 Daniel J. Wikenheiser
Daniel J. Wikenheiser Jason S. Stumhofer
Jason S. Stumhofer- Department of Microbiology and Immunology, University of Arkansas for Medical Sciences, Little Rock, AR, USA
Over the last 15 years, the inducible T cell co-stimulator (ICOS) has been implicated in various immune outcomes, including the induction and regulation of Th1, Th2, and Th17 immunity. In addition to its role in directing effector T cell differentiation, ICOS has also been consistently linked with the induction of thymus-dependent (TD) antibody (Ab) responses and the germinal center (GC) reaction. ICOS co-stimulation, therefore, appears to play a complex role in dictating the course of adaptive immunity. In this article, we summarize the initial characterization of ICOS and its relationship with the related co-stimulatory molecule CD28. We then address the contribution of ICOS in directing an effector T cell response, and ultimately disease outcome, against various bacterial, viral, and parasitic infections. Next, we assess ICOS in the context of TD Ab responses, connecting ICOS signaling to follicular helper T cell differentiation and its role in the GC reaction. Finally, we address the link between ICOS and human autoimmune disorders and evaluate potential therapies aiming to mitigate disease progression by modulating ICOS signaling.
Introduction
During conventional T cell activation, interaction of the T cell receptor (TCR) with MHC class I or class II-peptide complexes initiates the cascade of T cell activation. However, an important secondary co-stimulatory signal must be delivered in concert with TCR stimulation in order to facilitate proper T cell activation (1). Previously, CD28 ligation had been shown to play a critical role in providing the “second signal” necessary to promote cellular proliferation and survival following T cell activation (2, 3), while CTLA-4 ligation served to mitigate this process (4), as recently reviewed elsewhere (5–7). The identification of the inducible T cell co-stimulator (ICOS) (8, 9) as a new member of the immunoglobulin (Ig) family of co-receptor molecules (8) led to a flurry of research regarding its role in adaptive immune responses.
First identified in humans (8) and shortly thereafter in mice (9), ICOS has significant homology to the co-stimulatory molecule CD28 and the immune-attenuator CTLA-4 (8, 10). Appropriately termed the inducible co-stimulator, ICOS is not constitutively expressed on resting T cells, but is instead rapidly induced following TCR cross-linking and/or CD28 co-stimulation (8, 11, 12). Along with CD28 and CTLA-4, ICOS is expressed on activated CD4 and CD8 T cells (13), suggesting that ICOS – analogous to CD28 and CTLA-4 activity – also regulates the adaptive T cell response. Thus, due to the large degree of homology between ICOS and other Ig family co-stimulatory molecules, in addition to T cell expression of ICOS, early research questioned whether ICOS played a homologous role to CD28 in the process of T cell activation and the initiation of cell-mediated adaptive immunity.
While a number of early studies evaluated the function of ICOS in T cell activation, proliferation, and differentiation, the predominant phenotype that emerged from the initial characterization of ICOS and ICOS ligand (ICOSL)-deficient mice revealed that this co-stimulatory molecule played a significant role in the generation of class-switched antibodies (Abs) against thymus-dependent (TD) antigens, which was attributed to a reduction in the number and size of germinal centers (GCs) in the spleen in the absence of ICOS signaling. Long-lived plasma cells (LLPCs) and memory B cells (MBCs) that have undergone class-switching and somatic hypermutation to increase Ab affinity are products of GC reactions; as these cell types and the Abs they produce are thought to be absolutely critical for maintaining life-long protection against pathogens following exposure or vaccination, or for contributing to the development of a number of autoimmune diseases, the most recent developments regarding the function of ICOS have focused on how ICOS–ICOSL interactions contribute to GC-derived Ab production.
Herein, we assess the relationship between CD28 and ICOS in regard to intracellular signaling events, and evaluate how these co-stimulatory molecules serve different roles in CD4+ T cell activation and proliferation. We then evaluate the influence of ICOS on the development of Th1 and Th2 type immunity in response to various bacterial, viral, and parasitic infections. Next, we discuss the early findings in Icos−/− and Icosl−/− mice that implicated ICOS in promoting TD antibody responses. We will then discuss more recent findings that suggest a role for ICOS signaling in the differentiation and maintenance of follicular helper T (Tfh) cells. To connect the function of ICOS during Tfh cell development with the humoral response as a whole, we will also assess how ICOS–ICOSL interactions modulate the GC reaction. Finally, a survey of potential therapeutic interventions regarding attenuation of ICOS signaling, and potential human considerations, is evaluated.
ICOS and CD28
Although similar in structure, ICOS and CD28 appear to play non-redundant roles in modulating the activation of T cells (14, 15). Accordingly, in contrast to CD80 and CD86, which interact with CD28 and CTLA-4 (4), ICOSL binds ICOS exclusively (12, 16, 17). Interestingly, ICOS lacks the specific MYPPPY motif present in CD28 and CTLA-4 that has been shown to be necessary for interaction with CD80 and CD86 (8, 18). These data suggest the single receptor–ligand pair ICOS:ICOSL is likely regulated in a fashion distinct from CTLA-4 and CD28, as ICOSL expression itself has been shown to be downregulated following interaction with ICOS (19). ICOSL expression is largely restricted to professional antigen-presenting cells (APCs), including B cells [in which ICOSL is regulated by BAFFR and non-canonical NFκB signaling (20)], macrophages, and dendritic cells (DCs) (12, 17, 21, 22), but is also expressed by certain endothelial cells (23) and lung epithelium (24).
Early research indicated that although ICOS and CD28 downstream signaling events were related, they were not identical (25) (Figure 1). For example, both ICOS and CD28 ligation induce the recruitment of class IA phosphatidylinositol 3-kinase (PI3K) (26, 27), a signaling molecule that leads to the production of membrane-bound phosphatidylinositol 3,4,5-trisphosphate (PIP3), culminating in the activation of Akt – a kinase known to promote cellular proliferation and survival (28). Similar to CD28 cross-linking, ICOS ligation can yield the recruitment of p50α (27) and p85α (29) regulatory subunits of PI3K, in conjunction with recruitment of the p110δ catalytic subunit (27, 29). However, ICOS ligation of activated CD4+ T cells was demonstrated to enhance production of PIP3 (27, 30) and induce stronger Akt phosphorylation than CD28 cross-linking (14, 27). Interestingly, the particular YxxM signaling motif YMFM found in the cytoplasmic tail of ICOS (YMNM in the case of CD28) (31) was responsible for preferential recruitment of the p50α subunit of PI3K (27), a regulatory subunit with inherently greater lipid kinase activity relative to p85α (32). Although ICOS signaling serves to specifically promote p50α recruitment and subsequent AKT activity, ICOS-mediated p85α recruitment has recently been shown to play a critical CD28-independent role in directing T helper cell effector fate by promoting the Tfh cell phenotype (29).
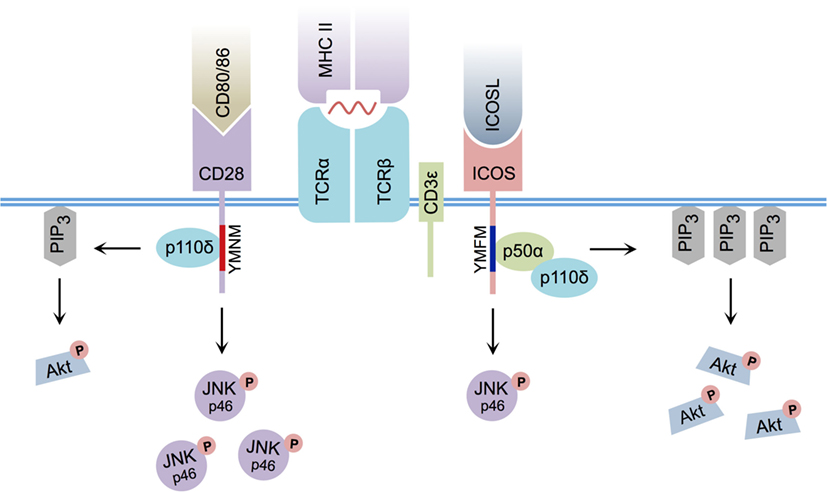
Figure 1. Differential CD28 and ICOS signal transduction. ICOS:ICOSL interaction induces PI3K p50α regulatory subunit recruitment via its YMFM signaling motif, resulting in strong PIP3 production and enhanced Akt phosphorylation. CD80/CD86:CD28 interaction induces less PIP3 production and weaker Akt phosphorylation. ICOS:ICOSL interaction induces less c-Jun N-terminal kinase (JNK) p46 phosphorylation relative to CD80/86:CD28 ligation.
Although slight differences in PI3K signaling can explain some of the differences between CD28 and ICOS signaling, it cannot fully account for the specific activity of ICOS signaling itself, as disruption of the ICOS–PI3K interaction or deletion of PI3K components from T cells does not result in a complete phenocopy of Icos−/− mice (15, 33, 34). Interestingly, differences in the pattern of MAPK activation between CD28 and ICOS have been reported, and ICOS co-stimulation has been shown to induce weaker phosphorylation of p46 c-Jun N-terminal kinase than CD28 (14). Furthermore, an additional intracellular signaling motif was recently identified in the cytoplasmic tail of ICOS that is not present in CD28. In response to simultaneous ICOS and CD3ε ligation, this cell-membrane proximal motif, termed IProx, was shown to recruit TBK1 (35), a member of the inhibitor of NF-κB kinase (IKK) family (36). While recruitment of TBK1 to the related co-stimulatory molecules CD28 and CTLA-4 was not observed following ligation with their respective ligands, ICOS:ICOSL-mediated recruitment of TBK1 was found to play an essential role in promoting the transition from an early Tfh cell to a GC-resident Tfh cell (35). As a whole, these data suggest that, although a commonality exists between CD28 and ICOS-mediated signal transduction, differences in downstream PI3K and MAPK signaling, as well as ICOS-specific TBK1 recruitment, likely contribute to their functional diversity.
ICOS and T Cell Proliferation
Due to the shared homology between the co-stimulatory molecules CD28 and ICOS, early research sought to characterize the role of ICOS in T cell activation and proliferation. Interestingly, ICOS-deficient T cells exhibited a proliferation defect in vitro when compared with wild-type CD4+ T cells (37, 38). Furthermore, when immunized with keyhole limpet hemocyanin (KLH) adsorbed to alum, lymph nodes from Icos−/− mice were found to be smaller in size when compared with wild-type mice, suggesting a similar proliferation defect in vivo (37). Similarly, during Trichuris muris and Toxoplasma gondii infection, CD4+ T cells isolated from Icos−/− mice exhibited defects in activation and proliferation, but not differentiation (39). Additionally, when TCR transgenic T cells specific for ovalbumin (OVA) were polarized under Th1 or Th2 conditions in vitro and then transferred into naive recipients, ICOS was found to be necessary for expansion of both subsets (40). However, when ICOS-deficient mice were immunized with KLH in complete Freund’s adjuvant (CFA), no defect in cellular activation or proliferation was observed (37). These conflicting results led researchers to assess IL-2 production, an important step in promoting T cell clonal expansion (41). In contrast to CD28 ligation, multiple research groups discovered ICOS cross-linking did not induce IL-2 expression (8, 11, 14, 25, 26, 37), and instead induced the production of the anti-inflammatory cytokine IL-10 in vitro (8, 42). Thus, the role of ICOS in promoting CD4+ T cell proliferation is likely independent of IL-2 signaling, and the molecular basis for the role of this co-stimulatory molecule in promoting T cell expansion remains unclear. It is quite possible that ICOS signaling delivers a unique pro-survival or expansion signal not provided by CD28, but this remains to be determined. Furthermore, as differences in CD4+ T cell expansion have not been reported in every immunization or infectious disease model, the nature of the adjuvant or pathogen, as well as the degree of inflammation induced, may dictate the necessity of ICOS in T cell activation and clonal expansion – a topic we will touch upon further in the next section.
ICOS and Infection
To better characterize the in vivo role of ICOS in the process of T cell differentiation during conditions relevant to human disease, a multitude of murine infection models, as well as strategies designed to disrupt ICOS signaling, have been investigated. As a whole, ICOS has been shown to regulate various T helper cell subsets during different infection scenarios, largely by promoting or inhibiting Th1 and Th2 immune responses (Table 1).
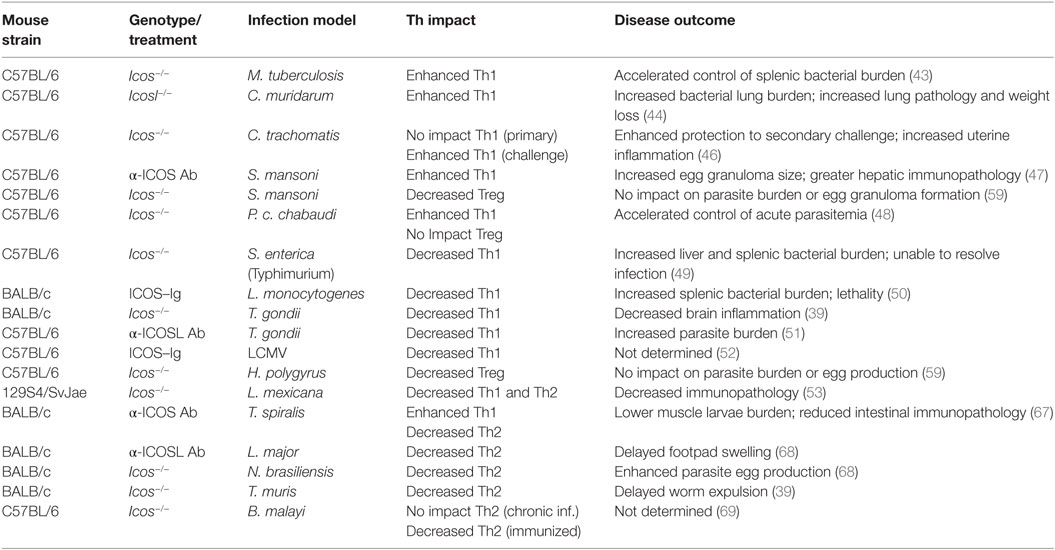
Table 1. Summary of Th impact and disease outcome in various infection models when ICOS signaling is disrupted.
Th1 Immunity
In the context of Mycobacterium tuberculosis infection, for example, mice lacking expression of ICOS exhibited evidence of enhanced Th1 immunity, producing a significantly greater number of CD4+IFN-γ+ T cells in the spleen and lungs during later stages of infection. Concomitantly, regulatory T cell (Treg) frequency was significantly reduced in ICOS-deficient mice in this infection model. In the end, ICOS deficiency led to enhanced control of M. tuberculosis infection in the spleen, but not the lungs (43).
ICOS ligand-deficient mice infected with Chlamydia muridarum also produced a significantly stronger Th1 response, with enhanced production of IFN-γ, IL-6, and TNF-α. Furthermore, the authors observed lower production of the anti-inflammatory cytokines IL-10 and TGF-β in ICOSL-deficient mice. Similar to M. tuberculosis infection, Icosl−/− mice infected with C. muridarum exhibited higher bacterial lung burden and showed greater evidence of lung pathology, as well as losing more body weight than wild-type control mice (44). Additional research has also linked ICOS-mediated PI3K signaling with the induction of Th17 responses during C. muridarum infection. In this study, transgenic mice harboring an ICOS signaling mutation (preventing PI3K from interacting with ICOS) mounted a defective Th17 response compared with wild-type mice, culminating in decreased control of bacterial burden in the lungs (45).
On the other hand, during Chlamydia trachomatis genital tract infection, Icos−/− mice controlled primary infection similarly to wild-type mice. However, following resolution of acute infection, ICOS-deficient mice exhibited increased cellular infiltrate and inflammation within the uterine horns. Surprisingly, Icos−/− mice showed enhanced protection to secondary C. trachomatis challenge, with a greater frequency of IFN-γ+ T cells observed in the uterus of Icos−/− mice (46).
In the case of Schistosoma mansoni infection, wild-type mice treated with α-ICOS neutralizing Abs developed larger egg granulomas and displayed evidence of enhanced hepatic immunopathology. Increased production of IFN-γ concomitant with decreased IL-10 secretion in α-ICOS Ab-treated mice suggests an enhanced Th1 response likely mediated the associated hepatic pathology (47).
In agreement with evidence of enhanced Th1 immunity in Icos−/− mice, during Plasmodium chabaudi chabaudi AS infection, ICOS served to dampen the Th1 response, as Icos−/− mice produced more IFN-γ secreting CD4+ and CD8+ T cells and had higher serum IFN-γ than wild-type mice. Additionally, enhanced expansion of CD25+T-bet+ effector CD4+ T cells was observed in ICOS-deficient mice. The augmented Th1 response observed in Icos−/− mice led to lower peak parasitemia and accelerated control of acute P. c. chabaudi AS infection relative to wild-type mice (48).
There are, however, examples in which ICOS appears to promote Th1 immunity, such as systemic Salmonella enterica (serovar Typhimurium) infection. In this model, ICOS-deficient mice were defective in CD4+ T cell IFN-γ production, despite having no defect in total CD4+ T cell activation. Accordingly, Icos−/− mice had substantially increased bacterial burden in the liver and spleen at days 14 and 21 post-infection compared with wild-type mice. In the end, Icos−/− mice were unable to fully resolve S. enterica infection, and continued to harbor bacteria in the spleen at day 36 post-infection (49).
In the same manner, disruption of ICOS signaling during oral Listeria monocytogenes infection via ICOS–Ig treatment led to decreased Th1 immunity and significantly increased splenic bacterial burden and lethality. Interestingly, in ICOS–Ig treated mice, CD4+ T cells were unable to produce IFN-γ in response to ex vivo stimulation with heat-killed Listeria, contrary to control-Ig treated mice. These data suggest the magnitude of the Th1 response was blunted when ICOS signaling was impaired (50).
In the context of a strong Th1-inducing parasite, such as T. gondii, ICOS and CD28 appear to play a redundant role in the induction of Th1 immunity. In the absence of CD28, mice were still capable of producing IFN-γ. However, disruption of ICOS signaling via α-ICOSL Ab treatment significantly reduced production of IFN-γ while simultaneously increasing parasite burden (51). In T. gondii-infected Icos−/− mice (with intact CD28 signaling), no defect in T cell-derived IFN-γ was observed. Instead, the absence of ICOS signaling decreased CD4+ T cell expansion during acute and chronic infection leading to reduced inflammation, but did not impact parasite burden, suggesting a redundant role concerning IFN-γ production but a unique role for ICOS in promoting T cell expansion (39). In contrast, disruption of ICOS signaling via ICOS–Ig fusion protein during LCMV infection diminished IFN-γ production, and this phenotype was potentiated in ICOS–Ig-treated Cd28−/− mice (52). These data suggest that, in response to LCMV infection, ICOS and CD28 may synergistically promote Th1 immunity.
Additionally, ICOS may simultaneously promote ongoing Th1 and Th2 immune responses. During cutaneous Leishmania mexicana infection, for example, researchers observed a slight reduction in IFN-γ and IL-4 production in Icos−/− mice at week 6 post-infection, while the production of both cytokines was substantially reduced in ICOS-deficient mice by week 12 post-infection. This reduced inflammation did not lead to an increase in parasite burden; in fact, lesions were initially slower to develop in Icos−/− mice, but by week 12 post-infection, lesions were similar to those observed in wild-type mice. Despite normal lesion formation, however, tissue damage was less severe in Icos−/− mice at week 12 post-infection compared with wild-type mice (53), suggesting that, in the case of chronic L. mexicana infection, ICOS signaling served to enhance immune-mediated pathology.
Although ICOS is seemingly capable of alternatively promoting or repressing Th1 responses under different infection conditions, these divergent phenotypes may in part be explained by ICOS-dependent Treg induction (54, 55). For instance, it is well established that CD4+ T cells from naive mice that express high levels of ICOS also produce the greatest amount of IL-10 (56), a cytokine often produced by Tregs. In a different study, administration of a toll-like receptor (TLR) 4 ligand enhanced the frequency of IL-10-producing ICOS+CD4+Foxp3+ Tregs in lung draining lymph nodes following birch pollen allergen nasal challenge, ameliorating the development of severe disease (57). Similarly, Icos−/− mice tolerized with intranasal OVA developed significantly fewer splenic and lung-resident CD4+Foxp3+ Tregs at day 6 post-challenge relative to wild-type mice (58). Together, these data connect ICOS expression with Treg functionality in models of allergic challenge, as well as during naive conditions. Additionally, ICOS has been directly linked with Treg induction during infection. Icos−/− mice infected with Heligmosomoides polygyrus or S. mansoni, for instance, exhibited delayed expansion and significantly fewer total CD4+Foxp3+ Tregs in the mediastinal lymph node and spleen, respectively, when compared with wild-type mice (59). Thus, the ability of ICOS to influence Treg induction further complicates its role in regulating CD4+ T cell differentiation, and suggests ICOS signaling may be mutually important in preventing immune-mediated pathology, as well as inducing pro-inflammatory CD4+ T cells. However, the regulatory function of ICOS is not limited to Treg cell induction and IL-10 production, as Icos−/− mice display an enhanced Th1 response after infection with P. c. chabaudi AS, despite similar numbers of splenic Tregs and IL-10 secreting CD4+ T cells as wild-type mice (48).
Altogether, these data strongly implicate ICOS in modulating Th1 immune responses. It is, however, difficult to isolate the precise contribution of ICOS in promoting or regulating Th1 immunity, as ICOS signaling likely influences T cell proliferation, as well as effector cell fate decisions, including Treg induction, simultaneously. As there is a degree of redundancy between CD28 and ICOS signaling, perhaps the integration of ICOS and CD28 co-stimulatory signals, as well as signals from other surface proteins, such as the TCR, cytokine receptors, and other co-stimulatory molecules, dictate the significance of ICOS:ICOSL signals in promoting or regulating T helper cell differentiation. As various pathogens are capable of influencing APC activation status by modulating CD80 (60), CD86, and ICOSL (61–64) surface expression, it is not unreasonable to suggest the relative ratio of ICOS:CD28 co-stimulation a CD4+ T cell receives varies greatly based on the infectious agent. The relevance of ICOS signaling in CD4+ helper T cell differentiation, therefore, may also be connected to the innate signaling events responsible for promoting APC maturation and subsequent upregulation of CD28 co-stimulatory ligands and ICOSL.
Th2 Immunity
One of the original hypotheses for the function of ICOS suggested that it played an important role in driving Th2 immunity, as ICOS expression is preferentially sustained on Th2 immune cell subsets (11, 26, 39), while in vitro disruption of ICOS signaling yields decreased IL-4 production (37–39, 65, 66). Likewise, in the context of Trichinella spiralis (67) and Leishmania major (68) infection, treatment of wild-type mice with anti-ICOS or anti-ICOSL Abs served to diminish the Th2 immune response, reducing the production of IL-4, IL-5, and IgE. However, during T. spiralis infection, anti-ICOS Ab treatment also simultaneously promoted TNF-α, IFN-γ, and IL-10 production. While disruption of ICOS signaling did not impact worm load in the intestine, it did substantially reduce immunopathology within the intestine itself. Interestingly, anti-ICOS Ab-treated mice showed significantly lower larvae burden in striated muscle, a site of secondary colonization, when compared with control mice (67).
Blunted Th2 immunity was also observed in Nippostrongylus brasiliensis-infected Icos−/− mice, in which parasite egg production was enhanced in the absence of ICOS (68). Similarly, disruption of ICOS signaling in Cd28−/− mice via ICOS–Ig treatment diminished IL-4 and IL-5, as well as IFN-γ and IL-10 production in N. brasiliensis-infected mice, but surprisingly did not hinder or delay worm expulsion (52).
Although ICOS appears to play an important role in promoting Th2 immunity, in the case of chronic Brugia malayi infection, Icos−/− mice were not defective in their ability to produce IL-4 or IgE compared with wild-type mice. Furthermore, eosinophil recruitment to the peritoneal cavity was not defective in the absence of ICOS. However, during primary immunization with B. malayi antigen, fewer IL-4 producing Th2 cells were identified in Icos−/− mice at day 10 post-immunization (69), suggesting that ICOS may be more important during the priming phase of Th2 immune responses.
In a similar fashion, although defective Th2 immunity was observed during acute Trichuris muris infection, Icos−/− mice produced more IL-13 and IL-5 at day 35 post-infection compared with wild-type mice, suggesting that Th2 immunity was slower to develop in the absence of ICOS. Worm burden data corroborate this idea, as Icos−/− mice continued to harbor worms at day 18 post-infection – a time-point at which wild-type mice had eliminated the parasite. However, by day 54 post-infection, Icos−/− mice had largely controlled worm burden, suggesting that the onset of Th2 immunity was delayed, but not absent, in Icos−/− mice (39). Similar to the studies with T. gondii-infected Icos−/− mice, the delayed Th2 response was attributed to reduced expansion of Th2 polarized CD4+ T cells.
Collectively, the development of Th2 immunity appears to be more critically reliant on ICOS signaling than Th1 immune responses. In all infection models reviewed herein, disruption of ICOS signaling led to poor CD4+ T cell Th2 polarization and diminished IL-4 production, which may or may not have been due to reduced expansion of CD4+ T cells in all cases. Similar to the results seen with infection models that induce a Th1 response, the absence of ICOS signaling did not completely suppress the development of Th2 immunity. In a number of circumstances, anti-worm Th2 immunity in Icos−/− mice was acutely defective, but ultimately played an important, albeit delayed, role in controlling worm burden. In the case of the worm infections described above, the late enhancement of the Th2 response coincided with the development of the larvae into adult worms, indicating changes in antigen or persistence of antigen may allow for the late expansion of the Th2 response in these models. Similarly, variation in antigen or antigen load may also account for the variable requirement of ICOS signaling in Th1 cell induction in previously discussed Th1 infection models. Nevertheless, the studies presented here indicate that ICOS co-stimulation plays an important role in either enhancing or regulating Th cell expansion and/or differentiation after infection.
ICOS Promotes TD Antibody Responses
Although ICOS can modulate Th1/Th2 differentiation during infection, early observations connected ICOS co-stimulation with Ab production. During the initial phenotypic characterization of ICOS, for instance, researchers noted that, within secondary lymphoid tissue, ICOS expression was largely restricted to GC light zones (8), an area within B cell follicles known to be populated by CD4+ T cells (70). To address the contribution of ICOS to Ab production, Icos−/− or Icosl−/− mice were immunized with a number of thymus-independent (TI) and TD antigens.
When Icosl−/− mice were immunized with the TI type I (TI-I) antigen nitrophenol (NP)-conjugated LPS no defect in the production of NP-specific IgM or IgG3 Abs was noted (66). Although a number of TLR agonists, including LPS, are capable of upregulating ICOSL expression on splenic B cells (64), the absence of ICOSL expression did not affect B cell activation in response to NP-LPS immunization. Likewise, immunization of Icosl−/− mice with the TI type II (TI-II) antigen NP-Ficoll yielded no defect in NP-specific IgM or IgG3 Abs relative to wild-type control mice. However, while immunization of Icosl−/− mice with the TD antigen NP21-conjugated chicken globulin (CG) adsorbed to alum led to no defect in NP-specific IgM Abs, a substantial decline in IgG1 and IgG2a production was observed (66). Similarly, following immunization with the TD antigen sheep red blood cells (SRBC), treatment of mice with α-ICOSL Abs did not impact production of SRBC-specific IgM, but significantly diminished IgG1, IgG2a, and IgG2b-specific Abs (71). Ultimately, SRBC-boosted Icos−/− mice (72) and NP21-CGG boosted Icosl−/− mice (66) failed to form secondary GCs when compared with wild-type mice.
In the case of the TI anti-polysaccharide response to whole Streptococcus pneumoniae immunization, no defect in IgG Ab production was observed in Icos−/− mice (73). Perhaps not surprisingly, given that TI antigens often induce abortive GCs and instead promote isotype-switched Ab production through an extrafollicular response (74, 75), this class-switched Ab was determined to be largely extrafollicular in nature. However, immunization of Icos−/− mice with S. pneumoniae cell-wall polysaccharide conjugated to PspA (an S. pneumoniae surface protein) yielded defective anti-polysaccharide IgG production relative to wild-type mice, indicating that direct conjugation of a TI antigen to protein results in a dependency on a GC reaction and ICOS signaling to generate isotype-switched Abs (73). Collectively, although ICOS did not appear to play an essential role in TI Ab production or IgM-specific Ab formation against protein or a haptenated protein antigen, production of TD class-switched Abs appeared to require intact ICOS signaling. Furthermore, ICOS performed an important function in mediating the secondary humoral response to previously encountered TD antigens.
To further interrogate the role of ICOS in TD humoral immunity, the Ab response to various model Ag-adjuvant combinations was assessed in ICOS-deficient mice. In the presence of either alum or CFA, for example, Icos−/− and Icosl−/− mice exhibited a profound defect in the IgG1-specific Ab response to KLH when compared with wild-type mice (37, 38, 65), with a similar reduction in GC formation (38, 65). In agreement with these data, immunization of Icos−/− mice with 2,4,6-trinitrophenol (TNP)-conjugated KLH in incomplete Freund’s adjuvant (lacking Mycobacterium cell-wall components) or alum yielded gross defects in the production of IgG2a and IgG1-specific TNP Abs, but did not diminish TNP-specific IgM production. Accordingly, GCs were smaller in size and number in Icos−/− mice compared with wild-type mice. However, when immunized with TNP–KLH in CFA, ICOS-deficient mice were not defective in the production of TNP-specific IgG isotype Abs, and subsequent GC size was not impaired, although a defect in the total number of GCs was observed in Icos−/− mice relative to wild-type mice (76).
Collectively, these data provide seemingly conflicting results concerning the necessity of ICOS in TD Ab responses. While the use of a strong inflammatory adjuvant, such as CFA, which contains components that can serve as TLR agonists, could explain why TNP-KLH can elicit a class-switched Ab response in the absence of ICOS, it does not explain why immunization with un-conjugated KLH in CFA cannot. Interestingly, only the IgG1, but not IgG2a, response against KLH was significantly reduced in Icos−/− and Icosl−/− mice after immunization with KLH in CFA (38, 65). These data suggest that, similar to the result of TNP-KLH in CFA immunization, the inclusion of CFA can result in the production of class-switched Abs of certain isotypes in the absence of ICOS signaling. As the Th2-associated cytokine IL-4 is required for isotype switching to IgG1, and ICOS signaling has been implicated in Th2 differentiation (37–39, 65, 66), the failure to induce a strong IgG1 response against KLH in CFA may be partially due to impaired Th2 development. In support of this idea, Icos−/− and Icosl−/− mice have been shown to have lower serum baseline concentrations of IgG1 (38, 65). Also, differences in the Ab response against KLH and TNP-KLH in CFA could be explained by the nature of the antigen itself (protein alone versus hapten–protein complex, respectively). As only the antibody response against the hapten was measured in the TNP-KLH in CFA immunization studies, the KLH-specific Ab response could be defective in the Icos−/− mice as well. Also, as IgG production was only assessed in the short-term for these particular studies (between days 7 and 14 post-immunization), it is possible extrafollicular isotype-switched Ab production could account for the TNP-specific IgG1 produced in Icos−/− mice immunized with TNP-KLH in CFA. In future studies, long-term maintenance of isotype-switched Ab titers, perhaps, may prove a more useful metric for assessing the role of ICOS in TD Ab production.
In connection to its role in promoting TD Ab responses, ICOS may also regulate Ab affinity maturation – an important function of the GC reaction. In addition to producing significantly lower amounts of NP-specific IgG1 Abs than wild-type mice following NP-conjugated OVA immunization, the NP4/NP23 IgG1 ratio was significantly lower in Icosl−/− mice at 6 weeks post-immunization, indicating a defect in Ab affinity maturation occurred in the absence of ICOS signaling. However, the NP4/NP23 IgG1 ratio did not differ significantly at 10 weeks post-immunization relative to wild-type mice, suggesting that between weeks 6 and 10 post-NP-OVA immunization affinity maturation proceeded in an ICOS-independent manner (65). As the absence of ICOS signaling does not completely prevent GC formation but rather reduces the size and number of GCs (77), perhaps the few GCs present in Icosl−/− mice are capable of slowly producing affinity-matured NP-specific IgG1, such that by 10 weeks post-immunization the NP4/NP23 IgG1 ratio is similar between wild-type and Icosl−/− mice. In contrast to this result, P. c. chabaudi AS-infected Icos−/− mice exhibited a profound defect in parasite-specific IgG affinity maturation at weeks 3 and 9 post-infection, suggesting that affinity maturation during murine Plasmodium infection is critically dependent on continuous ICOS signaling (48).
While these data provide conflicting results concerning the role of ICOS in promoting TD Ab responses, it ultimately indicates that ICOS is a critical component in the production of isotype-switched Abs, particularly affinity-matured ones. Because the nature of the adjuvant or immunogen – hapten, protein, or whole pathogen – vary widely across experiments, the precise role of ICOS in promoting these Ab responses is not immediately clear. Nonetheless, the presence of ICOS+ CD4+ T cells within GCs suggests an important role for this molecule in supporting the GC reaction. Subsequent research, therefore, has explored the link between ICOS and CD4+ T cells in the larger context of TD Ab responses.
ICOS and Tfh Cell Differentiation
Recently, ICOS has been directly implicated in the induction of a specific CD4+ T cell effector subset known as Tfh cells (15, 29, 71, 78–81). Within the GC, Tfh cells play a critical role in promoting the selection and survival of B cells expressing high-affinity B cell receptors, a process that ensures only B cells with the highest affinity for a given antigen differentiate into plasma cells and MBCs (70). The Tfh cell program of differentiation is critically reliant on Bcl6 expression (82, 83), a transcriptional co-repressor that antagonizes Blimp-1 – a transcription factor responsible for dictating non-Tfh CD4+ T cell effector programs. Successful repression of Blimp-1 by Bcl6 promotes the Tfh cell phenotype, and allows a pre-Tfh cell to upregulate additional molecules required for Tfh cell differentiation (84).
A critical component of the Tfh cell phenotype is expression of the chemokine homing receptor CXCR5, which binds CXCL13 produced by follicular DCs to promote the migration of Tfh cells to the B cell follicle. In addition to CXCR5, Tfh cells also express the co-inhibitory receptor PD-1, which serves to prevent excessive, unchecked proliferation. Tfh cells that have established residence within the GC light zone itself (GC Tfh cells) adopt a PD-1highCXCR5high phenotype, and are phenotypically distinct from non-GC-resident Tfh cells. Additionally, Tfh cell production of the cytokine IL-21 is a key component in promoting the GC reaction, as well as reinforcing the Tfh cell phenotype itself in an autocrine manner (84).
Although Tfh cells have been published on extensively in the last several years, the link between ICOS and CXCR5+ CD4+ T cells had been the subject of earlier research. For example, it had been previously demonstrated that following immunization with SRBCs (71) or type II collagen (78), treatment of mice with α-ICOSL Abs abrogated expression of CXCR5+ on CD4+ T cells. Additionally, during secondary SRBC immunization, α-ICOSL Ab treatment similarly prevented induction of CXCR5+ on CD4+ T cells when compared with control mice, suggesting that ICOS supports T cell CXCR5 expression during primary and secondary GC responses (71).
While these studies highlighted the importance of ICOS signaling in promoting CXCR5 expression and possibly the generation of Tfh cells, the source and timing requirements of ICOSL co-stimulation were not fully understood. Continued efforts to better understand how ICOS influences the Tfh cell program have yielded novel insights into the process of Tfh cell differentiation. For instance, a recent report demonstrated CD8α− DCs upregulate ICOSL expression following stimulation with polyI:C or LPS. This CD8α− DC subset was subsequently shown to be important for induction of Tfh cells in vitro and in vivo (85). Previous research had also investigated the role of ICOS in the early stages of Tfh cell induction during DC:T cell interactions following lymphocytic choriomeningitis virus (LCMV) infection. Transfer of Icos−/− LCMV-specific SMARTA CD4+ T cells into wild-type mice followed by LCMV infection resulted in a marked defect in Bcl6 and CXCR5 expression at days 3 and 4 post-infection relative to wild-type SMARTA T cells, suggesting that ICOS plays an essential role in the induction of Tfh cells. Furthermore, B cell-deficient μMT mice infected with LCMV were shown to have no defect in CD4+ T cell expression of Bcl6 or CXCR5 at day 3 post-infection, suggesting that B cells are not exclusively responsible for promoting early Tfh cell induction. However, by day 4 post-infection, Tfh cell frequency in μMT mice had declined significantly relative to wild-type mice, suggesting that interaction with B cells is necessary for maintenance of Tfh cells. Collectively, the authors concluded that ICOSL signaling from DCs is initially required for early Tfh cell priming, while cognate B cells provide a secondary source of ICOSL to reinforce the GC Tfh cell phenotype (79).
In agreement with a role for B cells in Tfh cell maintenance, B cell ICOSL expression was found to be essential for sustained Tfh cell:B cell engagement within the GC itself (86), suggesting that ICOS:ICOSL interactions increase the duration of T:B cell contact, consequently impacting affinity maturation, B cell survival, and plasma cell differentiation. In a different study, despite no defect in generating Bcl6+ pre-Tfh cells (early Tfh cells that have not yet entered the B cell follicle), when MHC-II expression was restricted to CD11chigh DCs, PD-1++ GC Tfh cells were unable to form following NP-OVA-alum immunization (87). Together, these data indicate that B cells deliver a critical ICOSL signal necessary to promote full GC Tfh cell maturation. Indeed, Tfh cells polarized in vitro in the presence of B cells produced substantially more IL-21, a cytokine linked to the GC reaction, than Tfh cells co-cultured with DCs alone (87). Furthermore, co-culture of wild-type or Icos−/− α-CD3ε-stimulated memory CD4+ T cells with ICOSL-expressing B cells revealed enhanced IL-21 mRNA synthesis in the presence of ICOS signaling (88), indicating that B cells support the production of IL-21 via ICOS:ICOSL interactions. Interestingly, while transfer of T cells alone into immunodeficient SCID mice was not sufficient for proper Tfh cell development following SRBC immunization, co-transfer of T and B cells yielded Tfh cell differentiation; administration of α-ICOSL Abs disrupted Tfh cell differentiation under these conditions (71). As a whole, these data suggest that cooperative DC and B cell ICOSL co-stimulation serves a central role in the process of Tfh cell induction and maintenance, respectively. Whether ICOSL signaling from other cell types within the GC, such as DCs or follicular DCs, is also necessary to maintain or promote Tfh cell polarization during the GC reaction is still unknown.
Inducible T cell co-stimulator signaling, however, may not be critical for initial Tfh cell differentiation under all circumstances. For instance, Icos−/− OT-II cells co-transferred with NP-specific B cells were not defective in Bcl6, c-Maf, or IL-21 expression following NP-OVA immunization. Nonetheless, Icos−/− T cells were unable to fully downregulate CCR7 – an essential step in assuming the GC Tfh cell phenotype – and upregulate CXCR5, resulting in an inability of ICOS-deficient T cells to migrate into the B cell follicle. Furthermore, ICOS signaling was required to maintain the GC reaction, as α-ICOSL Ab treatment on day 6 post-NP-OVA immunization led to a loss of Tfh cells and the GC itself (80).
Indeed, in the context of murine Plasmodium infection, ICOS appears to play a more substantial role in promoting Ab production late in the infection, suggesting that B cells may be the most important source of ICOSL stimulation for Tfh cell maintenance. Prior to day 6 post-infection, for example, Icos−/− CD4+ T cells were not defective in their ability to express the canonical Tfh cell markers Bcl6, PD-1, CXCR5, or IL-21. However, by week 3 post-infection, there was a substantial reduction in CD4+ T cells expressing these Tfh cell markers in Icos−/− mice, and the remaining Tfh cells were unable to form PD-1++CXCR5++ GC Tfh cells. The end result was a drastic decrease in the total number of splenic GCs in ICOS-deficient mice relative to wild-type mice. Quite surprisingly, Icos−/− mice were not defective in production of merozoite surface protein-1 42 kDa fragment (MSP-142)-specific IgM or IgG isotype Abs at week 3 post-infection. However, the GC defect in Icos−/− mice resulted in a significant decrease in both quantity and quality of parasite-specific Abs by week 6 post-infection (48). As a whole, the complexity of the Ab response to Plasmodium (89), coupled with the parasite’s ability to induce a strong extrafollicular Ab response, may help explain why ICOS is less critical in promoting early isotype-switched Ab production during P. c. chabaudi AS infection. Although initial Tfh cell induction was not impaired in Icos−/− mice, proper GC Tfh cell differentiation and Ab affinity maturation required intact ICOS signaling (48), providing further evidence that GC Tfh cell formation necessitates B cell-specific ICOSL co-stimulation.
There are additional circumstances in which ICOS:ICOSL signaling is not required for differentiation of Tfh cells or the establishment of GCs. For instance, in response to NP-OVA immunization in alum, when antigen is abundant and antigen-specific B cells are present in high frequency, Tfh cell differentiation, GC formation, and production of class-switched, affinity-matured NP-specific IgG1 Abs is not impaired when cognate B cells are deficient in ICOSL expression (90). These data suggest ICOS co-stimulation by cognate B cells is only required under conditions when antigen is limiting. This idea is supported by additional findings indicating that DCs can support Tfh cell differentiation in the presence of excess antigen, even when B cell antigen presentation is abolished, indicating that a unique B cell-specific signal is not required for Tfh cell development under all conditions. However, this study further demonstrated that Tfh cells needed to interact with cognate B cells to form GCs, as DC-Tfh cell interactions could not promote GC formation on their own (91). Interestingly, not only the interaction of T cells with APCs but also the duration of these interactions has been shown to influence differentiation of Tfh cells, recruitment of Tfh cells into GCs, and subsequent function of Tfh cells within the GC (77). As such, ICOS:ICOSL interactions may help facilitate and extend the length of these interactions, particularly with cognate B cells, when antigen is limiting.
While ICOS:ICOSL signaling between cognate T and B cells is critical for progression to and maintenance of the GC Tfh cell phenotype, B cells may also play a more unconventional role in promoting Tfh cell differentiation – potentially in the form of non-cognate help. After noting that Icos−/− OT-II CD4+ T cells expressed less CXCR5 and failed to migrate beyond the T:B cell border, researchers sought to determine if the lack of CXCR5 impacted follicular homing of Icos−/− pre-Tfh cells. However, forced CXCR5 or Bcl6 expression by Icos−/− OT-II cells did not enhance their ability to migrate beyond the T:B cell border into the B cell follicle. Strikingly, neither ICOSL expression by DCs or cognate B cells was found to facilitate proper follicular recruitment of early Tfh cells. Instead, ICOS:ICOSL interactions between T cells and non-cognate B cells facilitated migration of T cells into the GC. In the end, non-cognate B cells at the T:B cell border were found to provide an important ICOSL signal that led to T cell-intrinsic PI3K signaling, ultimately promoting T cell motility (92).
Relative to model antigen:adjuvant-based immunization strategies, the presence of an actively replicating pathogen dynamically alters the distribution and abundance of antigen available to the immune system. As such, infectious conditions add additional layers of complexity to the interpretation of data. While under certain circumstances, ICOS plays a critical role in priming Tfh cell induction, different inflammatory conditions and/or model systems appear to preclude the necessity of ICOS in promoting Tfh cell formation. However, as a whole, ICOS signaling critically supports GC Tfh cell functionality. Altogether, although ICOS clearly regulates the processes of Tfh cell differentiation, maintenance, and function, the particular timing of requisite ICOSL signal delivery appears to vary widely based on the model system employed.
ICOS Signaling and Tfh Cells
Mechanistically, the signals underlying Tfh cell differentiation are not yet fully understood. However, the specific contribution of ICOS to the process of Tfh cell induction has recently come under greater scrutiny (Figure 2). The significance of ICOS-mediated PI3K signaling, for example, had been well characterized and described previously (14, 27). A specific role for PI3K recruitment in the context of Tfh cell development was demonstrated by generating knock-in mice expressing a mutant ICOS receptor harboring a cytoplasmic tail incapable of recruiting PI3K while retaining the ability to promote Ca2+ mobilization in concert with TCR signaling. ICOS–PI3K-signaling mutant mice produced fewer splenic Tfh cells in response to NP-CGG plus alum immunization, and were unable to promote affinity maturation of NP-specific IgG following a secondary immunization when compared with wild-type mice. Furthermore, it was demonstrated that PI3K activity was required for promoting IL-21 and IL-4 production during in vitro T cell stimulation (15).
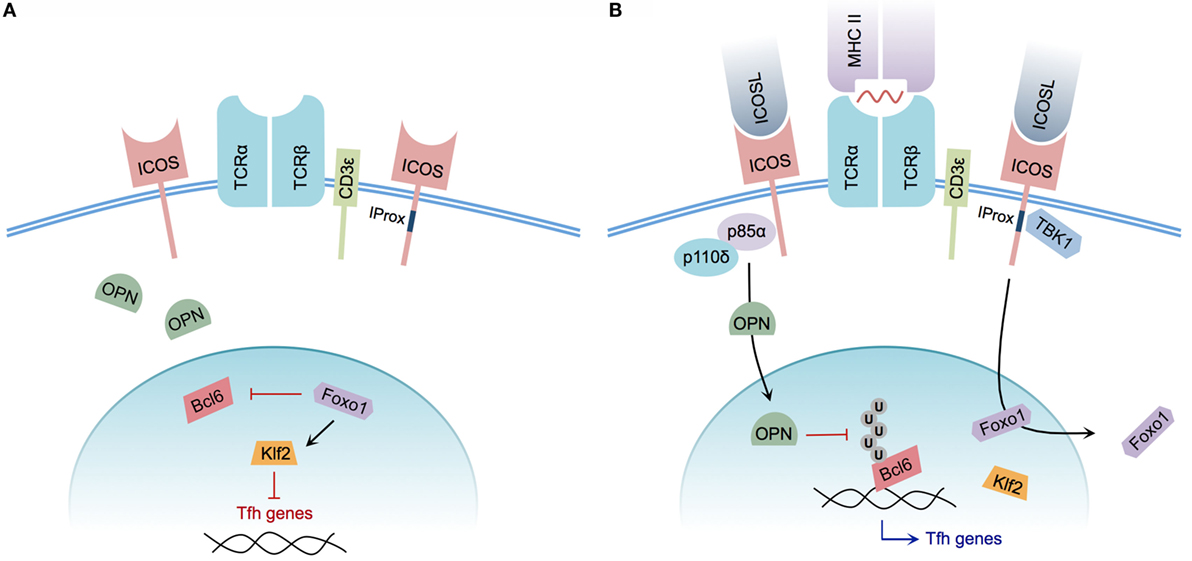
Figure 2. Model of ICOS-mediated Tfh cell differentiation. (A) CD4+ T cell without ICOS ligation. Osteopontin (OPN) is sequestered in the cytoplasm; Foxo1 represses Bcl6 while promoting Klf2-mediated repression of Tfh cell-associated genes. (B) CD4+ T cell with engaged TCR and ICOS co-stimulation. The p85α regulatory subunit of PI3K promotes OPN translocation to the nucleus, preventing ubiquitination and promoting the stability of Bcl6. Foxo1 is exported from the nucleus, relieving Klf2-mediated repression of Tfh cell genes. Integrated CD3ε and ICOS ligation induces recruitment of TBK1 to the intracellular IProx domain of ICOS, facilitating Tfh to GC Tfh transition.
In addition to the previously characterized roles of catalytic PI3K p110δ subunit recruitment to ICOS, the regulatory p85α subunit of PI3K plays additional roles in driving Tfh cell differentiation. Following ICOS ligation, p85α promotes translocation of osteopontin (OPN) (29), a regulatory protein expressed by activated T cells (93), to the cell nucleus. Within the nucleus, OPN promotes Bcl6 stability by disrupting Bcl6 ubiquitination, thereby reinforcing the Tfh phenotype (29).
Although ICOS-mediated PI3K activity plays an essential role in the process of Tfh cell differentiation, the newly identified ICOS signaling motif IProx has recently been linked to GC Tfh cell commitment. Using TCR Tg SMARTA CD4+ T cells expressing a mutant IProx motif within ICOS, Pedros and colleagues demonstrated that these Tg T cells were not defective in early Tfh cell differentiation at day 3 post-LCMV glycoprotein 61-conjugated KLH immunization. However, significant defects in PC, GC B cell, and gp61-KLH-specific isotype-switched Ab production were observed at day 10 post-immunization in mice receiving mutant SMARTA T cells. This IProx motif was further demonstrated to bind the serine–threonine kinase TBK1, and facilitate GC Tfh cell differentiation in concert with conventional ICOS–PI3K signaling. Interestingly, in vitro ligation of ICOS alone was insufficient to recruit TBK1 to the IProx motif. Successful ICOS–TBK1 interaction required simultaneous CD3ε and ICOS ligation, suggesting that both TCR and ICOS engagement are necessary for adoption of the GC Tfh phenotype (35). While TBK1 has been shown to be important for induction of type I interferon in innate immune cells (94, 95), its precise role and the additional members of the signaling pathway that promote GC Tfh cell differentiation remain to be defined.
Recently, following a transcriptome analysis designed to identify genes differentially regulated by ICOS, the expression of transcription factor Klf2 was found to be dramatically upregulated after disrupting ICOS signaling. In the absence of ICOS signaling, Klf2 served to decrease CXCR5 expression while increasing CCR7 expression, thereby reversing the Tfh cell phenotype. Ultimately, the decreased expression of Klf2 in Tfh cells was linked to ICOS-mediated repression of Foxo1 (80), a member of the FOXO family of transcription factors responsible for regulating T cell differentiation, as well as numerous other cellular processes (96). In agreement with initiation of the Tfh phenotype via Foxo1 repression, in vitro ICOS ligation mediated the nuclear to cytoplasmic translocation of Foxo1. Relief of Foxo1-mediated Bcl6 repression was subsequently shown to consequently promote Tfh cell differentiation. However, Foxo1-deficient CD4+ T cells were impaired in their ability to progress to a GC Tfh cell phenotype (81), indicating that ICOS signaling transiently inactivates Foxo1 to allow for initiation of Tfh cell differentiation, but that progression of Tfh cells to a GC Tfh cell phenotype is completed in a Foxo1-dependent manner.
ICOS and the Germinal Center Reaction
As ICOS co-stimulation plays a critical role in Tfh cell induction and adoption of the GC Tfh cell phenotype, research has also questioned the necessity of ICOS in the production of key GC products, such as MBCs and LLPCs. However, as the absence of ICOS or ICOSL in mice affects multiple steps in the development of T and B cell responses, these mutations cannot be assumed to only influence the GC itself (77). The impact of ICOS on the products of GC reactions, therefore, must be interpreted cautiously. Once thought to be derived solely from the GC reaction, recent evidence indicates that MBC production may not always require GC formation. For instance, early IgM+ MBC differentiation in response to TD antigens is largely GC independent (97). While there is evidence that some class-switched MBCs can be produced in the absence of a GC (98), the majority of class-switched MBC formation requires an active GC (99), indicating that ICOS could play an influential role in the production of isotype-switched MBCs. However, while inhibition of ICOS signaling in wild-type mice via α-ICOS Ab administration following NP-CG-alum immunization disrupted early GC formation, it did not impede generation or maintenance of NP-specific IgG1+ MBCs between days 10 and 70 post-immunization. However, NIP-binding MBCs from α-ICOS Ab-treated mice accumulated significantly fewer VH186.2 gene mutations relative to control mice between days 30 and 70 post-immunization (98). These studies provided three key insights into MBC production: first, they suggest that isotype-switched MBCs present in α-ICOS-treated mice were mostly likely derived independently of the GC. Second, that over time these early class-switched MBCs are replaced by MBCs derived from a GC reaction that have higher affinity for binding antigen. Finally, despite the lack of somatic hypermutation, the presence of IgG1+ NP-specific MBCs in the absence of ICOS signaling suggests that production of class-switched MBCs is not critically reliant on ICOS:ICOSL interactions.
Interestingly, in response to P. c. chabaudi AS infection, although Icos−/− mice produced fewer IgM− MBCs at week 9 post-infection relative to wild-type mice, class-switched MBC production was not fully abrogated in the absence of ICOS signaling (48). However, the source of these MBCs is still questionable. While the extrafollicular Ab response dominates the humoral response in P. c. chabaudi AS-infected Icos−/− mice, there is evidence that a small number of GCs are nevertheless formed in the absence of ICOS signaling. Therefore, the isotype-switched MBCs found in Icos−/− mice may have a GC origin. Meanwhile, during human systemic lupus erythematosus, ICOS:ICOSL interactions have been linked with aberrant, overproduction of class-switched MBCs (100). Also, human patients with deficiencies in ICOS have significantly reduced numbers of isotype-switched MBCs, but often have normal or slightly reduced numbers of mutated IgM+ MBCs (101). Together, these data indicate that early signaling events outside of ICOS:ICOSL interactions are sufficient to promote formation of both IgM+ and class-switched MBCs. Nevertheless, without ICOS signaling the number of MBCs generated is reduced, and the production of high-affinity isotype-switched MBCs is impaired, presumably due to disruption of the GC microenvironment.
By facilitating the process of Tfh cell development and promoting the subsequent GC reaction, ICOS:ICOSL signaling may also facilitate production of LLPCs. Interestingly, following DNP-KLH in alum immunization, Icos−/− mice were not defective in production of switched DNP-specific IgG or IgG1+ splenic and bone marrow-resident LLPCs at week 3 post-immunization when compared with wild-type mice. When re-challenged with DNP-KLH-alum at week 5 post-immunization, however, significantly fewer class-switched antibody-secreting cells (ASCs) were identified in the bone marrow of Icos−/− mice at day 5 post-secondary challenge (102), suggesting that secondary PC production is more reliant on ICOS:ICOSL signaling than primary ASC formation.
In the context of P. c. chabaudi AS infection, Icos−/− mice were not defective in the production of bone marrow-resident, isotype-switched parasite-specific (MSP-119) LLPCs at week 9 p.i., despite a decrease in the quantity and quality of serum MSP-119–IgG relative to wild-type mice at this time point. As Icos−/− mice developed dramatically fewer GCs throughout the course of P. c. chabaudi AS infection, it is striking that the number of bone marrow-resident ASCs was not significantly impaired (48), as LLPC production is thought to occur relatively late during the GC reaction (99). As an overall decrease in Ab avidity was noted in this study, it is quite possible that a large proportion of the parasite-specific LLPCs seen in the bone marrow of Icos−/− mice were derived from the extrafollicular response rather than the GC response, and had homed to the bone marrow relatively early after infection. It is also possible bone marrow-resident ASCs found in Icos−/− mice are not long-lived, and are instead continually replaced by splenic plasmablasts as the result of a perpetual extrafollicular response in the absence of GC formation.
Research has also questioned the role of ICOS in the secondary response to previously encountered antigens. Interestingly, disruption of ICOS signaling in wild-type mice via anti-ICOSL Ab during secondary (but not primary) challenge with heat-killed S. pneumoniae did not impact boosted PspA-specific IgG Ab titers, suggesting that ICOS is not critical for MBC activation (73). Similarly, following secondary influenza A infection, Icos−/− mice were defective in virus-specific IgG2a Ab production but were unimpaired in IgG1 production, harboring only a slight defect in total virus-neutralizing Ab at day 7 post-challenge relative to wild-type mice. Furthermore, splenocytes transferred from wild-type or Icos−/− mice previously infected with vesicular stomatitis virus (VSV) induced similar levels of virus-specific IgG in sub-lethally irradiated wild-type recipient mice at day 6 post-VSV infection (103).
However, wild-type mice immunized with phycoerythrin (PE) in alum produced more PE-specific B cells relative to Icos−/− mice following secondary PE challenge (102). Similarly, re-challenge of wild-type and Icosl−/− mice with NP-CG in alum resulted in higher antigen-specific IgM but lower IgG1 and IgG2a Ab production in Icosl−/− mice compared with wild-type mice (66). Although, when NP-primed B cells were isolated from mice treated with or without α-ICOS Ab at the onset of GC formation, no difference in reconstitution of the bone marrow anti-NP IgG1 response was seen upon re-challenge when these B cells were transferred into Rag-1−/− mice. However, the ratio of high-affinity NP-specific IgG1 ASCs in the bone marrow was significantly reduced in mice receiving NP-primed B cells from α-ICOS-treated mice (98). These results suggest that ICOS signaling can contribute to the secondary expansion of antigen-specific B cells, particularly by promoting production of isotype-switched high-affinity Abs. The divergent results reported here concerning the necessity of ICOS for MBC activation likely stem from dramatic differences in the source of antigen (i.e., active viral infection versus protein:adjuvant immunization). As alluded to previously, strong inflammatory stimuli, or in this case active viral infection, may overcome a necessity for ICOS signaling in secondary Ab responses.
Although certain aspects of the GC response may be more or less reliant on ICOS signaling depending upon the immunization or infection model in question, these data provide evidence of dysregulated GCs in the absence of ICOS signaling. These defective GC responses may stem from inadequate Tfh cell differentiation, as previously discussed, but could also arise from larger problems in GC architecture. For example, GC B cells isolated from Icos−/− mice immunized with NP-CGG exhibit lower expression of lymphotoxin αβ (LTαβ), a molecule upregulated on GC B cells and important in the regulation of the GC microenvironment (104). As LTαβ expression helps maintain a functional FDC network (in part by promoting FDC activation) (105), it is not surprising that impaired LTαβ expression was associated with diminished GC responses (104). Collectively, ICOS:ICOSL signaling promotes critical cellular processes necessary for the production of affinity-matured, class-switched MBCs and LLPCs, but may be less critically important for the initial generation of these cells per se.
ICOS and Memory T Cells
Inducible T cell co-stimulator–ICOSL interactions are important in determining the effector fate decisions of CD4+ T cells; however, defective ICOS signaling can also influence the production of memory CD4+ T cells. For instance, following priming of mice with Moloney murine leukemia virus envelope protein (H19-env)-pulsed DCs, Icos−/− CD4+ memory T cells did not expand as abundantly as wild-type memory T cells, despite being retained at similar numbers prior to H19-env immunization (102). Meanwhile, fewer central and effector CD4+ memory T cells were observed in Icosl−/− or α-ICOS Ab treated wild-type mice 4 weeks after Lm-2W1S infection (106) [attenuated L. monocytogenes secreting a chicken OVA-2W1S fusion protein (107)]. Similarly, long-term maintenance of M. tuberculosis-specific memory-like CD4+ T cells required ICOS expression (108). In a different study, mixed bone marrow chimeras revealed dramatically fewer Icos−/− LLOp:I-Ab-binding T cells (listeriolysin O-specific CD4+ T cells) adopted a central memory phenotype at day 20 post-Lm-2W (L. monocytogenes producing the 2W peptide and a fragment of listeriolysin O) infection relative to wild-type LLOp:I-Ab-binding T cells (109). Furthermore, human ICOS deficiency is associated with fewer circulating CD4+ memory T cells relative to healthy controls (110). Together, these data suggest ICOS promotes formation and maintenance of a stable population of CD4+ memory T cells, and in certain cases may influence their re-activation. However, whether memory CD4+ T cell ICOS expression has a direct effect on the ability of MBCs to differentiate into antibody-producing cells or participate in secondary GC reactions during a recall response remains to be determined.
Conclusion and Implications/Therapeutics
On the whole, ICOS co-stimulation supports a remarkable number of distinct processes during adaptive immune responses. By promoting the induction (15, 29, 71, 78–81), maintenance (29, 48, 79, 80, 87), and follicular homing of Tfh cells (86, 92), ICOS promotes TD Ab responses (37, 38, 65, 66, 71, 72, 76) and drives Ab affinity maturation in the GC reaction (15, 48, 65). Additionally, ICOS serves to enhance or dampen Th1 and Th2 inflammatory responses, depending on the given pathogen. Thus, because impaired ICOS signaling can yield such dramatically different disease outcomes in mice, the role of ICOS in regulating human disease states is of great importance.
In humans, homozygous ICOS deficiency results in common variable immunodeficiency (CVID) (111), a condition characterized by aberrantly low serum gammaglobulin concentration. As such, CVID typically results in frequent bacterial infections of the respiratory and digestive tracts (112). Perhaps not surprisingly, in cases of human ICOS deficiency, there is a dramatic reduction in the frequency of circulating CXCR5+ CD4+ T cells (113), the presence of which have been linked to active GCs in humans (114–116) and mice (114, 116). Collectively, defects in human ICOS expression correlate with defects in TD Ab production (111), circulating Tfh cells, and GC formation (113), thereby supporting a role for ICOS in human Tfh cell differentiation.
Although human Tfh cells express ICOS (84), a greater frequency of circulating Tfh cells isolated from systemic lupus erythematous (SLE) patients – a disease characterized by autoantibody production (117) – were found to express ICOS relative to healthy controls (116, 118). Interestingly, in vitro ICOS co-stimulation of peripheral T cells from patients with active SLE resulted in greatly enhanced IFN-γ production relative to normal controls, and ICOS ligation preferentially induced production of isotype-switched α-double-stranded DNA (α-dsDNA) Abs during co-culture with autologous B cells (118). Similarly, in the murine BXSB-Yaa model of SLE, splenic ICOS+ CD4+ T cell production of IL-21 was linked to renal disease and early mortality (119). In a different study, DC ICOSL expression was correlated with kidney nephritis and proteinuria, as well as kidney-infiltrating T cells, in MRL. Faslpr (lupus-prone) mice (120), further implicating ICOS in promoting tissue inflammation during active SLE. In fact, mutation of roquin – a ubiquitin ligase (121) responsible for post-transcriptionally regulating ICOS mRNA (122, 123) – results in ICOS over-expression on CD4+ T cells, thereby promoting spontaneous Tfh cell induction, GC formation, and autoantibody production. Interestingly, unimmunized sanroque roquin-mutant mice (Rc3h1san/san) develop an SLE-like disease, characterized by hypergammaglobulinemia and aberrant IL-21 production (121). As a whole, by way of enhancing autoantibody formation and promoting organ inflammation, these data strongly suggest that ICOS plays a direct role in promoting SLE disease progression in mice and humans.
Not surprisingly, therapeutic interventions designed to disrupt ICOS:ICOSL signaling have been evaluated in mice and humans. The human α-ICOSL antibody AMG 557, for example, proved efficacious in diminishing isotype-switched Ab production, but not IgM production, in SLE patients challenged nasally with KLH in a phase I clinical trial (124). AMG 557 was similarly effective in reducing the production of KLH-specific IgG Abs following intra-dermal immunization with KLH (followed by boost 4 weeks later) in patients diagnosed with mild SLE (125). The results of these studies are promising, and similar success has been achieved in murine model systems. For instance, treatment with α-ICOSL Ab minimized disease symptoms and decreased α-dsDNA Ab production in the NZB/NZW F1 model of SLE. Furthermore, in a model of rheumatoid arthritis, α-ICOSL Ab administration suppressed the production of collagen-specific isotype-switched Abs (78). Although these studies indicate neutralization of ICOSL can help ameliorate symptoms associated with SLE, the therapeutic potential of disrupting ICOS signaling may extend beyond TD humoral responses.
Recently, characterization of type 2 innate lymphoid cells (ILC2s), an innate immune cell capable of secreting Th2 type cytokines (126), has revealed that ILC2s express ICOS and ICOSL. Researchers identified a critical role for ICOS:ICOSL signaling in the induction of ILC2-mediated cytokine production that led to airway hyperreactivity (AHR), an indicator of asthma. Interestingly, transfer of human ILC2s into humanized Rag-deficient mice followed by disruption of ICOS signaling via α-ICOSL Ab treatment yielded decreased evidence of IL-33-induced AHR relative to mice receiving isotype control Ab (127). In part, this study suggests novel therapeutic options for AHR could be pioneered with α-ICOS or α-ICOSL Ab treatment.
Along a similar line, α-ICOS Ab therapy has been shown to improve cardiac allograft survival in rats (128–130). However, disruption of ICOS signaling following kidney transplantation in non-human primates was not efficacious in preventing graft rejection (131). Together, these data suggest that ICOS blockade may be more beneficial in combination with traditional drugs (i.e., cyclosporine) used to ameliorate graft-versus-host disease (GVHD). Future studies seeking to gain additional insight regarding the role of ICOS in GVHD could provide efficacious tools to treat chronic graft rejection in humans.
Modulation of ICOS signaling has the potential to mitigate disease severity for a number of human autoimmune disorders. Beyond its key role in isotype-switched Ab production, ICOS can also promote GVHD and potentiate symptoms associated with AHR. Although monoclonal Ab therapy designed to disrupt ICOS:ICOSL interactions appears to be a promising, efficacious treatment, the potential for unintended side effects related to global disruption of TD Ab responses may be unavoidable. Thus, future research concerning how ICOS:ICOSL signals alternatively facilitate Ab production, on the one hand, and T helper cell polarization, on the other, may allow tailoring of treatment options to target a specific disease modality with minimal off-target complications.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National Institutes of Health Grant P20-GM103625-Project 3 (JS) and AI116653 (JS).
References
1. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol (2002) 2:116–26. doi:10.1038/nri727
2. Gmünder H, Lesslauer W. A 45-kDa human T-cell membrane glycoprotein functions in the regulation of cell proliferative responses. Eur J Biochem (1984) 142:153–60. doi:10.1111/j.1432-1033.1984.tb08263.x
3. Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, et al. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci U S A (1989) 86:1333–7. doi:10.1073/pnas.86.4.1333
4. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13. doi:10.1016/1074-7613(94)90071-X
5. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity (2016) 44:973–88. doi:10.1016/j.immuni.2016.04.020
6. Attanasio J, Wherry EJ. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity (2016) 44:1052–68. doi:10.1016/j.immuni.2016.04.022
7. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44:955–72. doi:10.1016/j.immuni.2016.05.002
8. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Anagnostopoulos I, Kroczek RA. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397:263–6. doi:10.1038/16717
9. Mages HW, Hutloff A, Heuck C, Büchner K, Himmelbauer H, Oliveri F, et al. Molecular cloning and characterization of murine ICOS and identification of B7h as ICOS ligand. Eur J Immunol (2000) 30:1040–7. doi:10.1002/(SICI)1521-4141(200004)30:4<1040::AID-IMMU1040>3.0.CO;2-6
10. Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol (2003) 3:544–56. doi:10.1038/nri1131
11. McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol (2000) 165:5035–40. doi:10.4049/jimmunol.165.9.5035
12. Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature (1999) 402:827–32. doi:10.1038/45582
13. Watanabe M, Hara Y, Tanabe K, Toma H, Abe R. A distinct role for ICOS-mediated co-stimulatory signaling in CD4+ and CD8+ T cell subsets. Int Immunol (2005) 17:269–78. doi:10.1093/intimm/dxh206
14. Arimura Y, Kato H, Dianzani U, Okamoto T, Kamekura S, Buonfiglio D, et al. A co-stimulatory molecule on activated T cells, H4/ICOS, delivers specific signals in T(h) cells and regulates their responses. Int Immunol (2002) 14:555–66. doi:10.1093/intimm/dxf022
15. Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci U S A (2009) 106:20371–6. doi:10.1073/pnas.0911573106
16. Ling V, Wu PW, Finnerty HF, Bean KM, Spaulding V, Fouser LA, et al. Cutting edge: identification of GL50, a novel B7-like protein that functionally binds to ICOS receptor. J Immunol (2000) 164:1653–7. doi:10.4049/jimmunol.164.4.1653
17. Aicher A, Hayden-Ledbetter M, Brady WA, Pezzutto A, Richter G, Magaletti D, et al. Characterization of human inducible costimulator ligand expression and function. J Immunol (2000) 164:4689–96. doi:10.4049/jimmunol.164.9.4689
18. Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, et al. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med (1994) 180:2049–58. doi:10.1084/jem.180.6.2049
19. Watanabe M, Takagi Y, Kotani M, Hara Y, Inamine A, Hayashi K, et al. Down-regulation of ICOS ligand by interaction with ICOS functions as a regulatory mechanism for immune responses. J Immunol (2008) 180:5222–34. doi:10.4049/jimmunol.180.8.5222
20. Hu H, Wu X, Jin W, Chang M, Cheng X, Sun S-C. Noncanonical NF-kappaB regulates inducible costimulator (ICOS) ligand expression and T follicular helper cell development. Proc Natl Acad Sci U S A (2011) 108:12827–32. doi:10.1073/pnas.1105774108
21. Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity (1999) 11:423–32. doi:10.1016/S1074-7613(00)80117-X
22. Yoshinaga SK, Zhang M, Pistillo J, Horan T, Khare SD, Miner K, et al. Characterization of a new human B7-related protein: B7RP-1 is the ligand to the co-stimulatory protein ICOS. Int Immunol (2000) 12:1439–47. doi:10.1093/intimm/12.10.1439
23. Khayyamian S, Hutloff A, Büchner K, Gräfe M, Henn V, Kroczek RA, et al. ICOS-ligand, expressed on human endothelial cells, costimulates Th1 and Th2 cytokine secretion by memory CD4+ T cells. Proc Natl Acad Sci U S A (2002) 99:6198–203. doi:10.1073/pnas.092576699
24. Qian X, Agematsu K, Freeman GJ, Tagawa Y-I, Sugane K, Hayashi T. The ICOS-ligand B7-H2, expressed on human type II alveolar epithelial cells, plays a role in the pulmonary host defense system. Eur J Immunol (2006) 36:906–18. doi:10.1002/eji.200535253
25. Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U S A (2002) 99:11790–5. doi:10.1073/pnas.162359999
26. Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity (2000) 13:95–105. doi:10.1016/S1074-7613(00)00011-X
27. Fos C, Salles A, Lang V, Carrette F, Audebert S, Pastor S, et al. ICOS ligation recruits the p50alpha PI3K regulatory subunit to the immunological synapse. J Immunol (2008) 181:1969–77. doi:10.4049/jimmunol.181.3.1969
28. Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res (2012) 18:5856–64. doi:10.1158/1078-0432.CCR-12-0662
29. Leavenworth JW, Verbinnen B, Yin J, Huang H, Cantor H. A p85α-osteopontin axis couples the receptor ICOS to sustained Bcl-6 expression by follicular helper and regulatory T cells. Nat Immunol (2015) 16:96–106. doi:10.1038/ni.3050
30. Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol (2003) 171:166–74. doi:10.4049/jimmunol.171.1.166
31. Harada Y, Ohgai D, Watanabe R, Okano K, Koiwai O, Tanabe K, et al. A single amino acid alteration in cytoplasmic domain determines IL-2 promoter activation by ligation of CD28 but not inducible costimulator (ICOS). J Exp Med (2003) 197:257–62. doi:10.1084/jem.20021305
32. Inukai K, Funaki M, Anai M, Ogihara T, Katagiri H, Fukushima Y, et al. Five isoforms of the phosphatidylinositol 3-kinase regulatory subunit exhibit different associations with receptor tyrosine kinases and their tyrosine phosphorylations. FEBS Lett (2001) 490:32–8. doi:10.1016/S0014-5793(01)02132-9
33. Li J, Heinrichs J, Leconte J, Haarberg K, Semple K, Liu C, et al. Phosphatidylinositol 3-kinase-independent signaling pathways contribute to ICOS-mediated T cell costimulation in acute graft-versus-host disease in mice. J Immunol (2013) 191:200–7. doi:10.4049/jimmunol.1203485
34. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LMC, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol (2010) 185:4042–52. doi:10.4049/jimmunol.1001730
35. Pedros C, Zhang Y, Hu JK, Choi YS, Canonigo-Balancio AJ, Yates JR, et al. A TRAF-like motif of the inducible costimulator ICOS controls development of germinal center TFH cells via the kinase TBK1. Nat Immunol (2016) 17:825–33. doi:10.1038/ni.3463
36. Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene (1999) 18:6867–74. doi:10.1038/sj.onc.1203219
37. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature (2001) 409:97–101. doi:10.1038/35051100
38. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS is essential for effective T-helper-cell responses. Nature (2001) 409:105–9. doi:10.1038/35051113
39. Wilson EH, Zaph C, Mohrs M, Welcher A, Siu J, Artis D, et al. B7RP-1-ICOS interactions are required for optimal infection-induced expansion of CD4+ Th1 and Th2 responses. J Immunol (2006) 177:2365–72. doi:10.4049/jimmunol.177.4.2365
40. Smith KM, Brewer JM, Webb P, Coyle AJ, Gutierrez-Ramos C, Garside P. Inducible costimulatory molecule-B7-related protein 1 interactions are important for the clonal expansion and B cell helper functions of naive, Th1, and Th2 T cells. J Immunol (2003) 170:2310–5. doi:10.4049/jimmunol.170.5.2310
41. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science (1976) 193:1007–8. doi:10.1126/science.181845
42. Witsch EJ, Peiser M, Hutloff A, Büchner K, Dorner BG, Jonuleit H, et al. ICOS and CD28 reversely regulate IL-10 on re-activation of human effector T cells with mature dendritic cells. Eur J Immunol (2002) 32:2680–6. doi:10.1002/1521-4141(200209)32:9<2680::AID-IMMU2680>3.0.CO;2-6
43. Nouailles G, Day TA, Kuhlmann S, Loewe D, Dorhoi A, Gamradt P, et al. Impact of inducible co-stimulatory molecule (ICOS) on T-cell responses and protection against Mycobacterium tuberculosis infection. Eur J Immunol (2011) 41:981–91. doi:10.1002/eji.201040608
44. Kadkhoda K, Wang S, Joyee AG, Fan Y, Yang J, Yang X. Th1 cytokine responses fail to effectively control Chlamydia lung infection in ICOS ligand knockout mice. J Immunol (2010) 184:3780–8. doi:10.4049/jimmunol.0901384
45. Gao X, Gigoux M, Yang J, Leconte J, Yang X, Suh W-K. Anti-chlamydial Th17 responses are controlled by the inducible costimulator partially through phosphoinositide 3-kinase signaling. PLoS One (2012) 7:e52657. doi:10.1371/journal.pone.0052657
46. Marks E, Verolin M, Stensson A, Lycke N. Differential CD28 and inducible costimulatory molecule signaling requirements for protective CD4+ T-cell-mediated immunity against genital tract Chlamydia trachomatis infection. Infect Immun (2007) 75:4638–47. doi:10.1128/IAI.00465-07
47. Rutitzky LI, Ozkaynak E, Rottman JB, Stadecker MJ. Disruption of the ICOS-B7RP-1 costimulatory pathway leads to enhanced hepatic immunopathology and increased gamma interferon production by CD4 T cells in murine schistosomiasis. Infect Immun (2003) 71:4040–4. doi:10.1128/IAI.71.7.4040-4044.2003
48. Wikenheiser DJ, Ghosh D, Kennedy B, Stumhofer JS. The costimulatory molecule ICOS regulates host Th1 and follicular Th cell differentiation in response to Plasmodium chabaudi chabaudi AS Infection. J Immunol (2016) 196:778–91. doi:10.4049/jimmunol.1403206
49. Vidric M, Bladt AT, Dianzani U, Watts TH. Role for inducible costimulator in control of Salmonella enterica serovar Typhimurium infection in mice. Infect Immun (2006) 74:1050–61. doi:10.1128/IAI.74.2.1050-1061.2006
50. Mittrücker H-W, Kursar M, Köhler A, Yanagihara D, Yoshinaga SK, Kaufmann SHE. Inducible costimulator protein controls the protective T cell response against Listeria monocytogenes. J Immunol (2002) 169:5813–7. doi:10.4049/jimmunol.169.10.5813
51. Villegas EN, Lieberman LA, Mason N, Blass SL, Zediak VP, Peach R, et al. A role for inducible costimulator protein in the CD28-independent mechanism of resistance to Toxoplasma gondii. J Immunol (2002) 169:937–43. doi:10.4049/jimmunol.169.2.937
52. Kopf M, Coyle AJ, Schmitz N, Barner M, Oxenius A, Gallimore A, et al. Inducible costimulator protein (ICOS) controls T helper cell subset polarization after virus and parasite infection. J Exp Med (2000) 192:53–61. doi:10.1084/jem.192.1.53
53. Greenwald RJ, McAdam AJ, Van der Woude D, Satoskar AR, Sharpe AH. Cutting edge: inducible costimulator protein regulates both Th1 and Th2 responses to cutaneous leishmaniasis. J Immunol (2002) 168:991–5. doi:10.4049/jimmunol.168.3.991
54. Vermeiren J, Ceuppens JL, Van Ghelue M, Witters P, Bullens D, Mages HW, et al. Human T cell activation by costimulatory signal-deficient allogeneic cells induces inducible costimulator-expressing anergic T cells with regulatory cell activity. J Immunol (2004) 172:5371–8. doi:10.4049/jimmunol.172.9.5371
55. Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med (2002) 8:1024–32. doi:10.1038/nm745
56. Löhning M, Hutloff A, Kallinich T, Mages HW, Bonhagen K, Radbruch A, et al. Expression of ICOS in vivo defines CD4+ effector T cells with high inflammatory potential and a strong bias for secretion of interleukin 10. J Exp Med (2003) 197:181–93. doi:10.1084/jem.20020632
57. Shalaby KH, Jo T, Nakada E, Allard-Coutu A, Tsuchiya K, Hirota N, et al. ICOS-expressing CD4 T cells induced via TLR4 in the nasal mucosa are capable of inhibiting experimental allergic asthma. J Immunol (2012) 189:2793–804. doi:10.4049/jimmunol.1201194
58. Busse M, Krech M, Meyer-Bahlburg A, Hennig C, Hansen G. ICOS mediates the generation and function of CD4+CD25+Foxp3+ regulatory T cells conveying respiratory tolerance. J Immunol (2012) 189:1975–82. doi:10.4049/jimmunol.1103581
59. Redpath SA, van der Werf N, Cervera AM, MacDonald AS, Gray D, Maizels RM, et al. ICOS controls Foxp3(+) regulatory T-cell expansion, maintenance and IL-10 production during helminth infection. Eur J Immunol (2013) 43:705–15. doi:10.1002/eji.201242794
60. Agrewala JN, Kumar B, Vohra H. Potential role of B7-1 and CD28 molecules in immunosuppression in leprosy. Clin Exp Immunol (1998) 111:56–63. doi:10.1046/j.1365-2249.1998.00463.x
61. Bonato VL, Medeiros AI, Lima VM, Dias AR, Faccioliti LH, Silva CL. Downmodulation of CD18 and CD86 on macrophages and VLA-4 on lymphocytes in experimental tuberculosis. Scand J Immunol (2001) 54:564–73. doi:10.1046/j.1365-3083.2001.00996.x
62. Yao T, Mecsas J, Healy JI, Falkow S, Chien Y. Suppression of T and B lymphocyte activation by a Yersinia pseudotuberculosis virulence factor, yopH. J Exp Med (1999) 190:1343–50. doi:10.1084/jem.190.9.1343
63. Chaudhry A, Das SR, Hussain A, Mayor S, George A, Bal V, et al. The Nef protein of HIV-1 induces loss of cell surface costimulatory molecules CD80 and CD86 in APCs. J Immunol (2005) 175:4566–74. doi:10.4049/jimmunol.175.7.4566
64. Zhou Z, Hoebe K, Du X, Jiang Z, Shamel L, Beutler B. Antagonism between MyD88- and TRIF-dependent signals in B7RP-1 up-regulation. Eur J Immunol (2005) 35:1918–27. doi:10.1002/eji.200525971
65. Mak TW, Shahinian A, Yoshinaga SK, Wakeham A, Boucher L-M, Pintilie M, et al. Costimulation through the inducible costimulator ligand is essential for both T helper and B cell functions in T cell-dependent B cell responses. Nat Immunol (2003) 4:765–72. doi:10.1038/ni947
66. Wong S-C, Oh E, Ng C-H, Lam K-P. Impaired germinal center formation and recall T-cell-dependent immune responses in mice lacking the costimulatory ligand B7-H2. Blood (2003) 102:1381–8. doi:10.1182/blood-2002-08-2416
67. Scales HE, Ierna MX, Gutierrez-Ramos J-C, Coyle AJ, Garside P, Lawrence CE. Effect of inducible costimulator blockade on the pathological and protective immune responses induced by the gastrointestinal helminth Trichinella spiralis. Eur J Immunol (2004) 34:2854–62. doi:10.1002/eji.200324364
68. Miyahira Y, Akiba H, Ogawa S-H, Ishi T, Watanabe S, Kobayashi S, et al. Involvement of ICOS-B7RP-1 costimulatory pathway in the regulation of immune responses to Leishmania major and Nippostrongylus brasiliensis infections. Immunol Lett (2003) 89:193–9. doi:10.1016/S0165-2478(03)00137-8
69. Loke P, Zang X, Hsuan L, Waitz R, Locksley RM, Allen JE, et al. Inducible costimulator is required for type 2 antibody isotype switching but not T helper cell type 2 responses in chronic nematode infection. Proc Natl Acad Sci U S A (2005) 102:9872–7. doi:10.1073/pnas.0503961102
70. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
71. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol (2005) 175:2340–8. doi:10.4049/jimmunol.175.4.2340
72. Dong C, Temann UA, Flavell RA. Cutting edge: critical role of inducible costimulator in germinal center reactions. J Immunol (2001) 166:3659–62. doi:10.4049/jimmunol.166.6.3659
73. Chen Q, Cannons JL, Paton JC, Akiba H, Schwartzberg PL, Snapper CM. A novel ICOS-independent, but CD28- and SAP-dependent, pathway of T cell-dependent, polysaccharide-specific humoral immunity in response to intact Streptococcus pneumoniae versus pneumococcal conjugate vaccine. J Immunol (2008) 181:8258–66. doi:10.4049/jimmunol.181.12.8258
74. Toellner K-M, Jenkinson WE, Taylor DR, Khan M, Sze DM-Y, Sansom DM, et al. Low-level hypermutation in T cell-independent germinal centers compared with high mutation rates associated with T cell-dependent germinal centers. J Exp Med (2002) 195:383–9. doi:10.1084/jem.20011112
75. de Vinuesa CG, Cook MC, Ball J, Drew M, Sunners Y, Cascalho M, et al. Germinal centers without T cells. J Exp Med (2000) 191:485–94. doi:10.1084/jem.191.3.485
76. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. ICOS is critical for CD40-mediated antibody class switching. Nature (2001) 409:102–5. doi:10.1038/35051107
77. Good-Jacobson KL, Shlomchik MJ. Plasticity and heterogeneity in the generation of memory B cells and long-lived plasma cells: the influence of germinal center interactions and dynamics. J Immunol (2010) 185:3117–25. doi:10.4049/jimmunol.1001155
78. Hu Y-L, Metz DP, Chung J, Siu G, Zhang M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J Immunol (2009) 182:1421–8. doi:10.4049/jimmunol.182.3.1421
79. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi:10.1016/j.immuni.2011.03.023
80. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz L-J, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Krüppel-like factor 2. J Exp Med (2015) 212:217–33. doi:10.1084/jem.20141432
81. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai C-Y, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity (2015) 42:239–51. doi:10.1016/j.immuni.2015.01.017
82. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325:1006–10. doi:10.1126/science.1175870
83. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325:1001–5. doi:10.1126/science.1176676
84. Crotty S. T Follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41:529–42. doi:10.1016/j.immuni.2014.10.004
85. Shin C, Han J-A, Koh H, Choi B, Cho Y, Jeong H, et al. CD8α(-) dendritic cells induce antigen-specific T follicular helper cells generating efficient humoral immune responses. Cell Rep (2015) 11:1929–40. doi:10.1016/j.celrep.2015.05.042
86. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517:214–8. doi:10.1038/nature13803
87. Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, et al. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol (2011) 187:1091–5. doi:10.4049/jimmunol.1100853
88. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29:127–37. doi:10.1016/j.immuni.2008.06.001
89. Portugal S, Pierce SK, Crompton PD. Young lives lost as B cells falter: what we are learning about antibody responses in malaria. J Immunol (2013) 190:3039–46. doi:10.4049/jimmunol.1203067
90. Weinstein JS, Bertino SA, Hernandez SG, Poholek AC, Teplitzky TB, Nowyhed HN, et al. B cells in T follicular helper cell development and function: separable roles in delivery of ICOS ligand and antigen. J Immunol (2014) 192:3166–79. doi:10.4049/jimmunol.1302617
91. Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity (2010) 33:241–53. doi:10.1016/j.immuni.2010.07.015
92. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature (2013) 496:523–7. doi:10.1038/nature12058
93. Rittling SR, Singh R. Osteopontin in immune-mediated diseases. J Dent Res (2015) 94:1638–45. doi:10.1177/0022034515605270
94. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol (2003) 4:491–6. doi:10.1038/ni921
95. Sharma S, Tenoever BR, Grandvaux N, Zhou G-P, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science (2003) 300:1148–51. doi:10.1126/science.1081315
96. Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol (2012) 12:649–61. doi:10.1038/nri3278
97. Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med (2012) 209:597–606. doi:10.1084/jem.20111696
98. Inamine A, Takahashi Y, Baba N, Miyake K, Tokuhisa T, Takemori T, et al. Two waves of memory B-cell generation in the primary immune response. Int Immunol (2005) 17:581–9. doi:10.1093/intimm/dxh241
99. Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity (2016) 44:116–30. doi:10.1016/j.immuni.2015.12.004
100. Hutloff A, Büchner K, Reiter K, Baelde HJ, Odendahl M, Jacobi A, et al. Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis Rheum (2004) 50:3211–20. doi:10.1002/art.20519
101. Tangye SG, Good KL. Human IgM+CD27+ B cells: memory B cells or “memory” B cells? J Immunol (2007) 179:13–9. doi:10.4049/jimmunol.179.1.13
102. Mahajan S, Cervera A, MacLeod M, Fillatreau S, Perona-Wright G, Meek S, et al. The role of ICOS in the development of CD4 T cell help and the reactivation of memory T cells. Eur J Immunol (2007) 37:1796–808. doi:10.1002/eji.200636661
103. Bertram EM, Tafuri A, Shahinian A, Chan VSF, Hunziker L, Recher M, et al. Role of ICOS versus CD28 in antiviral immunity. Eur J Immunol (2002) 32:3376–85. doi:10.1002/1521-4141(200212)32:12<3376::AID-IMMU3376>3.0.CO;2-Y
104. Vu F, Dianzani U, Ware CF, Mak T, Gommerman JL. ICOS, CD40, and lymphotoxin beta receptors signal sequentially and interdependently to initiate a germinal center reaction. J Immunol (2008) 180:2284–93. doi:10.4049/jimmunol.180.4.2284
105. Myers RC, King RG, Carter RH, Justement LB. Lymphotoxin α1β2 expression on B cells is required for follicular dendritic cell activation during the germinal center response. Eur J Immunol (2013) 43:348–59. doi:10.1002/eji.201242471
106. Marriott CL, Carlesso G, Herbst R, Withers DR. ICOS is required for the generation of both central and effector CD4(+) memory T-cell populations following acute bacterial infection. Eur J Immunol (2015) 45:1706–15. doi:10.1002/eji.201445421
107. Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP, et al. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol (2010) 11:83–9. doi:10.1038/ni.1826
108. Moguche AO, Shafiani S, Clemons C, Larson RP, Dinh C, Higdon LE, et al. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J Exp Med (2015) 212:715–28. doi:10.1084/jem.20141518
109. Pepper M, Pagán AJ, Igyártó BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity (2011) 35:583–95. doi:10.1016/j.immuni.2011.09.009
110. Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, et al. Impaired CD4 and CD8 effector function and decreased memory T cell populations in ICOS-deficient patients. J Immunol (2009) 182:5515–27. doi:10.4049/jimmunol.0803256
111. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol (2003) 4:261–8. doi:10.1038/ni902
112. Di Renzo M, Pasqui AL, Auteri A. Common variable immunodeficiency: a review. Clin Exp Med (2004) 3:211–7. doi:10.1007/s10238-004-0027-2
113. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol (2006) 177:4927–32. doi:10.4049/jimmunol.177.7.4927
114. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39:770–81. doi:10.1016/j.immuni.2013.09.007
115. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1(+)CXCR3(-)CXCR5(+) memory Tfh Cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39:758–69. doi:10.1016/j.immuni.2013.08.031
116. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum (2010) 62:234–44. doi:10.1002/art.25032
117. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol (2015) 11:329–41. doi:10.1038/nrneph.2015.33
118. Kawamoto M, Harigai M, Hara M, Kawaguchi Y, Tezuka K, Tanaka M, et al. Expression and function of inducible co-stimulator in patients with systemic lupus erythematosus: possible involvement in excessive interferon-gamma and anti-double-stranded DNA antibody production. Arthritis Res Ther (2006) 8:R62. doi:10.1186/ar1928
119. Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A (2009) 106:1518–23. doi:10.1073/pnas.0807309106
120. Teichmann LL, Cullen JL, Kashgarian M, Dong C, Craft J, Shlomchik MJ. Local triggering of the ICOS coreceptor by CD11c(+) myeloid cells drives organ inflammation in lupus. Immunity (2015) 42:552–65. doi:10.1016/j.immuni.2015.02.015
121. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
122. Yu D, Tan AH-M, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature (2007) 450:299–303. doi:10.1038/nature06253
123. Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol (2010) 11:725–33. doi:10.1038/ni.1902
124. Sullivan B, Tsuji WH, Chindalore VL, Geppert TD, Rudinskaya A, Pardo P, et al. Administration of AMG 557, a human Anti-B7RP-1 (ICOSL) antibody, leads to the selective inhibition of Anti-KLH IgG responses in subjects with SLE: results of a phase 1 randomized, double-blind, placebo-controlled, sequential, rising, multiple-dose study [abstract]. Arthritis Rheum (2013) 65:1743.
125. Sullivan BA, Tsuji W, Kivitz A, Peng J, Arnold GE, Boedigheimer MJ, et al. Inducible T-cell co-stimulator ligand (ICOSL) blockade leads to selective inhibition of anti-KLH IgG responses in subjects with systemic lupus erythematosus. Lupus Sci Med (2016) 3:e000146. doi:10.1136/lupus-2016-000146
126. Walker JA, Barlow JL, McKenzie ANJ. Innate lymphoid cells – how did we miss them? Nat Rev Immunol (2013) 13:75–87. doi:10.1038/nri3349
127. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity (2015) 42:538–51. doi:10.1016/j.immuni.2015.02.007
128. Guillonneau C, Aubry V, Renaudin K, Séveno C, Usal C, Tezuka K, et al. Inhibition of chronic rejection and development of tolerogenic T cells after ICOS-ICOSL and CD40-CD40L co-stimulation blockade. Transplantation (2005) 80:546–54. doi:10.1097/01.TP.0000165429.57421.D6
129. Pan X-C, Guo L, Deng Y-B, Naruse K, Kimura H, Sugawara Y, et al. Further study of anti-ICOS immunotherapy for rat cardiac allograft rejection. Surg Today (2008) 38:815–25. doi:10.1007/s00595-007-3734-y
130. Guo L, Fujino M, Kimura H, Funeshima N, Kitazawa Y, Harihara Y, et al. Simultaneous blockade of co-stimulatory signals, CD28 and ICOS, induced a stable tolerance in rat heart transplantation. Transpl Immunol (2003) 12:41–8. doi:10.1016/S0966-3274(03)00016-9
Keywords: T cell activation, co-stimulatory molecules, antibody production, parasitic and bacterial infection
Citation: Wikenheiser DJ and Stumhofer JS (2016) ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 7:304. doi: 10.3389/fimmu.2016.00304
Received: 06 May 2016; Accepted: 27 July 2016;
Published: 10 August 2016
Edited by:
Rene De Waal Malefyt, Merck Research Laboratories, Palo Alto, USAReviewed by:
António Gil Castro, University of Minho, PortugalKarin Schilbach, University of Tübingen, Germany
Copyright: © 2016 Wikenheiser and Stumhofer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason S. Stumhofer, anN0dW1ob2ZlckB1YW1zLmVkdQ==