 Esmaeil Mortaz1,2,3
Esmaeil Mortaz1,2,3 Payam Tabarsi4Davod Mansouri2Adnan Khosravi2Johan Garssen3,5Aliakbar Velayati6Ian M. Adcock7*
Payam Tabarsi4Davod Mansouri2Adnan Khosravi2Johan Garssen3,5Aliakbar Velayati6Ian M. Adcock7*
- 1Department of Immunology, Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 2Chronic Respiratory Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 3Division of Pharmacology, Faculty of Science, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Utrecht, Netherlands
- 4Clinical Tuberculosis and Epidemiology Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 5Nutricia Research Centre for Specialized Nutrition, Utrecht, Netherlands
- 6Mycobacteriology Research Center (MRC), National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 7Cell and Molecular Biology Group, Airways Disease Section, Faculty of Medicine, National Heart and Lung Institute, Imperial College London, London, UK
The life span of patients with primary and secondary immunodeficiency is increasing due to recent improvements in therapeutic strategies. While the incidence of primary immunodeficiencies (PIDs) is 1:10,000 births, that of secondary immunodeficiencies are more common and are associated with posttransplantation immune dysfunction, with immunosuppressive medication for human immunodeficiency virus or with human T-cell lymphotropic virus infection. After infection, malignancy is the most prevalent cause of death in both children and adults with (PIDs). PIDs more often associated with cancer include common variable immunodeficiency (CVID), Wiskott–Aldrich syndrome, ataxia-telangiectasia, and severe combined immunodeficiency. This suggests that a protective immune response against both infectious non-self-(pathogens) and malignant self-challenges (cancer) exists. The increased incidence of cancer has been attributed to defective elimination of altered or “transformed” cells and/or defective immunity towards cancer cells. The concept of aberrant immune surveillance occurring in PIDs is supported by evidence in mice and from patients undergoing immunosuppression after transplantation. Here, we discuss the importance of PID defects in the development of malignancies and the current limitations associated with molecular pathogenesis of these diseases and emphasize the need for further knowledge of how specific mutations can modulate the immune system to alter immunosurveillance and thereby play a key role in the etiology of malignancies in PID patients.
Introduction
The earliest evidence that individuals with primary immunodeficiencies (PIDs) develop cancer was reported in 1963 (1, 2). An increasing number of reports subsequently indicated that subjects with primary abnormalities of the immune system are at a high risk for developing cancer (3–6) and PID biology represents a rapidly developing area of clinical immunology. PIDs are inherited disorders of the immune system in which at least one, and often more, immune component is decreased, missing, or has an inappropriate function. PIDs comprise more than 130 different heterogeneous disorders that affect various aspects of immune development and function as well as the morphology of the immune system (7–10). PIDs are rare with a prevalence in the United States of 1:1200 live births (11), and the overall risk for children with PIDs of developing malignancy is estimated at 4–25% (12, 13). The type of malignancy that is seen is highly dependent on the precise PID, the age of the patient, and probably viral infection indicating that different pathogenic mechanisms may be implicated in each case (12).
As stated above, PIDs were originally described as rare, to only occur in infants and young children, and to be associated with severe clinical symptoms. However, advances in gene sequencing technologies, such as whole exome sequencing, have revealed that they are much more common than originally appreciated and are present in older children, adolescents, and adults, and can present with relatively mild clinical disease in some patients (14, 15).
PIDs are classified according to which component of the immune system is primarily involved. Defects in adaptive immune responses include antibody deficiency syndromes, combined immunodeficiencies (CIDs), and severe combined immunodeficiency (SCID) (16), whereas defects in innate immunity comprise disorders of phagocytes, and toll-like receptor (TLR)-mediated signaling and complement (17). All these forms of disease are characterized by increased susceptibility to recurrent infections and/or severe infections with the susceptibility to specific pathogens dependent upon the nature of the specific immune defect (18).
Immune dysregulation is present in some forms of PIDs, while other forms of PID are more complex and immunodeficiency represents only a part of the phenotype (immunodeficiency syndromes) (19, 20). The early detection of patients with PID is critically important as effective therapy is available for virtually all of the different disorders but is most beneficial when instituted before target organ damage has occurred (e.g., in the lung) by infection or by autoimmunity (21). Similarly, early recognition of primary immunodeficiency may lead to a precise genetic diagnosis, which in turn may be important to the family in planning their future reproductive options (22).
Genomic instability due to defective DNA repair processes and other unknown mechanisms in PID patients leads to an enhanced risk of cancer. We have reviewed here the etiology, pathogenesis, clinical, and laboratory features of each of the various categories of PIDs and highlighted their association with various malignancies.
The Link between PIDs and Malignancies
Primary immunodeficiencies are occasionally referred to as inborn errors of immunity, inherited immunodeficiencies, heritable immune defects, or congenital immunodeficiencies. In all cases, it is explicit or implicit that the primary lesions are present in the germline, whether due to a de novo mutation or due to a mutation inherited from a parent carrying the mutation. It has also become evident that a normal cellular and clinical phenotype may be restored in some patients with PIDs when somatic mutations occur in the hematopoietic cell lineage (23, 24). As predicted by Burnet, there is an increased predisposition to malignancy in patients with severe primary immunodeficiency (1, 13, 25, 26).
PIDs are a heterogeneous group of rare disorders characterized by impaired humoral and/or cell-mediated immunity, in the absence of any recognized cause such as drug treatment or human immunodeficiency virus (HIV). The most common PID syndromes are common variable immunodeficiency (CVID) with a 25-year mortality rate of 24% mostly due to lymphoma (18%), chronic pulmonary disease (11%), and X-linked agammaglobulinemia (XLA), which accounts for between 80 and 90% of all cases (21, 27, 28). This compares with the survival rate of 92% (males) and 94% (females) within the general population (27). There is an enhanced incidence of several cancers including lymphoma and of stomach, breast, bladder, and cervical epithelial cancers in these patients with CVID associated with the presence of defective humoral immunity (12, 13, 25, 26). This highlights the importance of an effective immune response against infection in the prevention of oncogenesis.
Common Variable Immune Deficiency
Common variable immune deficiency (CVID) is characterized by recurrent sino-pulmonary infections, autoimmune disorders, and granulomatous disease with an increased risk of malignancy (12- to 18-fold greater than in the general population) (29–34). CVID affects both children and adults, with an estimated prevalence of 1:25–50,000 (35, 36). The hallmark of CVID is hypogammaglobulinemia due to impaired B cell differentiation. CVID is heterogeneous, with most patients suffering mainly from infections, while others are particularly prone to non-infectious complications. These differences correlate with the particular B cell phenotype present in each patient (37).
An increased risk of malignancy particularly that of lymphoma and gastric cancer is associated with CVID (38–41). Indeed, malignancy is recognized as one of the five clinical CVID phenotypes proposed by Chapel and colleagues (38). In this classification, patients with polyclonal lymphadenopathy were shown to have fivefold increased risk of lymphoid malignancy (38). Lymphoid malignancy generally occurs late in disease and in patients with pre-existing polyclonal lymphocytic infiltration (38). However, lymphoma can appear in young pediatric CVID patients in the absence of previous polyclonal lymphadenopathy (42).
Lymphoma is one of the more severe complications of CVID, but the driver(s) of this increased risk are unclear, although it seems to be multifactorial with the interplay of genetics, immune dysregulation, and chronic infectious agents including non-oncogenic and oncogenic viruses such as Epstein–Barr virus (EBV) being important (43). This increase in infection, therefore, may be a critical driver of malignancy in patients with CVID. CVID-associated lymphomas are more likely to be of B cell origin with a predominance of non-Hodgkin lymphoma (NHL), and these usually occur in the fourth to seventh decades of life and are rarely seen in children. NHL is frequently extranodal and usually EBV-negative (43), and although the parotid gland, sinuses, orbital cavity, and stomach can be affected, the majority of cases affects the lung (44, 45).
Hyper-IgE Syndrome (Job’s Syndrome)
Job’s syndrome is a complex CID disease that was first described in 1966 and named in reference to the Biblical character Job who was “smote with sore boils” (46). The description of the syndrome was refined after the realisation that in addition to the occurrence of recurrent boils, eczema, and pneumonias, these patients also had very high serum IgE levels (47). Two distinct entities have been recognized: the classical hyper-IgE syndrome, which is inherited in an autosomal-dominant pattern [autosomal-dominant hyper-IgE syndrome (AD-HIES)], and the autosomal recessive hyper-IgE syndrome. The vascular, skeletal, and connective tissue problems seen in AD-HIES reflect the multisystem status of the disorder (48, 49).
A mutation in the signal transducer and the activator of the transcription 3 (STAT3) gene has been identified in the majority of AD-HIES patients, which results in impaired Th17 cell differentiation and downregulation of antimicrobial responses (50–55). Chronic mucocutaneous candidiasis (CMC) patients with a dominant-negative STAT3 mutation present with a decreased number of central memory CD4+ and CD8+ lymphocytes and an increased number of naive T cells (56). Due to this loss of memory T cells, patients with HIES are predisposed to develop varicella zoster virus reactivation and prolonged EBV viremia. The impaired IL-17 + T cell differentiation and function is a plausible explanation for the susceptibility of HIES patients to CMC (51, 57). However, such patients are also susceptible to infections by pyogenic bacteria, especially Staphylococcus aureus causing recurrent skin and lung infections due to a failure of Th17 CD4+ cells to recruit neutrophils to the site of infection and to upregulate antimicrobial peptides (58).
Reduced levels of IL-17 are linked to mucocutaneous infections with Candida albicans in man (59–63) and to enhanced susceptibility to Candida and Klebsiella infections in mice (64, 65). Th17 cells are also important for IL-22 secretion, which is critical for beta-defensin production and subsequent protection against S. aureus infection in individuals with eczema (66). This suggests that the S. aureus-infected abscesses seen in AD-HIES are due to reduced beta-defensin production secondary to the lack of Th17 cells. Beta defensins are also expressed in the lung and might in part explain the susceptibility to pneumonias (67).
The reason why AD-HIES subjects have a very high serum IgE levels is unknown, although this has been linked to impaired IL-21 signaling (68). However, human B cells differ from those in mice in that IL-21 acts synergistically with IL-4 to increase IgE production in man (69), whereas IgE levels in serum are increased in IL-21R- and IL-21-deficient mice (68, 70). Therefore, the lack of IL-21 might be associated with less, not more, IgE in man. The combined effect of IL-4 and IL-21 on IgE secretion was also unrelated to alterations in expression of their receptors because neither IL-4 nor IL-21 enhanced the level of CD40L-induced IL-21R or IL-4R expression on B cells. It is possible that the increased IgE reflects a lack of suppression of IL-21 and/or IL-4 by IL-10 or IFNγ. It is noteworthy that this cardinal feature of AD-HIES has not yet been clearly explained.
A similar, but distinct, syndrome was reported in 2004, which was characterized by extremely elevated serum IgE levels, severe eczema, and recurrent bacterial and viral skin infections as well as by sino-pulmonary infections (71). In comparison to AD-HIES, these individuals lack the somatic features such as the characteristic faces, scoliosis, and the failure of baby teeth to exfoliate. In addition, although pneumonias occur in autosomal recessive hyper IgE syndrome (AR-HIES), pneumatoceles do not form.
Autosomal recessive hyper IgE syndrome has a much higher rate of coetaneous viral infections such as molluscum contagiosum, herpes simplex, and varicella infections. They also have frequent neurologic disease, ranging from facial paralysis to hemiplegia often due to CNS vasculitis. Mortality is high in younger patients with AR-HIES with sepsis being more frequent than in AD-HIES. Two other autosomal recessive forms of HIES are caused by mutations in the gene for tyrosine kinase 2 (Tyk2). Kilic et al. (72) described a patient with Tyk2 deficiency who displayed none of the other three cardinal features of HIES: atopic dermatitis and eczema, staphylococcal infections of the skin and lung, or high serum IgE concentrations; his highest recorded IgE concentration being 218 IU/mL at the age of 16 years. A diagnosis of Tyk2 deficiency should be contemplated in patients with BCG clinical disease, particularly in the presence of unusually severe herpes virus infection, even in the absence of cardinal features of HIES. This diagnosis may also apply to patients with other mycobacterial diseases, whether they are tuberculous or atypical, as well as those with infections caused by other intramacrophage pathogens, such as Salmonella and Brucella.
A related syndrome is seen in patients with defects in the gene dedicator of cytokinesis (DOCK) 8 (73–78). Patients with DOCK8 mutations have elevated IgE levels and the presence of allergy, eczema, cutaneous viruses, and malignancies (79). Contrary to its initial description as a form of hyper-IgE syndrome, DOCK8 deficiency can be regarded as a CID disease that features eczema and elevated IgE much like the Wiskott–Aldrich syndrome (WAS) (80). On the other hand, phosphoglucomutase 3 (PGM3) deficiency is not associated with cold abscesses, CMC, retained childhood dentition, and joint hypermobility that are seen very commonly in patients with STAT3 mutations. PGM3 deficiency can present with elevated IgE, atopic dermatitis, bronchiectasis, and scoliosis, which help to distinguish this disorder from the two best-known diseases with elevated IgE: STAT3 and DOCK8 deficiencies. Although PGM3-deficient patients occasionally have viral skin infections, they were not as prevalent as in DOCK8 deficiency. Primary neurocognitive deficits are seen in PGM3 deficiency but not in either STAT3 or DOCK8 deficiencies (81).
In the context of this review, HIES patients have a much higher risk of developing aggressive B cell lymphomas, which may be linked to abnormalities in STAT3/IL-21-dependent differentiation of B cells into plasma cells with the possible involvement of T follicular helper cells (82–84). B-cells are also important in the formation, progression, and metastases of many other cancers including lung cancer and melanoma (85, 86). Viral infection in AD-HIES and AR-HIES is associated with an increased risk of cancer over and above that seen with PIDs alone, suggesting that the inability of these patients to mount an efficient immune response to infection may allow pre-malignant cells to grow unchecked and develop into cancer. The role of Th17 and memory T-cells in this process needs to be better defined.
Chronic Mucocutaneous Candidiasis and CMC Disease
Several PIDs typically characterized by CMC and impaired IL-17-mediated immunity have recently been identified (87). CMC in such patients may be part of a complex clinical phenotype exemplified by a dominant-negative STAT3 deficiency, interleukin (IL)-12Rβ1 and IL-12p40 deficiencies, and autoimmune polyendocrine syndrome type 1 (APS-1) (57, 88–90). In a subgroup of PIDs, a predisposition to superficial candidiasis may be the only, or at least the dominant, characteristic of the underlying genetic disorder [chronic mucocutaneous candidiasis disease (CMCD)] (91). Inborn errors of IL-17-mediated immunity lead to CMC in humans, and an autosomal-dominant mutation in IL-17F and autosomal recessive mutations in IL-17RA or IL17-RC predispose patients to complete CMC. In contrast, the heterozygous STAT1 mutations associated with CMC are gain-of-function (92–94).
Candida albicans is the most common species isolated from patients with CMC. Candidal infections develop and persist, usually beginning during infancy but sometimes during early adulthood. The fungus may cause mouth infections (thrush) and infections of the scalp, skin, and nails. Membranes lining the mouth, eyelids, digestive tract, and vagina may also be infected. In infants, the first symptoms are often difficult to treat thrush and diaper rash. The disorder may cause one or more nails to thicken, crack, and become discolored. A disfiguring rash may cover the face and scalp. The rash is crusted and thick and may ooze and when present on the scalp may cause hair to fall out (95).
Th17 cytokines are important for the prevention of infection with Candida colonizing the mucocutaneous surfaces (57, 61, 96). The differentiation, expansion, and maintenance of human IL-17-producing T cells is regulated by a set of cytokines including IL-1β, IL-6, IL-21, IL-23, and TGF-β along with the transcription factors STAT3, retinoic acid-related orphan receptor (ROR)-γt, and interferon regulatory factor 4 (IRF4) (48, 91, 97–100). However, alternative mechanisms to explain the unique vulnerability to infection of the skin, mucous membranes, and the lung in HIES patients may be required.
Chronic mucocutaneous candidiasis is associated with squamous cell cancers of the esophagus and of the oral cavity (95). This provides a link between the site of repeated or recurrent infection and a defect in the Th17 response. These cancers are also seen in patients with autosomal recessive autoimmune polyendocrinopathy syndrome type I (AR APS-I), which is caused by mutations in the protein autoimmune regulator (AIRE), who are not susceptible to any other infectious disease (91). The distinct mechanisms that underpin the increased incidence of esophagal cancer in CMC patients require further study (101).
Familial Hemophagocytic Lymphohistiocytosis
The rare autosomal recessive disorder termed familial hemophagocytic lymphohistiocytosis (FHL) is due to defects in natural killer (NK) and cytotoxic T lymphocytes (CTLs), resulting in a multisystem inflammatory disease with persistent fever and hepatosplenomegaly associated with cytopenias and blood metabolic alterations. Type 2 FHL (FHL2) is linked to mutations in the PRF1 gene encoding human perforin, which represent 13–58% of the cases reported to date depending on the ethnicity of the patient (102–108). To date, more than 70 mutations in PRF1 have been described in FHL2. Although most of these are missense mutations, it is unclear how they affect protein expression and function (109–111). Studies of missense mutations from patient-derived cells and cell lines are limited by the infrequent occurrence of individuals with homozygous mutations.
Overall, there is a striking similarity between the biologic changes induced by proinflammatory cytokines and the clinical and laboratory findings in FHL (112). The proinflammatory cytokines soluble IL-2 receptor, IL-6, interferon-γ, and TNF-α are reported to be commonly elevated in FHL (112–114). More recent studies indicated elevated plasma levels of IL-12 and IL-10 in these patients. The former stimulates the production of Th1 cytokines, and the latter suppresses Th1 responses. In contrast, the levels of the Th2-stimulating cytokine IL-4 were not increased (115). It is reasonable to assume, therefore, that most of the symptoms, signs, and the laboratory alterations in FHL patients are mediated by proinflammatory cytokines.
Mutations in the PRF1 gene result in a reduced ability of immune cells to perform their essential immunosurveillance roles, particularly against spontaneous lymphomas, which accounts for the enhanced risk of lymphoma in FHL patients. Furthermore, a third of the patients with interleukin-10 receptor (IL-10R) deficiency, which results in abnormal Th1 responses and a loss of immunosurveillance capacity, develop B-cell lymphomas in the first decade of their life. The lymphomas uniformly contained amplifications of c-rel, activation of inflammatory nuclear factor κB (NF-κB)-induced target genes, and defective intratumoral CD81 T-cell tumor immunosurveillance (116).
IgA Deficiency
Selective IgA deficiency is one of the most prevalent PID subtypes with an estimated prevalence of ~1:600 (117). Although selective IgA deficiency is asymptomatic in many patients, clinical manifestations include respiratory and GI infections, atopy, autoimmune diseases, and lymphoid and GI malignancies, which occur later in life (118). Since IgA deficiency and CVID share a common genetic basis, they may be considered as a spectrum ranging from the mild reductions in IgA levels to severe deficiencies of multiple antibodies in CVID (119). The progression from mild or asymptomatic selective IgA deficiency to CVID is possible and, if noted, should prompt initiation of Ig replacement therapy to prevent infectious and pulmonary complications (120). Patients with selective IgA deficiency have a high incidence of gastrointestinal cancers, highlighting the immunoprotective role of IgA against this type of malignancy.
Patients with X-linked immunodeficiency with hyper-IgM, caused by mutations in the CD40 ligand, have a high incidence of tumors of the pancreas and liver (121). Interestingly, a primary cutaneous marginal zone lymphoma (PCMZL) with the sequential development of nodal marginal zone lymphoma has been recently reported in a patient with selective immunoglobulin A deficiency (122). This is in addition to a previous report of a γ/δ type abdominal T-cell non-Hodgkin’s lymphoma in a patient with selective IgA deficiency (123).
DNA Repair Disorders
A number of rare PIDs are the result of genetic defects in DNA repair that also lead to immune deficits and susceptibility to malignancy (124, 125).
Ataxia-Telangiectasia
Ataxia-telangiectasia (AT) is an autosomal recessive disorder and is characterized by progressive cerebellar ataxia presenting in infancy, oculocutaneous telangiectasia, and dysarthria (126, 127). Cells from patients with AT have a reduced ability to activate cell cycle checkpoints, which is seen particularly following radiation exposure for example (γ-irradiation or radiomimetic agents) because of mutations in the AT mutated (ATM) gene. The ATM gene normally acts as a sensor of double-stranded DNA breakage (124), and the predisposition to leukemia is thought to be related to excessive production of DNA translocations (126).
Not surprisingly, therefore, AT has the highest risk for malignancy of any PID (126, 128), and the overall incidence of cancer among patients with AT is as high as 40%. Leukemias and lymphomas are particularly prevalent, appearing in AT patients with rates 70- to 500-fold and 200- to 750-fold, respectively, higher than in the general population (127). Interestingly, there is an increased risk of breast cancer among heterozygote AT carriers, who are otherwise generally healthy with the greatest risk in elderly AT patients who are smokers (129). The ability of immune checkpoint blockers to treat a number of different cancers also indicates a key role for defects in immune function being critical for the development of cancer.
Nijmegen Breakage Syndrome
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive disorder closely related to AT that, although it occurs worldwide, it has a greater prevalence in people of Central and Eastern European descent (130). The defective NBS gene (NBS1, nibrin, or p95) is a component within the same pathway as ATM and both proteins are part of a multi-subunit complex involved in correcting radiation-induced chromosomal aberrations (124, 131). Immune deficiency is generally severe and characterized by both humoral, such as agammaglobulinemia, IgA deficiency, and IgG2 and IgG4 deficiency, and cellular immunity, including lymphopenia, decreased CD31+ and CD41+ T helper cells, and a decreased CD41:CD81 T suppressor cell ratio (132). The physical features of NBS are typified by microcephaly, short stature, and “bird-like” faces (131, 132).
Not surprisingly, therefore, NBS exhibits many similarities to AT, including humoral and T-cell defects, radiosensitivity, and chromosomal instability, which are linked to immune dysfunction and to a high risk of malignancy.
Wiskott–Aldrich Syndrome
Wiskott–Aldrich syndrome is a rare X-linked primary immunodeficiency characterized by microthrombocytopenia, eczema, recurrent infections, and an increased incidence of autoimmunity and malignancies. The disease is caused by mutations in the WAS gene, which is expressed only in hematopoietic cells (133). The overall incidence of cancer in WAS patients is unclear, although 18% of WAS subjects in a small study were reported as having malignant lymphoma (134). In all but one patient, the diagnosis of lymphoma was made ante-mortem and located predominantly in extranodal sites or within the brain. There was no involvement of peripheral lymph nodes (134). A higher incidence of NHL has also been described in WAS patients (135).
Phagocyte Disorders
Severe congenital neutropenia (SCN) is a heterogeneous disorder due to the arrest of myelopoiesis maturation at the promyelocyte/myelocyte stage, resulting in neutropenia with systemic neutrophil blood counts <0.5 × 109/L. Patients with SCN present with recurrent, severe infections during infancy and persistent neutropenia. A total of 50–60% of cases are associated with mutations in the elastase (ELA) 2 gene, and >50 unique mutations have been identified. Other common genetic defects associated with SCN include biallelic mutations in the hematopoietic cell-specific Lyn substrate 1 (HCLS1)-associated protein X-1 (HAX1) or in glucose-6-phosphatase, catalytic, 3 (G6PC3) genes. For most patients, daily treatment with granulocyte colony-stimulating factor (G-CSF) results in elevated blood neutrophil counts and reduces the risk of infection (136).
Heterozygous mutations in GATA2 resulting in GATA2 deficiency is a recently described disorder of hematopoiesis, lymphatics, and immunity. GATA2 is a zinc finger transcription factor essential for differentiation of immature hematopoietic cells (137). In addition, GATA2 regulates phagocytosis by alveolar macrophages (138), and GATA2 overexpression in alveolar macrophages increases phagocytic activity up to threefold (139).
Patients with GATA2 mutations present with numerous diagnoses and symptoms including MDS, AML, chronic myelomonocytic leukemia (CMML), severe viral, disseminated mycobacterial and invasive fungal infections, pulmonary arterial hypertension, warts, panniculitis, human papillomavirus (HPV)-positive tumors, EBV-positive tumors, venous thrombosis, lymphedema, sensorineural hearing loss, miscarriage, and hypothyroidism (140).
A very high rate of leukemia is seen in subjects with SCN. This is predominantly AML, but subjects may also suffer ALL, CML, and bi-phenotypic leukemia (141). Point mutations in the colony-stimulating factor 3 receptor (granulocyte) (GCSFR or CSF3R) gene are seen in bone marrow cells from most SCN patients who develop leukemia, suggesting that such mutations are highly predictive, although not essential, for malignant transformation in these subjects (142). CSF3R mutations are very specific for patients with SCN and are not found in patients with primary AML or other forms of chronic neutropenia requiring long-term treatment with G-CSF (142). These data emphasize the critical role of immune neutrophils in immunosurveillance of pre-cancerous cells.
Defects in the Immune Response in Pid Patients Drives Malignancy
The data presented above suggest that a defective immune response in PID patients is associated with a greater incidence of cancer independent of whether the defect is primary or secondary. For example, lower serum Ig levels are associated with malignancies, highlighting the importance immunodeficiency in the predisposition to neoplasia (143). This risk is amplified by the enhanced susceptibility to viral infections in patients with PIDs (144). Defective immunosurveillance is undoubtedly a major factor in the risk of developing cancer, and this is seen most evidently in cells with a strong antigenic potential that have undergone viral induction. Supporting this concept is the fact that lymphoma, an immune system-related malignancy, is the most common cancer subtype in immunodeficient patients (12, 145). NHL and Hodgkin’s disease (HD) account for 48.6 and 10%, respectively, of the malignancies seen in patients with PIDs according to the Immunodeficiency Cancer Registry (ICR) database (124, 146). The overall risk for cancer developing in children with PID is estimated to range from 4 to 25% (12, 147). Finally, EBV infection in WAS patients induces lymphoma (135, 148), and the occurrence of lymphoproliferative disorders (LPDs) in patients with PID has been documented for nearly 40 years.
Mutations in the X-linked inhibitor of apoptosis (XIAP) are associated with a rare primary immunodeficiency. XIAP is an anti-apoptotic molecule but is also important in many other pathways, including control of innate immunity and in the negative regulation of inflammation (149). Loss of XIAP results in reduced NOD ligand-induced proinflammatory cytokine expression in both mouse and man (150–152). Furthermore, the ability of the pattern recognition receptor dectin-1 to recognize fungal β-glucan from fungi is modulated by XIAP (153). Under these conditions, XIAP regulated β-glucan-induced NF-κB and MAPK activation, cytokine production, and phagocytosis via ubiquitination of BCL10. The T-cell receptor (TCR) pathway activation of NF-κB also requires BCL10 and XIAP (151).
X-linked inhibitor of apoptosis deficiency is mostly reported in young boys who can be affected during the first few months of life, with symptoms being most severe in the youngest patients. The most frequent clinical manifestations are HLH (54%), recurrent splenomegaly (57%), and IBD (26%). Viral infection is often the trigger for HLH, with EBV (60% of cases) with cytomegalovirus (CMV) and human herpes virus 6 (HHV-6) being the most important, although symptoms may occur without an identified infectious agent (149).
These PIDs are a heterogeneous group of genetically determined disorders, which give rise to a number of diverse and variable LPDs. The susceptibility of developing LPD is associated with the type of PID presence, but accurate quantification of this risk is difficult since PIDs are rare. Analysis of case reports provides risk estimates of 0.7–15% (154). The extent to which primary immunodeficiency in man leads to increased cancer development is therefore unclear and generally relies on gene-targeted murine studies (155). For example, recombination-activating gene 2 (RAG2)-deficient mice that lack both T and B cells are more susceptible to spontaneous and carcinogen-induced carcinomas (156), while mice lacking γδT cells are highly susceptible to cutaneous carcinogenesis (157).
In contrast, interferon-α/β (IFN-α/β) and IFN-γ protect mice against both spontaneous and carcinogen-induced cancers (156, 158–160). Moreover, perforin, which is used by cytotoxic lymphocytes to kill target cells and is defective in FHL, is important for the surveillance of spontaneous lymphoma in mice (161).
Collectively, the human and mouse data reveal a consistent association between primary immunodeficiency and an increased incidence of cancers. Overall, two major mechanisms appear to be important for this link, prevention of infection and immunosurveillance and elimination of pre-malignant cells, and these are described in more detail below.
Insufficient Functioning of the Immune System Allows Infection and Prevents Effective Immunosurveillance
The immune system confers protection against viral and bacterial pathogens and parasitic worms. Although evidence exists for the immune system targeting invading cancers, there are less data demonstrating the immunological eradication of pre-cancerous lesions in man and thereby preventing cancer (143, 162). For example, although organ transplant recipients who are treated with immunosuppressive drugs are more prone to cancer development (163, 164), the majority of posttransplantation lymphomas was associated with EBV infection (165). Thus, most of the lymphomas reported in PID patients are likely to be secondary events resulting from reduced antiviral immunity, rather than a direct effect of reduced antitumor immunity.
The HIV1 virus causes acquired immunodeficiency by selectively infecting and killing CD4+ T cells, and this is associated with an elevated risk of cancer. These cancers are generally linked to oncogenic viruses such as Kaposi sarcoma [caused by human herpes virus 8 (HHV8)], Hodgkin’s lymphoma and NHL (EBV), anal and cervical cancer (human papilloma virus), and liver cancer (hepatitis B and C viruses). Kaposi sarcoma, NHL, and cervical cancer are particularly frequent and are considered as acquired immunodeficiency syndrome (AIDS)-defining cancers (166). Blockade of the immune check-point molecules CTLA-4, PD-1, or PDL1 is of value in these cancers by potentiating the patient’s own immune response (167).
Elimination of pre-malignant cells can occur, and the importance of immune system in defending against cancers is exemplified by lymphocytes bearing the receptor killer cell lectin-like receptor subfamily K, member 1 (KLRK1), which are able to recognize and eliminate stressed pre-malignant cells (168). Self-proteins, such as MICA and MICB, are ligands for KLRK1 as are six different ULBP proteins that are poorly expressed in normal resting cells but whose cell surface expression is upregulated after treatment with DNA-damaging agents such as ionizing radiation and UV light (169, 170). Many freshly isolated lung, breast, kidney, ovary, prostate, colon, and liver carcinomas express MICA and MICB (171). The expression of these KLRK1 ligands is also induced by oncogenic growth factors acting through their receptors such as the epidermal growth factor receptor (EGFR) (172). The EGFR pathway is frequently dysregulated in human cancer, and EGFR activation may regulate the immunological visibility of stressed pre-malignant cells (169). KLRK1 knockout mice develop prostate adenocarcinomas and B cell lymphomas (173), and polymorphisms in the KLRK1 gene are associated with the susceptibility of developing liver and cervix cancers (174, 175).
In summary, the data from both man and mice suggest that the expression of stress-induced endogenous molecules associated with cell transformation may be used by the immune system to recognize and eliminate pre-malignant cells.
Role of Infectious Agents in Driving Malignancy
The data to date suggest that the increase in viral infection seen in PIDs patients further enhanced the susceptibility to cancer, although the precise mechanisms for this remain unclear. The impact of infectious agents on cancer risk has been investigated in CVID patients. High HHV8 copy numbers are present in the malignant lymph node of CVID patients with granulomas (176). In addition, there is a 13-fold increase in CD8+ T cells specific for CMV-derived peptides in CVID patients compared with controls (177). Clinical observations indicate that an exaggerated T cell response to CMV may exacerbate enteropathy in CVID patients (147). Moreover, Helicobacter pylori infection and pernicious anemia are risk predictors for gastric cancer in CVID patients and in the general population (42).
Epstein–Barr virus is an important co-factor for the development of CVID and can affect the host, either primarily during an immunocompromised state or following reactivation during a disease flare (12, 178). The range of lymphoid tumors linked with EBV includes HD, angioimmunoblastic lymphadenopathy, lymphomatoid granulomatosis, some forms of HIV-associated lymphoma, and primary central nervous system lymphomas. It is paradoxical that a ubiquitous virus which causes a generally benign and self-limiting infective illness of childhood or early adult life may also be responsible for such aggressive tumors. Further studies are required to delineate the mechanism(s) of EBV and other viral infections on cancer susceptibility.
Prognosis and Perspectives
We have described here how dysfunctions in the immune system of patients with PIDs can predispose patients to malignancies. Early treatment of the immunodeficiencies in some of these patients, before end organ damage has occurred, may reduce the risk of cancer, but clearly a greater understanding of the pathways responsible for this increased risk is needed. This may lead to the development of novel therapeutic agents that prevent cancer. In addition to the critical role in protection from infectious non-self-pathogens, the immune system is also important in modulating malignant self-challenges (cancer). Numerous immune cells are involved in cancer surveillance and prevention including those of the adaptive (T and B cells) and innate (NK and macrophages) immune systems, and all of these are defective to a greater or lesser extent in patients with PID. However, our knowledge of the precise mechanisms by which the immune system fights cancer, particularly in relation to infection by non-oncogenic viruses, remains rudimentary at best, although an inaccurate antitumor immune response may occur (179). There is no inherent difference in cancer treatment response in patients with PID compared to in non-immunodeficient patients (124). However, since patients with PIDs often have disseminated tumors that require systemic cytotoxic therapy which is poorly tolerated, there is an increased risk for infection and end-organ damage. New drugs are needed for these patients directed against the oncogenic pathways linked to each PID, but in the meantime, effective and early treatment of the immune defects present in each PID may reduce the associated risk of cancer.
The causative associations between immune factors and oncologic processes can be a pathological two-way street. For example, immunocompetent patients with cancer can develop immune suppression secondary to the cancer, from chemotherapy or from posttransplant immunosuppressive therapy (180). This is particularly evident for B-cell chronic lymphocytic leukemia and associated hypogammaglobulinemia. As in patients with PID, infection control is paramount. Immunoglobulin replacement therapy has been shown to reduce the frequency of bacterial infections in patients with chronic lymphocytic leukemia (181–184) and NHL (182), and this may impact upon the subsequent risk of malignancy.
In conclusion, the increased incidence of cancer in patients with some types of PID is due to the inherent dysregulation of the immune response present and/or exposure to an infectious organism (Figure 1). It is only through greater understanding of how the specific mutations in each patient relate to and modify oncogenic pathways that we will prevent the development of these malignancies.
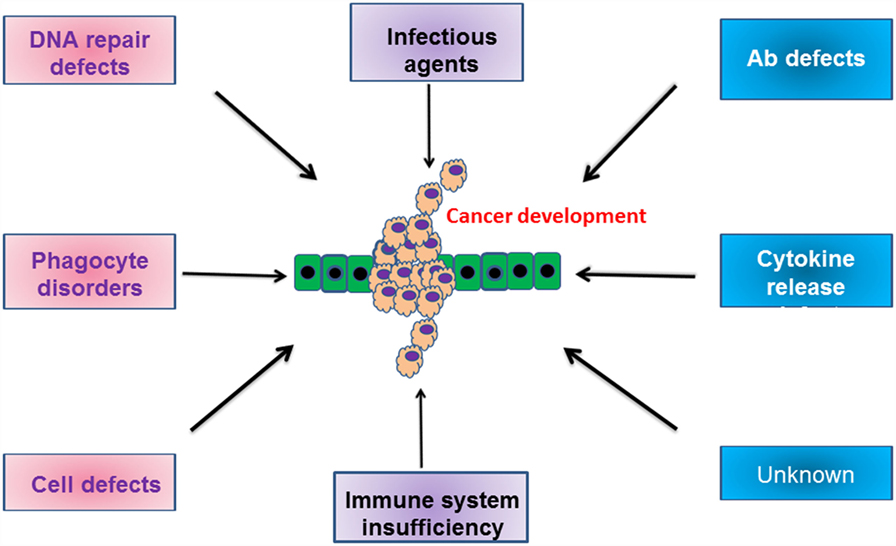
Figure 1. Schematic cartoon indicating the various defects in humoral and cellular compartments, which drive the development of malignancies. For more information, please refer to the text.
Author Contributions
All authors contributed in writing and approval of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
IA is supported by Wellcome Trust grant 093080/Z/10/Z. This project was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. The views expressed in this publication are those of the authors(s) and not necessarily those of the NHS, The National Institute for Health Research or the Department of Health.
Abbreviations
ADA, adenosine deaminase; ADAM17, a disintegrin and metalloproteinase 17; AD-HIES, autosomal-dominant hyper-IgE syndrome; AICDA, activation-induced cytidine deaminase; AIDS, acquired immunodeficiency syndrome; AIRE, autoimmune regulator; APS, autoimmune polyendocrine syndrome; AR-HIES, autosomal recessive hyper-IgE syndrome; ATM, ataxia-telangiectasia mutated; BAFF-R, B-cell-activating factor receptor; BLNK, B-cell linker; BTK, B-cell tyrosine kinase; CD, cluster of differentiation; CD40L, CD40 ligand; CGD, chronic granulomatous disease; CIDs, combined immunodeficiencies; CMC, chronic mucocutaneous candidiasis; CMCD, chronic mucocutaneous candidiasis disease; CMV, cytomegalovirus; CN, congenital neutropenia; CTLs, cytotoxic T lymphocytes; CVID, common variable immunodeficiency; CYBA, cytochrome b alpha; CYBB, cytochrome b beta; DOCK8, dedicator of cytokinesis 8; EBV, Epstein–Barr virus; ELA, elastase; FHL, familial hemophagocytic lymphohistiocytosis; G6PC3, glucose-6-phosphatase, catalytic, 3; G-CSF, granulocyte colony-stimulating factor; GF, growth factor; HAX1, hematopoietic cell-specific Lyn substrate 1 (HCLS1)-associated protein X-1; HD, Hodgkin’s disease; HHV8, human herpes virus 8; HSE, herpes simplex encephalitis; ICOS, inducible co-stimulator; ICR, Immunodeficiency Cancer Registry; IFNGR, IFN γ receptor; IGHMM, Ig heavy chain mu; IGLL1, Ig light chain lambda; IL12R, IL-12 receptor; IL2RG, IL-2 receptor gamma; IL7R, IL-7 receptor; IRF4, interferon regulatory factor 4; JAK, janus-associated kinase; LPDs, lymphoproliferative disorders; MAPBP, mitogen-activated protein kinase-binding protein; MSMD, Mendelian susceptibility to mycobacterial disease; NBS, Nijmegen breakage syndrome; NCF, neutrophil cytosolic factor; NHL, non-Hodgkin lymphoma; NK, natural killer; PGM3, phosphoglucomutase 3; PIDs, primary immunodeficiencies; PRF, perforin; RAG, recombinase-activating gene; RMRP, RNA component of mitochondrial RNA processing endoribonuclease; RORγt, retinoic acid-related orphan receptor γt; SCN, severe congenital neutropenia; STAT, signal transducer and activator of transcription; STX11, syntaxin; TACI, transmembrane activator and CAML interactor (calcium modulator); Th17, IL-17 secreting T helper cell; TLR, toll-like receptor; TNFRSF13B, TNF receptor soluble factor; Tyk2, tyrosine kinase 2; UNG, uracil N-glycosylase; WAS, Wiskott–Aldrich syndrome; WASP, Wiskott–Aldrich syndrome protein; XIAP, X-linked inhibitor of apoptosis; XLA, X-linked agammaglobulinemia.
References
1. Page AR, Hansen AE, Good RA. Occurrence of leukemia and lymphoma in patients with agammaglobulinemia. Blood (1963) 21(2):197–206.
2. Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics (1958) 21:526–54.
3. Gatti RA, Good RA. Occurrence of malignancy in immunodeficiency diseases. Cancer (1971) 28:89–98. doi: 10.1002/1097-0142(197107)28:1<89::AID-CNCR2820280117>3.0.CO;2-Q
4. Waldmantn A, Strober W, Blaeser M. Immunodeficiency disease and malignancy: various immunologic deficiencies of man and the role of immune processes in the control of malignant disease. Ann Intern Med (1972) 77:605–28. doi:10.7326/0003-4819-77-4-605
5. Sedgwick RP, Boder E. Ataxia-telangiectasia. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. Amsterdam: North-Holland (1972). p. 267–339.
6. Kersey JH, Spectorb D, Good RA. Immunodeficiency and cancer. In: Weinhouse S, Klein G, editors. Advances in Cancer Research (Vol. 18), New York: Academic Press (1973). p. 75–8.
7. Madkaikar M, Mishra A, Ghosh K. Diagnostic approach to primary immune deficiency disorders. Indian Pediatr (2013) 50:579–86. doi:10.1007/s13312-013-0171-4
8. Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol (2007) 120:776–94. doi:10.1016/j.jaci.2007.08.053
9. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
10. Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. J Clin Immunol (2015) 35:696–726. doi:10.1007/s10875-015-0201-1
11. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol (2007) 27:497–502. doi:10.1007/s10875-007-9103-1
12. Mueller BU, Pizzo PA. Cancer in children with primary or secondary immunodeficiencies. J Pediatr (1996) 126:1–10. doi:10.1016/S0022-3476(95)70491-4
13. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res (2008) 28(2B):1263–9.
14. Tabarsi P, Marjani M, Mansouri N, Farnia P, Boisson-Dupuis S, Bustamante J, et al. Lethal tuberculosis in a previously healthy adult with IL-12 receptor deficiency. J Clin Immunol (2011) 31:537–9. doi:10.1007/s10875-011-9523-9
15. Mansouri D, Adimi P, Mirsaedi M, Mansouri N, Tabarsi P, Amiri M, et al. Primary immune deficiencies presenting in adults: seven years of experience from Iran. J Clin Immunol (2005) 25:385–91. doi:10.1007/s10875-005-4124-0
16. McCusker C, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol (2011) 7:S11. doi:10.1186/1710-1492-7-S1-S11
17. Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev (2003) 16:597–621. doi:10.1128/CMR.16.4.597-621.2003
18. Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev (2009) 22:396–414. doi:10.1128/CMR.00001-09
19. Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2011) 2:54. doi:10.3389/fimmu.2011.00054
20. Slatter MA, Gennery AR. Clinical immunology review series: an approach to the patient with recurrent infections in childhood. Clin Exp Immunol (2008) 152:389–96. doi:10.1111/j.1365-2249.2008.03641.x
21. Bonilla FA, Geha RS. Primary immunodeficiency diseases. J Allergy Clin Immunol (2003) 111:571–81. doi:10.1067/mai.2003.86
22. Ballow M. Primary immunodeficiency disorders: antibody deficiency. J Allergy Clin Immunol (2002) 109:581–91. doi:10.1067/mai.2002.122466
23. Casanova JL, Fieschi C, Zhang SY, Abel L. Revisiting human primary immunodeficiencies. J Intern Med (2008) 264:115–27. doi:10.1111/j.1365-2796.2008.01971.x
24. Corthay A. Does the immune system naturally protect against cancer? Front Immunol (2014) 5:197. doi:10.3389/fimmu.2014.00197
25. Kinlen LJ, Webster AD, Bird AG, Haile R, Peto J, Soothill JF, et al. Prospective study of cancer in patients with hypogammaglobulinemia. Lancet (1985) 1:263–6. doi:10.1016/S0140-6736(85)91037-2
26. van der Meer JW, Weening RS, Schellekens PT, van Munster IP, Nagengast FM. Colorectal cancer in patients with X-linked agammaglobulinemia. Lancet (1993) 341:1439–40. doi:10.1016/0140-6736(93)90883-I
27. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol (1999) 92:34–48. doi:10.1006/clim.1999.4725
28. Saiki O, Ralph P, Cunningham-Rundles C, Good RA. Three distinct stages of B-cell defects in common varied immunodeficiency. Proc Natl Acad Sci U S A (1982) 79:6008–12. doi:10.1073/pnas.79.19.6008
29. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol (2002) 130:495–500. doi:10.1046/j.1365-2249.2002.02004.x
30. Mohammadinejad P, Aghamohammadi A, Abolhassani H, Sadaghiani MS, Abdollahzade S, Sadeghi B, et al. Pediatric patients with common variable immunodeficiency: long-term follow-up. J Investig Allergol Clin Immunol (2012) 22(3):208–14.
31. Ogershok PR, Hogan MB, Welch JE, Corder WT, Wilson NW. Spectrum of illness in pediatric common variable immunodeficiency. Ann Allergy Asthma Immunol (2006) 97:653–6. doi:10.1016/S1081-1206(10)61096-4
32. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol (2007) 27:308–16. doi:10.1007/s10875-007-9075-1
33. Knight AK, Cunningham-Rundles C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun Rev (2006) 5:156–9. doi:10.1016/j.autrev.2005.10.002
34. Brandt D, Gershwin ME. Common variable immune deficiency and autoimmunity. Autoimmun Rev (2006) 5:465–70. doi:10.1016/j.autrev.2006.03.010
35. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119:1650–7. doi:10.1182/blood-2011-09-377945
36. Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet (2008) 372:489–502. doi:10.1016/S0140-6736(08)61199-X
37. Aghamohammadi A, Farhoudi A, Moin M, Rezaei N, Kouhi A, Pourpak Z, et al. Clinical and immunological features of 65 Iranian patients with common variable immunodeficiency. Clin Diagn Lab Immunol (2005) 12(7):825–32.
38. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112:277–86. doi:10.1182/blood-2007-11-124545
39. Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, Lieberman P. Incidence of cancer in 98 patients with common varied immunodeficiency. J Clin Immunol (1987) 7:294–9. doi:10.1007/BF00915550
40. Dhalla F, Silva SP, Lucas M, Travis S, Chapel H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin Exp Immunol (2011) 165:1–7. doi:10.1111/j.1365-2249.2011.04384.x
41. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood (2010) 116:1228–34. doi:10.1182/blood-2010-03-272351
42. Baron-Ruiz I, Martin-Mateos MA, Plaza-Martin AM, Giner-Munoz MT, Piquer M. Lymphoma as presentation of common variable immunodeficiency. Allergol Immunopathol (2009) 37:51–3. doi:10.1016/S0301-0546(09)70254-X
43. Chua I, Quinti I, Grimbacher B. Lymphoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics. Curr Opin Hematol (2008) 15:368–74. doi:10.1097/MOH.0b013e328302c7b6
44. Aghamohammadi A, Parvaneh N, Tirgari F, Mahjoob F, Movahedi M, Gharagozlou M, et al. Lymphoma of mucosa-associated lymphoid tissue in common variable immunodeficiency. Leuk Lymphoma (2006) 47:343–6. doi:10.1080/10428190500285285
45. Cunningham-Rundles C, Cooper DL, Duffy TP, Strauchen J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am J Hematol (2002) 69:171–8. doi:10.1002/ajh.10050
46. Davis SD, Schaller J, Wedgwood RJ. Job’s syndrome. Recurrent, “cold” staphylococcal abscesses. Lancet (1966) 1:1013–5. doi:10.1016/S0140-6736(66)90119-X
47. Buckley RH, Wray BB, Belmaker EZ. Extreme hyper immunoglobulinemia E and undue susceptibility to infection. Pediatrics (1972) 49:59–70.
48. Buckley RH. The hyper IgE syndrome. Clin Rev Allergy Immunol (2001) 20:139–54. doi:10.1385/CRIAI:20:1:139
49. Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) (2012) 91:1–19. doi:10.1097/MD.0b013e31825f95b9
50. Milner JD. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature (2008) 452:773–6. doi:10.1038/nature06764
51. Beaucoudrey LD, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17 producing T cells. J Exp Med (2008) 205:1543–50. doi:10.1084/jem.20080321
52. Jiao H, Tóth B, Erdos M, Fransson I, Rákóczi E, Balogh I, et al. Novel and recurrent STAT3 mutations in hyper-IgE syndrome patients from different ethnic groups. Mol Immunol (2008) 46:202–6. doi:10.1016/j.molimm.2008.07.001
53. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448:1058–62. doi:10.1038/nature06096
54. Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol (2008) 122:181–7. doi:10.1016/j.jaci.2008.04.037
55. Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med (2008) 205:1551–7. doi:10.1084/jem.20080218
56. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity (2011) 35:806–18. doi:10.1016/j.immuni.2011.09.016
57. Meloni A, Willcox N, Meager A, Atzeni M, Wolff AS, Husebye ES, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocrinol Metab (2012) 97:1114–24. doi:10.1210/jc.2011-2461
58. Dong C. Th17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol (2008) 8:337–48. doi:10.1038/nri2295
59. Gaffen SL, Jain R, Garg AV, Cua DJ. IL-23-IL-17 immune axis: discovery, mechanistic understanding, and clinical testing. Nat Rev Immunol (2014) 14:585–600. doi:10.1038/nri3707
60. Eyerich E, Eyerich K, Cavani A, Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol (2010) 31:354–61. doi:10.1016/j.it.2010.06.004
61. Sparber F, LeibundGut-Landmann S. Interleukin 17-mediated host defense against Candida albicans. Pathogens (2015) 4:606–19. doi:10.3390/pathogens4030606
62. Whibley N, Gaffen SL. Brothers in arms: Th17 and Treg responses in Candida albicans immunity. PLoS Pathog (2014) 10:e1004456. doi:10.1371/journal.ppat.1004456
63. Conti HR, Whibley N, Coleman BM, Garg AV, Jillian Jaycox R, Gaffen SL. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS One (2015) 10(4):e0122807. eCollection 2015 doi:10.1371/journal.pone.0122807
64. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 medicates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14:275–81. doi:10.1038/nm1710
65. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis (2004) 190:624–31. doi:10.1086/422329
66. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med (2002) 347:1151–60. doi:10.1056/NEJMoa021481
67. Kao CY, Chen Y, Thai P, Wachi S, Kim C, Tam L, et al. IL-17 markedly up-regulates betadefensin-2 expression in human airway epithelium via JAK and NF-kB signaling pathways. J Immunol (2004) 173:3482–91. doi:10.4049/jimmunol.173.5.3482
68. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science (2002) 298:1630–4. doi:10.1126/science.1077002
69. Avery DT, Ma CS, Bryant VL, Santner-Nanan B, Nanan R, Wong M, et al. STAT3 is required for IL-21 induced secretion of IgE from human naive B cells. Blood (2008) 112:1784–93. doi:10.1182/blood-2008-02-142745
70. Shang XZ, Ma KY, Radewonuk J, Li J, Song XY, Griswold DE, et al. IgE isotype switch and IgE production are enhanced in IL-21-deficient but not IFN-gamma-deficient mice in a Th2-biased response. Cell Immunol (2006) 241:66–74. doi:10.1016/j.cellimm.2006.07.011
71. Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr (2004) 144:93–9. doi:10.1016/S0022-3476(03)00449-9
72. Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, Kreins AY, Grant AV, Abel L, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr (2012) 160:1055–7. doi:10.1016/j.jpeds.2012.01.056
73. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med (2009) 361:2046–55. doi:10.1056/NEJMoa0905506
74. Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR–MyD88 signaling to B cell activation. Nat Immunol (2012) 13:612–20. doi:10.1038/ni.2305
75. Randall KL, Chan SS, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med (2011) 208:2305–20. doi:10.1084/jem.20110345
76. Davi SD, Schaller J, Wedgwood RJ. Job’s syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet (1966) 1:1013–5.
77. Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am (2008) 28:277–91. doi:10.1016/j.iac.2008.01.005
78. Szczawinska-Poplonyk A, Kycler Z, Pietrucha B, Heropolitanska-Pliszka E, Breborowicz A, Gerreth K. The hyperimmunoglobulin E syndrome – clinical manifestation diversity in primary immune deficiency. Orphanet J Rare Dis (2011) 6:76. doi:10.1186/1750-1172-6-76
79. Randall KL, Lambe T, Goodnow CC, Cornall RJ. The essential role of DOCK8 in humoral immunity. Dis Markers (2010) 29:141–50. doi:10.3233/DMA-2010-0739
80. Su HC. DOCK8 (dedicator of cytokinesis 8) deficiency. Curr Opin Allergy Clin Immunol (2010) 10:515–20. doi:10.1097/ACI.0b013e32833fd718
81. Zhang Y. Autosomal recessive PGM3 mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol (2014) 133:1400–9. doi:10.1016/j.jaci.2014.02.013
82. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med (2010) 207:155–71. doi:10.1084/jem.20091706
83. Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity (2012) 36:515–28. doi:10.1016/j.immuni.2012.03.016
84. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med (2013) 368:161–70. doi:10.1056/NEJMra1202117
85. Forte G, Sorrentino R, Montinaro A, Luciano A, Adcock IM, Maiolino P, et al. Inhibition of CD73 improves B cell-mediated anti-tumor immunity in a mouse model of melanoma. J Immunol (2012) 189:2226–33. doi:10.4049/jimmunol.1200744
86. Sorrentino R, Morello S, Forte G, Montinaro A, De Vita G, Luciano AP, et al. B cells contribute to the antitumor activity of CpG-oligodeoxynucleotide in a mouse model of metastatic lung carcinoma. Am J Respir Crit Care Med (2011) 183:1369–79. doi:10.1164/rccm.201010-1738OC
87. Maródi L. Mucocutaneous candidiasis. In: Sullivan KE, Stiehm RE, editors. Stiehm’s Immunodeficiencies. Amsterdam: Elsevier Inc. (2014). p. 775–802.
88. Mogensen TH. STAT3 and the Hyper-IgE syndrome: clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAKSTAT (2013) 2:e23435. doi:10.4161/jkst.23435
89. Ouederni M, Sanal O, Ikinciogullari A, Ouederni M, Sanal O, Ikinciogullari A, et al. Clinical features of Candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis (2014) 58:204–13. doi:10.1093/cid/cit722
90. Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, et al. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from30 kindreds. Medicine (Baltimore) (2013) 92:109–22. doi:10.1097/MD.0b013e31828a01f9
91. Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol (2012) 12:616–22. doi:10.1097/ACI.0b013e328358cc0b
92. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science (2011) 332:65–8. doi:10.1126/science.1200439
93. Zheng J, van de Veerdonk FL, Crossland KL, Smeekens SP, Chan CM, Shehri TA, et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol (2015) 45:2834–46. doi:10.1002/eji.201445344
94. Mizoguchi Y, Tsumura M, Okada S, Hirata O, Minegishi S, Hyakuna N, et al. STAT1 gain-of-function in patients with chronic mucocutaneous candidiasis can be detected by the excessive phosphorylation of STAT1 in peripheral blood monocytes. Blood (2014) 124:4111.
95. Delsing CE, Bleeker-Rovers CP, van de Veerdonk FL, Tol JJ, van der Meer WM, Kullberg BJ, et al. Association of esophageal candidiasis and squamous cell carcinoma. Med Mycol Case Rep (2012) 1:5–8. doi:10.1016/j.mmcr.2012.02.003
96. Cypowyj S, Picard C, Maródi L, Casanova JL, Puel A. Immunity to infection in IL-17-deficient mice and humans. Eur J Immunol (2012) 42:2246–54. doi:10.1002/eji.201242605
97. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
98. Maródi L, Cypowyj S, Toth B, Chernyshova L, Puel A, Casanova JL. Molecular mechanisms of mucocutaneous immunity against Candida and Staphylococcus species. J Allergy Clin Immunol (2012) 130:1019–27. doi:10.1016/j.jaci.2012.09.011
99. Maródi L, Cypowyj S, Casanova JL, Puel A. The role of human IL-17 immunity in fungal disease. Curr Fungal Infect Rep (2013) 7:132–7. doi:10.1007/s12281-013-0131-4
100. Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev (2005) 203:244–50. doi:10.1111/j.0105-2896.2005.00228.x
101. Rosa DD, Pasqualotto AC, Denning DW. Chronic mucocutaneous candidiasis and oesophageal cancer. Med Mycol (2008) 46:85–91. doi:10.1080/13693780701616023
102. Goransdotter EK, Fadeel B, Nilsson-Ardnor S, Söderhäll C, Samuelsson A, Janka G, et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet (2001) 68:590–7. doi:10.1086/318796
103. Trizzino A, Zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genotype–phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet (2008) 45:15–21. doi:10.1136/jmg.2007.052670
104. Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood (2002) 99:61–6. doi:10.1182/blood.V99.1.61
105. Feldmann J, Le Deist F, Ouachee-Chardin M, Certain S, Alexander S, Quartier P, et al. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol (2002) 117:965–72. doi:10.1046/j.1365-2141.2002.03534.x
106. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med (2012) 63:233–46. doi:10.1146/annurev-med-041610-134208
107. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol (2006) 6:940–52. doi:10.1038/nri1983
108. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286:1957–9. doi:10.1126/science.286.5446.1957
109. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood (2011) 118:5794–8. doi:10.1182/blood-2011-07-370148
110. Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood (2014) 124:1331–4. doi:10.1182/blood-2014-05-573105
111. Mhatre S, Madkaikar M, Desai M, Ghosh K. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis (FHL) patients in India. Blood Cells Mol Dis (2015) 54:250–7. doi:10.1016/j.bcmd.2014.11.023
112. Henter JI, Elinder G, Söder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood (1991) 78:2918–22.
113. Komp DM, McNamara J, Buckley P. Elevated soluble interleukin-2 receptor in childhood hemophagocytic syndromes. Blood (1989) 73:2128–32.
114. Imashuku S, Ikushima S, Esumi N, Todo S, Saito M. Serum levels of interferon-gamma, cytotoxic factor and soluble interleukin-2 receptor in childhood hemophagocytic syndromes. Leuk Lymphoma (1991) 3:287–92. doi:10.3109/10428199109107916
115. Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood (1997) 89:4100–3.
116. Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian predisposition to B cell lymphoma caused by IL-10R deficiency. Blood (2013) 122:3713–22. doi:10.1182/blood-2013-06-508267
117. Filipovich AH, Mathur A, Kamat D, Kersey JH, Shapiro RS. Lymphoproliferative disorders and other tumors complicating immunodeficiencies. Immunodeficiency (1994) 5:91–112.
118. Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol (2005) 94:S1–63. doi:10.1016/S1081-1206(10)61142-8
119. Schroeder HW Jr, Zhu ZB, March RE, Campbell RD, Berney SM, Nedospasov SA, et al. Susceptibility locus for IgA deficiency and common variable immunodeficiency in the HLA-DR3, 2B8, 2A1 haplotypes. Mol Med (1998) 4:72–86.
120. Aghamohammadi A, Mohammadi J, Parvaneh N, Rezaei N, Moin M, Espanol T, et al. Progression of selective IgA deficiency to common variable immunodeficiency. Int Arch Allergy Immunol (2008) 147:87–92. doi:10.1159/000135694
121. Hayward AR, Levy J, Facchetti F, Notarangelo L, Ochs HD, Etzioni A, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol (1997) 158:977–83.
122. Wobser M, Kerstan A, Kneitz H, Goebeler M, Kunzmann V, Rosenwald A, et al. Primary cutaneous marginal zone lymphoma with sequential development of nodal marginal zone lymphoma in a patient with selective immunoglobulin A deficiency. J Cutan Pathol (2013) 40:1035–41. doi:10.1111/cup.12230
123. Ott MM, Ott G, Klinker H, Trunk MJ, Katzenberger T, Müller-Hermelink HK. Abdominal T-cell non-Hodgkin’s lymphoma of the gamma/delta type in a patient with selective immunoglobulin A deficiency. Am J Surg Pathol (1998) 22:500–6. doi:10.1097/00000478-199804000-00017
124. Gross TG, Shiramizu B. Lymphoproliferative disorders and malignancies related to immunodeficiencies. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. Philadelphia, PA: Lippincott Williams & Wilkins (2006). p. 748–67.
125. de Miranda NF, Björkman A, Pan-Hammarström Q. DNA repair: the link between primary immunodeficiency and cancer. Ann N Y Acad Sci (2011) 1246:50–63. doi:10.1111/j.1749-6632.2011.06322.x
126. Taylor AMR, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood (1996) 87:423–38.
127. Sandoval C, Swift M. Hodgkin disease in ataxis-telangiectasia patients with poor outcomes. Med Pediatr Oncol (2003) 40:162–6. doi:10.1002/mpo.10251
128. Heintz N. Ataxia telangiectasia: cell signaling, cell death and the cell cycle. Curr Opin Neurol (1996) 9:137–40. doi:10.1097/00019052-199604000-00014
129. Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst (1986) 77:89–92.
130. Dembowska-Baginska B, Perek D, Brozyna A, Wakulinska A, Olczak-Kowalczyk D, Gladkowska-Dura M, et al. Non-Hodgkin lymphoma (NHL) in children with Nijmegen breakage syndrome (NBS). Pediatr Blood Cancer (2009) 52:186–90. doi:10.1002/pbc.21789
131. Online Mendelian Inheritance in Man (OMIM) database. Johns Hopkins University. Nijmegen Breakage Syndrome. (2009). Available from: http://www.omim.org/entry/613078
132. van der Burgt I, Chrzanowska KH, Smeets D, Weemaes C. Nijmegen breakage syndrome. J Med Genet (1996) 33:153–6. doi:10.1136/jmg.33.2.153
133. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci (2013) 1285:26–43. doi:10.1111/nyas.12049
134. Cotelingam JD, Witebsky FG, Hsu SM, Blaese RM, Jaffe ES. Malignant lymphoma in patients with the Wiskott-Aldrich syndrome. Cancer Invest (1985) 3:515–22. doi:10.3109/07357908509039813
135. Kroft SH, Finn WG, Singleton TP, Ross CW, Sheldon S, Schnitzer B. Follicular large cell lymphoma with immunoblastic features in a child with Wiskott-Aldrich syndrome: an unusual immunodeficiency-related neoplasm not associated with Epstein-Barr virus. Am J Clin Pathol (1998) 110:95–9. doi:10.1093/ajcp/110.1.95
136. Singh VK, Newmana VL, Seed TM. Colony-stimulating factors for the treatment of the hematopoietic component of the acute radiation syndrome (H-ARS): a review. Cytokine (2015) 71:22–37. doi:10.1016/j.cyto.2014.08.003
137. Bresnick EH, Katsumura KR, Lee HY, Johnson KD, Perkins AS. Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res (2012) 40:5819–31. doi:10.1093/nar/gks281
138. Tang X, Lasbury ME, Davidson DD, Bartlett MS, Smith JW, Lee CH. Downregulation of GATA-2 transcription during Pneumocystis carinii infection. Infect Immun (2015) 68:4720–4. doi:10.1128/IAI.68.8.4720-4724.2000
139. Lasbury ME, Tang X, Durant PJ, Lee CH. Effect of transcription factor GATA-2 on phagocytic activity of alveolar macrophages from Pneumocystis carinii-infected hosts. Infect Immun (2003) 71:4943–52. doi:10.1128/IAI.71.9.4943-4952.2003
140. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood (2014) 123:809–21. doi:10.1182/blood-2013-07-515528
141. Welte K, Zeidler C. Severe congenital neutropenia. Hematol Oncol Clin North Am (2009) 23:307–20. doi:10.1016/j.hoc.2009.01.013
142. Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: results of a long-term survey. Blood (2007) 109:93–9. doi:10.1182/blood-2006-02-004275
143. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res (1970) 13:1–27. doi:10.1159/000386035
144. Umaimainthan P, Rickinson AB. Primary immunodeficiencies and the control of Epstein–Barr virus infection. Ann N Y Acad Sci (2015) 1356:22–44. doi:10.1111/nyas.12937
145. Jonkman-Berk BM, van den Berg JM, Ten Berge IJ, Bredius RG, Driessen GJ, Dalm VA, et al. Primary immunodeficiencies in the Netherlands: national patient data demonstrate the increased risk of malignancy. Clin Immunol (2015) 156:154–62. doi:10.1016/j.clim.2014.10.003
146. Kersey JH, Shapiro RS, Filipovich AH. Relationship of immunodeficiency to lymphoid malignancy. Pediatr Infect Dis J (1988) 7:S10–2. doi:10.1097/00006454-198805001-00003
147. Filipovich AH, Mathur A, Kamat D, Shapiro RS. Primary immunodeficiencies: genetic risk factors for lymphoma. Cancer Res (1992) 52:5465–7.
148. Penn I, Hammond W, Brettschneider L, Starzl TE. Malignant lymphomas in transplantation patients. Transplant Proc (1969) 1:106–12.
149. Latour S, Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol (2015) 39:115–23. doi:10.1016/j.semcdb.2015.01.015
150. Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A (2009) 106:14524–9. doi:10.1073/pnas.0907131106
151. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell (2012) 46:1–13. doi:10.1016/j.molcel.2012.04.014
152. Damgaard RB, Fiil BK, Speckmann C, Yabal M, Stadt UZ, Bekker-Jensen S, et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol Med (2013) 5:1278–95. doi:10.1002/emmm.201303090
153. Hsieh WC, Chuang YT, Chiang IH, Hsu SC, Miaw SC, Lai MZ. Inability to resolve specific infection generates innate immunodeficiency syndrome in Xiap−/− mice. Blood (2014) 124:2847–57. doi:10.1182/blood-2014-03-564609
154. Oertel S, Reiss H. Immunosurveillance, Immunodeficiency and Lymphoproliferations. Heidelberg: Springer (2002). 159 p.
155. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117:1137–46. doi:10.1172/JCI31405
156. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFN gamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410:1107–11. doi:10.1038/35074122
157. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by gamma delta T cells. Science (2001) 294:605–9. doi:10.1126/science.1063916
158. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6:722–9. doi:10.1038/ni1213
159. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferongamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A (1998) 95:7556–61. doi:10.1073/pnas.95.13.7556
160. Street SE, Trapani JA, MacGregor D, Smyth MJ. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J Exp Med (2002) 196:129–34. doi:10.1084/jem.20020063
161. Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med (2000) 192:755–60. doi:10.1084/jem.192.5.755
162. Thomas L. Discussion. In: Lawrence HS, editor. Cellular and Humoral Aspects of the Hypersensitive States. New York: Hoeber-Harper (1959). p. 529–32.
163. Calne RY, White DJ, Thiru S, Evans DB, McMaster P, Dunn DC, et al. Cyclosporin A in patients receiving renal allografts from cadaver donors. Lancet (1978) 2:1323–7. doi:10.1016/S0140-6736(78)91970-0
164. Hoover R, Fraumeni JF. Risk of cancer in renal-transplant recipients. Lancet (1973) 2:55–7. doi:10.1016/S0140-6736(73)93256-X
165. List AF, Greco FA, Vogler LB. Lympho proliferative diseases in immuno-compromised hosts: the role of Epstein-Barr virus. J Clin Oncol (1987) 5:1673–89.
166. Clifford GM, Polesel J, Rickenbach M, DalMaso L, Keiser O, Kofler A, et al. Cancer risk in the Swiss HIV Cohort Study: associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J Natl Cancer Inst (2005) 97:425–32. doi:10.1093/jnci/dji072
167. Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett (2014) 588:368–76. doi:10.1016/j.febslet.2013.10.015
168. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi:10.1126/science.285.5428.727
169. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A (1996) 93:12445–50. doi:10.1073/pnas.93.22.12445
170. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436:1186–90. doi:10.1038/nature03884
171. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A (1999) 96:6879–84. doi:10.1073/pnas.96.12.6879
172. Vantourout P, Willcox C, Turner A, Swanson CM, Haque Y, Sobolev O, et al. Immunological visibility: posttranscriptional regulation of human NKG2D ligands by the EGF receptor pathway. Sci Transl Med (2014) 6:231–49. doi:10.1126/scitranslmed.3007579
173. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28:571–80. doi:10.1016/j.immuni.2008.02.016
174. Chen D, Juko-Pecirep I, Hammer J, Ivansson E, Enroth S, Gustavsson I, et al. Genome-wide association study of susceptibility loci for cervical cancer. J Natl Cancer Inst (2013) 105:624–33. doi:10.1093/jnci/djt051
175. Melum E, Karlsen TH, Schrumpf E, Bergquist A, Thorsby E, Boberg KM, et al. Cholangiocarcinoma in primary sclerosing cholangitis is associated with NKG2D polymorphisms. Hepatology (2008) 47:90–6. doi:10.1002/hep.21964
176. Wheat WH, Cool CD, Morimoto Y, Rai PR, Kirkpatrick CH, Lindenbaum BA, et al. Possible role of human herpes virus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med (2005) 202:479–84. doi:10.1084/jem.20050381
177. Raeiszadeh M, Kopycinski J, Paston SJ, Diss T, Lowdell M, Hardy GAD, et al. The T cell response to persistent herpes virus infections in common variable immunodeficiency. Clin Exp Immunol (2006) 146:234–42. doi:10.1111/j.1365-2249.2006.03209.x
178. Rezk SA, Weiss LM. Epstein-Barr virus-associated lymphoproliferative disorders. Hum Pathol (2007) 38:1293–304. doi:10.1016/j.humpath.2007.05.020
179. Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science (2014) 343:152–7. doi:10.1126/science.1246886
180. Filipovich AH, Heinitz KJ, Robison LL, Frizzera G. The Immunodeficiency Cancer Registry. A research resource. Am J Pediatr Hematol Oncol (1987) 9:183–4. doi:10.1097/00043426-198722000-00017
181. A Randomized, Controlled Clinical Trial Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia. Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia. N Engl J Med (1988) 319:902–7.
182. Griffiths H, Brennan V, Lea J, Bunch C, Lee M, Chapel H. Crossover study of immunoglobulin replacement therapy in patients with low-grade B-cell tumors. Blood (1989) 73:366–8.
183. Gamm H, Huber C, Chapel H, Lee M, Ries F, Dicato MA. Intravenous immune globulin in chronic lymphocytic leukemia. Clin Exp Immunol (1994) 97:17–20.
Keywords: primary immunodeficiency, malignancy
Citation: Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A and Adcock IM (2016) Cancers Related to Immunodeficiencies: Update and Perspectives. Front. Immunol. 7:365. doi: 10.3389/fimmu.2016.00365
Received: 22 June 2016; Accepted: 05 September 2016;
Published: 20 September 2016
Edited by:
Guzide Aksu, Ege University, TurkeyReviewed by:
Elham Hossny, Ain Shams University, EgyptEsther De Vries, Tilburg University, Netherlands
Copyright: © 2016 Mortaz, Tabarsi, Mansouri, Khosravi, Garssen, Velayati and Adcock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian M. Adcock, aWFuLmFkY29ja0BpbXBlcmlhbC5hYy51aw==