 Stavroula Masouridi-Levrat
Stavroula Masouridi-Levrat Federico Simonetta
Federico Simonetta Yves Chalandon*
Yves Chalandon*
- Division of Hematology, Department of Medical Specialties, Faculty of Medicine, Geneva University Hospitals, University of Geneva, Geneva, Switzerland
Bone marrow failure (BMF) syndromes are severe complications of allogeneic hematopoietic stem cell transplantation (allo-HSCT). In this paper, we distinguish two different entities, the graft failure (GF) and the poor graft function (PGF), and we review the current understanding of the interactions between the immune and hematopoietic compartments in these conditions. We first discuss how GF occurs as the result of classical alloreactive immune responses mediated by residual host cellular and humoral immunity persisting after conditioning and prevented by host and donor regulatory T cells. We next summarize the current knowledge about the contribution of inflammatory mediators to the development of PGF. In situations of chronic inflammation complicating allo-HSCT, such as graft-versus-host disease or infections, PGF seems to be essentially the result of a sustained impairment of hematopoietic stem cells (HSC) self-renewal and proliferation caused by inflammatory mediators, such as interferon-γ (IFN-γ) and tumor necrosis factor-α, and of induction of apoptosis through the Fas/Fas ligand pathway. Interestingly, the production of inflammatory molecules leads to a non-MHC restricted, bystander inhibition of hematopoiesis, therefore, representing a promising target for immunological interventions. Finally, we discuss immune-mediated impairment of bone marrow microenvironment as a potential mechanism hampering hematopoietic recovery. Better understanding of immunological mechanisms responsible for BMF syndromes after allo-HSCT may lead to the development of more efficient immunotherapeutic interventions.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is standard of care for many hematological diseases, and sustained engraftment of donor stem cells represents a fundamental prerequisite for a good outcome. Bone marrow failure (BMF) syndromes remain severe complications of allo-HSCT associated with considerable morbidity and mortality, highlighting the need for therapeutic manipulation. In this paper, we review the current understanding of the complex interactions between the immune and hematopoietic compartments in BMF syndromes after allo-HSCT.
Two entities with different clinical and physiopathological features can be distinguished: the graft failure (GF) and the poor graft function (PGF). Unfortunately, in a large number of studies, there is not a clear distinction between GF and PGF. Other frequent limitations of the literature are the lack of separation between primary and secondary cases and the heterogeneity of definition criteria. Primary GF is characterized by the lack of donor engraftment and is defined as never having achieved absolute neutrophil count ≥0.5 × 109/L lasting for at least three consecutive days without evidence of disease relapse (Table 1). Thus, in case of myeloablative conditioning, the patient never recovers from the neutropenia making second allo-HSCT the only therapeutic option; while in case of reduced intensity conditioning (RIC), an autologous recovery may occur. Secondary GF refers to the loss of a previously functioning graft (1) associated with loss of full donor chimerism. Therefore, in both primary and secondary GF, chimerism status is either mixed, with the presence of recipient cells, or full recipient. GF incidence ranges from 3.8 to 5.6% (1–3) and varies significantly according to different transplant settings. Factors associated with increased risk for GF are HLA-mismatched grafts, RIC regimens (4), bone marrow (BM) grafts, low stem cell dose, non-malignant disorders, major ABO incompatibility, female donor grafts for male recipients (1–3), myeloproliferative disease (5), and disease status at transplantation (3). In contrast to GF, PGF is characterized by the presence of a full donor chimerism status (Table 1) as donor engraftment is achieved and sustained (6). Graft function can be poor either as a result of incomplete hematological recovery (primary PGF) or of a decrease of blood counts after prompt recovery (secondary PGF) (7). It is diagnosed in patients with two or three cytopenic lineages, hypoplastic/aplastic BM and full donor chimerism (Table 1). PGF occurs in 5–27% of patients (7) and it is associated with several post allo-HSCT conditions, such as infections, mostly viral, use of myelotoxic drugs, and graft-versus-host disease (GvHD) (8). Of note, GvHD, a common post allo-HSCT complication with an overall incidence up to 59% (9), represents the major risk factor for developing PGF (10) either in the context of the recently identified entity of BM GvHD (11, 12), or more usually, without specifically targeting BM. Thus, cytopenias are observed in ~40% of GvHD patients (13), while PGF is also seen in the absence of GvHD.

Table 1. Clinical characteristics of graft failure and poor graft function.
Immunological Basis of Graft Failure
Graft failure occurs as the result of a classical alloreactive immune response mediated by residual host immunity persisting after the conditioning regimen (Figure 1A). Residual host T cells are considered the most prominent effector cells mediating rejection (14). Importantly, T-cell-mediated graft rejection can occur in both HLA-mismatched (15) and HLA-matched (16) settings, in the latter case as a result of responses directed against minor histocompatibility antigens (MiHA). The male-specific H-Y antigen represents a good example of a target MiHA involved in graft rejection (17, 18), increasing the risk of GF in sex mismatched transplantations (2, 19). The molecular pathways involved in T-cell-mediated graft rejection are still not completely defined and are probably multiple and somehow redundant as ablation of perforin, FasL, tumor necrosis factor receptor-1 (TNFR-1), and of tumor necrosis factor ligands TRAIL, TWEAK, and TL1A fails to prevent rejection (20, 21). Conversely, donor cytotoxic T cells are well known to mediate a facilitative effect on HSC engraftment (22–24) while graft T-cell depletion is associated with increased incidence of GF (25–27).
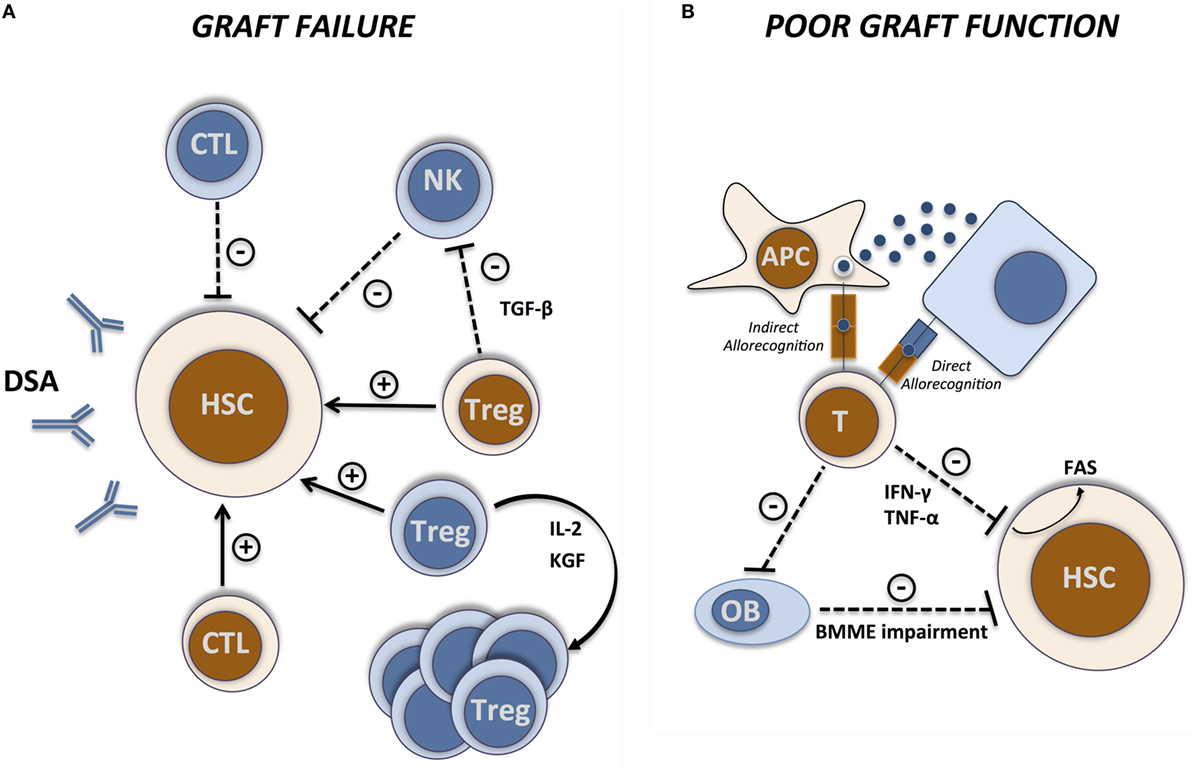
Figure 1. Immunological basis of GF and PGF. Immunological interactions between host (blue) and donors (brown) cells during (A) graft failure and (B) poor graft function. Arrows indicate facilitating effects and dashed lines indicate inhibitory effects. CTL, cytotoxic T lymphocyte; APC, antigen-presenting cell; OB, osteoblast; BMME, bone marrow microenvironment.
Residual host natural killer (NK) cells can also eliminate donor HSC as demonstrated in early studies in animal models (28–30). NK-mediated graft rejection, mainly observed in MHC-mismatched transplantations, is the result of a “missing-self recognition,” when the inhibitory receptors expressed on donor NK cells, belonging to the Ly49 family in mice and to the killer immunoglobulin-like receptors (KIRs) in humans, fail to recognize their cognate MHC class I molecule on host cells (31). Graft rejection by host NK cells is, at least partially, dependent on perforin-mediated cytotoxicity (32, 33). Similar to donor T cells, donor NK cells can facilitate HSC engraftment. In mice, adoptive transfer of donor NK cells facilitates HSC engraftment (34, 35), at least partly by abrogating the resistance of residual host effector immune cells to the graft. In humans, a similar effect has been observed in the HLA-haploidentical setting as transplantation with KIR ligand incompatible cells in the graft-versus-host direction conferred a significant protection from graft rejection in patients with acute myeloid leukemia (36).
The role of antibody-mediated rejection after allo-HSCT is much more controversial (37, 38). Recent studies demonstrate that the presence of donor-specific HLA antibodies (DSA), resulting in most cases from previous transfusion-induced sensitization, is associated with GF in HLA-mismatched (39–42) and haploidentical (42, 43) transplantations. Nevertheless, it is not clear whether DSA can actually mediate graft rejection or if they are surrogate markers for the cellular immunity that causes graft rejection. Recently published data suggest that integrated humoral and cellular immunity recognizing the same alloantigen of the donor can mediate graft rejection in DSA-positive patients undergoing HLA-mismatched cord blood allo-HSCT (44). Furthermore, patients displaying pretransplant donor-specific antibodies directed against CD34+/VEGFR-2+ endothelial progenitor stem cells presented a higher risk of GF, providing additional evidence supporting antibody-mediated rejection (45).
CD4+CD25+ regulatory T cells (Tregs) are critical immunomodulatory cells participating to immune and hematopoietic compartment interactions. In a syngeneic setting, Treg ablation enhanced early post-transplant hematopoiesis while co-administration of Tregs at time of transplantation inhibited myelopoiesis (46). Conversely, in the allogeneic setting, an engraftment facilitating effect of both host and donor Tregs seems to exist. Host Tregs prevent GF as their prior removal using anti-CD25 mAbs strongly enhanced allogeneic BM rejection in mice (47). Accordingly, adoptive transfer of host-type Tregs improved durable engraftment of allogeneic BM grafts (48–50). Interestingly, high resolution in vivo imaging showed marked co-localization of HSCs with host Tregs on the endosteal surface in the calvarial and trabecular BM, while after Treg depletion HSCs were lost, suggesting a direct effect of Tregs in HSC niches generation and maintenance (51). This effect seems to be essentially dependent on IL-10 production by Treg as IL-10 blockade by monoclonal antibodies or the use of Tregs isolated from IL-10 KO mice prevent the Treg-mediated protection of HSCs (51).
Donor Tregs seem as well to exert an engraftment facilitating effect without causing GvHD. In a fully MHC-mismatched BMT murine model, the co-transplantation of donor Tregs into sublethally conditioned recipients resulted in decreased early rejection of hematopoietic progenitors and improved long-term donor chimerism without inducing GvHD (52). The precise mechanisms through which host and donor Tregs exert their function on hematopoiesis are still incompletely understood. Transforming growth factor-β (TGF-β), a key mediator of Treg function, seems to play a role in Treg-mediated inhibition of IL-3-induced colony-forming units (46). Similarly, TGF-β is also involved in Treg engraftment facilitation as anti-TGF-β mAb treatment prior to allo-HSCT led to a significant increase in NK cell-mediated graft rejection, suggesting that Tregs mediate NK cell suppression through TGF-β (47). A major limitation of Treg-based therapies comes from the difficulties to isolate them from peripheral blood in sufficient amounts for adoptive transfer. Use of freshly isolated or ex vivo expanded third-party Tregs (53) or pharmacological approaches to induce Treg expansion in vivo represent potential alternatives. Administration of IL-2/anti-IL-2 mAb complexes to RIC conditioned mice early after MHC-matched allogeneic HSCT induces a strong expansion of host Tregs that efficiently facilitate early and long-term engraftment (54). Of note, in the absence of prior cytoreductive treatment, IL-2/anti-IL-2 complexes administration failed to promote BM engraftment as its effect was extended to several lymphocytes populations (55). In addition to IL-2, other molecules inducing in vivo expansion of Tregs display a potential to improve engraftment, including keratinocyte growth factor (KGF). KGF facilitated engraftment in an MHC-matched HSCT murine model by increasing the frequency of Tregs and enhancing their in vivo immunosuppression ability (56). Importantly KGF lost its ability to improve engraftment in Scurfy mice that lack Tregs.
Mesenchymal stem cells (MSCs) are stromal-derived multipotent progenitors displaying immune-modulatory properties of potential interest in HSCT [recently reviewed in Ref. (57)]. Several pilot studies performed in humans reported accelerated hematopoietic engraftment when MSCs were co-transplanted with HSC (58, 59), suggesting a potential for MSCs in preventing (58–61) and treating (58) GF.
Immunological Basis of Poor Graft Function
While GF/rejection derives from “classical” alloreactive immune responses, PGF is the consequence of more complex and less well-defined interactions between the immune system and the hematopoietic compartment (Figure 1B). Important insights into the alloreactive immune responses involved in BMF syndromes first came from murine models of GvHD or aplastic anemia. Parental lymph node (LN) cells infusion into hybrid F1 with MHC-mismatch either sublethally irradiated (62) or not (63) induced a drastic and cell dose-dependent reduction of hematopoietic progenitors and stem cells. Marrow aplasia was associated with massive infiltration by T cells, mainly of the CD8+ compartment (62). The observation of strongly increased interferon-γ (IFN-γ) concentrations in these models (62, 63) pointed to IFN-γ produced by alloreactive T cells as the main effector molecule mediating BMF in this setting. Similar results were reported in a MHC class II-disparate, CD4-dependent model of GvHD in which BM aplasia was associated with massive infiltration by CD4+ T cells secreting IFN-γ (64). Interestingly, IFN-γ producing CD4+ T cells in this model expressed high levels of CXCR4 at their surface, suggesting a preferential BM homing potential. The relevance of IFN-γ as a key mediator of PGF in humans is supported by the observation of higher proportions of IFN-γ producing CD4+ and CD8+ T cells and decreased proportions of IL-4-producing T cells in BM from PGF patients, resulting in a shift of the IFN-γ/IL-4 ratio toward a type-1 immune response (65). IFN-γ and IL-4 levels in BM plasma were consistent with cellular results. The formal demonstration of the impact of IFN-γ in alloreactive PGF came from the observation that CD4+ T cells isolated from IFN-γKO mice adoptively co-transferred along with T-cell-depleted BM cells into sublethally irradiated recipients failed to induce the BM aplasia observed when WT CD4+ T cells were employed (66). Conversely, administration of IFN-γ receptor-1 deficient (IFN-γ-RKO) BM cells resulted in reduced severity of BMF (64). The use of IFN-γ-RKO recipients, in which extremely high, supraphysiological concentrations of IFN-γ are reached as a consequence of accumulation of IFN-γ produced by alloreactive T cells (67) or after inflammatory stimulation (68), provides complimentary information. In the MHC-matched, mHAg-mismatched C3H.SW > C57BL/6 transplantation model, donor HSCs were exposed to high levels of IFN-γ produced by alloreactive T cells leading to their decreased proliferation and finally to BM aplasia (67). A similar inhibition of HSCs was reported even when donor HSCs were transferred into syngenic IFN-γ-RKO mice in which extremely high levels of IFN-γ were induced through Mycobacterium bovis bacillus Calmette–Guérin (BCG) infection (68). Interestingly, the inhibitory effect of IFN-γ on hematopoiesis finally leads to lack of HSC engraftment despite the absence of alloreactive responses given the syngenic setting. Accordingly, using the already discussed BMF model induced by MHC-mismatch LN cells infusion, Chen and coworkers showed that co-transplantation of BM cells from BMF mice and from syngeneic healthy mice led to the destruction of hematopoietic progenitor stem and stromal cells from fully compatible healthy donors as “innocent bystanders” and not as targets (62). These observations suggest that, once elevated levels of IFN-γ are established, a non-HLA restricted inhibitory action is exerted on HSCs.
IFN-γ is a well-known negative regulator of HSC as demonstrated since earliest in vitro studies in both murine (69, 70) and human (71, 72) cells. IFN-γ seems to facilitate HSC programmed cell death either directly (73) or through induction of caspase-1, TRAIL (74), and FAS (75). Moreover, IFN-γ inhibits HSC self-renewal modulating expression of key cell-cycle genes, such as cyclin D1 (67, 76), Myc (67), and p57 (76). Notably, IFN-γ-mediated cyclin D1 suppression seems to essentially affect the immature LSK (Lin−Sca-1+c-kit+) hematopoietic stem/progenitor cell (HSPC) subset but not mature Lin+ cells (67). Finally, IFN-γ interferes with thrombopoietin-induced phosphorylation of signal transducer and activator of transcription-5 (STAT-5) in HSCs through modulation of suppressor of cytokine signaling (SOCS) 1 (76), further participating to inhibition of HSC proliferation through repression of STAT-5 target genes.
Collectively, these results suggest that in situations of chronic inflammation post allo-HSCT, such as GvHD or viral infections, PGF is, at least partially, the result of a sustained impairment of HSC self-renewal and proliferation caused by chronic IFN-γ exposure. In mice, IFN-γ blockade by administration of anti-IFN-γ neutralizing antibodies significantly improved cytopenias reversing or preventing BM aplasia in all aforementioned BMF models in which this strategy was tested (62, 63, 67, 68). IFN-γ, therefore, probably represents the most promising target for immunotherapeutic interventions in PGF. Targeted treatment with anti-IFN-γ blocking antibodies (NI-0501) is currently under clinical evaluation in primary hemophagocytic lymphohistiocytosis (NCT01818492), another setting of immune-mediated BMF.
Tumor necrosis factor-α (TNF-α) represents another inflammatory cytokine that, if overexpressed, may participate in post-allo-HSCT PGF mediation. Accumulating evidence has been established regarding the inhibitory role of TNF-α in clonogenic progenitor cells proliferation in vitro (77, 78). Incubation of LSK murine HSPC with TNF-α has been shown to upregulate Fas expression and to inhibit colony formation in vitro by reducing the size – but not the number – of proliferative clones (79), suggesting that HSCs can undergo a limited number of cell divisions before becoming sensitive to TNF-induced growth inhibition. Transplantation of these cells into lethally irradiated mice resulted in reduced short-term engraftment and long-term reconstituting activity, showing the impact of TNF-α on HSC function (79). Similarly, in a xenograft model of human CD34+CD38− cells treated with TNF-α in vitro and transplanted into NOD–SCID mice, their repopulating ability was dramatically compromised. A negative effect of TNF-α on HSC maintenance by enhancing their differentiation rather than self-renewal has been suggested (80). However, there is no general agreement about the in vivo role of TNF-α in HSC regulation. In BMF murine models, treatment with anti-TNF-α mAbs resulted in significantly prolongation of animal survival although less impressive than using anti-IFN-γ mAbs treatment (63). Furthermore, using mice deficient for either Tnfrsf1a receptor or Tnfrf1b receptor or both, a TNF-mediated suppression of HSCs activity was shown (81). Of note, it was dependent on both receptors as Tnfrsf1aKO and Tnfrsf1bKO BM cells presented a greater ability to long-term reconstitute myeloid and lymphoid cell lineages in myeloablated WT recipients compared to WT BM cells. This advantage was more pronounced when Tnfrsf1-doubleKO BM cells were used (81). It was also shown that in vivo administration of TNF targets actively cycling rather than quiescent HSCs, finding of relevance in the early post-allo-HSCT setting where HSCs are recruited from quiescence to active proliferation. Conversely, a positive role of TNF-α in HSC engraftment and function has been reported in models of purified HSC transplantation into allogeneic and syngeneic mice in which small numbers of BM-derived CD8+/TCR− cells have been shown to act as graft facilitating cells through a TNF-α-dependent mechanism (82).
In addition to soluble inflammatory mediators, substantial evidence implicates the Fas/Fas ligand pathway in the post-allo-HSCT PGF and mainly in the myelosuppressive effect of GvHD. Fas is expressed at CD34+ HSC surface (75, 83) and its expression is modulated by several factors, including IFN-γ and TNF-α (75). HSC exposure to agonist anti-Fas antibodies inhibits hematopoiesis by inducing HSC apoptosis (75, 83) and enhancing the effects of IFN-γ and TNF-α (75). In vivo, the Fas/FasL pathway has been shown to be relevant for BMF associated with cytomegalovirus infection (84) and GvHD (62, 63, 67, 83). In GvHD models, the use of FasL-defective (gld) donor cells prevented BM aplasia (83). Similarly, blockade of Fas/FasL interaction by the use of anti-FasL blocking antibodies significantly reduced cytopenias and BM aplasia (67, 83).
Hematopoiesis depends on special BM microenvironments known as “niches” in which HSCs reside as well as on the functional cross-talk between HSCs and these niches (85). BM endosteal, perivascular and vascular endothelial cells as well as osteoblasts have a fundamental role in the maintenance of HSCs by providing signals that regulate cell self-renewal, differentiation, and quiescence in mice (86). Two prospective nested case-control studies provided evidence that endosteal, perivascular cells, and endothelial progenitor cells are dramatically reduced in patients with primary and secondary PGF, suggesting that an impaired BM microenvironment hampers hematopoietic recovery after allo-HSCT (87, 88). The etiology of BM microenvironment impairment and how this leads to PGF remain unclear. Studies of BM GvHD in murine models identified osteoblasts as a major target for GvHD. Donor effector CD4+, and to a lesser extent CD8+, T cells caused early destruction of osteoblasts leading to severe impairment of B lymphopoiesis with dramatically decreased numbers of B-cell precursors and decreased expression of transcriptional factors essential for B lymphopoiesis, such as E2A and PAX5 (11). Surprisingly enough, there were no differences in serum levels of major inflammatory cytokines thought to be involved in GvHD, such as IL-1, IFN-γ, and TNF-α, suggesting that they are dispensable for CD4+ T cell-mediated osteoblast destruction. These findings were recently confirmed in allo-HSCT patients with PGF associated with chronic GvHD in which loss of osteoblasts, decreased numbers of B cells and an increased CD4/CD8 ratio were shown (89).
Conclusion
Bone marrow failure syndromes, namely GF and PGF, can develop after allo-HSCT and are associated with significant morbidity and mortality. In the present paper, we summarized the distinct immune-pathological mechanisms underlying GF and PGF and we discussed evidence identifying several inflammatory molecules as crucial mediators in their pathogenesis. Some preclinical evidence obtained in murine studies suggest that therapeutic interventions blocking these inflammatory mediators may represent a promising strategy to prevent and even reverse these severe complications.
Author Contributions
SM-L wrote the manuscript and designed the table and the figure. FS and YC critically discussed the work and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
FS is supported by grants from the Geneva University Hospitals’ Clinical Research Center and the Dubois-Ferrière-Dinu-Lipatti Foundation.
References
1. Olsson R, Remberger M, Schaffer M, Berggren D, Svahn B, Mattsson J, et al. Graft failure in the modern era of allogeneic hematopoietic SCT. Bone Marrow Transplant (2013) 48(4):537–43. doi:10.1038/bmt.2012.239
2. Olsson RF, Logan BR, Chaudhury S, Zhu X, Akpek G, Bolwell BJ, et al. Primary graft failure after myeloablative allogeneic hematopoietic cell transplantation for hematologic malignancies. Leukemia (2015) 29(8):1754–62. doi:10.1038/leu.2015.75
3. Cluzeau T, Lambert J, Raus N, Dessaux K, Absi L, Delbos F, et al. Risk factors and outcome of graft failure after HLA matched and mismatched unrelated donor hematopoietic stem cell transplantation: a study on behalf of SFGM-TC and SFHI. Bone Marrow Transplant (2016) 51(5):687–91. doi:10.1038/bmt.2015.351
4. Delgado J, Thomson K, Russell N, Ewing J, Stewart W, Cook G, et al. Results of alemtuzumab-based reduced-intensity allogeneic transplantation for chronic lymphocytic leukemia: a British Society of Blood and Marrow Transplantation Study. Blood (2006) 107(4):1724–30. doi:10.1182/blood-2005-08-3372
5. Ballen KK, Woolfrey AE, Zhu X, Ahn KW, Wirk B, Arora M, et al. Allogeneic hematopoietic cell transplantation for advanced polycythemia vera and essential thrombocythemia. Biol Blood Marrow Transplant (2012) 18(9):1446–54. doi:10.1016/j.bbmt.2012.03.009
6. Au W, Chan E, Lie A, Liang R, Leung A, Ma S, et al. Poor engraftment after allogeneic bone marrow transplantation: role of chimerism analysis in treatment and outcome. Ann Hematol (2003) 82(7):410–5. doi:10.1007/s00277-003-0676-3
7. Larocca A, Piaggio G, Podestà M, Pitto A, Bruno B, Di Grazia C, et al. Boost of CD34+-selected peripheral blood cells without further conditioning in patients with poor graft function following allogeneic stem cell transplantation. Haematologica (2006) 91(7):935–40.
8. Nakamae H, Storer B, Sandmaier BM, Maloney DG, Davis C, Corey L, et al. Cytopenias after day 28 in allogeneic hematopoietic cell transplantation: impact of recipient/donor factors, transplant conditions and myelotoxic drugs. Haematologica (2011) 2011:044966. doi:10.3324/haematol.2011.044966
9. Jagasia M, Arora M, Flowers ME, Chao NJ, McCarthy PL, Cutler CS, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood (2012) 119(1):296–307. doi:10.1182/blood-2011-06-364265
10. Peralvo J, Bacigalupo A, Pittaluga P, Occhini D, Van Lint M, Frassoni F, et al. Poor graft function associated with graft-versus-host disease after allogeneic marrow transplantation. Bone Marrow Transplant (1987) 2(3):279–85.
11. Shono Y, Ueha S, Wang Y, Abe J, Kurachi M, Matsuno Y, et al. Bone marrow graft-versus-host disease: early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood (2010) 115(26):5401–11. doi:10.1182/blood-2009-11-253559
12. Szyska M, Na I-K. Bone marrow GvHD after allogeneic hematopoietic stem cell transplantation. Front Immunol (2016) 7:118. doi:10.3389/fimmu.2016.00118
13. Lee K, Lee J, Choi S, Kim S, Seol M, Lee Y, et al. Failure of trilineage blood cell reconstitution after initial neutrophil engraftment in patients undergoing allogeneic hematopoietic cell transplantation–frequency and outcomes. Bone Marrow Transplant (2004) 33(7):729–34. doi:10.1038/sj.bmt.1704428
14. Nakamura H, Gress RE. Graft rejection by cytolytic T cells. Transplantation (1990) 49:453–8. doi:10.1097/00007890-199002000-00042
15. Kernan NA, Flomenberg N, Dupont B, O’Reilly RJ. Graft rejection in recipients of t-cell-depleted hla-nonidentical marrow transplants for leukemia: identification of host-derived antidonor allocytotoxic T lymphocytes 1. Transplantation (1987) 43(6):842–7. doi:10.1097/00007890-198743060-00014
16. Voogt P, Fibbe W, Marijt W, Veenhof W, Hamilton M, Zwaan F, et al. Rejection of bone-marrow graft by recipient-derived cytotoxic T lymphocytes against minor histocompatibility antigens. Lancet (1990) 335(8682):131–4. doi:10.1016/0140-6736(90)90003-N
17. Vogt M, De Paus R, Voogt P, Willemze R, Falkenburg J. DFFRY codes for a new human male-specific minor transplantation antigen involved in bone marrow graft rejection. Blood (2000) 95(3):1100–5.
18. Vogt MH, Goulmy E, Kloosterboer FM, Blokland E, de Paus RA, Willemze R, et al. UTY gene codes for an HLA-B60-restricted human male-specific minor histocompatibility antigen involved in stem cell graft rejection: characterization of the critical polymorphic amino acid residues for T-cell recognition. Blood (2000) 96(9):3126–32.
19. Stern M, Passweg JR, Locasciulli A, Socié G, Schrezenmeier H, Békássy AN, et al. Influence of donor/recipient sex matching on outcome of allogeneic hematopoietic stem cell transplantation for aplastic anemia. Transplantation (2006) 82(2):218–26. doi:10.1097/01.tp.0000226156.99206.d1
20. Komatsu M, Mammolenti M, Jones M, Jurecic R, Sayers TJ, Levy RB. Antigen-primed CD8+ T cells can mediate resistance, preventing allogeneic marrow engraftment in the simultaneous absence of perforin-, CD95L-, TNFR1-, and TRAIL-dependent killing. Blood (2003) 101(10):3991–9. doi:10.1182/blood-2002-09-2859
21. Zimmerman Z, Shatry A, Deyev V, Podack E, Mammolenti M, Blazar BR, et al. Effector cells derived from host CD8 memory T cells mediate rapid resistance against minor histocompatibility antigen-mismatched allogeneic marrow grafts without participation of perforin, Fas ligand, and the simultaneous inhibition of 3 tumor necrosis factor family effector pathways. Biol Blood Marrow Transplant (2005) 11(8):576–86. doi:10.1016/j.bbmt.2005.05.006
22. Lapidot T, Faktorowich Y, Lubin I, Reisner Y. Enhancement of T-cell-depleted bone marrow allografts in the absence of graft-versus-host disease is mediated by CD8+ CD4-and not by CD8-CD4+ thymocytes. Blood (1992) 80(9):2406–11.
23. Martin PJ. Donor CD8 cells prevent allogeneic marrow graft rejection in mice: potential implications for marrow transplantation in humans. J Exp Med (1993) 178(2):703–12. doi:10.1084/jem.178.2.703
24. Gandy KL, Domen J, Aguila H, Weissman IL. CD8+ TCR+ and CD8+ TCR− cells in whole bone marrow facilitate the engraftment of hematopoietic stem cells across allogeneic barriers. Immunity (1999) 11(5):579–90. doi:10.1016/S1074-7613(00)80133-8
25. Patterson J, Prentice H, Brenner M, Gilmore M, Janossy G, Ivory K, et al. Graft rejection following HLA matched T-lymphocyte depleted bone marrow transplantation. Br J Haematol (1986) 63(2):221–30. doi:10.1111/j.1365-2141.1986.tb05544.x
26. Kernan NA, Bordignon C, Heller G, Cunningham I, Castro-Malaspina H, Shank B, et al. Graft failure after T-cell-depleted human leukocyte antigen identical marrow transplants for leukemia: I. Analysis of risk factors and results of secondary transplants. Blood (1989) 74(6):2227–36.
27. Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, Van Bekkum DW, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood (1991) 78(8):2120–30.
28. Kiessling R, Hochman PS, Haller O, Shearer G, Wigzell H, Cudkowicz G. Evidence for a similar or common mechanism for natural killer cell activity and resistance to hemopoietic grafts. Eur J Immunol (1977) 7(9):655–63. doi:10.1002/eji.1830070915
29. Raff RF, Deeg HJ, Loughran TJ, Graham TC, Aprile JA, Sale GE, et al. Characterization of host cells involved in resistance to marrow grafts in dogs transplanted from unrelated DLA-nonidentical donors. Blood (1986) 68(4):861–8.
30. Murphy WJ, Kumar V, Bennett M. Rejection of bone marrow allografts by mice with severe combined immune deficiency (SCID). Evidence that natural killer cells can mediate the specificity of marrow graft rejection. J Exp Med (1987) 165(4):1212–7. doi:10.1084/jem.165.4.1212
31. Sun K, Alvarez M, Ames E, Barao I, Chen M, Longo DL, et al. Mouse NK cell–mediated rejection of bone marrow allografts exhibits patterns consistent with Ly49 subset licensing. Blood (2012) 119(6):1590–8. doi:10.1182/blood-2011-08-374314
32. Bennett M, Taylor PA, Austin M, Baker MB, Schook LB, Rutherford M, et al. Cytokine and cytotoxic pathways of NK cell rejection of class I-deficient bone marrow grafts: influence of mouse colony environment. Int Immunol (1998) 10(6):785–90. doi:10.1093/intimm/10.6.785
33. Taylor MA, Ward B, Schatzle JD, Bennett M. Perforin- and Fas-dependent mechanisms of natural killer cell-mediated rejection of incompatible bone marrow cell grafts. Eur J Immunol (2002) 32(3):793–9. doi:10.1002/1521-4141(200203)32:3<793::AID-IMMU793>3.0.CO;2-M
34. Murphy WJ, Bennett M, Kumar V, Longo DL. Donor-type activated natural killer cells promote marrow engraftment and B cell development during allogeneic bone marrow transplantation. J Immunol (1992) 148(9):2953–60.
35. Murphy WJ, Keller JR, Harrison CL, Young HA, Longo DL. Interleukin-2-activated natural killer cells can support hematopoiesis in vitro and promote marrow engraftment in vivo. Blood (1992) 80(3):670–7.
36. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi:10.1126/science.1068440
37. Barge AJ, Johnson G, Witherspoon R, Torok-Storb B. Antibody-mediated marrow failure after allogeneic bone marrow transplantation. Blood (1989) 74(5):1477–80.
38. Taylor PA, Ehrhardt MJ, Roforth MM, Swedin JM, Panoskaltsis-Mortari A, Serody JS, et al. Preformed antibody, not primed T cells, is the initial and major barrier to bone marrow engraftment in allosensitized recipients. Blood (2007) 109(3):1307–15. doi:10.1182/blood-2006-05-022772
39. Spellman S, Bray R, Rosen-Bronson S, Haagenson M, Klein J, Flesch S, et al. The detection of donor-directed, HLA-specific alloantibodies in recipients of unrelated hematopoietic cell transplantation is predictive of graft failure. Blood (2010) 115(13):2704–8. doi:10.1182/blood-2009-09-244525
40. Cutler C, Kim HT, Sun L, Sese D, Glotzbecker B, Armand P, et al. Donor-specific anti-HLA antibodies predict outcome in double umbilical cord blood transplantation. Blood (2011) 118(25):6691–7. doi:10.1182/blood-2011-05-355263
41. Ciurea SO, Thall PF, Wang X, Wang SA, Hu Y, Cano P, et al. Donor-specific anti-HLA Abs and graft failure in matched unrelated donor hematopoietic stem cell transplantation. Blood (2011) 118(22):5957–64. doi:10.1182/blood-2011-06-362111
42. Yoshihara S, Maruya E, Taniguchi K, Kaida K, Kato R, Inoue T, et al. Risk and prevention of graft failure in patients with preexisting donor-specific HLA antibodies undergoing unmanipulated haploidentical SCT. Bone Marrow Transplant (2012) 47(4):508–15. doi:10.1038/bmt.2011.131
43. Ciurea SO, de Lima M, Cano P, Korbling M, Giralt S, Shpall EJ, et al. High risk of graft failure in patients with anti-HLA antibodies undergoing haploidentical stem cell transplantation. Transplantation (2009) 88(8):1019. doi:10.1097/TP.0b013e3181b9d710
44. Hanajiri R, Murata M, Sugimoto K, Murase M, Sakemura R, Goto T, et al. Integration of humoral and cellular HLA-specific immune responses in cord blood allograft rejection. Bone Marrow Transplant (2015) 50(9):1187–94. doi:10.1038/bmt.2015.119
45. Nordlander A, Mattsson J, Sundberg B, Sumitran-Holgersson S. Novel antibodies to the donor stem cell population CD34+/VEGFR-2+ are associated with rejection after hematopoietic stem cell transplantation. Transplantation (2008) 86(5):686–96. doi:10.1097/TP.0b013e3181820333
46. Urbieta M, Barao I, Jones M, Jurecic R, Panoskaltsis-Mortari A, Blazar BR, et al. Hematopoietic progenitor cell regulation by CD4+CD25+ T cells. Blood (2010) 115(23):4934–43. doi:10.1182/blood-2009-04-218826
47. Barao I, Hanash AM, Hallett W, Welniak LA, Sun K, Redelman D, et al. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T cells. Proc Natl Acad Sci U S A (2006) 103(14):5460–5. doi:10.1073/pnas.0509249103
48. Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med (2008) 14(1):88–92. doi:10.1038/nm1688
49. Pilat N, Klaus C, Gattringer M, Jaeckel E, Wrba F, Golshayan D, et al. Therapeutic efficacy of polyclonal tregs does not require rapamycin in a low-dose irradiation bone marrow transplantation model. Transplantation (2011) 92(3):280–8. doi:10.1097/TP.0b013e3182241133
50. Müller AM, Poyser J, Küpper NJ, Burnett C, Ko RM, Kohrt HE, et al. Donor hematopoiesis in mice following total lymphoid irradiation requires host T-regulatory cells for durable engraftment. Blood (2014) 123(18):2882–92. doi:10.1182/blood-2013-10-530212
51. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature (2011) 474(7350):216–9. doi:10.1038/nature10160
52. Hanash AM, Levy RB. Donor CD4+ CD25+ T cells promote engraftment and tolerance following MHC-mismatched hematopoietic cell transplantation. Blood (2005) 105(4):1828–36. doi:10.1182/blood-2004-08-3213
53. Steiner D, Brunicki N, Blazar BR, Bachar-Lustig E, Reisner Y. Tolerance induction by third-party “off-the-shelf” CD4+CD25+ Treg cells. Exp Hematol (2006) 34(1):66–71. doi:10.1016/j.exphem.2005.10.011
54. Shatry A, Levy RB. In situ activation and expansion of host tregs: a new approach to enhance donor chimerism and stable engraftment in major histocompatibility complex-matched allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2009) 15(7):785–94. doi:10.1016/j.bbmt.2009.03.011
55. Mahr B, Unger L, Hock K, Pilat N, Baranyi U, Schwarz C, et al. IL-2/α-IL-2 complex treatment cannot be substituted for the adoptive transfer of regulatory T cells to promote bone marrow engraftment. PLoS One (2016) 11(1):e0146245. doi:10.1371/journal.pone.0146245
56. Bruinsma M, van Soest PL, Leenen PJ, Lowenberg B, Cornelissen JJ, Braakman E. Keratinocyte growth factor improves allogeneic bone marrow engraftment through a CD4+Foxp3+ regulatory T cell-dependent mechanism. J Immunol (2009) 182(12):7364–9. doi:10.4049/jimmunol.0803253
57. Zhao K, Liu Q. The clinical application of mesenchymal stromal cells in hematopoietic stem cell transplantation. J Hematol Oncol (2016) 9(1):1–9. doi:10.1186/s13045-016-0276-z
58. Le Blanc K, Samuelsson H, Gustafsson B, Remberger M, Sundberg B, Arvidson J, et al. Transplantation of mesenchymal stem cells to enhance engraftment of hematopoietic stem cells. Leukemia (2007) 21(8):1733–8. doi:10.1038/sj.leu.2404777
59. Ball LM, Bernardo ME, Roelofs H, Lankester A, Cometa A, Egeler RM, et al. Cotransplantation of ex vivo–expanded mesenchymal stem cells accelerates lymphocyte recovery and may reduce the risk of graft failure in haploidentical hematopoietic stem-cell transplantation. Blood (2007) 110(7):2764–7. doi:10.1182/blood-2007-04-087056
60. Lazarus HM, Koc ON, Devine SM, Curtin P, Maziarz RT, Holland HK, et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant (2005) 11(5):389–98. doi:10.1016/j.bbmt.2005.02.001
61. MacMillan ML, Blazar BR, DeFor TE, Wagner JE. Transplantation of ex-vivo culture-expanded parental haploidentical mesenchymal stem cells to promote engraftment in pediatric recipients of unrelated donor umbilical cord blood: results of a phase I-II clinical trial. Bone Marrow Transplant (2008) 43(6):447–54. doi:10.1038/bmt.2008.348
62. Chen J, Lipovsky K, Ellison FM, Calado RT, Young NS. Bystander destruction of hematopoietic progenitor and stem cells in a mouse model of infusion-induced bone marrow failure. Blood (2004) 104(6):1671–8. doi:10.1182/blood-2004-03-1115
63. Bloom ML, Wolk AG, Simon-Stoos KL, Bard JS, Chen J, Young NS. A mouse model of lymphocyte infusion-induced bone marrow failure. Exp Hematol (2004) 32(12):1163–72. doi:10.1016/j.exphem.2004.08.006
64. Chewning JH, Zhang W, Randolph DA, Swindle CS, Schoeb TR, Weaver CT. Allogeneic Th1 cells home to host bone marrow and spleen and mediate IFNgamma-dependent aplasia. Biol Blood Marrow Transplant (2013) 19(6):876–87. doi:10.1016/j.bbmt.2013.03.007
65. Wang Y-T, Kong Y, Song Y, Han W, Zhang Y-Y, Zhang X-H, et al. Increased type 1 immune response in the bone marrow immune microenvironment of patients with poor graft function after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2016) 22(8):1376–82. doi:10.1016/j.bbmt.2016.04.016
66. Welniak LA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Opposing roles of interferon-gamma on CD4+ T cell-mediated graft-versus-host disease: effects of conditioning. Biol Blood Marrow Transplant (2000) 6(6):604–12. doi:10.1016/S1083-8791(00)70025-5
67. Delisle JS, Gaboury L, Belanger MP, Tasse E, Yagita H, Perreault C. Graft-versus-host disease causes failure of donor hematopoiesis and lymphopoiesis in interferon-gamma receptor-deficient hosts. Blood (2008) 112(5):2111–9. doi:10.1182/blood-2007-12-130534
68. Rottman M, Soudais C, Vogt G, Renia L, Emile J-F, Decaluwe H, et al. IFN-γ mediates the rejection of haematopoietic stem cells in IFN-γR1-deficient hosts. PLoS Med (2008) 5(1):e26. doi:10.1371/journal.pmed.0050026
69. Zoumbos N, Djeu J, Young N. Interferon is the suppressor of hematopoiesis generated by stimulated lymphocytes in vitro. J Immunol (1984) 133(2):769–74.
70. Yu J-M, Emmons RV, Hanazono Y, Sellers S, Young NS, Dunbar CE. Expression of interferon-γ by stromal cells inhibits murine long-term repopulating hematopoietic stem cell activity. Exp Hematol (1999) 27(5):895–903. doi:10.1016/S0301-472X(99)00009-0
71. Snoeck H-W, Van Bockstaele D, Nys G, Lenjou M, Lardon F, Haenen L, et al. Interferon gamma selectively inhibits very primitive CD342+ CD38-and not more mature CD34+ CD38+ human hematopoietic progenitor cells. J Exp Med (1994) 180(3):1177–82. doi:10.1084/jem.180.3.1177
72. Yang L, Dybedal I, Bryder D, Nilsson L, Sitnicka E, Sasaki Y, et al. IFN- negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol (2005) 174(2):752–7. doi:10.4049/jimmunol.174.2.752
73. Selleri C, Sato T, Anderson S, Young NS, Maciejewski JP. Interferon-γ and tumor necrosis factor-α suppress both early and late stages of hematopoiesis and induce programmed cell death. J Cell Physiol (1995) 165(3):538–46. doi:10.1002/jcp.1041650312
74. Zeng W, Miyazato A, Chen G, Kajigaya S, Young NS, Maciejewski JP. Interferon-γ-induced gene expression in CD34 cells: identification of pathologic cytokine-specific signature profiles. Blood (2006) 107(1):167–75. doi:10.1182/blood-2005-05-1884
75. Maciejewski J, Selleri C, Anderson S, Young N. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood (1995) 85(11):3183–90.
76. de Bruin AM, Demirel Ö, Hooibrink B, Brandts CH, Nolte MA. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood (2013) 121(18):3578–85. doi:10.1182/blood-2012-05-432906
77. Jacobsen FW, Rothe M, Rusten L, Goeddel DV, Smeland EB, Veiby OP, et al. Role of the 75-kDa tumor necrosis factor receptor: inhibition of early hematopoiesis. Proc Natl Acad Sci U S A (1994) 91(22):10695–9. doi:10.1073/pnas.91.22.10695
78. Zhang Y, Harada A, Bluethmann H, Wang J, Nakao S, Mukaida N, et al. Tumor necrosis factor (TNF) is a physiologic regulator of hematopoietic progenitor cells: increase of early hematopoietic progenitor cells in TNF receptor p55-deficient mice in vivo and potent inhibition of progenitor cell proliferation by TNF alpha in vitro. Blood (1995) 86(8):2930–7.
79. Bryder D, Ramsfjell V, Dybedal I, Theilgaard-Mönch K, Högerkorp C-M, Adolfsson J, et al. Self-renewal of multipotent long-term repopulating hematopoietic stem cells is negatively regulated by Fas and tumor necrosis factor receptor activation. J Exp Med (2001) 194(7):941–52. doi:10.1084/jem.194.7.941
80. Dybedal I, Bryder D, Fossum A, Rusten LS, Jacobsen SEW. Tumor necrosis factor (TNF)–mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood (2001) 98(6):1782–91. doi:10.1182/blood.V98.6.1782
81. Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med (2011) 208(8):1563–70. doi:10.1084/jem.20110752
82. Rezzoug F, Huang Y, Tanner MK, Wysoczynski M, Schanie CL, Chilton PM, et al. TNF-α is critical to facilitate hemopoietic stem cell engraftment and function. J Immunol (2008) 180(1):49–57. doi:10.4049/jimmunol.180.1.49
83. Mori T, Nishimura T, Ikeda Y, Hotta T, Yagita H, Ando K. Involvement of Fas-mediated apoptosis in the hematopoietic progenitor cells of graft-versus-host reaction-associated myelosuppression. Blood (1998) 92(1):101–7.
84. Mori T, Ando K, Tanaka K, Ikeda Y, Koga Y. Fas-mediated apoptosis of the hematopoietic progenitor cells in mice infected with murine cytomegalovirus. Blood (1997) 89(10):3565–73.
85. Scadden DT. The stem-cell niche as an entity of action. Nature (2006) 441(7097):1075–9. doi:10.1038/nature04957
86. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature (2012) 481(7382):506–10. doi:10.1038/nature10738
87. Kong Y, Chang YJ, Wang YZ, Chen YH, Han W, Wang Y, et al. Association of an impaired bone marrow microenvironment with secondary poor graft function after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2013) 19(10):1465–73. doi:10.1016/j.bbmt.2013.07.014
88. Kong Y, Wang Y, Hu Y, Han W, Chang Y, Zhang X, et al. The bone marrow microenvironment is similarly impaired in allogeneic hematopoietic stem cell transplantation patients with early and late poor graft function. Bone Marrow Transplant (2016) 51(2):249–55. doi:10.1038/bmt.2015.229
89. Shono Y, Shiratori S, Kosugi-Kanaya M, Ueha S, Sugita J, Shigematsu A, et al. Bone marrow graft-versus-host disease: evaluation of its clinical impact on disrupted hematopoiesis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20(4):495–500. doi:10.1016/j.bbmt.2013.12.568
Keywords: bone marrow failure, graft failure, poor graft function, HSCT
Citation: Masouridi-Levrat S, Simonetta F and Chalandon Y (2016) Immunological Basis of Bone Marrow Failure after Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 7:362. doi: 10.3389/fimmu.2016.00362
Received: 30 June 2016; Accepted: 02 September 2016;
Published: 16 September 2016
Edited by:
Delmiro Fernandez-Reyes, University College London, UKReviewed by:
Hideki Nakano, National Institute of Environmental Health Sciences, USAShi Yue, University of Southern California, USA
Copyright: © 2016 Masouridi-Levrat, Simonetta and Chalandon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stavroula Masouridi-Levrat, c3RhdnJvdWxhLm1hc291cmlkaUBoY3VnZS5jaA==;
Yves Chalandon, eXZlcy5jaGFsYW5kb25AaGN1Z2UuY2g=