 Abigail Basson
Abigail Basson Ashley Trotter
Ashley Trotter Alex Rodriguez-Palacios
Alex Rodriguez-Palacios Fabio Cominelli
Fabio Cominelli- 1Digestive Health Research Institute, Case Western Reserve University, Cleveland, OH, USA
- 2University Hospitals Case Medical Center, Cleveland, OH, USA
Numerous reviews have discussed gut microbiota composition changes during inflammatory bowel diseases (IBD), particularly Crohn’s disease (CD). However, most studies address the observed effects by focusing on studying the univariate connection between disease and dietary-induced alterations to gut microbiota composition. The possibility that these effects may reflect a number of other interconnected (i.e., pantropic) mechanisms, activated in parallel, particularly concerning various bacterial metabolites, is in the process of being elucidated. Progress seems, however, hampered by various difficult-to-study factors interacting at the mucosal level. Here, we highlight some of such factors that merit consideration, namely: (1) the contribution of host genetics and diet in altering gut microbiome, and in turn, the crosstalk among secondary metabolic pathways; (2) the interdependence between the amount of dietary fat, the fatty acid composition, the effects of timing and route of administration on gut microbiota community, and the impact of microbiota-derived fatty acids; (3) the effect of diet on bile acid composition, and the modulator role of bile acids on the gut microbiota; (4) the impact of endogenous and exogenous intestinal micronutrients and metabolites; and (5) the need to consider food associated toxins and chemicals, which can introduce confounding immune modulating elements (e.g., antioxidant and phytochemicals in oils and proteins). These concepts, which are not mutually exclusive, are herein illustrated paying special emphasis on physiologically inter-related processes.
Introduction
Inflammatory bowel diseases (IBD) are chronic, inflammatory disorders of the gastrointestinal tract that develop as a result of a deregulation of the T cell-mediated immune responses toward the intestinal bacteria. Attenuated response to pathogen recognition and clearance facilitated by impaired mucus barrier function, as a result of a bacterially driven, aberrant immune-mediated response in genetically susceptible hosts, are all characteristic of IBD (1–4). The inflammatory response is a critical component to host survival, particularly during infection; however, the specific nature of immune response, which is initiated by the host to eradicate the infectious invader, depends on the type of pathogen, and thus factors such as pathogen localization (intracellular, extracellular) and endotoxins contained within pathogens [e.g., lipopolysaccharide (LPS), and lipoteichoic acid] or exotoxins secreted by pathogens (e.g., Staphylococcus aureus, Streptococcus pyogenes) (5, 6), may have direct and indirect consequences on the severity and maintenance of host inflammatory responses to pathogenic and commensal bacteria (7). Intracellular pathogens and all viruses activate professional phagocytes and generation of cytotoxic T-lymphocytes, whereas pathogen-specific antibodies are required for extracellular pathogens.
To date, it is unclear to what extent the inflammatory response in IBD is due to host inflammatory reactivity to gut local microbial molecules, or whether the changes observed in the gut microbiota (dysbiosis) are a consequence reflecting the level of intestinal inflammaton. Host tissue damage is an indirect consequence of the natural cascades initiated by host inflammatory responses, which may derive from exposure to uncontrolled concentrations of LPS. Toll-like receptor 4 (TLR4), stimulated by soluble LPS, activates intracellular signaling that results in nuclear factor-kappa beta (NF-kB) activation, and subsequent production of interleukin-1beta (IL-1β), tumor necrosis factor beta (TNFβ), interleukin-6 (IL-6), and nitric oxide synthase (iNOS) (8). Nitric oxide (NO) is synthesized from amino acid l-arginine, via nitric oxide synthases (NOS), can be generated by the inducible isoform of NOS (NOS-2 or iNOS) through a Ca2+-independent pathway (9). Herein, we present a synopsis of metabolic examples where mucosal interactions between genetics, diet, and microbiome may be relevant in triggering, modulating, or alleviating inflammation in so-called IBD from data available in murine models and in humans affected particularly with CD.
Genetics and Gut Microbiota
Theoretically, a healthy symbiotic host–microbe relationship is necessary for the normal development of gut mucosal immunity to maintain intestinal homeostasis and prevent excessive uncontrolled periods of local inflammation (10). The gut microbiota also prevents the colonization and virulence of pathogens, while promoting epithelial-barrier function, partly by promoting the renewal of epithelial cells. Host genetics and gene expression modulate immune mechanism of microbial molecular pattern recognition to influence the diversity and functionality of local microbiota (11). Since the discovery of the nucleotide oligomerization domain (NOD2) gene, numerous gene loci associated with abnormal innate immune responses (CARD15/NOD2, TLR4, CARD9, RAGE), differentiation of Th17-lymphocytes (IL-23R, JAK2, STAT3, CCR6, ICOSLG), autophagy (ATG16L1, IRGM, LRRK2, DEFB2/hBD2, SCL11A1), maintenance of epithelial barrier function (IBD5, DLG5, PTGER4, ITLN1, DMBT1, XBP1), and the initiation of secondary immune response (HLA-region, TNFSF15/TL1A, IRF5, PTPN2, PTPN22, NKX2-3, IL-12B, IL-18RAP, MST1) have been recognized for their role in CD pathogenesis (1, 2, 12–16). The discovery of novel susceptibility variants continues to grow (17, 18). Thus genetic alterations may influence immunity by either suppressing or promoting pathogenic microbial blooms, in turn, affecting epithelial-barrier integrity, host intestinal immunity and inflammation, all converging to regulate transient periods of susceptibility to IBD flare-ups.
Immune Cells and Gut Microbiota
Throughout the lifetime of an individual, the diversity and the composition of the microbiome are subject to change not only as a function of age but also of diet, environment (hygiene and demographics), and lifestyle (19, 20). The immunologic reactiveness to dietary and microbial antigens locally is primarily tolerogenic; maintained by the innate immune system ability to recognize antigen patterns, which determines the level and direction of T cell reactivity (21–23). Intestinal macrophages sense and respond to intestinal microorganisms through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) (24). T helper (Th) cells originally divided into Th1 and Th2 subsets, have the Th1 cells as prime mediators of immunity to extracellular pathogens, due to their ability to secrete proinflammatory cytokine interferon gamma (INFy), which activates local and systemic macrophages (25). Complementary T cell subsets, proinflammatory Th17 (IL-17-secreting CD4+ T cell subset) and anti-inflammatory (tolerogenic to self-antigen and commensal bacteria) T regulatory cells (Tregs) (forkhead box P3; Foxp3) contribute to that balance (26, 27).
In the gut, Tregs function to suppress the proliferation and effector functions of other T cells, with imbalances between Treg and Th17 cells playing an intricate role in T cell-mediated inflammatory disorders, and also microbial immunity (26). Although Tregs and Th17 cells differentiate from the same T cell precursor (naïve T cells) pool, in murine models, it is the transforming growth factor (TGF-β) in the presence of retinoic acid, which drives differentiation to Tregs, whereas Th17 cells result from collective activity of TGF-β and IL-6, which in humans also necessitates IL-21 (28–31).
CD4+ T cells orchestrate pro- and anti-inflammatory immune responses, but this balance depends on naive CD4+ T cells differentiation into functionally distinct regulatory or effector subsets in secondary lymphoid organs (i.e., spleen, lymph nodes) (32). New data have expanded the CD4+ T cell differentiation framework to include Foxp3-independent activation of the CD4+ T cell regulatory axis via IL-27, a differentiation factor for regulatory type 1 cells (Tr1), a major class of IL-10 producing CD4+ T cells with important immunosuppressive functions, which lack Foxp3 expression (33). Folicular T helper cells (Tfh) also influence the CD4+ T cell balance by migrating from the T cell area to the B cell follicle via CCR7 downregulation and concurrent expression of homing chemokine receptor CXCR5 (34, 35), a crosstalk promoted by Tfh production of IL-21 through transcription factor BCL-6, which also promotes the Th1/Th17 profile (32). The BCL2 pathway has been implicated in STAT factors induced by IL-6 that in turn promotes IL-21 and TNFα production, and Th1-differentiated cells can adopt a Tfh-like phenotype by interacting with STAT proteins and downregulating BLIMP1 in the presence of IL-2 (36). Finally, discovery of the Th9 phenotype, via IL-9 produced by the transcription factor PU.1, and also the Th22 phenotype, via IL-22 produced by the aryl hydrocarbon receptor (AhR), suggests that CD4+ T cell populations are highly heterogeneous in nature. Delineation of this wide biological complexity is now commanding the implementation of computational modeling (37–39) as novel tools to describe the differentiation process of immune cell types, simulate their interplay between intricate pathways in context to immune-mediated disorders and also diet, synthesize, and advance novel hypotheses (32).
Computational Biology and Mathematical Modeling
If microbial dynamics are host specific, mathematical-based predictive strategies could be used for interventions modulating microbiota. Conversely, some interactions between gut microbial communities may be found universal, i.e., consistent across hosts. Knowledge of such parameters could be combined across different studies making it useful for the development of common mathematical models (40). An example of such development includes the elegant use of dissimilarity-overlap curve (DOC) tests, which have been useful to assess, for instance, whether microbial communities within a specific body part have the same underlying dynamics across individuals (41), and whether subjects with reccurent Clostridiun difficile infection have comparable gut microbial behavior before and after fecal material transplantation (FMT) (41).
Modeling systems enabled for the study of mucosal and nutritional immunology have been recently reviewed (32, 39). In brief, primary examples of successfully implemented modeling approaches are the Modeling Immunity to Enteric Pathogens project (MIEP) and the Nutritional Immunology and Molecular Medicine Laboratory. In an elaborate series of experiments that included computational-based drug design methods, biochemical and in vivo studies (42, 43), MIEP identified the lanthionine sythetase component cyclase-like protein (LANCL2), a molecular target of abscisic acids, a plant phytohormone with insulin-sensitizing (44–46) and immunomodulatory actions (47–49), as a target for drug development against inflammatory, immune-mediated, and metabolic disease (50). Following the validation of these results in experimental IBD mouse models, MIEP formulated these preliminary results into advanced machine-learning algorithms to design a Phase III clinical in silico trial comprised of synthetically generated CD patients (51). Other notable MIEP-based acheivements include the development of validated computational models of CD4+ T cell differentiation and function (32, 43, 52), and the characterization of CD4+ T cell (via IL-21) (43) and mucosal immune responses to Helicobacter pylori infection (53).
Gut Microbiota Changes in IBD
Several reviews have discussed the role of microbiota on gut immunology, and how genetically encoded mechanisms can shape the composition of the intestinal microbiome (11). Discussions are available on the role of the human gut microbiome in the pathogenesis of IBD, and on the emerging patterns of reduced microbial diversity and imbalances (i.e., “dysbiosis”) that occurs in IBD (10, 54), and in twins discordant for CD (20, 55–59).
Such patterns include a reduction in the number of several microbial species within the phylum Firmicutes, specifically the Clostridium clusters XIVa and IV as observed in CD, while Bacillus spp increase (10, 54, 58, 60–62). As a commensal species, Clostridium clusters XIVa and IV induce colonic Tregs (63), preventing the development of excessive inflammation, a process that seems facilitated by high local concentrations of TGF-β (64). Proteobacteria, particularly the presence of Escherichia coli (mainly AIEC) is higher in IBD (65–67), compared with healthy controls (59, 68). Enterobacteriacae are one of the families consistently increased within the Proteobacteria phylum in IBD, while reductions in Bifidobacterium, Lactobacillus, and Ruminococcaceae (particularly the butyrate-producing genus Faecalibacterum) are decreased within the Bacteroidetes phylum in ileal CD (69). Faecalibacterum prausnitzii has anti-inflammatory properties, and low abundances have being associated with higher risk for repeated CD surgery (70, 71). Differences in fungal (72, 73) and viral diversity (74) are also observed in CD patients, with more bacteriophages especially in non-ulcerated mucosa samples of CD patients (75).
Animal studies illustrate the role of genetic–virus interaction in CD. In conventional mice, the genetic–virus interaction between ATG16L1 and norovirus infection is required for CD-like onset (76), whereas mice lacking Dectin1 had increased susceptibility to chemically induced colitis due to altered responses to indigenous fungi, namely Candida tropicalis, which is also common in the stools of humans (77). Interestingly, a polymorphism in Dectin1 (CLEC7A) was associated with severe ulcerative colitis (UC) in humans (77, 78). Metagenomics have also identified functional differences in up to 12% of metabolic pathways of active IBD patients compared to controls, despite only the 2% genus-level changes observed in stool and intestinal biopsies specimens (69). No metabolic studies are available in mice affected by spontaneous forms of IBD (79, 80).
Intestinal and Adiposity Constrains on Microbial Abundance
Microbial colonization occurs from mouth to anus, although density and composition varies by location, intestinal transit rate, host secretions, environment, availability of dietary substrates, and intestinal structures. The acidic environment in the stomach limits bacterial growth mostly to H. pylori, but bacterial species progressively increase within the ileum, which is mainly colonized by Enterobacteria, Bacteriodes, and Clostridia (81). The colon (lowest pH) provides a favorable environment for anaerobic microbial growth where organisms harvest energy via host-derived nutrients from secreted mucin (mucus) and fermentation of non-digested dietary fibers. This results in the production of short chain fatty acids (SCFAs), acetate, propionate and butyrate, and also the gasses hydrogen (H2), carbon dioxide (CO2), methane (CH4), and hydrogen sulfide (H2S) (81). These factors alter intestinal motility and inflammation, mechanisms that can influence locally the mucosa or the enteric nervous and muscle system (82–84).
Despite the fact that more than sixty phyla exist in the bacterial world, in fecal samples of healthy human volunteers (85–87), two phyla, Firmicutes (~65%) and Bacteroidetes (~25%) (88), comprise most of the microbial species detected, suggesting that microbiota composition is subject to strong constraints (81). The remaining species belong to the phyla Actinobacteria (e.g., Bifidobacterium spp.), Proteobacteria (e.g., E. coli), Verrucomicrobia (e.g., Akkermansia muciniphilia), Fusobacteria, and Cyanobacteria (89), all capable of mediating metabolic conditions (obesity, diabetes) and local intestinal inflammation. Based on health status and diet, fecal metagenomic data have been used since 2010 (90) to classify human intestinal phenotypes into three broadly defined “enterotypes.” The hallmark for differentiation is genus dominance of Prevotella, Bacteroides, or Ruminococcus (91), but Ruminococcus can be subdivided by genus abundance of methane-producing Methanobrevibacter (92). Despite progress in this field, it is increasingly evident that intestinal microbial pattern phenotyping is rather more complex.
Body mass as surrogate for cumulative adiposity also seems to correlate with gut microbiota diversity and richness, although microbiome meta-analysis indicates that early findings are largely inconsistent. More predictability (less study-to-study variability) has been identified for IBDs. Individually, studies suggest that obesity correlates with an increased Firmicutes: Bacteroidetes ratio at the phylum level (93–99), and that FMT shifts the recipient gut microbiota profile based on the obesity-associated phenotype of the donor, both in humans and animals (100–102). Adipose tissue has been established to have a pro-inflammatory role (103, 104) via its function as endocrine tissue, secreting proteins, such as leptin, cytokines and chemokines (105). However, significant differences exist between subcutaneous and visceral fat tissue in how gene expression is modulated (106–111), particularly of inflammatory pathways (109, 110, 112–115). Transcriptomics have shown that dietary interventions for weight-loss returns the equilibrium between pro- and anti-inflammatory factors by macrophages of visceral adipose tissue origin, in that production of pro-inflammatory cytokines (e.g., tumor necrosis factor alpha; TNFα) reduce, while anti-inflammatory molecules (IL-10 and IL1-Ra) increase (108, 116, 117). Plasma levels of these proteins seem to reflect dietary interventions concerning weight loss or weight gain (118), with weight loss being induced by a very-low calorie diet and alterations in expression of dozens of genes (108), alterations expected to result in changes of local microbial abundance.
Compared to non-obese counterparts, one metagenome study found that obese humans share an inflammatory enterotype, having a higher prevalence of IBD-associated Bacteroides genus and R. gnavus species (119–122). Consequently, sub-division of microbiota diversity has been proposed, on the basis of bacterial genome diversity, termed “low gene count” and “high gene count” (119). Key metabolites associated with low gene count bacteria include modules for degradation of aromatic amino acids, β-glucuronide degradation, and dissimilatory nitrate reduction (direct reduction from nitrate to ammonium), all of which have deleterious effects on intestinal mucosa. There is now evidence that Type-2 Diabetes in humans, a condition often accompanied by overweight and obesity, is associated with microbial shifts portrayed by decreased Roseburia intestinalis, F. prausnitzii, and Akkermansia municiphila (123–126). Identified for its protective effects, A. mucinphila is a novel mucin-degrading bacterial species, which vastly colonize the mucus layer localized at the epithelial surface of the ileum and colon (127). Higher levels of the bacterium positively correlate to glucagon-like peptide 2 (GLP-2) levels, involved in gut barrier function (128). A number of murine studies have shown prebiotic (non-digestible carbohydrates) feeding favors A. mucinphila, improved gut barrier function, mucus layer thickness, and locally produced antimicrobials, including regenerating islet-derived 3-gamma (Reg3y) proteins (128–133). Strong evidence also exists to link the gut virome (early infections with enteric viruses) to the growing incidence of Type-1 diabetes in humans. Primarily driven by autoimmunity againt pancreatic cells, such enteric viral hypothesis, based on the type of virus and host genetics, can also intriguingly be protective (134–136).
Obviously, our understanding of host-regulated microbial abundances, the gut mucosa, and local virome is limited (Figure 1). Secretory IgA seems to be one increasingly understood mechanism to modulate bacterial abundance (137–140). Other multivariable interactions need to be elucidated to enable the use of therapeutic strategies to decrease the severity of IBD and metabolic conditions characterized by microbe-driven pro-inflammatory responses.
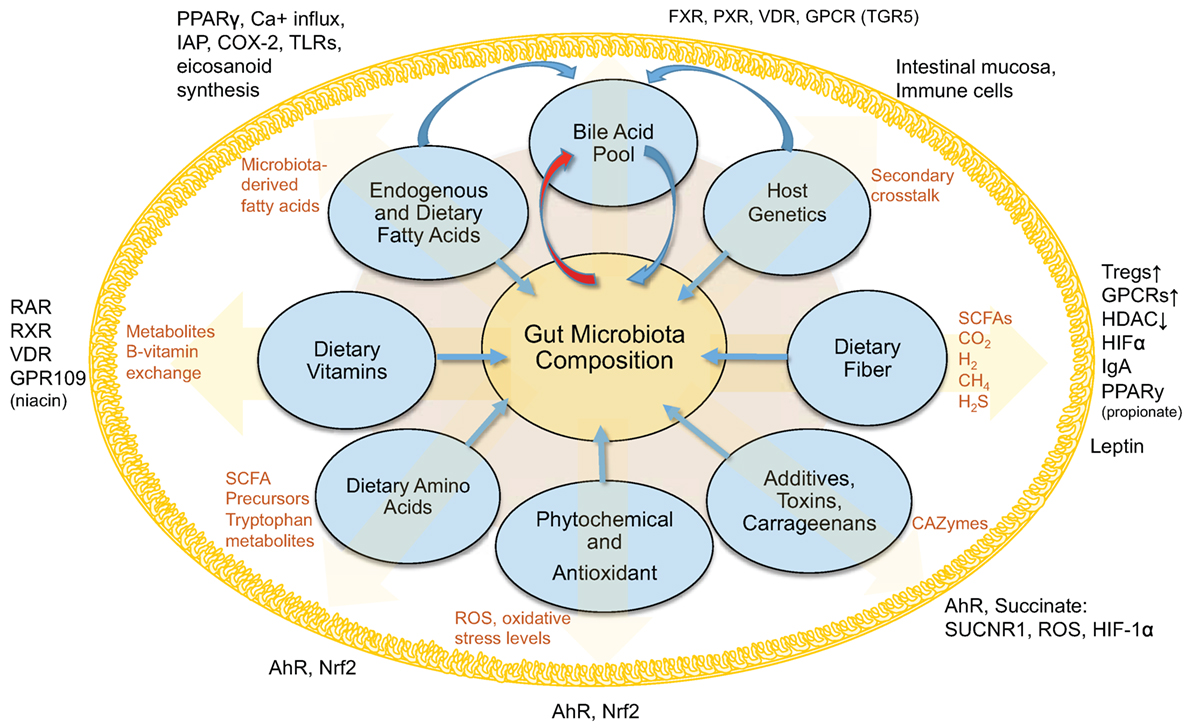
Figure 1. Interactions between diet, gut microbiota, and the host within the intestinal lumen. Figure depicts an overview of the interconnected (pantropic) interactions between diet, gut microbiota and the host within the intestinal lumen.
Dietary Fatty Acids
Clinically known as triglycerides, fats are the storage form of fatty acids, which differ in chemical structure. Depending on chemical structure, fatty acids influence membrane structure and fluidity (141, 142) and many are directly involved in intracellular signaling pathways, including receptor operated calcium channels (143), a key component in intracellular free calcium concentrations (144).
Short-Chain Fatty Acids
Of all components of the human diet, fats are generally believed to be associated with worsening of symptoms in IBD (145), despite the fact that dietary fats comprise diverse kinds of fatty acids [the basic molecules of fat, linear chains of carbon (C) surrounded by hydrogen]. Fatty acids can be classified as short-chain (<6 carbons, SCFA), medium-chain (6–12, MCFA), long-chain (12–21, LCFA), or very long chain (>22, VLCFA) fatty acids. Fatty acids also differ in the presence/absence of double carbon-to-carbon C:C bonds. Those without double C:C bonds are called saturated fatty acids (SFA), whereas one double bond is unsaturated (UFA), and more than one is polyunsaturated (PUFA). Most SCFAs are naturally saturated and are bacterially derived from dietary fibers, not fats. Dietary fibers come from the indigestible part of plant foods and substantially contribute to fecal SCFA concentrations, namely acetic acid (C2:0), propionic acid (C3:0), and butyric acid (C4:0), traditionally referred to by their conjugate bases as acetate, propionate (also known as propanoate), and butyrate (also known as butanoate) in the salt form.
Depending on the chemical structure, fatty acids may be absorbed directly into the blood via the portal vein and transported to the liver, or actively via chylomicrons by transport mechanisms where they could interfere with gene expression, metabolic pathways of microbial metabolism, or community composition. LCFAs are absorbed as small bile-covered micelles by enterocytes in regions of the small intestine, although the portal route has been described (146). Once inside intestinal cells, micelles are re-esterified to chylomicrons, a mixture of lipids, proteins, fat-soluble vitamins and cholesterol, surrounded by a lipoprotein coat. Chylomicrons (lipoproteins) are first transported to the lymphatic system, and then the thoracic system, where they are carried to several tissues, including the liver. Because of their shorter chain length, MCFAs can be absorbed by mucosal cells without esterification and directly transported to the liver (bypassing lymphatic system), where they are metabolized into CO2, ketones, and acetate. The exception is lauric acid (C12:0), which undergoes similar digestion as LCFAs. By comparison, most SCFAs are generated and utilized within the gut, contributing to host immune responses, by regulating Tregs numbers and function (136, 147–152) via putative epigenetic Treg-associated transcription factors (153, 154). Only a small percent of SCFAs exist in the gut as unionized forms and can be absorbed via passive diffusion across the cell membrane, or actively in an ionized state mediated by receptors abundantly expressed in the intestinal tract (i.e., monocarboxylate transporter 1, MCT-1; and sodium-coupled monocarboxylate transporter 1, SMCT-1) (155, 156). Alternatively, some acetate and propionate enter the portal blood to the liver, to be used in gluconeogenesis (155).
Regulation of colonic Tregs by SCFAs depends on the type of SCFA. Propionate and acetate mediate colonic migration of extraintestinal Tregs by upregulating G-protein coupled receptors (GPCRs), namely GPR15 in a GPR43-dependant manner, whereas butyrate plays a central role in de novo generation of colonic Tregs (157) by facilitating naive CD4+ T cell differentiation into Tregs via histone H3 acetylation in the promoter and CNS3 enhancer regulatory regions of Fox3p gene (149), or by altering the phenotype of dendritic cells (DCs) and inducing Treg differentiation via GPR109a activation (158–160). Histone deacetylates (HDACs) inhibition is also characteristic of butyrate, with HDAC9 inhibition shown to efficiently increase the proliferative and functional capabilities of Tregs through increased Fox3p expression (151, 155, 161). In addition, butyrate-mediated HDAC inhibition may facilitate the anti-inflammatory response by active suppression of pro-inflammatory cytokine production in innate cells such as macrophages and DCs, mediated through modulation of NF-kB (150, 162). Of note, in vitro administration of butyrate to human Tregs moderately decreased Treg proliferation, but increased their ability to inhibit T cell proliferation through a CTLA-4-mediated mechanism (152), with the inhibitory activities of butyrate on HDAC shown to stimulate effector CD4+ T cells, independently of Tregs (163). These observations underscore the crucial role of bacteria-derived metabolites in the development of the immune system, locally and systemically.
Non-digested fibers and proteins, or amino acids (glycine, threonine, glutamate, lysine, ornithine, and aspartate) derived from microbial fermentation, can be precursors of SCFA in the colon (164–168). Exception may include branched chain amino acids (BCAAs), valine, leucine, and isoleucine (169). Anaerobic bacterial utilization can produce acetate, which can then be used by other bacteria to generate butyrate (168). Threonine can also be used for production of propionate (170, 171). In addition to the SCFA mentioned, lactate is important because bacterial groups compete for lactate and its utilization or production by certain bacteria may result in the production of hydrogen sulfide and other SCFA.
Gut Microbiota and Polysaccharide Utilization
Disease-related variability of the microbiome composition is important because the production of SCFA is highly dependent on microbial metabolism of host and dietary polysaccharides (“glycans”), their mucosal absorption, and local immunomodulation. Figure 2 depicts an overview of gut microbiota interactions in SCFA production and degradation, including substrates associated with bacterial utilization. Bacteroides possess collectively more hydrolase genes for utilization of polysaccharides of dietary and host origin (172–175). This is important in mucosal immunology because germ-free (GF) murine colitis models colonized with specific commensal bacteria demonstrated localized bacterial adaptation to host IgA-mediated responses via switching capsular polysaccharides, preventing opsonization and mucosal clearance (176). Bacteria can also adapt to chronic T cell-mediated inflammation by downregulating genes involved in their growth, while inducing growth of bacterial genes involved in metabolism of host-derived products of innate immunity, such as in NO (177). An example is Bacteroides thetaiotaomicron, implicated in the pathogenesis of human and murine models of IBD (178), which has genes for utilization of host-derived polysaccharides, when dietary sources are lacking during disease (179, 180). Patients with CD often have decreased levels of butyrate, and other SCFAs, consistent with the decreases in SCFA-producing Firmicutes as seen in taxonomic profiling studies (69, 181). In line with reduced SCFA production in IBD (181), metagenomic and metaproteomic studies also confirm decreased microbial gene expression for butanoate (butyrate) and propanoate (propionate) metabolism in patients with ileal CD (69), and that increased mucin (a polysaccharide-rich host mucosal secretion) degradation is inversely related to the abundance of Firmicutes in active CD (181, 182). To illustrate the complex dynamic interactions in the gut mucosa, the beneficial properties of SCFA are minimized when the colonic concentrations of H2S increases as byproduct of bacterial fermentation, which inhibits host DNA repair (183, 184).
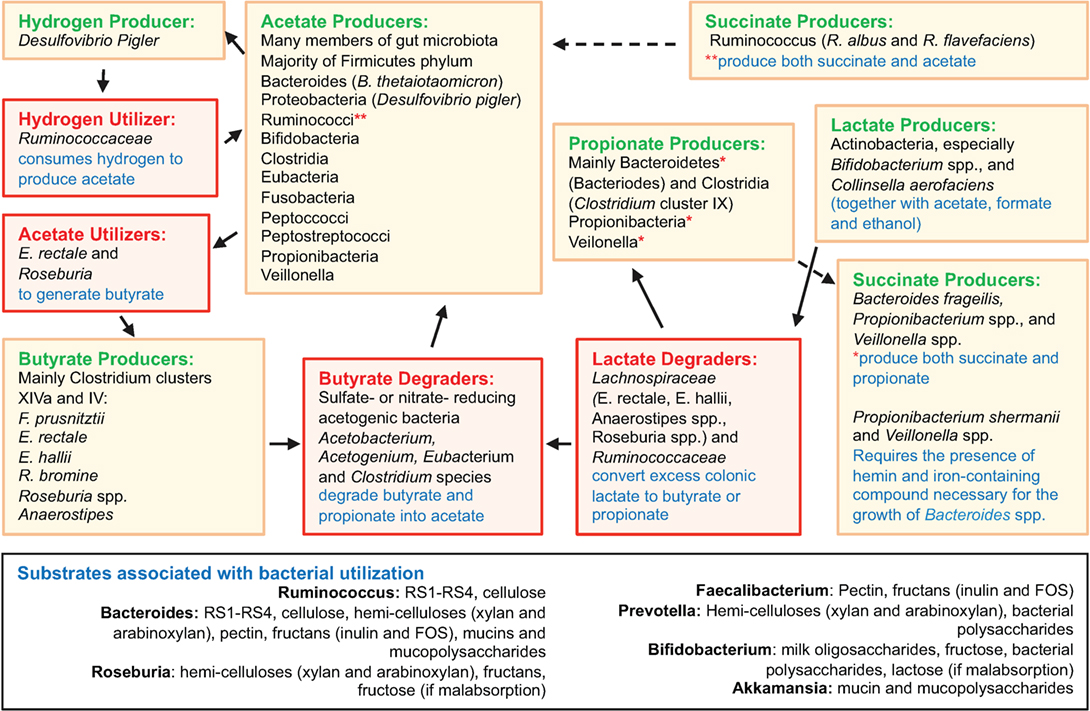
Figure 2. Overview of gut microbiota interactions in SCFA production and degradation. Figure depicts an overview of gut microbiota interactions in SCFA production and degradation, including substrates associated with bacterial utilization. Compared to Bacteroides spp., the Firmicutes phylum encompasses fewer genes for polysaccharide degradation, implying this phylum plays a vital role in nutrition metabolic pathways. Complex interactions also exist among intestinal fungi and dietary fibers (185). Compiled from Ref. (69, 164–168, 174, 175, 183, 185–206).
Role of SCFA in Mucosal Immunity
There are two major mechanisms of action by which SCFAs modulate host biological responses; namely, by direct inhibition of HDACs to directly regulate anti-inflammatory gene expression (207–210); and by acting as signaling molecules, activation of select GPCRs. As ligands, SCFAs bind with variable affinity to GPCRs, namely GPR43, GPR41 (renamed free fatty acid receptor 2 and 3; FFA2 and FFA3, respectively) (136, 211–213), and GPR109A (HCAR2; receptor for butyrate) (158, 211, 212) (Table 1).
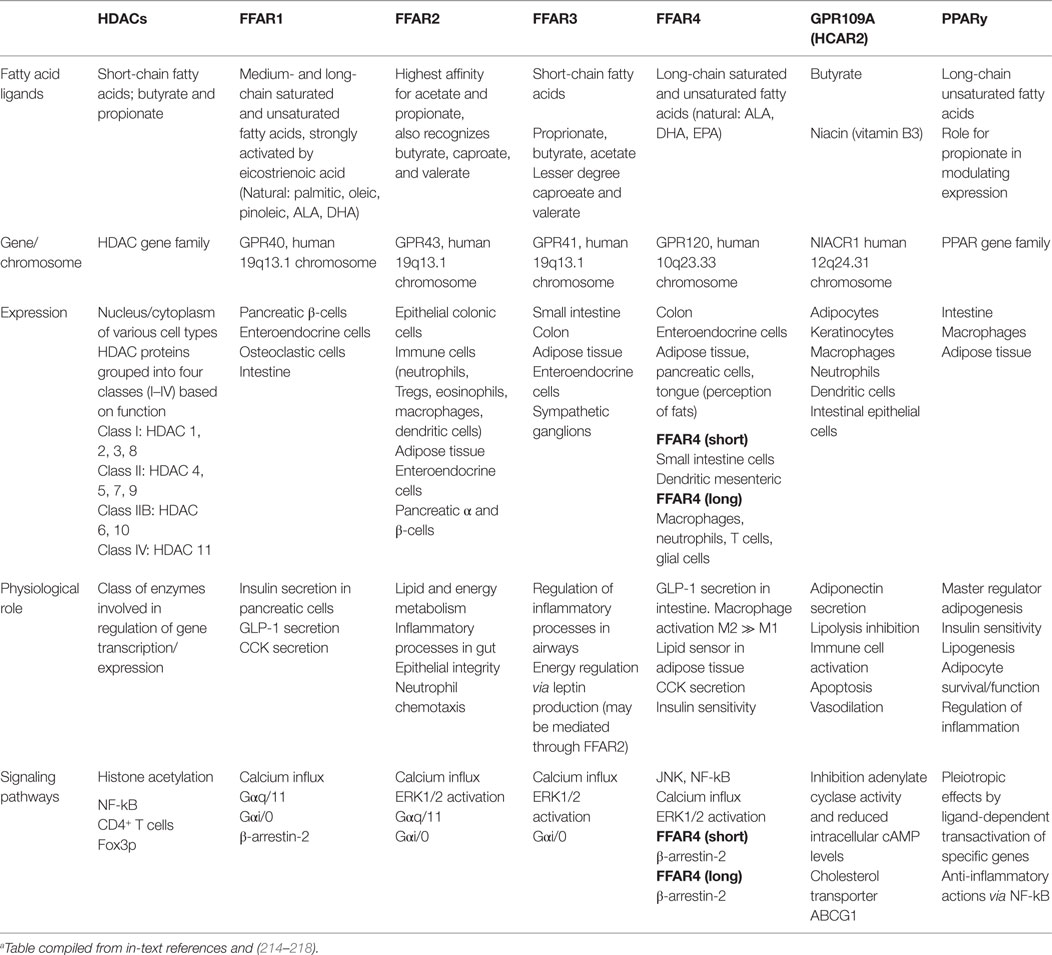
Table 1. Overview of fatty acid receptor ligands.a
Acetate, propionate, caproate (caproic acid; C6:0), and valerate (valeric acid; C5:0) are all recognized by FFA2/GPR43, and while acetate and propionate are the strongest activators, FFA2/GPR43 is the primary receptor for acetate (212, 219). FFA3/GPR41 has arguably higher affinity for propionate than for acetate and butyrate (155, 211–213, 219, 220). Caproate and valerate are recognized by FFA3/GPR41, but to a lesser extent (221). FFA2/GPR43 and FFA3/GPR41 are expressed extensively throughout the small intestine and colon (211, 222, 223), but have also been detected in organs outside of the gut. FFA2/GPR43 mRNA is present in adipose, skeletal, heart, and spleen tissues (211, 223, 224), being expressed on eosinophils, basophils (212), monocytes, neutrophils, DCs (212, 225), and mucosal mast cells (226).
SCFA-FFA2/GPR43 interactions profoundly affect inflammatory responses. Marked amplifications of inflammation in the intestine and other organs are observed in FFA2/GPR43-deficient mice, while GF mice expressing little or no SCFAs exhibit similar dysregulation of inflammatory responses (227). SCFA-induced FFA3/GPR41 expression is a potent inducer of leptin in adipose tissue, but this may be mediated via FFA2/GPR43 (228). Leptin functions to regulate body weight, hunger, satiety and body temperature, with increased expression in adipocytes in vitro after treatment with acetate and propionate (228–230); propionate studies have shown adipose human mRNA leptin stimulation in vivo (228). Interestingly, an overexpression of FFA2/GPR43 was reported in mice fed a high-fat diet, but supplementation of inulin-type fructans (class of dietary fibers that resist digestion), counteracted these effects, including peroxisome proliferator-activated receptor gamma (PPARy)-related adipogenesis (231). PPARy is a nuclear receptor involved in innate immune pathways, and PPARy deregulation has been associated with defective antimicrobial response against Candida albicans, Bacteroides fragilis, Entercoccus faecalis, and E. coli (232). Recent evidence also implicated propionate in modulating PPARy expression (208).
Microbial fermentation of dietary fibers associated with butyrate, propionate, and acetate is summarized in Table 2. Overall, SCFAs help maintain epithelial integrity by promoting mucus production (233, 234), by suppressing or altering pathways that produce various pro-inflammatory cytokines, chemokines (155) and adhesion molecules (235), or by interfering with inflammasome cytokines IL-8 and IL-22 (236). Butyrate appears to be the most potent with regard to host immunity in that it can actively suppress NF-kB formation (237, 238), repress production of TNFα, IL-6, and NO (162, 238, 239), and inhibit macrophage migration induced by bacterial LPS (240). Colonic epithelial cells also use butyrate as a main energy source (241, 242) and have been shown to promote “physiological hypoxia” by increasing colonic epithelial cell oxygen consumption (243), which in turn, supports gut barrier function through hypoxia-inducible factor (HIF) (244). Antibiotic therapy diminishes this effect, lowering both epithelial anaerobic metabolism and luminal SCFA concentrations, resulting in HIF destabilization and barrier dysfunction (243).
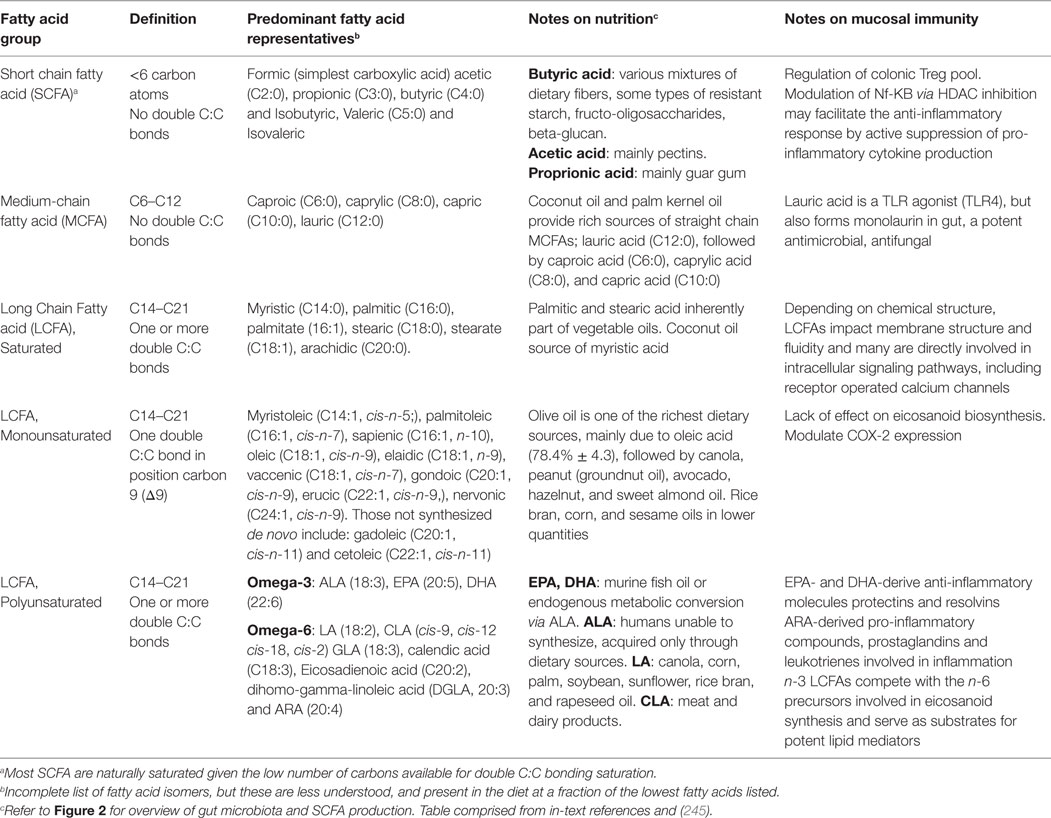
Table 2. Overview of fatty acids and features.
Long-Chain Fatty Acids
Monounsaturated Fatty Acids
Long-chain monounsaturated fatty acids (LCMUFAs; MUFAs) are largely represented by fatty acids formed by introducing a double bond in position carbon 9 (Δ9) counting from the carboxyl carbon. Major sources of long-chain monounsaturated fatty acids (e.g., olive oil) (245) are listed in Table 2.
In the clinical setting, olive oil-based n-9 lipid emulsions, administered parenterally, are considered an “immunoneutral” energy source and thus are used in combination with PUFAs for the therapeutic management of inflammatory illness (246). Epidemiologically, incorporation of MUFA-rich foods (e.g., olive oil), a cornerstone of the Mediterranean diet, is well-recognized for its beneficial effects on cardiovascular health (247), and because of its lack of effect on eicosanoid biosynthesis (248, 249), oleic acid is commonly used as the control fatty acid for PUFAs in dietary intervention trials. Notably however, several pieces of evidence demonstrate that olive oils (n-9) can modulate COX-2 expression (250), thus providing a possible mechanism by which various dietary oils, rich in n-3 and n-6 PUFAs, or n-9 MUFAs influence chronic inflammation. Olive oil has been shown to exert inhibitory effects on COX-2 expression in IL-10−/− mice, thereby decreasing the risk of neoplasia associated with chronic colitis (251). One study demonstrated a close clustering of colonic gene and protein expression profiles between two mouse genotypes, IL-10−/− and C57BL/6J, fed either AIN-76A diet (LA-rich; corn oil) or an oleic acid-enriched modified AIN-76A diet (252). Comparing the top five pathways between the oleic acid and LA diet within the same mouse genotype revealed nine gene expression changes, representing six unique genes within IL-10 mice, and within the C57BL/6J oleic acid-fed mice, 32 unique genes increased in expression when compared to the LA-fed C57BL/6J mice. Proteomics revealed more colonic protein abundance changes within the IL-10−/− mice (16 proteins) than in C57BL/6J mice (7 proteins), with colonic protein changes (IL-10−/− or C57 mice) associated with a number of metabolic (e.g., energy, carbohydrate, and lipid metabolism) and signaling processes (e.g., immune, apoptotic, cytoskeletal), as well as pronounced increase in lipid metabolism protein levels, such as fatty acid binding proteins FABP4 and FABP6, and those related to cell structure assembly and signaling (252). The effects of n-9 oleic acid on host inflammatory responses, in some circumstances, may also be more indirect in nature. In one mouse model of Type 2 Diabetes (T2DM) pre-treatment with oleic acid reversed the inhibitory effects of TNFα on insulin production, while in vivo, oleic acid-treated cells resulted in elevated translocation of the PPAR-activated receptor transcription factor to the nucleus (253).
Polyunsaturated Fatty Acids
Long-chain polyunsaturated fatty acids (LCPUFAs; PUFAs) are classified into omega-3 (n-3) and omega-6 (n-6) families (Table 2). The LCPUFA n-3 family includes alpha-linoleic acid (ALA; 18:3, n-3), and the most powerful LCPUFAs, eicosapentanoic acid (EPA; 20:5, n-3), and docosahexanenoic acid (DHA; 22:6, n-3), which can be directly acquired primarily in fish oils, or through endogenous metabolic conversion via ALA, their 18-carbon precursor. However, humans are unable to synthesize ALA. EPA- and DHA-derived compounds can result in derived molecules referred to as protectins and resolvins, potent anti-inflammatory mediators with distinct pathways of action (254–257) and specific-binding receptors (e.g., ChemR23, leukotriene B4 receptor 1, GPR32, PPARy, GPR120) (258, 259). Both EPA and DHA can generate “resolution-phase interaction products” (resolvins) with the DHA-derived D-series resolvin, 17-HDHA (17-hydroxy-docosahexaenoic acid), and the EPA-derived E-series resolvin, 18-HEPE (18-hydroxy-eicosapentaenoic acid), characterized as the central pathway precursors and metabolites (260). Omega-6 LCPUFAs includes linoleic acid (LA; 18:2, n-6), gamma-linoleic acid (GLA; 18:3, a product of LA metabolism in the body), conjugated linoleic acid (CLA), and arachidonic acid (ARA; 20:4, n-6), as well as the ARA-derived pro-inflammatory eicosanoids, prostaglandins (PG; includes PTE2 and PGD2), and leukotrienes (LT; includes LTB4 and LTE4). Biological functions of these eicosanoids include; vascular permeability and chemotactism in immune cells, transmigration of leukocytes and neutrophils into inflamed intestinal tissue involving adhesion molecules, and initiation of acute inflammation (261). Specifically, LTE4 upregulates COX-2 expression resulting in PGD2 generation in mast cells by a PPARy-dependent mechanism (262). Of note, increased levels of PGs with excessive production of other ARA-derived eicosanoids are observed in inflamed IBD mucosa, correlating with disease activity (263).
The effect of fatty acids in inflammation has been studied pre-clinically at various doses using primarily mouse models. Dextran Sulfate Sodium (DSS) colitis is a widely used murine model of IBD as it diminishes murine colonic regeneration eliciting inflammatory cell infiltration into colonic mucosa. Among DSS models, the Fat 1 mouse model of endogenously increased n-3 PUFA, uniquely characterized by the de novo n-3 PUFA synthesis from n-6 PUFA, has served to prove the anti-inflammatory effects of n-3 PUFAs (264–268). This model is important because of its ability to address molecular events underlying the effect of n-3 fatty acids in the absence of restricted dietary interventions, which may introduce potential confounding factors. There is however growing evidence that dietary n-3 PUFAs (DHA, EPA) also impair host immunity, increase B-cell function, and inherently modulate humoral responses (269), although fatty acid composition and dosage govern these responses. For instance, lower dietary concentrations of n-3 LCPUFA (1% as; fish oil, EPA, DHA) are protective by inhibiting pro-inflammatory cytokines (270–272), whereas higher concentrations (6–8%) exacerbated DSS colitis (6, 251, 273). These LCPUFAs are competitive antagonists of lipoxygenase (LOX) and cyclooxygenase (COX), key enzymes in the biosynthesis of prostaglandins via ARA oxidation (260, 274, 275). n-3 LCPUFAs also compete with the n-6 precursors involved in the synthesis of eicosanoids (276, 277) and serve as substrates for potent lipid mediators and significant changes in the lipidome and eicosanome are seen in the presence of high fatty acid abundance (260, 274).
Genetics Determine Binary Inflammatory Effect of Fat Diets and Microbiome
In addition to fatty acid composition and dosage, host genetics (genomic background or single gene mutations) contribute to LCPUFA-mediated mechanisms. Comparing two mouse models of disease may illustrate such clinical dichotomy. Compared to a n-6 unsaturated and a saturated fat diet, a diet rich in n-3 PUFA (8% fish oil, EPA, DHA), which worsens DSS colitis (two 7-day cycles) in C57BL/6 mice, ameliorates the disease and decreased MAdCAM-1 expression in a mouse model of CD-like ileitis (SAMP1/YitFc, SAMP1) (278). With respect to PPARy signaling, SAMP1 mice exhibit complex-, age-, and tissue-specific regulation (79), with PPARy staining extending only from the base to the tip of intestinal villi, remaining absent in the epithelial crypts, independent of age (79). In SAMP1, treatment with PPARy agonist roglitazone has no beneficial effects on the severity of ileitis (79). However, feeding n-3 to C57BL/6 mice decreased adiponectin and colitis severity, but the effect was canceled by pioglitazone, a PPARy agonist, suggesting the mechanism involves PPARy (278). Perhaps one of the most important studies demonstrating how host genetics interacts with diet, obesity traits, global gene expression, and gut microbiota composition in response to a high-fat/high-sucrose diet tested >100 inbred mouse strains (279) and revealed that high-fat/high-sucrose feeding promoted a wide, strain-specific variation in the gut microbiome, depending on mouse strain, and identified an association between a region of chromosome 3 (contains three amylase genes), with a significant enrichment of Enterobacteriaceae on the high-fat/high-sucrose diet. Previously, a study of 65 species-level phylotypes correlating differences between the gut microbiome and diet revealed that genetics and diet account for 12 and 57% of total microbiome variation (280). The authors also noted an increase in sulfate-reducing Desulfovibrionaceae in animals with impaired glucose tolerance.
Human studies support interactions between genetic background/ancestry, dietary LCPUFA intake, plasma/tissue fatty acids, and microbiome (260, 281–286). For instance, monozygotic (identical) twins have more similar microbiomes that dizygotic twins or unrelated individuals (98, 287–290). The role of host diet–gene interactions is also modulated by epigenetic effects; hypothetical effects have been proposed for n-3 LCPUFAs on cell proliferation, survival, and immunity (291–293). Maternal exposure to LCPUFA is known to alter the gut microbiota in offspring; n-3 fish oil increases Bacteroidetes; other fish oil diets increase pathobionts linked to altered immunity, namely B. wadsworthia, Enterococcus faecium, and B. fragilis, and n-6 safflower oil increases Firmicutes (294–296). PUFA (i.e., n-6) exposure can also induce epigenetic protective effects (transferrable to GF mice via FMT) in young animals against DSS colitis (297).
PUFA and Microbial Inhibition
While some studies support in vitro and in vivo antibacterial effects of LCPUFAs (e.g., liposomal LA formulations against H. pylori), others indicate adverse effects (298–305). Of interest, bacterially produced LCPUFAs may also exert anti-microbial effects, for instance, Bifidobacteria can produce C18:3 and 18:4 conjugated fatty acids effective in inhibiting gastrointestinal pathogens, including methicillin-resistant S. aureus (306). Yeasts, fungi, microalgae, and Thraustochytrids can also produce LCPUFAs, implying that microbiota structure influences fatty acid composition in the gut, and systemically if absorbed (5, 307). The anti-inflammatory, immunomodulatory effects of n-3 fatty acids on host inflammation that attenuate tissue damage, inflammation, and improve survival against Gram-negative bacteria, may equally function to reduce the generation of cell-mediated immunity, diminishing host resistance to gram-positive intracellular pathogens.
Overall, the bacterial response a LCPUFA induces (i.e., bacterial survival, dysbiosis) (251, 270, 273, 308–310), depends on the type of infectious agent (6, 260), diversity and richness of gut microbiota (311), and the n-3:n-6 dietary LCPUFA ratio (312, 313). In C57BL/6J mice fed with high-fat diets for 5 weeks (high corn oil, n-6 PUFA; corn oil + fish oil; supplemented n-3 PUFA; and low fat control, n-6 PUFA; 5% corn oil) and infected with Citrobacter rodentium to induce colitis, both high-fat diets reduced Bacteroides spp. and increased Clostridia spp., with a concomitant reduction in Clostridium coccides in the fish oil-supplemented mice (309). Supplementation with n-3 PUFA reversed the n-6 PUFA-induced dysbiosis by reducing Enterobacteria and Segmented Filamentous Bacteria, while increasing Lactobacillus spp., Bifidobacteria spp., and E. faecium. Clinically, mice in n-3 PUFA had higher sepsis and mortality [higher serum LPS-binding protein, TNFα and IL-5, and reduced intestinal alkaline phosphatase (AP)] (309), while n-6 PUFA exhibited lower disease severity. With transcriptomics, EPA and ARA (arachidonic) enriched AIN-76A diets (fat free + 1% corn oil + either 3.7% ARA, EPA, or oleic acid) modulates the colonic gene expression patterns following inoculation of mice with either complex intestinal microflora or pure bacterial cultures (E. faecalis and E. faecium, in C57BL/6J IL-10−/− mice) (314).
Another example of interactions between fatty acids and flora are CLA, which are characterized by the presence of conjugated double bonds with cis or trans configuration representing positional and geometric isomers of LA (cis-9, cis-12, cis-18, and cis-2). In humans, the primary source is dairy and ruminant meat. In ruminants, the source of CLA is endogenous bio-hydrogenation of LA, via fatty acid intermediates; stearic acid (18:0 CLA isomers), and vacceinic acid (trans-11-C18:1, which can be desaturated into rumenic acid; cis-9, trans-11-C18:2) (315–320). Similar pathways exist for the gut microbiota, with some endogenously producing CLA (321–323). Diets enriched with CLA may promote Bacteroidetes/Prevotella and Akkermansia muciniphila in C57BL/6J mice (324). The three major bacterial genera Bifidobacteria spp. (325, 326), Lactobacillus spp. (327), and Roseburia spp. (322), can produce CLA and vaccenic acid, and co-administration of LA with Bifidobacterium breve was shown to increase fatty acid tissue composition of rumenic acid (cis-9, trans-11–18:2 CLA) in mice (328). Thus, CLA acts as a metabolite precursor for specific microbes, which may modulate gut barrier function. Together, this section highlights that host responses to a microbial agent are highly dependent on the dietary fat intake.
Medium-Chain Fatty Acids
Medium-chain fatty acids (MCFA) are fatty acids found in medium-chain triglycerides (MCTs), saturated fats comprised of a glycerol and three fatty acid chains, two or three of which are 6–12 carbon atoms in length. MCTs are primarily gathered into straight chain (unbranched) fatty acids, but side chain (branched) fatty acids (e.g., nonanoic acid) also exist. Sources of MCFAs are listed in Table 2. Of note, lauric acid (C12:0) is the main antibacterial and antiviral substance found in breastmilk and in the gut is enzymatically broken down to form monolaurin, a monoester with profound anti-microbial and anti-fungal properties against lipid-coated bacteria, including inactivation of H. pylori and Listeria monocytogenes (329).
Evidence indicates that MCFAs, mainly lauric acid (C12:0), act as “non-microbial” agonists to TLR4 triggering inflammation pathways similar to gram-negative-derived LPS (330–333). High-fat SFA intake modifies gut microbiota with an overproduction of LPS and endotoxemia enhancing TLR4 activation (333, 334). The oxidative stress caused by this dietary metabolic endotoxemia produces oxidative molecules, which activate inflammation via CD36–TLR4–TLR6 and CD14–TLR4–MD2 (334–337), with MyD88-dependent and independent pathways (338) promoting expression of transcription factor NF-kB and pro-inflammatory COX-2, TNFα, IL-1β, IL-6, IL-8, IL-12, INFy, MIP1-α/β/2, MCP1, VCAM1, and RANTES (330, 335, 339–342).
In IL-10−/− mice, a subset of colonic mononuclear phagocytes that express MyD88 and signal through TLR ligands are the initiators of colitis (343), and studies demonstrated that partial replacement of dietary LCPUFAs with MCTs (lauric acid: 28 g/100 g total fat) decreased the severity of colitis, CD3+ intraepithelial lymphocytes (by increasing apoptosis), and pro-inflammatory cytokines IL-6 and INFy, in IL-10−/− mice (344). Under specific pathogen-free conditions for mRNA assessment of TLR-2 and TLR-9, gram-positive sensors of commensal intestinal bacteria occur solely through a MyD88 pathway, instead of TLR4 (i.e., gram-negative bacteria) (344). Assessment of TLR-2 and TLR-9 gene expression as sensors of gram-positive bacteria via MyD88 showed that the MCT diet decreased TLR-9 while TLR-2 was unaltered (344).
One MCT (i.e., MCFA) formulation, largely comprised of caprylic acid (97.8%, C8:0), has been shown to reduce intestinal inflammation and exert stronger anti-inflammatory effects than LCTs (i.e., LCFA) in a trinitrobenzene sulfonic acid (TNB)-induced ileitis in rats (345, 346). Intraileal injection of TNBS with the MCT formulation tended to reduce the levels of mucosal TNFα and LTB4, with levels 72% of the corresponding value in the LCT group (74.2% linoleic, C18:2; 14.8% oleic, C18:1) (345, 346). Using the same model, it was shown that gavaging as little as 1.5 ml of the MCT formulation can mediate polymophonuclear activation and mucosal infiltration, including that the LCT formulation (1.5 ml) promoted greater proinflammatory activity (345, 346).
Sphingolipids
Sphingolipids, present in small amounts in most foods, are a class of lipids containing a backbone of 18 carbon amino-alcohols that includes sphingosine, and are synthesized in the endoplasmic reticulum from non-sphingolipid precursors (347). In a Western diet, complex sphingolipids (sphingomyelin, cerebrosides, gangliosides, glycosphingolipids) can reach 0.3–0.4 g per day, with dairy, eggs, meat, and soy being rich sources, followed by cereals, vegetables, pulses, and fruits (348, 349). Animal products contain all complex sphingolipids, whereas plants contain mostly cerebrosides and structurally diverse glycosyol inositol phosphoceramides (347).
Intestinal sphingolipid digestion is catalyzed by the three isoforms of sphingomyelinase (SMase) and ceramidase (CDase), each compartmentalized in the gut depending on local and mucosal pH (acidic, A-SMase and A-CDase; neutral, N-SMase and N-CDase; alkaline, Alk-SMase and Alk-CDase), with A-SMase mainly localized in highly proliferating crypt cells, especially in small intestine, and Alk-SMase primarily in the brush border of the mid-intestine. N-CDase (acidic) exhibits the highest activity in the presence of bile salts, but direct sphingomyelin and cerebroside absorption cannot be excluded (350). In the gut, complex sphingolipids are part of intestinal membranes, which play an important role in regulating digestion, absorption, and protecting the mucosa (351, 352). Selective abundance of sphingomyelin and glycosylceramide (GlcCer) is characteristic to enterocytes in the human small intestine, with high levels associated with selective enrichment and localization of several microbial species in the apical membrane of villous cells, paralleling the constant differentiation of mucosal cell throughout the crypt-villus axis (352, 353). Abnormal sphingolipid metabolism and composition patterns have been linked to inflammatory responses, as well as abnormal intestinal permeability during IBD via abnormal intracellular tight junctions (351).
Intestinal metabolism, dietary catabolism and “de novo” synthesis, involves multiple enzymes, signaling pathways, and metabolites such as ceramide, sphingosine, and derivatives ceramide-1-phosphate (C1P) and sphingosine-1phosphate (S1P) (351, 354). Increases in ceramide, sphingomyelin and their 1-phosphorylated derivatives C1 and S1P with decreased GlcCer is believed to contribute to IBD progression (347, 352, 354, 355). These metabolites modulate eicosanoid production important in inflammation by exerting synergistic effects (of C1P and S1P) in the activation of cPLA2-α and COX-2, with C1P and S1P involved in PGE2 production (356–362). Simple sphingolipid intermediates (sphingolipids/sphingoids) mediate cell survival, proliferation, differentiation and apoptosis, whereas the dietary metabolite of sphingomyelin, ceramide, mediates intestinal epithelial cell apoptosis by capthesis D activation, which impairs absorptive and mucosal barrier (363). At the cell-to-cell adhesion level, sphyngolipids can modulate tight junctions; dietary gangliosides induce claudin-1 expression, whereas GlcCer inhibits occludin degradation (364, 365). Together, the sphyngolipid metabolism can be modified by diets rich in fibers (psyllium) and fats (366), tetrahydroxyfavone and luteolin (367, 368) (fruits, vegetables, medicinal herbs), and probiotic bacteria (VSL#3) (369), which regulates mucosal inflammation (351).
Bile Acids
Bile acids are steroids predominantly found in the bile of mammals and conjugated with taurine or glycine by the cytochrome P450 enzyme, cholesterol 7α-hydroxylase (CYP7A1) in the liver to form bile salts. Primary bile acids, cholic acid (CA) and chenodeoxycholic acids (CDCA)/ursodeoxycholic acid (UDCA), are those synthesized in the liver while secondary bile acids, deoxycholic acid (DCA) and lithocholic acid (LCA), result from bacterial 7α/β–dihydroxylation in the colon, primarily via the genus Clostridium, including C. scindens, C. hiranonis, C. hylemonae (Clostridium cluster XVIa), and C. sordelli (Clostridium cluster XI) (370). Bile acids differentially activate the nuclear receptors, farsenoid X receptor (FXR), pregane X receptor (PXR), and the vitamin D receptor (VDR), the latter a feedback mechanism that represses bile acid synthesis (371, 372). Specific bile acids also serve as natural ligands for TGR5, a GPCR highly expressed in gallbladder epithelial cells, shown to influence bile acids, intestinal motility, the immune system, and energy and glucose homeostasis by increasing GLP-1 production (371, 373, 374).
In humans, bile acid pool size and composition are important elements in regulating microbial community structure, and low levels have been significantly correlated with gram-negative bacterial dysbiosis, including potent producers of LPS (375), while favoring reductions in microbial species involved in secondary bile acid formation (e.g., Clostridium genus) (375–378). Bile acid feeding (CA) in rats resulted in significant inhibition of Bacteroidetes and Actinobacteria, with significant phylum-level expansion of Firmicutes, including members of Clostridium Cluster XIVa closely related to DCA-producing 7α-dehydroxylating species (379, 380). In one diet-related model of colitis, mice fed a AIN-93G diet supplemented with 0.2% deoxycholate (8 months), colonic inflammation and histological changes correlated (over time) with the increases or reductions in gene expression central to epithelial barrier function, inflammation, oxidative stress, cell proliferation/cell cycle/DNA repair, and related processes (381).
Bile holds clear antimicrobial activity, both indirectly, through FXR-induced antimicrobial peptides, and directly, through bacteriostatic functions on intestinal microbes, although some bile-tolerant microbes not only exist, but thrive in the presence of bile acids (382–384). Devkota et al. produced the first paper demonstrating that a Western-based diet containing high amounts of a particular saturated fat source (milk fat), enhanced development of colitis in IL-10−/− mice via a specific molecule, taurocholic acid (382), correlating with a significant bloom in Bilophila wadsworthia, a sulfite-reducing bacterim and member of Deltaproteobacteria. This bile acid-resistant bacterium expresses enzymes, such as bile salt hydrolase (BSH), which facilitates bile salt deconjugation (385, 386). Other BSH-expressing-microbiota includes some pathobionts (e.g., E.coli and L. monocytogenes), members of the gut commensals (e.g., Lactobacillus and Bifdobacterium spp.), and members of the Bacteroides genus, such as B. fragilis (383, 385, 386), although the latter yields BSH shown to “attack” the taurine conjugates of dihydroxy bile acids more readily than trihydroxy taurine conjugates (385). One study demonstrated that the antioxidant, tempol targets BSH-producing Lactobacilli, inducing a near inversion of the Firmicutes to Bacteroides ratio (387). In a follow-up set of studies by Devkota and colleagues, a milk-fat diet supplemented with 5% n-3 fish oil completely inhibited blooms of B.wadsworthia, seemingly mediated shifts in bile acid composition (388).
These findings hold important clinical implications for patients with IBD considering several studies report increased sulfate-reducing bacteria (e.g., Desulfovibrio) in fecal and mucosal biopsies (389–392), and metagenomics observations of increased functionality characteristic of auxotrophic and pathobiont bacteria, namely decreased biosynthesis of amino acids, and increased sulfate transport (particularly in ileal CD), correlating with increased expression of the amino acid transporter genes involved in metabolism of cysteine, a sulfur-containing amino acid (183, 186). These organisms have a reduced ability to produce their own nutrients, but rather transport them from sites of inflammation and tissue destruction where they are readily available. Nitrate-derived products generated from the inflamed gut can be utilized by Enterobacteriaceae, particularly E.coli, to out-compete commensals which require fermentation substrates (184). Mesalamine treatment has been associated with decreases in Escherichia (69), and found to inhibit fecal sulfide production in UC patients (393).
Alkaline Phosphatases
In addition to bile acids favoring the absorption of dietary fats, AP, are important enzymes produced by most host cells (especially epithelial cells) that assist in the absorption and transport of fatty acids. APs are encoded by a large family of genes ubiquitously expressed in multiple tissues. Mice AP isozymes are encoded by five loci, one of which, the Akp3, encodes for the duodenal-specific IAP isozome (dIAP) (394). Human AP isozymes are encoded by four different genes, three of which are tissue-specific alkaline phosphatase (TSAP) (i.e., expression restricted to intestine, placenta, germ cells), while the fourth, tissue non-specific alkaline phosphatase (TNAP), is expressed in bone, kidney, liver, and other tissues. In human hepatocytes, bile acids increase TNAP activity (395), and its secretion in bile (396), suggesting TNAP exerts inhibitory effects on bile secretion (394, 397). The liver is the major LPS-removing organ, and one important function of TNAP is to dephosphorylate endotoxins, such as LPS (398, 399). By contrast, murine TNAP, encoded by Akp2, is not expressed by mouse hepatocytes (400).
Intestinal alkaline phosphatase (IAP) is involved in fatty acid absorption and in a rate-limiting step of fatty acid transport in the gut, modulated via Akp6 IAP isozyme (global IAP or gIAP) and FAT/CD36 expression levels, a phosphorylated fatty acid translocase facilitating the transport of LCFAs into cells (401–404). As a brush border enzyme, IAP protects gut barrier function and the detoxification of bacterial LPS, through dephosphorylation (405). Compared to WT mice, Akp3−/− mice (i.e., dIAP-deficient) exhibited considerably different and fewer types of aerobic and anaerobic microbes in their stools, whereas IAP supplementation restored growth of commensal bacteria, but inhibited growth of Salmonella typhimurium (406). Interestingly, oral administration of IAP had a protective effect against DSS-induced colitis (four cycles of 2% DSS ad libitum for 7 days) in Akp3−/− mice, attenuating disease in both Akp3−/− mice and WT mice (407).
Dietary Amino Acids
Tryptophan
Dietary tryptophan is an essential amino acid found in fish and cruciferous vegetables (broccoli, cauliflower, and cabbage) that can activate the AhR, mostly through the formation and metabolism of tryptophan-derived metabolites. The AhR is a basic helix-loop-helix/Per-Arnt-Sim (bHLH-PAS) transcription factor containing several modular domains: N-terminal bHLH domain (DNA binding); two PAS domains (PAS A and PAS B) required for ARNT dimerization; and a transactivation domain of the C-terminus consisting of acidic, glutamine Q-rich and proline/serine/threonine (P/S/T)-rich regions, responsible for gene transcription, and protein–protein interactions with co-regulator proteins (408, 409). Expressed by epithelial, immune and some tumor cells, the AhR modulates host immunity, and protects against extracellular pathogens at epithelial sites by promoting functional differentiation of interleukin-17-producing Th17 cells of the lamina propria (410–414). AhR activation is also required for IL-22 production (Th17 cells) (415–417) and promotes IL-22 expression in group 3 innate lymphoid cells (ILC3) (418). IL-22 secretion stimulates production of antimicrobial peptides (Reg3β and Reg3y) (419, 420) and mucus (421, 422), crucial factors in maintaining epithelial integrity.
The first tryptophan-derived AhR mechanism involves the catabolism of tryptophan by the rate-limiting enzyme indoleamine 2,3-dioxygenase in the kynurenine pathway, thereby producing l-kinurenine, an AhR ligand (423, 424) associated with promoting Tregs (425) and DCs (426). Indoleamine 2,3-dioxygenase produced by alternatively activated macrophages (427) inhibits intracellular pathogens such as Toxoplasma and Chlamydia via INFy, and has anti-proliferative effects on tumor cells (428, 429), although depletion of l-tryptophan attenuates these effects (430). Rosmarinic acid, a caffeic acid ester found in various plants, can inhibit indoleamine 2,3-dioxygenase expression because of its COX-inhibiting properties (431). COX-2 inhibitors have a similar effect in that they downregulate indoleamine 2,3-dioxygenase production, leading to reduced kynurenine levels (432, 433). Of interest, tryptophan-derived kynurenic acid also acts as a signaling molecule for certain GPCRs (434, 435), and the protein structural motif of AhR is comparable to HIF/HIF-1α receptors (bHLH-PAS family) (436), thus illustrating the important relationship between environmental signals and host cellular responses (437, 438).
The second tryptophan-derived mechanism occurs through tryptophan metabolism as an energy source by Lactobacilli (L. reuteri, L. johnsonii), to produce indole-3-aldeyde, an AhR activator that induces IL-22 transcription (439). These interactions generate microbiota resistance to C. albicans colonization and are protective against mucosal inflammation (439). AhR-deficient mice exhibit shifts in microbial composition (increased Bacteroidetes), marked by increased epithelial permeability and colitis severity, whereas diets enriched in AhR ligands partially reversed these effects (416). Another dietary-derived AhR ligand, indole-3-carbinol (I3C), a component of the same cruciferous vegetables as tryptophan, also alters gut microbiota composition (416, 422, 440), implying that AhR ligands, including the receptor itself, mediates microbiota profiles. Finally, the tryptophan-derived lipophilic molecule, 6-formylindolo(3,2-b) carbazole (FIZC), a high-affinity AhR ligand, elicits increased activation of natural killer cells, increased INFy production and cytolitic activity (441, 442).
The AhR recognizes tryptophan and many other ligands derived from food antigens, phytochemicals [flavonoids (443)], polyphenols (444) [quercetin (445, 446), curcumin (447), resveratrol; all AhR antagonists (444)], ARA products (PGs and lipoxins A4), and natural chemicals, including those derived from dietary bacterial metabolites (19). For example, Propionibacterium freudenreichi, a bacterium from Swiss-type cheese, produces vitamin K2 precursor, 1,4-dihydroxy-2-naphtholic acid, to activate AhR in vivo, increasing antimicrobial peptide synthesis in mice (448). Other bacterial AhR ligands control antibacterial responses against Mycobacterium tuberculosis and Pseudomonsa aeruginosa (449). Numerous in vitro and in vitro studies have shown an interaction between AhR and retinoic acid signaling pathways (i.e., RXR), including AhR binding to some synthetic retinoids (450). Environmental pollutants, particularly 2, 3, 7, 8 tetrachlorodibenxo-para-dioxin (TCDD) (Dioxins), and polycyclic aromatic hydrocarbons (PAHs), such as those from tobacco smoke, are also AhR activators (451), although AhR binding by a xenobiotic- or a dioxin-response element (XRE/DRE) in the promotor region of genes requires activation by a ligand (452, 453). Notably, AhR pathway activation through exposure to environmental compounds alters the synthesis, catabolism, transport, and excretion of retinoic acid. The AhR-mediated effect on retinoid homeostasis may upregulate or downregulate gene expression (450), and can be enhanced by VDR presence, given its interaction with other receptors (e.g., RARs, RXRs) (19).
Aryl hydrocarbon receptor signaling pathways are sensitive to oxidative stress. Crosstalk between other pathways can indirectly activate AhR signaling, inhibiting the metabolic turnover endogenous AhR ligands. This interaction can mediate mucosal oxidative stress by regulating cytochrome P450 monooxygenase 1 protein enzymes (CYP1s) involved in drug metabolism, with its antagonistic effects on the estrogen receptor extensively described (409, 452, 453). In addition, the AhR works in close concert with the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (454–456), a master regulator of antioxidant responses that protects cells against reactive oxygen species (ROS) by inducing expression of many cytoprotective molecules (e.g., enzymes) (457). Oxidative stress by Nrf2 interacts with the AhR pathway. For example, the AhR ligand TCDD directly upregulates Nrf2 and phase II liver metabolizing enzymes via the multiple copies of XRE/DRE in promotor regions of human Nrf2 genes (458). AhR/ARNT/Nrf2 can also engage in the mutual binding of XRE/DREs (458). Intriguingly, TCDD-induced AhR activation impairs Th2-type immunity (459–465), whereas TCDD activation of Nrf2 overall promotes CD4+ T cells toward Th2 differentiation, induces Th2-derived cytokines IL-4, IL-5, and IL-13, subsequently suppressing INF-y production (466–471).
Signals or stresses generated from host microbiota also have the potential to activate Nrf2 cross-talk with AhR pathways, influencing metabolic processes. Increased oxidative stress has been described in high-fat, diet-induced obesity (472); however Nrf2 KO mice fed a high-fat diet (3 and 6 months), gain significantly less weight over time, have higher insulin sensitivity and more glucose tolerance compared to WT mice (472, 473). The exact role of Nrf2 in adipogenic pathways remains controversial; some reports indicate Nrf2 deletion impairs adipogenesis through PPARy (472), whereas others indicate Nrf2-induced activation of AhR can inhibits adipogenesis (474). These discrepancies may be attributed to differences in cellular models or glutathione potential (i.e., ROS levels) as a result of Nrf2 expression levels (455).
Glutamine
Glutamine is another important amino acid that mediates mucosal metabolism, and Nrf2 target genes are involved in glutathione synthesis (475). The Nrf2 pathway impacts glutamine and glutamate metabolism, although glutamine concentrations may not necessarily define cellular glutamate levels (476, 477). Glutamine is one of the most abundant non-essential amino acids in the body, serving as a source of nitrogen in the citric acid cycle and many anabolic processes (478, 479). Its importance in the immune system lies in the support of different pathways, such as cytokine secretion and T cell proliferation, and that immune cells transform glutamine to glutamate in high rates (480). Obese rats fed glutamine have decreased TNFα and IL-6 in serum and peripheral tissues (481, 482), but little is known on the effects of l-glutamine on gut microbiota. In humans, a study of overweight and obese volunteers (BMI ≥25 kg/m2) showed that l-glutamine (30 g) supplementation Veillonella genera is positively correlated with higher levels of epithelial inflammation and the occurrence of and colitis (120, 483). Glutamine supplementation decreased the Firmicutes to Bacteroidetes ratio (from 0.85 to 0.57) and reduced Actinobacteria, compared to l-alanine supplementation (484).
Hydrosoluble Vitamins
B Complex
The vitamin B complex includes niacin (B3), riboflavin (B2), cobalamin (B12), biotin (B8), folate (B9), thiamin (B1), pantothenate (B5), and pyridoxine (B7) (485). The main source of Vitamins B for animals and humans is the gut microbiota, but the gut microbial gene profiles present in all hosts is highly variable, and some microorganisms lack essential biosynthetic genes. To compensate for such deficiencies microbes exchange B-vitamin molecules with other microbes (486). In short, vitamin B-deficient genomes are grouped in three types. Most gut Actinobacteria can produce only niacin, pyridoxine, and thiamin; Some Firmicutes and Actinobacteria lack all genetic pathways, except for niacin; and some Firmicutes and Proteobacteria lack all pathways except those for biotin and folate (486).
Niacin
Niacin (vitamin B3) is directly involved in GPR109A activation (known as Niacin1 receptor), because aside from butyric acid (C4:0) (158), the receptor also interacts with the metabolite nicotinic acid, an end product of tryptophan degradation (487). Activation of GPR109A by nicotinic acid induces Treg development and expression of anti-inflammatory molecules within macrophages and DCs (158). Complementary relationships in the synthesis pathways between microbiota have been described (486). For instance, cobalamin (vitamin B12) synthesis pathways, which are missing in several Bacteroidetes and Proteobacteria, are present in other Firmicutes, whereas those Firmicutes lacked pathways held by the respective Bacteroidetes and Proteobacteria. Among the Firmicutes, some members of the Clostridia class are cobalamin producers, whereas all Lactobacillales are non-producers, with the exception of some L. reuteri and Bacillus strain (L. monocytogenes 1/2a F6854) (486). Interestingly, three non-redundant vitamin B12 receptors are expressed by B. thetaiotaomicron, giving it competitive advantage for certain B12 analogs (488).
Riboflavin
Riboflavin production by the human gut microbiota has been associated with activation of T cells, and riboflavin precursors selectively activate mucosal-associated invariant T cells (MAIT) by their presentation via the major histocompatibility complex (MHC)-related protein MR1 (489, 490). Riboflavin is also used as a major redox mediator for extracellular transfer by the butyrate-producing and main acetate-consuming bacterium F. prausnitzzi, and in vitro, flavins (such as riboflavin), in combination with cysteine, are a prerequisites for promoting the growth of this strictly anaerobic bacterium, however glutathione, a primary thiol-containing antioxidant in the gut, was found to facilitate this process (491, 492). Riboflavin synthesis has been reported to be enabled in the genomes of half Firmicutes and most Bacteroidetes, Fusobacteria, and Proteobacteria tested. Conversely, most Actinobacteria cannot produce it, but compensating have (RibU) riboflavin transporters to utilize available preformed riboflavin, as do the non-producing Firmicutes (486).
Increased glutathione transport and riboflavin metabolism is observed in patients with UC (69), whereas marked reductions in F. prausnitzii, with concomitant increases in E.coli abundance is observed in patients with ileal CD (59, 68). In healthy volunteers, oral riboflavin supplementation (100 mg) preferentially increased F. prausnitzzi and Roseburia, and reduced Enterobacteriaceae (E. coli and relatives) abundance (493). Following the cessation of riboflavin supplementation, F. prausnitzzi numbers dropped significantly in all volunteers. Of interest, the essential roles for folate metabolism are missing in all studied F. prausnitzii genomes, and while growth of F. prausnitzii A2-165 has not been found require folic acid (494), it has been suggested that folate may enhance growth of this bacterium. Taken together, perturbations to the gut microbiome may impact individual B-vitamin requirements, and deficiencies in one or more B-vitamins, may impact the gut microbiome leading to proliferation of inflammation-promoting organisms (486). The therapeutic use of B-vitamins could serve as a novel strategy to modulate microbial physiology.
Choline
Choline can be synthesized in small amounts by the liver, and is an important member of the B-vitamin-complex (495, 496). Phosphatidylcholine is a type of phospholipid in lecithin, and structurally, phosphatidylcholine is comprised of a SFA, an unsaturated fatty acid, a glycerol, and a quaternary ammonium compound that comprises a choline group. Carnitine itself is a quaternary ammonium compound biosynthesized from the amino acids lysine and methionine, and the choline group within carnitine is structurally similar to that found in phosphatidylcholine (496). Dietary sources of phosphatidylcholine and carnitine include animal-based foods such as red meat, eggs (yolk), milk and certain fish, which provide significant sources of choline in the body. In a metabolomics-based dietary intervention study, foods rich in phosphatidylcholine and carnitine resulted in the catabolism of choline via a microbiota-dependent mechanism to form: trimethylamine (TMA), and betaine (trimethyglyciene; TMG) (497), a molecule structurally similar to the amino acid glycine, but with three methyl groups (498). In the liver, TMA is oxidized into trimethylamine N-oxide (TMAO) (499), a small molecule strongly associated with promoting inflammatory-based atherosclerosis (500, 501). Most of the bacteria that express genes encoding for TMA lyases are found in three of the four major phyla of the human gut microbiome (502). In apoliprotein E-deficient mice fed a diet with l-carnitine, the promotion of atherosclerosis was also mediated by a microbiota-dependent mechanism (501).
While higher plasma levels of l-carnitine, in association with TMAO, are positively correlated with cardiovascular events in humans, intriguingly, omnivores produce more TMAO from dietary l-carnitine than vegans or vegetarians (501). One plausible explanation for these observed differences can be in underlying archaeal lineages within the human gut microbiome. For instance, certain strains of methanogens, such as Methanomassiliicoccus luminyensis and Methanosarcina barkeri strictly use methyl-based compounds, including TMA, as substrates for methanogenesis to enable their growth, which can deplete TMA levels (503). M. luminyensis is a hydrogen-dependent organism and member of the novel archaeal group Methanomassiliicoccales, and like other methanogens, capable of using methanol for methanogenesis (504). However, one remarkable feature of M.luminyensis is their ability to encode a rare proteinogenetic amino acid pyrrolysine (Pyl), a unique characteristic shared only by a limited number of other bacteria and some of the family Methanosarcinaceae (504). This is a truly distinguishing characteristic because methylotropic methanogenesis of methylated amines, such as TMA, monomethylamine, and dimethylamine can only occur in the presence of pyrrolysine in the active catalytic site (505). Furthermore, studies exploring the abundance and activity of archaeal taxa have identified differences in organism groups between human populations based on geography and dietary habits (e.g., salt-fermented seafood) (504, 506–511). The detection of Archaea in the vaginal flora of pregnant women also indicates critical mother-to-child “microbial inheritance” (503, 512).
Lipid-Soluble Vitamins
Vitamin A
Vitamin A is an essential dietary component that includes the group of unsaturated nutritional organic compounds retinol, retinal, retinoic acid, several pro-vitamin A carotenoids, and also beta carotene. Retinol is derived in the small intestine from retinyl palmitate, a vitamin A ester found in foods of animal origin, and in the body, can be converted to either retinal, or irreversibly to retinoic acid (496). The effects of vitamin A are employed via its genomic actions on a specific group of nuclear receptors, namely RXR and RAR (for which three isoforms exist). By comparison, retinoic acid participates in the activation of T cell lymphocytes (513–515), and CD103+ DCs migration from the lamina propria to mesenteric lymph nodes to promote Treg generation (516), and thereby extract non-pathogenic and pathogenic luminal bacteria (e.g., Salmonella typhimurium) by means of dendrite extension into the intestinal lumen (517). The lamina propria is also enriched with the IL-22-producing, IL3 and Th17 cells (mostly small intestine), and retinoic acid promotes enhanced resistance to experimental colitis by stimulating T cell production of IL-22 (518). Retinoic acid is also needed in utero for development of lymphoid tissue inducer cells (LTi) (519), a subset of retinoic acid receptor-related orphan receptor (ROR)yt-dependent ILC3s, and an important contributor to innate immune development in intestinal tissues (520). In murine models, levels of vitamin A exposure during pregnancy influence the size of lymph nodes and Peyer’s patches, and in doing so determine immune competence in the next generation adult offspring (519, 521).
Vitamin D
The VDR is abundantly expressed in both intestinal and all immune cells (522–525), with several lines of evidence implicating vitamin D deficiency, or downregulation of the VDR, in the pathogenesis and severity of experimental IBD (522, 524, 526–528). This process is now, in part, attributed to the barrier-protecting role of VDR signaling in maintaining epithelial integrity and its effect on gut microbiota composition (529). While the bioactive forms of vitamin D (1,25[OH]2D and 1,25[OH]2D3) activate the VDR to transcribe (or repress) at least 913 genes (530), VDR expression differs between genders, with females possessing an extra site of VDR gene expression (endometrium) (531). In addition, the bioactive forms of vitamin D are known to robustly increase the expression of some (e.g., NOD2) (532) – but not all – CD-associated susceptibility genes (533, 534), suggesting its immunomodulatory activity to be vastly heterogeneous in nature. Recent evidence demonstrated that the anti-inflammatory effect of certain lactobacilli is via NOD2-mediated signaling (535), and that oral supplementation with Lactobacillus reuteri NCIMB 30242 can elicit increased concentrations of circulating 25(OH)D (536).
Under normal circumstances, a series of tightly controlled feedback pathways, namely the hepatic microsomal or mitochondrial enzyme 25-hydroxylase (catalyzed by CYP24A1) and the renal mitochondrial enzyme 1α-hydroxylase (catalyzed by CYP27B1), regulate the production and serum concentrations of 25(OH)D and 1,25(OH)2D, respectively, and in turn, VDR function (537, 538). The production of CYP24A1 is controlled by the VDR, whereas CYP27B1 production is controlled by immune-specific responses, with several cell types involved in innate and adaptive immune responses expressing CYP27B1 (538–541). However, intestinal bacteria can also regulate intestinal expression of CYP27B1 (542), with VSL#3 treatment shown to induce VDR expression and activity in the host (543). The VDR plays a central role in regulating proteins involved in intracellular microbial recognition, namely TLRs and the anti-microbial proteins, human beta-defensin-2 (hBD-2) and cathelicidin, the latter known to progressively decline during CD over time (544, 545). Notably, mouse cells do not express the same cathelicidin gene as humans, and cellular response to active vitamin D differs, since murine cathelicidin (Cramp) lack the vitamin D response element (VDRE) (546).
Various microbes are able to evade the innate immune response by secreting VDR antagonists, allowing microbial survival within the cell, and modify human gene expression (547, 548). During pathogen-induced VDR dysregulation, VDR production of CYP24A1 decreases, resulting in low serum concentrations of 25(OH)D, consistent with the suboptimal vitamin D status commonly observed in CD patients (537). Over time, VDR activity, as well as the immune system, becomes increasingly compromised, and new pathogens are acquired by the host (548). There is also evidence that intestinal VDR can modulate hepatic bile acid synthesis, and that phytochemicals (e.g., curcumin) (549), and other dietary metabolites (e.g., butyrate) (550, 551), can bind to the VDR, thereby activating intestinal VDR target genes, including CYP3A family members (552–554). The likelihood of a VDR-dysregulating microbial community in immune-mediated disorders is strengthened by data from VDR/CYB27B1-deficient mice, in which unregulated intestinal inflammation results in an environment conducive to phylum-level bacterial expansion within Proteobacteria, particularly species from the Desulfovibrionaceae family, with corresponding bacterial number reductions in Lachnospiraceae and Firmicutes phylum (555). One recent study demonstrated that VDR deficiency (VDR−/− mice) enriched Eggerthella (genus of Actinobacteria implicated in UC/CD) (556) in cecal stool, but depleted Alistipes and Odoribacter, whereas a depletion of Lactobacillus with markedly increased Clostridium and Bacteroides levels was observed in fecal stool (compared to WT mice) (557). Loss of VDR function also resulted in local enrichment related to synthesis and degradation of metabolites, namely fatty acids, glycans and LPS (557). Overall, taxonomic shifts such as these are thought to contribute to changes in the microbiome during inflammation, and similar alterations have been described in IBD patients compared with healthy controls (121, 289, 558–561). Patients treated with VDR antagonist (olmesartan) in conjunction with bacteriostatic antibiotics experience exacerbation of disease and immunopathology (562), indicating that VDR intricately mediates immune function, in part through gut microbiota.
The Complexity of Pantropic Interactions Between Microbes and Diet
Macronutrients
Animal models using an isocaloric comparison between high-fat and low-fat diets require macronutrient displacement of carbohydrates by fats. Thus, the possibility exists that pro-inflammatory states are enhanced by a high-fat low-carbohydrate diet, which may lack dietary fibers needed to produce SCFAs. Few studies have investigated whether altering the dietary ratios of protein, carbohydrate, and fiber influences host response to a dietary fatty acid and/or bioactive compound (e.g., prebiotics, probiotics). For instance, the beneficial, albeit species-specific effect, conferred by a probiotic to the host may be related to its fermentation processes, and release of bioactive molecules; it is possible that fermentation products and bioactive molecules released by probiotic bacteria are dependent on the substrates available to them within the gut. Conceptually, this can apply to some, if not all, intestinal bacteria, but this is best studied in probiotics. The Lactobacillus helveticus R0052 is known for modulating inflammation and gut microbiota structure in IL-10−/− mice (563, 564). However, outcomes depend on the diet consumed, mouse genotype and the presence of active inflammation, the latter correlates with gut microbiome alterations (565). IL-10−/− (129/SvEv) mice fed a standard mouse chow (29% protein, 55% carbohydrates, 13% fat; 3.8 kcal/g) or a Western style-diet (85% basal mix, non-hydrogenated lard, flax oil, sunflower oil; 28% protein, 49% refined carbohydrate, 33% fat; 4.2 kcal/g), with or without L. helveticus R0052 for 21 days under SPF conditions (565), showed L. helveticus-mediated reversal of chronic microscopic lesions in IL-10−/− mice fed the Western diet, and major increase of IL-1β on regular chow. The L. helveticus also reversed diet-induced increase of Proteobacteria abundance (E.coli, Salmonella) and increased Lentospirae.
Dietary fibers (pectin, guar gum, a mixture of both, or fiber-free) are capable of mediating the pro-inflammatory potential of dietary lipids (566). Conventional rats fed a high-fat diet primarily reduced SCFA formation (compared to low-fat) with the guar gum group having significantly higher abundance of Bacteroidetes compared to other groups after 6 weeks (566). The fiber fiber-free group fed a high-fat had significantly higher abundance of Akkermansia, and higher MCP-1 concentrations to that of the guar gum group fed a high-fat diet for 2 and 4 weeks (566). In rats, high-cholesterol diets (AIN76A w/w 1% cholesterol), with soy in combination with fiber (psyllium, resistant maltodextrin and chicory powder), caused notable rise in abundance of Bacteroides spp., and reduction of Firmicutes: Bacteroidetes ratio, whereas soy alone increased the ratio partly due to a significant increase in Lactobacillus, Coproccus, and Blautia spp. (all Firmicutes) (567). Other studies have shown that potato-resistant starch attenuates the detrimental effects of a meat-based diet in mice by upregulating genes pivotal in colonic barrier function and promoting higher numbers of beneficial Lactobacillus spp. (568, 569).
High-fat diets combined with high-sugar increased E.coli (AIEC) populations in transgenic carcinoembryonic antigen-related cell adhesion molecule 6 (CEABAC10) mice; leading to increased intestinal permeability, induction of NOD2 and TLR5 transcription, and TNFα secretion (570). By comparison, in mice fed a high-fat diet for 10 weeks and treated with coffee at 20 g/L with zero calorie sweetener aspartame added for taste (60 mg/L), coffee consumption not only attenuated the increase in Firmicutes: Bacteroidetes ratio and Clostridium Cluster XI but also resulted in increased levels of Enterobacteria (571). Coffee also increased serum levels of aromatic and circulating SCFAs, while lowering BCAAs. Clearly, confounding effects may exist associated with adding aspartame as sweetener to the mix. However in another study, in which DSS-colitis-induced mice lacking expression of TLR5 by either intestinal epithelial cells [TLR5(DeltaIEC)] or DCs [TLR5(DeltaDC)] were examined for basal phenotypes in response to a high-fat diet and pathobiont challenge, microbiota composition was found to cluster more closely according to genotype than housing (572).
Phytochemicals
Approximately 90–95% of total dietary polyphenols reach the colon unabsorbed (573), and co-administration of quercetin was shown to enhance the anti-inflammatory properties of n-3 PUFA in a DSS-colitis mouse model (574), implying that gut microbiota can indirectly influence (enhance or suppress) the effects of n-3 PUFA via the metabolism of phytochemicals available in the gut. Conversely, many functional foods, particularly those containing polyphenols and polyphenolic compounds, have the ability to influence the composition and metabolic activity of gut microbiota, although these outcomes are dose-dependent, and intimately reflect the chemical structure and form in which bioactive compounds are provided (i.e., whole foods vs. single bioactive), as this affects bioavailability, and derived physiological effects (575, 576). For example, distinct patterns in the prebiotic effects of four saponin-rich herbal teas (ginseng, red ginseng, notoginseng and gynostemma pentaphyllum) occur on gut microbiota composition in C57BL/6 mice (577). The effects of notoginseng were most prominent on Lactobacillus, although both red ginseng and gynostemma pentaphyllum increased abundance of F. prausnitzii, whereas gynostemma pentaphyllum resulted in strongest enhancement of Bifidobacterium at the species level (577). Another powerful phenolic compound, ellagic acid (ellagitannins) present in foods such as pomegranate, raspberries, blackberries, and strawberries has been recognized for modulating intestinal inflammation (578–582) and provides a rich source for ellagitannin gut microbiota metabolites-urolithins (583–586).
Dietary Emulsifiers
Dietary emulsifiers also impact gut microbiota community structure, with maltodextrin widely demonstrated for its ability to impair anti-bacterial cellular responses, reduce epithelial-barrier defense and directly affect multiple E.coli strains, including the promotion of AIEC colonization and cellular adhesion (587). Other additives include carboxymethycellulose (CMC or E466), polyscorbate-80 (P80 or E433), carrageenan, and xanthan gum. In an elaborate series of 12-week experiments, perturbations in IL-10−/−, TLR5−/− and wild-type C57BL/6 control mice gut microbiota composition were noted following exposure to CMC and P80 (dosages: 1, 0.1, 0.5%) (588–590). Both emulsifiers markedly reduced microbial diversity in IL-10−/− mice, while fostering expansion of Proteobacteria and Verrucomimicobia phyla, particularly Akkermansia muciniphila (591, 592). In IL-10−/− mice (not TLR5−/− mice), emulsifier-induced colitis correlated with enrichments in Biophila and Helicobacter, consistent with previous observations in IL-10−/− mice (382, 593). Intriguingly, CMC and P80 also lead to alterations in both bile acid and fecal SCFA levels, including reductions in butyrate (593). It is important to note however, that dosages used were much higher than the practical human equivalent of 25 mg/kg body weight per day (594) and equated to a daily intake of 150,000 mg in a 60 kg adult. In addition, sodium sulfite was used as the control material for the emulsifiers. Sodium sulfite is added to various food and beverages (concentrations from 0.03 to 1.5 g/kg) as a preservative and as an antioxidant (595), but unlike emulsifiers, sodium sulfites are large hygroscopic molecules, not easily digested, which may increase fecal bulk (596). Sodium sulfite has been shown to impact composition and yield of fecal fiber, affect granular structure of resistant starches (597), and prevent the formation of lignans, correlating with lower recoveries of fecal neutral detergent fibers (598). Furthermore, after ingestion of sulfite-containing beverages, nitrite present in saliva from the oral cavity mixes with sulfite, and the mixture of salivary nitrate can then be transformed into NO with the gastric juice of the stomach (595).
Derived from several species of red seaweed (Rhodophyceae), carrageenans are various polysaccharides that differ in amount and distribution of sulfate groups, and widely used in processed foods to improve product viscosity and enhance texture of food products. Carrageenans are known activators of the B-cell leukemia/lymphoma (Bcl10)-mediated TLR4 signaling pathways, and Bcl10 has been implicated as a key signaling molecule in most, but not all, carrageenan-initiated inflammatory pathways in epithelial cells (599). In animal models, ingestion of carrageenans predictably induce epithelial inflammation, with development of ulcerations and inflammatory infiltrates; however, inflammation is not suppressed in a GF environment, suggesting that colonic inflammation is not entirely bacterially driven (600–605). On the other hand, specific microflora, namely the two Lactobacillus species L. casei and L. acidophilus, exerted anti-inflammatory effects on carrageenan-induced inflammatory responses, significantly downregulating pro-inflammatory cytokine pathways IL-6 and TNFα, while upregulating IL-10 (606). One study demonstrated that continual (25 weeks) prophylactic dietary supplemention with Aquamin, a mineral extract obtained from red algae Lithothamnion corallioides, ameliorated the serverity of spontaneous colitis in IL-10−/− mice on C57BL/6J background, but had no affect on IL-10−/− mice on a BALB/c background (607).
The unique structure of major matrix polysaccharides in the cell walls of red algae, the most common red seaweed polysaccharides present in carrageenans, requires a distinct set of gut microbiome-encoded-enzymes known as carbohydrate active enzymes, or “CAZymes” (608, 609). CAZymes are predominantly encoded in genomes of marine microbes (610, 611), but absent in the human genome, thus it was thought that humans cannot digest or absorb carrageenans (612). However, metagenomics studies have revealed genes encoding for CAZymes in members of the genus Bacteroides (e.g., B. plebeius), allowing additional sources of energy and nitrogen through bacterial degradation of specific carbohydrates (612, 613). This evolutionary adaptation appears to result from horizontal gene transfer, in that genes encoding for CAZymes which metabolize marine red algae, can be acquired by specific bacterial taxa in the human gut microbiome, from microbes living outside the gut, for instance, marine-associated bacteria (610, 612). Extrinsically acquired genes and their functions in the human gut microbiome warrants further investigations, considering the rising consumption of seaweeds, such as nori (species of red algae genus Porphyra), traditionally used to prepare sushi meals.
Succinic Acid
Succinic acid, an industrial food and beverage additive that regulates acidity, is also an end product of carbohydrate fermentation that acts as an inflammatory signal in immune cells to induce IL-1β through HIF-1α (transcription factor induced by hypoxia), a downstream target of succinate (614, 615). Succinate has the ability to stimulate ROS (616, 617), and succinate accumulation in immune cells acts as an inflammatory signal for macrophages, via HIF-1α (614). HIF-1α activation attenuates Treg development, induces IL-17 production and increases RORyt transcription, favoring differentiation of T lymphocytes into pro-inflammatory Th17 cells (618). Succinate is also a ligand for the succinate-receptor 1 (SUCNR1; formally GPCR91) expressed on DCs (615) and can enhance both pro-inflammatory cytokine (TNFα and Il-1β) production and the antigen presentation capacity of DCs, thereby inducing adaptive immune responses (615, 619). In the colonic mucosa of rats, succinic acid lead to reduced crypt size and inhibition of epithelial cell proliferation rate (620). Notably, leptin is also a well-known HIF-1α-inducible modulator (621), with HIF-1α overexpression observed in obese adipose tissue, and reduction during weight loss (118).
A Concluding Remark on Dietary Complexity
Clearly, animal models investigating the effects of single nutrient components are indispensable for our mechanistic understandings of the gut microbiota. Thus, animals serve as a fundamental preclinical resource to understand the human gut microbiome. However, as the research continues to evolve, it is becoming clear that several basic diet-mediated pathways interact, and thus increasing efforts should be made to account for them to unveil novel mechanisms of sustained (diet/microbial-driven) inflammation. The gut microbial community is also dependent on host genetics. In order to appropriately translate the experimental data into clinically viable recommendations and dosages, which are suitable for IBD, diet could be regarded as an “entity” and a sum of all individual components.
Author Contributions
Drafting and concepts of the manuscript: AB. Editing and concepts: AR-P and FC. Editing of Final Manuscript: AT. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the reseach was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial research support for this publication comes from the National Institute of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health (NIH) under Award number P30DK097948 as part of the Digestive Diseases Research Core Centers program, as well as NIH awards DK091222, DK042191, DK055812, and DK097948 to FC. Special thanks go to the Crohn’s & Colitis Foundation of America for their generous support in the form of a Career Development Award to ARP in study the genetics of inflammatory bowel disease in mice. AB would like to acknowledge Mr. and Mrs. Raffner for their support.
References
1. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411:603–6. doi:10.1038/35079114
2. Hugot J, Chamaillard M, Zouali H, Lesage S, Cézard J, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
3. Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clinl Pract Gastroenterol Hepatol (2006) 3:390–407. doi:10.1038/ncpgasthep0528
4. Engel MA, Neurath MF. New pathophysiological insights and modern treatment of IBD. J Gastroenterol (2010) 45:571–83. doi:10.1007/s00535-010-0219-3
5. Anderson M, Fritsche KL. (n-3) Fatty acids and infectious disease resistance. J Nutr (2002) 132:3566–76.
6. Fenton JI, Hord NG, Ghosh S, Gurzell EA. Immunomodulation by dietary long chain omega-3 fatty acids and the potential for adverse health outcomes. Prostaglandins Leukot Essent Fatty Acids (2013) 89:379–90. doi:10.1016/j.plefa.2013.09.011
7. Buttó LF, Haller D. Dysbiosis in intestinal inflammation: cause or consequence. Int J Med Microbiol (2016). doi:10.1016/j.ijmm.2016.02.010
8. Park BS, Song DH, Kim HM, Choi B, Lee H, Lee J. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature (2009) 458:1191–5. doi:10.1038/nature07830
9. Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, et al. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science (1992) 256:225–8. doi:10.1126/science.1373522
10. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut (2006) 55:205–11. doi:10.1136/gut.2005.073817
11. Jacobs JP, Braun J. Immune and genetic gardening of the intestinal microbiome. FEBS Lett (2014) 588:4102–11. doi:10.1016/j.febslet.2014.02.052
12. Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet (2004) 36:471–5. doi:10.1038/ng1339
13. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2006) 39:207–11. doi:10.1038/ng1954
14. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314:1461–3. doi:10.1126/science.1135245
15. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39:596–604. doi:10.1038/ng2032
16. Zaahl MG, Winter TA, Warnich L, Kotze MJ. The –237C – >T promoter polymorphism of the SLC11A1 gene is associated with a protective effect in relation to inflammatory bowel disease in the South African population. Int J Colorectal Dis (2006) 21:402–8. doi:10.1007/s00384-005-0019-z
17. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42:1118–25. doi:10.1038/ng.717
18. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi:10.1038/nature11582
19. Veldhoen M, Ferreira C. Influence of nutrient-derived metabolites on lymphocyte immunity. Nat Med (2015) 21:709–18. doi:10.1038/nm.3894
20. Sheehan D, Moran C, Shanahan F. The microbiota in inflammatory bowel disease. J Gastroenterol (2015) 50:495–507. doi:10.1007/s00535-015-1064-1
21. Bisgaard H, Bønnelykke K, Stokholm J. Immune-mediated diseases and microbial exposure in early life. Clin Exp Allergy (2014) 44:475–81. doi:10.1111/cea.12291
22. Niessner M, Volk B. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT-PCR). Clin Exp Immunol (1995) 101:428–35. doi:10.1111/j.1365-2249.1995.tb03130.x
23. Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology (2008) 134:577–94. doi:10.1053/j.gastro.2007.11.059
24. Delgado M, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ (2009) 16:976–83. doi:10.1038/cdd.2009.40
25. Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res (2008) 87:817–28. doi:10.1177/154405910808700908
26. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
27. Suri-Payer E, Fritzsching B. Regulatory T cells in experimental autoimmune disease. Semin Immunopathol (2006) 28:3–16. doi:10.1007/s00281-006-0021-8
28. Mucida D, Cheroutre H. TGFbeta and retinoic acid intersect in immune-regulation. Cell Adh Migr (2007) 1:142–4. doi:10.4161/cam.1.3.5062
29. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448:480–3. doi:10.1038/nature05969
30. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger BTGF. β in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
31. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
32. Carbo A, Hontecillas R, Andrew T, Eden K, Mei Y, Hoops S, et al. Computational modeling of heterogeneity and function of CD4 T cells. Front Cell Dev Biol (2014) 2:31. doi:10.3389/fcell.2014.00031
33. Pot C, Apetoh L, Kuchroo VK. Type 1 regulatory T cells (Tr1) in autoimmunity. Semin Immunol (2011) 23(3):202–8. doi:10.1016/j.smim.2011.07.005
34. Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med (1999) 190:1123–34. doi:10.1084/jem.190.8.1123
35. Hardtke S, Ohl L, Forster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood (2005) 106:1924–31. doi:10.1182/blood-2004-11-4494
36. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192:1545–52. doi:10.1084/jem.192.11.1545
37. Lyons J, Herring CA, Banerjee A, Simmons AJ, Lau KS. Multiscale analysis of the murine intestine for modeling human diseases. Integr Biol (2015) 7:740–57. doi:10.1039/c5ib00030k
38. Mei Y, Abedi V, Carbo A, Zhang X, Lu P, Philipson C, et al. Multiscale modeling of mucosal immune responses. BMC Bioinformatics (2015) 16:S2. doi:10.1186/1471-2105-16-S12-S2
39. Verma M, Hontecillas R, Abedi V, Leber A, Tubau-Juni N, Philipson C, et al. Modeling-enabled systems nutritional immunology. Front Nutr (2016) 3:5. doi:10.3389/fnut.2016.00005
40. Faust K, Raes J. Host-microbe interaction: rules of the game for microbiota. Nature (2016) 534:182–3. doi:10.1038/534182a
41. Bashan A, Gibson TE, Friedman J, Carey VJ, Weiss ST, Hohmann EL, et al. Universality of human microbial dynamics. Nature (2016) 534:259–62. doi:10.1038/nature18301
42. Lu P, Bevan DR, Lewis SN, Hontecillas R, Bassaganya-Riera J. Molecular modeling of lanthionine synthetase component C-like protein 2: a potential target for the discovery of novel type 2 diabetes prophylactics and therapeutics. J Mol Model (2011) 17:543–53. doi:10.1007/s00894-010-0748-y
43. Carbo A, Olivares-Villagomez D, Hontecillas R, Bassaganya-Riera J, Chaturvedi R, Piazuelo MB, et al. Systems modeling of the role of interleukin-21 in the maintenance of effector CD4+ T cell responses during chronic Helicobacter pylori infection. MBio (2014) 5:e1213–4. doi:10.1128/mBio.01243-14
44. Guri AJ, Misyak SA, Hontecillas R, Hasty A, Liu D, Si H, et al. Abscisic acid ameliorates atherosclerosis by suppressing macrophage and CD4 T cell recruitment into the aortic wall. J Nutr Biochem (2010) 21:1178–85. doi:10.1016/j.jnutbio.2009.10.003
45. Guri AJ, Hontecillas R, Bassaganya-Riera J. Abscisic acid ameliorates experimental IBD by downregulating cellular adhesion molecule expression and suppressing immune cell infiltration. Clin Nutr (2010) 29:824–31. doi:10.1016/j.clnu.2010.02.009
46. Guri AJ, Hontecillas R, Si H, Liu D, Bassaganya-Riera J. Dietary abscisic acid ameliorates glucose tolerance and obesity-related inflammation in db/db mice fed high-fat diets. Clin Nutr (2007) 26:107–16. doi:10.1016/j.clnu.2006.07.008
47. Bassaganya-Riera J, Guri AJ, Lu P, Climent M, Carbo A, Sobral BW, et al. Abscisic acid regulates inflammation via ligand-binding domain-independent activation of peroxisome proliferator-activated receptor gamma. J Biol Chem (2011) 286:2504–16. doi:10.1074/jbc.M110.160077
48. Hontecillas R, Roberts PC, Carbo A, Vives C, Horne WT, Genis S, et al. Dietary abscisic acid ameliorates influenza-virus-associated disease and pulmonary immunopathology through a PPARγ-dependent mechanism. J Nutr Biochem (2013) 24:1019–27. doi:10.1016/j.jnutbio.2012.07.010
49. Magnone M, Ameri P, Salis A, Andraghetti G, Emionite L, Murialdo G, et al. Microgram amounts of abscisic acid in fruit extracts improve glucose tolerance and reduce insulinemia in rats and in humans. FASEB J (2015) 29:4783–93. doi:10.1096/fj.15-277731
50. Carbo A, Hontecillas R, Cooper J, Gandour RD, Ehrich M, Bassaganya-Riera J. Mo1691 lanthionine synthetase C-like receptor 2 (LANCL2): a novel therapeutic target for inflammatory bowel disease. Gastroenterology (2015) 148:S–686–S–687. doi:10.1016/S0016-5085(15)32321-0
51. Abedi V, Lu P, Hontecillas R, Verma M, Vess G, Philipson CW, et al. Phase III placebo-controlled, randomized clinical trial with synthetic Crohn’s disease patients to evaluate treatment response. Computational Modeling-Based Discovery of Novel Classes of Anti-Inflammatory Drugs that Target Lanthionine Synthetase C-Like Protein. Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology – Systems & Applications. (Elsevier/MK) (Vol. 2) (2015). 79 p.
52. Carbo A, Hontecillas R, Kronsteiner B, Viladomiu M, Pedragosa M, Lu P, et al. Systems modeling of molecular mechanisms controlling cytokine-driven CD4 T cell differentiation and phenotype plasticity. PLoS Comput Biol (2013) 9:e1003027. doi:10.1371/journal.pcbi.1003027
53. Carbo A, Bassaganya-Riera J, Pedragosa M, Viladomiu M, Marathe M, Eubank S, et al. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PLoS One (2013) 8:e73365. doi:10.1371/journal.pone.0073365
54. Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe (2008) 3:417–27. doi:10.1016/j.chom.2008.05.001
55. Wright EK, Kamm MA, Teo SM, Inouye M, Wagner J, Kirkwood CD. Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: a systematic review. Inflamm Bowel Dis (2015) 21:1219–28. doi:10.1097/MIB.0000000000000382
56. Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin Immunopathol (2015) 37:47–55. doi:10.1007/s00281-014-0454-4
57. Li J, Butcher J, Mack D, Stintzi A. Functional impacts of the intestinal microbiome in the pathogenesis of inflammatory bowel disease. Inflamm Bowel Dis (2015) 21:139–53. doi:10.1097/MIB.0000000000000215
58. Dicksved J, Halfvarson J, Rosenquist M, Järnerot G, Tysk C, Apajalahti J, et al. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J (2008) 2:716–27. doi:10.1038/ismej.2008.37
59. Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis (2009) 15:653–60. doi:10.1002/ibd.20783
60. Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, et al. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis (2006) 12:106–11. doi:10.1097/01.MIB.0000200323.38139.c6
61. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A (2007) 104:13780–5. doi:10.1073/pnas.0706625104
62. Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflamm Bowel Dis (2008) 14:858–67. doi:10.1002/ibd.20392
63. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331:337–41. doi:10.1126/science.1198469
64. Josefowicz SZ, Lu L, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
65. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology (2004) 127:412–21. doi:10.1053/j.gastro.2004.04.061
66. Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology (2011) 140:1720–8.e3. doi:10.1053/j.gastro.2011.01.054
67. Martinez-Medina M, Aldeguer X, Lopez-Siles M, González-Huix F, López-Oliu C, Dahbi G, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis (2009) 15:872–82. doi:10.1002/ibd.20860
68. Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, et al. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol (2014) 52:398–406. doi:10.1128/JCM.01500-13
69. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol (2012) 13:R79. doi:10.1186/gb-2012-13-9-r79
70. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A (2008) 105:16731–6. doi:10.1073/pnas.0804812105
71. Sokol H, Seksik P, Furet J, Firmesse O, Nion-Larmurier I, Beaugerie L, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis (2009) 15:1183–9. doi:10.1002/ibd.20903
72. Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe (2007) 2:119–29. doi:10.1016/j.chom.2007.06.010
73. Ott SJ, Kühbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, et al. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol (2008) 43:831–41. doi:10.1080/00365520801935434
74. Pérez-Brocal V, García-López R, Vázquez-Castellanos JF, Nos P, Beltrán B, Latorre A, et al. Study of the viral and microbial communities associated with Crohn’s disease: a metagenomic approach. Clin Transl Gastroenterol (2013) 4:e36. doi:10.1038/ctg.2013.9
75. Lepage P, Colombet J, Marteau P, Sime-Ngando T, Dore J, Leclerc M. Dysbiosis in inflammatory bowel disease: a role for bacteriophages? Gut (2008) 57:424–5. doi:10.1136/gut.2007.134668
76. Cadwell K, Patel KK, Maloney NS, Liu T, Ng AC, Storer CE, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell (2010) 141:1135–45. doi:10.1016/j.cell.2010.05.009
77. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science (2012) 336:1314–7. doi:10.1126/science.1221789
78. Haritunians T, Taylor KD, Targan SR, Dubinsky M, Ippoliti A, Kwon S, et al. Genetic predictors of medically refractory ulcerative colitis. Inflamm Bowel Dis (2010) 16:1830–40. doi:10.1002/ibd.21293
79. Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis (2011) 17:2566–84. doi:10.1002/ibd.21638
80. Jones-Hall YL, Grisham MB. Immunopathological characterization of selected mouse models of inflammatory bowel disease: comparison to human disease. Pathophysiology (2014) 21:267–88. doi:10.1016/j.pathophys.2014.05.002
81. Cultrone A, Tap J, Lapaque N, Doré J, Blottière HM. Metagenomics of the human intestinal tract: from who is there to what is done there. Curr Opin Food Sci (2015) 4:64–8. doi:10.1016/j.cofs.2015.05.011
82. Scheppach W. Effects of short chain fatty acids on gut morphology and function. Gut (1994) 35:S35–8. doi:10.1136/gut.35.1_Suppl.S35
83. Schley P, Field C. The immune-enhancing effects of dietary fibres and prebiotics. Br J Nutr (2002) 87:S221–30. doi:10.1079/BJNBJN/2002541
84. de Giorgio R, Blandizzi C. Targeting enteric neuroplasticity: diet and bugs as new key factors. Gastroenterology (2010) 138:1663–6. doi:10.1053/j.gastro.2010.03.022
85. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science (2005) 308:1635–8. doi:10.1126/science.1110591
86. Suau A, Bonnet R, Sutren M, Goden JJ, Gibson GR, Collins MD, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol (1999) 65:4799–807.
87. Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol (2002) 39:33–9. doi:10.1111/j.1574-6941.2002.tb00904.x
88. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science (2008) 320:1647–51. doi:10.1126/science.1155725
89. Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J (2011) 5:220–30. doi:10.1038/ismej.2010.118
90. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464:59–65. doi:10.1038/nature08821
91. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature (2011) 473:174–80. doi:10.1038/nature09944
92. Levitt MD, Furne JK, Kuskowski M, Ruddy J. Stability of human methanogenic flora over 35 years and a review of insights obtained from breath methane measurements. Clin Gastroenterol Hepatol (2006) 4:123–9. doi:10.1016/j.cgh.2005.11.006
93. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A (2005) 102:11070–5. doi:10.1073/pnas.0504978102
94. Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A (2009) 106:2365–70. doi:10.1073/pnas.0812600106
95. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature (2006) 444:1027–131. doi:10.1038/nature05414
96. Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut (2010) 59:1635–42. doi:10.1136/gut.2010.215665
97. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi:10.1038/4441022a
98. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature (2009) 457:480–4. doi:10.1038/nature07540
99. Parekh PJ, Balart LA, Johnson DA. The influence of the gut microbiome on obesity, metabolic syndrome and gastrointestinal disease. Clin Transl Gastroenterol (2015) 6:e91. doi:10.1038/ctg.2015.16
100. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med (2009) 1:6ra14. doi:10.1126/scitranslmed.3000322
101. Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology (2012) 143:913–6.e7. doi:10.1053/j.gastro.2012.06.031
102. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341:1241214. doi:10.1126/science.1241214
103. Hass DJ, Brensinger CM, Lewis JD, Lichtenstein GR. The impact of increased body mass index on the clinical course of Crohn’s disease. Clin Gastroenterol Hepatol (2006) 4:482–8. doi:10.1016/j.cgh.2005.12.015
104. Blain A, Cattan S, Beaugerie L, Carbonnel F, Gendre J, Cosnes J. Crohn’s disease clinical course and severity in obese patients. Clin Nutr (2002) 21:51–7. doi:10.1054/clnu.2001.0503
105. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab (2004) 89:2548–56. doi:10.1210/jc.2004-0395
106. Van Harmelen V, Reynisdottir S, Eriksson P, Thorne A, Hoffstedt J, Lonnqvist F, et al. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes (1998) 47:913–7. doi:10.2337/diabetes.47.6.913
107. Motoshima H, Wu X, Sinha MK, Hardy VE, Rosato EL, Barbot DJ, et al. Differential regulation of adiponectin secretion from cultured human omental and subcutaneous adipocytes: effects of insulin and rosiglitazone. J Clin Endocrinol Metab (2002) 87:5662–7. doi:10.1210/jc.2002-020635
108. Clement K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J (2004) 18:1657–69. doi:10.1096/fj.04-2204com
109. Poitou C, Coussieu C, Rouault C, Coupaye M, Cancello R, Bedel J, et al. Serum amyloid A: a marker of adiposity-induced low-grade inflammation but not of metabolic status. Obesity (2006) 14:309–18. doi:10.1038/oby.2006.40
110. Yang R, Lee M, Hu H, Pollin TI, Ryan AS, Nicklas BJ, et al. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med (2006) 3:e287. doi:10.1371/journal.pmed.0030287
111. Poitou C, Viguerie N, Cancello R, De Matteis R, Cinti S, Stich V, et al. Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia (2005) 48:519–28. doi:10.1007/s00125-004-1654-6
112. Ziccardi P, Nappo F, Giugliano G, Esposito K, Marfella R, Cioffi M, et al. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation (2002) 105:804–9. doi:10.1161/hc0702.104279
113. Ballantyne GH, Gumbs A, Modlin IM. Changes in insulin resistance following bariatric surgery and the adipoinsular axis: role of the adipocytokines, leptin, adiponectin and resistin. Obes Surg (2005) 15:692–9. doi:10.1381/0960892053923789
114. Taleb S, Lacasa D, Bastard JP, Poitou C, Cancello R, Pelloux V, et al. Cathepsin S, a novel biomarker of adiposity: relevance to atherogenesis. FASEB J (2005) 19(11):1540–2. doi:10.1096/fj.05-3673fje
115. Taleb S, Cancello R, Poitou C, Rouault C, Sellam P, Levy P, et al. Weight loss reduces adipose tissue cathepsin S and its circulating levels in morbidly obese women. J Clin Endocrinol Metab (2006) 91:1042–7. doi:10.1210/jc.2005-1601
116. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes (2005) 54:2277–86. doi:10.2337/diabetes.54.8.2277
117. Tilg H, Hotamisligil GS. Nonalcoholic fatty liver disease: cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology (2006) 131:934–45. doi:10.1053/j.gastro.2006.05.054
118. Clément K, Langin D. Regulation of inflammation-related genes in human adipose tissue. J Intern Med (2007) 262:422–30. doi:10.1111/j.1365-2796.2007.01851.x
119. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature (2013) 500:541–6. doi:10.1038/nature12506
120. Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol (2005) 43:3380–9. doi:10.1128/JCM.43.7.3380-3389.2005
121. Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut (2011) 60:631–7. doi:10.1136/gut.2010.223263
122. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A (2010) 107:14691–6. doi:10.1073/pnas.1005963107
123. Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. Gut microbiota and metabolic syndrome. World J Gastroenterol (2014) 20:16079–94. doi:10.3748/wjg.v20.i43.16079
124. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature (2012) 490:55–60. doi:10.1038/nature11450
125. Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One (2010) 5:e9085. doi:10.1371/journal.pone.0009085
126. Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature (2013) 498:99–103. doi:10.1038/nature12198
127. Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol (2004) 54:1469–76. doi:10.1099/ijs.0.02873-0
128. Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes (2011) 60:2775–86. doi:10.2337/db11-0227
129. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A (2013) 110:9066–71. doi:10.1073/pnas.1219451110
130. Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut (2014) 63:727–35. doi:10.1136/gutjnl-2012-303839
131. Tachon S, Zhou J, Keenan M, Martin R, Marco ML. The intestinal microbiota in aged mice is modulated by dietary resistant starch and correlated with improvements in host responses. FEMS Microbiol Ecol (2013) 83:299–309. doi:10.1111/j.1574-6941.2012.01475.x
132. Everard A, Lazarevic V, Gaïa N, Johansson M, Ståhlman M, Backhed F, et al. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J (2014) 8:2116–30. doi:10.1038/ismej.2014.45
133. Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, Svenson KL, et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe (2015) 17:72–84. doi:10.1016/j.chom.2014.11.010
134. Precechtelova J, Borsanyiova M, Sarmirova S, Bopegamage S. Type I diabetes mellitus: genetic factors and presumptive enteroviral etiology or protection. J Pathog (2014) 2014:738512. doi:10.1155/2014/738512
135. Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (2010) 18:190–5. doi:10.1038/oby.2009.167
136. Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature (2012) 489:242–9. doi:10.1038/nature11552
137. Pabst O, Cerovic V, Hornef M. Secretory IgA in the coordination of establishment and maintenance of the microbiota. Trends Immunol (2016) 37(5):287–96. doi:10.1016/j.it.2016.03.002
138. Macpherson AJ, Köller Y, McCoy KD. The bilateral responsiveness between intestinal microbes and IgA. Trends Immunol (2015) 36:460–70. doi:10.1016/j.it.2015.06.006
139. Buttó LF, Schaubeck M, Haller D. Mechanisms of microbe–host interaction in Crohn’s disease: dysbiosis vs. pathobiont selection. Front Immunol (2015) 6:Article555. doi:10.3389/fimmu.2015.00555
140. Shapiro JM, Cho JH, Sands BE, LeLeiko NS. Bridging the gap between host immune response and intestinal dysbiosis in inflammatory bowel disease: does immunoglobulin A mark the spot? Clin Gastroenterol Hepatol (2015) 13:842–6. doi:10.1016/j.cgh.2015.02.028
141. Stubbs C, Smith A. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim Biophys Acta (1984) 779:89. doi:10.1016/0304-4157(84)90005-4
142. Brenner RR. Effect of unsaturated acids on membrane structure and enzyme kinetics. Prog Lipid Res (1984) 23:69–96. doi:10.1016/0163-7827(84)90008-0
143. Chow SC, Ansotegui IJ, Jondal M. Inhibition of receptor-mediated calcium influx in T cells by unsaturated non-esterified fatty acids. Biochem J (1990) 267:727–32. doi:10.1042/bj2670727
144. Teitelbaum JE, Walker WA. Review: the role of omega 3 fatty acids in intestinal inflammation. J Nutr Biochem (2001) 12:21–32. doi:10.1016/S0955-2863(00)00141-8
145. Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol (2011) 106:563–73. doi:10.1038/ajg.2011.44
146. McDonald GB, Saunders DR, Weidman M, Fisher L. Portal venous transport of long-chain fatty acids absorbed from rat intestine. Am J Physiol (1980) 239:G141–50.
147. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi:10.1126/science.1241165
148. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature (2013) 500:232–6. doi:10.1038/nature12331
149. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi:10.1038/nature12721
150. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, Liu H, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
151. Lucas JL, Mirshahpanah P, Haas-Stapleton E, Asadullah K, Zollner TM, Numerof RP. Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell Immunol (2009) 257:97–104. doi:10.1016/j.cellimm.2009.03.004
152. Akimova T, Ge G, Golovina T, Mikheeva T, Wang L, Riley JL, et al. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3 Tregs. Clin Immunol (2010) 136:348–63. doi:10.1016/j.clim.2010.04.018
153. Kamada N, Seo S, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13:321–35. doi:10.1038/nri3430
154. Zeissig S, Blumberg RS. Life at the beginning: perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nat Immunol (2014) 15:307–10. doi:10.1038/ni.2847
155. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol (2014) 121:91–119. doi:10.1016/B978-0-12-800100-4.00003-9
156. Iwanaga T, Takebe K, Kato I, Karaki S, Kuwahara A. Cellular expression of monocarboxylate transporters (MCT) in the digestive tract of the mouse, rat, and humans, with special reference to slc5a8. Biomed Res (2006) 27:243–54. doi:10.2220/biomedres.27.243
157. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol (2014) 16:1024–33. doi:10.1111/cmi.12308
158. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40:128–39. doi:10.1016/j.immuni.2013.12.007
159. Kim SV, Xiang WV, Kwak C, Yang Y, Lin XW, Ota M, et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science (2013) 340:1456–9. doi:10.1126/science.1237013
160. Furusawa Y, Obata Y, Hase K. Commensal microbiota regulates T cell fate decision in the gut. Semin Immunopathol (2015) 37:17–25. doi:10.1007/s00281-014-0455-3
161. Tao R, de Zoeten EF, Özkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13:1299–307. doi:10.1038/nm1652
162. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients (2011) 3:858–76. doi:10.3390/nu3100858
163. Fontenelle B, Gilbert K. n-butyrate anergized effector CD4 T cells independent of regulatory T cell generation or activity. Scand J Immunol (2012) 76:457–63. doi:10.1111/j.1365-3083.2012.02740.x
164. Neis EP, Dejong CH, Rensen SS. The role of microbial amino acid metabolism in host metabolism. Nutrients (2015) 7:2930–46. doi:10.3390/nu7042930
165. Mortensen PB, Holtug K, Bonnén H, Clausen MR. The degradation of amino acids, proteins, and blood to short-chain fatty acids in colon is prevented by lactulose. Gastroenterology (1990) 98:353–60.
166. Rasmussen HS, Holtug K, Mortensen P. Degradation of amino acids to short-chain fatty acids in humans: an in vitro study. Scand J Gastroenterol (1988) 23:178–82. doi:10.3109/00365528809103964
167. Nordgaard I, Mortensen PB, Langkilde AM. Small intestinal malabsorption and colonic fermentation of resistant starch and resistant peptides to short-chain fatty acids. Nutrition (1995) 11:129–37.
168. Barker H. Amino acid degradation by anaerobic bacteria. Annu Rev Biochem (1981) 50:23–40. doi:10.1146/annurev.bi.50.070181.000323
169. Elsden SR, Hilton MG. Volatile acid production from threonine, valine, leucine and isoleucine by clostridia. Arch Microbiol (1978) 117:165–72. doi:10.1007/BF00402304
170. Davila A, Blachier F, Gotteland M, Andriamihaja M, Benetti P, Sanz Y, et al. Re-print of “Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host”. Pharmacol Res (2013) 69:114–26. doi:10.1016/j.phrs.2013.01.003
171. Davila A, Blachier F, Gotteland M, Andriamihaja M, Benetti P, Sanz Y, et al. Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharmacol Res (2013) 68:95–107. doi:10.1016/j.phrs.2012.11.005
172. Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem (2009) 284:24673–7. doi:10.1074/jbc.R109.022848
173. Salyers AA, Vercellotti JR, West SE, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol (1977) 33:319–22.
174. El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol (2013) 11:497–504. doi:10.1038/nrmicro3050
175. Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J (2012) 6:1535–43. doi:10.1038/ismej.2012.4
176. Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe (2007) 2:328–39. doi:10.1016/j.chom.2007.09.013
177. Hansen JJ, Huang Y, Peterson DA, Goeser L, Fan T, Chang EB, et al. The colitis-associated transcriptional profile of commensal Bacteroides thetaiotaomicron enhances adaptive immune responses to a bacterial antigen. PLoS One (2012) 7:e42645. doi:10.1371/journal.pone.0042645
178. Edwards L, Lucas M, Edwards E, Torrente F, Heuschkel R, Klein N, et al. Aberrant response to commensal Bacteroides thetaiotaomicron in Crohn’s disease: an ex vivo human organ culture study. Inflamm Bowel Dis (2011) 17:1201–8. doi:10.1002/ibd.21501
179. Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science (2005) 307:1955–9. doi:10.1126/science.1109051
180. Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell (2010) 141:1241–52. doi:10.1016/j.cell.2010.05.005
181. Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One (2012) 7:e49138. doi:10.1371/journal.pone.0049138
182. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut (2014) 63:1275–83. doi:10.1136/gutjnl-2013-304833
183. Attene-Ramos MS, Nava GM, Muellner MG, Wagner ED, Plewa MJ, Gaskins HR. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ Mol Mutagen (2010) 51:304–14. doi:10.1002/em.20546
184. Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science (2013) 339:708–11. doi:10.1126/science.1232467
185. Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One (2013) 8:e66019. doi:10.1371/journal.pone.0066019
186. Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol (2012) 3:448. doi:10.3389/fphys.2012.00448
187. Chassard C, Lacroix C. Carbohydrates and the human gut microbiota. Curr Opin Clin Nutr Metab Care (2013) 16:453–60. doi:10.1097/MCO.0b013e3283619e63
188. Asano I, Ikeda Y, Fujii S, Iino H. Effects of mannooligosaccharides from coffee on microbiota and short chain fatty acids in rat cecum. Food Sci Technol Res (2004) 10:273–7. doi:10.3136/fstr.10.273
189. Hosseini E, Grootaert C, Verstraete W, Van de Wiele T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr Rev (2011) 69:245–58. doi:10.1111/j.1753-4887.2011.00388.x
190. Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci (2008) 1125:171–89. doi:10.1196/annals.1419.019
191. Macfarlane GT, Gibson GR. Carbohydrate fermentation, energy transduction and gas metabolism in the human large intestine. In: Mackie R, White B, editors. Gastrointestinal Microbiology. New York, NY: Chapman and Hall (1997). p. 269–318.
192. Moore W, Cato EP, Holdeman LV. Ruminococcus bromii sp. n. and emendation of the description of Ruminococcus Sijpestein. Int J Syst Bacteriol (1972) 22:78–80. doi:10.1099/00207713-22-2-78
193. Robert C, Bernalier-Donadille A. The cellulolytic microflora of the human colon: evidence of microcrystalline cellulose-degrading bacteria in methane-excreting subjects. FEMS Microbiol Ecol (2003) 46:81–9. doi:10.1016/S0168-6496(03)00207-1
194. Westermann P, Ahring BK, Mah RA. Acetate production by methanogenic bacteria. Appl Environ Microbiol (1989) 55:2257–61.
195. Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A (2009) 106:5859–64. doi:10.1073/pnas.0901529106
196. Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes (2012) 3:289–306. doi:10.4161/gmic.19897
197. Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol (2000) 66:1654–61. doi:10.1128/AEM.66.4.1654-1661.2000
198. Duncan SH, Flint HJ. Proposal of a neotype strain (A1-86) for Eubacterium rectale. Request for an opinion. Int J Syst Evol Microbiol (2008) 58:1735–6. doi:10.1099/ijs.0.2008/004580-0
199. Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol (2004) 70:5810–7. doi:10.1128/AEM.70.10.5810-5817.2004
200. Duncan SH, Aminov RI, Scott KP, Louis P, Stanton TB, Flint HJ. Proposal of Roseburia faecis sp. nov., Roseburia hominis sp. nov. and Roseburia inulinivorans sp. nov., based on isolates from human faeces. Int J Syst Evol Microbiol (2006) 56:2437–41. doi:10.1099/ijs.0.64098-0
201. Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA: acetate CoA-transferase gene. Environ Microbiol (2010) 12:304–14. doi:10.1111/j.1462-2920.2009.02066.x
202. Marquet P, Duncan SH, Chassard C, Bernalier-Donadille A, Flint HJ. Lactate has the potential to promote hydrogen sulphide formation in the human colon. FEMS Microbiol Lett (2009) 299:128–34. doi:10.1111/j.1574-6968.2009.01750.x
203. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54:2325–40. doi:10.1194/jlr.R036012
204. Ragsdale SW, Pierce E. Acetogenesis and the Wood–Ljungdahl pathway of CO(2) fixation. Biochim Biophys Acta (2008) 1784:1873–98. doi:10.1016/j.bbapap.2008.08.012
205. Guilloteau P, Martin L, Eeckhaut V, Ducatelle R, Zabielski R, Van Immerseel F. From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr Res Rev (2010) 23:366–84. doi:10.1017/S0954422410000247
206. Reichardt N, Duncan SH, Young P, Belenguer A, Leitch CM, Scott KP, et al. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J (2014) 8:1323–35. doi:10.1038/ismej.2014.14
207. Blottiere HM, Buecher B, Galmiche J, Cherbut C. Molecular analysis of the effect of short-chain fatty acids on intestinal cell proliferation. Proc Nutr Soc (2003) 62:101–6. doi:10.1079/PNS2002215
208. Lukovac S, Belzer C, Pellis L, Keijser BJ, de Vos WM, Montijn RC, et al. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio (2014) 5:e01438–14. doi:10.1128/mBio.01438
209. Kendrick SF, O’Boyle G, Mann J, Zeybel M, Palmer J, Jones DE, et al. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology (2010) 51:1988–97. doi:10.1002/hep.23572
210. Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, et al. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res (2008) 28:321–8. doi:10.1016/j.nutres.2008.02.012
211. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem (2003) 278:11312–9. doi:10.1074/jbc.M211609200
212. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem (2003) 278:25481–9. doi:10.1074/jbc.M301403200
213. Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA 2 R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun (2003) 303:1047–52. doi:10.1016/S0006-291X(03)00488-1
214. Elangovan S, Pathania R, Ramachandran S, Ananth S, Padia RN, Lan L, et al. The niacin/butyrate receptor GPR109A suppresses mammary tumorigenesis by inhibiting cell survival. Cancer Res (2014) 74:1166–78. doi:10.1158/0008-5472.CAN-13-1451
215. Milligan G, Alvarez-Curto E, Watterson K, Ulven T, Hudson B. Characterizing pharmacological ligands to study the long-chain fatty acid receptors GPR40/FFA1 and GPR120/FFA4. Br J Pharmacol (2015) 172:3254–65. doi:10.1111/bph.12879
216. Alvarez-Curto E, Milligan G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol (2016). doi:10.1016/j.bcp.2016.03.017
217. Wanders D, Judd R. Future of GPR109A agonists in the treatment of dyslipidaemia. Diabetes Obes Metab (2011) 13:685–91. doi:10.1111/j.1463-1326.2011.01400.x
218. Yuan G, Chen X, Li D. Modulation of peroxisome proliferator-activated receptor gamma (PPAR γ) by conjugated fatty acid in obesity and inflammatory bowel disease. J Agric Food Chem (2015) 63:1883–95. doi:10.1021/jf505050c
219. Hudson BD, Tikhonova IG, Pandey SK, Ulven T, Milligan G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J Biol Chem (2012) 287:41195–209. doi:10.1074/jbc.M112.396259
220. Byrne C, Chambers E, Morrison D, Frost G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int J Obes (2015) 39:1331–9. doi:10.1038/ijo.2015.84
221. Tazoe H, Otomo Y, Karaki S, Kato I, Fukami Y, Terasaki M, et al. Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed Res (2009) 30:149–56. doi:10.2220/biomedres.30.149
222. Nøhr MK, Pedersen MH, Gille A, Egerod KL, Engelstoft MS, Husted AS, et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology (2013) 154:3552–64. doi:10.1210/en.2013-1142
223. Li G, Su H, Zhou Z, Yao W. Identification of the porcine G protein-coupled receptor 41 and 43 genes and their expression pattern in different tissues and development stages. PLoS One (2014) 9:e97342. doi:10.1371/journal.pone.0097342
224. Masui R, Sasaki M, Funaki Y, Ogasawara N, Mizuno M, Iida A, et al. G protein-coupled receptor 43 moderates gut inflammation through cytokine regulation from mononuclear cells. Inflamm Bowel Dis (2013) 19:2848–56. doi:10.1097/01.MIB.0000435444.14860.ea
225. Cox MA, Jackson J, Stanton M, Rojas-Triana A, Bober L, Laverty M, et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J Gastroenterol (2009) 15:5549–57. doi:10.3748/wjg.15.5549
226. Karaki S, Tazoe H, Hayashi H, Kashiwabara H, Tooyama K, Suzuki Y, et al. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J Mol Histol (2008) 39:135–42. doi:10.1007/s10735-007-9145-y
227. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461:1282–6. doi:10.1038/nature08530
228. Xiong Y, Miyamoto N, Shibata K, Valasek MA, Motoike T, Kedzierski RM, et al. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc Natl Acad Sci U S A (2004) 101:1045–50. doi:10.1073/pnas.2637002100
229. Hong Y, Nishimura Y, Hishikawa D, Tsuzuki H, Miyahara H, Gotoh C, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology (2005) 146:5092–9. doi:10.1210/en.2005-0545
230. Zaibi MS, Stocker CJ, O’Dowd J, Davies A, Bellahcene M, Cawthorne MA, et al. Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett (2010) 584:2381–6. doi:10.1016/j.febslet.2010.04.027
231. Dewulf EM, Cani PD, Neyrinck AM, Possemiers S, Van Holle A, Muccioli GG, et al. Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARγ-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J Nutr Biochem (2011) 22:712–22. doi:10.1016/j.jnutbio.2010.05.009
232. Peyrin-Biroulet L, Beisner J, Wang G, Nuding S, Oommen ST, Kelly D, et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc Natl Acad Sci U S A (2010) 107:8772–7. doi:10.1073/pnas.0905745107
233. Wrzosek L, Miquel S, Noordine ML, Bouet S, Joncquel Chevalier-Curt M, Robert V, et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol (2013) 11:61. doi:10.1186/1741-7007-11-61
234. Burger-van Paassen N, Vincent A, Puiman P, Van Der Sluis M, Bouma J, Boehm G, et al. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: implications for epithelial protection. Biochem J (2009) 420:211–9. doi:10.1042/BJ20082222
235. Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-κB activity in a human colonic epithelial cell line. Gastroenterology (2000) 118:724–34. doi:10.1016/S0016-5085(00)70142-9
236. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
237. Fung KY, Cosgrove L, Lockett T, Head R, Topping DL. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br J Nutr (2012) 108:820–31. doi:10.1017/S0007114512001948
238. Ni YF, Wang J, Yan XL, Tian F, Zhao JB, Wang YJ, et al. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir Res (2010) 11:33. doi:10.1186/1465-9921-11-33
239. Park J, Lee E, Lee J, Kim W, Kim H. Anti-inflammatory effects of short chain fatty acids in IFN-γ-stimulated RAW 264.7 murine macrophage cells: involvement of NF-κB and ERK signaling pathways. Int Immunopharmacol (2007) 7:70–7. doi:10.1016/j.intimp.2006.08.015
240. Maa M, Chang MY, Hsieh M, Chen Y, Yang C, Chen Z, et al. Butyrate reduced lipopolysaccharide-mediated macrophage migration by suppression of Src enhancement and focal adhesion kinase activity. J Nutr Biochem (2010) 21:1186–92. doi:10.1016/j.jnutbio.2009.10.004
241. Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut (2000) 46:493–9. doi:10.1136/gut.46.4.493
242. Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol (2006) 40:235–43. doi:10.1097/00004836-200603000-00015
243. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe (2015) 17:662–71. doi:10.1016/j.chom.2015.03.005
244. Cushing K, Alvarado DM, Ciorba MA. Butyrate and mucosal inflammation: new scientific evidence supports clinical observation. Clin Transl Gastroenterol (2015) 6:e108. doi:10.1038/ctg.2015.34
245. Kostik V, Memeti S, Bauer B. Fatty acid composition of edible oils and fats. J Hyg Eng Des (2013) 4:112–6.
246. Laviano A, Rossi F. Lipid emulsions in parenteral nutrition: does one size fits all? SAJCN (2010) 23:S8–10. doi:10.1080/16070658.2010.11734260
247. Serra-Majem L, Roman B, Estruch R. Scientific evidence of interventions using the Mediterranean diet: a systematic review. Nutr Rev (2006) 64:S27–47. doi:10.1301/nr.2006.feb.S27-S47
248. James MJ, Gibson RA, Cleland LG. Dietary polyunsaturated fatty acids and inflammatory mediator production. Am J Clin Nutr (2000) 71(1 Suppl):343S–8S.
249. McEntee MF, Ziegler C, Reel D, Tomer K, Shoieb A, Ray M, et al. Dietary n-3 polyunsaturated fatty acids enhance hormone ablation therapy in androgen-dependent prostate cancer. Am J Pathol (2008) 173:229–41. doi:10.2353/ajpath.2008.070989
250. Babcock TA, Novak T, Ong E, Jho DH, Helton WS, Espat NJ. Modulation of lipopolysaccharide-stimulated macrophage tumor necrosis factor-α production by ω-3 fatty acid is associated with differential cyclooxygenase-2 protein expression and is independent of interleukin-10. J Surg Res (2002) 107:135–9. doi:10.1016/S0022-4804(02)96498-X
251. Hegazi RA, Saad RS, Mady H, Matarese LE, O’Keefe S, Kandil HM. Dietary fatty acids modulate chronic colitis, colitis-associated colon neoplasia and COX-2 expression in IL-10 knockout mice. Nutrition (2006) 22:275–82. doi:10.1016/j.nut.2005.06.006
252. Knoch B, Barnett MP, Cooney J, McNabb WC, Barraclough D, Laing W, et al. Dietary oleic acid as a control fatty acid for polyunsaturated fatty acid intervention studies: a transcriptomics and proteomics investigation using interleukin-10 gene-deficient mice. Biotechnol J (2010) 5:1226–40. doi:10.1002/biot.201000066
253. Vassiliou EK, Gonzalez A, Garcia C, Tadros JH, Chakraborty G, Toney JH. Oleic acid and peanut oil high in oleic acid reverse the inhibitory effect of insulin production of the inflammatory cytokine TNF-α both in vitro and in vivo systems. Lipids Health Dis (2009) 8:25. doi:10.1186/1476-511X-8-25
254. Weylandt KH, Chiu C, Gomolka B, Waechter SF, Wiedenmann B. Omega-3 fatty acids and their lipid mediators: towards an understanding of resolvin and protectin formation. Prostaglandins Other Lipid Mediat (2012) 97:73–82. doi:10.1016/j.prostaglandins.2012.01.005
255. Weylandt KH, Kang JX. Rethinking lipid mediators. Lancet (2005) 366:618–20. doi:10.1016/S0140-6736(05)67119-X
256. Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest (2005) 115:2774–83. doi:10.1172/JCI25420
257. Mukherjee PK, Marcheselli VL, de Rivero Vaccari JC, Gordon WC, Jackson FE, Bazan NG. Photoreceptor outer segment phagocytosis attenuates oxidative stress-induced apoptosis with concomitant neuroprotectin D1 synthesis. Proc Natl Acad Sci U S A (2007) 104:13158–63. doi:10.1073/pnas.0705963104
258. Im D. Omega-3 fatty acids in anti-inflammation (pro-resolution) and GPCRs. Prog Lipid Res (2012) 51:232–7. doi:10.1016/j.plipres.2012.02.003
259. Calder PC. Marine omega-3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance. Biochim Biophys Acta (2015) 1851:469–84. doi:10.1016/j.bbalip.2014.08.010
260. Weylandt KH, Serini S, Chen YQ, Su HM, Lim K, Cittadini A, et al. Omega-3 polyunsaturated fatty acids: the way forward in times of mixed evidence. Biomed Res Int (2015) 2015:143109. doi:10.1155/2015/143109
261. Marion-Letellier R, Savoye G, Ghosh S. Polyunsaturated fatty acids and inflammation. IUBMB Life (2015) 67:659–67. doi:10.1002/iub.1428
262. Li Y, Kanaji N, Wang X, Sato T, Nakanishi M, Kim M, et al. Prostaglandin E 2 switches from a stimulator to an inhibitor of cell migration after epithelial-to-mesenchymal transition. Prostaglandins Other Lipid Mediat (2015) 116:1–9. doi:10.1016/j.prostaglandins.2014.10.003
263. Masoodi M, Pearl DS, Eiden M, Shute JK, Brown JF, Calder PC, et al. Altered colonic mucosal polyunsaturated fatty acid (PUFA) derived lipid mediators in ulcerative colitis: new insight into relationship with disease activity and pathophysiology. PLoS One (2013) 8:e76532. doi:10.1371/journal.pone.0076532
264. Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature (2004) 427:504–504. doi:10.1038/427504a
265. Hudert CA, Weylandt KH, Lu Y, Wang J, Hong S, Dignass A, et al. Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proc Natl Acad Sci U S A (2006) 103:11276–81. doi:10.1073/pnas.0601280103
266. Monk JM, Jia Q, Callaway E, Weeks B, Alaniz RC, McMurray DN, et al. Th17 cell accumulation is decreased during chronic experimental colitis by (n-3) PUFA in Fat-1 mice. J Nutr (2012) 142:117–24. doi:10.3945/jn.111.147058
267. Nowak J, Weylandt KH, Habbel P, Wang J, Dignass A, Glickman JN, et al. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis (2007) 28:1991–5. doi:10.1093/carcin/bgm166
268. Jia Q, Lupton JR, Smith R, Weeks BR, Callaway E, Davidson LA, et al. Reduced colitis-associated colon cancer in Fat-1 (n-3 fatty acid desaturase) transgenic mice. Cancer Res (2008) 68:3985–91. doi:10.1158/0008-5472.CAN-07-6251
269. Teague H, Fhaner CJ, Harris M, Duriancik DM, Reid GE, Shaikh SR. n-3 PUFAs enhance the frequency of murine B-cell subsets and restore the impairment of antibody production to a T-independent antigen in obesity. J Lipid Res (2013) 54:3130–8. doi:10.1194/jlr.M042457
270. Turk HF, Monk JM, Fan YY, Callaway ES, Weeks B, Chapkin RS. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing. Am J Physiol Cell Physiol (2013) 304:C905–17. doi:10.1152/ajpcell.00379.2012
271. Whiting CV, Bland PW, Tarlton JF. Dietary n-3 polyunsaturated fatty acids reduce disease and colonic proinflammatory cytokines in a mouse model of colitis. Inflamm Bowel Dis (2005) 11:340–9. doi:10.1097/01.MIB.0000164016.98913.7c
272. Campos F, Waitzberg D, Habr-Gama A, Logullo A, Noronha I, Jancar S, et al. Impact of parenteral n-3 fatty acids on experimental acute colitis. Br J Nutr (2002) 87:S83–8. doi:10.1079/BJN2001460
273. Matsunaga H, Hokari R, Kurihara C, Okada Y, Takebayashi K, Okudaira K, et al. Omega-3 fatty acids exacerbate DSS-induced colitis through decreased adiponectin in colonic subepithelial myofibroblasts. Inflamm Bowel Dis (2008) 14:1348–57. doi:10.1002/ibd.20491
274. Fischer R, Konkel A, Mehling H, Blossey K, Gapelyuk A, Wessel N, et al. Dietary omega-3 fatty acids modulate the eicosanoid profile in man primarily via the CYP-epoxygenase pathway. J Lipid Res (2014) 55:1150–64. doi:10.1194/jlr.M047357
275. Tabbaa M, Golubic M, Roizen MF, Bernstein AM. Docosahexaenoic acid, inflammation, and bacterial dysbiosis in relation to periodontal disease, inflammatory bowel disease, and the metabolic syndrome. Nutrients (2013) 5:3299–310. doi:10.3390/nu5083299
276. Lauritsen K, Laursen LS, Bukhave K, Rask-Madsen J. In vivo profiles of eicosanoids in ulcerative colitis, Crohn’s colitis, and Clostridium difficile colitis. Gastroenterology (1988) 95:11–7. doi:10.1016/0016-5085(88)90284-3
277. Vilaseca J, Salas A, Guarner F, Rodriguez R, Martinez M, Malagelada JR. Dietary fish oil reduces progression of chronic inflammatory lesions in a rat model of granulomatous colitis. Gut (1990) 31:539–44. doi:10.1136/gut.31.5.539
278. Hokari R, Matsunaga H, Miura S. Effect of dietary fat on intestinal inflammatory diseases. J Gastroenterol Hepatol (2013) 28:33–6. doi:10.1111/jgh.12252
279. Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab (2013) 17:141–52. doi:10.1016/j.cmet.2012.12.007
280. Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J (2010) 4:232–41. doi:10.1038/ismej.2009.112
281. Kantor ED, Lampe JW, Peters U, Vaughan TL, White E. Long-chain omega-3 polyunsaturated fatty acid intake and risk of colorectal cancer. Nutr Cancer (2014) 66:716–27. doi:10.1080/01635581.2013.804101
282. Habermann N, Ulrich CM, Lundgreen A, Makar KW, Poole EM, Caan B, et al. PTGS1, PTGS2, ALOX5, ALOX12, ALOX15, and FLAP SNPs: interaction with fatty acids in colon cancer and rectal cancer. Genes Nutr (2013) 8:115–26. doi:10.1007/s12263-012-0302-x
283. Fradet V, Cheng I, Casey G, Witte JS. Dietary omega-3 fatty acids, cyclooxygenase-2 genetic variation, and aggressive prostate cancer risk. Clin Cancer Res (2009) 15:2559–66. doi:10.1158/1078-0432.CCR-08-2503
284. Simopoulos AP. Genetic variants in the metabolism of omega-6 and omega-3 fatty acids: their role in the determination of nutritional requirements and chronic disease risk. Exp Biol Med (Maywood) (2010) 235:785–95. doi:10.1258/ebm.2010.009298
285. Gago-Dominguez M, Castelao JE, Sun CL, Van Den Berg D, Koh WP, Lee HP, et al. Marine n-3 fatty acid intake, glutathione S-transferase polymorphisms and breast cancer risk in post-menopausal Chinese women in Singapore. Carcinogenesis (2004) 25:2143–7. doi:10.1093/carcin/bgh230
286. Porenta SR, Ko YA, Gruber SB, Mukherjee B, Baylin A, Ren J, et al. Interaction of fatty acid genotype and diet on changes in colonic fatty acids in a Mediterranean diet intervention study. Cancer Prev Res (Phila) (2013) 6:1212–21. doi:10.1158/1940-6207.CAPR-13-0131
287. Stewart JA, Chadwick VS, Murray A. Investigations into the influence of host genetics on the predominant eubacteria in the faecal microflora of children. J Med Microbiol (2005) 54:1239–42. doi:10.1099/jmm.0.46189-0
288. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486:222–7. doi:10.1038/nature11053
289. Lepage P, Häsler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology (2011) 141:227–36. doi:10.1053/j.gastro.2011.04.011
290. Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, et al. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J (2013) 7:707–17. doi:10.1038/ismej.2012.146
291. Sadli N, Ackland ML, De Mel D, Sinclair AJ, Suphioglu C. Effects of zinc and DHA on the epigenetic regulation of human neuronal cells. Cell Physiol Biochem (2012) 29:87–98. doi:10.1159/000337590
292. Amarasekera M, Noakes P, Strickland D, Saffery R, Martino DJ, Prescott SL. Epigenome-wide analysis of neonatal CD4 T-cell DNA methylation sites potentially affected by maternal fish oil supplementation. Epigenetics (2014) 9:1570–6. doi:10.4161/15592294.2014.983366
293. Jacobson K, Mundra H, Innis SM. Intestinal responsiveness to experimental colitis in young rats is altered by maternal diet. Am J Physiol Gastrointest Liver Physiol (2005) 289:G13–20. doi:10.1152/ajpgi.00459.2004
294. Gibson D, Gill S, Brown K, Tasnim N, Ghosh S, Innis S, et al. Maternal exposure to fish oil primes offspring to harbor intestinal pathobionts associated with altered immune cell balance. Gut Microbes (2015) 6:24–32. doi:10.1080/19490976.2014.997610
295. Ghosh S, Molcan E, DeCoffe D, Dai C, Gibson DL. Diets rich in n-6 PUFA induce intestinal microbial dysbiosis in aged mice. Br J Nutr (2013) 110:515–23. doi:10.1017/S0007114512005326
296. Innis SM, Dai C, Wu X, Buchan AM, Jacobson K. Perinatal lipid nutrition alters early intestinal development and programs the response to experimental colitis in young adult rats. Am J Physiol Gastrointest Liver Physiol (2010) 299:G1376–85. doi:10.1152/ajpgi.00258.2010
297. Nagy-Szakal D, Mir SA, Harris RA, Dowd SE, Yamada T, Lacorazza HD, et al. Loss of n-6 fatty acid induced pediatric obesity protects against acute murine colitis. FASEB J (2015) 29:3151–9. doi:10.1096/fj.14-267690
298. Hazell SL, Graham DY. Unsaturated fatty acids and viability of Helicobacter (Campylobacter) pylori. J Clin Microbiol (1990) 28:1060–1.
299. Thompson L, Cockayne A, Spiller RC. Inhibitory effect of polyunsaturated fatty acids on the growth of Helicobacter pylori: a possible explanation of the effect of diet on peptic ulceration. Gut (1994) 35:1557–61. doi:10.1136/gut.35.11.1557
300. Khulusi S, Ahmed H, Patel P, Mendall M, Northfield T. The effects of unsaturated fatty acids on Helicobacter pylori in vitro. J Med Microbiol (1995) 42:276–82. doi:10.1099/00222615-42-4-276
301. Petschow BW, Batema RP, Ford LL. Susceptibility of Helicobacter pylori to bactericidal properties of medium-chain monoglycerides and free fatty acids. Antimicrob Agents Chemother (1996) 40:302–6.
302. Bergsson G, Steingri´msson Ó, Thormar H. Bactericidal effects of fatty acids and monoglycerides on Helicobacter pylori. Int J Antimicrob Agents (2002) 20:258–62. doi:10.1016/S0924-8579(02)00205-4
303. Sun CQ, O’Connor CJ, Roberton AM. Antibacterial actions of fatty acids and monoglycerides against Helicobacter pylori. FEMS Immunol Med Microbiol (2003) 36:9–17. doi:10.1016/S0928-8244(03)00008-7
304. Correia M, Michel V, Matos AA, Carvalho P, Oliveira MJ, Ferreira RM, et al. Docosahexaenoic acid inhibits Helicobacter pylori growth in vitro and mice gastric mucosa colonization. PLoS One (2012) 7:e35072. doi:10.1371/journal.pone.0035072
305. Jung SW, Lee SW. The antibacterial effect of fatty acids on Helicobacter pylori infection. Korean J Intern Med (2016) 31:30–5. doi:10.3904/kjim.2016.31.1.30
306. Hennessy AA, Ross RP, Devery R, Stanton C. Bifidobacterially produced, C18: 3 and C18: 4 conjugated fatty acids exhibit in vitro anti-carcinogenic and anti-microbial activity. Eur J Lipid Sci Technol (2016). doi:10.1002/ejlt.201500424
307. Béligon V, Christophe G, Fontanille P, Larroche C. Microbial lipids as potential source to food supplements. Curr Opin Food Sci (2016) 7:35–42. doi:10.1016/j.cofs.2015.10.002
308. Woodworth HL, McCaskey SJ, Duriancik DM, Clinthorne JF, Langohr IM, Gardner EM, et al. Dietary fish oil alters T lymphocyte cell populations and exacerbates disease in a mouse model of inflammatory colitis. Cancer Res (2010) 70:7960–9. doi:10.1158/0008-5472.CAN-10-1396
309. Ghosh S, DeCoffe D, Brown K, Rajendiran E, Estaki M, Dai C, et al. Fish oil attenuates omega-6 polyunsaturated fatty acid-induced dysbiosis and infectious colitis but impairs LPS dephosphorylation activity causing sepsis. PLoS One (2013) 8:e55468. doi:10.1371/journal.pone.0055468
310. Chapkin RS, Davidson LA, Ly L, Weeks BR, Lupton JR, McMurray DN. Immunomodulatory effects of (n-3) fatty acids: putative link to inflammation and colon cancer. J Nutr (2007) 137(1 Suppl):200S–4S.
311. Hekmatdoost A, Feizabadi MM, Djazayery A, Mirshafiey A, Eshraghian MR, Yeganeh SM, et al. The effect of dietary oils on cecal microflora in experimental colitis in mice. Indian J Gastroenterol (2008) 27(5):186–9.
312. Tyagi A, Kumar U, Santosh VS, Reddy S, Mohammed SB, Ibrahim A. Partial replacement of dietary linoleic acid with long chain n-3 polyunsaturated fatty acids protects against dextran sulfate sodium-induced colitis in rats. Prostaglandins Leukot Essent Fatty Acids (2014) 91:289–97. doi:10.1016/j.plefa.2014.09.003
313. Tyagi A, Kumar U, Reddy S, Santosh VS, Mohammed SB, Ehtesham NZ, et al. Attenuation of colonic inflammation by partial replacement of dietary linoleic acid with α-linolenic acid in a rat model of inflammatory bowel disease. Br J Nutr (2012) 108:1612–22. doi:10.1017/S0007114511007197
314. Roy N, Barnett M, Knoch B, Dommels Y, McNabb W. Nutrigenomics applied to an animal model of Inflammatory Bowel Diseases: transcriptomic analysis of the effects of eicosapentaenoic acid-and arachidonic acid-enriched diets. Mutat Res (2007) 622:103–16. doi:10.1016/j.mrfmmm.2007.04.003
315. Jenkins T, Wallace R, Moate P, Mosley E. Board-invited review: recent advances in biohydrogenation of unsaturated fatty acids within the rumen microbial ecosystem. J Anim Sci (2008) 86:397–412. doi:10.2527/jas.2007-0588
316. Bauman D, Baumgard L, Corl B, Griinari DJ. Biosynthesis of conjugated linoleic acid in ruminants. J Anim Sci (2000) 77:1–15. doi:10.2134/jas2000.77E-Suppl1f
317. Ferlay A, Bernard L, Rouel J, Doreau M. Diet, rumen biohydrogenation and nutritional quality of cow and goat milk fat. Eur J Lipid Sci Technol (2007) 109:828–55. doi:10.1002/ejlt.200700080
318. Griinari JM, Corl BA, Lacy SH, Chouinard PY, Nurmela KV, Bauman DE. Conjugated linoleic acid is synthesized endogenously in lactating dairy cows by Delta(9)-desaturase. J Nutr (2000) 130:2285–91.
319. Turpeinen AM, Mutanen M, Aro A, Salminen I, Basu S, Palmquist DL, et al. Bioconversion of vaccenic acid to conjugated linoleic acid in humans. Am J Clin Nutr (2002) 76:504–10.
320. Santora JE, Palmquist DL, Roehrig KL. Trans-vaccenic acid is desaturated to conjugated linoleic acid in mice. J Nutr (2000) 130:208–15.
321. Devillard E, McIntosh FM, Duncan SH, Wallace RJ. Metabolism of linoleic acid by human gut bacteria: different routes for biosynthesis of conjugated linoleic acid. J Bacteriol (2007) 189:2566–70. doi:10.1128/JB.01359-06
322. McIntosh FM, Shingfield KJ, Devillard E, Russell WR, Wallace RJ. Mechanism of conjugated linoleic acid and vaccenic acid formation in human faecal suspensions and pure cultures of intestinal bacteria. Microbiology (2009) 155:285–94. doi:10.1099/mic.0.022921-0
323. Barrett E, Ross RP, Fitzgerald GF, Stanton C. Rapid screening method for analyzing the conjugated linoleic acid production capabilities of bacterial cultures. Appl Environ Microbiol (2007) 73:2333–7. doi:10.1128/AEM.01855-06
324. Chaplin A, Parra P, Serra F, Palou A. Conjugated linoleic acid aupplementation under a high-fat diet modulates stomach protein expression and Iitestinal microbiota in adult mice. PLoS One (2015) 10:e0125091. doi:10.1371/journal.pone.0125091
325. Coakley M, Ross R, Nordgren M, Fitzgerald G, Devery R, Stanton C. Conjugated linoleic acid biosynthesis by human-derived Bifidobacterium species. J Appl Microbiol (2003) 94:138–45. doi:10.1046/j.1365-2672.2003.01814.x
326. Rosberg-Cody E, Ross RP, Hussey S, Ryan CA, Murphy BP, Fitzgerald GF, et al. Mining the microbiota of the neonatal gastrointestinal tract for conjugated linoleic acid-producing bifidobacteria. Appl Environ Microbiol (2004) 70:4635–41. doi:10.1128/AEM.70.8.4635-4641.2004
327. Lee H, Park J, Seok S, Baek M, Kim D, Lee K, et al. Human originated bacteria, Lactobacillus rhamnosus PL60, produce conjugated linoleic acid and show anti-obesity effects in diet-induced obese mice. Biochim Biophys Acta (2006) 1761:736–44. doi:10.1016/j.bbalip.2006.05.007
328. Wall R, Ross RP, Shanahan F, O’Mahony L, O’Mahony C, Coakley M, et al. Metabolic activity of the enteric microbiota influences the fatty acid composition of murine and porcine liver and adipose tissues. Am J Clin Nutr (2009) 89:1393–401. doi:10.3945/ajcn.2008.27023
329. Lieberman S, Enig MG, Preuss HG. A review of monolaurin and lauric acid: natural virucidal and bactericidal agents. Altern Complement Ther (2006) 12:310–4. doi:10.1089/act.2006.12.310
330. Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem (2003) 278:37041–51. doi:10.1074/jbc.M305213200
331. Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem (2009) 284:27384–92. doi:10.1074/jbc.M109.044065
332. Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res (2003) 44:479–86. doi:10.1194/jlr.M200361-JLR200
333. Rocha D, Caldas A, Oliveira L, Bressan J, Hermsdorff H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis (2016) 244:211–5. doi:10.1016/j.atherosclerosis.2015.11.015
334. Lucas K, Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol Neurobiol (2013) 48:190–204. doi:10.1007/s12035-013-8425-7
335. Di Gioia M, Zanoni I. Toll-like receptor co-receptors as master regulators of the immune response. Mol Immunol (2015) 63:143–52. doi:10.1016/j.molimm.2014.05.008
336. Oury C. CD36: linking lipids to the NLRP3 inflammasome, atherogenesis and atherothrombosis. Cell Mol Immunol (2014) 11:8–10. doi:10.1038/cmi.2013.48
337. Botham KM, Wheeler-Jones CP. Postprandial lipoproteins and the molecular regulation of vascular homeostasis. Prog Lipid Res (2013) 52:446–64. doi:10.1016/j.plipres.2013.06.001
338. Moreira APB, Texeira TFS, Ferreira AB, Peluzio Mdo C, Alfenas Rde C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr (2012) 108:801–9. doi:10.1017/S0007114512001213
339. de Vries MA, Klop B, Eskes SA, van der Loos TL, Klessens-Godfroy FJ, Wiebolt J, et al. The postprandial situation as a pro-inflammatory condition. Clín Investig Arterioscler (2014) 26:184–92. doi:10.1016/j.arteri.2014.02.007
340. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res (2014) 42:D199–205. doi:10.1093/nar/gkt1076
341. Manna SK, Aggarwal BB. Wortmannin inhibits activation of nuclear transcription factors NF-κB and activated protein-1 induced by lipopolysaccharide and phorbol ester. FEBS Lett (2000) 473:113–8. doi:10.1016/S0014-5793(00)01501-5
342. Hwang D. Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways. FASEB J (2001) 15:2556–64. doi:10.1096/fj.01-0432com
343. Hoshi N, Schenten D, Nish SA, Walther Z, Gagliani N, Flavell RA, et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat Commun (2012) 3:1120. doi:10.1038/ncomms2113
344. Mane J, Pedrosa E, Loren V, Ojanguren I, Fluvia L, Cabre E, et al. Partial replacement of dietary (n-6) fatty acids with medium-chain triglycerides decreases the incidence of spontaneous colitis in interleukin-10-deficient mice. J Nutr (2009) 139:603–10. doi:10.3945/jn.108.101170
345. Ohta N, Tsujikawa T, Nakamura T, Andoh A, Sasaki M, Bamba T. A comparison of the effects of medium-and long-chain triglycerides on neutrophil stimulation in experimental ileitis. J Gastroenterol (2003) 38:127–33. doi:10.1007/s005350300021
346. Tsujikawa T, Ohta N, Nakamura T, Satoh J, Uda K, Ihara T, et al. Medium-chain triglycerides modulate ileitis induced by trinitrobenzene sulfonic acid. J Gastroenterol Hepatol (1999) 14:1166–72. doi:10.1046/j.1440-1746.1999.02024.x
347. Schmelz EM, Zhou H, Roberts PC. Dietary sphingolipids in colon cancer prevention. In: Yusuf H, Chiara L, Cungui M, Lina MO, editors. Bioactive Sphingolipids in Cancer Biology and Therapy. Switzerland: Springer (2015). p. 161–88.
348. Vesper H, Schmelz EM, Nikolova-Karakashian MN, Dillehay DL, Lynch DV, Merrill AH Jr. Sphingolipids in food and the emerging importance of sphingolipids to nutrition. J Nutr (1999) 129:1239–50.
349. Yunoki K, Ogawa T, Ono J, Miyashita R, Aida K, Oda Y, et al. Analysis of sphingolipid classes and their contents in meals. Biosci Biotechnol Biochem (2008) 72:222–5. doi:10.1271/bbb.70463
350. Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM, et al. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J Biol Chem (2006) 281:7324–31. doi:10.1074/jbc.M508382200
351. Abdel Hadi L, Di Vito C, Riboni L. Fostering inflammatory bowel disease: sphingolipid strategies to join forces. Mediators Inflamm (2016) 2016:3827684. doi:10.1155/2016/3827684
352. Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature (2014) 510:58–67. doi:10.1038/nature13475
353. Michael Danielsen E, Hansen GH. Lipid raft organization and function in brush borders of epithelial cells (Review). Mol Membr Biol (2006) 23:71–9. doi:10.1080/09687860500445604
354. Hannun YA. Sphingolipid-Mediated Signal Transduction. Germany: Springer Science & Business Media (2013).
355. Furuya H, Choi S, Obeid LM, Kawamori T, Snider AJ. Colon cancer: the role of sphingolipid metabolic enzymes. In: Yusuf H, Chiara L, Cungui M, Lina MO, editors. Bioactive Sphingolipids in Cancer Biology and Therapy. Switzerland: Springer (2015). p. 141–59.
356. Pettus BJ, Kitatani K, Chalfant CE, Taha TA, Kawamori T, Bielawski J, et al. The coordination of prostaglandin E2 production by sphingosine-1-phosphate and ceramide-1-phosphate. Mol Pharmacol (2005) 68:330–5.
357. Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J Biol Chem (2003) 278:38206–13. doi:10.1074/jbc.M304816200
358. Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J (2003) 17:1411–21. doi:10.1096/fj.02-1038com
359. Nakamura H, Hirabayashi T, Shimizu M, Murayama T. Ceramide-1-phosphate activates cytosolic phospholipase A2α directly and by PKC pathway. Biochem Pharmacol (2006) 71:850–7. doi:10.1016/j.bcp.2005.12.027
360. Subramanian P, Stahelin RV, Szulc Z, Bielawska A, Cho W, Chalfant CE. Ceramide 1-phosphate acts as a positive allosteric activator of group IVA cytosolic phospholipase A2 alpha and enhances the interaction of the enzyme with phosphatidylcholine. J Biol Chem (2005) 280:17601–7. doi:10.1074/jbc.M414173200
361. Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem (2004) 279:11320–6. doi:10.1074/jbc.M309262200
362. Nakamura H, Murayama T. The role of sphingolipids in arachidonic acid metabolism. J Pharmacol Sci (2014) 124:307–12. doi:10.1254/jphs.13R18CP
363. Fischbeck A, Leucht K, Frey-Wagner I, Bentz S, Pesch T, Kellermeier S, et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut (2011) 60:55–65. doi:10.1136/gut.2009.201988
364. Park EJ, Thomson AB, Clandinin MT. Protection of intestinal occludin tight junction protein by dietary gangliosides in lipopolysaccharide-induced acute inflammation. J Pediatr Gastroenterol Nutr (2010) 50:321–8. doi:10.1097/MPG.0b013e3181ae2ba0
365. Kawada C, Hasegawa T, Watanabe M, Nomura Y. Dietary glucosylceramide enhances tight junction function in skin epidermis via induction of claudin-1. Biosci Biotechnol Biochem (2013) 77:867–9. doi:10.1271/bbb.120874
366. Cheng Y, Ohlsson L, Duan R. Psyllium and fat in diets differentially affect the activities and expressions of colonic sphingomyelinases and caspase in mice. Br J Nutr (2004) 91:715–23. doi:10.1079/BJN20041107
367. Hadi LA, Di Vito C, Marfia G, Ferraretto A, Tringali C, Viani P, et al. Sphingosine kinase 2 and ceramide transport as key targets of the natural flavonoid luteolin to induce apoptosis in colon cancer cells. PLoS One (2015) 10:e0143384. doi:10.1371/journal.pone.0143384
368. Lopez-Lazaro M. Distribution and biological activities of the flavonoid luteolin. Mini Rev Med Chem (2009) 9:31–59. doi:10.2174/138955709787001712
369. Soo I, Madsen KL, Tejpar Q, Sherbaniuk R, Cinque B, Marzio LD, et al. VSL# 3 probiotic upregulates intestinal mucosal alkaline sphingomyelinase and reduces inflammation. Can J Gastroenterol (2008) 22:237–42. doi:10.1155/2008/520383
370. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res (2006) 47:241–59. doi:10.1194/jlr.R500013-JLR200
371. de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metabol (2013) 17:657–69. doi:10.1016/j.cmet.2013.03.013
372. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol (2014) 30:332–8. doi:10.1097/MOG.0000000000000057
373. Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J (2006) 25:1419–25. doi:10.1038/sj.emboj.7601049
374. Eloranta JJ, Kullak-Ublick GA. The role of FXR in disorders of bile acid homeostasis. Physiology (Bethesda) (2008) 23:286–95. doi:10.1152/physiol.00020.2008
375. Bajaj JS, Ridlon JM, Hylemon PB, Thacker LR, Heuman DM, Smith S, et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointest Liver Physiol (2012) 302:G168–75. doi:10.1152/ajpgi.00190.2011
376. Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol (2013) 58:949–55. doi:10.1016/j.jhep.2013.01.003
377. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol (2014) 60:940–7. doi:10.1016/j.jhep.2013.12.019
378. Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, et al. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol (2012) 303:G675–85. doi:10.1152/ajpgi.00152.2012
379. Islam KS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology (2011) 141:1773–81. doi:10.1053/j.gastro.2011.07.046
380. Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes (2013) 4:382–7. doi:10.4161/gmic.25723
381. Bernstein H, Holubec H, Bernstein C, Ignatenko N, Gerner E, Dvorak K, et al. Unique dietary-related mouse model of colitis. Inflamm Bowel Dis (2006) 12:278–93. doi:10.1097/01.MIB.0000209789.14114.63
382. Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/-mice. Nature (2012) 487:104–8. doi:10.1038/nature11225
383. Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev (2005) 29:625–51. doi:10.1016/j.femsre.2004.09.003
384. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A (2006) 103:3920–5. doi:10.1073/pnas.0509592103
385. Masuda N. Deconjugation of bile salts by Bacteroides and Clostridium. Microbiol Immunol (1981) 25:1–11. doi:10.1111/j.1348-0421.1981.tb00001.x
386. Begley M, Hill C, Gahan CG. Bile salt hydrolase activity in probiotics. Appl Environ Microbiol (2006) 72:1729–38. doi:10.1128/AEM.72.3.1729-1738.2006
387. Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun (2013) 4:2384. doi:10.1038/ncomms3384
388. Devkota S, Chang EB. Interactions between diet, bile acid metabolism, gut microbiota, and inflammatory bowel diseases. Dig Dis (2015) 33:351–6. doi:10.1159/000371687
389. Gibson G, Cummings J, Macfarlane G. Growth and activities of sulphate-reducing bacteria in gut contents of healthy subjects and patients with ulcerative colitis. FEMS Microbiol Lett (1991) 86:103–11. doi:10.1111/j.1574-6941.1991.tb01742.x
390. Zinkevich VV, Beech IB. Screening of sulfate-reducing bacteria in colonoscopy samples from healthy and colitic human gut mucosa. FEMS Microbiol Ecol (2000) 34:147–55. doi:10.1111/j.1574-6941.2000.tb00764.x
391. Loubinoux J, Mory F, Pereira IA, Le Faou AE. Bacteremia caused by a strain of Desulfovibrio related to the provisionally named Desulfovibrio fairfieldensis. J Clin Microbiol (2000) 38:931–4.
392. Rao VA. The prebiotic properties of oligofructose at low intake levels. Nutr Res (2001) 21:843–8. doi:10.1016/S0271-5317(01)00284-6
393. Pitcher MC, Beatty ER, Cummings JH. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut (2000) 46:64–72. doi:10.1136/gut.46.1.64
394. Buchet R, Millán JL, Magne D. Multisystemic functions of alkaline phosphatases. In: Millan JL, editor. Methods in Molecular Biology: Phosphatase Modulators. New York: Springer (2013). p. 27–51.
395. Hatoff DE, Hardison WG. Bile acids modify alkaline phosphatase induction and bile secretion pressure after bile duct obstruction in the rat. Gastroenterology (1981) 80:666–72.
396. Hatoff DE, Hardison WG. Bile acid-dependent secretion of alkaline phosphatase in rat bile. Hepatology (1982) 2:S433–9. doi:10.1002/hep.1840020407
397. Alvaro D, Benedetti A, Marucci L, Delle Monache M, Monterubbianesi R, Di Cosimo E, et al. The function of alkaline phosphatase in the liver: regulation of intrahepatic biliary epithelium secretory activities in the rat. Hepatology (2000) 32:174–84. doi:10.1053/jhep.2000.9078
398. Tuin A, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. On the role and fate of LPS-dephosphorylating activity in the rat liver. Am J Physiol Gastrointest Liver Physiol (2006) 290:G377–85. doi:10.1152/ajpgi.00147.2005
399. Poelstra K, Bakker WW, Klok PA, Kamps JA, Hardonk MJ, Meijer DK. Dephosphorylation of endotoxin by alkaline phosphatase in vivo. Am J Pathol (1997) 151:1163–9.
400. Linder CH, Englund UH, Narisawa S, Millán JL, Magnusson P. Isozyme profile and tissue-origin of alkaline phosphatases in mouse serum. Bone (2013) 53:399–408. doi:10.1016/j.bone.2012.12.048
401. Narisawa S, Hoylaerts MF, Doctor KS, Fukuda MN, Alpers DH, Millan JL. A novel phosphatase upregulated in Akp3 knockout mice. Am J Physiol Gastrointest Liver Physiol (2007) 293:G1068–77. doi:10.1152/ajpgi.00073.2007
402. Glickman R, Alpers D, Drummey G, Isselbacher K. Increased lymph alkaline phosphatase after fat feeding: effects of medium chain triglycerides and inhibition of protein synthesis. Biochim Biophys Acta (1970) 201:226–35. doi:10.1016/0304-4165(70)90296-5
403. Narisawa S, Huang L, Iwasaki A, Hasegawa H, Alpers DH, Millan JL. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol Cell Biol (2003) 23:7525–30. doi:10.1128/MCB.23.21.7525-7530.2003
404. Lynes M, Narisawa S, Millan JL, Widmaier EP. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am J Physiol Regul Integr Comp Physiol (2011) 301:R1738–47. doi:10.1152/ajpregu.00235.2011
405. Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S, et al. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci U S A (2008) 105:3551–6. doi:10.1073/pnas.0712140105
406. Malo MS, Alam SN, Mostafa G, Zeller SJ, Johnson PV, Mohammad N, et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut (2010) 59:1476–84. doi:10.1136/gut.2010.211706
407. Ramasamy S, Nguyen DD, Eston MA, Nasrin Alam S, Moss AK, Ebrahimi F, et al. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis (2011) 17:532–42. doi:10.1002/ibd.21377
408. Reen RK, Cadwallader A, Perdew GH. The subdomains of the transactivation domain of the aryl hydrocarbon receptor (AhR) inhibit AhR and estrogen receptor transcriptional activity. Arch Biochem Biophys (2002) 408:93–102. doi:10.1016/S0003-9861(02)00518-0
409. Kumar MB, Ramadoss P, Reen RK, Vanden Heuvel JP, Perdew GH. The Q-rich subdomain of the human Ah receptor transactivation domain is required for dioxin-mediated transcriptional activity. J Biol Chem (2001) 276:42302–10. doi:10.1074/jbc.M104798200
410. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A (2008) 105:9721–6. doi:10.1073/pnas.0804231105
411. Conti HR, Peterson AC, Brane L, Huppler AR, Hernandez-Santos N, Whibley N, et al. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J Exp Med (2014) 211:2075–84. doi:10.1084/jem.20130877
412. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med (2009) 206:299–311. doi:10.1084/jem.20081463
413. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science (2011) 332:65–8. doi:10.1126/science.1200439
414. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld J, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453:106–9. doi:10.1038/nature06881
415. Kiss EA, Vonarbourg C. Aryl hydrocarbon receptor: a molecular link between postnatal lymphoid follicle formation and diet. Gut Microbes (2012) 3:577–82. doi:10.4161/gmic.21865
416. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell (2011) 147:629–40. doi:10.1016/j.cell.2011.09.025
417. Dumoutier L, Van Roost E, Ameye G, Michaux L, Renauld JCIL-TIF. /IL-22: genomic organization and mapping of the human and mouse genes. Genes Immun (2000) 1:488–94. doi:10.1038/sj.gene.6363716
418. Qiu J, Guo X, Zong-ming EC, He L, Sonnenberg GF, Artis D, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity (2013) 39:386–99. doi:10.1016/j.immuni.2013.08.002
419. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14:282–9. doi:10.1038/nm1720
420. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334:255–8. doi:10.1126/science.1209791
421. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39:372–85. doi:10.1016/j.immuni.2013.08.003
422. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334:1561–5. doi:10.1126/science.1214914
423. Terakara M, Fukuwatari T, Sano M, Nakao N, Sasaki R, Fukuoka S, et al. Establishment of true niacin deficiency in quinolinic acid phosphoribosyltransferase knockout mice. J Nutr (2012) 142:148–53. doi:10.3945/jn.112.167569
424. Terakata M, Fukuwatari T, Kadota E, Sano M, Kanai M, Nakamura T, et al. The niacin required for optimum growth can be synthesized from l-tryptophan in growing mice lacking tryptophan-2,3-dioxygenase. J Nutr (2013) 143:1046–51. doi:10.3945/jn.113.176875
425. Munn DH, Mellor AL. Indoleamine 2, 3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34:137–43. doi:10.1016/j.it.2012.10.001
426. Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol (2000) 164:3596–9. doi:10.4049/jimmunol.164.7.3596
427. Murray HW, Szuro-Sudol A, Wellner D, Oca MJ, Granger AM, Libby DM, et al. Role of tryptophan degradation in respiratory burst-independent antimicrobial activity of gamma interferon-stimulated human macrophages. Infect Immun (1989) 57:845–9.
428. Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J (1991) 5:2516–22.
429. Thomas SM, Garrity LF, Brandt CR, Schobert CS, Feng GS, Taylor MW, et al. IFN-gamma-mediated antimicrobial response. Indoleamine 2,3-dioxygenase-deficient mutant host cells no longer inhibit intracellular Chlamydia spp. or Toxoplasma growth. J Immunol (1993) 150:5529–34.
430. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197–203. doi:10.1038/nature10491
431. Lee HJ, Jeong Y, Lee T, Jung ID, Lee JS, Lee C, et al. Rosmarinic acid inhibits indoleamine 2, 3-dioxygenase expression in murine dendritic cells. Biochem Pharmacol (2007) 73:1412–21. doi:10.1016/j.bcp.2006.12.018
432. Cesario A, Rocca B, Rutella S. The interplay between indoleamine 2, 3-dioxygenase 1 (IDO1) and cyclooxygenase (COX)-2 in chronic inflammation and cancer. Curr Med Chem (2011) 18:2263–71. doi:10.2174/092986711795656063
433. Johnson BA, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2, 3 dioxygenase pathway in immunotherapy. Immunotherapy (2009) 1:645–61. doi:10.2217/IMT.09.21
434. Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem (2006) 281:22021–8. doi:10.1074/jbc.M603503200
435. Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov (2008) 7:339–57. doi:10.1038/nrd2518
436. Card PB, Erbel PJ, Gardner KH. Structural basis of ARNT PAS-B dimerization: use of a common beta-sheet interface for hetero-and homodimerization. J Mol Biol (2005) 353:664–77. doi:10.1016/j.jmb.2005.08.043
437. McIntosh BE, Hogenesch JB, Bradfield CA. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol (2010) 72:625–45. doi:10.1146/annurev-physiol-021909-135922
438. Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix–loop–helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol (2004) 36:189–204. doi:10.1016/S1357-2725(03)00211-5
439. Romani L, Zelante T, Luca AD, Iannitti RG, Moretti S, Bartoli A, et al. Microbiota control of a tryptophan–AhR pathway in disease tolerance to fungi. Eur J Immunol (2014) 44:3192–200. doi:10.1002/eji.201344406
440. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A (1991) 88:9543–7. doi:10.1073/pnas.88.21.9543
441. Shin JH, Zhang L, Murillo-Sauca O, Kim J, Kohrt HE, Bui JD, et al. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A (2013) 110:12391–6. doi:10.1073/pnas.1302856110
442. Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol (2009) 30:447–54. doi:10.1016/j.it.2009.06.005
443. Wang H, Yeh C, Iwamoto T, Satsu H, Shimizu M, Totsuka M. Dietary flavonoid naringenin induces regulatory T cells via an aryl hydrocarbon receptor mediated pathway. J Agric Food Chem (2012) 60:2171–8. doi:10.1021/jf204625y
444. Mohammadi-Bardbori A, Bengtsson J, Rannug U, Rannug A, Wincent E. Quercetin, resveratrol, and curcumin are indirect activators of the aryl hydrocarbon receptor (AHR). Chem Res Toxicol (2012) 25:1878–84. doi:10.1021/tx300169e
445. Ciolino H, Daschner P, Yeh G. Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem J (1999) 340:715–22. doi:10.1042/bj3400715
446. Ross JA, Kasum CM. Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu Rev Nutr (2002) 22:19–34. doi:10.1146/annurev.nutr.22.111401.144957
447. Ciolino HP, Daschner PJ, Wang TT, Yeh GC. Effect of curcumin on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Biochem Pharmacol (1998) 56:197–206. doi:10.1016/S0006-2952(98)00143-9
448. Fukumoto S, Toshimitsu T, Matsuoka S, Maruyama A, Oh-oka K, Takamura T, et al. Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol Cell Biol (2014) 92:460–5. doi:10.1038/icb.2014.2
449. Moura-Alves P, Faé K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature (2014) 512:387–92. doi:10.1038/nature13684
450. Murphy KA, Quadro L, White LA. The intersection between the aryl hydrocarbon receptor (AhR)- and retinoic acid-signaling pathways. Vitam Horm (2007) 75:33–67. doi:10.1016/S0083-6729(06)75002-6
451. Beischlag TV, Morales JL, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr (2008) 18:207–50. doi:10.1615/CritRevEukarGeneExpr.v18.i3.20
452. Safe S, Wormke M. Inhibitory aryl hydrocarbon receptor-estrogen receptor α cross-talk and mechanisms of action. Chem Res Toxicol (2003) 16:807–16. doi:10.1021/tx034036r
453. Callero MA, Loaiza-Pérez AI. The role of aryl hydrocarbon receptor and crosstalk with estrogen receptor in response of breast cancer cells to the novel antitumor agents benzothiazoles and aminoflavone. Int J Breast Cancer (2011) 2011:923250. doi:10.4061/2011/923250
454. Bardbori AM. Influence of oxidative stress on aryl hydrocarbon receptor signaling. Karolinska Institutet (2013). Available from: http://hdl.handle.net/10616/41558
455. Chartoumpekis DV, Kensler TW. New player on an old field; the keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr Diabetes Rev (2013) 9:137–45. doi:10.2174/157339913805076490
456. Murakami S, Motohashi H. Roles of NRF2 in cell proliferation and differentiation. Free Radic Biol Med (2015) 88:168–78. doi:10.1016/j.freeradbiomed.2015.06.030
457. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci (2014) 39:199–218. doi:10.1016/j.tibs.2014.02.002
458. Hankinson O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch Biochem Biophys (2005) 433:379–86. doi:10.1016/j.abb.2004.09.031
459. Fujimaki H, Nohara K, Kobayashi T, Suzuki K, Eguchi-Kasai K, Tsukumo S, et al. Effect of a single oral dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin on immune function in male NC/Nga mice. Toxicol Sci (2002) 66:117–24. doi:10.1093/toxsci/66.1.117
460. Nohara K, Fujimaki H, Tsukumo S, Inouye K, Sone H, Tohyama C. Effects of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) on T cell-derived cytokine production in ovalbumin (OVA)-immunized C57Bl/6 mice. Toxicology (2002) 172:49–58. doi:10.1016/S0300-483X(01)00582-0
461. Ito T, Inouye K, Fujimaki H, Tohyama C, Nohara K. Mechanism of TCDD-induced suppression of antibody production: effect on T cell-derived cytokine production in the primary immune reaction of mice. Toxicol Sci (2002) 70:46–54. doi:10.1093/toxsci/70.1.46
462. Inouye K, Pan X, Imai N, Ito T, Takei T, Tohyama C, et al. T cell-derived IL-5 production is a sensitive target of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD). Chemosphere (2005) 60:907–13. doi:10.1016/j.chemosphere.2005.01.014
463. Kato Y, Negishi T, Furusako S, Mizuguchi K, Mochizuki H. An orally active Th1/Th2 balance modulator, M50367, suppresses Th2 differentiation of naıve Th cell in vitro. Cell Immunol (2003) 224:29–37. doi:10.1016/j.cellimm.2003.07.001
464. Luebke RW, Copeland CB, Daniels M, Lambert AL, Gilmour MI. Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol Sci (2001) 62:71–9. doi:10.1093/toxsci/62.1.71
465. Negishi T, Kato Y, Ooneda O, Mimura J, Takada T, Mochizuki H, et al. Effects of aryl hydrocarbon receptor signaling on the modulation of TH1/TH2 balance. J Immunol (2005) 175:7348–56. doi:10.4049/jimmunol.175.11.7348
466. Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest (2006) 116:984–95. doi:10.1172/JCI25790
467. Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci (2010) 114:237–46. doi:10.1093/toxsci/kfp274
468. Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol (2008) 181:680–9. doi:10.4049/jimmunol.181.1.680
469. Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, et al. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci (2008) 104:218–27. doi:10.1093/toxsci/kfn079
470. Williams MA, Rangasamy T, Bauer SM, Killedar S, Karp M, Kensler TW, et al. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol (2008) 181:4545–59. doi:10.4049/jimmunol.181.7.4545
471. Rockwell CE, Zhang M, Fields PE, Klaassen CD. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol (2012) 188:1630–7. doi:10.4049/jimmunol.1101712
472. Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Sykiotis GP, et al. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes (2011) 60:2465–73. doi:10.2337/db11-0112
473. Pi J, Leung L, Xue P, Wang W, Hou Y, Liu D, et al. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem (2010) 285:9292–300. doi:10.1074/jbc.M109.093955
474. Alexander DL, Ganem LG, Fernandez-Salguero P, Gonzalez F, Jefcoate CR. Aryl-hydrocarbon receptor is an inhibitory regulator of lipid synthesis and of commitment to adipogenesis. J Cell Sci (1998) 111(Pt 22):3311–22.
475. Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2 – an update. Free Radic Biol Med (2014) 66:36–44. doi:10.1016/j.freeradbiomed.2013.02.008
476. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell (2012) 22:66–79. doi:10.1016/j.ccr.2012.05.016
477. Murugaiyan J, Rockstroh M, Wagner J, Baumann S, Schorsch K, Trump S, et al. Benzo [a] pyrene affects Jurkat T cells in the activated state via the antioxidant response element dependent Nrf2 pathway leading to decreased IL-2 secretion and redirecting glutamine metabolism. Toxicol Appl Pharmacol (2013) 269:307–16. doi:10.1016/j.taap.2013.03.032
479. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol (2007) 178:93–105. doi:10.1083/jcb.200703099
480. Novak F, Heyland DK, Avenell A, Drover JW, Su X. Glutamine supplementation in serious illness: a systematic review of the evidence. Crit Care Med (2002) 30:2022–9. doi:10.1097/00003246-200209000-00011
481. Greenfield JR, Farooqi IS, Keogh JM, Henning E, Habib AM, Blackwood A, et al. Oral glutamine increases circulating glucagon-like peptide 1, glucagon, and insulin concentrations in lean, obese, and type 2 diabetic subjects. Am J Clin Nutr (2009) 89:106–13. doi:10.3945/ajcn.2008.26362
482. Prada P, Hirabara S, De Souza C, Schenka A, Zecchin H, Vassallo J, et al. l-glutamine supplementation induces insulin resistance in adipose tissue and improves insulin signalling in liver and muscle of rats with diet-induced obesity. Diabetologia (2007) 50:1949–59. doi:10.1007/s00125-007-0723-z
483. Keighley MR, Arabi Y, Dimock F, Burdon DW, Allan RN, Alexander-Williams J. Influence of inflammatory bowel disease on intestinal microflora. Gut (1978) 19:1099–104. doi:10.1136/gut.19.12.1099
484. de Souza AZ, Zambom AZ, Abboud KY, Reis SK, Tannihão F, Guadagnini D, et al. Oral supplementation with l-glutamine alters gut microbiota of obese and overweight adults: a pilot study. Nutrition (2015) 31:884–9. doi:10.1016/j.nut.2015.01.004
485. Anonymous. Handbook of Vitamins. Boca Raton, FL: CRC Press Taylor & Francis Group (2007). 233 p.
486. Magnusdottir S, Ravcheev D, de Crécy-Lagard V, Thiele I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front Genet (2015) 6:148. doi:10.3389/fgene.2015.00148
487. Horwitt MK, Harper AE, Henderson LM. Niacin-tryptophan relationships for evaluating niacin equivalents. Am J Clin Nutr (1981) 34:423–7.
488. Degnan PH, Barry NA, Mok KC, Taga ME, Goodman AL. Human gut microbes use multiple transporters to distinguish vitamin B 12 analogs and compete in the gut. Cell Host Microbe (2014) 15:47–57. doi:10.1016/j.chom.2013.12.007
489. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature (2012) 491:717–23. doi:10.1038/nature11605
490. Birkinshaw RW, Kjer-Nielsen L, Eckle SB, McCluskey J, Rossjohn J. MAITs, MR1 and vitamin B metabolites. Curr Opin Immunol (2014) 26:7–13. doi:10.1016/j.coi.2013.09.007
491. Khan MT, Duncan SH, Stams AJ, van Dijl JM, Flint HJ, Harmsen HJ. The gut anaerobe Faecalibacterium prausnitzii uses an extracellular electron shuttle to grow at oxic–anoxic interphases. ISME J (2012) 6:1578–85. doi:10.1038/ismej.2012.5
492. Khan MT, Browne WR, van Dijl JM, Harmsen HJ. How can Faecalibacterium prausnitzii employ riboflavin for extracellular electron transfer? Antioxid Redox Signal (2012) 17:1433–40. doi:10.1089/ars.2012.4701
493. Sadabad MS, Khan MT, van Dijl JM, Harmsen HJ. Riboflavin Increases the Abundance of Faecalibacterium prausnitzii and Roseburia in Fecal Samples of Healthy Volunteers and Positively Affects the Overall Microbial Balance in the Gut. European Patent Application No. 12190948.5 (2015). Available from: http://www.rug.nl/research/portal/files/16269641/Chapter_3_.pdf
494. Heinken A, Khan MT, Paglia G, Rodionov DA, Harmsen HJ, Thiele I. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J Bacteriol (2014) 196:3289–302. doi:10.1128/JB.01780-14
495. Duric M, Sivanesan S, Bakovic M. Phosphatidylcholine functional foods and nutraceuticals: a potential approach to prevent non-alcoholic fatty liver disease. Eur J Lipid Sci Technol (2012) 114:389–98. doi:10.1002/ejlt.201100350
497. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature (2011) 472:57–63. doi:10.1038/nature09922
498. Al-Waiz M, Mikov M, Mitchell S, Smith R. The exogenous origin of trimethylamine in the mouse. Metabolism (1992) 41:135–6. doi:10.1016/0026-0495(92)90140-6
499. Lang D, Yeung C, Peter R, Ibarra C, Gasser R, Itagaki K, et al. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: selective catalysis by FMO3. Biochem Pharmacol (1998) 56:1005–12. doi:10.1016/S0006-2952(98)00218-4
500. Tang WW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med (2013) 368:1575–84. doi:10.1056/NEJMoa1109400
501. Koeth RA, Wang Z, Levison BS, Buffa JA, Sheehy BT, Britt EB, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med (2013) 19:576–85. doi:10.1038/nm.3145
502. Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio (2015) 6:e02481. doi:10.1128/mBio.02481-14
503. Brugère J, Borrel G, Gaci N, Tottey W, O’Toole PW, Malpuech-Brugère C. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes (2014) 5:5–10. doi:10.4161/gmic.26749
504. Horz H. Archaeal lineages within the human microbiome: absent, rare or elusive? Life (2015) 5:1333–45. doi:10.3390/life5021333
505. Gaci N, Borrel G, Tottey W, O’Toole PW, Brugère J. Archaea and the human gut: new beginning of an old story. World J Gastroenterol (2014) 20:16062–78. doi:10.3748/wjg.v20.i43.16062
506. Nava GM, Carbonero F, Ou J, Benefiel AC, O’Keefe SJ, Gaskins HR. Hydrogenotrophic microbiota distinguish native Africans from African and European Americans. Environ Microbiol Rep (2012) 4:307–15. doi:10.1111/j.1758-2229.2012.00334.x
507. Nam Y, Chang H, Kim K, Roh SW, Kim M, Jung M, et al. Bacterial, archaeal, and eukaryal diversity in the intestines of Korean people. J Microbiol (2008) 46:491–501. doi:10.1007/s12275-008-0199-7
508. Tyakht AV, Kostryukova ES, Popenko AS, Belenikin MS, Pavlenko AV, Larin AK, et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun (2013) 4:2469. doi:10.1038/ncomms3469
509. Pitt P, de Bruijn KM, Beeching MF, Goldberg E, Blendis LM. Studies on breath methane: the effect of ethnic origins and lactulose. Gut (1980) 21:951–4. doi:10.1136/gut.21.11.951
510. Segal I, Walker AR, Lord S, Cummings JH. Breath methane and large bowel cancer risk in contrasting African populations. Gut (1988) 29:608–13. doi:10.1136/gut.29.5.608
511. Marchand LL, Wilkens LR, Harwood P, Cooney RV. Breath hydrogen and methane in populations at different risk for colon cancer. Int J Cancer (1993) 55:887–90. doi:10.1002/ijc.2910550603
512. Dridi B, Raoult D, Drancourt M. Archaea as emerging organisms in complex human microbiomes. Anaerobe (2011) 17:56–63. doi:10.1016/j.anaerobe.2011.03.001
513. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity (2011) 35:13–22. doi:10.1016/j.immuni.2011.07.002
514. Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, et al. Essential role for retinoic acid in the promotion of CD4 T cell effector responses via retinoic acid receptor alpha. Immunity (2011) 34:435–47. doi:10.1016/j.immuni.2011.03.003
515. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science (2014) 343:432–7. doi:10.1126/science.1247606
516. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204:1757–64. doi:10.1084/jem.20070590
517. Farache J, Koren I, Milo I, Gurevich I, Kim K, Zigmond E, et al. Luminal bacteria recruit CD103 dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity (2013) 38:581–95. doi:10.1016/j.immuni.2013.01.009
518. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med (2013) 210:1117–24. doi:10.1084/jem.20121588
519. van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature (2014) 508:123–7. doi:10.1038/nature13158
520. Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science (2013) 342:1243–6. doi:10.1126/science.1243364
521. Liu X, Cui T, Li Y, Wang Y, Wang Q, Li X, et al. Vitamin A supplementation in early life enhances the intestinal immune response of rats with gestational vitamin A deficiency by increasing the number of immune cells. PLoS One (2014) 9:e114934. doi:10.1371/journal.pone.0114934
523. Cantorna MT, Zhu Y, Froicu M, Wittke A. Vitamin D status, 1, 25-dihydroxyvitamin D3, and the immune system. Am J Clin Nutr (2004) 80:S1717–20.
524. Lim W, Hanauer SB, Li YC. Mechanisms of disease: vitamin D and inflammatory bowel disease. Nat Clin Prac Gastroenterol Hepatol (2005) 2:308–15. doi:10.1038/ncpgasthep0215
525. Garg M, Lubel J, Sparrow M, Holt S, Gibson P. Review article: vitamin D and inflammatory bowel disease – established concepts and future directions. Aliment Pharmacol Ther (2012) 36:324–44. doi:10.1111/j.1365-2036.2012.05181.x
526. Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1, 25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr (2000) 130:2648–52.
527. Cantorna MT, Mahon BD. Mounting evidence for vitamin D as an environmental factor affecting autoimmune disease prevalence. Exp Biol Med (2004) 229:1136–42.
528. Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol (2003) 17:2386–92. doi:10.1210/me.2003-0281
529. Li YC, Chen Y, Du J. Critical roles of intestinal epithelial vitamin D receptor signaling in controlling gut mucosal inflammation. J Steroid Biochem Mol Biol (2015) 148:179–83. doi:10.1016/j.jsbmb.2015.01.011
530. Wang T, Tavera-Mendoza LE, Laperriere D, Libby E, Burton MacLeod N, Nagai Y, et al. Large-scale in silico and microarray-based identification of direct 1, 25-dihydroxyvitamin D3 target genes. Mol Endocrinol (2005) 19:2685–95. doi:10.1210/me.2005-0106
531. Vigano P, Lattuada D, Mangioni S, Ermellino L, Vignali M, Caporizzo E, et al. Cycling and early pregnant endometrium as a site of regulated expression of the vitamin D system. J Mol Endocrinol (2006) 36:415–24. doi:10.1677/jme.1.01946
532. Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, et al. Vitamin d-directed rheostatic regulation of monocyte antibacterial responses. J Immunol (2009) 182:4289–95. doi:10.4049/jimmunol.0803736
533. Wang TT, Dabbas B, Laperriere D, Bitton AJ, Soualhine H, Tavera-Mendoza LE, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem (2010) 285:2227–31. doi:10.1074/jbc.C109.071225
534. Verway M, Behr MA, White JH. Vitamin D, NOD2, autophagy and Crohn’s disease. Expert Rev Clin Immunol (2010) 6:505–8. doi:10.1586/eci.10.31
535. Macho Fernandez E, Valenti V, Rockel C, Hermann C, Pot B, Boneca IG, et al. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut (2011) 60:1050–9. doi:10.1136/gut.2010.232918
536. Jones ML, Martoni CJ, Prakash S. Oral supplementation with probiotic L. reuteri NCIMB 30242 increases mean circulating 25-hydroxyvitamin D: a post hoc analysis of a randomized controlled trial. J Clin Endocrinol Metab (2013) 98:2944–51. doi:10.1210/jc.2012-4262
537. Marshall TG. Vitamin D discovery outpaces FDA decision making. Bioessays (2008) 30:173–82. doi:10.1002/bies.20708
538. Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol (2014) 5:1–15. doi:10.3389/fphys.2014.00151
539. Bhalla AK, Amento E, Serog B, Glimcher L. 1, 25-Dihydroxyvitamin D3 inhibits antigen-induced T cell activation. J Immunol (1984) 133:1748–54.
540. Adorini L, Penna G. Dendritic cell tolerogenicity: a key mechanism in immunomodulation by vitamin D receptor agonists. Hum Immunol (2009) 70:345–52. doi:10.1016/j.humimm.2009.01.016
541. Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol (2010) 10:482–96. doi:10.1016/j.coph.2010.04.001
542. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science (2006) 311:1770–3. doi:10.1126/science.1123933
543. Appleyard CB, Cruz ML, Isidro AA, Arthur JC, Jobin C, De Simone C. Pretreatment with the probiotic VSL#3 delays transition from inflammation to dysplasia in a rat model of colitis-associated cancer. Am J Physiol Gastrointest Liver Physiol (2011) 301:G1004–13. doi:10.1152/ajpgi.00167.2011
544. Wang TT, Tavera-Mendoza LE, Laperriere D, Libby E, MacLeod NB, Nagai Y, et al. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol Endocrinol (2005) 19:2685–95. doi:10.1210/me.2005-0106
545. Auvynet C, Rosenstein Y. Multifunctional host defense peptides: antimicrobial peptides, the small yet big players in innate and adaptive immunity. FEBS J (2009) 276:6497–508. doi:10.1111/j.1742-4658.2009.07360.x
546. Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J (2005) 19:1067–77. doi:10.1096/fj.04-3284com
547. Albert PJ, Proal AD, Marshall TG. Vitamin D: the alternative hypothesis. Autoimmun Rev (2009) 8:639–44. doi:10.1016/j.autrev.2009.02.011
548. Proal AD, Albert PJ, Marshall TG. The human microbiome and autoimmunity. Curr Opin Rheumatol (2013) 25:234–40. doi:10.1097/BOR.0b013e32835cedbf
549. Bartik L, Whitfield GK, Kaczmarska M, Lowmiller CL, Moffet EW, Furmick JK, et al. Curcumin: a novel nutritionally derived ligand of the vitamin D receptor with implications for colon cancer chemoprevention. J Nutr Biochem (2010) 21:1153–61. doi:10.1016/j.jnutbio.2009.09.012
550. Plöger S, Stumpff F, Penner GB, Schulzke J, Gäbel G, Martens H, et al. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann N Y Acad Sci (2012) 1258:52–9. doi:10.1111/j.1749-6632.2012.06553.x
551. Gaschott T, Werz O, Steinmeyer A, Steinhilber D, Stein J. Butyrate-induced differentiation of Caco-2 cells is mediated by vitamin D receptor. Biochem Biophys Res Commun (2001) 288:690–6. doi:10.1006/bbrc.2001.5832
552. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science (2002) 296:1313–6. doi:10.1126/science.1070477
553. Schmidt DR, Holmstrom SR, Fon Tacer K, Bookout AL, Kliewer SA, Mangelsdorf DJ. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem (2010) 285:14486–94. doi:10.1074/jbc.M110.116004
554. MacDonald RS, Wagner K. Influence of dietary phytochemicals and microbiota on colon cancer risk. J Agric Food Chem (2012) 60:6728–35. doi:10.1021/jf204230r
555. Ooi JH, Li Y, Rogers CJ, Cantorna MT. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr (2013) 143:1679–86. doi:10.3945/jn.113.180794
556. Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol (2010) 59:1114–22. doi:10.1099/jmm.0.021170-0
557. Jin D, Wu S, Zhang Y, Lu R, Xia Y, Dong H, et al. Lack of vitamin D receptor causes dysbiosis and changes the functions of the murine intestinal microbiome. Clin Ther (2015) 37:996–1009.e7. doi:10.1016/j.clinthera.2015.04.004
558. Walker AW. Intestinal microbiology and ecology in inflammatory bowel disease. In: Baumgaut D, editor. Crohn’s Disease and Ulcerative Colitis. United States: Springer (2012). p. 85–95.
559. Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut (2003) 52:237–42. doi:10.1136/gut.52.2.237
560. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe (2014) 15:382–92. doi:10.1016/j.chom.2014.02.005
561. Leach S, Mitchell H, Eng W, Zhang L, Day A. Sustained modulation of intestinal bacteria by exclusive enteral nutrition used to treat children with Crohn’s disease. Aliment Pharmacol Ther (2008) 28:724–33. doi:10.1111/j.1365-2036.2008.03796.x
562. Perez T. Bacteria induced vitamin D receptor dysfunction in autoimmune disease: theoretical and practical implications for interpretation of serum vitamin D metabolite levels. In: Anonymous. 6th International Congress on Autoimmunity; Porto, Portugal (2008).
563. Madsen KL, Doyle JS, Jewell LD, Tavernini MM, Fedorak RN. Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology (1999) 116:1107–14. doi:10.1016/S0016-5085(99)70013-2
564. Mencarelli A, Distrutti E, Renga B, D’Amore C, Cipriani S, Palladino G, et al. Probiotics modulate intestinal expression of nuclear receptor and provide counter-regulatory signals to inflammation-driven adipose tissue activation. PLoS One (2011) 6:e22978. doi:10.1371/journal.pone.0022978
565. Ohland CL, Kish L, Bell H, Thiesen A, Hotte N, Pankiv E, et al. Effects of Lactobacillus helveticus on murine behavior are dependent on diet and genotype and correlate with alterations in the gut microbiome. Psychoneuroendocrinology (2013) 38:1738–47. doi:10.1016/j.psyneuen.2013.02.008
566. Jakobsdottir G, Xu J, Molin G, Ahrné S, Nyman M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS One (2013) 8:e80476. doi:10.1371/journal.pone.0080476
567. Lee S, Han HW, Yim SY. Beneficial effects of soy milk and fiber on high cholesterol diet-induced alteration of gut microbiota and inflammatory gene expression in rats. Food Funct (2015) 6:492–500. doi:10.1039/c4fo00731j
568. Paturi G, Nyanhanda T, Butts CA, Herath TD, Monro JA, Ansell J. Effects of potato fiber and potato-resistant starch on biomarkers of colonic health in rats fed diets containing red meat. J Food Sci (2012) 77:H216–23. doi:10.1111/j.1750-3841.2012.02911.x
569. Paturi G, Bentley-Hewitt KL, Butts CA, Nyanhanda T, Monro JA, Ansell J. Dietary combination of potato resistant starch and red meat up-regulates genes involved in colonic barrier function of rats. Int J Food Sci Tech (2013) 48:2441–6. doi:10.1111/ijfs.12208
570. Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, et al. Western diet induces dysbiosis with increased E. coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut (2014) 63:116–24. doi:10.1136/gutjnl-2012-304119
571. Cowan TE, Palmnäs MS, Yang J, Bomhof MR, Ardell KL, Reimer RA, et al. Chronic coffee consumption in the diet-induced obese rat: impact on gut microbiota and serum metabolomics. J Nutr Biochem (2014) 25:489–95. doi:10.1016/j.jnutbio.2013.12.009
572. Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology (2014) 147:1363–77.e17. doi:10.1053/j.gastro.2014.08.033
573. Cardona F, Andrés-Lacueva C, Tulipani S, Tinahones FJ, Queipo-Ortuño MI. Benefits of polyphenols on gut microbiota and implications in human health. J Nutr Biochem (2013) 24:1415–22. doi:10.1016/j.jnutbio.2013.05.001
574. Camuesco D, Comalada M, Concha A, Nieto A, Sierra S, Xaus J, et al. Intestinal anti-inflammatory activity of combined quercitrin and dietary olive oil supplemented with fish oil, rich in EPA and DHA (n-3) polyunsaturated fatty acids, in rats with DSS-induced colitis. Clin Nutr (2006) 25:466–76. doi:10.1016/j.clnu.2005.12.009
575. Duda-Chodak A, Tarko T, Satora P, Sroka P. Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: a review. Eur J Nutr (2015) 54:325–41. doi:10.1007/s00394-015-0852-y
576. Duda-Chodak A. The inhibitory effect of polyphenols on human gut microbiota. J Physiol Pharmacol (2012) 63:497–503.
577. Chen L, Tai WC, Hsiao WW. Dietary saponins from four popular herbal tea exert prebiotic-like effects on gut microbiota in C57BL/6 mice. J Funct Foods (2015) 17:892–902. doi:10.1016/j.jff.2015.06.050
578. Wang LS, Kuo CT, Stoner K, Yearsley M, Oshima K, Yu J, et al. Dietary black raspberries modulate DNA methylation in dextran sodium sulfate (DSS)-induced ulcerative colitis. Carcinogenesis (2013) 34:2842–50. doi:10.1093/carcin/bgt310
579. Rosillo MA, Sánchez-Hidalgo M, Cárdeno A, Aparicio-Soto M, Sánchez-Fidalgo S, Villegas I, et al. Dietary supplementation of an ellagic acid-enriched pomegranate extract attenuates chronic colonic inflammation in rats. Pharmacol Res (2012) 66:235–42. doi:10.1016/j.phrs.2012.05.006
580. Larrosa M, González-Sarrías A, Yáñez-Gascón MJ, Selma MV, Azorín-Ortuño M, Toti S, et al. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J Nutr Biochem (2010) 21:717–25. doi:10.1016/j.jnutbio.2009.04.012
581. Mandalari G, Bisignano C, Genovese T, Mazzon E, Wickham MS, Paterniti I, et al. Natural almond skin reduced oxidative stress and inflammation in an experimental model of inflammatory bowel disease. Int Immunopharmacol (2011) 11:915–24. doi:10.1016/j.intimp.2011.02.003
582. Kanodia L, Borgohain M, Das S. Effect of fruit extract of Fragaria vesca L. on experimentally induced inflammatory bowel disease in albino rats. Indian J Pharmacol (2011) 43:18–21. doi:10.4103/0253-7613.75660
583. Gonzalez-Barrio R, Edwards CA, Crozier A. Colonic catabolism of ellagitannins, ellagic acid, and raspberry anthocyanins: in vivo and in vitro studies. Drug Metab Dispos (2011) 39:1680–8. doi:10.1124/dmd.111.039651
584. Piwowarski JP, Granica S, Zwierzyńska M, Stefańska J, Schopohl P, Melzig MF, et al. Role of human gut microbiota metabolism in the anti-inflammatory effect of traditionally used ellagitannin-rich plant materials. J Ethnopharmacol (2014) 155:801–9. doi:10.1016/j.jep.2014.06.032
585. Truchado P, Larrosa M, García-Conesa MT, Cerdá B, Vidal-Guevara ML, Tomás-Barberán FA, et al. Strawberry processing does not affect the production and urinary excretion of urolithins, ellagic acid metabolites, in humans. J Agric Food Chem (2011) 60:5749–54. doi:10.1021/jf203641r
586. Tulipani S, Urpi-Sarda M, García-Villalba R, Rabassa M, LÓpez-Uriarte P, BullÓ M, et al. Urolithins are the main urinary microbial-derived phenolic metabolites discriminating a moderate consumption of nuts in free-living subjects with diagnosed metabolic syndrome. J Agric Food Chem (2012) 60:8930–40. doi:10.1021/jf301509w
587. Nickerson KP, Chanin R, McDonald C. Deregulation of intestinal anti-microbial defense by the dietary additive, maltodextrin. Gut Microbes (2015) 6:78–83. doi:10.1080/19490976.2015.1005477
588. Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell (1993) 75:263–74. doi:10.1016/0092-8674(93)80068-P
589. Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest (2007) 117:3909–12. doi:10.1172/JCI33084
590. Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature (2015) 519:92–6. doi:10.1038/nature14232
591. Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe (2012) 12:139–52. doi:10.1016/j.chom.2012.07.004
592. Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD – what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol (2012) 9:219–30. doi:10.1038/nrgastro.2012.14
593. Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12-and gamma interferon-dependent mechanism. Infect Immun (1998) 66:5157–66.
594. Joint FAO/WHO Expert Committee on Food Additives, World Health Organization. Evaluation of Certain Food Additives and Contaminants: Sixty-First Report of the Joint FAO/WHO Expert Committee on Food Additives (Vol 61). Geneva: World Health Organization (2004).
595. Takahama U, Hirota S. Effects of the food additive sulfite on nitrite-dependent nitric oxide production under Conditions simulating the mixture of saliva and gastric juice. J Agric Food Chem (2012) 60:1102–12. doi:10.1021/jf2049257
596. Weil, Sandler SR. Sulfur compounds. In: Kroschwitz JI, editor. Kirk-Othmer Concise Encylclopedia of Chemical Technology. New York: John Wiley & Sons, Inc (1999). 1937 p.
597. Ahmed M, Akter MS, Eun J. Peeling, drying temperatures, and sulphite-treatment affect physicochemical properties and nutritional quality of sweet potato flour. Food Chem (2010) 121:112–8. doi:10.1016/j.foodchem.2009.12.015
598. Marlett JA, Johnson EJ. Composition of fecal fiber from human subjects. J Nutr (1985) 115:650–60.
599. Borthakur A, Bhattacharyya S, Anbazhagan AN, Kumar A, Dudeja PK, Tobacman JK. Prolongation of carrageenan-induced inflammation in human colonic epithelial cells by activation of an NFκB-BCL10 loop. Biochim Biophys Acta (2012) 1822:1300–7. doi:10.1016/j.bbadis.2012.05.001
600. Nickerson KP, McDonald C. Crohn’s disease-associated adherent-invasive Escherichia coli adhesion is enhanced by exposure to the ubiquitous dietary polysaccharide maltodextrin. PLoS One (2012) 7:e52132. doi:10.1371/journal.pone.0052132
601. Borthakur A, Bhattacharyya S, Dudeja PK, Tobacman JK. Carrageenan induces interleukin-8 production through distinct Bcl10 pathway in normal human colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol (2007) 292:G829–38. doi:10.1152/ajpgi.00380.2006
602. Bhattacharyya S, Gill R, Chen ML, Zhang F, Linhardt RJ, Dudeja PK, et al. Toll-like receptor 4 mediates induction of the Bcl10-NFkappaB-interleukin-8 inflammatory pathway by carrageenan in human intestinal epithelial cells. J Biol Chem (2008) 283:10550–8. doi:10.1074/jbc.M708833200
603. Tobacman JK. Review of harmful gastrointestinal effects of carrageenan in animal experiments. Environ Health Perspect (2001) 109:983–94. doi:10.1289/ehp.01109983
604. IARC Working Group on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Carrageenan. IARC Monogr Eval Carcinog Risk Chem Hum (1983) 31:79–94.
605. Bhattacharyya S, Xue L, Devkota S, Chang E, Morris S, Tobacman JK. Carrageenan-induced colonic inflammation is reduced in Bcl10 null mice and increased in IL-10-deficient mice. Mediators Inflamm (2013) 2013:13. doi:10.1155/2013/397642
606. Amdekar S, Roy P, Singh V, Kumar A, Singh R, Sharma P. Anti-inflammatory activity of Lactobacillus on carrageenan-induced paw edema in male wistar rats. Int J Inflam (2012) 2012:6. doi:10.1155/2012/752015
607. Aviello G, Amu S, Saunders SP, Fallon PG. A mineral extract from red algae ameliorates chronic spontaneous colitis in IL-10 deficient mice in a mouse strain dependent manner. Phytother Res (2014) 28:300–4. doi:10.1002/ptr.4989
608. Michel C, Macfarlane G. Digestive fates of soluble polysaccharides from marine macroalgae: involvement of the colonic microflora and physiological consequences for the host. J Appl Bacteriol (1996) 80:349–69. doi:10.1111/j.1365-2672.1996.tb03230.x
609. Michel G, Nyval-Collen P, Barbeyron T, Czjzek M, Helbert W. Bioconversion of red seaweed galactans: a focus on bacterial agarases and carrageenases. Appl Microbiol Biotechnol (2006) 71:23–33. doi:10.1007/s00253-006-0377-7
610. Hehemann J, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature (2010) 464:908–12. doi:10.1038/nature08937
611. Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G. Environmental and gut bacteroidetes: the food connection. Front Microbiol (2011) 2:93. doi:10.3389/fmicb.2011.00093
612. Hehemann JH, Kelly AG, Pudlo NA, Martens EC, Boraston AB. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc Natl Acad Sci U S A (2012) 109:19786–91. doi:10.1073/pnas.1211002109
613. Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol (2011) 9:e1001221. doi:10.1371/journal.pbio.1001221
614. Tannahill G, Curtis A, Adamik J, Palsson-McDermott E, McGettrick A, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi:10.1038/nature11986
615. Mills E, O’Neill LAJ. Succinate: a metabolic signal in inflammation. Trends Cell Biol (2014) 24:313–20. doi:10.1016/j.tcb.2013.11.008
616. Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol (2008) 28:718–31. doi:10.1128/MCB.01338-07
617. Kietzmann T, Görlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol (2005) 16:474–86. doi:10.1016/j.semcdb.2005.03.010
618. Dang EV, Barbi J, Yang H, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
619. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol (2008) 9:1261–9. doi:10.1038/ni.1657
620. Inagaki A, Ichikawa H, Sakata T. Inhibitory effect of succinic acid on epithelial cell proliferation of colonic mucosa in rats. J Nutr Sci Vitaminol (2007) 53:377–9. doi:10.3177/jnsv.53.377
Keywords: inflammatory bowel disease, Crohn’s disease, diet, gut microbiota, fatty acids, mucosal, inflammation
Citation: Basson A, Trotter A, Rodriguez-Palacios A and Cominelli F (2016) Mucosal Interactions between Genetics, Diet, and Microbiome in Inflammatory Bowel Disease. Front. Immunol. 7:290. doi: 10.3389/fimmu.2016.00290
Received: 10 May 2016; Accepted: 19 July 2016;
Published: 02 August 2016
Edited by:
Adria Carbo, Biotherapeutics Inc., USACopyright: © 2016 Basson, Trotter, Rodriguez-Palacios and Cominelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abigail Basson, YXhiODYwQGNhc2UuZWR1;
Fabio Cominelli, ZnhjOTBAY2FzZS5lZHU=