 Anke Redeker
Anke Redeker Ramon Arens
Ramon Arens- Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, Leiden, Netherlands
Adoptive cellular therapy (ACT) is a form of immunotherapy whereby antigen-specific T cells are isolated or engineered, expanded ex vivo, and transferred back to patients. Clinical benefit after ACT has been obtained in treatment of infection, various hematological malignancies, and some solid tumors; however, due to poor functionality and persistence of the transferred T cells, the efficacy of ACT in the treatment of most solid tumors is often marginal. Hence, much effort is undertaken to improve T cell function and persistence in ACT and significant progress is being made. Herein, we will review strategies to improve ACT success rates in the treatment of cancer and infection. We will deliberate on the most favorable phenotype for the tumor-specific T cells that are infused into patients and on how to obtain T cells bearing this phenotype by applying novel ex vivo culture methods. Moreover, we will discuss T cell function and persistence after transfer into patients and how these factors can be manipulated by means of providing costimulatory signals, cytokines, blocking antibodies to inhibitory molecules, and vaccination. Incorporation of these T cell stimulation strategies and combinations of the different treatment modalities are likely to improve clinical response rates further.
Introduction
During the recent years, immunotherapy has emerged to be a powerful and potentially curative therapy for the treatment of various types of cancer and recurrent viral diseases. Adoptive cellular therapy (ACT) is a form of immunotherapy that involves the ex vivo isolation and expansion of antigen-specific T cells for adoptive transfer back to patients (1, 2). Although clinical benefit has been obtained in treatment of hematologic malignancies and melanoma, the efficacy of ACT in the treatment of most solid tumors is thus far limited since transferred T cells fail to function and persist in vivo. This is in sharp contrast to clinical results obtained with patients treated by ACT for virus-associated malignancies and recurrent viral infections. Here, sustained presence of functional virus-specific T cells is observed, even up to 9 years post-infusion (3–5). This prolonged presence of transferred virus-specific T cells translates into a high clinical response rate that is being observed in patients that are treated with these cells. The superior efficacy obtained with ACT in the treatment of viral infections and virus-associated malignancies compared to the treatment of most solid cancers can be attributed to several factors, including tolerance to tumor-associated antigens (TAAs) and inhibition of tumor-specific T cells due to the suppressive tumor environment. Moreover, also the necessity for extensive culturing of tumor-specific T cells to obtain sufficient numbers for infusion into patients greatly influences the quality of the T cells and, hence, persistence and anti-tumor efficacy in vivo. In addition to lessons that can be learned from studying T cells in a setting of viral infection, valuable lessons can also be learned by critical evaluation of results obtained with current protocols and, importantly, by improving our understanding of the underlying mechanisms. In this review, we will focus on current protocols of adoptive T cell therapy in cancer treatment, and discuss the various attempts to improve the clinical success rate of ACT by aiming to advance the quality of the infused T cells through delivery of costimulatory signals and cytokines, blocking of inhibitory signals and vaccination. As such, these developments are of interest for ACT improvement in cancer but also for other complicated T cell-dependent treatment modalities.
Approaches of Act
One form of ACT involves expansion and infusion of natural T cells isolated from autologous tumors. Generation of tumor-infiltrating lymphocyte (TIL) cultures is performed by first culturing resected tumor fragments or tumor single-cell suspensions in medium containing IL-2 for 3–5 weeks followed by a rapid expansion protocol (REP) involving the activation of TILs using an anti-CD3 monoclonal antibody in the presence of irradiated peripheral blood mononuclear cells (PBMC) and IL-2 (2, 6, 7). Systemic administration of TILs to patients with advanced stage melanoma has mounted high and durable responses that resulted in objective clinical responses in >50% of the patients and complete regression in up to 24% of the patients (6, 8–10). However, such results have only been described for ACT in melanoma patients and not for other solid tumors. This is probably due to the high mutational load in melanoma giving rise to neoepitopes, which can serve as neoantigens facilitating tumor recognition by T cells (11–14). The stability of this neoantigen expression, however, is altering upon ACT demonstrating a dynamic interaction of the transferred T cells with their targets and advocates for ACT procedures inducing a broad tumor-specific T cell response (15).
In most medical centers, lymphodepletion before transfer is a standard part of the treatment (16). However, ~50% of the patients experience side effects of this pretreatment, which are mostly infection related, i.e., neutropenia and bacteremia (10). There is some evidence that alternative approaches can overcome the necessity to pre-condition the patient, e.g., selection of particular T cell clones, tailoring tumor-specific T cells to produce IL-12 or administration of low dose IFN-α (17–19). Another hurdle in ACT for solid tumors is the failure to successfully isolate TILs or expand TILs to sufficient numbers. In ACT for melanoma patients, these procedures are usually very efficacious, showing a success rate of more than 50% (7, 8). However, TILs harboring sufficient anti-tumor activity can rarely be generated from tumors other than melanoma and, moreover, for other types of cancer, adequate amounts of surgical/bioptic material is often not available (20).
One strategy to circumvent these limitations is genetic engineering of autologous T cells by lentiviral or retroviral transduction to express TCRs that recognize TAAs. Although a promising clinical response rate of 30% was observed in a clinical trial for melanoma patients using a high-affinity HLA-A0201-restricted MART-1 TCR, in 29 out of 36 patients severe off-target toxicity was seen in the skin, ears, and eyes as destruction of melanocytes also occurred at these sites (21). In a clinical trial where myeloma and melanoma patients were infused with autologous engineered T cells expressing an affinity-enhanced TCR against MAGE-A3, the first two patients died of cardiogenic shock. This severe cardiac toxicity was due to recognition of a MAGE-A3-unrelated protein expressed by normal cardiac tissue (22). This off-target activity is likely caused by the fact that an affinity-enhanced TCR was used instead of the low-affinity parental TCR against MAGE-A3. Thus, a major drawback of this approach is the (sometimes unidentified) expression of target antigens on healthy tissue resulting in unwanted cross-reactivity. Nevertheless, certain antigens, such as cancer-testis antigens (CTAs), do form an attractive target since they are expressed by a variety of tumor types, but usually not by adult tissue, with the exception of germline cells on which HLA class I and II is not expressed. In clinical trials, targeting of the CTA NY-ESO-1 antigen, 61% of synovial cell carcinoma patients and 55% of the melanoma patients demonstrated objective clinical responses without signs of off-target toxicity (23). Another report showed even an 80% response rate in multiple myeloma (24). Although TCR transduction allows the generation of tumor-specific T cells without the necessity to isolate TILs, a major limitation of this approach is the HLA-restriction. For example, transduction of a TCR that recognizes its antigen in the context of HLA-A*0201 is only functional in patients with the same HLA type.
An alternative approach to obtain T cells with anti-tumor reactivity without the complication of HLA-restriction is genetic engineering of T cells to express chimeric antigen receptors (CARs). CARs are constructed by linking an antigen-binding domain, usually a single-chain variable fragment (scFv), to an intracellular T cell signaling domain such as CD3-ζ (first generation CAR), and currently also including one or two costimulatory domains (second/third-generation CAR). The specific binding of CAR T cells occurs, thus, in a non-MHC-restricted fashion, yet antigen-binding results in T cell activation. The most impressive clinical results so far have been obtained using CAR T cells targeting CD19 in patients suffering from B cell malignancies (25–30). Mixing defined populations of CD4+ and CD8+ CAR T cells recognizing both CD19 further improved this therapy (31, 32). However, since all CD19-expressing cells are targeted using this approach, also non-malignant B cells are depleted. The drawback that healthy cells expressing the antigen are also targeted by CAR T cells has also been reported for CARs directed to her2/neu and carboxy-anhydrase-IX (33, 34). In addition to this on-target off-tumor effect, acute anaphylaxis and tumor lysis syndrome (TLS) occurs frequently after CAR T cell therapy, but most often observed is cytokine release syndrome (CRS) (35–37). It has been suggested that the incidence and severity of the CAR T cell-mediated toxicity is related to tumor burden and T cell infusion dose. To minimize toxicity in patients with a high tumor burden, treatment with a low T cell dose may be required (30, 32, 35, 37). Other strategies to overcome these adverse events include addition of a suicide gene (e.g., HSV-TK and iCasp9), whereby transferred cells can be selectively eliminated, and the generation of CAR T cells with dual specificity whereby each CAR targets a different tumor antigen and only engaging of both results in proper T cell activation and effector function (38–44).
Clinical trials using CAR T cells targeting other antigens than CD19 have, thus far, only shown limited anti-tumor efficacy. In a trial wherein neuroblastoma patients received CAR T cells recognizing the extracellular domain of L1-CAM, present on neuroblastoma cells, two out of six patients showed a clinical response (34). However, in two other trials, using CAR T cells specific for carboxy-anhydrase-IX and alpha-folate receptor to treat renal cell carcinoma and ovarian cancer, respectively, no clinical responses were observed (45, 46). The limited success for these CAR T cells may in part be due to antigen-independent CAR signaling due to clustering of CAR scFvs resulting in their early exhaustion. This tonic CAR signaling is observed for several CARs, except the CD19 CAR (47). Incorporation of the endodomain of 4-1BB (CD137), a costimulatory member of the TNF receptor (TNFR) superfamily, rather than a CD28 domain ameliorates this induction of exhaustion (47, 48). In addition, novel targets for CAR T cell therapy for solid tumors are on their way, which may have high clinical potential. For example, it was recently reported that CAR T cells can be engineered to target aberrantly glycosylated antigens on MUC1, which is expressed by multiple cancers, thereby providing a potential broad application (49).
Although CAR T cells and TCR transgenic T cells are favorable cancer treatment modalities, they usually target a single tumor antigen, which increases the chance of tumor escape (50, 51), and limits eradication of the often very heterogeneous tumors. The use of a combination of CARs with different antigen specificity or bispecific CARs could prevent antigen escape (52).
Quality of Anti-Tumor T Cells
A major challenge in ACT is to obtain sufficient numbers of tumor-specific T cells for infusion into patients and, importantly, since durable clinical responses profoundly depend on persistence of the infused T cells, the transferred cells should have the capacity to persist long-term in vivo (53). Several reports suggest that the relative contribution to long-term persistence of T cells mainly comprises the least effector-differentiated memory T cells: central memory T cells (Tcm) and T memory stem cells (Tscm) (54). Tcm and Tscm circulate in the lymphoid organs and are endowed with an excellent expansion potential upon antigenic challenge as opposed to more differentiated memory T cells. Effector and effector memory T cells (Teff/Tem) home to tissues and respond to antigen with immediate effector function as compared to Tscm/Tcm, but have a reduced regenerative capacity (55). In addition, Tem in humans can be subdivided into cells that are either CD45RA− or cells that re-express CD45RA+. The re-expressing cells, termed Temra, are thought to be the most differentiated memory cells, as these cells have low proliferative capacity, strong cytotoxic potential, and a higher susceptibility to apoptosis (56).
Tscm have the capacity to differentiate into Tcm and Tem, and display a superior potential to self-renew as evidenced by a positive correlation of the amount of infused Tscm with early expansion after transfer and absolute numbers of long-term persisting cells (57–59). However, very low numbers of Tscm are found in the periphery and extensive expansion would be required, which likely results in loss of memory potential (60, 61). The limitation of low natural frequencies can be bypassed by targeting the Wnt/β-catenin pathway in naive cells that results in arrested Teff differentiation and promotion of memory-like CD8+ T cells with Tscm features. Although targeting the Wnt signaling pathway appears to be an effective method to promote stemness and inhibit differentiation, this may restrict the proliferation and function; hence, further research is required for its suitability to improve ACT (62). An alternative method to generate sufficient Tscm is a procedure whereby human naive T cells are activated by CD3/CD28 engagement and culturing in the presence of IL-7, IL-15, and IL-21 (63, 64). Another approach currently being explored is based on inhibition of the Akt-signaling pathway during the ex vivo expansion of tumor-specific T cells resulting in the induction of early memory-like cells (65, 66). The advantage of this approach is that the ex vivo proliferation is not strongly inhibited and sufficient numbers of cells can be obtained for treatment. However, the role of Akt in T cell differentiation and metabolism needs to be further validated in order to determine if Akt inhibition could potentially be used in ACT protocols. Thus, although it is clear that Tscm have excellent stemness properties and much effort is being made to optimize isolation and expansion protocols, there are still some major hurdles and it is, therefore, not feasible yet to use these cells routinely for adoptive cell therapy.
A recent report demonstrates an alternative approach in which TCR transgenic CD8+ T cells were successfully reprogrammed into induced pluripotent stem (iPS) cells using a Sendai virus vector. After transfer into melanoma-bearing mice, iPS-derived T cells mediated potent anti-tumor activity. Nevertheless, their anti-tumor activity and persistence were comparable with their non-reprogrammed counterparts (67). Importantly, the Busch laboratory convincingly showed in mice that also Tcm have stemness and long-term persistence potential after transfer. Actually, both naive T cells and Tcm cells were highly efficient in inducing epitope-specific T cell populations during serial single cell adoptive transfers (68). Also, infused Tcm clones in monkeys and humans have shown to have the capacity to mount long-term persistent clonotypes, and furthermore CD19 CAR T cells derived from Tcm have superior anti-tumor effects (31, 59, 69, 70).
In the current point of view, both Tscm and Tcm seem to be bona fide T cell subsets to use in ACT. Moreover, also naive T cell subsets have the potential to establish long-term persistence allowing for prolonged anti-tumor activity (71, 72). However, these less-differentiated T cell subsets are not per definition superior in all tumor eradication settings. In cases of solid tumors where the level of tumor-antigen presentation by antigen-presenting cells in lymphoid organs is low, these T cell subsets may not be activated sufficiently to exit the lymphoid organs and invade the tumor to exert their anti-tumor effects. One strategy to overcome this hurdle is increasing the level of antigen presentation in the lymphoid organs by vaccination, which results in appropriate T cell stimulation (as will be discussed later). Another approach is co-infusion of Teff and Tem cells. These cells have direct effector function and have (extralymphoid) tissue migrating properties leading to tumor destruction (55, 73–75). Consequently, this may also lead to sufficient activation of the co-transferred Tcm/Tscm, which enables long-term anti-tumor immunity.
Ex Vivo Expansion Protocols and Costimulation
The expansion protocols that are currently used to expand TILs or generate engineered tumor-specific T cells often mount expanded T cell pools with a highly differentiated phenotype that have lost CD28 expression, decreased expression of the costimulatory TNFR family member CD27 and more susceptibility to activation-induced cell death (AICD) (76–78). Approaches to obtain sufficient numbers of TILs with a favorable phenotype or to reprogram TILs or TCR engineered T cells to the preferred phenotype during ex vivo expansion include manipulation of critical costimulatory and cytokine signaling pathways. Costimulatory signals can be provided via agonistic antibodies and artificial APCs (aAPCs), of which the latter can either be cell-based or non-cell-based (79). An advantage of non-cell-based aAPCs over cell-based aAPCs is that they can be engineered to be magnetic, which makes removal of the cells before infusion of the T cell product straightforward. Also bio-degradable particles can be designed of which removal is not necessary. One of the costimulatory pathways known to be critical for priming T cells, the CD28 pathway, is currently implicated in ACT protocols for ex vivo expansion and transduction. Another candidate is 4-1BB, which is expressed on activated T cells and upon triggering enhances T cell responses by promoting proliferation, survival, and effector function, and by regulating the suppressive potential of regulatory T cells (Tregs) (80). Comparison of aAPCs providing costimulation via CD28 or 4-1BB showed that signaling through 4-1BB preferably expands memory CD8+ T cells, whereas CD28 costimulation favors expansion of naive cells. In addition, the CD8+ T cells that received 4-1BB signals displayed improved cytolytic function (81). Interestingly, enhanced 4-1BB costimulation through an agonistic antibody has been shown to rescue expression of CD27 and CD28 on post-REP CD8+ TILs, improved expansion of CD8+ T cells, and increased responsiveness to antigenic re-stimulation and increased expression of the CD127 (IL-7Rα) (82, 83). Also, when combined with a potent TCR trigger, signaling through 4-1BB induces prominent upregulation of CD25 (IL-2Rα) and IL-2 (84). Thus, while generating tumor-specific T cell pools from naive cells, 4-1BB triggering could promote the generation of T cells capable of expanding upon secondary challenge. Another costimulatory molecule that could potentiate ex vivo culturing of tumor-specific T cells is CD27. The interaction of CD27 with its ligand CD70 has been shown to be important for IL-2-mediated T cell activation and in vitro activation of human T cells with anti-CD3 in the presence of an agonistic CD27 antibody showed comparable expansion potential as stimulation through 4-1BB (85). On the other hand, in vitro experiments have shown that in a co-culture of naive CD4+ T cells with CD70 expressing tumor cells, Tregs accumulate because of increased IL-2 production by non-Treg CD4+ cells (86). Other costimulatory members of the TNFR superfamily include OX40 (CD134), HVEM, and GITR, and agonistic antibodies targeting these molecules could also potentially be used to improve REP cultures. OX40 has been described to promote T cell expansion and survival, the latter probably by regulating BCL-2 and BCL-xL expression (80, 87, 88). It has been shown that ligation of OX40 increases expression of IL-7Rα on antigen-specific CD8+ T cells, which leads to enhanced survival and accumulation upon IL-7 signaling, and combining OX40 and 4-1BB costimulation further enhanced this effect (89). Thus, to further improve the ex vivo culturing procedure, targeting of two or more costimulatory pathways simultaneously can be taken into consideration. Importantly, although the signal strength that is delivered to the T cells should be robust enough for proliferation, it should not result in an overall terminal differentiation of the T cells. An alternative approach is to make combinations of an agonistic antibody with cytokines that prevent overt differentiation, as will be discussed hereafter. To be able to select the most favorable agonist–cytokine combinations, it would be highly recommendable to expand our knowledge regarding the effect on the expression of cytokines and cytokine receptors by targeting the costimulatory pathways simultaneously.
Ex Vivo Expansion and Cytokines
An alternate strategy to boost cultured T cells and modulate the phenotype is via cytokine-mediated signals. The common-gamma chain (γc)-cytokine IL-2 is long been known to massively expand T cells, and high doses of IL-2 have been used to establish and expand ACT T cell cultures for more than 20 years (90). Enforced expression of IL-2 by the T cells themselves results also in prolonged survival in vitro and maintains the tumor specificity and function (91, 92). However, IL-2 can promote differentiation of T cells (93, 94), which may lead to an unfavorable phenotype for ACT usage. So strategies to optimize ex vivo T cell cultures for ACT involving the (co-)use of alternative cytokines are fully explored. Next to IL-2, other γc-cytokines, such as IL-7, IL-15, and IL-21, have been described to play a role in memory T cell formation, proliferation, and survival, yet result in a lower degree of T cell differentiation but are still able to enhance anti-tumor responses (95–99). Also IL-12 and IFN-α, non-γc-cytokines, hold promise to enhance the efficacy of ACT. IL-12 has been shown to play an essential role in T cell differentiation and memory formation and IFN-α is important in driving memory cell development (100–102). Use of these cytokines in ex vivo T cell cultures present a promising moiety to yield T cells with an improved capacity to respond (103, 104). T cells forced to secrete IL-12 benefited also of this cytokine during culture (105).
In particular combinations of cytokines have shown encouraging results. In expansion, protocols using naive T cells as a starting source, different combinations of IL-2, IL-7, IL-15, and IL-21 have proven to efficiently expand T cells and result in populations expressing early-differentiation markers, such as CD27, CD28, CD62L, and CD127 (64, 106–108). It has also been shown that CAR T cells can be efficiently expanded using a protocol devoid of IL-2, using CD3 and CD28 stimulation in the presence of IL-7 and IL-15 (109). Interestingly, also in TIL cultures and TCR/CAR engineered T cell cultures that are established in the presence of anti-CD3 and IL-2 and usually display a substantial degree of differentiation, cytokine cocktails were able to establish T cell populations with a less-differentiated phenotype (76, 110, 111). This suggests that cytokine cocktails can be used to reprogram late differentiated T cells. Besides delivery of cytokine-mediated signals via cytokine supplementation to the cultures, aAPCs can be designed to express cytokines or T cells can be triggered or engineered to produce abundant cytokines themselves (99, 112–114). The advantage of aAPCs is that they can be designed to simultaneously provide costimulation and cytokine-mediated signals (112, 115). Irrespective of the cytokine delivery manner, more research is required to pinpoint the amount, ideal timing, and combination of cytokines for ex vivo cultures (108, 110). Moreover, requirements for establishing cultures from naive T cells or previously primed T cells are likely to be different. For example, addition of IL-21 causes naive T cells to significantly expand, while memory T cells fail to do so (98). Nevertheless, in both subsets, IL-21 signaling increases CD28 expression.
In Vivo Costimulation
Another approach to improve ACT is by enhancing the T cell expansion and function after transfer. The essentiality of costimulatory pathways has been demonstrated in experimental settings of adoptive transfers showing, e.g., that CD27- and CD28-mediated costimulatory signals are important for expansion of both naive and memory CD8+ T cells upon transfer (116, 117). To study the benefit of augmenting costimulatory pathways in patients after T cell infusion, preclinical studies and clinical trials have been performed exploring the use of agonistic antibodies against TNFR superfamily members 4-1BB, OX40, GITR, CD27, HVEM, and CD40. The promise of these molecules in cancer immunotherapy has been reviewed recently (118–120).
Besides expansion, an additional beneficial effect delivered by costimulation is induction of T cells with the capacity to produce IL-2 that acts in an autocrine manner (84, 121). In contrast to exogenous IL-2, either provided in vitro or in vivo, autocrine IL-2 seems to be highly beneficial for both the (secondary) expansion potential and survival of CD8 T cells (113, 122). However, in order to be able to optimize ACT protocols further and minimize the chance of severe adverse side effects as observed in the Phase I clinical trial with anti-CD28 and to a lesser extent in the Phase II study with the anti-4-1BB antibody Urelumab (NCT00612664), which was associated with a high incidence of severe hepatitis, a better understanding of the underlying mechanisms by which these antibodies exert their effects is crucial (123, 124). In preclinical models, agonistic 4-1BB contributes to tumor regression by promoting survival and avoiding AICD of CD8+ T cells and more importantly in the context of this review; in models using OVA-expressing tumors, it has been demonstrated that a combination of agonistic 4-1BB antibody and transfer of OVA-specific CD8+ T cells significantly improves tumor control (125, 126). Whether combining ACT and 4-1BB agonists enhances anti-tumor activity in humans has not yet been assessed, but when used as a monotherapy, 4-1BB antibodies seem to have some anti-tumor activity. Although two 4-1BB agonists have already been used in clinical trials, only recently more insight into the mechanisms by which anti-tumor effect is exerted, is obtained, and it has become clear that at least in preclinical models systemic 4-1BB activation induces a phenotype of CD4+ and CD8+ T cells that is characterized by high expression of the T-box transcription factor Eomes, KLRG1+, and high cytotoxic capacity (125, 127–129). KLRG1 marks Tem and Teff cells and as already mentioned above, ACT of Tem and Teff cells combined with less-differentiated cells might be beneficial. Furthermore, agonistic 4-1BB antibody treatment correlated with decreased expression of the inhibitory receptors programmed death-1 (PD-1) and Lag3.
OX40 signaling can enhance T cell differentiation and survival via effects on IL-2 and IL-7-mediated signaling, and via increasing the anti-apoptotic molecules Bcl-2 and Bcl-xL (130) Essentially, providing OX40 triggering augmented anti-tumor activity in a preclinical model of adoptive T cell transfer mediated by both CD4+ and CD8+ T cells (131). Conflicting results are reported on whether Treg responses are inhibited or promoted by OX40, which is most likely due to differences in dose and/or timing of OX40 ligation, and may depend on the model/setting (132). Although unraveling the precise mechanism of OX40 agonists remains a challenge, anti-OX40 has already been used in a Phase I clinical trial for patients with metastatic solid malignancies, albeit not in ACT settings (133). Results were promising, and indicated enhanced proliferation of CD4+ and CD8+ T cells that coincided with regression of at least one metastatic lesion in 12 out of 30 patients. Tregs in the tumor showed a higher expression of OX40 compared to peripheral blood Tregs.
Explored as well, albeit to a lesser extent are agonistic CD27 antibodies (134, 135). Promising results were reported in preclinical models, and are likely related to improved CD27-mediated T cell expansion, survival, and function (77, 136–138). Conversely, it has also been reported that CD27 signaling can increase survival of Tregs in vivo and thereby promote tumor progression (86). The Teff:Treg ratio in the tumor has been suggested to determine whether CD27 agonist will promote or diminish tumor control (139).
An indirect way to improve the efficacy of transferred T cells is via administration of agonistic antibodies to CD40 resulting in activation of APCs, such as dendritic cells (DCs) (120). Consequent upregulation of costimulatory molecules on the APCs then provide the necessary stimulatory signals to activate tumor-specific T cells. In addition to DCs, CD40 antibodies also activate other myeloid cells (140) and the activity can also depend on complement-mediated cytotoxicity (CMC) or antibody-dependent cell-mediated cytotoxicity (ADCC), or even be immune effector independent when CD40 is expressed on tumor cells (141–146). Important to note is that CD40 triggering in malignant cells is able to promote tumor cell proliferation leading to tumor progression (147, 148). Likely depending on one or more of the above-described mechanisms, targeting of CD40 has already been proven a promising strategy in several preclinical models and clinical trials against solid cancer (120, 149–154). Also in preclinical models of ACT, agonistic CD40 antibodies promote tumor-specific T cell expansion and enhanced anti-tumor activity (155, 156). Thus, clinical effectiveness in ACT has potential given that CD40 antibody-associated toxicity is managed (152, 157). So far three agonistic CD40 antibodies, which differ in their agonistic activity, have been tested in clinical trials. The strongest agonistic antibody, CP-870.893, is a humanized antibody of an IgG2 isotype. Human IgG2 antibodies typically interact marginally with Fc receptors and are, therefore, not very effective mediators of CMC and ADCC (158). Nevertheless, this antibody is a potent activator of macrophages and DCs and can thereby mediate T cell-dependent anti-tumor immune responses, which suggests that it has the potential to enhance ACT. The two other antibodies that have been tested in the clinic (i.e., Dacetuzumab and ChiLob 7/4), displaying less agonistic activity compared to CP-870.89, are of an IgG1 isotype and, hence, are more potent mediators of CMC and ADCC, making them less suitable for combinations with T cell transfer (120). Adverse effects that were observed after CD40 antibody treatment include CRS and liver damage (120). Targeting of CD40 is also possible by imposed expression of CD40L (CD154) on the transferred T cells. In an experimental model, CD19-specific CAR/CD40L T cells displayed increased cytotoxicity and enhanced tumor eradication (159).
Also currently under investigation in clinical trials is an agonistic antibody against GITR. In multiple animal models of cancer, this antibody has proven to exert anti-tumor immune responses by providing costimulatory signals to T cells and skewing the balance between induced Treg and TH9 cell differentiation in favor of TH9 (160–162). In a preclinical adoptive transfer setting, agonistic GITR antibody has shown to increase the polyfunctionality of the transferred T cells and reduce the frequency of Tregs in the tumor, resulting in tumor regression (163). Repetitive doses of a GITR agonist is, however, potentially toxic (164).
Taken together, considerable progress has been made in dissecting the mechanisms by which agonistic antibodies to costimulatory molecules exert their anti-tumor effects but further unraveling is required to be able to implement this therapy into patients receiving ACT. Importantly, in case of treatment with such powerful agonists, also the mechanisms underlying the adverse immune-mediated side effects require attention. Undoubtedly, the effects of agonistic antibody administration are often multifaceted thereby making it challenging to predict treatment outcome. Attempts to minimize the chance of antibody-induced toxicity could include pretreatment with corticosteroids and local administration of agonistic antibodies (151, 152, 154).
As mentioned before, second-generation CAR T cells contain a costimulatory domain placed in series with CD3ζ and thereby costimulation is provided per definition upon target recognition. Since it is well appreciated that T cells require costimulation for proper activation, it is not surprising that incorporation of costimulatory domains advanced CAR T cell treatment. Alike for providing costimulation by agonistic antibodies, the choice of the costimulatory signaling domain influences CAR T cell functionality and persistence, i.e., by differential regulation of down-stream signaling expression. Most extensively explored are CAR T cells with incorporated CD28 and 4-1BB signaling domains and although treatment with both CAR T cells have resulted in clear clinical responses, comparisons showed prolonged persistence and ameliorated exhaustion of CAR T cells using the 4-1BB domain (47, 165, 166). Alternative signaling domains that have been integrated include domains of CD27, ICOS, and OX40 (165, 167–169). In third-generation CAR T cells, attempts to further enhance anti-tumor activity and long-term persistence rely on incorporation of two costimulatory domains. So far, combinations of CD28 with 4-1BB and CD28 with OX40 have shown to be promising, resulting in T cells having potent effector functions and improved capacity to persist long term (168, 170). In a small pilot trial, a CD20-specific CAR with CD28 and 4-1BB costimulatory domains has been tested in four relapsed indolent B cell and mantle cell lymphoma patients and the data suggest improved CAR T cell persistence (171).
In addition to improving T cell function by triggering costimulatory pathways, inhibitory pathways can be blocked and this strategy, also known as immune checkpoint blockade, has led to significant clinical advances in cancer immunotherapy (172–174). Several reports show that combinations of ACT and blockade of inhibitory molecules, such as CTL-associated antigen 4 (CTLA-4) and PD-1, have the potency to augment anti-tumor efficacy and increase T cell persistence (175–179). An alternative method in which PD-1-mediated inhibition was turned into CD28-mediated costimulation by generating PD-1–CD28 fusion receptors was also effective in ACT (180). Nevertheless, although targeting of either costimulatory or inhibitory pathways for the benefit of ACT may improve anti-tumor responses, for achieving greater clinical response rates, combinations of the two might be required and are currently under investigation. So far this approach has yielded encouraging results as evidenced by inhibition of tumor growth in preclinical settings, including ACT cancer models (181–185). Decreased tumor progression coincided with an increase in Teffs and a decrease in Tregs and myeloid suppressor cells at tumor sites. This shift from a more immunosuppressive to a more immunostimulatory tumor environment might explain the potent effects of these antibody combinations.
Cytokines In Vivo
Adoptive cellular therapy using TILs generally includes in vivo administration of high-dose IL-2 to improve proliferation and survival of the transferred TILs. Unfortunately, exogenous IL-2 treatment has two major drawbacks; it is often associated with severe toxicity and can promote Treg proliferation. It has been reported that the number of doses of IL-2 that are administered after adoptive TIL transfer is positively correlated with Treg reconstitution after lymphodepletion and, furthermore, that the degree of Treg reconstitution is inversely correlated with the patient’s response to treatment (186).
Attempts to circumvent IL-2-induced toxicity and Treg stimulation have been made. A straightforward measurement is by reduction of the IL-2 administration (187). Tailoring CD8+ T cells to augmented autocrine IL-2 production seems an alternative promising manner, which increases the availability of IL-2 to the right cell without promoting Treg proliferation. This can be achieved using retroviral or lentiviral transduction and this would especially be feasible in situations where transduction is already required. For instance, in case of generating TCR engineered T cells, but in fact also TILs can be transduced in the same manner. Recently, we have shown in a preclinical model that CD8+ T cells cultured in the presence of IL-7 and IL-15 that are forced to overexpress IL-2 display improved persistence and expansion potential after transfer and subsequent vaccination (113). Consequently, this heightened anti-viral and anti-tumor immunity in vivo compared to mock transduced cells. Notably, after in vivo secondary challenge, the cells with elevated autocrine IL-2 efficiently re-expanded yet also expressed IL-7Rα, suggesting that although these cells underwent prolonged IL-2 signaling, they still seem to be of a less-differentiated phenotype, which may be related to the transduction procedure in the presence of IL-7 and IL-15. In addition, we did not observe any alterations in Treg homeostasis (113). ACT with human T cells overexpressing IL-2 has also been explored yielding promising results with respect to longevity but large clinical studies should be performed to determine if IL-2 over-expressing T cells result in clinical benefit (44, 92). Likely, ACT approaches with IL-2+ T cells are most successful when they are combined with vaccination given the prominent role of autocrine IL-2 production for secondary expansion of CD8+ T cells (113, 122). As discussed before, another strategy to circumvent exogenous IL-2 administration is to provide agonistic antibodies to costimulatory receptors that promote autocrine IL-2 production in T cells.
Other cytokines than IL-2 have also been explored to enhance ACT-mediated T cell responses. IL-7 and IL-15 are crucial cytokines for lymphoid homeostasis by playing an important role in orchestrating the survival of naive and memory T cells and memory cell differentiation (188, 189). Increased availability of IL-7 and IL-15 has been shown to be an important mechanism by which a lymphodepleting regimen improves the engraftment of the adoptively transferred T cells and, hence, the success of ACT (190–192). Preclinical ACT models, in which the effect of exogenous IL-7 and IL-15 on tumor outgrowth has been tested, demonstrated that both cytokines can improve tumor control, including in vaccinated lymphodepleted or immunodeficient hosts (193–195). Recently, a phase I clinical trial has been conducted to determine safety, adverse event profiles, and the maximum tolerated dose of rhIL-15 in humans (196). Patients with metastatic melanoma and metastatic renal cancer were infused with different doses of IL-15 (0.3/1.0/3.0 μg/kg/day) for 12 consecutive days and this treatment regimen resulted in markedly altered homeostasis of mainly NK cells, γδ cells, and to somewhat lesser extent of memory CD8+ T cells. No clinical responses according to the RECIST criteria (197), which includes the persistence of the cells after transfer, were observed and the maximum tolerated dose was determined to be the lowest used dose. Because of clinical toxicity caused by strong cytokine production, the authors stated that rhIL-15 is too difficult to administer intravenously and suggest developing alternative dosing strategies and new trials to assess this are being conducted (196). IL-7 administration is tolerated better in humans, but anti-tumor efficacy requires further evaluation (198, 199).
As for IL-2, systemic IL-15-mediated toxicity might be circumvented by tailoring tumor-specific T cells to express IL-15. In that way, the IL-15-mediated effects are likely confined to the tumor environment, eluding systemic toxicity. In experimental models, it was shown that IL-15-expressing CD8+ T cells improve anti-tumor activity (95), and human IL-15 secreting cells perform also well in vivo (43, 200, 201).
The more recently discovered member of the γc cytokine family, IL-21, has also been explored as an anti-cancer treatment and IL-21 monotherapy of thymoma and melanoma in mice has shown to result in improved CD8+ T cell-mediated anti-tumor responses with augmented long-term survival (193, 202–204). IL-21 treatment prolonged persistence of endogenous and adoptively transferred tumor-specific transgenic CD8+ T cells, which was mainly attributed to IL-21-mediated promotion of survival (202). Additionally, it has been shown that IL-21 is able to potentiate tumor-specific antibody responses, which resulted in complement-mediated tumor cell lysis (204). Combination of cytokines involving IL-21 demonstrated further enhancement of anti-tumor immunity compared to IL-21 as a monotherapy. In a study wherein mice were challenged with B16F10 melanoma, treatment by adoptive transfer of transgenic tumor-specific CD8+ T cells, combined administration of IL-21 and IL-2 and vaccination resulted in higher absolute numbers of circulating tumor-specific T cells and improved tumor-free survival compared to therapy with IL-2 or IL-21 alone (203). In addition, in a model using murine B16 melanoma cells that were transfected to secrete IL-21, it was shown that local presence of IL-21 can also promote anti-tumor immunity by preventing IL-2-mediated Treg induction (205). Experiments in the B16 model has shown that mixing IL-21 with IL-15 improves expansion of transferred tumor-antigen-specific CD8+ T cells and enhances tumor control after vaccination (106). Clinical trials using IL-21 as a single agent in melanoma and renal cell carcinoma show that this cytokine is well tolerated and favorable clinical responses have been observed as evidenced by patients in which disease stabilized (206, 207). To our knowledge, the anti-tumor effect of IL-21 in human ACT has not been addressed yet. A study by Markley and Sadelain showed that forced expression of IL-7 and IL-21 by CD8+ T cells resulted in improved rejection of systemic lymphoma compared to T cells that overexpressed IL-2 or IL-15 (99). However, in these experiments, vaccination was not provided post-transfer, which may have resulted in improved expansion of the transferred IL-2+ cells.
Anti-tumor efficacy of some cytokines that do not belong to the γc cytokine family has been explored as well. In preclinical models, administration of IL-12 caused tumor regression and promoted survival of tumor-bearing animals. This provided the rationale for applying IL-12 treatment in a clinical setting, however, translation into the clinic was hindered by severe toxicity (208, 209). To be able to explore IL-12 as a treatment modality, multiple attempts have been made to design a safe IL-12-based treatment, including different administration schedules and routes and intratumoral delivery (210). Several reports explored the therapeutic efficacy of tumor-specific T cells designed to express IL-12. In experimental settings, T cells modified to produce IL-12 improved tumor eradication (19, 101, 211–213). Promising results were obtained using IL-12 gene transduced human TILs. Here, different doses of IL-12-producing TILs were infused into metastatic melanoma patients and higher doses resulted in better clinical responses (214). Nevertheless, IL-12 transduced TILs did not persist for more than a month. In addition, higher cell doses led to severe adverse side effects attributable to the secreted IL-12 (214). The lack of persistence of the transferred T cells that is observed might be due to a negative feedback loop that involves IL-10 production to limit ongoing T cell activation (215). So far, these reports suggest that ACT with IL-12-producing T cells has potential but improving the longevity of the cells and measures to prevent IL-12-mediated toxicity are vital for further application.
IFN-α is known for its direct anti-tumor effect and is currently a frequently used cytokine for the treatment of cancer. It has also been recognized that IFN-α promotes T cell activation, survival, expansion, and memory formation through activation and differentiation of DCs (216). The mechanism by which IFN-α mediates memory formation is suggested to be mediated by enhancement of IL-15 presentation of DCs to T cells (217). Moreover, IFN-α can also affect T cell expansion directly. This so-called signal 3 further amplifies the signals T cells receive via the TCR (signal 1) and costimulatory receptors (signal 2) (218–220). However, due to severe adverse events caused by IFN-α administration, treatment is discontinued in up to 50% of the cases. Frequently observed symptoms of IFN-α-related toxicities include “flulike” symptoms, fatique, anorexia, and nausea (18, 221). To improve tolerability of IFN-α treatment, a polyethylene glycol (peg) moiety was added, resulting in a longer half-life. This allowed for less-frequent administration and, hence, less toxicity. Accordingly, discontinuation rates of IFN-α treatment were lower when a pegylated form was used (up to ~25%). To further enhance the anti-tumor effect of cytokines in ACT, blocking antibodies to inhibitory receptors can be co-administered, e.g., IL-21 and CTLA-4 (222).
Overall, we conclude that although combining ACT with cytokine treatment seems a promising approach, as hold true for treatments that trigger costimulatory pathways, great care must be taken in applying cytokine therapy. The immune effects are not solely confined to the infused T cells and affect many other cells, frequently leading to severe systemic toxicities.
Vaccination Provided Post Transfer
Therapeutic cancer vaccines have the potential to mediate clinical benefit, even as a monotherapy, providing the rational to consider it as an approach to improve ACT (223). Preclinical tumor models provided the insight that vaccination can improve ACT and strategies that have been used to aid anti-tumor efficacy in ACT include vaccination with viruses encoding tumor antigens, long peptides, peptide-pulsed DCs, and DNA vaccination (224–229). Vaccination predominantly seems to improve anti-tumor responses by enhancing tumor infiltration, persistence, and IFN-γ production of adoptively transferred T cells. Also in clinical trials, the potential of vaccination to enhance ACT has been explored, but thus far clinical success is marginal (230–233). An encouraging approach by Rapoport and colleagues showed that in the setting of autologous HSC transplantation for multiple myeloma pre-transplant vaccination, adoptive transfer of in vivo vaccine-primed T cells and subsequent vaccinations led to significant improvement of immunity in patients that would otherwise suffer from severe immunodeficiency due to high-dose chemotherapy (233). Since then, similar strategies have been applied in anti-cancer treatment in patients; autologous vaccine-primed lymphocytes were expanded ex vivo and adoptively transferred accompanied by vaccinations. Using this strategy, promising results have been obtained with respect to enhancement of the tumor-specific T cell response, but clinical activity remains to be further validated (234–237). More recently, two preclinical reports showed enhancement of CAR T cell-mediated anti-tumor responses by vaccination. Both studies were conducted using bispecific T cells targeting CMV and CD19, and vaccination consisted of the CMV peptide pp65 presented by either T cells or CD40L and OX40L expressing K562 cells (238, 239). Compared to mice receiving vaccination with an irrelevant peptide, control of tumor cell growth was improved and this coincided with increased frequencies of CAR–CMV–CTLs, suggesting that CMVpp65 stimulation expanded the bispecific T cells efficiently.
Thus vaccination as a modality to enhance ACT has so far not been explored in great detail and clinical trials using this approach have so far not yielded outstanding results. One of the reasons for this might be that the responsiveness of the adoptively transferred T cells to the vaccine is poor. As aforementioned pointed out, vaccination may be best suitable for less-differentiated T cells producing IL-2. Another possible explanation is that the immunosuppressive tumor environment is hampering T cell activation (240). Strategies to improve vaccination in the context of ACT include combination with peritumoral administration of TLR ligands and TLR-based adjuvants (226, 241).
Additional of great importance is the antigen that is used for vaccination. Often TAAs are used for vaccination. The advantage of this approach is that it is broadly applicable as it allows treatment of most patients with a certain tumor type. However, the specific T cell response toward the TAA can be blunted by central tolerance mechanisms. By contrast, T cells reacting to neoantigens expressed by tumors are not centrally tolerized (240). However, these antigens harbor unique mutations in a patient and, thus, targeting these antigens would require the production of personalized vaccines. This is a topic of intense interest and future studies should resolve the feasibility of such approaches.
Taken together, it is clear that strategies to improve ACT by vaccination need to be optimized and it seems that vaccination as a single modality to enhance this treatment is not sufficient.
Concluding Remarks
Currently multiple clinically approved immunostimulatory antibodies and cytokines are available that target a multitude of receptors expressed by T cells (Figure 1). It is expected that the agents targeting these receptors as well as the number of receptors that are targeted will increase in the coming years. The anti-tumor activity and persistence of infused T cells is highly dependent on the costimulatory pathways that are triggered after T cell transfer and on the expressed cytokine receptors. Unfortunately, the question as to which of the many T cell-stimulating pathways need to be activated during ACT to attain T cells that exert a superior anti-tumor effect and are able to persist long-term has no unanimous answer. Ex vivo culture methods should be designed in such way that the expression of the appropriate receptors on particular T cell subsets is induced, and this holds true for (autocrine) cytokine production as well. To predict the expression pattern, a detailed understanding of the regulation of these receptors is essential. Once this question has been addressed, the next challenge would be to make sure that the transferred T cells remain functional longitudinally, which involves likely a certain degree of heterogeneity of T cell subsets expressing various costimulatory and cytokine receptors.
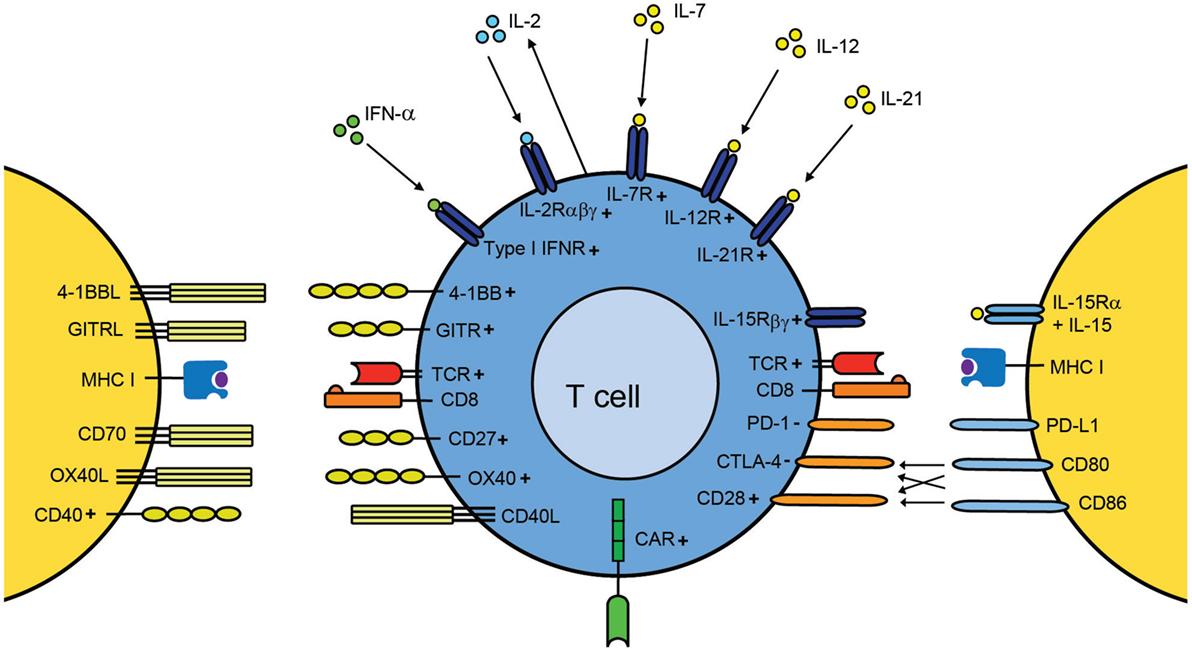
Figure 1. A T cell centric view of improving adoptive T cell therapy by provision of T cell stimulation signals. Upon transfer of ex vivo-expanded disease-specific T cells into the host, these cells recognize their cognate antigen in the context of MHC molecules via their TCR. Amplification of costimulatory signals (e.g., via agonistic antibodies) can be used to additionally stimulate T cells. Antibodies against CD28, CD27, 4-1BB, OX40, and GITR have been evaluated in preclinical models and clinical trials for their capacity to enhance T cell function. Targeting of costimulatory receptors can also be used during the ex vivo expansion of the T cells. In case of CAR T cells, recognition occurs via the CAR, a chimeric TCR that already provides a certain degree of costimulation. Blockade of inhibitory molecules, such as CTLA-4 and PD-1 by antibodies after transfer, counteracts suppressed T cells thereby improving T cell activity. In addition, inflammatory cytokines are able to provide signals for enhancing expansion, differentiation, and migration. Cytokines, such as IFN-α, IL-2, IL-7, IL-12, IL-15, and IL-21, have shown to have the capacity to enhance T cell efficacy either during ex vivo culturing or after adoptive transfer. Autocrine production of IL-2 is a vital property for secondary population expansion, and enhancing autocrine IL-2 is a promising way to improve T cell therapies. The + and − symbols indicate positive and negative signaling, respectively.
Moreover, it is important to keep in mind that the effect of delivery of antibodies or cytokines to patients in order to improve survival, accumulation, and anti-tumor efficacy of the transferred T cells is not confined to the transferred T cells alone, but can affect also host cells bearing the appropriate receptor, potentially resulting in severe toxicity. Due to these toxicity issues, the overall results of using cytokines and agonistic antibodies against immune costimulators may have been modest with respect to the anti-tumor activities in clinical trials. It is conceivable that also by enhancing the quality of engineered T cells (including CAR T cells), which generally already recognize their targets with good affinity, the provision of (additional) costimulation or cytokines might potentiate cross-reactivity or toxicity. An approach to circumvent treatment-related adverse effects includes local administration or specific targeting to the tumor site (152, 226, 242). Furthermore, combinations of treatment modalities are likely to reduce the dose that is required for clinical responses and this might avoid severe adverse effects. On the other hand, certain combinations, even at lower concentrations might result in unexpected toxicity. In addition to strategies to enhance ACT that have been discussed in this review, i.e., by providing costimulation, blocking inhibitory molecules, cytokines, and/or vaccination, the T cell quality can be further enhanced by changing the tumor microenvironment to induce a more favorable milieu for T cells. Those strategies, which are beyond the scope of this review to be discussed in detail, include therapies counteracting myeloid-derived suppressor cells (MDSCs), neutralization of tumor acidity, chemotherapy, inhibition of IDO, and treatment with antibodies against immune suppressive cytokines (243–248). Finally, as discussed vaccination provided post transfer as an approach to enhance the efficiency of ACT is promising, yet such a combined treatment requires substantial effort to make it clinically successful.
Taken together, ACT holds its promise as an effective anti-cancer treatment but improvement is required. Concluding from the aforementioned discussion, the inclusion of T cell costimulation and cytokines should be an integral part for optimization of ACT protocols. In addition, combinations with other immune therapies, such as vaccination are expected to further improve the clinical success rates of ACT.
Author Contributions
AR and RA contributed equally to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Els Verdegaal for critically reading the manuscript.
Funding
This work was supported by a Dutch Cancer Society grant (KWF UL2015-7817) awarded to RA.
References
1. Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol (2014) 32:189–225. doi:10.1146/annurev-immunol-032713-120136
2. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi:10.1126/science.aaa4967
3. Gerdemann U, Katari UL, Papadopoulou A, Keirnan JM, Craddock JA, Liu H, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther (2013) 21(11):2113–21. doi:10.1038/mt.2013.151
4. Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, Martinez C, et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med (2014) 6(242):242ra83. doi:10.1126/scitranslmed.3008825
5. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood (2010) 115(5):925–35. doi:10.1182/blood-2009-08-239186
6. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
7. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother (2003) 26(4):332–42. doi:10.1097/00002371-200307000-00005
8. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res (2010) 16(9):2646–55. doi:10.1158/1078-0432.CCR-10-0041
9. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res (2012) 18(24):6758–70. doi:10.1158/1078-0432.CCR-12-1177
10. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol (2016) 34(20):2389–97. doi:10.1200/JCO.2016.66.7220
11. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi:10.1038/nature12477
12. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439–42. doi:10.1200/JCO.2012.47.7521
13. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344(6184):641–5. doi:10.1126/science.1251102
14. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi:10.1038/nm.3161
15. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature (2016) 536(7614):91–5. doi:10.1038/nature18945
16. Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol (2006) 6(5):383–93. doi:10.1038/nri1842
17. Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med (2008) 358(25):2698–703. doi:10.1056/NEJMoa0800251
18. Verdegaal EM, Visser M, Ramwadhdoebe TH, van der Minne CE, van Steijn JA, Kapiteijn E, et al. Successful treatment of metastatic melanoma by adoptive transfer of blood-derived polyclonal tumor-specific CD4+ and CD8+ T cells in combination with low-dose interferon-alpha. Cancer Immunol Immunother (2011) 60(7):953–63. doi:10.1007/s00262-011-1004-8
19. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119(18):4133–41. doi:10.1182/blood-2011-12-400044
20. Aranda F, Vacchelli E, Obrist F, Eggermont A, Galon J, Herve Fridman W, et al. Trial watch: adoptive cell transfer for anticancer immunotherapy. Oncoimmunology (2014) 3:e28344. doi:10.4161/onci.28344
21. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114(3):535–46. doi:10.1182/blood-2009-03-211714
22. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122(6):863–71. doi:10.1182/blood-2013-03-490565
23. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res (2015) 21(5):1019–27. doi:10.1158/1078-0432.CCR-14-2708
24. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med (2015) 21(8):914–21. doi:10.1038/nm.3910
25. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3(95):95ra73. doi:10.1126/scitranslmed.3002842
26. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. doi:10.1056/NEJMoa1103849
27. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. doi:10.1126/scitranslmed.aac5415
28. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi:10.1056/NEJMoa1215134
29. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3
30. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. doi:10.1056/NEJMoa1407222
31. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi:10.1038/leu.2015.247
32. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126(6):2123–38. doi:10.1172/JCI85309
33. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi:10.1038/mt.2010.24
34. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24(13):e20–2. doi:10.1200/JCO.2006.05.9964
35. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124(2):188–95. doi:10.1182/blood-2014-05-552729
36. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6(224):224ra25. doi:10.1126/scitranslmed.3008226
37. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi:10.1182/blood-2016-04-703751
38. Ciceri F, Bonini C, Marktel S, Zappone E, Servida P, Bernardi M, et al. Antitumor effects of HSV-TK-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood (2007) 109(11):4698–707. doi:10.1182/blood-2006-05-023416
39. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi:10.1056/NEJMoa1106152
40. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32(5):1059–70. doi:10.1007/s10875-012-9689-9
41. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol (2013) 31(1):71–5. doi:10.1038/nbt.2459
42. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res (2013) 1(1):43–53. doi:10.1158/2326-6066.CIR-13-0008
43. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24(6):1160–70. doi:10.1038/leu.2010.75
44. Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood (2007) 110(8):2793–802. doi:10.1182/blood-2007-02-072843
45. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther (2007) 15(4):825–33. doi:10.1038/sj.mt.6300104
46. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20 Pt 1):6106–15. doi:10.1158/1078-0432.CCR-06-1183
47. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi:10.1038/nm.3838
48. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res (2015) 3(4):356–67. doi:10.1158/2326-6066.CIR-14-0186
49. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44(6):1444–54. doi:10.1016/j.immuni.2016.05.014
50. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A (2002) 99(25):16168–73. doi:10.1073/pnas.242600099
51. Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol (2006) 24(31):5060–9. doi:10.1200/JCO.2006.07.1100
52. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YYT. Cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res (2016) 4(6):498–508. doi:10.1158/2326-6066.CIR-15-0231
53. Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev (2014) 257(1):264–76. doi:10.1111/imr.12135
54. Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother (2012) 35(9):651–60. doi:10.1097/CJI.0b013e31827806e6
55. Arens R, Schoenberger SP. Plasticity in programming of effector and memory CD8 T-cell formation. Immunol Rev (2010) 235(1):190–205. doi:10.1111/j.0105-2896.2010.00899.x
56. Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood (2003) 101(11):4260–6. doi:10.1182/blood-2002-11-3577
57. Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med (2015) 7(273):ra13–13. doi:10.1126/scitranslmed.3010314
58. Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Maillard SA, et al. Long-lasting stem cell–like memory CD8+ T cells with a naïve-like profile upon yellow fever vaccination. Sci Transl Med (2015) 7(282):ra48–48. doi:10.1126/scitranslmed.aaa3700
59. Oliveira G, Ruggiero E, Stanghellini MT, Cieri N, D’Agostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med (2015) 7(317):317ra198. doi:10.1126/scitranslmed.aac8265
60. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi:10.1038/nm.2446
61. Lugli E, Gattinoni L, Roberto A, Mavilio D, Price DA, Restifo NP, et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc (2013) 8(1):33–42. doi:10.1038/nprot.2012.143
62. Tiemessen MM, Baert MR, Kok L, van Eggermond MC, van den Elsen PJ, Arens R, et al. T cell factor 1 represses CD8+ effector T cell formation and function. J Immunol (2014) 193(11):5480–7. doi:10.4049/jimmunol.1303417
63. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood (2013) 121(4):573–84. doi:10.1182/blood-2012-05-431718
64. Alvarez-Fernandez C, Escriba-Garcia L, Vidal S, Sierra J, Briones J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J Transl Med (2016) 14(1):214. doi:10.1186/s12967-016-0973-y
65. van der Waart AB, van de Weem NM, Maas F, Kramer CS, Kester MG, Falkenburg JH, et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood (2014) 124(23):3490–500. doi:10.1182/blood-2014-05-578583
66. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res (2015) 75(2):296–305. doi:10.1158/0008-5472.CAN-14-2277
67. Saito H, Okita K, Chang AE, Ito F. Adoptive transfer of CD8+ T cells generated from induced pluripotent stem cells triggers regressions of large tumors along with immunological memory. Cancer Res (2016) 76(12):3473–83. doi:10.1158/0008-5472.CAN-15-1742
68. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity (2014) 41(1):116–26. doi:10.1016/j.immuni.2014.05.018
69. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest (2008) 118(1):294–305. doi:10.1172/JCI32103
70. Wang A, Chandran S, Shah SA, Chiu Y, Paria BC, Aghamolla T, et al. The stoichiometric production of IL-2 and IFN-gamma mRNA defines memory T cells that can self-renew after adoptive transfer in humans. Sci Transl Med (2012) 4(149):149ra20. doi:10.1126/scitranslmed.3004306
71. Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A (2009) 106(41):17469–74. doi:10.1073/pnas.0907448106
72. Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood (2011) 117(3):808–14. doi:10.1182/blood-2010-05-286286
73. van Duikeren S, Fransen MF, Redeker A, Wieles B, Platenburg G, Krebber WJ, et al. Vaccine-induced effector-memory CD8+ T cell responses predict therapeutic efficacy against tumors. J Immunol (2012) 189(7):3397–403. doi:10.4049/jimmunol.1201540
74. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi:10.1146/annurev.immunol.22.012703.104702
75. Huster KM, Stemberger C, Busch DH. Protective immunity towards intracellular pathogens. Curr Opin Immunol (2006) 18(4):458–64. doi:10.1016/j.coi.2006.05.008
76. Li Y, Liu S, Hernandez J, Vence L, Hwu P, Radvanyi L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J Immunol (2010) 184(1):452–65. doi:10.4049/jimmunol.0901101
77. Huang J, Kerstann KW, Ahmadzadeh M, Li YF, El-Gamil M, Rosenberg SA, et al. Modulation by IL-2 of CD70 and CD27 expression on CD8+ T cells: importance for the therapeutic effectiveness of cell transfer immunotherapy. J Immunol (2006) 176(12):7726–35. doi:10.4049/jimmunol.176.12.7726
78. Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol (2005) 175(10):7046–52. doi:10.4049/jimmunol.175.10.7046
79. Butler MO, Hirano N. Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol Rev (2014) 257(1):191–209. doi:10.1111/imr.12129
80. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9(4):271–85. doi:10.1038/nri2526
81. Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol (2007) 179(7):4910–8. doi:10.4049/jimmunol.179.7.4910
82. Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One (2013) 8(4):e60031. doi:10.1371/journal.pone.0060031
83. Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber JS, et al. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J Immunother (2011) 34(3):236–50. doi:10.1097/CJI.0b013e318209e7ec
84. Oh HS, Choi BK, Kim YH, Lee DG, Hwang S, Lee MJ, et al. 4-1BB signaling enhances primary and secondary population expansion of CD8+ T cells by maximizing autocrine IL-2/IL-2 receptor signaling. PLoS One (2015) 10(5):e0126765. doi:10.1371/journal.pone.0126765
85. Ramakrishna V, Sundarapandiyan K, Zhao B, Bylesjo M, Marsh HC, Keler T. Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J Immunother Cancer (2015) 3:37. doi:10.1186/s40425-015-0080-2
86. Claus C, Riether C, Schurch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res (2012) 72(14):3664–76. doi:10.1158/0008-5472.CAN-11-2791
87. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity (2001) 15(3):445–55. doi:10.1016/S1074-7613(01)00191-1
88. Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J Immunol (2005) 175(6):3534–41. doi:10.4049/jimmunol.175.6.3534
89. Lee SJ, Rossi RJ, Lee SK, Croft M, Kwon BS, Mittler RS, et al. CD134 costimulation couples the CD137 pathway to induce production of supereffector CD8 T cells that become IL-7 dependent. J Immunol (2007) 179(4):2203–14. doi:10.4049/jimmunol.179.4.2203
90. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192(12):5451–8. doi:10.4049/jimmunol.1490019
91. Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol (2001) 167(11):6356–65. doi:10.4049/jimmunol.167.11.6356
92. Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S, et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther (2008) 19(5):496–510. doi:10.1089/hum.2007.0171
93. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity (2010) 32(1):91–103. doi:10.1016/j.immuni.2009.11.010
94. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32(1):79–90. doi:10.1016/j.immuni.2009.11.012
95. Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A (2004) 101(7):1969–74. doi:10.1073/pnas.0307298101
96. Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity (2001) 14(2):105–10. doi:10.1016/S1074-7613(01)00093-0
97. Jicha DL, Mule JJ, Rosenberg SA. Interleukin 7 generates antitumor cytotoxic T lymphocytes against murine sarcomas with efficacy in cellular adoptive immunotherapy. J Exp Med (1991) 174(6):1511–5. doi:10.1084/jem.174.6.1511
98. Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol (2005) 175(4):2261–9. doi:10.4049/jimmunol.175.4.2261
99. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood (2010) 115(17):3508–19. doi:10.1182/blood-2009-09-241398
100. Diaz-Montero CM, El Naggar S, Al Khami A, El Naggar R, Montero AJ, Cole DJ, et al. Priming of naive CD8+ T cells in the presence of IL-12 selectively enhances the survival of CD8+CD62Lhi cells and results in superior anti-tumor activity in a tolerogenic murine model. Cancer Immunol Immunother (2008) 57(4):563–72. doi:10.1007/s00262-007-0394-0
101. Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res (2010) 70(17):6725–34. doi:10.1158/0008-5472.CAN-10-0735
102. Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood (2009) 113(22):5516–25. doi:10.1182/blood-2008-11-188458
103. Rubinstein MP, Su EW, Suriano S, Cloud CA, Andrijauskaite K, Kesarwani P, et al. Interleukin-12 enhances the function and anti-tumor activity in murine and human CD8(+) T cells. Cancer Immunol Immunother (2015) 64(5):539–49. doi:10.1007/s00262-015-1655-y
104. Hervas-Stubbs S, Mancheno U, Riezu-Boj JI, Larraga A, Ochoa MC, Alignani D, et al. CD8 T cell priming in the presence of IFN-alpha renders CTLs with improved responsiveness to homeostatic cytokines and recall antigens: important traits for adoptive T cell therapy. J Immunol (2012) 189(7):3299–310. doi:10.4049/jimmunol.1102495
105. Wagner HJ, Bollard CM, Vigouroux S, Huls MH, Anderson R, Prentice HG, et al. A strategy for treatment of Epstein-Barr virus-positive Hodgkin’s disease by targeting interleukin 12 to the tumor environment using tumor antigen-specific T cells. Cancer Gene Ther (2004) 11(2):81–91. doi:10.1038/sj.cgt.7700664
106. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med (2005) 201(1):139–48. doi:10.1084/jem.20041057
107. Wolfl M, Merker K, Morbach H, Van Gool SW, Eyrich M, Greenberg PD, et al. Primed tumor-reactive multifunctional CD62L+ human CD8+ T cells for immunotherapy. Cancer Immunol Immunother (2011) 60(2):173–86. doi:10.1007/s00262-010-0928-8
108. Albrecht J, Frey M, Teschner D, Carbol A, Theobald M, Herr W, et al. IL-21-treated naive CD45RA+ CD8+ T cells represent a reliable source for producing leukemia-reactive cytotoxic T lymphocytes with high proliferative potential and early differentiation phenotype. Cancer Immunol Immunother (2011) 60(2):235–48. doi:10.1007/s00262-010-0936-8
109. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy (2015) 17(4):487–95. doi:10.1016/j.jcyt.2014.12.002
110. Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother (2013) 62(4):727–36. doi:10.1007/s00262-012-1378-2
111. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123(24):3750–9. doi:10.1182/blood-2014-01-552174
112. Santegoets SJ, Turksma AW, Suhoski MM, Stam AG, Albelda SM, Hooijberg E, et al. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J Transl Med (2013) 11:37. doi:10.1186/1479-5876-11-37
113. Redeker A, Welten SP, Baert MR, Vloemans SA, Tiemessen MM, Staal FJ, et al. The quantity of autocrine IL-2 governs the expansion potential of CD8+ T cells. J Immunol (2015) 195(10):4792–801. doi:10.4049/jimmunol.1501083
114. Steenblock ER, Wrzesinski SH, Flavell RA, Fahmy TM. Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy. Expert Opin Biol Ther (2009) 9(4):451–64. doi:10.1517/14712590902849216
115. Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem (2011) 286(40):34883–92. doi:10.1074/jbc.M111.276329
116. Welten SP, Redeker A, Franken KL, Benedict CA, Yagita H, Wensveen FM, et al. CD27-CD70 costimulation controls T cell immunity during acute and persistent cytomegalovirus infection. J Virol (2013) 87(12):6851–65. doi:10.1128/JVI.03305-12
117. Welten SP, Redeker A, Franken KL, Oduro JD, Ossendorp F, Cicin-Sain L, et al. The viral context instructs the redundancy of costimulatory pathways in driving CD8(+) T cell expansion. Elife (2015) 4:e07486. doi:10.7554/eLife.07486
118. Schaer DA, Hirschhorn-Cymerman D, Wolchok JD. Targeting tumor-necrosis factor receptor pathways for tumor immunotherapy. J Immunother Cancer (2014) 2:7. doi:10.1186/2051-1426-2-7
119. Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol (2015) 42(4):640–55. doi:10.1053/j.seminoncol.2015.05.014
120. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res (2013) 19(5):1035–43. doi:10.1158/1078-0432.CCR-12-2064
121. Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J Exp Med (1998) 187(11):1849–62. doi:10.1084/jem.187.11.1849
122. Feau S, Arens R, Togher S, Schoenberger SP. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat Immunol (2011) 12(9):908–13. doi:10.1038/ni.2079
123. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355(10):1018–28. doi:10.1056/NEJMoa063842
124. Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol (2010) 37(5):508–16. doi:10.1053/j.seminoncol.2010.09.008
125. Weigelin B, Bolanos E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci U S A (2015) 112(24):7551–6. doi:10.1073/pnas.1506357112
126. Lin GH, Liu Y, Ambagala T, Kwon BS, Ohashi PS, Watts TH. Evaluating the cellular targets of anti-4-1BB agonist antibody during immunotherapy of a pre-established tumor in mice. PLoS One (2010) 5(6):e11003. doi:10.1371/journal.pone.0011003
127. May KF Jr, Chen L, Zheng P, Liu Y. Anti-4-1BB monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific CD8+ T cells. Cancer Res (2002) 62(12):3459–65.
128. Song C, Sadashivaiah K, Furusawa A, Davila E, Tamada K, Banerjee A. Eomesodermin is required for antitumor immunity mediated by 4-1BB-agonist immunotherapy. Oncoimmunology (2014) 3(1):e27680. doi:10.4161/onci.27680
129. Curran MA, Geiger TL, Montalvo W, Kim M, Reiner SL, Al-Shamkhani A, et al. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J Exp Med (2013) 210(4):743–55. doi:10.1084/jem.20121190
130. Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol (2007) 179(11):7244–53. doi:10.4049/jimmunol.179.11.7244
131. Kjaergaard J, Peng L, Cohen PA, Drazba JA, Weinberg AD, Shu S. Augmentation versus inhibition: effects of conjunctional OX-40 receptor monoclonal antibody and IL-2 treatment on adoptive immunotherapy of advanced tumor. J Immunol (2001) 167(11):6669–77. doi:10.4049/jimmunol.167.11.6669
132. So T, Lee SW, Croft M. Immune regulation and control of regulatory T cells by OX40 and 4-1BB. Cytokine Growth Factor Rev (2008) 19(3–4):253–62. doi:10.1016/j.cytogfr.2008.04.003
133. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res (2013) 73(24):7189–98. doi:10.1158/0008-5472.CAN-12-4174
134. He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol (2013) 191(8):4174–83. doi:10.4049/jimmunol.1300409
135. Vitale LA, He LZ, Thomas LJ, Widger J, Weidlick J, Crocker A, et al. Development of a human monoclonal antibody for potential therapy of CD27-expressing lymphoma and leukemia. Clin Cancer Res (2012) 18(14):3812–21. doi:10.1158/1078-0432.CCR-11-3308
136. Ochsenbein AF, Riddell SR, Brown M, Corey L, Baerlocher GM, Lansdorp PM, et al. CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J Exp Med (2004) 200(11):1407–17. doi:10.1084/jem.20040717
137. Peperzak V, Xiao Y, Veraar EA, Borst J. CD27 sustains survival of CTLs in virus-infected nonlymphoid tissue in mice by inducing autocrine IL-2 production. J Clin Invest (2010) 120(1):168–78. doi:10.1172/JCI40178
138. Arens R, Schepers K, Nolte MA, van Oosterwijk MF, van Lier RA, Schumacher TN, et al. Tumor rejection induced by CD70-mediated quantitative and qualitative effects on effector CD8+ T cell formation. J Exp Med (2004) 199(11):1595–605. doi:10.1084/jem.20031111
139. Riether C, Schurch C, Ochsenbein AF. Modulating CD27 signaling to treat cancer. Oncoimmunology (2012) 1(9):1604–6. doi:10.4161/onci.21425
140. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331(6024):1612–6. doi:10.1126/science.1198443
141. Ranheim EA, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med (1993) 177(4):925–35. doi:10.1084/jem.177.4.925
142. Funakoshi S, Longo DL, Beckwith M, Conley DK, Tsarfaty G, Tsarfaty I, et al. Inhibition of human B-cell lymphoma growth by CD40 stimulation. Blood (1994) 83(10):2787–94.
143. Szocinski JL, Khaled AR, Hixon J, Halverson D, Funakoshi S, Fanslow WC, et al. Activation-induced cell death of aggressive histology lymphomas by CD40 stimulation: induction of bax. Blood (2002) 100(1):217–23. doi:10.1182/blood.V100.1.217
144. Hirano A, Longo DL, Taub DD, Ferris DK, Young LS, Eliopoulos AG, et al. Inhibition of human breast carcinoma growth by a soluble recombinant human CD40 ligand. Blood (1999) 93(9):2999–3007.
145. von Leoprechting A, van der Bruggen P, Pahl HL, Aruffo A, Simon JC. Stimulation of CD40 on immunogenic human malignant melanomas augments their cytotoxic T lymphocyte-mediated lysis and induces apoptosis. Cancer Res (1999) 59(6):1287–94.
146. de Vos S, Forero-Torres A, Ansell SM, Kahl B, Cheson BD, Bartlett NL, et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J Hematol Oncol (2014) 7:44. doi:10.1186/1756-8722-7-44
147. Castillo R, Mascarenhas J, Telford W, Chadburn A, Friedman SM, Schattner EJ. Proliferative response of mantle cell lymphoma cells stimulated by CD40 ligation and IL-4. Leukemia (2000) 14(2):292–8. doi:10.1038/sj.leu.2401664
148. Burington B, Yue P, Shi X, Advani R, Lau JT, Tan J, et al. CD40 pathway activation status predicts response to CD40 therapy in diffuse large B cell lymphoma. Sci Transl Med (2011) 3(74):74ra22. doi:10.1126/scitranslmed.3001620
149. Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med (1999) 5(7):774–9. doi:10.1038/10495
150. Todryk SM, Tutt AL, Green MH, Smallwood JA, Halanek N, Dalgleish AG, et al. CD40 ligation for immunotherapy of solid tumours. J Immunol Methods (2001) 248(1–2):139–47. doi:10.1016/S0022-1759(00)00349-5
151. van Mierlo GJ, den Boer AT, Medema JP, van der Voort EI, Fransen MF, Offringa R, et al. CD40 stimulation leads to effective therapy of CD40(-) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc Natl Acad Sci U S A (2002) 99(8):5561–6. doi:10.1073/pnas.082107699
152. Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJ. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res (2011) 17(8):2270–80. doi:10.1158/1078-0432.CCR-10-2888
153. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res (2013) 19(22):6286–95. doi:10.1158/1078-0432.CCR-13-1320
154. Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and biological effects of an agonist anti-CD40 antibody: a cancer research UK phase I study. Clin Cancer Res (2015) 21(6):1321–8. doi:10.1158/1078-0432.CCR-14-2355
155. Liu C, Lewis CM, Lou Y, Xu C, Peng W, Yang Y, et al. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J Immunother (2012) 35(3):276–82. doi:10.1097/CJI.0b013e31824e7f43
156. Ryan CM, Staveley-O’Carroll K, Schell TD. Combined anti-CD40 conditioning and well-timed immunization prolongs CD8+ T cell accumulation and control of established brain tumors. J Immunother (2008) 31(9):906–20. doi:10.1097/CJI.0b013e318189f155
157. Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol (2007) 25(7):876–83. doi:10.1200/JCO.2006.08.3311
158. Richman LP, Vonderheide RH. Role of crosslinking for agonistic CD40 monoclonal antibodies as immune therapy of cancer. Cancer Immunol Res (2014) 2(1):19–26. doi:10.1158/2326-6066.CIR-13-0152
159. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther (2015) 23(4):769–78. doi:10.1038/mt.2015.4
160. Kim IK, Kim BS, Koh CH, Seok JW, Park JS, Shin KS, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat Med (2015) 21(9):1010–7. doi:10.1038/nm.3922
161. Tone M, Tone Y, Adams E, Yates SF, Frewin MR, Cobbold SP, et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci U S A (2003) 100(25):15059–64. doi:10.1073/pnas.2334901100
162. Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, et al. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol (2004) 34(3):613–22. doi:10.1002/eji.200324804
163. Imai N, Ikeda H, Tawara I, Wang L, Wang L, Nishikawa H, et al. Glucocorticoid-induced tumor necrosis factor receptor stimulation enhances the multifunctionality of adoptively transferred tumor antigen-specific CD8+ T cells with tumor regression. Cancer Sci (2009) 100(7):1317–25. doi:10.1111/j.1349-7006.2009.01179.x
164. Murphy JT, Burey AP, Beebe AM, Gu D, Presta LG, Merghoub T, et al. Anaphylaxis caused by repetitive doses of a GITR agonist monoclonal antibody in mice. Blood (2014) 123(14):2172–80. doi:10.1182/blood-2013-12-544742
165. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood (2012) 119(3):696–706. doi:10.1182/blood-2011-03-344275
166. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther (2009) 17(8):1453–64. doi:10.1038/mt.2009.83
167. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol (2013) 6:33. doi:10.1186/1756-8722-6-33
168. Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer (2011) 129(12):2935–44. doi:10.1002/ijc.25960
169. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol (2004) 172(1):104–13. doi:10.4049/jimmunol.172.1.104
170. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A (2009) 106(9):3360–5. doi:10.1073/pnas.0813101106
171. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood (2012) 119(17):3940–50. doi:10.1182/blood-2011-10-387969
172. Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest (2015) 125(9):3377–83. doi:10.1172/JCI80012
173. Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front Immunol (2015) 6:418. doi:10.3389/fimmu.2015.00418
174. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov (2015) 14(8):561–84. doi:10.1038/nrd4591
175. Ishikawa T, Adachi S, Okayama T, Kokura S, Mizushima K, Doi T, et al. Cytotoxic T lymphocyte-associated antigen 4 inhibition increases the antitumor activity of adoptive T-cell therapy when carried out with naive rather than differentiated T cells. Oncol Rep (2015) 33(5):2545–52. doi:10.3892/or.2015.3815
176. Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O’Brien S, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res (2016) 22(2):436–47. doi:10.1158/1078-0432.CCR-15-1070
177. Berrien-Elliott MM, Jackson SR, Meyer JM, Rouskey CJ, Nguyen TL, Yagita H, et al. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res (2013) 73(2):605–16. doi:10.1158/0008-5472.CAN-12-2179
178. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19(20):5636–46. doi:10.1158/1078-0432.CCR-13-0458
179. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol (2013) 190(9):4899–909. doi:10.4049/jimmunol.1300271
180. Kobold S, Grassmann S, Chaloupka M, Lampert C, Wenk S, Kraus F, et al. Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J Natl Cancer Inst (2015) 107(8):djv146. doi:10.1093/jnci/djv146
181. Buchan SL, Manzo T, Flutter B, Rogel A, Edwards N, Zhang L, et al. OX40- and CD27-mediated costimulation synergizes with anti-PD-L1 blockade by forcing exhausted CD8+ T cells to exit quiescence. J Immunol (2015) 194(1):125–33. doi:10.4049/jimmunol.1401644
182. Guo Z, Wang X, Cheng D, Xia Z, Luan M, Zhang S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS One (2014) 9(2):e89350. doi:10.1371/journal.pone.0089350
183. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi:10.1158/0008-5472.CAN-13-1550
184. Kocak E, Lute K, Chang X, May KF Jr, Exten KR, Zhang H, et al. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res (2006) 66(14):7276–84. doi:10.1158/0008-5472.CAN-05-2128
185. Watanabe A, Hara M, Chosa E, Nakamura K, Sekiya R, Shimizu T, et al. Combination of adoptive cell transfer and antibody injection can eradicate established tumors in mice – an in vivo study using anti-OX40mAb, anti-CD25mAb and anti-CTLA4mAb. Immunopharmacol Immunotoxicol (2010) 32(2):238–45. doi:10.3109/08923970903222355
186. Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, et al. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood (2012) 119(24):5688–96. doi:10.1182/blood-2011-10-386482
187. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL-2 regimen. Clin Cancer Res (2016) 22(15):3734–45. doi:10.1158/1078-0432.CCR-15-1879
188. Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol (2003) 3(4):269–79. doi:10.1038/nri1052
189. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol (2006) 24:657–79. doi:10.1146/annurev.immunol.24.021605.090727
190. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med (2005) 202(7):907–12. doi:10.1084/jem.20050732
191. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol (2008) 26(32):5233–9. doi:10.1200/JCO.2008.16.5449
192. Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood (2011) 117(6):1888–98. doi:10.1182/blood-2010-10-310599
193. Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res (2011) 17(16):5343–52. doi:10.1158/1078-0432.CCR-11-0503
194. Roychowdhury S, May KF Jr, Tzou KS, Lin T, Bhatt D, Freud AG, et al. Failed adoptive immunotherapy with tumor-specific T cells: reversal with low-dose interleukin 15 but not low-dose interleukin 2. Cancer Res (2004) 64(21):8062–7. doi:10.1158/0008-5472.CAN-04-1860
195. Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res (2014) 20(1):131–9. doi:10.1158/1078-0432.CCR-13-1016
196. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi:10.1200/JCO.2014.57.3329
197. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer (2009) 45(2):228–47. doi:10.1016/j.ejca.2008.10.026
198. Rosenberg SA, Sportes C, Ahmadzadeh M, Fry TJ, Ngo LT, Schwarz SL, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother (2006) 29(3):313–9. doi:10.1097/01.cji.0000210386.55951.c2
199. Sportes C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK, et al. Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin Cancer Res (2010) 16(2):727–35. doi:10.1158/1078-0432.CCR-09-1303
200. Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol (2005) 175(11):7226–34. doi:10.4049/jimmunol.175.11.7226
201. Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood (2007) 109(12):5168–77. doi:10.1182/blood-2006-06-029173
202. Moroz A, Eppolito C, Li Q, Tao J, Clegg CH, Shrikant PA. IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: comparative evaluation of IL-2, IL-15, and IL-21. J Immunol (2004) 173(2):900–9. doi:10.4049/jimmunol.173.2.900
203. He H, Wisner P, Yang G, Hu HM, Haley D, Miller W, et al. Combined IL-21 and low-dose IL-2 therapy induces anti-tumor immunity and long-term curative effects in a murine melanoma tumor model. J Transl Med (2006) 4:24. doi:10.1186/1479-5876-4-24
204. Iuchi T, Teitz-Tennenbaum S, Huang J, Redman BG, Hughes SD, Li M, et al. Interleukin-21 augments the efficacy of T-cell therapy by eliciting concurrent cellular and humoral responses. Cancer Res (2008) 68(11):4431–41. doi:10.1158/0008-5472.CAN-07-5530
205. Kim-Schulze S, Kim HS, Fan Q, Kim DW, Kaufman HL. Local IL-21 promotes the therapeutic activity of effector T cells by decreasing regulatory T cells within the tumor microenvironment. Mol Ther (2009) 17(2):380–8. doi:10.1038/mt.2008.249
206. Thompson JA, Curti BD, Redman BG, Bhatia S, Weber JS, Agarwala SS, et al. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J Clin Oncol (2008) 26(12):2034–9. doi:10.1200/JCO.2007.14.5193
207. Davis ID, Brady B, Kefford RF, Millward M, Cebon J, Skrumsager BK, et al. Clinical and biological efficacy of recombinant human interleukin-21 in patients with stage IV malignant melanoma without prior treatment: a phase IIa trial. Clin Cancer Res (2009) 15(6):2123–9. doi:10.1158/1078-0432.CCR-08-2663
208. Cohen J. IL-12 deaths: explanation and a puzzle. Science (1995) 270(5238):908. doi:10.1126/science.270.5238.908a
209. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood (1997) 90(7):2541–8.
210. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ (2015) 22(2):237–46. doi:10.1038/cdd.2014.134
211. Galvan DL, O’Neil RT, Foster AE, Huye L, Bear A, Rooney CM, et al. Anti-tumor effects after adoptive transfer of IL-12 transposon-modified murine splenocytes in the OT-I-melanoma mouse model. PLoS One (2015) 10(10):e0140744. doi:10.1371/journal.pone.0140744
212. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res (2012) 18(6):1672–83. doi:10.1158/1078-0432.CCR-11-3050
213. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology (2015) 4(3):e994446. doi:10.4161/2162402X.2014.994446
214. Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res (2015) 21(10):2278–88. doi:10.1158/1078-0432.CCR-14-2085
215. Meyaard L, Hovenkamp E, Otto SA, Miedema F. IL-12-induced IL-10 production by human T cells as a negative feedback for IL-12-induced immune responses. J Immunol (1996) 156(8):2776–82.
216. Bracci L, La Sorsa V, Belardelli F, Proietti E. Type I interferons as vaccine adjuvants against infectious diseases and cancer. Expert Rev Vaccines (2008) 7(3):373–81. doi:10.1586/14760584.7.3.373
217. Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol (2001) 167(3):1179–87. doi:10.4049/jimmunol.167.3.1179
218. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med (2005) 202(5):637–50. doi:10.1084/jem.20050821
219. Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol (2006) 176(8):4525–9. doi:10.4049/jimmunol.176.8.4525
220. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol (2005) 174(8):4465–9. doi:10.4049/jimmunol.174.8.4465
221. Silver RT, Woolf SH, Hehlmann R, Appelbaum FR, Anderson J, Bennett C, et al. An evidence-based analysis of the effect of busulfan, hydroxyurea, interferon, and allogeneic bone marrow transplantation in treating the chronic phase of chronic myeloid leukemia: developed for the American Society of Hematology. Blood (1999) 94(5):1517–36.
222. Chapuis AG, Lee SM, Thompson JA, Roberts IM, Margolin KA, Bhatia S, et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J Exp Med (2016) 213(7):1133–9. doi:10.1084/jem.20152021
223. Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest (2015) 125(9):3401–12. doi:10.1172/JCI80009
224. Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med (2003) 198(4):569–80. doi:10.1084/jem.20030590
225. Lou Y, Wang G, Lizee G, Kim GJ, Finkelstein SE, Feng C, et al. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo. Cancer Res (2004) 64(18):6783–90. doi:10.1158/0008-5472.CAN-04-1621
226. Ly LV, Sluijter M, Versluis M, Luyten GP, van der Burg SH, Melief CJ, et al. Peptide vaccination after T-cell transfer causes massive clonal expansion, tumor eradication, and manageable cytokine storm. Cancer Res (2010) 70(21):8339–46. doi:10.1158/0008-5472.CAN-10-2288
227. Jorritsma A, Bins AD, Schumacher TN, Haanen JB. Skewing the T-cell repertoire by combined DNA vaccination, host conditioning, and adoptive transfer. Cancer Res (2008) 68(7):2455–62. doi:10.1158/0008-5472.CAN-07-5254
228. Teshima T, Liu C, Lowler KP, Dranoff G, Ferrara JL. Donor leukocyte infusion from immunized donors increases tumor vaccine efficacy after allogeneic bone marrow transplantation. Cancer Res (2002) 62(3):796–800.
229. Parviz M, Chin CS, Graham LJ, Miller C, Lee C, George K, et al. Successful adoptive immunotherapy with vaccine-sensitized T cells, despite no effect with vaccination alone in a weakly immunogenic tumor model. Cancer Immunol Immunother (2003) 52(12):739–50. doi:10.1007/s00262-003-0405-8
230. Poschke I, Lovgren T, Adamson L, Nystrom M, Andersson E, Hansson J, et al. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Cancer Immunol Immunother (2014) 63(10):1061–71. doi:10.1007/s00262-014-1575-2
231. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res (2014) 20(9):2457–65. doi:10.1158/1078-0432.CCR-13-3017
232. Sloan AE, Dansey R, Zamorano L, Barger G, Hamm C, Diaz F, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurg Focus (2000) 9(6):e9. doi:10.3171/foc.2000.9.6.10
233. Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med (2005) 11(11):1230–7. doi:10.1038/nm1310
234. Chang AE, Li Q, Jiang G, Sayre DM, Braun TM, Redman BG. Phase II trial of autologous tumor vaccination, anti-CD3-activated vaccine-primed lymphocytes, and interleukin-2 in stage IV renal cell cancer. J Clin Oncol (2003) 21(5):884–90. doi:10.1200/JCO.2003.08.023
235. Szmania S, Gnjatic S, Tricot G, Stone K, Zhan F, Moreno A, et al. Immunization with a recombinant MAGE-A3 protein after high-dose therapy for myeloma. J Immunother (2007) 30(8):847–54. doi:10.1097/CJI.0b013e318158fcff
236. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang HB, Cai L, et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood (2011) 117(3):788–97. doi:10.1182/blood-2010-08-299396
237. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Xu YY, Kalos M, et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin Cancer Res (2014) 20(5):1355–65. doi:10.1158/1078-0432.CCR-13-2817
238. Wang X, Wong CW, Urak R, Mardiros A, Budde LE, Chang WC, et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin Cancer Res (2015) 21(13):2993–3002. doi:10.1158/1078-0432.CCR-14-2920
239. Caruana I, Weber G, Ballard BC, Wood MS, Savoldo B, Dotti G. K562-derived whole-cell vaccine enhances antitumor responses of CAR-redirected virus-specific cytotoxic T lymphocytes in vivo. Clin Cancer Res (2015) 21(13):2952–62. doi:10.1158/1078-0432.CCR-14-2998
240. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer (2016) 16(4):219–33. doi:10.1038/nrc.2016.16
241. Kohlmeyer J, Cron M, Landsberg J, Bald T, Renn M, Mikus S, et al. Complete regression of advanced primary and metastatic mouse melanomas following combination chemoimmunotherapy. Cancer Res (2009) 69(15):6265–74. doi:10.1158/0008-5472.CAN-09-0579
242. Xiao H, Peng Y, Hong Y, Huang L, Guo ZS, Bartlett DL, et al. Local administration of TLR ligands rescues the function of tumor-infiltrating CD8 T cells and enhances the antitumor effect of lentivector immunization. J Immunol (2013) 190(11):5866–73. doi:10.4049/jimmunol.1203470
243. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res (2016) 76(6):1381–90. doi:10.1158/0008-5472.CAN-15-1743
244. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34(3):137–43. doi:10.1016/j.it.2012.10.001
245. Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL, et al. TGF-beta blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget (2015) 6(40):43005–15. doi:10.18632/oncotarget.5656
246. Hsu FT, Chen TC, Chuang HY, Chang YF, Hwang JJ. Enhancement of adoptive T cell transfer with single low dose pretreatment of doxorubicin or paclitaxel in mice. Oncotarget (2015) 6(42):44134–50. doi:10.18632/oncotarget.6628
247. Hosoi A, Matsushita H, Shimizu K, Fujii S, Ueha S, Abe J, et al. Adoptive cytotoxic T lymphocyte therapy triggers a counter-regulatory immunosuppressive mechanism via recruitment of myeloid-derived suppressor cells. Int J Cancer (2014) 134(8):1810–22. doi:10.1002/ijc.28506
Keywords: adoptive cell therapy, T cells, costimulation, cytokines, vaccination
Citation: Redeker A and Arens R (2016) Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front. Immunol. 7:345. doi: 10.3389/fimmu.2016.00345
Received: 17 June 2016; Accepted: 24 August 2016;
Published: 06 September 2016
Edited by:
Christopher M. Snyder, Thomas Jefferson University, USACopyright: © 2016 Redeker and Arens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anke Redeker, YS5yZWRla2VyQGx1bWMubmw=;
Ramon Arens, ci5hcmVuc0BsdW1jLm5s