GS, Jayne DR, Jennette JC, et al. Granulomatosis with polyangiitis (Wegener’s): an alternative name for Wegener’s granulomatosis. Arthritis Rheum (2011) 63(4):863–4. doi:10.1002/art.30286
3. Gadola SD, Gross WL. Vasculitis in 2011: the renaissance of granulomatous inflammation in AAV. Nat Rev Rheumatol (2012) 8(2):74–6.
4. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi:10.1002/art.37715
5. Allenbach Y, Seror R, Pagnoux C, Teixeira L, Guilpain P, Guillevin L. High frequency of venous thromboembolic events in Churg-Strauss syndrome, Wegener’s granulomatosis and microscopic polyangiitis but not polyarteritis nodosa: a systematic retrospective study on 1130 patients. Ann Rheum Dis (2009) 68(4):564–7. doi:10.1136/ard.2008.099051
6. Stassen PM, Derks RP, Kallenberg CG, Stegeman CA. Venous thromboembolism in ANCA-associated vasculitis – incidence and risk factors. Rheumatology (Oxford) (2008) 47(4):530–4. doi:10.1093/rheumatology/ken035
7. Flossmann O, Berden A, de Groot K, Hagen C, Harper L, Heijl C, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis (2011) 70(3):488–94. doi:10.1136/ard.2010.137778
8. Kallenberg CG. Pathogenesis of ANCA-associated vasculitides. Ann Rheum Dis (2011) 70(Suppl 1):i59–63. doi:10.1136/ard.2010.138024
9. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest (2002) 110(7):955–63. doi:10.1172/JCI0215918
10. Little MA, Al-Ani B, Ren S, Al-Nuaimi H, Leite M Jr, Alpers CE, et al. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS One (2012) 7(1):e28626. doi:10.1371/journal.pone.0028626
11. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A (1990) 87(11):4115–9. doi:10.1073/pnas.87.11.4115
12. Radford DJ, Savage CO, Nash GB. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum (2000) 43(6):1337–45. doi:10.1002/1529-0131(200006)43:6<1337::AID-ANR16>3.0.CO;2-M
13. Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol (2007) 170(1):52–64. doi:10.2353/ajpath.2007.060573
14. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol (2009) 20(2):289–98. doi:10.1681/ASN.2008050497
15. Tomasson G, Grayson PC, Mahr AD, Lavalley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis – a meta-analysis. Rheumatology (Oxford) (2012) 51(1):100–9. doi:10.1093/rheumatology/ker280
16. Kemna MJ, Damoiseaux J, Austen J, Winkens B, Peters J, van Paassen P, et al. ANCA as a predictor of relapse: useful in patients with renal involvement but not in patients with nonrenal disease. J Am Soc Nephrol (2015) 26(3):537–42. doi:10.1681/ASN.2013111233
17. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi:10.1126/science.1092385
18. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176(2):231–41. doi:10.1083/jcb.200606027
19. Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE (2007) 2007(379):e11. doi:10.1126/stke.3792007pe11
20. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog (2009) 5(10):e1000639. doi:10.1371/journal.ppat.1000639
21. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi:10.1126/scitranslmed.3001201
22. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191(3):677–91. doi:10.1083/jcb.201006052
23. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ (2011) 18(4):581–8. doi:10.1038/cdd.2011.1
24. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16(11):1438–44. doi:10.1038/cdd.2009.96
25. Kraaij T, Tengstrom FC, Kamerling SW, Pusey CD, Scherer HU, Toes RE, et al. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmun Rev (2016) 15(6):577–84. doi:10.1016/j.autrev.2016.02.018
26. Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, et al. Classical ROS-dependent and early/rapid ROS-independent release of neutrophil extracellular traps triggered by Leishmania parasites. Sci Rep (2015) 5:18302. doi:10.1038/srep18302
27. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185(12):7413–25. doi:10.4049/jimmunol.1000675
28. Leffler J, Gullstrand B, Jonsen A, Nilsson JA, Martin M, Blom AM, et al. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res Ther (2013) 15(4):R84. doi:10.1186/ar4264
29. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5(178):178ra40. doi:10.1126/scitranslmed.3005580
30. Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol (2005) 66(11):1146–54. doi:10.1016/j.humimm.2005.11.003
31. Dwivedi N, Upadhyay J, Neeli I, Khan S, Pattanaik D, Myers L, et al. Felty’s syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum (2012) 64(4):982–92. doi:10.1002/art.33432
32. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther (2014) 16(3):R122. doi:10.1186/ar4579
33. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi:10.1038/nm.4027
34. Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi:10.1038/nm.1959
35. Yoshida M, Sasaki M, Sugisaki K, Yamaguchi Y, Yamada M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin Kidney J (2013) 6(3):308–12. doi:10.1093/ckj/sft048
36. Tang S, Zhang Y, Yin SW, Gao XJ, Shi WW, Wang Y, et al. Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin Exp Immunol (2015) 180(3):408–18. doi:10.1111/cei.12589
37. O’Sullivan KM, Lo CY, Summers SA, Elgass KD, McMillan PJ, Longano A, et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int (2015) 88(5):1030–46. doi:10.1038/ki.2015.202
38. Nakazawa D, Tomaru U, Yamamoto C, Jodo S, Ishizu A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front Immunol (2012) 3:333. doi:10.3389/fimmu.2012.00333
39. Flint SM, Savage CO. Anti-LAMP-2 autoantibodies in ANCA-associated pauci-immune glomerulonephritis. J Am Soc Nephrol (2012) 23(3):378–80. doi:10.1681/ASN.2012010065
40. Abreu-Velez AM, Smith JG Jr, Howard MS. Presence of neutrophil extracellular traps and antineutrophil cytoplasmic antibodies associated with vasculitides. N Am J Med Sci (2009) 1(6):309–13.
41. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood (2012) 120(15):3007–18. doi:10.1182/blood-2012-03-416156
42. Imamoto T, Nakazawa D, Shida H, Suzuki A, Otsuka N, Tomaru U, et al. Possible linkage between microscopic polyangiitis and thrombosis via neutrophil extracellular traps. Clin Exp Rheumatol (2014) 32(1):149–50.
43. Soderberg D, Kurz T, Motamedi A, Hellmark T, Eriksson P, Segelmark M. Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology (Oxford) (2015) 54(11):2085–94. doi:10.1093/rheumatology/kev217
44. Surmiak MP, Hubalewska-Mazgaj M, Wawrzycka-Adamczyk K, Szczeklik W, Musial J, Sanak M. Circulating mitochondrial DNA in serum of patients with granulomatosis with polyangiitis. Clin Exp Immunol (2015) 181(1):150–5. doi:10.1111/cei.12628
45. Wang H, Sha LL, Ma TT, Zhang LX, Chen M, Zhao MH. Circulating level of neutrophil extracellular traps is not a useful biomarker for assessing disease activity in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS One (2016) 11(2):e0148197. doi:10.1371/journal.pone.0148197
46. Surmiak M, Hubalewska-Mazgaj M, Wawrzycka-Adamczyk K, Szczeklik W, Musial J, Brzozowski T, et al. Neutrophil-related and serum biomarkers in granulomatosis with polyangiitis support extracellular traps mechanism of the disease. Clin Exp Rheumatol (2016) 34(3 Suppl 97):98–104.
47. Pepper RJ, Hamour S, Chavele KM, Todd SK, Rasmussen N, Flint S, et al. Leukocyte and serum S100A8/S100A9 expression reflects disease activity in ANCA-associated vasculitis and glomerulonephritis. Kidney Int (2013) 83(6):1150–8. doi:10.1038/ki.2013.2
48. Bruchfeld A, Wendt M, Bratt J, Qureshi AR, Chavan S, Tracey KJ, et al. High-mobility group box-1 protein (HMGB1) is increased in antineutrophilic cytoplasmatic antibody (ANCA)-associated vasculitis with renal manifestations. Mol Med (2011) 17(1–2):29–35. doi:10.2119/molmed.2010.00132
49. Ohlsson S, Wieslander J, Segelmark M. Increased circulating levels of proteinase 3 in patients with anti-neutrophilic cytoplasmic autoantibodies-associated systemic vasculitis in remission. Clin Exp Immunol (2003) 131(3):528–35. doi:10.1046/j.1365-2249.2003.02083.x
50. Wibisono D, Csernok E, Lamprecht P, Holle JU, Gross WL, Moosig F. Serum HMGB1 levels are increased in active Wegener’s granulomatosis and differentiate between active forms of ANCA-associated vasculitis. Ann Rheum Dis (2010) 69(10):1888–9. doi:10.1136/ard.2009.119172
51. Haubitz M, Schulzeck P, Schellong S, Schulze M, Koch KM, Brunkhorst R. Complexed plasma elastase as an in vivo marker for leukocyte activation in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum (1997) 40(9):1680–4. doi:10.1002/art.1780400918
52. Henshaw TJ, Malone CC, Gabay JE, Williams RC Jr. Elevations of neutrophil proteinase 3 in serum of patients with Wegener’s granulomatosis and polyarteritis nodosa. Arthritis Rheum (1994) 37(1):104–12. doi:10.1002/art.1780370116
53. Ohlsson S, Hellmark T, Pieters K, Sturfelt G, Wieslander J, Segelmark M. Increased monocyte transcription of the proteinase 3 gene in small vessel vasculitis. Clin Exp Immunol (2005) 141(1):174–82. doi:10.1111/j.1365-2249.2005.02819.x
54. Wang C, Gou SJ, Chang DY, Yu F, Zhao MH, Chen M. Association of circulating level of high mobility group box 1 with disease activity in antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Care Res (Hoboken) (2013) 65(11):1828–34. doi:10.1002/acr.22187
55. Baslund B, Petersen J, Permin H, Wiik A, Wieslander J. Measurements of proteinase 3 and its complexes with alpha 1-proteinase inhibitor and anti-neutrophil cytoplasm antibodies (ANCA) in plasma. J Immunol Methods (1994) 175(2):215–25. doi:10.1016/0022-1759(94)90364-6
56. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol (2012) 189(6):2689–95. doi:10.4049/jimmunol.1201719
57. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7(2):e32366. doi:10.1371/journal.pone.0032366
58. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett (2010) 584(14):3193–7. doi:10.1016/j.febslet.2010.06.006
59. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187(1):538–52. doi:10.4049/jimmunol.1100450
60. Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol (2015) 181(3):518–27. doi:10.1111/cei.12654
61. Kumar SV, Kulkarni OP, Mulay SR, Darisipudi MN, Romoli S, Thomasova D, et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J Am Soc Nephrol (2015) 26(10):2399–413. doi:10.1681/ASN.2014070673
62. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107(36):15880–5. doi:10.1073/pnas.1005743107
63. Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol (2015) 67(10):2780–90. doi:10.1002/art.39239
64. Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis (2014) 73(10):1854–63. doi:10.1136/annrheumdis-2013-203430
65. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi:10.1084/jem.20112322
66. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12(12):2074–88. doi:10.1111/jth.12710
67. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126(2):242–6. doi:10.1182/blood-2015-01-624023
68. Ma YH, Ma TT, Wang C, Wang H, Chang DY, Chen M, et al. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res Ther (2016) 18:2. doi:10.1186/s13075-015-0903-z
69. Nakahira K, Hisata S, Choi AM. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid Redox Signal (2015) 23(17):1329–50. doi:10.1089/ars.2015.6407
70. Abdgawad M, Pettersson A, Gunnarsson L, Bengtsson AA, Geborek P, Nilsson L, et al. Decreased neutrophil apoptosis in quiescent ANCA-associated systemic vasculitis. PLoS One (2012) 7(3):e32439. doi:10.1371/journal.pone.0032439
71. Grayson PC, Carmona-Rivera C, Xu L, Lim N, Gao Z, Asare AL, et al. Neutrophil-related gene expression and low-density granulocytes associated with disease activity and response to treatment in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol (2015) 67(7):1922–32. doi:10.1002/art.39153
72. Kettritz R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clin Exp Immunol (2012) 169(3):220–8. doi:10.1111/j.1365-2249.2012.04615.x
73. Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, et al. Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest (2013) 123(4):1773–83. doi:10.1172/JCI65292
74. Ohlsson SM, Ohlsson S, Soderberg D, Gunnarsson L, Pettersson A, Segelmark M, et al. Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti-neutrophil cytoplasmic antibodies. Clin Exp Immunol (2014) 176(3):363–72. doi:10.1111/cei.12301
75. Witko-Sarsat V, Lesavre P, Lopez S, Bessou G, Hieblot C, Prum B, et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol (1999) 10(6):1224–33.
76. Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol (1994) 95(2):244–50. doi:10.1111/j.1365-2249.1994.tb06518.x
77. Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest (2010) 120(9):3209–19. doi:10.1172/JCI40034
78. Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol (2014) 25(5):990–7. doi:10.1681/ASN.2013060606
79. Yoshida M, Yamada M, Sudo Y, Kojima T, Tomiyasu T, Yoshikawa N, et al. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology (Carlton) (2016) 21(7):624–9. doi:10.1111/nep.12736
80. Nakazawa D, Tomaru U, Suzuki A, Masuda S, Hasegawa R, Kobayashi T, et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum (2012) 64(11):3779–87. doi:10.1002/art.34619
81. Cui Z, Zhao MH, Segelmark M, Hellmark T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int (2010) 78(6):590–7. doi:10.1038/ki.2010.198
82. Patry YC, Trewick DC, Gregoire M, Audrain MA, Moreau AM, Muller JY, et al. Rats injected with syngenic rat apoptotic neutrophils develop antineutrophil cytoplasmic antibodies. J Am Soc Nephrol (2001) 12(8):1764–8.
83. Wada N, Mukai M, Kohno M, Notoya A, Ito T, Yoshioka N. Prevalence of serum anti-myeloperoxidase antineutrophil cytoplasmic antibodies (MPO-ANCA) in patients with Graves’ disease treated with propylthiouracil and thiamazole. Endocr J (2002) 49(3):329–34. doi:10.1507/endocrj.49.329
84. Balavoine AS, Glinoer D, Dubucquoi S, Wemeau JL. Antineutrophil cytoplasmic antibody-positive small-vessel vasculitis associated with antithyroid drug therapy: how significant is the clinical problem? Thyroid (2015) 25(12):1273–81. doi:10.1089/thy.2014.0603
85. Panda S, Ding JL. Natural antibodies bridge innate and adaptive immunity. J Immunol (2015) 194(1):13–20. doi:10.4049/jimmunol.1400844
86. Carlsson M, Eriksson L, Pressler T, Kornfalt R, Mared L, Meyer P, et al. Autoantibody response to BPI predict disease severity and outcome in cystic fibrosis. J Cyst Fibros (2007) 6(3):228–33. doi:10.1016/j.jcf.2006.10.005
87. Flores-Suarez LF, Cabiedes J, Villa AR, van der Woude FJ, Alcocer-Varela J. Prevalence of antineutrophil cytoplasmic autoantibodies in patients with tuberculosis. Rheumatology (Oxford) (2003) 42(2):223–9. doi:10.1093/rheumatology/keg066
88. Cornely OA, Hauschild S, Weise C, Csernok E, Gross WL, Salzberger B, et al. Seroprevalence and disease association of antineutrophil cytoplasmic autoantibodies and antigens in HIV infection. Infection (1999) 27(2):92–6. doi:10.1007/BF02560504
89. Jansen TL, van Houte D, de Vries T, Wolthuis A. ANCA seropositivity in HIV: a serological pitfall. Neth J Med (2005) 63(7):270–4.
90. Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med (2008) 14(10):1088–96. doi:10.1038/nm.1874
91. Pendergraft WF III, Preston GA, Shah RR, Tropsha A, Carter CW Jr, Jennette JC, et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med (2004) 10(1):72–9. doi:10.1038/nm968
92. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi:10.1038/nm1565
93. Tanaka K, Koike Y, Shimura T, Okigami M, Ide S, Toiyama Y, et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One (2014) 9(11):e111888. doi:10.1371/journal.pone.0111888
94. Jung CJ, Yeh CY, Hsu RB, Lee CM, Shun CT, Chia JS. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation (2015) 131(6):571–81. doi:10.1161/CIRCULATIONAHA.114.011432
95. Mahr A, Batteux F, Tubiana S, Goulvestre C, Wolff M, Papo T, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol (2014) 66(6):1672–7. doi:10.1002/art.38389
96. Ying CM, Yao DT, Ding HH, Yang CD. Infective endocarditis with antineutrophil cytoplasmic antibody: report of 13 cases and literature review. PLoS One (2014) 9(2):e89777. doi:10.1371/journal.pone.0089777
97. Murdoch DR, Corey GR, Hoen B, Miro JM, Fowler VG Jr, Bayer AS, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med (2009) 169(5):463–73. doi:10.1001/archinternmed.2008.603
Keywords: neutrophil extracellular traps, ANCA-associated vasculitis, ANCA, NET, small-vessel vasculitis, NET remnants
Citation: Söderberg D and Segelmark M (2016) Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front. Immunol. 7:256. doi: 10.3389/fimmu.2016.00256
Received: 30 April 2016; Accepted: 15 June 2016;
Published: 30 June 2016
Edited by:
Mariana Julieta Kaplan, National Institutes of Health, USAReviewed by:
Angelo A. Manfredi, Vita-Salute San Raffaele University, ItalyPeter Korsten, University Medical Center Göttingen, Germany
Copyright: © 2016 Söderberg and Segelmark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Söderberg, ZGFuaWVsLnNvZGVyYmVyZ0BsaXUuc2U=