Abstract
Neutrophil extracellular traps (NETs) arise from the release of granular and nuclear contents of neutrophils in the extracellular space in response to different classes of microorganisms, soluble factors, and host molecules. NETs are composed by decondensed chromatin fibers coated with antimicrobial granular and cytoplasmic proteins, such as myeloperoxidase, neutrophil elastase (NE), and α-defensins. Besides being expressed on NET fibers, NE and MPO also regulate NET formation. Furthermore, histone deimination by peptidylarginine deiminase 4 (PAD4) is a central step to NET formation. NET formation has been widely demonstrated to be an effective mechanism to fight against invading microorganisms, as deficiency in NET release or dismantling NET backbone by bacterial DNases renders the host susceptible to infections. Therefore, the primary role of NETs is to prevent microbial dissemination, avoiding overwhelming infections. However, an excess of NET formation has a dark side. The pathogenic role of NETs has been described for many human diseases, infectious and non-infectious. The detrimental effect of excessive NET release is particularly important to lung diseases, because NETs can expand more easily in the pulmonary alveoli, causing lung injury. Moreover, NETs and its associated molecules are able to directly induce epithelial and endothelial cell death. In this regard, massive NET formation has been reported in several pulmonary diseases, including asthma, chronic obstructive pulmonary disease, cystic fibrosis, respiratory syncytial virus bronchiolitis, influenza, bacterial pneumonia, and tuberculosis, among others. Thus, NET formation must be tightly regulated in order to avoid NET-mediated tissue damage. Recent development of therapies targeting NETs in pulmonary diseases includes DNA disintegration with recombinant human DNase, neutralization of NET proteins, with anti-histone antibodies and protease inhibitors. In this review, we summarize the recent knowledge on the pathophysiological role of NETs in pulmonary diseases as well as some experimental and clinical approaches to modulate their detrimental effects.
Introduction
Neutrophils are key players in microbial killing, being the first immune cells to achieve the site of injury or infection (1). Therefore, neutrophils act as the first line of defense against microorganisms through phagocytosis, release of reactive oxygen species (ROS), and degranulation (2). Aside from these traditional mechanisms, neutrophils are also able to extrude DNA lattices, called neutrophil extracellular traps (NETs), which entrap and facilitate the killing of bacteria, fungi, protozoa, and even viruses (3–8). NETs are composed of decondensed chromatin fibers coated with antimicrobial proteins, such as histones, neutrophil elastase (NE), myeloperoxidase (MPO), and α-defensins (3, 7). Besides being expressed on NET fibers, NE and MPO also regulate NET formation (9). Differently, the participation of NADPH oxidase-derived ROS in NET release seems to be a matter of time of stimulation. While ROS are required to NET generation in time points beyond 1 h after stimulation (10, 11), a very rapid process (5–30 min) of NET extrusion has been reported to be ROS-independent in response to Staphylococcus aureus and Candida albicans (12, 13). Furthermore, histone deimination by peptidylarginine deiminase 4 (PAD4) is a central step to NET formation (14). Additionally, the release of these DNA threads requires autophagy and activation of p38 MAPK and the Raf-MEK-ERK signaling pathways (15–17). However, it is important to keep in mind that the specific cell components and signaling cascades may vary depending on the stimulus (18).
The primary role of NETs is to prevent microbial dissemination because of its stringy structure, and to kill pathogens due to the high local concentrations of antimicrobial molecules (19). However, these attributes make NETs potentially detrimental to the host. The pathogenic role of NETs has been described for many human diseases, infectious and non-infectious (20), being particularly important to lung diseases. Netting neutrophils in the lung tissue are able to disturb microcirculation and NETs produced in the pulmonary alveoli can expand easily, filling the lungs, as is the case for cystic fibrosis (CF) (19, 21). Therefore, NET formation must be tightly regulated. In this review, we summarize the recent knowledge on the pathophysiological role of NETs in pulmonary diseases as well as some experimental and clinical approaches to modulate their detrimental effects.
Cystic Fibrosis
Cystic fibrosis is a fatal hereditary disorder resulting from mutations in the CF transmembrane conductance regulator (CFTR) anion channel (22). This anion channel is responsible for the transport of chloride ions across the epithelial layer of the airways, which is necessary for the production of thin, freely flowing mucus. Therefore, the lungs of CF patients produce large amounts of thick mucus, leading to an obstruction of the airways and colonization by bacteria (23). Typically, CF infants are rapidly colonized by Haemophilus influenzae or S. aureus, or both. Over time, Pseudomonas aeruginosa represents the main bacterial pathogen infecting CF lungs (23, 24). Due to these frequent infections, there is a massive neutrophil infiltration to the lungs and development of chronic inflammation (25, 26). The chronic and progressive lung disease accounts for morbidity and mortality of CF patients (25).
Cystic fibrosis sputum constituents include DNA, NE, MPO, and other neutrophil proteins (27), as it has been shown that bronchoalveolar lavage fluid (BAL) from CF infants presented high concentrations of DNA, which correlated with neutrophil numbers in BAL (28). However, the great amounts of extracellular DNA in CF sputum were considered to be from necrotic neutrophils and lung tissues (29). More recently, several studies have demonstrated that NETs and NET-associated proteins are present in CF sputum (30–35). Marcos and coworkers quantified free DNA levels in airway fluid from CF patients and found that those patients with poor pulmonary function presented higher levels of extracellular DNA compared to patients with mild lung disease (36), indicating that the accumulation of NET–DNA in the airways contributes to airflow obstruction in CF. Moreover, analysis of CF sputum samples revealed that elevated levels of macrophage migration inhibitory factor (MIF), a potent pro-inflammatory cytokine, correlated with poor pulmonary function, and MIF was able to induce NET formation (33). Although many of the microorganisms that colonize CF airways have been shown to induce NET formation directly (4, 6, 12, 37, 38), pro-inflammatory cytokines and neutrophil chemokines present in CF lungs are also able to stimulate NET release (30, 33), thus perpetuating the inflammation.
Neutrophil recruitment and NET production in the lungs would be key events to fight against invading microorganisms, but their mission accomplishment is profoundly compromised in CF airways as patients often suffer chronic infections. Together with the failure in killing the bacteria, the excessive release of extracellular DNA accounts for biofilm formation by P. aeruginosa, and NETs act as a proinflammatory component of biofilms (39). Furthermore, over the time of infection in CF airways, P. aeruginosa is able to acquire resistance to NET-mediated killing (38), probably due to its hypermutability, a well-described mechanism for P. aeruginosa adaptation within CF lungs (40–42). In addition, it has been recently demonstrated that sub-inhibitory concentrations of LL-37, a NET component, triggers P. aeruginosa mutagenesis in chronic infections (43). Interestingly, P. aeruginosa triggers the release of the eicosanoid hepoxilin A3 by lung epithelial cells, which induces neutrophil transepithelial migration and is a natural inducer of NET formation (44, 45). Thus, the excessive release of NETs coated with proteases, together with the colonizing bacteria may worsen pulmonary inflammation and dysfunction. Besides NETs being able to directly induce endothelial and epithelial cell death in vitro through histones (46), MPO and NE expressed on NET fibers could exacerbate lung pathology through the destruction of connective tissue and degradation of endothelial cell matrix heparan sulfate proteoglycan (47, 48). Moreover, it has been shown that NE cleaves host proteins at the site of inflammation (49). Additionally, histones are highly cytotoxic to endothelial cells in vitro and are lethal in mice (50). Altogether, these findings highlight the need to target the massive NET release in CF.
The current therapy to improve CF symptoms is the administration of recombinant human DNase I (pulmozyme/dornase alpha) (51). DNase inhalation is one of the successful treatments for CF, as it improves lung function and reduces infectious exacerbations (52); however, it is not effective for all CF patients. Therefore, alternative therapeutic options are desired. Dubois and colleagues have demonstrated that DNase administration to CF sputum dramatically increased its elastase activity (53). Thus, the combined administration of DNase and an elastase inhibitor could be useful to avoid the devastating effects of excessive proteases in CF lungs. There are also candidate drugs to inhibit NET release, such as chloroquine and PAD4 inhibitors, however neither of these molecules has been evaluated in animal models of CF.
Asthma
Asthma is a chronic heterogeneous inflammatory disorder of the airways characterized by airway inflammation and reversible airflow obstruction (54–56). Asthmatic subjects present periods of stable condition that alternate with severe episodes of exacerbations, leading to the impairment of lung function (57). Asthma symptoms include recurrent wheezing, coughing, and shortness of breath (55). This very complex disease is caused by multiple environmental factors that act in combination with hundreds of susceptibility genes (55). Asthma has been seen for a long time as an eosinophilic disease (56); however, in recent years, it has become evident that some asthmatics have a prominent neutrophilic inflammation in the lungs (58). Patients with neutrophilic asthma usually present a severe form of the disease that does not respond to the classical treatment with glucocorticoids (59, 60). In addition, glucocorticoid administration to neutrophilic asthmatics could aggravate lung inflammation, since glucocorticoids can prolong neutrophil survival (61). It has been described that neutrophils recruited to the lungs of atopic asthmatic patients generated NETs colocalized with elastase (62). In some patients, the number of neutrophils and NET-releasing neutrophils exceeded the number of eosinophils in the lungs. In this study, Dworski and colleagues also demonstrated that eosinophils infiltrating the airways of atopic asthmatics were able to release eosinophil extracellular traps (EETs), which colocalized with eosinophil granule proteins, such as major basic protein (MBP) and eosinophil cationic protein (ECP). Similar to the first study reporting the release of EETs from viable eosinophils (63), the DNA actively released by eosinophils in asthmatic lungs was from mitochondrial origin, and not nuclear (62). Interestingly, allergen challenge did not increase EET or NET formation in the airways of asthmatic subjects (62). Thus, what would be the role of EETs and/or NETs in the pathogenesis of asthma? And what would be the cause of EET/NET release in asthma? Taking into consideration the high concentrations of proteases anchored in extracellular DNA traps, one can assume that these enzymes could contribute to epithelial and endothelial cell damage, a hallmark of asthma. On the other hand, the formation of DNA lattices could protect the host against possible infections secondary to cell damage. Currently, these and many other questions regarding DNA traps formation in allergic diseases are still open for debate. More recently, it has been demonstrated that eosinophils from asthmatic mice release EETs decorated with eosinophil peroxidase (EPO) with no signs of cell death (64), indicating that DNA release is an active process. In addition, recombinant human DNase treatment of asthmatic mice improves lung resistance and decreases oxidative stress in the lungs, providing a potential antioxidant effect on asthma (65, 66). Accordingly, the combined use of recombinant human DNase therapy together with the current treatments (such as inhaled glucocorticoids) for severe acute asthma may prove effective in decreasing sputum viscosity, as it has been shown in specific case reports (67, 68).
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD) is a progressive disorder of the airways characterized by persistent neutrophilic inflammation (69, 70). The disease develops following long-term exposure to external stresses, such as inhaled tobacco smoke (71–73). COPD patients are affected by recurrent bacterial and respiratory viral infections, which represent the main causes of exacerbations in these subjects. Exacerbations are associated with increased upper and lower airway and systemic inflammation (74). Patients with severe COPD present large amounts of airway neutrophils when stable, and these numbers further increase during exacerbations, which may be due to the high expression of neutrophil chemokines and chemokine receptors in airway mucosa (75). Furthermore, NE is expressed in the airway mucosa of COPD patients during severe exacerbations (75) and has a proinflammatory role by inducing the secretion of IL-8 in COPD (76). It is noteworthy that IL-8 is a potent NET inducer (3, 77). These features make COPD lungs more likely to be filled by NETs. Indeed, confocal microscopy analysis has shown that sputum from exacerbated COPD patients presents extracellular DNA, frequently entangled with bacteria (78), characterizing NETs. Moreover, NETs are present not only during COPD acute exacerbations, but also in the lungs of patients with stable disease (79–81). There is a clear correlation between the abundance of NETs in the sputum of COPD patients and disease severity – over 90% of exacerbated COPD subjects presented large amounts of NETs in their sputum compared to 45% of stable COPD subjects. In addition, the very large quantities of NETs directly correlate with the severity of airflow limitation in these patients (79). Why NETs are produced in excess in COPD and what would be the trigger for NET release are unsolved issues. Under physiological conditions, NETs would be degraded by endogenous nucleases and cleared by alveolar macrophages (82). However, COPD subjects present lower numbers of alveolar macrophages (81) and these macrophages are defective in phagocytosis (83), which may explain the persistence of NETs in the airways. Nonetheless, a recent interesting study has shown that the outcome of the interaction of macrophages and netting neutrophils depend on macrophage phenotype. M2 macrophages in contact with netting neutrophils helped to perpetuate an inflammatory response, while M1 macrophages initially released extracellular DNA and thereafter degraded DNA in a caspase-activated DNase-dependent manner (84). These findings highlight a phenotype-dependent mechanism of macrophage regulation of NET release, which reinforce the argument that a prolonged exposure to NETs may favor the development of autoimmunity. The exact role of NETs in COPD pathogenesis is uncertain, but the need for developing novel diagnostic and therapeutic strategies is clear. The treatment for COPD is very difficult, as anti-inflammatory drugs are ineffective. The most successful current treatment for COPD is long-acting bronchodilators, but no therapy reduces the progression or inhibits the inflammation (85). As NETs were implicated in disease worsening, selective inhibitors of NET formation or NET-associated proteins (such as NE, MPO, histones) may prove valuable in improving the clinical picture of the disease.
Tuberculosis
Tuberculosis (TB) remains a major health problem for humankind. Annually, there are approximately nine million new cases and 1.5 million deaths caused by the disease (86). This chronic bacterial infection is caused by Mycobacterium tuberculosis and affects the lungs, promoting huge morbidity and mortality rates (86, 87). M. tuberculosis is usually transmitted by tiny droplets from cough or sneeze of an infected subject. Once in the lungs, the bacilli is phagocytosed and killed by alveolar macrophages. However, M. tuberculosis developed strategies to survive inside the macrophages. Therefore, the infection develops as a latent infection, inducing granuloma formation in the lung parenchyma. Consequently, the subject remains healthy while harboring dormant bacteria (87, 88). The key factor for the maintenance of latent TB infection is the equilibrium between the bacteria and the host immune response. TB reactivation is achieved when the immune response decreases and cannot restraint bacteria growth, inducing cell death and an increase in granulomatous lesions, as a result of inflammatory cell recruitment (88). Clinical symptoms of TB are caused by a severe impairment of lung function and by substantial morphological alterations in the lung parenchyma (87).
Although macrophages are generally viewed as the main cells involved in harboring M. tuberculosis, a growing body of evidence shows that neutrophils are rapidly recruited to infected lungs and can serve as bacterial reservoirs. Additionally, neutrophils were identified as the main immune cell type in sputum and BAL from active TB patients (89). Furthermore, human neutrophils are able to phagocytose M. tuberculosis in vitro, but fail to kill the bacilli (90). Neutrophils have been assigned to play both protective and pathological roles during active TB (91–93). As a part of their role in TB pathogenesis, neutrophils have been shown to release NETs coated with NE and histones when stimulated by two genotypes of M. tuberculosis (H37Rv and M. canetti). NETs were able to trap mycobacteria but not to kill them (94). This lack of killing ability of NETs may favor lung destruction in active TB. Another study has found matrix metalloproteinase-8 (MMP-8) expressed on NET fibers induced by M. tuberculosis in vitro. In addition, induced sputum from TB patients had increased amounts of NETs compared to healthy subjects and MMP-8 secretion correlated to lung tissue destruction in these patients (95). The effect of M. tuberculosis on NET induction might be mediated by the early secretory antigen-6 (ESAT-6), a protein secreted by M. tuberculosis, responsible for the escape of mycobacteria from phagosome to cytoplasm of cells (96), as ESAT-6 induces the production of NETs colocalized with MPO (97). ESAT-6 is also secreted in large quantities in the extracellular space and therefore can interact with immune cells to stimulate them and facilitate the maintenance of chronic inflammation in the lungs of TB patients (98). Importantly, neutrophils release high levels of calprotectin (S100A8/A9) within lung granulomas of patients with active TB (99), which are constituents of NETs (100). The release of calprotectin in TB could be related to NET formation, as neutrophil cytoplasmic proteins can attach to DNA fibers before being released. Urban and coworkers have shown that calprotectin can be released from neutrophils in two ways: bound to NETs and unbound (100). This could be the case for calprotectin release in the lungs of TB subjects; however whether M. tuberculosis induces the formation of NETs expressing calprotectin remains to be determined.
Tuberculosis is a curable disease, although the treatment is difficult, since it can take several months (6–9 months) and has different drug regimens. Currently, the first line anti-TB drugs include isoniazid, rifampin, ethambutol, and pyrazinamide, among others when necessary, according to CDC (Centers for Disease Control and Prevention – http://www.cdc.gov/tb/topic/treatment/). Moreover, new therapies aiming to improve the treatment outcomes, shorten the duration of treatment, and reduce lung pathology in TB patients were described (101). However, no therapeutic approach aimed to specifically regulate the deleterious effects of NETs in TB lungs was reported.
Bacterial Pneumonia
The most common type of bacterial pneumonia is community-acquired pneumonia (CAP). CAP remains a burden worldwide, being responsible for approximately 3.5 million deaths annually (102). A total of 20–60% of CAP patients require hospitalization due to disease severity, including children under age 5 years (102, 103). The etiology of CAP is variable, depending partly on the diagnostic tools used in the population studied. Among all bacteria, Streptococcus pneumoniae (S. pneumoniae) is the most frequently identified cause of CAP, with high morbidity and mortality rates, but H. influenzae is also an important etiologic agent of CAP (102, 104).
Once bacterial infection is established in the lungs, neutrophils are massively recruited to the infection site, inducing a prominent inflammatory response. The clinical outcome in CAP depends on the balance between the inflammatory response and pathogen clearance (102). In this sense, neutrophils actively producing NETs during CAP might lead to potential collateral damage to the lungs. Indeed, three different strains of S. pneumoniae (serotypes 3, 4, and 19F) were able to induce pulmonary NET formation in mice, which correlated with the histopathologic severity. In addition, the pneumococcal capsule directly contributes to excessive NET release that paralleled with pneumonia severity in mice (105). The mechanism of NET induction by S. pneumoniae seems to be mediated by the pneumococcal protein α-enolase, which binds to myoblast antigen 24.1D5 on neutrophil surface and stimulates NET generation (106). However, S. pneumoniae appears to have evolved strategies to counteract NET-mediated killing. In an elegant study, Beiter and colleagues have demonstrated that S. pneumoniae expresses EndA, a membrane-localized endonuclease able to degrade NETs in vitro and to promote spreading of bacteria from the upper airways to the lungs and from the lungs to the bloodstream of mice. Additionally, mutant bacteria lacking EndA infect the upper airways but fail to disseminate to the lungs and bloodstream (107). Moreover, EndA is secreted into the culture medium during pneumococcal cell growth and rapidly dismantle DNA in NETs, being required for full virulence of S. pneumoniae during lung infection (108). Corroborating with these studies, streptococcal endonuclease has been previously implicated in disease progression (109). Besides EndA, streptococcal cells hold other important mechanisms to protect them from NET trapping and killing, such as a positive charge on their surfaces as a result of capsule expression and lipoteichoic acid d-alanylation (110). Thus, it seems that NETs released during S. pneumoniae infection function only to damage lung tissue, instead of having a bactericidal activity. The evidence that NETs released in response to bacterial infections can trap and inactivate viruses (8, 111) points out the utmost importance of NETs during co-infections in vivo. On the other hand, secondary pneumococcal infection following primary influenza intensified NET formation, but NETs did not show any bactericidal activity, only worsening lung pathogenesis (112). Altogether, these findings suggest that the nature of NET trigger is fundamental to the clearance of subsequent infections.
Non-typeable H. influenzae (which lacks a capsule) is an important cause of pneumonia, mainly in subjects with chronic bronchitis and COPD (113), and the persistence of NETs could worsen lung inflammation in these subjects. Viable and heat-killed H. influenzae induces NET release in vitro, in a mechanism possibly mediated by lipooligosaccharide binding to TLR-4 and Myeloid Differentiation Primary Response (MyD)-88, an adaptor protein necessary to TLR-4 signaling. Interestingly, bacteria are not killed by NET proteins and survive within NETs (114). Accordingly, it has been recently demonstrated that these bacteria evolved to express specific molecules, peroxiredoxin–glutaredoxin and catalase, which allow them to resist to host oxidants and to survive within NETs in vivo (115). In addition, non-typeable H. influenzae populations survive in biofilm communities in the airway surface, and NETs constitute an integral part of these biofilms (116). Astoundingly, it has been reported a fatal case of non-typeable H. influenzae infection with severe pneumonia and bacteremia in an adult found to have large amounts of NETs expressing NE and histone H3 in his sputum (117). This case highlights the association between excessive NET generation and severe respiratory infection and sepsis. More recently, it has been shown that besides NETs, non-typeable H. influenzae is also able to induce macrophage extracellular traps (METs) expressing MMP-12 (118). MMP-12 has been implicated as a key factor for protease imbalance and emphysema. Therefore, the release of METs together with NETs may have a detrimental role during emphysema, pneumonia, and COPD. Importantly, DNase was effective to dismantle non-typeable H. influenzae-induced MET and NET formation (118), which could be used as a short-term adjunctive therapy to avoid the injurious effects of these extracellular traps and associated proteases during pneumonia and other lung diseases.
Respiratory Syncytial Virus Bronchiolitis
Respiratory Syncytial Virus (RSV) is the leading cause of acute bronchiolitis in children under age 2 years (119). Throughout the winter, RSV causes a significant number of hospitalizations, resulting in a huge burden to communities worldwide (119, 120). Due to the high infectivity of RSV, almost 70% of all children are infected with the virus during the first year of life, and by age 3, practically all children will have experienced at least one infection with this virus (121, 122). The clinical symptoms of RSV bronchiolitis include labored breathing, coughing, and wheezing (123). Microscopically, there is a massive neutrophil recruitment to the airways of infected children – these cells comprise for approximately 80% of infiltrated cells (124–127). Once in the airways, RSV is able to activate neutrophils, inducing degranulation and IL-8 secretion (128), and also to inhibit neutrophil apoptosis, through phosphoinositide 3-kinase (PI3K) and nuclear factor-κB (NF-κB)-dependent mechanisms (129). This body of evidence suggests that neutrophils may play a significant role in disease pathogenesis.
Aside from the mechanisms mentioned above, we have recently demonstrated that RSV particles and one of its membrane-bound glycoproteins are capable of inducing NET formation by human neutrophils (130). RSV Fusion protein mediates the fusion of virus with the host cell and it is essential for viral replication both in vivo and in vitro (131), being considered the primary target for vaccine and antiviral drug development. RSV F protein induces the release of NETs coated with MPO and NE through Toll-like receptor (TLR)-4 activation. Moreover, F protein stimulates ROS generation and MAPK phosphorylation, and these signaling pathways are necessary to F protein-induced NET formation (130). Data in the literature regarding the role of NETs in viral diseases are conflicting (132). We hypothesized that the excessive production of NETs could fill the lungs and impair lung function, worsening inflammation in young children and babies affected by RSV infection. Indeed, analysis of bronchoalveolar fluid cytology samples from children with severe RSV lower respiratory tract infection revealed the presence of NETs expressing NE and citrullinated histone 3 (citH3) (133). Furthermore, the infection of calves with bovine RSV induced an extensive release of NETs colocalized with dense cellular plugs containing shed epithelial cells and large amounts of neutrophils, which obstructed the airways (133). These recent studies indicate that NETs contribute to the airway obstruction and immunopathology observed in children and animals infected with RSV.
Despite extensive research efforts, there is no RSV vaccine currently available. Nevertheless, monoclonal antibodies targeting the RSV fusion protein have been developed and they passively protect against RSV challenge in an animal model and reduce the severity of infection in premature and newborn babies (134, 135). However, the humanized monoclonal antibody against RSV F protein is only used in high-risk groups, such as preterm infants and those suffering from cardiovascular diseases or immunosuppression (134). In addition, ribavirin is an antiviral drug used to treat severe RSV bronchiolitis due to its anti-replicative activity, but it presents a high cost and is administered only to high-risk infants (136). Moreover, the use of recombinant human DNase in the management of severe RSV bronchiolitis has been previously reported. The administration of nebulized DNase to young babies with complicated bronchiolitis was able to immediately improve the clinical signs and chest radiograph, and even led to the resolution of atelectasis (137, 138). In contrast, in infants with mild RSV bronchiolitis, recombinant DNase therapy did not reduce the length of hospital stay or the duration of supplemental oxygen (139). Thus, DNase seems to be a useful therapeutic option in the treatment of infants who develop atelectasis due to severe RSV bronchiolitis.
Influenza Virus Infection
Influenza A virus is responsible for regular outbreaks, whose severity may vary among the population. While the influenza pandemic that started with the Spanish flu in 1918 killed approximately 50 million people worldwide, the pandemic influenza A H1N1 2009 virus has affected more than 214 countries and caused nearly 18,449 deaths (140, 141). The clinical features of influenza infection include fever and upper respiratory symptoms, such as cough, runny nose, and sore throat (141). To date, there is little information about clinical complications of influenza A infection, but they appear to be similar to those of seasonal influenza, including sinusitis, otitis media, pneumonia, bronchiolitis, seizures, toxic shock syndrome, and secondary bacterial pneumonia with or without sepsis. Among subjects with high risk for complications are those at extremes of age and those with pre-existing medical conditions (141).
The characteristic feature of acute lung inflammation following influenza virus infection is the excessive infiltration of neutrophils in the lungs (142, 143), and CXCR2 seems to be the major receptor mediating neutrophil recruitment during this infection (144). Neutrophils have been demonstrated to play both protective and detrimental roles during influenza virus infection (143, 145, 146). Among the harmful roles played by neutrophils is the excessive production of NETs in the lungs of animals infected with influenza A H1N1 virus. NETs expressing histones and MMP-9 were found entangled with alveoli, causing increased alveolar capillary damage and obstruction of the small airways, thus confirming the link of these DNA lattices with lung damage (146). Furthermore, NET formation stimulated by influenza A infection is dependent on histone deimination by PAD4 (147). In addition, NET release induced by influenza virus is potentiated by the cathelicidin LL-37 (148), which has been shown to facilitate the formation of NETs (149). Paradoxically, the antimicrobial protein expressed on NETs, α-defensin-1, is able to directly inhibit influenza replication through the inhibition of protein kinase C (PKC) in infected cells (150); however the expression of α-defensins on NETs induced by this virus has yet to be demonstrated. The expression of α-defensins on NETs could inactivate the virions sequestered in NET fibers and consequently prevent them from reaching the target cells in the lungs. Thus, although antimicrobial proteins expressed on NETs have the ability to inactivate the virus and to prevent spreading, they are also able to inflict damage to host cells and tissues due to their cytotoxic properties.
Currently, influenza treatment relies on the administration of two groups of antiviral drugs, the adamantanes and neuraminidase inhibitors. Zanamivir and oseltamivir are neuraminidase inhibitors active against both influenza A and B, and are approved for the prevention and treatment of influenza in the United States. Supportive care of uncomplicated cases of influenza includes administration of fluids and rest (141). To date, there is no study describing the effect of DNase treatment on the outcome of influenza infection in animal models.
Transfusion-Related Acute Lung Injury
Transfusion-related acute lung injury (TRALI) is a serious complication of blood transfusion (whole blood or blood components) that develops within 6 h of transfusion and is characterized by hypoxemia, respiratory distress, and pulmonary infiltrates (151, 152). Currently, TRALI is the most important cause of transfusion-related morbidity and mortality (152). Histological analysis revealed lung edema, capillary leucostasis, and massive neutrophil infiltration (153). TRALI development requires the presence of antileukocyte antibodies in the transfused product, and antineutrophil antibodies have been linked to the most severe cases of TRALI (154). These antibodies activate recipient’s neutrophils, inducing their sequestration in the pulmonary capillaries and consequently tissue injury (155).
In an elegant study, Thomas and coworkers have found NET biomarkers (DNA, nucleosomes and MPO) in the serum of patients with documented TRALI (156). In addition, in a fatal case of TRALI neutrophils with decondensed nuclei were detected in lung vessels together with abundant extracellular histones and MPO (157). In a mouse model of TRALI, DNA streaks colocalizing with citrullinated histone H3 were found in alveoli outside blood vessels (156). Moreover, platelets also accumulate in the lungs of mice with TRALI, being required for injury development (158). In this model, platelets were shown to induce NET formation during TRALI (157). As a vicious cycle, histones expressed on NETs may activate platelets (159), which in turn induce further NET release, promoting coagulation and thrombi formation in the lungs. Accordingly, the pretreatment of mice with a histone-blocking antibody decreased lung edema, lung vascular permeability, and even mortality. This treatment also reduced NET generation detected in plasma, indicating that extracellular histones may help to spread NETs in the body (157). Furthermore, intranasal administration of DNase provided several benefits to mice undergoing TRALI, such as improvement of blood oxygenation, reduction in lung edema and vascular permeability, impairment of NET formation, and platelet sequestration in the lungs (156, 157). These studies support the argument that NETs are formed and play a critical role in the pathogenesis of TRALI and may be a promising target for therapeutic approaches.
Mechanical Ventilation
Mechanical ventilation is a supportive intervention and a key feature of intensive care for patients with acute respiratory failure, including those with severe RSV bronchiolitis, pneumonia, or influenza infection (160, 161). However, it can be potentially injurious to the ventilated lung, inducing the so-called ventilator-associated lung injury (VALI), which contributes to morbidity and mortality in those patients (162). Furthermore, animal models of acute lung injury have been developed and characterize an experimental insult to a normal lung and therefore were named ventilator-induced lung injury (VILI) (162).
Neutrophils have been implicated as central cells in the pathogenesis of both VALI and VILI. It has been described that the early phase of VILI involves the release of several pro-inflammatory cytokines and chemokines, whereas the late phase is characterized by the infiltration of a lung-marginated neutrophil pool (163, 164). However, more recently Choudhury and coworkers demonstrated that injurious mechanical ventilation induced a prominent neutrophil recruitment to the lung at the very early stage of VILI, before the development of physiological signs of lung injury. The infiltration of neutrophils in the course of VILI was dependent on L-selectin engagement but independent of CD18 (165), indicating that immune mechanisms mediate neutrophil recruitment and activation during mechanical ventilation. Moreover, lung-derived soluble mediators appear to have a pathogenic role in an isolated perfused lung model of VILI (166). In line with this evidence, the chemokine receptor CXCR2 and its ligands, CXCL1 (KC) and CXCL2/3 (MIP-2), were shown to play a significant role in mediating neutrophil recruitment and promoting lung inflammation in VILI (167). Accordingly, short periods of mechanical ventilation in preterm infants induce an overproduction of the pro-inflammatory cytokines TNF and IL-1β, neutrophil chemokines IL-8 and MCP-1, and MMP-9 (168, 169). These inflammatory mediators may work together to induce a massive neutrophil infiltration to ventilated lungs and to stimulate NET release in response to mechanical ventilation in those patients. So far, IL-8, TNF, and IL-1β were shown to promote NET release in different experimental settings (10, 170, 171). In fact, excessive NET formation has been recently implicated in the pathogenesis of VILI. A double-hit model of intratracheal LPS challenge followed by high tidal mechanical ventilation induced a prominent lung injury in mice, with high amounts of NETs, decreased lung compliance and release of pro-inflammatory cytokines (172). The mechanism of NET formation during VILI seems to rely on the simultaneous engagement of G protein-coupled receptors (GPCR) and Mac-1 (CD11b), by the platelet-derived CCL5/CXCL4 heterodimer and a β2-integrin ligand, respectively (173). Surprisingly, these two studies showed opposing results regarding the role of NETs during VILI. Rossaint and coworkers found that DNase treatment of mice after induction of VILI was protective, as treated mice showed an improved gas exchange and reduced NET markers in the blood; whereas Yildiz and colleagues did not find a significant impact of DNase treatment on lung injury induced by VILI. There is at least one possible explanation for these differences: in the study of Yildiz and colleagues, the lungs of mice were already filled with neutrophils at the early stage of VILI due to LPS instillation, which could not be counteracted by DNase. Whereas in the study of Rossaint and coworkers, neutrophils infiltrated the lungs in the course of VILI, in this case a sterile inflammation. Although the outcome of DNase treatment in VILI is an issue for debate, there is no doubt that excessive NET formation accounts for the pathogenesis of acute lung injury.
Other Pulmonary Diseases and NETs
Besides the pulmonary diseases aforementioned, there are other disorders or syndromes affecting the lungs, in which NETs may play harmful roles as well.
Acute lung injury following severe sepsis is a common clinical consequence with significant morbidity and mortality rates (174), as the lung is the most sensitive target organ during systemic inflammation (175). Czaikoski and collaborators have recently shown that NETs are produced systemically in mice with cecal ligation and puncture (CLP) model of sepsis. The excessive release of NETs was directly correlated to heart, liver, and lung injury, as rhDNase plus antibiotics treatment of septic mice drastically decreased organ damage (176). Additionally to extracellular DNA measurement, NETs were observed in alveolar spaces and pulmonary capillaries of septic mice (177). Furthermore, higher concentrations of cell-free NETs were present in the serum of septic patients who developed severe acute respiratory distress syndrome (ARDS) compared to healthy controls (176), extending the experimental observations in mice to the clinical setting. Mechanistically, platelet TLR-4 is essential for NET induction within hepatic sinusoids and pulmonary capillaries of septic mice (178). Interestingly, NETs retained their integrity under flow conditions and were able to trap bacteria in septic blood. Therefore, platelets may serve as a platform for neutrophil activation and NET production, which can trap and kill pathogens but also induce disseminated organ injury during severe sepsis (178).
Another lung disorder featuring neutrophil-induced injury is interstitial lung disease (ILD). Actually, ILD are a group of diffuse parenchymal lung disorders characterized by pulmonary fibrosis. ILD can be frequently associated with a specific environmental exposure or an underlying connective tissue disease (179). Activated neutrophils were found increased in BAL from patients with idiopathic pulmonary fibrosis and were associated with early mortality (180). Interestingly, patients with ILD complications due to autoimmunity showed elevated levels of circulating cell-free NETs and plasma LL-37 (a NET component), together with a decreased DNase activity (181), suggesting that the prolonged exposure to NETs is involved in the pathogenesis of ILD. In vitro, NETs have been demonstrated to promote the activation of lung fibroblasts and differentiation into myofibroblast phenotype. Moreover, these fibrotic effects were significantly decreased after degradation of NETs with DNase (182). Consistently, these findings were supported by the detection of NETs in close proximity to alpha-smooth muscle actin-expressing fibroblasts in biopsies from patients with fibrotic ILD (182). This effect is very likely to be mediated by NE, since NE directed both lung fibroblast proliferation and myofibroblast differentiation in vitro (183). In addition, a NE inhibitor attenuated pulmonary fibrosis induced by bleomycin in mice via inhibition of TGF-β1 and inflammatory cell recruitment to the lungs (184). Altogether, these studies point to a key role of NETs in the development of ILD of different etiologies.
Conclusion
Neutrophil extracellular traps formation by activated neutrophils has a crucial role in host defense against microorganisms, as deficiency in NET release or dismantling NET backbone by bacterial DNases render the host susceptible to disseminated and lethal infections (107, 185). Moreover, aggregated NETs have been shown to limit sterile inflammation by degrading cytokines and chemokines via serine proteases (186). However, an excess or persistence of NET release is potentially injurious to host organs and cells, leading to worsening or perpetuation of many diseases. The pathogenic effects of excessive NET production is especially important in pulmonary diseases due to lung architecture itself, which may favor the spreading of DNA fibers, consequently enhancing tissue damage and impairing lung function (Figure 1). The mechanisms underlying NET production and the boundaries between the beneficial and detrimental effects of NETs during disease state are still to be unveiled. To date, recombinant human DNase is the only treatment targeting NETs approved for a small number of pulmonary disorders. Nevertheless, a long-term DNase therapy presents side effects to patients. Hence, the quest for an ideal therapy targeting NETs and its associated proteins continues to be a challenge for scientists around the globe.
Figure 1
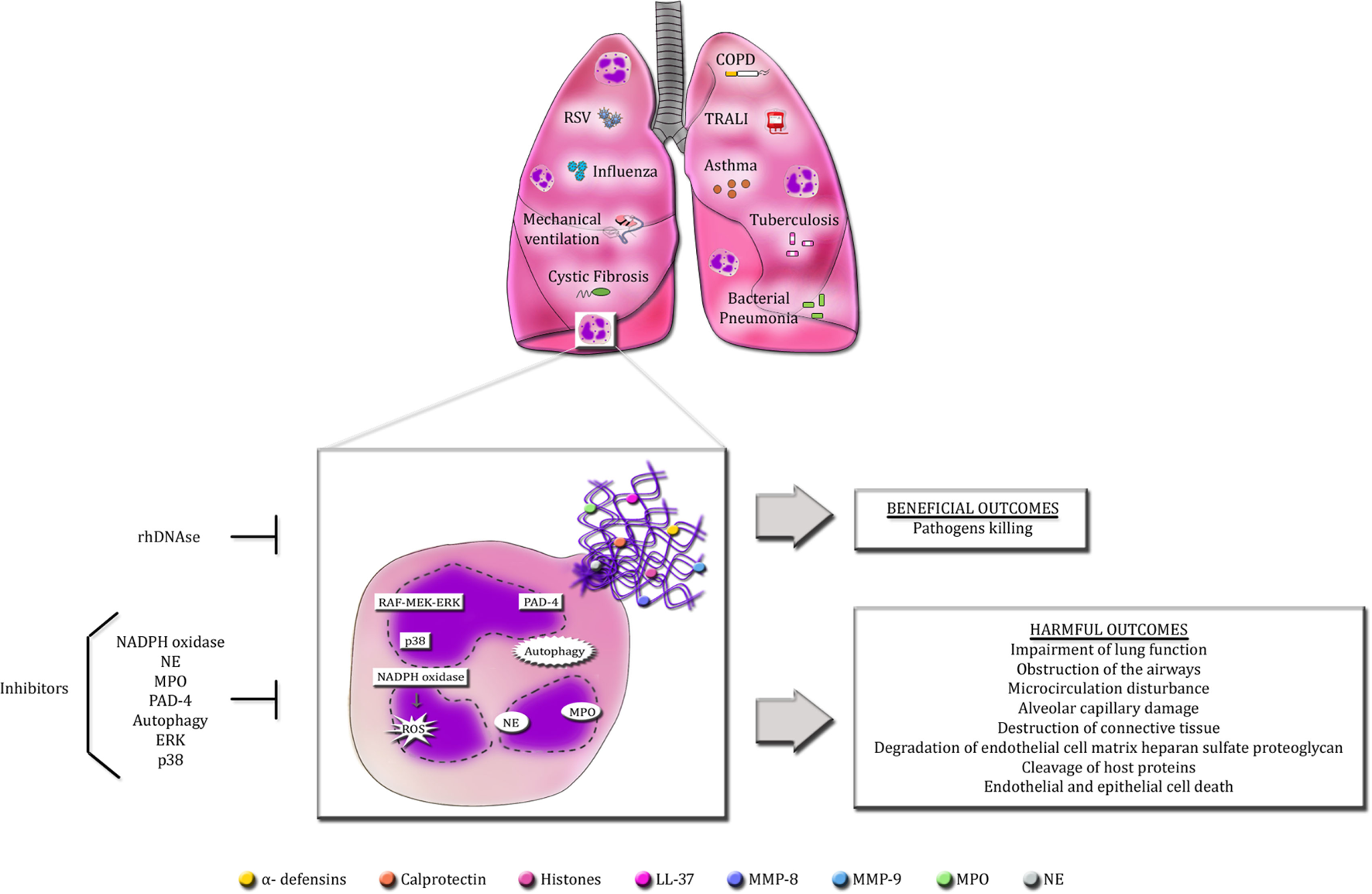
Overview of the beneficial and detrimental roles of NETs in pulmonary diseases. Infectious and non-infectious pulmonary diseases cause the massive infiltration of neutrophils into the lungs. Activated neutrophils release an excess of NETs in the airways. The production of NETs requires the activation of specific signaling pathways described so far, such as raf-MEK-ERK and p38 MAPK, PAD-4, autophagy and NADPH oxidase-induced ROS generation. Additionally, NE and MPO also regulate NET formation. Accordingly, selective inhibitors of these signaling pathways are able to abolish or decrease NET release. The primary goal of NETs is to protect the host from invading microorganisms through their sticky nature and the high concentrations of antimicrobial proteins. However, these characteristics make NETs potentially detrimental to host cells and tissues. Excessive NET formation enhances mucus viscosity, filling the lungs, and impairing lung function. NET proteins are highly cytotoxic and can induce endothelial and epithelial cell death and cause the disruption of host proteins and cellular matrix.
Statements
Author contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work has not been funded by any agency.
Acknowledgments
We thank Stéfanie P. Muraro for support with figure, Natália Jaeger and Rafael S. Czepielewski for critical reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Nathan C . Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6:173–82.10.1038/nri1785
2
Segal AW . How neutrophils kill microbes. Annu Rev Immunol (2005) 23:197–223.10.1146/annurev.immunol.23.021704.115653
3
Brinkmann V Reichard U Goosmann C Fauler B Uhlemann Y Weiss DS et al Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5.10.1126/science.1092385
4
Urban CF Reichard U Brinkmann V Zychlinsky A . Neutrophils extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol (2006) 8:668–76.10.1111/j.1462-5822.2005.00659.x
5
Guimarães-Costa AB Nascimento MTC Froment G Soares RPP Morgado FN Conceição-Silva F et al Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci U S A (2009) 106:6748–53.10.1073/pnas.0900226106
6
McCormick A Heesemann L Wagener J Marcos V Hartl D Loeffler J et al NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect (2010) 12:928–36.10.1016/j.micinf.2010.06.009
7
Saitoh T Komano J Saitoh Y Misawa T Takahama M Kozaki T et al Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe (2012) 12:109–16.10.1016/j.chom.2012.05.015
8
Jenne CN Wong CHY Zemp FJ McDonald B Rahman MM Forsyth PA et al Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe (2013) 13:169–80.10.1016/j.chom.2013.01.005
9
Papayannopoulos V Metzler KD Hakkim A Zychlinsky A . Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191:677–91.10.1083/jcb.201006052
10
Fuchs TA Abed U Goosmann C Hurwitz R Schulze I Wahn V et al Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41.10.1083/jcb.200606027
11
Kirchner T Möller S Klinger M Solbach W Laskay T Behnen M . The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators Inflamm (2012) 2012:849136.10.1155/2012/849136
12
Pilsczek FH Salina D Poon KKH Fahey C Yipp BG Sibley CD et al A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185:7413–25.10.4049/jimmunol.1000675
13
Byrd AS O’Brien XM Johnson CM Lavigne LM Reichner JS . An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol (2013) 190:4136–48.10.4049/jimmunol.1202671
14
Li P Li M Lindberg MR Kennett MJ Xiong N Wang Y . PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207:1853–62.10.1084/jem.20100239
15
Remijsen Q Vanden Berghe T Wirawan E Asselbergh B Parthoens E De Rycke R et al Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21:290–304.10.1038/cr.2010.150
16
Hakkim A Fuchs TA Martinez NE Hess S Prinz H Zychlinsky A et al Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol (2011) 7:75–7.10.1038/nchembio.496
17
Keshari RS Verma A Barthwal MK Dikshit M . Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J Cell Biochem (2012) 114:532–40.10.1002/jcb.24391
18
Parker H Dragunow M Hampton MB Kettle AJ Winterbourn CC . Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol (2012) 92:841–9.10.1189/jlb.1211601
19
Brinkmann V Zychlinsky A . Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol (2007) 5:577–82.10.1038/nrmicro1710
20
Kaplan MJ Radic M . Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol (2012) 189:2689–95.10.4049/jimmunol.1201719
21
Cheng OZ Palaniyar N . NET balancing: a problem in inflammatory lung diseases. Front Immunol (2013) 4:1.10.3389/fimmu.2013.00001
22
Collins FS Drumm ML Cole JL Lockwood WK Vande Woude GF Iannuzzi MC . Construction of a general human chromosome jumping library, with application to cystic fibrosis. Science (1987) 235:1046–9.10.1126/science.2950591
23
O’Sullivan BP Freedman SD . Cystic fibrosis. Lancet (2009) 373:1891–904.10.1016/S0140-6736(09)60327-5
24
Lynch SV Bruce KD . The cystic fibrosis airway microbiome. Cold Spring Harb Perspect Med (2013) 3:a009738.10.1101/cshperspect.a009738
25
Cohen TS Prince A . Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med (2012) 18:509–19.10.1038/nm.2715
26
Ciofu O Hansen CR Hoiby N . Respiratory bacterial infections in cystic fibrosis. Curr Opin Pulm Med (2013) 19:251–8.10.1097/MCP.0b013e32835f1afc
27
Ratjen F . Recent advances in cystic fibrosis. Paediatr Respir Rev (2008) 9:144–8.10.1016/j.prrv.2008.01.004
28
Kirchner KK Wagener JS Khan TZ Copenhaver SC Accurso FJ . Increased DNA levels in bronchoalveolar lavage fluid obtained from infants with cystic fibrosis. Am J Respir Crit Care Med (1996) 154:1426–9.10.1164/ajrccm.154.5.8912759
29
Lethem MI James SL Marriot C Burke JF . The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. Eur Respir J (1990) 3:19–23.
30
Marcos V Zhou Z Yildirim AO Bohla A Hector A Vitkov L et al CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med (2010) 16:1018–23.10.1038/nm.2209
31
Papayannopoulos V Staab D Zychlinsky A . Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNAse therapy. PLoS One (2011) 6:e28526.10.1371/journal.pone.0028526
32
Manzenreiter R Kienberger F Marcos V Schilcher K Krautgartner WD Obermayer A et al Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J Cyst Fibros (2012) 11:84–92.10.1016/j.jcf.2011.09.008
33
Dwyer M Shan Q D’Ortona S Maurer R Mitchell R Olesen H et al Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J Innate Immun (2014) 6:765–79.10.1159/000363242
34
Yoo D Floyd M Winn M Moskowitz SM Rada B . NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol Lett (2014) 160:186–94.10.1016/j.imlet.2014.03.003
35
Marcos V Zhou Z Yildirim AÖ Bohla A Hector A Vitkov L et al Retraction: CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med (2010) 16:1018–23.10.1038/nm.2209
36
Marcos V Zhou-Suckow Z Yildirim AÖ Bohla A Hector A Vitkov L et al Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediators Inflamm (2015) 2015:408935.10.1155/2015/408935
37
Bruns S Kniemeyer O Hasenberg M Aimanianda V Nietzsche S Thywissen A et al Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog (2010) 6:e1000873.10.1371/journal.ppat.1000873
38
Young RL Malcolm KC Kret JE Caceres SM Poch KR Nichols DP et al Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One (2011) 6:e23637.10.1371/journal.pone.0023637
39
Fuxman Bass JI Russo DM Gabelloni ML Geffner JR Giordano M Catalano M et al Extracellular DNA: a major proinflammatory component of Pseudomonas aeruginosa biofilms. J Immunol (2010) 184:6386–95.10.4049/jimmunol.0901640
40
Oliver A Canton R Campo P Baquero F Blazquez J . High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science (2000) 288:1252–4.10.1126/science.288.5469.1251
41
Smith EE Buckley DG Wu Z Saenphimmachak C Hoffman LR D’Argenio DA et al Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A (2006) 103:8487–92.10.1073/pnas.0602138103
42
Rahman S Gadjeva M . Does NETosis contribute to the bacterial pathoadaptation in cystic fibrosis?Front Immunol (2014) 5:378.10.3389/fimmu.2014.00378
43
Limoli DH Rockel AB Host KM Jha A Kopp BT Hollis T et al Cationic antimicrobial peptides promote microbial mutagenesis and pathoadaptation in chronic infections. PLoS Pathog (2014) 10:e1004083.10.1371/journal.ppat.1004083
44
Hurley BP Siccardi D Mrsny RJ McCormick BA . Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J Immunol (2004) 173:5712–20.10.4049/jimmunol.173.9.5712
45
Douda DN Grasemann H Pace-Asciak C Palaniyar N . A lipid mediator hepoxilin A3 is a natural inducer of neutrophil extracellular traps in human neutrophils. Mediators Inflamm (2015) 2015:520871.10.1155/2015/520871
46
Saffarzadeh M Juenemann C Queisser MA Lochnit G Barreto G Galuska SP et al Neutrophils extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7:e32366.10.1371/journal.pone.0032366
47
Klebanoff SJ Kinsella MG Wight TN . Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am J Pathol (1993) 143:907–17.
48
Logters T Margraf S Altrichter J Cinatl J Mitzner S Windolf J et al The clinical value of neutrophil extracellular traps. Med Microbiol Immunol (2009) 198:211–9.10.1007/s00430-009-0121-x
49
Fujie K Shinguh Y Inamura N Yasumitsu R Okamoto M Okuhara M . Release of neutrophil elastase and its role in tissue injury in acute inflammation: effect of the elastase inhibitor, FR134043. Eur J Pharmacol (1999) 374:117–25.10.1016/S0014-2999(99)00268-X
50
Xu J Zhang X Pelayo R Monestier M Ammollo CT Semeraro F et al Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15:1318–21.10.1038/nm.2053
51
Konstan MW Wagener JS Pasta DJ Millar SJ Jacobs JR Yegin A et al Clinical use of dornase alpha is associated with a slower rate of FEV1 decline in cystic fibrosis. Pediatr Pulmonol (2011) 46:545–53.10.1002/ppul.21388
52
Fuchs HJ Borowitz DS Christiansen DH Morris EM Nash ML Ramsey BW et al Effect of aerosolized recombinant human DNAse on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The pulmozyme study group. N Engl J Med (1994) 331:637–42.10.1056/NEJM199409083311003
53
Dubois AV Gauthier A Bréa D Varaigne F Diot P Gauthier F et al Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am J Respir Cell Mol Biol (2012) 47:80–6.10.1165/rcmb.2011-0380OC
54
Locksley RM . Asthma and allergic inflammation. Cell (2010) 140:777–83.10.1016/j.cell.2010.03.004
55
Kim HY DeKruyff RH Umetsu DT . The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol (2010) 11:577–84.10.1038/ni.1892
56
Lambrecht BN Hammad H . The immunology of asthma. Nat Immunol (2015) 16:45–56.10.1038/ni.3049
57
Vestbo J Hurd SS Agustí AG Jones PW Vogelmeier C Anzueto A et al Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med (2013) 187:347–65.10.1164/rccm.201204-0596PP
58
Deckers J Madeira FB Hammad H . Innate immune cells in asthma. Trends Immunol (2013) 34:540–7.10.1016/j.it.2013.08.004
59
Haldar P Pavord ID . Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol (2007) 119:1043–52.10.1016/j.jaci.2007.02.042
60
Holgate ST . Trials and tribulations in identifying new biologic treatments for asthma. Trends Immunol (2012) 33:238–46.10.1016/j.it.2012.02.003
61
Cox G . Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J Immunol (1995) 154:4719–25.
62
Dworski R Simon H-U Hoskins A Yousefi S . Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol (2011) 127:1260–6.10.1016/j.jaci.2010.12.1103
63
Yousefi S Gold JA Andina N Lee JJ Kelly AM Kozlowski E et al Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med (2008) 14:949–53.10.1038/nm.1855
64
Cunha AA Porto BN Nuñez NK Souza RG Vargas MHM Silveira JS et al Extracellular DNA traps in bronchoalveolar fluid from a murine eosinophilic pulmonary response. Allergy (2014) 69:1696–700.10.1111/all.12507
65
Cunha AA Nuñez NK Souza RG Vargas MHM Silveira JS Antunes GL et al Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp Lung Res (2016) 42:66–74.10.3109/01902148.2016.1143537
66
da Cunha AA Nuñez NK Souza RG Vargas MHM Silveira JS Antunes GL et al Recombinant human deoxyribonuclease attenuates oxidative stress in a model of eosinophilic pulmonary response in mice. Mol Cell Biochem (2016) 413:47–55.10.1007/s11010-015-2638-1
67
Greally P . Human recombinant DNAse for mucus plugging in status asthmaticus. Lancet (1995) 346:1423–4.10.1016/S0140-6736(95)92436-1
68
Puterman AS Weinberg EG . rhDNAse in acute asthma. Pediatr Pulmonol (1997) 23:316–7.
69
Baraldo S Turato G Badin C Bazzan E Beghé B Zuin R et al Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax (2004) 59:308–12.10.1136/thx.2003.012146
70
Singh D Edwards L Tal-Singer R Rennard S . Sputum neutrophils as a biomarker in COPD: findings from the ECLIPSE study. Respir Res (2010) 11:77–89.10.1186/1465-9921-11-77
71
Botelho FM Gaschler GJ Kianpour S Zavitz CCJ Trimble NJ Nikota JK et al Innate immune processes are sufficient for driving cigarette smoke-induced inflammation in mice. Am J Respir Cell Mol Biol (2009) 42:394–403.10.1165/rcmb.2008-0301OC
72
Gaschler GJ Skrtic M Zavitz CCJ Lindahl M Onnervik P Murphy TF et al Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med (2009) 179:666–75.10.1164/rccm.200808-1306OC
73
Zhang X Zheng H Zhang H Ma W Wang F Liu C et al Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provked by cigarette smoke. Cytokine (2011) 56:717–25.10.1016/j.cyto.2011.09.010
74
Wedzicha JA Seemungal TAR . COPD exacerbations: defining their cause and prevention. Lancet (2007) 370:786–96.10.1016/S0140-6736(07)61382-8
75
Qiu Y Zhu J Bandi V Atmar RL Hattotuwa K Guntupalli KK et al Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2003) 168:968–75.10.1164/rccm.200208-794OC
76
Ungurs MJ Sinden NJ Stockley RA . Progranulin is a substrate for neutrophil-elastase and proteinase-3 in the airway and its concentration correlates with mediators of airway inflammation in COPD. Am J Physiol Lung Cell Mol Physiol (2013) 306:L80–7.10.1152/ajplung.00221.2013
77
Gupta AK Giaglis S Hasler P Hahn S . Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS One (2014) 9:e97088.10.1371/journal.pone.0097088
78
Obermayer A Stoiber W Krautgartner W-D Klappacher M Kofler B Steinbacher P et al New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PLoS One (2014) 9:e97784.10.1371/journal.pone.0097784
79
Grabcanovic-Musija F Obermayer A Stoiber W Krautgartner W-D Steinbacher P Winterberg N et al Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res (2015) 16:59.10.1186/s12931-015-0221-7
80
Pedersen F Marwitz S Holz O Kirsten A Bahmer T Waschki B et al Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir Med (2015) 109:1360–2.10.1016/j.rmed.2015.08.008
81
Wright TK Gibson PG Simpson JL McDonald VM Wood LG Baines KJ . Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology (2016) 21:467–75.10.1111/resp.12730
82
Farrera C Fadeel B . Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol (2013) 191:2647–56.10.4049/jimmunol.1300436
83
Hodge S Hodge G Scicchitano R Reynolds PN Holmes M . Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol (2003) 81:289–96.10.1046/j.1440-1711.2003.t01-1-01170.x
84
Nakazawa D Shida H Kusunoki Y Miyoshi A Nishio S Tomaru U et al The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun (2016) 67:19–28.10.1016/j.jaut.2015.08.018
85
Barnes PJ . Frontrunners in novel pharmacotherapy of COPD. Curr Opin Pharmacol (2008) 8:300–7.10.1016/j.coph.2008.03.001
86
World Health Organization. Global tuberculosis report 2015. 20th ed. Geneva: World Health Organization (2015). Available from: http://www.who.int/tb/publications/global_report/en/
87
Dorhoi A Kaufmann SH . Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J Immunol (2015) 45:2191–202.10.1002/eji.201545493
88
Amaral EP Lasunskaia EB D’Império-Lima MR . Innate immunity in tuberculosis: how the sensing of mycobacteria and tissue damage modulates macrophage death. Microbes Infect (2016) 18:11–20.10.1016/j.micinf.2015.09.005
89
Eum SY Kong JH Hong MS Lee YJ Kim JH Hwang SH et al Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest (2010) 137:122–8.10.1378/chest.09-0903
90
Corleis B Korbel D Wilson R Bylund J Chee R Schaible UE . Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol (2012) 14:1109–21.10.1111/j.1462-5822.2012.01783.x
91
Dorhoi A Iannaccone M Maertzdorf J Nouailles G Weiner J III Kaufmann SH . Reverse translation in tuberculosis: neutrophils provide clues for understanding development of active disease. Front Immunol (2014) 5:36.10.3389/fimmu.2014.00036
92
Lowe DM Redford PS Wilkinson RJ O’Garra A Martineau AR . Neutrophils in tuberculosis: friend or foe?Trends Immunol (2012) 33:14–25.10.1016/j.it.2011.10.003
93
Dallenga T Schaible UE . Neutrophils in tuberculosis – first line of defence or booster of disease and targets for host-directed therapy?Pathog Dis (2016) 74:ftw012.10.1093/femspd/ftw012
94
Ramos-Kichik V Mondragón-Flores R Mondragón-Castelán M Gonzalez-Pozos S Muñiz-Hernandez S Rojas-Espinosa O et al Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (2009) 89:29–37.10.1016/j.tube.2008.09.009
95
Ong CWM Elkington PT Brilha S Ugarte-Gil C Tome-Esteban MT Tezera LB et al Neutrophil-derived MMP-8 drives AMPK dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog (2015) 11:e1004917.10.1371/journal.ppat.1004917
96
Houben D Demangel C van Ingen J Perez J Baldeon L Abdallah AM et al ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol (2012) 14:1287–98.10.1111/j.1462-5822.2012.01799.x
97
Francis RJ Butler RE Stewart GR . Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis (2014) 5:e1474.10.1038/cddis.2014.394
98
Orme IM . A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis (Edinb) (2014) 94:8–14.10.1016/j.tube.2013.07.004
99
Gopal R Monin L Torres D Slight S Mehra S McKenna KC et al S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med (2013) 188:1137–46.10.1164/rccm.201304-0803OC
100
Urban CF Ermert D Schmid M Abu-Abed U Goosmann C Nacken W et al Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog (2009) 5:e1000639.10.1371/journal.ppat.1000639
101
Wallis RS Maeurer M Mwaba P Chakaya J Rustomjee R Migliori GB et al Tuberculosis – advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect Dis (2016) 16:e34–46.10.1016/S1473-3099(16)00070-0
102
Rendon A Rendon-Ramirez EJ Rosas-Taraco AG . Relevant cytokines in the management of community-acquired pneumonia. Curr Infect Dis Rep (2016) 18:10.10.1007/s11908-016-0516-y
103
McCulloh RJ Patel K . Recent developments in pediatric community-acquired pneumonia. Curr Infect Dis Rep (2016) 18:14.10.1007/s11908-016-0521-1
104
Pletz MW Rohde GG Welte T Kolditz M Ott S . Advances in the prevention, management, and treatment of community-acquired pneumonia. F1000Res (2016) 5:300.10.12688/f1000research.7657.1
105
Moorthy AN Rai P Jiao H Wang S Tan KB Qin L et al Capsules of virulent pneumococcal serotypes enhance formation of neutrophil extracellular traps during in vivo pathogenesis of pneumonia. Oncotarget (2016) 7:19327–40.10.18632/oncotarget.8451
106
Mori Y Yamaguchi M Terao Y Hamada S Ooshima T Kawabata S . α-enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem (2012) 287:10472–81.10.1074/jbc.M111.280321
107
Beiter K Wartha F Albiger B Normak S Zychlinsky A Henriques-Normak B . An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16:401–7.10.1016/j.cub.2006.01.056
108
Zhu L Kuang Z Wilson BA Lau GW . Competence-independent activity of pneumococcal EndA mediates degradation of extracellular DNA and NETs and is important for virulence. PLoS One (2013) 8:e70363.10.1371/journal.pone.0070363
109
Sumby P Barbian KD Gardner DJ Whitney AR Welty DM Long RD et al Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A (2005) 102:1679–84.10.1073/pnas.0406641102
110
Wartha F Beiter K Albiger B Fernebro J Zychlinsky A Normak S et al Capsule and d-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol (2007) 9:1162–71.10.1111/j.1462-5822.2006.00857.x
111
Short KR von Kockritz-Blickwede M Langereis JD Chew KY Job ER Armitage CW et al Antibodies mediate formation of neutrophil extracellular traps in the middle ear and facilitate secondary pneumococcal otitis media. Infect Immun (2014) 82:364–70.10.1128/IAI.01104-13
112
Narayana Moorthy A Narasaraju T Rai P Perumalsamy R Tan KB Wang S et al In vivo and in vitro studies on the role of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol (2013) 4:56.10.3389/fimmu.2013.00056
113
Murphy TF Brauer AL Schiffmacher AT Sethi S . Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2004) 170:266–72.10.1164/rccm.200403-354OC
114
Juneau RA Pang B Weimer KED Armbruster CE Swords WE . Nontypeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infect Immun (2011) 79:431–8.10.1128/IAI.00660-10
115
Juneau RA Pang B Armbruster CE Murrah KA Perez AC Swords WE . Peroxiredoxin-glutaredoxin and catalase promote resistance of nontypeable Haemophilus influenzae 86-028NP to oxidants and survival within neutrophil extracellular traps. Infect Immun (2014) 83:239–46.10.1128/IAI.02390-14
116
Hong W Juneau R Pang B Swords WE . Survival of bacterial biofilms within neutrophil extracellular traps promotes nontypeable Haemophilus influenzae persistence in the chinchilla model for otitis media. J Innate Immun (2009) 1:215–24.10.1159/000205937
117
Hamaguchi S Seki M Yamamoto N Hirose T Matsumoto N Irisawa T et al Case of invasive nontypeable Haemophilus influenzae respiratory tract infection with a large quantity of neutrophil extracellular traps in sputum. J Inflamm Res (2012) 5:137–40.10.2147/JIR.S39497
118
King PT Sharma R O’Sullivan K Selemidis S Lim S Radhakrishna N et al Nontypeable Haemophilus influenzae induces sustained lung oxidative stress and protease expression. PLoS One (2015) 10:e0120371.10.1371/journal.pone.0120371
119
Stott EJ Taylor G . Respiratory syncytial virus. Brief review. Arch Virol (1985) 84:1–52.10.1007/BF01310552
120
Nair H Nokes DJ Gessner BD Dherani M Madhi SA Singleton RJ et al Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet (2010) 375:1545–55.10.1016/S0140-6736(10)60206-1
121
Mejías A Chávez-Bueno S Jafri HS Ramilo O . Respiratory syncytial virus infections: old challenges and new opportunities. Pediatr Infect Dis J (2005) 24:S189–97.10.1097/01.inf.0000188196.87969.9a
122
González PA Bueno SM Carreño LJ Riedel CA Kalergis AM . Respiratory syncytial virus infection and immunity. Rev Med Virol (2012) 22:230–44.10.1002/rmv.1704
123
Lay MK González PA León MA Céspedes PF Bueno SM Riedel CA et al Advances in understanding respiratory syncytial virus infection in airway epithelial cells and consequential effects on the immune response. Microbes Infect (2013) 15:230–42.10.1016/j.micinf.2012.11.012
124
Everard ML Milner AD . The respiratory syncytial virus and its role in acute bronchiolitis. Eur J Pediatr (1992) 151:638–51.10.1007/BF01957564
125
Everard ML Swarbrick A Wrightham M McIntyre J Dunkley C James PD et al Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child (1994) 71:428–32.10.1136/adc.71.5.428
126
McNamara PS Ritson P Selby A Hart CA Smyth RL . Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch Dis Child (2003) 88:922–6.10.1136/adc.88.10.922
127
Geerdink RJ Pillay J Meyaard L Bont L . Neutrophils in respiratory syncytial virus infection: a target for asthma prevention. J Allergy Clin Immunol (2015) 136:838–47.10.1016/j.jaci.2015.06.034
128
Jaovisidha P Peeples ME Brees AA Carpenter LR Moy JN . Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J Immunol (1999) 163:2816–20.
129
Lindemans CA Coffer PJ Schellens IM de Graaff PM Kimpen JL Koenderman L . Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J Immunol (2006) 176:5529–37.10.4049/jimmunol.176.9.5529
130
Funchal GA Jaeger N Czepielewski RS Machado MS Muraro SP Stein RT et al Respiratory syncytial virus fusion protein promotes TLR-4–dependent neutrophil extracellular trap formation by human neutrophils. PLoS One (2015) 10:e0124082.10.1371/journal.pone.0124082
131
Karron RA Buonagurio DA Georgiu AF Whitehead SS Adamus JE Clements-Mann ML et al Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc Natl Acad Sci U S A (1997) 94:13961–6.10.1073/pnas.94.25.13961
132
Jenne CN Kubes P . Virus-induced NETs – critical component of host defense or pathogenic mediator?PLoS Pathog (2015) 11:e1004546.10.1371/journal.ppat.1004546
133
Cortjens B de Boer OJ de Jong R Antonis AFG Piñeros YSS Lutter R et al Neutrophils extracellular traps cause airway obstruction during respiratory syncytial virus disease. J Pathol (2015) 238:401–11.10.1002/path.4660
134
The IMpact-RSV Study Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics (1998) 102:531–7.10.1542/peds.102.3.531
135
Johnson S Oliver C Prince GA Hemming VG Pfarr DS Wang SC et al Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis (1997) 176:1215–24.10.1086/514115
136
Ventre K Randolph AG . Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database Syst Rev (2007) 1:CD000181.10.1002/14651858.CD000181.pub3
137
Merkus PJ de Hoog M van Gent R de Jongste JC . DNAse treatment for atelectasis in infants with severe respiratory syncytial virus bronchiolitis. Eur Respir J (2001) 18:734–7.
138
Nasr SZ Strouse PJ Soskolne E Maxvold NJ Garver KA Rubin BK et al Efficacy of recombinant human deoxyribonuclease I in the hospital management of respiratory syncytial virus bronchiolitis. Chest (2001) 120:203–8.10.1378/chest.120.1.203
139
Boogaard R Hulsmann AR van Veen L Vaessen-Verbene AA Yap YN Sprij AJ et al Recombinant human deoxyribonuclease in infants with respiratory syncytial virus bronchiolitis. Chest (2007) 131:788–95.10.1378/chest.06-2282
140
Morens DM Fauci AS . The 1918 influenza pandemic: insights for the 21st century. J Infect Dis (2007) 195:1018–28.10.1086/511989
141
Rewar S Mirdha D Rewar P . Treatment and prevention of pandemic H1N1. Ann Glob Health (2015) 81:645–53.10.1016/j.aogh.2015.08.014
142
Perrone LA Plowden JK García-Sastre A Katz JM Tumpey TM . H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog (2008) 4:e1000115.10.1371/journal.ppat.1000115
143
Tate D Brooks AG Reading PC . The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir Res (2008) 9:57.10.1186/1465-9921-9-57
144
Wareing MD Shea AL Inglis CA Dias PB Sarawar SR . CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol (2007) 20:369–78.10.1089/vim.2006.0101
145
Tate MD Deng YM Jones JE Anderson GP Brooks AG Reading PC . Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol (2009) 183:7441–50.10.4049/jimmunol.0902497
146
Narasaraju T Yang E Samy RP Ng HH Poh WP Liew A-A et al Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol (2011) 179:199–210.10.1016/j.ajpath.2011.03.013
147
Hemmers S Teijaro JR Arandjelovic S Mowen KA . PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One (2011) 6:e22043.10.1371/journal.pone.0022043
148
Tripathi S Verma A Kim E-J White MR Hartshorn KL . LL-37 modulates human neutrophil responses to influenza A virus. J Leukoc Biol (2014) 96:931–8.10.1189/jlb.4A1113-604RR
149
Neumann A Berends ET Nerlich A Molhoek EM Gallo RL Meerloo T et al The antimicrobial peptide LL-37 facilitates the formation of neutrophil extracellular traps. Biochem J (2014) 464:3–11.10.1042/BJ20140778
150
Salvatore M García-Sastre A Ruchala P Lehrer RI Chang T Klotman ME . α-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J Infect Dis (2007) 196:835–43.10.1086/521027
151
Looney MR Gilliss BM Matthay MA . Pathophysiology of transfusion-related acute lung injury. Curr Opin Hematol (2010) 17:418–23.10.1097/MOH.0b013e32833c07d3
152
Álvarez P Carrasco R Romero-Dapueto C Castillo RL . Transfusion-related acute lung injury (TRALI): current concepts. Open Respir Med J (2015) 9:92–6.10.2174/1874306401509010092
153
Bux J Sachs UJ . The pathogenesis of transfusion-related acute lung injury. Br J Haematol (2007) 136:788–99.10.1111/j.1365-2141.2007.06492.x
154
Davoren A Curtis BR Shulman IA Mohrbacher AF Bux J Kwiatkowska BJ et al TRALI due to granulocyte-agglutinating human neutrophil antigen-3a (5b) alloantigens in donor plasma: a report of 2 fatalities. Transfusion (2003) 43:641–5.10.1046/j.1537-2995.2003.00374.x
155
Silliman CC Curtis BR Kopko PM Khan SY Kelher MR Schuller RM et al Donor antibodies to HNA-3a implicated in TRALI reactions prime neutrophils and cause PMN-mediated damage to human pulmonary microvascular endothelial cells in a two-event in vitro model. Blood (2007) 109:1752–5.10.1182/blood-2006-05-025106
156
Thomas GM Carbo C Curtis BR Martinod K Mazo IB Schatzberg D et al Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood (2012) 119:6335–43.10.1182/blood-2012-01-405183
157
Caudrillier A Kessenbrock K Gilliss BM Nguyen JX Marques MB Monestier M et al Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122:2661–71.10.1172/JCI61303
158
Looney MR Nguyen JX Hu Y Van Ziffle JA Lowell CA Matthay MA . Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest (2009) 119:3450–61.10.1172/JCI38432
159
Semeraro F Ammollo CT Morrissey JH Dale GL Friese P Esmon NL et al Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61.10.1182/blood-2011-03-343061
160
Karcz M Vitkus A Papadakos PJ Schwaiberger D Lachmann B . State-of-the-art mechanical ventilation. J Cardiothorac Vasc Anesth (2012) 26:486–506.10.1053/j.jvca.2011.03.010
161
Drysdale SB Green CA Sande CJ . Best practice in the prevention and management of paediatric respiratory syncytial virus infection. Ther Adv Infect Dis (2016) 3:63–71.10.1177/2049936116630243
162
Oeckler RA Hubmayr RD . Ventilator-associated lung injury: a search for better therapeutic targets. Eur Respir J (2007) 30:1216–26.10.1183/09031936.00104907
163
Imai Y Kawano T Iwamoto S Nakagawa S Takata M Miyasaka K . Intratracheal anti-tumor necrosis factor-alpha antibody attenuates ventilator-induced lung injury in rabbits. J Appl Physiol (1985) (1999) 87:510–5.
164
Doyle NA Bhagwan SD Meek BB Kutkoski GJ Steeber DA Tedder TF et al Neutrophil margination, sequestration, and emigration in the lungs of L-selectin-deficient mice. J Clin Invest (1997) 99:526–33.10.1172/JCI119189
165
Choudhury S Wilson MR Goddard ME O’Dea KP Takata M . Mechanisms of early pulmonary neutrophil sequestration in ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol (2004) 287:L902–10.10.1152/ajplung.00187.2004
166
Jaecklin T Engelberts D Otulakowski G O’Brodovich H Post M Kavanagh BP . Lung-derived soluble mediators are pathogenic in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2011) 300:L648–58.10.1152/ajplung.00305.2010
167
Belperio JA Feane MP Burdick MD Londhe V Xue YY Li K et al Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest (2002) 110:1703–16.10.1172/JCI0215849
168
Bohrer B Silveira RC Neto EC Procianoy RS . Mechanical ventilation of newborns infant changes in plasma pro- and anti-inflammatory cytokines. J Pediatr (2010) 156:16–9.10.1016/j.jpeds.2009.07.027
169
Bose CL Laughon MM Allred EN O’Shea TM Van Marter LJ Ehrenkranz RA et al Systemic inflammation associated with mechanical ventilation among extremely preterm infants. Cytokine (2013) 61:315–22.10.1016/j.cyto.2012.10.014
170
Wang Y Li M Stadler S Correll S Li P Wang D et al Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184:205–13.10.1083/jcb.200806072
171
Mitroulis I Kambas K Chrysanthopoulou A Skendros P Apostolidou E Kourtzelis I et al Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One (2011) 6:e29318.10.1371/journal.pone.0029318
172
Yildiz C Palaniyar N Otulakowski G Khan MA Post M Kuebler WM et al Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology (2015) 122:864–75.10.1097/ALN.0000000000000605
173
Rossaint J Herter JM Van Aken H Napirei M Döring Y Weber C et al Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood (2014) 123:2573–84.10.1182/blood-2013-07-516484
174
Sadowitz B Roy S Gatto LA Habashi N Nieman G . Lung injury induced by sepsis: lessons learned from large animal models and future directions for treatment. Expert Rev Anti Infect Ther (2011) 9:1169–78.10.1586/eri.11.141
175
Seeley EJ Matthay MA Wolters PJ . Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol (2012) 303:L355–63.10.1152/ajplung.00069.2012
176
Czaikoski PG Mota JM Nascimento DC Sônego F Castanheira FV Melo PH et al Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One (2016) 11:e0148142.10.1371/journal.pone.0148142
177
Tanaka K Koike Y Shimura T Okigami M Ide S Toiyama Y et al In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One (2014) 9:e111888.10.1371/journal.pone.0111888
178
Clark SR Ma AC Tavener SA McDonald B Goodarzi Z Kelly MM et al Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9.10.1038/nm1565
179
Bagnato G Harari S . Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev (2015) 24:102–14.10.1183/09059180.00003214
180
Kolahian S Fernandez IE Eickelberg O Hartl D . Immune mechanisms in pulmonary fibrosis. Am J Respir Cell Mol Biol (2016).10.1165/rcmb.2016-0121TR
181
Zhang S Shu X Tian X Chen F Lu X Wang G . Enhanced formation and impaired degradation of neutrophil extracellular traps in dermatomyositis and polymyositis: a potential contributor to interstitial lung disease complications. Clin Exp Immunol (2014) 177:134–41.10.1111/cei.12319
182
Chrysanthopoulou A Mitroulis I Apostolidou E Arelaki S Mikroulis D Konstantinidis T et al Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol (2014) 233:294–307.10.1002/path.4359
183
Gregory AD Kliment CR Metz HE Kim KH Kargl J Agostini BA et al Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol (2015) 98:143–52.10.1189/jlb.3HI1014-493R
184
Takemasa A Ishii Y Fukuda T . A neutrophil elastase inhibitor prevents bleomycin-induced pulmonary fibrosis in mice. Eur Respir J (2012) 40:1475–82.10.1183/09031936.00127011
185
Meng W Paunel-Görgülü A Flohé S Hoffmann A Witte I MacKenzie C et al Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care (2012) 16:R137.10.1186/cc11442
186
Schauer C Janko C Munoz LE Zhao Y Kienhöfer D Frey B et al Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med (2014) 20:511–7.10.1038/nm.3547
Summary
Keywords
neutrophil, neutrophil extracellular traps, NETs, pulmonary diseases, lung infection, respiratory infection, bacteria, viruses
Citation
Porto BN and Stein RT (2016) Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing?. Front. Immunol. 7:311. doi: 10.3389/fimmu.2016.00311
Received
15 June 2016
Accepted
02 August 2016
Published
15 August 2016
Volume
7 - 2016
Edited by
Marko Radic, University of Tennessee, USA
Reviewed by
Patrizia Rovere Querini, Vita-Salute San Raffaele University, Italy; Anna M. Blom, Lund University, Sweden
Updates
Copyright
© 2016 Porto and Stein.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bárbara Nery Porto, barbara.porto@pucrs.br
Specialty section: This article was submitted to Molecular Innate Immunity, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.