 Samira Asgari1,2
Samira Asgari1,2 Paul J. McLaren3,4Jane Peake5Melanie Wong6Richard Wong7Istvan Bartha1,2
Paul J. McLaren3,4Jane Peake5Melanie Wong6Richard Wong7Istvan Bartha1,2 Joshua R. Francis8,9Katia Abarca10
Joshua R. Francis8,9Katia Abarca10 Kyra A. Gelderman11
Kyra A. Gelderman11 Philipp Agyeman12Christoph Aebi12
Philipp Agyeman12Christoph Aebi12 Christoph Berger13
Christoph Berger13 Jacques Fellay1,2*†
Jacques Fellay1,2*† Luregn J. Schlapbach5,12,14*† and The Swiss Pediatric Sepsis Study
Luregn J. Schlapbach5,12,14*† and The Swiss Pediatric Sepsis Study
- 1Global Health Institute, School of Life Sciences, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
- 2Swiss Institute of Bioinformatics, Lausanne, Switzerland
- 3National HIV and Retrovirology Laboratory, Public Health Agency of Canada, Winnipeg, MB, Canada
- 4Department of Medical Microbiology and Infectious Diseases, University of Manitoba, Winnipeg, MB, Canada
- 5Lady Cilento Children’s Hospital, Brisbane, QLD, Australia
- 6Children’s Hospital Westmead, Sydney, NSW, Australia
- 7Pathology Queensland Central Laboratory, Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
- 8Menzies School of Health Research, Charles Darwin University, Darwin, NT, Australia
- 9Royal Darwin Hospital, Darwin, NT, Australia
- 10Departamento de Enfermedades Infecciosas e Inmunología Pediátrica, Escuela de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile
- 11Sanquin Diagnostic Services, Amsterdam, Netherlands
- 12Department of Pediatrics, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland
- 13University Children’s Hospital Zurich, Zurich, Switzerland
- 14Paediatric Critical Care Research Group (PCCRG), Mater Research, University of Queensland, Brisbane, QLD, Australia
One out of three pediatric sepsis deaths in high income countries occur in previously healthy children. Primary immunodeficiencies (PIDs) have been postulated to underlie fulminant sepsis, but this concept remains to be confirmed in clinical practice. Pseudomonas aeruginosa (P. aeruginosa) is a common bacterium mostly associated with health care-related infections in immunocompromised individuals. However, in rare cases, it can cause sepsis in previously healthy children. We used exome sequencing and bioinformatic analysis to systematically search for genetic factors underpinning severe P. aeruginosa infection in the pediatric population. We collected blood samples from 11 previously healthy children, with no family history of immunodeficiency, who presented with severe sepsis due to community-acquired P. aeruginosa bacteremia. Genomic DNA was extracted from blood or tissue samples obtained intravitam or postmortem. We obtained high-coverage exome sequencing data and searched for rare loss-of-function variants. After rigorous filtrations, 12 potentially causal variants were identified. Two out of eight (25%) fatal cases were found to carry novel pathogenic variants in PID genes, including BTK and DNMT3B. This study demonstrates that exome sequencing allows to identify rare, deleterious human genetic variants responsible for fulminant sepsis in apparently healthy children. Diagnosing PIDs in such patients is of high relevance to survivors and affected families. We propose that unusually severe and fatal sepsis cases in previously healthy children should be considered for exome/genome sequencing to search for underlying PIDs.
Introduction
Despite worldwide reduction in childhood mortality, sepsis remains one of the leading causes of childhood deaths. Importantly, 35–50% of pediatric sepsis deaths occur in previously healthy children (1–3). These children often develop non-specific symptoms suggestive of a viral respiratory infection, followed by sudden deterioration and rapid progression to shock and multisystem organ failure. In addition to pathogen virulence factors, such as streptococcal Toxic Shock Toxin (4), rare genetic variants causing a primary immunodeficiency (PID) may underlie fulminant sepsis. Such variants have high effect sizes and are usually kept at low frequencies in the population due to purifying selection (5, 6). Previous studies searching for PID in children with bacterial sepsis were limited to conventional immunological testing (5). Using high-throughput sequencing studies on individuals with extreme phenotypes, who are most likely to be informative from a genetic point of view, has been demonstrated to be very powerful in Mendelian diseases but has not been reported in patient cohorts with invasive bacterial infections (6).
Pseudomonas aeruginosa (P. aeruginosa) is an aerobic Gram-negative bacterium commonly found in environment. This opportunistic pathogen causes invasive infections in immunosuppressed and hospitalized patients and represents a major cause of health care-related infections. In contrast, sepsis due to community-acquired P. aeruginosa is extremely rare in apparently healthy children, and carries very high mortality (7, 8), and may be the first manifestation of an underlying PID. Indeed, a few case reports have already described the identification of PIDs in children with P. aeruginosa sepsis (9, 10). Here, we used exome sequencing and bioinformatic analysis to identify genetic variants conferring extreme susceptibility to P. aeruginosa in a cohort of 11 previously healthy children.
Materials and Methods
Patients
Children below 60 months with community-acquired blood-culture positive P. aeruginosa sepsis were eligible. Children with any comorbidities (prematurity, congenital malformations, previous surgery, immunosuppression, known immunodeficiency, and chronic diseases) and children who had been exposed to broad-spectrum intravenous antibiotics were excluded. This study was carried out in accordance with the recommendations of the ethics committees of participating centers (Kantonale Ethikkommission Bern, KEK Ref Nr 029/11, Bern, Switzerland; Mater Health Services HREC13/MHS/4, Brisbane, QLD, Australia). Parents/guardians of all included patients gave written informed consent in accordance with the Declaration of Helsinki. Patients were recruited prospectively and retrospectively in hospital databases and infectious diseases networks. Whenever possible, DNA was also collected from the parents.
DNA Extraction and Exome Sequencing
Genomic DNA was extracted from whole blood (N = 8), frozen skin biopsy or lymphatic tissue (N = 2), or paraffin-fixed histology slides (N = 1). Exome sequencing libraries were prepared using Agilent SureSelect (V5, 50.4 Mb). Cluster generation was performed using Illumina TruSeq PE Cluster Kit v5 reagents. Libraries were sequenced as 100 bp long, paired-end reads on Illumina HiSeq 2500 using TruSeq SBS Kit v5 reagents.
Short Read Alignment
Sequencing reads were processed using CASAVA v1.82. Reads were aligned to the human reference genome hg19 using BWA (11, 12) v0.6.2. PCR duplicates were removed using Picard 1.27-1 (http://picard.sourceforge.net/).
Variant Calling
We used genome analysis toolkit (GATK) (13, 14) version 3.1-1 to call single nucleotide variants (SNVs) and small insertion and deletions (indels) from duplicate-marked bam files. We used HaplotypeCaller for multi-sample variant calling on all samples followed by GATK best practice to call the variants and included only variants that were flagged as PASS by GATK in subsequent analysis.
Variant Effect Prediction
We used SnpEff (15) version 4.1B to predict the functional impact of variants. As variants can have several predicted effects, we only considered the most severe effect for each variant. The effects in decreasing order of severity are frameshift and in-frame for indels and stop-gain, splice site-disrupting, non-synonymous, synonymous, intronic, UTR, non-coding exon, and intergenic for SNVs. Variants were annotated as putative loss-of-function (LoF) if they were stop-gain or splice site-disrupting SNVs or frameshift indels mapping to the first 95% of coding region, or larger deletions removing either the first exon or more than 50% of the protein-coding sequence of the affected transcript.
Variant Frequency Estimation
We checked the minor allele frequency (MAF) of all previously described variants in the following datasets: the NHLBI exome sequencing project (ESP, N = 6503) (16), the 1000 genomes project phase 2 (1KG, N = 876) (17) and the UK10K project (UK10K, N = 3621) (18), the Exome Aggregation Consortium database (ExAC, N = 60,706) (19), and a set of 533 in-house control exomes.
Identification of Potentially Causal Variants in Parent–Child Trios
We restricted analyses to non-synonymous and LoF exonic variants with MAF <1% in ESP, 1KG, UK10K, ExAC, and in-house control exomes. We analyzed each family separately assuming autosomal recessive, autosomal dominant, and X-linked recessive inheritance models. Only variants with >10× coverage in both the parents and the offspring were included in the de novo analysis. We assumed full penetrance for the potentially causal variants and given the fatality of the phenotype did not consider a mutation as potentially causal if it was present in the parents, any of the abovementioned databases or in the in-house control exomes in the same zygosity form (i.e., heterozygous or homozygous) as in our patients.
Identification of Potentially Causal Variants in Individual Patients
We restricted analyses to non-synonymous and LoF exonic variants with MAF <1% in ESP, 1KG, UK10K, ExAC, and in-house control exomes. We analyzed each individual separately assuming autosomal recessive and X-linked recessive inheritance models. We assumed full penetrance for the potentially causal variants and given the fatality of the phenotype did not consider a mutation as potentially causal if it was present in any of the abovementioned databases or in the in-house control exomes in the same zygosity form (i.e., heterozygous or homozygous) as in our patients.
Targeted Search in Primary Immunodeficiency Genes
We ran a targeted search for rare (MAF <1%), non-synonymous, and LoF variants in a list of 252 known PID genes. This list includes known genes in 50 PID syndromes compiled by The International Union of Immunological Societies (IUIS) Expert Committee in 2015 (20) and 3 newly published PID genes since the latest release of IUIS till April 2016 (Table 1).

Table 1. New PID genes discovered since the latest report of IUIS in 2015 till April 2016.
Gene-Annotation Enrichment Analysis
We used the database for annotation, visualization, and integrated discovery (DAVID) (24, 25) v6.7 with default options and highest classification stringency for functional annotation clustering of PID genes carrying rare, non-synonymous variants. We used hypergeometric test for assessing the significance of gene enrichment results.
C9 Reconstitution Experiment
Serum of a patient heterozygous for a complement C9 variant was diluted with GVB++ in a series starting at 1:10 to 1:640 in 1:2 steps. The same was done with a serum pool as a control. This pool consisted of serum from over 300 healthy donors. Forty microliters of these dilutions were pipetted in duplo in a round-bottom 96-wells plate. To these wells, 40 μl of GVB++ was added to the samples and the blanc, to the positive control 40 μl of 1.7% saponin was added (100% lysis). Ten microliters of purified C9 (Quidel) or GVB++ were added (end conc 50 μg/ml). Next, in all wells, 150 μl of EA’s were added. EA’s are sheep erythrocytes (Hatunalab, Sweden) coated with an optimal dose of Amboceptor (Rabbit-anti-Sheep erythrocyte; Dade Behring) brought to a concentration of 0.25 × 108 cells/ml. This was incubated for 1 h at 37°C while agitating. After incubation, the plate was centrifuged for 5 min at 2000 rpm. Fifty microliters of each supernatant were pipetted to a flat-bottom 96-wells plate and diluted with 150 μl GVB++. Extinction was measured at 415 nm using a spectrophotometer. The percentage of lysis was calculated as follows:
GVB++ buffer consisted of 2 mM 5,5 di-ethylbarbituurzuur (Genfarma), 1.15 mM Na 5,5 di-ethylbarbituraat (Bufa), 96 mM NaCl (Merck), 0.5 mM CaCl (Merck), and 0.18 mM MgCl2 (Merck).
Results
Clinical Presentation
We identified 11 previously healthy children with community-acquired P. aeruginosa bacteremia from whom DNA could be obtained. Child–parent DNA trios were available in seven affected families. The presenting age ranged from 6 months to 4 years, and 7/11 (64%) of patients were male (Table 2). All patients had never been admitted to hospital or exposed to intravenous antibiotics prior to presenting with P. aeruginosa sepsis. Nine patients had developed respiratory symptoms within 72 h of leading to hospital admission, one presented with Ecthyma gangrenosum (Figure 1), and eight (73%) died. Three children died in the emergency department shortly after presentation, and all deaths occurred within 48 h of hospital admission. Postmortem examination reports were available in seven cases. In four, P. aeruginosa grew in high quantities from nasal and oral swabs, tracheal secretions, and lungs. Viral co-infections were found in five deceased patients, including human herpes virus-6, parainfluenza virus-3, human metapneumovirus, respiratory syncytial virus, and varicella virus. One surviving patient presented with acute abdomen, and P. aeruginosa bacteremia was thought to result from intra-abdominal perforation. Another surviving patient was diagnosed with urosepsis and P. aeruginosa grew in urine and blood. The third surviving patient presented with recurrent parainfectious neutropenia. Six patients received aminoglycosides and/or anti-Pseudomonas beta-lactam antibiotics during sepsis, including all three survivors.
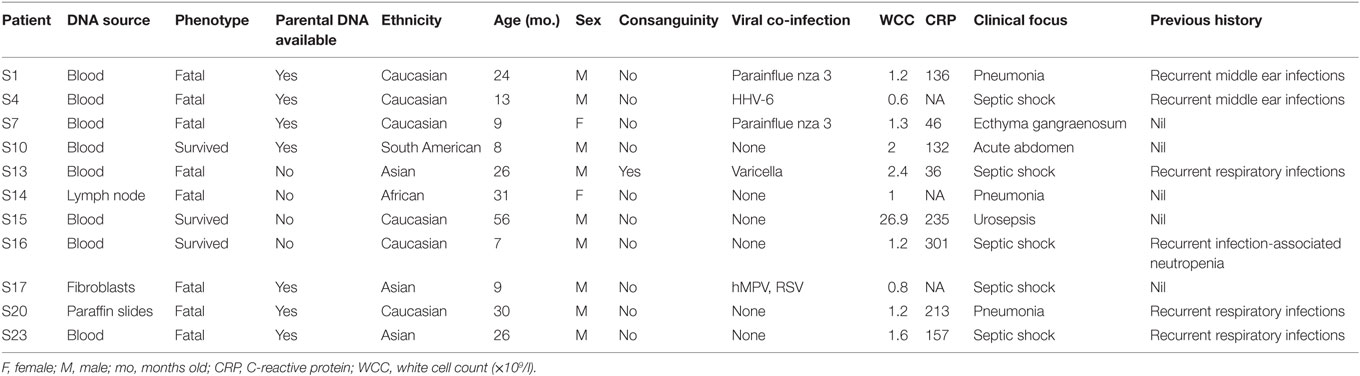
Table 2. Demographic and clinical characteristics of included children with community-acquired Pseudomonas aeruginosa septicemia.
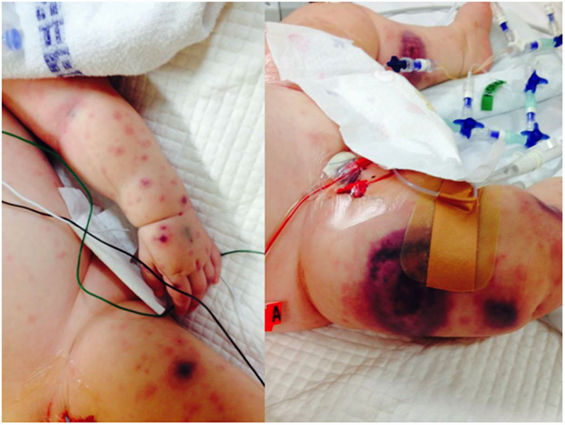
Figure 1. Ecthyma gangrenosum, a highly suggestive rapidly progressive purpuric skin lesion seen in a minority of children with P. aeruginosa bacteremia, is shown in one of the study patients (with permission from parents).
Exome Sequencing and Short Read Alignment
For each sample, 95% of reads passing filtering criteria were unique (not marked as duplicate); 97% of unique reads could be aligned to the human reference genome hg19. The mean on-bait coverage was 66×, with 95% of target bases achieving at least 10× coverage and 76% achieving at least 30× coverage (Table S1 in Supplementary Material). In 118 of the PID genes, at least one exon had an average coverage of <2×. Further investigation of these low-coverage intervals showed that the majority of them are overlapping with untranslated regions (UTRs).
Variant Calling and Variant Annotation
A total of 115,604 SNVs and 10,814 indels passed GATK quality control, including 28,795 synonymous variants, 28,284 non-synonymous variants, 713 in-frame indels, 660 frameshift indels, 283 stop-gained, and 151 splice-site variants (Tables S2 and S3 in Supplementary Material).
Variant Analysis
Thirty-eight rare, non-synonymous coding variants (MAF <1%) were observed in the 11 patients after testing for different inheritance models (Table 3), among which 12 were never seen in the same zygosity form (heterozygous or homozygous) in publicly available databases (Table 4). In addition to the above 38 variants, we performed a targeted search for rare, non-synonymous coding variants in 252 previously known PID genes and found 76 variants in 61 genes (Table 5). Functional classification of these 61 genes using DAVID highlighted the complement pathway as the most enriched cluster (DAVID enrichment score: 15.73). In total, 14 of the 28 complement pathway genes present in the list of 252 known PID genes carried at least one rare, non-synonymous coding variant in our cohort (hypergeometric probability: p(x > 14) = 0.003).
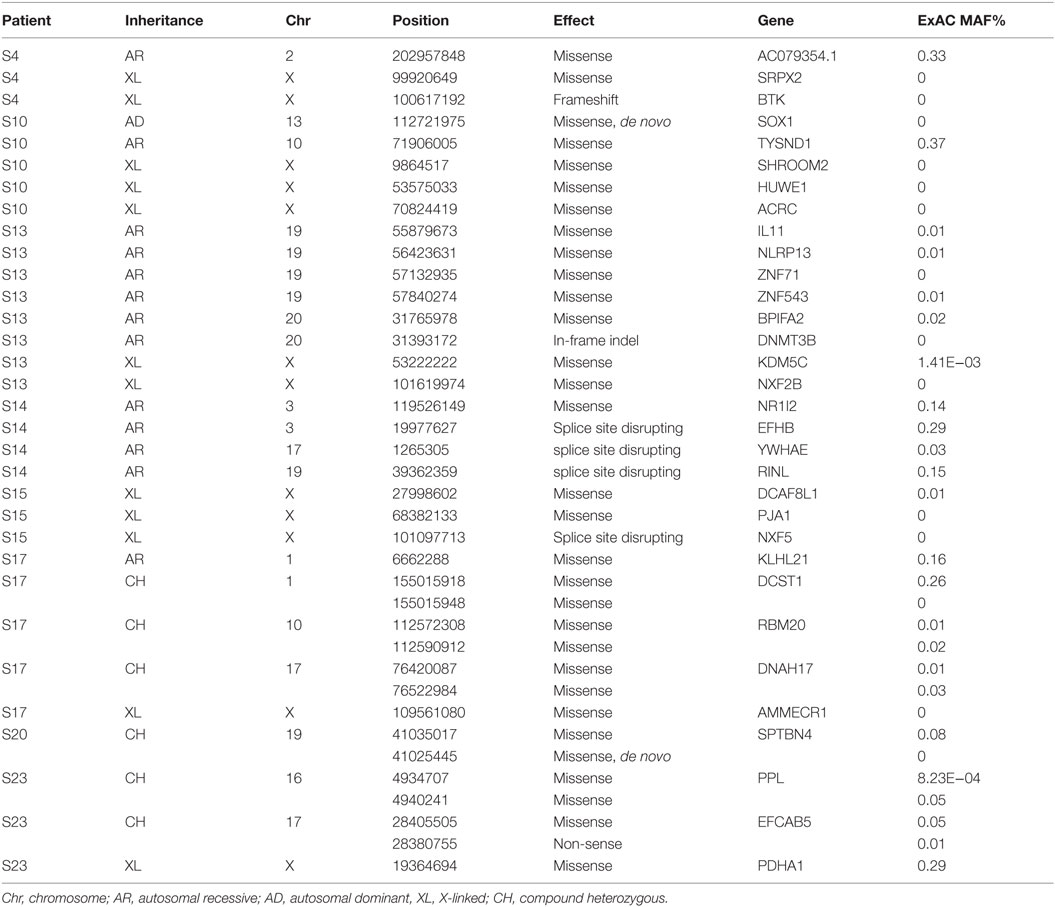
Table 3. 38 rare (MAF <1% in ESP, 1KP, UK10K, ExAC, and in-house controls) non-synonymous and putative LoF variants found in our 11 patients using different inheritance models (see Materials and Methods for details).
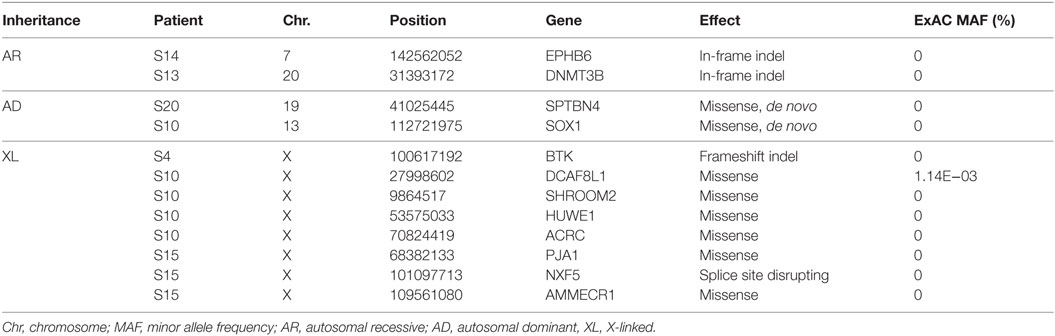
Table 4. Potentially causal variants (see Materials and Methods for details) identified in 6 of the 11 previously healthy children with community-acquired Pseudomonas aeruginosa septicemia.
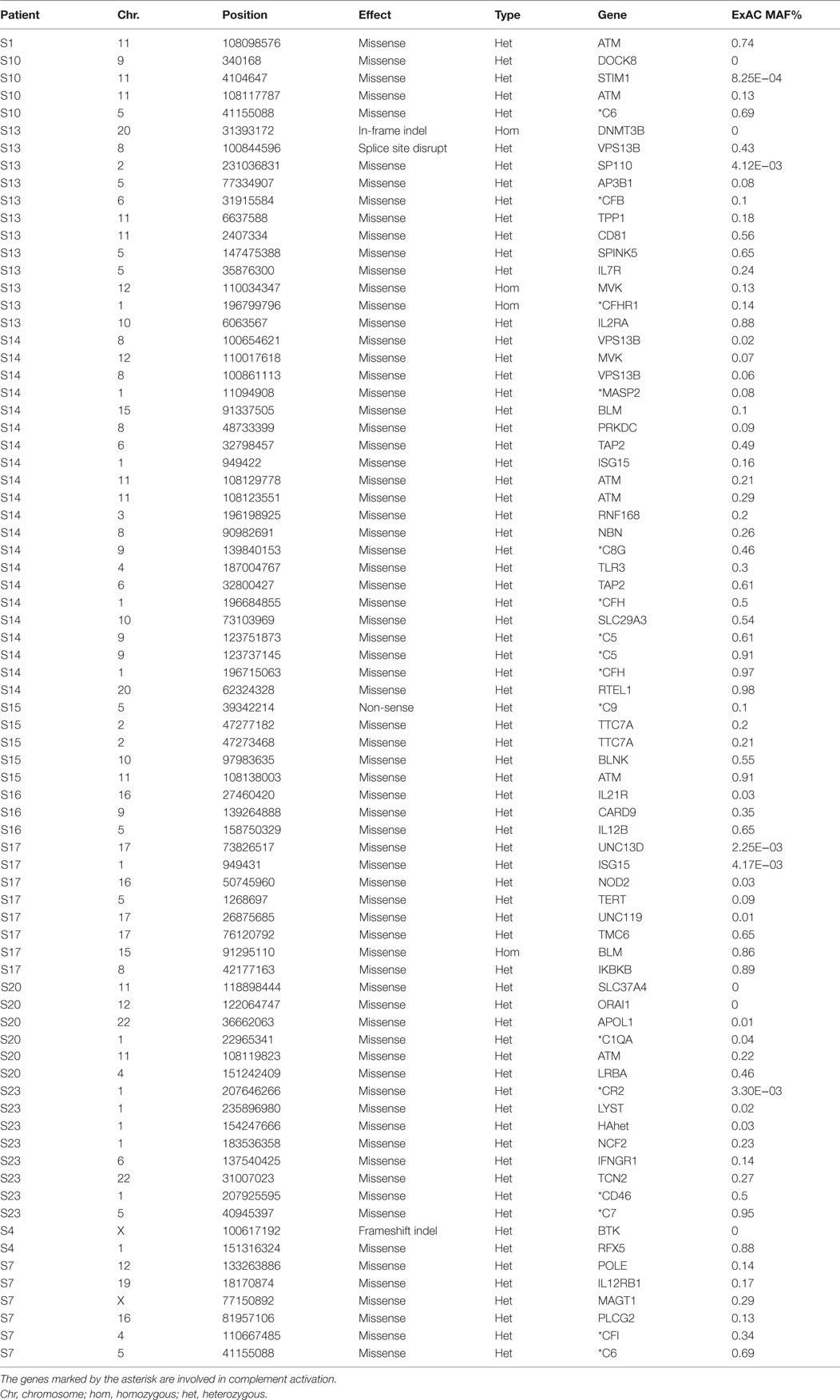
Table 5. 76 rare (ExAC MAF <1%) non-synonymous and putative LoF variants found in known 252 PID genes in our 11 patients.
In one patient (patient S4) who died of fulminant septic shock, we identified a novel single-base insertion on the X chromosome (C > CT, chromosome 20, position 100617191) leading to a frameshift in the Bruton agammaglobulinemia tyrosine kinase (BTK) gene. Other than a middle ear infection during infancy, the previous history, growth, development, and vaccination history of the patient had been unremarkable. Taqman genotyping and clinical genetic testing confirmed presence of mutation in the deceased patient and revealed a de novo occurrence in the mother, while other family members were healthy. BTK plays a crucial role in B-cell development. Postmortem serological testing confirmed the absence of immunoglobulins, consistent with BTK LoF: IgG < 1.3 g/l (4.22–11.9); IgA < 0.2 g/l (0.2–1.58); IgM < 0.2g/l (0.48–1.9).
A novel homozygous deletion spanning 6 bp on chromosome 20 (ACTCGAG > A, X chromosome, position 31393171) was observed in a 2-year-old boy with consanguineous parents (patient S13), leading to an in-frame deletion in a conserved region of the catalytic domain of the DNMT3B gene. Previously described missense mutations in the same protein domain are known to cause immunodeficiency-centromeric instability-facial anomalies (ICF) syndrome (26), a rare disease characterized by variable immunodeficiency and recurrent infections with mild facial abnormalities. The fatal septicemia was preceded by uncomplicated varicella, and the patient had a history of recurrent mild respiratory and upper airway infections that had been attributed to poor health conditions. The patient had normal T and B cell counts but reduced immunoglobulin levels, consistent with ICF syndrome: IgG 0.16 g/l (4.22–11.9); IgA <0.06 g/l (0.2–1.58); IgM 0.05 g/l (0.48–1.9).
We also observed a rare, known (MAF = 0.001 in ExAC) heterozygous stop-gain variant in the complement C9, on chromosome 5 (G > T, chromosome 5, position 39342214) in a male patient (patient S15) with severe Pseudomonas septicemia and recurrent parainfectious neutropenia. The patient survived without sequelae. Complement reconstitution assay with classical or terminal pathway proteins (C1–C9) showed normal lytic activity (Figure 2), suggesting that this variant is unlikely to be causal to increased susceptibility to P. aeruginosa.
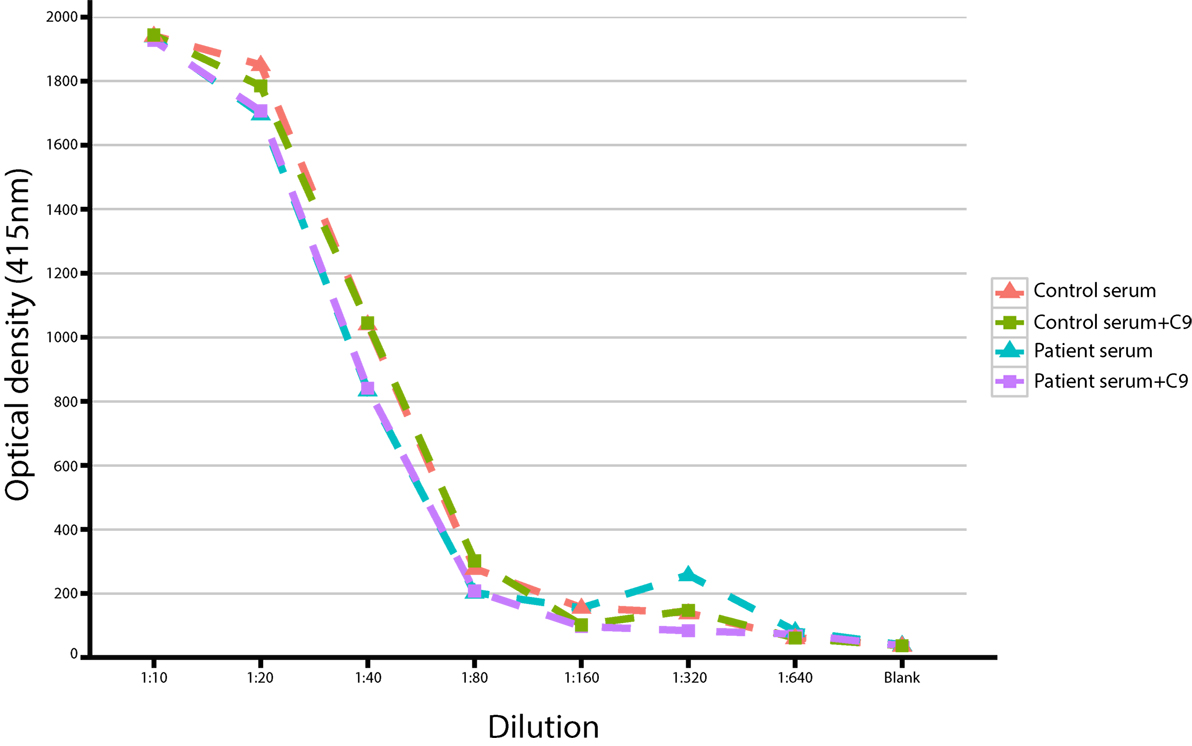
Figure 2. Titration of serum with/without C9 in a study patient with a heterozygous stop-gain variant in the C9 gene.
Discussion
We studied P. aeruginosa sepsis in previously healthy children as a model disease defining an extreme phenotype of fulminant sepsis. Fulminant sepsis in children is associated with high mortality, and the majority of deaths occur within hours of presentation (27). The disease burden is highest in children under 5 years of age (3, 7). We hypothesized that rare genetic variants with high effect sizes could explain life-threatening susceptibility to P. aeruginosa in children without known risk factors or comorbidities. Mendelian disorders of immunity have previously been shown to result in phenotypes with unusual susceptibility to bacterial infection, such as pseudomonal and pneumococcal infections in patients with IRAK-4 deficiency (10, 28). We used exome sequencing to explore the genetic cause of susceptibility to P. aeruginosa in a carefully selected group of previously healthy children with no familial history of immunodeficiency, who developed community-acquired P. aeruginosa septicemia. By systematic search for fully penetrant, rare, non-synonymous, and LoF variants in exonic regions, we identified 12 potentially causal variants including two novel variants in known PID genes: X-linked agammaglobulinemia due to a novel BTK mutation and ICF immunodeficiency syndrome due to a novel DNMT3B mutation. In addition, we performed a targeted search for rare, non-synonymous, and LoF variants in known PID genes and observed an enrichment of such variants among genes that are part of the complement pathway. One patient carried a heterozygous, stop-gain variant in the C9 gene, which has been associated with recurrent meningitis (29, 30). However, functional testing of patient samples showed normal lytic activity, demonstrating the need for strict validation of potentially causal mutations, even in genes with plausible biological links to the study phenotype. Variants conferring life-threatening susceptibility to common infections in children below reproductive age are subjected to strong purifying selection and will therefore only be found at very low frequencies (31–33). Out of the 12 potentially causal variants described in this study, 11 were novel and 1 was a hemizygous variant present in 1 individual (in heterozygous form) in ExAC.
Previous case reports describing P. aeruginosa sepsis in children with known PIDs, such as Wiskott–Aldrich syndrome, X-linked agammaglobulinemia, cyclic neutropenia, and IRAK-4/MyD-88 deficiency (8, 10, 22, 34), were based on conventional, candidate–gene-driven immunological testing. Ecthyma gangrenosum is present in a minority of patients and has been described as the presenting sign of an underlying PID, such as cyclic neutropenia, chronic granulomatous disease, or hypogammaglobulinemia (35–37). The range of PID types identified in these previous reports indicates that multiple distinct genetic defects have to be considered when investigating such clinical presentations. Importantly, classic PID screening may fail to identify new mutations in known PID genes (38) and will miss causal mutations in genes without an established link with PIDs. Failure to diagnose rare underlying PID in a child presenting with sepsis may result in potentially devastating consequences for survivors, undiagnosed siblings, and their families. High-throughput sequencing, in particular exome sequencing, has proved highly successful in the clinical diagnosis of suspected monogenic conditions in the pediatric population (39–41). This was recently illustrated by Record et al., who used exome sequencing in a girl with P. aeruginosa sepsis and found a homozygous LoF mutation in MAP3K9 (MKL1), a gene that was not known to cause PID (22).
A strength of the present cohort is that the case definition required significant growth of community-acquired P. aeruginosa in blood culture in previously healthy children presenting with signs and symptoms of sepsis, thereby constituting an extreme phenotype. The majority of children included in this study presented with fulminant sepsis and septic shock due to Pseudomonas sepsis. In the cases where postmortem examinations were performed, extensive growth of P. aeruginosa was found in several tissues, suggesting overwhelming bacterial infection. In agreement with other studies, we observed that several sepsis patients had viral co-infections (3). The role of viral co-infections in the pathogenesis of childhood bacterial sepsis is poorly understood and may include facilitated bacterial invasion due to respiratory epithelial disruption and increased host susceptibility during viremia (42). Immunological investigations in children presenting with fulminant sepsis can be extremely challenging, as severe leukopenia, coagulopathy, multi-organ failure, and fluid resuscitation often lead to severe alterations of cell counts, immunoglobulin levels, and complement levels. In addition, a proportion of patients die prior admission to an intensive care unit, or present with out-of-hospital cardiac arrest or sudden infant death syndrome (43). To date, there are no widely accepted guidelines to inform pediatricians, emergency and intensive care physicians, and pathologists about indications for specific immunological investigations in previously healthy children presenting with fulminant or fatal sepsis (44). Given the high fatality in our cohort, with several cases recruited considerable time after death of the patient, our study highlights the value of performing exome sequencing in this population using blood or tissue containing DNA. However, the lack of viable host cells in deceased patients may represent a major limitation toward functional validation of potentially causal variants. Elucidating the role of these variants in novel genes that may confer a PID phenotype will therefore require independent validation in other patients or cohorts.
While it is well known that PIDs can be responsible for severe bacterial infections, little is known about the proportion of children with invasive infections suffering from PID (5). In our cohort, defects in known PID genes were found in 25% of fatal cases. This is comparable to a recent study using exome sequencing in 50 patients with common variable immunodeficiency where exome sequencing identified disease-causing mutations in 30% of cases (45). While non-genetic factors, including variation in pathogen virulence and secondary neutropenia, may be responsible for the remaining cases, we cannot rule out that mutations in genes not previously associated with PID are at least partially involved. Sequencing of additional family members, sequencing of more cases with the same phenotype, and functional characterization of new candidate genes and variants will be required to identify such genetic factors. Furthermore, whole-genome sequencing is needed to explore the non-coding variants, large structural variants, and also exonic variants that are not be well-covered using exome sequencing.
In conclusion, this study provides proof of concept that exome sequencing allows the identification of rare genetic variants responsible for fulminant sepsis in children in whom immunodeficiency had not been previously suspected. Given the decreasing cost of exome and genome sequencing (39, 40, 46), we propose considering host DNA sequencing as a possible diagnostic procedure for apparently healthy children presenting with unusually severe or fatal sepsis.
Author Notes
Swiss Pediatric Sepsis Study Group: Klara Posfay-Barbe, Department of Pediatrics, University Hospitals of Geneva; Eric Giannoni, Service of Neonatology, Lausanne University Hospital, University of Lausanne, Lausanne; Christoph Aebi, Philipp Agyeman, Bendicht P. Wagner, and Luregn J. Schlapbach, Department of Pediatrics, Inselspital, Bern University Hospital, University of Bern, Switzerland; Ulrich Heininger, Infectious Diseases and Vaccinology, University of Basel Children’s Hospital, Basel; Gabriel Konetzny, Children’s Hospital Aarau; Alex Donas, Martin Stocker, Children’s Hospital Lucerne; Antonio Leone, Paul Hasters, Department of Neonatology, University Hospital Zurich; Anita Niederer-Loher, Christian Kahlert, Children’s Hospital of Eastern Switzerland St. Gallen; Walter Baer, Children’s Hospital Chur; Christa Relly, Christoph Berger, University Children’s Hospital Zurich, Switzerland.
Author Contributions
LS was responsible for the design of the study, patient recruitment and data acquisition, analysis, interpretation, and drafting of the manuscript. SA and JF were involved in the study design, genomic analyses, interpretation, and drafting of the manuscript. PM and IB were involved in genomic analyses, helped revise the manuscript, and approved the final version of the manuscript. JP, MW, RW, JRF, KA, KG, PA, CA, and CB were involved in study design, performed patient recruitment, were involved in revising the manuscript, and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all participating families for their support. We would like to thank Marc Vipond, Jane Armes, Deon Venter (Mater Pathology, Mater Health Services, Brisbane, QLD, Australia), Jonas Bjorkman (Pathology Queensland, Queensland Health, Brisbane, QLD, Australia), and Andrew Williams (The Children’s Hospital at Westmead Immunology Laboratory) for help in sample logistics and DNA extraction.
Funding
This study was supported by grants from the Mater Foundation, the Mater Medical Research Institute, the Intensive Care Foundation of Australia and New Zealand, the Swiss National Science Foundation (342730_153158), the Swiss Society of Intensive Care, the Bangerter Foundation, the Vinetum and Borer Foundation, and the Foundation for the Health of Children and Adolescents. JF is the recipient of an SNF Professorship from the Swiss National Science Foundation (PP00P3_133703). The funding sources did not have any role in the design of the study, the analyses, the writing of the manuscript, or the decision to submit it for publication.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00357
References
1. Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med (2003) 167:695–701. doi:10.1164/rccm.200207-682OC
2. Ladhani S, Pebody RG, Ramsay ME, Lamagni TL, Johnson AP, Sharland M. Continuing impact of infectious diseases on childhood deaths in England and Wales, 2003-2005. Pediatr Infect Dis J (2010) 29:310–3. doi:10.1097/INF.0b013e3181d73322
3. Schlapbach LJ, Straney L, Alexander J, MacLaren G, Festa M, Schibler A, et al. Mortality related to invasive infections, sepsis, and septic shock in critically ill children in Australia and New Zealand, 2002-13: a multicentre retrospective cohort study. Lancet Infect Dis (2015) 15:46–54. doi:10.1016/S1473-3099(14)71003-5
4. Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis (2009) 9:281–90. doi:10.1016/S1473-3099(09)70066-0
5. Gaschignard J, Levy C, Chrabieh M, Boisson B, Bost-Bru C, Dauger S, et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin Infect Dis (2014) 59:244–51. doi:10.1093/cid/ciu274
6. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet (2010) 42:30–5. doi:10.1038/ng.499
7. Huang Y-CC, Lin T-YY, Wang C-HH. Community-acquired Pseudomonas aeruginosa sepsis in previously healthy infants and children: analysis of forty-three episodes. Pediatr Infect Dis J (2002) 21:1049–52. doi:10.1097/01.inf.0000036089.63240.37
8. Wong SN, Tam AY, Yung RW, Kwan EY, Tsoi NN. Pseudomonas septicaemia in apparently healthy children. Acta Paediatr Scand (1991) 80:515–20. doi:10.1111/j.1651-2227.1991.tb11895.x
9. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) (2010) 89:403–25. doi:10.1097/MD.0b013e3181fd8ec3
10. Stergiopoulou T, Walsh TJ, Seghaye M-CC, Netea MG, Casanova J-LL, Moutschen M, et al. Deficiency of interleukin-1 receptor-associated kinase 4 presenting as fatal Pseudomonas aeruginosa bacteremia in two siblings. Pediatr Infect Dis J (2015) 34:299–300. doi:10.1097/INF.0000000000000550
11. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (2009) 25:1754–60. doi:10.1093/bioinformatics/btp324
12. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (2010) 26:589–95. doi:10.1093/bioinformatics/btp698
13. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res (2010) 20:1297–303. doi:10.1101/gr.107524.110
14. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics (2013) 43:11.10.1–33. doi:10.1002/0471250953.bi1110s43
15. Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (2012) 6:80–92. doi:10.4161/fly.19695
16. Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP). Seattle, WA (2016). Available from: http://evs.gs.washington.edu/EVS/
17. Abecasis GRR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature (2010) 467:1061–73. doi:10.1038/nature09534
18. Geihs M, Yan Y, Walter K, Huang J, Memari Y, Min JL, et al. An interactive genome browser of association results from the UK10K cohorts project. Bioinformatics (2015) 31:4029–31. doi:10.1093/bioinformatics/btv491
19. Exome Aggregation Consurtium (ExAC). Cambridge, MA (2016). Available from: http://exac.broadinstitute.org
20. Picard C, Al-Herz W, Bousfiha A, Casanova J-LL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol (2015) 35:696–726. doi:10.1007/s10875-015-0201-1
21. Aavikko M, Kaasinen E, Nieminen JK, Byun M, Donner I, Mancuso R, et al. Whole-genome sequencing identifies STAT4 as a putative susceptibility gene in classic kaposi sarcoma. J Infect Dis (2015) 211:1842–51. doi:10.1093/infdis/jiu667
22. Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood (2015) 126:1527–35. doi:10.1182/blood-2014-12-611012
23. Andersen LL, Mørk N, Reinert LS, Kofod-Olsen E, Narita R, Jørgensen SE, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med (2015) 212:1371–9. doi:10.1084/jem.20142274
24. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc (2009) 4:44–57. doi:10.1038/nprot.2008.211
25. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res (2009) 37:1–13. doi:10.1093/nar/gkn923
26. Weemaes CM, van Tol MJ, Wang J, van Ostaijen-ten Dam MM, van Eggermond MC, Thijssen PE, et al. Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur J Hum Genet (2013) 21(11):1219–25. doi:10.1038/ejhg.2013.40
27. Cvetkovic M, Lutman D, Ramnarayan P, Pathan N, Inwald DP, Peters MJ. Timing of death in children referred for intensive care with severe sepsis: implications for interventional studies. Pediatr Crit Care Med (2015) 16:410–7. doi:10.1097/PCC.0000000000000385
28. Picard C, Puel A, Bonnet M, Ku C-LL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science (2003) 299:2076–9. doi:10.1126/science.1081902
29. Zoppi M, Weiss M, Nydegger UE, Hess T, Späth PJ. Recurrent meningitis in a patient with congenital deficiency of the C9 component of complement. First case of C9 deficiency in Europe. Arch Intern Med (1990) 150:2395–9. doi:10.1001/archinte.150.11.2395
30. Witzel-Schlömp K, Späth PJ, Hobart MJ, Fernie BA, Rittner C, Kaufmann T, et al. The human complement C9 gene: identification of two mutations causing deficiency and revision of the gene structure. J Immunol (1997) 158:5043–9.
31. Alcaïs A, Abel L, Casanova J-LL. Human genetics of infectious diseases: between proof of principle and paradigm. J Clin Invest (2009) 119:2506–14. doi:10.1172/JCI38111
32. Loeb M. Host genomics in infectious diseases. Infect Chemother (2013) 45:253–9. doi:10.3947/ic.2013.45.3.253
33. Casanova J-LL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A (2015) 112:E7128–37. doi:10.1073/pnas.1521651112
34. Baro M, Marín MA, Ruiz-Contreras J, de Miguel SF, Sánchez-Díaz I. Pseudomonas aeruginosa sepsis and ecthyma gangrenosum as initial manifestations of primary immunodeficiency. Eur J Pediatr (2004) 163:173–4. doi:10.1007/s00431-003-1381-x
35. Ng W, Tan CL, Yeow V, Yeo M, Teo SH. Ecthyma gangrenosum in a patient with hypogammaglobulinemia. J Infect (1998) 36:331–5. doi:10.1016/S0163-4453(98)94603-5
36. Prindaville B, Nopper AJ, Lawrence H, Horii KA. Chronic granulomatous disease presenting with ecthyma gangrenosum in a neonate. J Am Acad Dermatol (2014) 71:e44–5. doi:10.1016/j.jaad.2013.12.038
37. Flinn A, McDermott M, Butler KM. A child with septic shock and purpura. JAMA Pediatr (2016) 170:391–2. doi:10.1001/jamapediatrics.2015.2466
38. Chou J, Ohsumi TK, Geha RS. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiencies. Curr Opin Allergy Clin Immunol (2012) 12:623–8. doi:10.1097/ACI.0b013e3283588ca6
39. Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med (2016). doi:10.1038/gim.2016.1
40. Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr (2015) 3:67. doi:10.3389/fped.2015.00067
41. Conley ME, Casanova J-LL. Discovery of single-gene inborn errors of immunity by next generation sequencing. Curr Opin Immunol (2014) 30:17–23. doi:10.1016/j.coi.2014.05.004
42. Jansen AG, Sanders EA, van der Ende A, van Loon AM, Hoes AW, Hak E. Invasive pneumococcal and meningococcal disease: association with influenza virus and respiratory syncytial virus activity? Epidemiol Infect (2008) 136:1448–54. doi:10.1017/S0950268807000271
43. Weber MA, Klein NJ, Hartley JC, Lock PE, Malone M, Sebire NJ. Infection and sudden unexpected death in infancy: a systematic retrospective case review. Lancet (2008) 371:1848–53. doi:10.1016/S0140-6736(08)60798-9
44. Randolph AG, McCulloh RJ. Pediatric sepsis: important considerations for diagnosing and managing severe infections in infants, children, and adolescents. Virulence (2014) 5:179–89. doi:10.4161/viru.27045
45. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova J-LL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol (2016) 7:220. doi:10.3389/fimmu.2016.00220
Keywords: bacteremia, sepsis, child, Pseudomonas, primary immunodeficiency, exome sequencing
Citation: Asgari S, McLaren PJ, Peake J, Wong M, Wong R, Bartha I, Francis JR, Abarca K, Gelderman KA, Agyeman P, Aebi C, Berger C, Fellay J, Schlapbach LJ and The Swiss Pediatric Sepsis Study (2016) Exome Sequencing Reveals Primary Immunodeficiencies in Children with Community-Acquired Pseudomonas aeruginosa Sepsis. Front. Immunol. 7:357. doi: 10.3389/fimmu.2016.00357
Received: 11 August 2016; Accepted: 01 September 2016;
Published: 20 September 2016
Edited by:
Andrew Gennery, Newcastle University, UKCopyright: © 2016 Asgari, McLaren, Peake, Wong, Wong, Bartha, Francis, Abarca, Gelderman, Agyeman, Aebi, Berger, Fellay, Schlapbach and The Swiss Pediatric Sepsis Study. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacques Fellay, amFjcXVlcy5mZWxsYXlAZXBmbC5jaA==;
Luregn J. Schlapbach, bC5zY2hsYXBiYWNoQHVxLmVkdS5hdQ==
†Jacques Fellay and Luregn J. Schlapbach contributed equally to this work.