 Günther Schönrich
Günther Schönrich Martin J. Raftery
Martin J. Raftery- Institute of Medical Virology, Helmut-Ruska-Haus, Charité − Universitätsmedizin Berlin, Berlin, Germany
Neutrophils are the most numerous immune cells. Their importance as the first line of defense against bacterial and fungal pathogens is well described. In contrast, the role of neutrophils in controlling viral infections is less clear. Bacterial and fungal pathogens can stimulate neutrophils extracellular traps (NETs) in a process called NETosis. Although NETosis has previously been described as a special form of programmed cell death, there are forms of NET production that do not end with the demise of neutrophils. As an end result of NETosis, genomic DNA complexed with microbicidal proteins is expelled from neutrophils. These structures can kill pathogens or at least prevent their local spread within host tissue. On the other hand, disproportionate NET formation can cause local or systemic damage. Only recently, it was recognized that viruses can also induce NETosis. In this review, we discuss the mechanisms by which NETs are produced in the context of viral infection and how this may contribute to both antiviral immunity and immunopathology. Finally, we shed light on viral immune evasion mechanisms targeting NETs.
Introduction
As the first line of defense against invading pathogens, neutrophils have a broad arsenal of antimicrobial functions (1). For example, activated neutrophils release granules containing antimicrobial molecules and produce reactive oxygen species (ROS) by oxidative burst. An alternative antimicrobial function of neutrophils is based on a special type of programmed cell death called NETosis that is distinct from apoptosis and necrosis (2, 3). During NETosis, the nuclei of neutrophils lose their characteristic shape, and chromatin decondensation takes place (4). Subsequently, the membranes of the nucleus and the granules disintegrate, allowing the mixing of their content. Finally, neutrophils release neutrophil extracellular traps (NETs). NETs are net-like structures that are composed of chromatin and endowed with granule proteins. They bind to, entrap, and often kill certain pathogens. NETs are released particularly in response to large microbial structures that cannot be easily phagocytosed such as Candida albicans hyphae and Mycobacterium bovis aggregates (5).
Classical NETosis requires the generation of ROS by NADPH oxidase. However, mitochondrial ROS production in the absence of a functional NADPH oxidase is sufficient to trigger NETosis (6). Moreover, a very rapid and ROS-independent form of NETosis is triggered by Staphylococcus aureus (7). Thus, depending on the stimulus NADPH is not always required for NET formation (8). Similar to necrosis and apoptosis, there are different forms of NETosis (9, 10). For example, it has been observed that NET formation can occur without concomitant neutrophil death (7, 11–14). The physiological and pathological meanings of these different NETosis forms still have to be elucidated.
Only recently, it was recognized that NETs are also generated during viral infection (15–17). Evidence is accumulating that neutrophils play a role in antiviral immune responses (18). These virus-induced NETs can both control the virus and damage the host (19). In this review, we focus our attention on the physiological and pathological relevance of virus-induced NETosis.
Viral NET Induction
Many viruses stimulate neutrophils in vitro directly to produce NETs at low levels (20). Some of these viruses can be detected inside neutrophils, but there is no direct evidence that they establish productive infection in this cell type (20–23). This suggests that pattern recognition receptors (PRRs) expressed on the surface or in endosomes of neutrophils play an essential role in NETosis (Figure 1). For example, neutrophils sense HIV-1 by endosomal PRRs that detect viral nucleic acids, i.e., toll-like receptor (TLR) 7 and TLR8, and subsequently undergo NETosis (17). The fusion protein of respiratory syncytial virus (RSV) induces NETosis through TLR4 (24). NET formation induced by hantaviruses is mediated by signaling through β2 integrins (20). Influenza virus A can also stimulate neutrophils directly to release NETs; however, the molecules involved have not been defined (25). Surprisingly, influenza A virus-induced NETs do not protect against secondary bacterial infection (26). Thus, virus-induced NETs differ structurally and functionally from those generated during bacterial infection. In line with this view, the protein content of NETs depends on the type of NET-inducing stimulus (27).
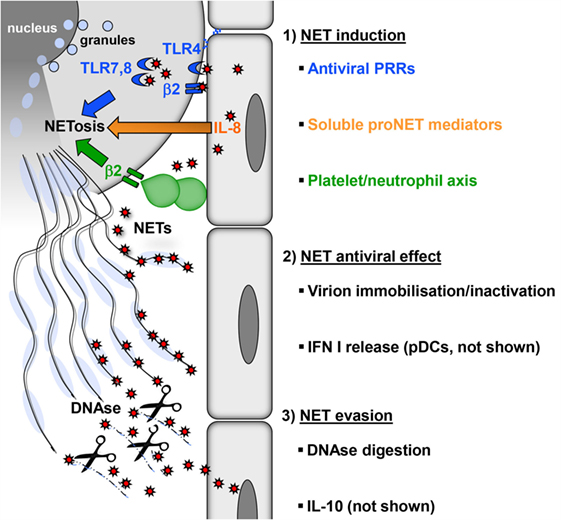
Figure 1. Induction, antiviral effect, and viral evasion of NETs. (1) Formation of NETs is induced directly by virions (red) through PRRs (blue) expressed by neutrophils on the surface (TLR4, β2 integrins) or in endosomes (TLR7, 8) or indirectly by proinflammatory mediators (e.g., IL-8), which are released from virus-infected cells (orange). In addition, viral activation of the platelet/neutrophil axis can trigger NETosis (green). As a consequence, granules fuse with the nucleus, which subsequently loses its characteristic lobulated shape and ruptures. Finally, neutrophils rupture releasing sticky strings of NETs. (2) NETs have antiviral effects by immobilizing or inactivating free virions, thereby preventing viral spread. NETs also potentiate the release of type I interferon by pDC (not shown), thus increasing the resistance of local cells to further infection. (3) Digestion of the DNA backbone by DNases releases trapped virions. These virions, if not already inactivated, opsonized, or degraded, can attempt to infect further cells. Moreover, viruses can interfere with NETosis by inducing cellular IL-10 or by expressing viral IL-10 homologs (not shown).
In the context of viral infection, neutrophils can switch on antiviral effector programs other than NETosis, such as release of antiviral agents or phagocytosis, and can even become apoptotic (18). At the moment, it is unclear how neutrophils decide between these different responses. Possibly, not a single PRR but rather as-yet undefined combinations of neutrophilic PRRs determine the antiviral mode of action of neutrophils. Moreover, only a proportion of cells undergo NETosis, suggesting that only a special neutrophil subtype or maturation stage is susceptible to NETosis induction (4).
Viruses also induce NETosis indirectly without engaging PRRs expressed by neutrophils (Figure 1). The inflammatory milieu created by virus-infected endothelial and epithelial cells contains cytokines and chemokines such as interleukin-8 (IL-8) that trigger NETosis (3, 28, 29). In addition, type I interferon (IFN) is produced in large amounts during viral infections and primes neutrophils for NET formation (30). There is also evidence that platelets play an important role in antiviral defense (31). Platelet activation is frequently observed during viral infections. For example, single-stranded RNA viruses from the family Picornaviridae activate platelets through TLR7. This is important for reducing viral titers and increasing the survival of the host (32, 33). Activated platelets form aggregates with neutrophils and in this process stimulate NETosis (34) (Figure 1). On the molecular level, this NET-inducing aggregation has been attributed to surface molecules: CD41 on activated platelets interacts with CD11b, a β2 integrin, on neutrophils. Other infection models have also shown that platelet–neutrophil interactions through β2 integrins induce NET formation (11, 35, 36). Massive activation of the platelet/neutrophil axis and subsequent NET-based clearance mechanisms may represent an emergency strategy of the host in the face of systemically multiplying viruses. This reaction is followed by a drop in platelet counts, which is observed in many viral infections, e.g., viral hemorrhagic fever (VHF) caused by hantaviruses (37, 38). In fact, the degree of platelet loss correlates with the severity of virus-induced disease and determines the clinical outcome (39–41).
Antiviral Activity of NETs
Although virus induction of NET formation is now well established, it is less clear how NETs contribute to antiviral immunity. In a mouse model of poxvirus infection, induction of NETs with LPS prior to infection strongly reduced the number of virus-infected liver cells and this protective effect was reversed by DNase treatment (34). There are direct mechanisms by which NETs develop antiviral activity (Figure 1). First of all, the web-like chromatin backbone of NETs can bind to and immobilize viral particles, in part by electrostatic attraction, thereby mechanically preventing virus spreading (17). Histones are enriched in positively charged amino acids and can attach to negatively charged viral envelope. For example, the core histones H3 and H4 induce aggregation of seasonal influenza A particles and may inactivate HIV-1 (17, 42). Intriguingly, extracellular histones also reduce HIV-1 transcription (43). Finally, histone H1 binds to noroviruses, the most common cause of viral gastroenteritis and prevents their attachment to intestinal cells (44). Second, attached to the chromatin backbone of NETs are antimicrobial molecules such as myeloperoxidase (MPO), cathelicidins, and α-defensin. They have a proven antiviral activity against both enveloped and non-enveloped viruses and can inactivate viral particles (45).
NETs components also indirectly contribute to antiviral immunity by stimulating antiviral effector mechanisms executed by other immune cells. For example, histones and high mobility group box-1 (HMGB1) proteins act as danger-associated molecular patterns (DAMPs) that trigger release of proinflammatory cytokines and chemokines by other immune cells (46). This process is self-limiting as under high neutrophil densities NETs build aggregates that in turn degrade cytokines and chemokines (47). NETs also activate plasmacytoid dendritic cells (pDCs) through TLRs (48–50). pDCs have a key function in antiviral immunity by releasing high amounts of type I IFN (51). In fact, NETs can be enriched in oxidized mitochondrial DNA which is very efficient in inducing a type I IFN response (52). Finally, NETs could increase antiviral adaptive immunity by reducing the activation threshold of T lymphocytes (53).
Viral NET Evasion
Viruses are known for their extraordinary capacity to evade immune control mechanisms. There are also viral mechanisms that counteract NET formation (Figure 1). For example, HIV-1 envelope glycoprotein stimulates DCs to produce cellular IL-10 through DC-SIGN (17). IL-10 is an immunosuppressive cytokine that also inhibits TLR-induced ROS production (54). It is quite often produced in the context of viral infections suggesting that more viruses exploit IL-10 as a means of NET evasion (55, 56). In the genome of several large DNA viruses IL-10, homologs have been found including ubiquitous human pathogens such as human cytomegalovirus (HCMV) and Epstein–Barr virus (EBV) (57, 58). As these virus-encoded IL-10 molecules shape the function and cell death of immune cells, they may also modulate NETosis similar to cellular IL-10 (59, 60). Dengue virus (DENV) serotype-2 can arrest NET formation at a ROS-independent late stage by interfering with glucose uptake (61, 62). Finally, latency-associated nuclear antigen 1 encoded by Kaposi’s sarcoma-associated herpesvirus (KSHV) impairs expression of NET-stimulating cellular IL-8 (63).
Some bacteria, such as streptococci, express DNase to degrade NETs (64–66). Herpesviruses also encode proteins that have DNAse activity. These viral molecules process and package the replicated viral genome into the capsid (67). If released from virus-infected cells, they could degrade NETs, thereby remobilizing NET-entrapped virions.
Taken together, virus-induced NETs help to control viral dissemination by several direct and indirect mechanisms, whereas at the same time viral evasion mechanisms target NET formation to minimize the antiviral NET effect and immunopathology.
Role of NETs in Viral Pathogenesis
As for all effective immune responses against pathogens, NETosis may also result in immunopathology. Unbalanced NET formation is associated with pathological conditions such as respiratory distress, autoimmune disease, and thrombosis (68). NETs are directly cytotoxic to epithelial and endothelial cells (69, 70) as well as hepatocytes (71). They contain several components such as histones that are antimicrobial but at the same time can cause tissue damage and other pathological abnormalities including thrombosis (72). Moreover, NETs can occlude secretory ducts or small airways, thereby driving inflammation (73, 74). Other components of NETs such as HMGB1 may also play a detrimental role in virus-associated disease (75).
There is evidence supporting the concept that local NET deposits contribute to viral immunopathology. NETs have been detected in bronchoalveolar lavage fluid from children with severe RSV infection of the lower respiratory tract (76). Dense plugs occluding the small airways in RSV-infected calves contain NETs (76). Moreover, in a mouse model of influenza pneumonia, NET formation was observed in areas of alveolar-capillary damage in the lung (16). On the other hand, mice deficient in peptidylarginine deiminase 4 (PAD4) were as efficient in controlling influenza virus and showed similar survival as wild-type mice (77). This result suggests that NETs do not play an important role in individual antiviral immunity and virus-induced pathology because PAD4 deiminates histone H3 and H4 and is required for NET formation. The different outcomes of these studies may be due to different virus and mouse strains used. In line with this view, neutrophils from different mouse strains undergo NETosis with different efficiency (78). Furthermore, the influence of NETs on viral dissemination was not addressed in these studies. If virus-induced NET deposits represent an important pathogenic factor treatments that alleviate NET-induced pathological manifestations such as DNase should ease symptoms of virus-associated disease (79). Clinical or radiological improvement after DNase treatment of infants with virus-associated bronchiolitis was observed in some clinical trials (80, 81) but not in others (82). Thus, further studies have to elucidate the precise pathogenic role of virus-induced NET deposits in the lung and explore the efficiency of anti-NET treatment.
NETs start to circulate in detectable amounts in the serum if the NET degradation and clearance machinery of the host is overwhelmed. This systemic NET overflow has severe direct and indirect adverse effects. First, NETs can damage directly endothelial cells lining the interior face of the blood vessels cells (69, 70). Second, NET overflow drives autodestructive processes as components of NETs act as neo self-antigens and induce autoantibodies. In fact, a number of molecules that have been identified as important targets in autoimmune diseases (e.g., dsDNA, histones, MPO, vimentin, and enolase) are actually NET components. Accordingly, NETs have been connected to systemic pathology associated with disease entities such as small vessel-vasculitis, systemic lupus erythematosus (SLE), disseminated intravascular coagulation, rheumatoid arthritis, and preeclampsia (83, 84).
Systemic NET overflow may result from clearance deficiency or increased NET production. For example, sera from a subpopulation of SLE patients show decreased DNase I activity and NET degradation (85, 86). Another enzyme that could prevent systemic NET overflow is DNASE1L3. It is released by DCs and macrophages and digests microparticle-associated chromatin, thereby preventing SLE (87). In those individuals who are deficient in NET-degrading enzymes even viruses with a relatively weak NETs-stimulatory capacity could drive NET-associated systemic pathology (Figure 2). NET formation represents a plausible link between viruses and systemic autoimmune disease. Supporting this idea, viral infections are associated with transient autoantibody production and are known to mimic SLE, induce SLE onset, or trigger lupus flares (88–90).
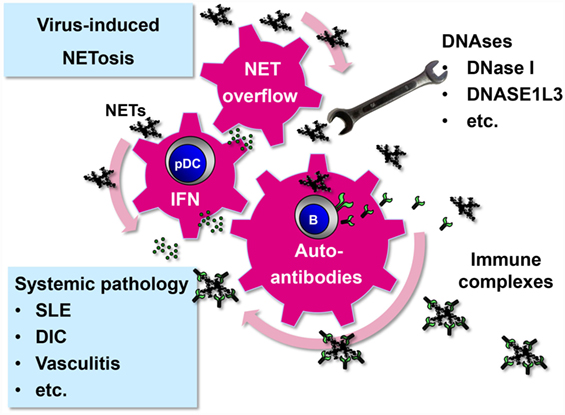
Figure 2. Systemic pathology driven by virus-induced NET formation. Virus-induced NETs may start to circulate and become systemic under certain circumstances. First, systemic infection with viruses that have a strong NET-stimulatory capacity, such as hantaviruses, may overwhelm intact NET-degrading function of DNAses (20). Second, persistent viruses with low NET-inducing capacity, such as herpesviruses, may produce systemic NET excess if DNAse activity is compromised. As a result of NET overflow, self-reactive memory B cells are stimulated to release autoantibodies after binding and internalizing NET components through their B cell receptor (91). NETs are enriched in oxidized mitochondrial DNA inducing a strong inflammatory response (52). NETs stimulate pDCs to release type I IFN that adds momentum to the vicious cycle by further activating and expanding autoreactive B cells (48–50). Immune complexes are formed which not only cause systemic pathology as observed in several disease entities such as SLE but also promote the autoimmune process by driving a positive feedback loop.
Transient systemic NET overflow due to increased NET formation without noticeable deficiency in DNase activity can occur during infection with hantaviruses (20) (Figure 2). Neutrophils play an antiviral role during VHF caused by hantaviruses (92–94). These zoonotic pathogens belong to the family Bunyaviridae and infect humans after transmission via inhalation of aerosolized urine, saliva, and feces from chronically infected rodents, their natural hosts. In humans, they can induce severe pulmonary and renal dysfunction as well as intravascular coagulation and hemorrhagic shock (95). Hantaviruses replicate in endothelial cells, their main target cells, without causing programed cell death in vitro. This suggests that immunopathological mechanisms such as those driven by NETs contribute to Hantavirus-associated pathogenesis (94, 96). In hantavirus-infected patients, high levels of circulating NETs are detected (20). In accordance, increased amounts of cell-free DNA (97) and histones (98) are found in the circulation of hantavirus-infected individuals. The cytotoxic effects of NETs may significantly contribute to hantavirus-associated pathology. In line with this view, histones have been shown to increase thrombin generation and intravascular coagulation (99, 100). They also upregulate the permeability of the endothelial barrier (101). Finally, NETs can induce the formation of autoantibodies that may contribute to the systemic pathology observed during hantavirus-associated disease (20).
Another form of VHF is caused by DENV. DENV is transmitted between humans by Aedes mosquitoes and poses a threat to roughly two billion people (102). There is no evidence as yet for a strong direct NET-stimulatory effect of DENV particles in vitro (61). Nevertheless, in vivo DENV-infected cells could stimulate NETosis indirectly by secreting the viral non-structural protein 1 (NS1). NS1 activates uninfected cells including endothelial cells via TLR4 (103, 104). Subsequently, activated endothelial cells could drive neutrophils into NETosis (69, 79). Moreover, NS1 could activate platelets via TLR4 which in turn stimulate neutrophils to undergo NETosis (105). Finally, IL-8 is produced by human endothelial cells in response to DENV (29) and is known to drive NETosis (3). In accordance, high levels of IL-8 and elastase, a key component of NETs, are found in DENV patients and correlate with disease severity (106).
These pathological effects explain why NET formation as part of an antiviral defense strategy is a double-edged sword. The host may benefit from NETs deposited precisely in the area of infection, thereby immobilizing or even neutralizing virus and killing virus-infected cells. This benefit may turn into disaster if NET formation is too widespread creating NET deposits in healthy tissue. As a consequence, too many uninfected host cells in the neighborhood of the infected areal may come under “friendly fire” resulting in considerable collateral tissue damage. Local NET-associated pathology may become systemic, if the NET degradation machinery (DNase activity) is impaired, or if the viral NET-stimulatory capacity is too strong. Such an unbalanced NET formation results in NET overflow. Under this condition, autoimmune phenomena are triggered that could result in systemic pathology (Figure 2).
Concluding Remarks
It is now evident that most pathogens, including viruses, can stimulate neutrophils to undergo NETosis. Although much smaller than bacteria, fungi, or parasites, viral particles do not seem to slip through NETs but rather become immobilized. Whether these viral particles are inactivated as well is a moot point, as long as they are ensnared by NETs, they represent no threat. However, an increasing number of studies indicate that a disproportionate virus-induced NET release can contribute to damage, locally as well as systemically. It will be important to explore the mechanisms that control NET formation in the context of viral infections. On the basis of this knowledge, it could be possible to prevent NET-assisted control of viruses becoming a Pyrrhic victory.
Author Contributions
Both authors contributed to the conception, writing, and editing of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (Support code SCHO/9-1).
References
1. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe (2014) 15:526–36. doi:10.1016/j.chom.2014.04.011
2. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol (1996) 59:229–40.
3. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi:10.1126/science.1092385
4. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi:10.1083/jcb.200606027
5. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15:1017–25. doi:10.1038/ni.2987
6. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A (2015) 112:2817–22. doi:10.1073/pnas.1414055112
7. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185:7413–25. doi:10.4049/jimmunol.1000675
8. Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol (2012) 92:841–9. doi:10.1189/jlb.1211601
9. Yipp BG, Kubes P. NETosis: how vital is it? Blood (2013) 122:2784–94. doi:10.1182/blood-2013-04-457671
10. Desai J, Mulay SR, Nakazawa D, Anders HJ. Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell Mol Life Sci (2016) 73:2211–9. doi:10.1007/s00018-016-2195-0
11. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi:10.1038/nm1565
12. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ (2009) 16:1438–44. doi:10.1038/cdd.2009.96
13. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18:1386–93. doi:10.1038/nm.2847
14. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol (2013) 190:4136–48. doi:10.4049/jimmunol.1202671
15. Wardini AB, Guimaraes-Costa AB, Nascimento MT, Nadaes NR, Danelli MG, Mazur C, et al. Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol (2010) 91:259–64. doi:10.1099/vir.0.014613-0
16. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol (2011) 179:199–210. doi:10.1016/j.ajpath.2011.03.013
17. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe (2012) 12:109–16. doi:10.1016/j.chom.2012.05.015
18. Galani IE, Andreakos E. Neutrophils in viral infections: current concepts and caveats. J Leukoc Biol (2015) 98:557–64. doi:10.1189/jlb.4VMR1114-555R
19. Jenne CN, Kubes P. Virus-induced NETs – critical component of host defense or pathogenic mediator? PLoS Pathog (2015) 11:e1004546. doi:10.1371/journal.ppat.1004546
20. Raftery MJ, Lalwani P, Krautkrämer E, Peters T, Scharffetter-Kochanek K, Kruger R, et al. beta2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J Exp Med (2014) 211:1485–97. doi:10.1084/jem.20131092
21. Orenstein JM. In vivo cytolysis and fusion of human immunodeficiency virus type 1-infected lymphocytes in lymphoid tissue. J Infect Dis (2000) 182:338–42. doi:10.1086/315640
22. Gerna G, Baldanti F, Revello MG. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum Immunol (2004) 65:381–6. doi:10.1016/j.humimm.2004.02.009
23. Halfhide CP, Flanagan BF, Brearey SP, Hunt JA, Fonceca AM, McNamara PS, et al. Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J Infect Dis (2011) 204:451–8. doi:10.1093/infdis/jir280
24. Funchal GA, Jaeger N, Czepielewski RS, Machado MS, Muraro SP, Stein RT, et al. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS One (2015) 10:e0124082. doi:10.1371/journal.pone.0124082
25. Tripathi S, Verma A, Kim EJ, White MR, Hartshorn KL. LL-37 modulates human neutrophil responses to influenza A virus. J Leukoc Biol (2014) 96:931–8. doi:10.1189/jlb.4A1113-604RR
26. Narayana Moorthy A, Narasaraju T, Rai P, Perumalsamy R, Tan KB, Wang S, et al. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol (2013) 4:56. doi:10.3389/fimmu.2013.00056
27. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5:178ra140. doi:10.1126/scitranslmed.3005580
28. Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest (1995) 96:549–57. doi:10.1172/JCI118067
29. Huang YH, Lei HY, Liu HS, Lin YS, Liu CC, Yeh TM. Dengue virus infects human endothelial cells and induces IL-6 and IL-8 production. Am J Trop Med Hyg (2000) 63:71–5.
30. Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem (2004) 279:44123–32. doi:10.1074/jbc.M405883200
31. Assinger A. Platelets and infection – an emerging role of platelets in viral infection. Front Immunol (2014) 5:649. doi:10.3389/fimmu.2014.00649
32. Koupenova M, Vitseva O, Mackay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood (2014) 124:791–802. doi:10.1182/blood-2013-11-536003
33. Negrotto S, Jaquenod De Giusti C, Rivadeneyra L, Ure AE, Mena HA, Schattner M, et al. Platelets interact with coxsackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. J Thromb Haemost (2015) 13:271–82. doi:10.1111/jth.12782
34. Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe (2013) 13:169–80. doi:10.1016/j.chom.2013.01.005
35. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122:2661–71. doi:10.1172/JCI61303
36. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi:10.1016/j.chom.2012.06.011
37. Zapata JC, Cox D, Salvato MS. The role of platelets in the pathogenesis of viral hemorrhagic fevers. PLoS Negl Trop Dis (2014) 8:e2858. doi:10.1371/journal.pntd.0002858
38. Connolly-Andersen AM, Sundberg E, Ahlm C, Hultdin J, Baudin M, Larsson J, et al. Increased thrombopoiesis and platelet activation in hantavirus-infected patients. J Infect Dis (2015) 212:1061–9. doi:10.1093/infdis/jiv161
39. Rasche FM, Uhel B, Kruger DH, Karges W, Czock D, Hampl W, et al. Thrombocytopenia and acute renal failure in Puumala hantavirus infections. Emerg Infect Dis (2004) 10:1420–5. doi:10.3201/eid1008.031069
40. Krautkramer E, Zeier M, Plyusnin A. Hantavirus infection: an emerging infectious disease causing acute renal failure. Kidney Int (2013) 83:23–7. doi:10.1038/ki.2012.360
41. Latus J, Schwab M, Tacconelli E, Pieper FM, Wegener D, Rettenmaier B, et al. Acute kidney injury and tools for risk-stratification in 456 patients with hantavirus-induced nephropathia epidemica. Nephrol Dial Transplant (2015) 30:245–51. doi:10.1093/ndt/gfu319
42. Hoeksema M, Tripathi S, White M, Qi L, Taubenberger J, Van Eijk M, et al. Arginine-rich histones have strong antiviral activity for influenza A viruses. Innate Immun (2015) 21:736–45. doi:10.1177/1753425915593794
43. Kozlowski HN, Lai ET, Havugimana PC, White C, Emili A, Sakac D, et al. Extracellular histones identified in crocodile blood inhibit in vitro HIV-1 infection. AIDS (2016) 30:2043–52. doi:10.1097/QAD.0000000000001159
44. Tamura M, Natori K, Kobayashi M, Miyamura T, Takeda N. Inhibition of attachment of virions of Norwalk virus to mammalian cells by soluble histone molecules. Arch Virol (2003) 148:1659–70. doi:10.1007/s00705-003-0143-4
45. Gwyer Findlay E, Currie SM, Davidson DJ. Cationic host defence peptides: potential as antiviral therapeutics. BioDrugs (2013) 27:479–93. doi:10.1007/s40259-013-0039-0
46. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol (2011) 187:2626–31. doi:10.4049/jimmunol.1003930
47. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med (2014) 20:511–7. doi:10.1038/nm.3547
48. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra20. doi:10.1126/scitranslmed.3001201
49. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra19. doi:10.1126/scitranslmed.3001180
50. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187:538–52. doi:10.4049/jimmunol.1100450
51. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15:471–85. doi:10.1038/nri3865
52. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi:10.1038/nm.4027
53. Tillack K, Breiden P, Martin R, Sospedra M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J Immunol (2012) 188:3150–9. doi:10.4049/jimmunol.1103414
54. Chang J, Kunkel SL, Chang CH. Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci U S A (2009) 106:18327–32. doi:10.1073/pnas.0905815106
55. Ng CT, Oldstone MB. IL-10: achieving balance during persistent viral infection. Curr Top Microbiol Immunol (2014) 380:129–44. doi:10.1007/978-3-662-43492-5_6
56. Shen P, Fillatreau S. Suppressive functions of B cells in infectious diseases. Int Immunol (2015) 27:513–9. doi:10.1093/intimm/dxv037
57. Hsu DH, De Waal Malefyt R, Fiorentino DF, Dang MN, Vieira P, De Vries J, et al. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science (1990) 250:830–2. doi:10.1126/science.2173142
58. Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad Sci U S A (2000) 97:1695–700. doi:10.1073/pnas.97.4.1695
59. Raftery M, Muller A, Schonrich G. Herpesvirus homologues of cellular genes. Virus Genes (2000) 21:65–75. doi:10.1023/A:1008184330127
60. Raftery MJ, Wieland D, Gronewald S, Kraus AA, Giese T, Schonrich G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J Immunol (2004) 173:3383–91. doi:10.4049/jimmunol.173.5.3383
61. Moreno-Altamirano MM, Rodriguez-Espinosa O, Rojas-Espinosa O, Pliego-Rivero B, Sanchez-Garcia FJ. Dengue virus serotype-2 interferes with the formation of neutrophil extracellular traps. Intervirology (2015) 58:250–9. doi:10.1159/000440723
62. Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology (2015) 145:213–24. doi:10.1111/imm.12437
63. Li X, Liang D, Lin X, Robertson ES, Lan K. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen reduces interleukin-8 expression in endothelial cells and impairs neutrophil chemotaxis by degrading nuclear p65. J Virol (2011) 85:8606–15. doi:10.1128/JVI.00733-11
64. Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A (2005) 102:1679–84. doi:10.1073/pnas.0406641102
65. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16:401–7. doi:10.1016/j.cub.2006.01.056
66. Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med (2007) 13:981–5. doi:10.1038/nm1612
67. Martinez R, Sarisky RT, Weber PC, Weller SK. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol (1996) 70:2075–85.
68. Sorensen OE, Borregaard N. Neutrophil extracellular traps – the dark side of neutrophils. J Clin Invest (2016) 126:1612–20. doi:10.1172/JCI84538
69. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett (2010) 584:3193–7. doi:10.1016/j.febslet.2010.06.006
70. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7:e32366. doi:10.1371/journal.pone.0032366
71. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62:600–14. doi:10.1002/hep.27841
72. Hoeksema M, Van Eijk M, Haagsman HP, Hartshorn KL. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol (2016) 11:441–53. doi:10.2217/fmb.15.151
73. Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med (2010) 16:1018–23. doi:10.1038/nm.2209
74. Leppkes M, Maueroder C, Hirth S, Nowecki S, Gunther C, Billmeier U, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun (2016) 7:10973. doi:10.1038/ncomms10973
75. Wang H, Ward MF, Fan XG, Sama AE, Li W. Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol (2006) 19:3–9. doi:10.1089/vim.2006.19.3
76. Cortjens B, De Boer OJ, De Jong R, Antonis AF, Sabogal Pineros YS, Lutter R, et al. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J Pathol (2016) 238:401–11. doi:10.1002/path.4660
77. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One (2011) 6:e22043. doi:10.1371/journal.pone.0022043
78. Ermert D, Urban CF, Laube B, Goosmann C, Zychlinsky A, Brinkmann V. Mouse neutrophil extracellular traps in microbial infections. J Innate Immun (2009) 1:181–93. doi:10.1159/000205281
79. Saffarzadeh M, Preissner KT. Fighting against the dark side of neutrophil extracellular traps in disease: manoeuvres for host protection. Curr Opin Hematol (2013) 20:3–9. doi:10.1097/MOH.0b013e32835a0025
80. Merkus PJ, De Hoog M, Van Gent R, De Jongste JC. DNase treatment for atelectasis in infants with severe respiratory syncytial virus bronchiolitis. Eur Respir J (2001) 18:734–7.
81. Nasr SZ, Strouse PJ, Soskolne E, Maxvold NJ, Garver KA, Rubin BK, et al. Efficacy of recombinant human deoxyribonuclease I in the hospital management of respiratory syncytial virus bronchiolitis. Chest (2001) 120:203–8. doi:10.1378/chest.120.1.203
82. Enriquez A, Chu IW, Mellis C, Lin WY. Nebulised deoxyribonuclease for viral bronchiolitis in children younger than 24 months. Cochrane Database Syst Rev (2012) 11:CD008395. doi:10.1002/14651858.CD008395.pub2
83. Hahn S, Giaglis S, Hoesli I, Hasler P. Neutrophil NETs in reproduction: from infertility to preeclampsia and the possibility of fetal loss. Front Immunol (2012) 3:362. doi:10.3389/fimmu.2012.00362
84. Grayson PC, Kaplan MJ. At the bench: neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol (2016) 99:253–64. doi:10.1189/jlb.5BT0615-247R
85. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107:9813–8. doi:10.1073/pnas.0909927107
86. Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188:3522–31. doi:10.4049/jimmunol.1102404
87. Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell (2016) 166:88–101. doi:10.1016/j.cell.2016.05.034
88. Hansen KE, Arnason J, Bridges AJ. Autoantibodies and common viral illnesses. Semin Arthritis Rheum (1998) 27:263–71. doi:10.1016/S0049-0172(98)80047-4
89. Ramos-Casals M. Viruses and lupus: the viral hypothesis. Lupus (2008) 17:163–5. doi:10.1177/0961203307086268
90. Ramos-Casals M, Cuadrado MJ, Alba P, Sanna G, Brito-Zeron P, Bertolaccini L, et al. Acute viral infections in patients with systemic lupus erythematosus: description of 23 cases and review of the literature. Medicine (Baltimore) (2008) 87:311–8. doi:10.1097/MD.0b013e31818ec711
91. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature (2002) 416:603–7. doi:10.1038/416603a
92. Koma T, Yoshimatsu K, Nagata N, Sato Y, Shimizu K, Yasuda SP, et al. Neutrophil depletion suppresses pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by hantavirus infection in C.B-17 SCID mice. J Virol (2014) 88:7178–88. doi:10.1128/JVI.00254-14
93. Kobak L, Raftery MJ, Voigt S, Kuhl AA, Kilic E, Kurth A, et al. Hantavirus-induced pathogenesis in mice with a humanized immune system. J Gen Virol (2015) 96:1258–63. doi:10.1099/vir.0.000087
94. Schonrich G, Kruger DH, Raftery MJ. Hantavirus-induced disruption of the endothelial barrier: neutrophils are on the payroll. Front Microbiol (2015) 6:222. doi:10.3389/fmicb.2015.00222
95. Kruger DH, Schonrich G, Klempa B. Human pathogenic hantaviruses and prevention of infection. Hum Vaccin (2011) 7:685–93. doi:10.4161/hv.7.6.15197
96. Lalwani P, Raftery MJ, Kobak L, Rang A, Giese T, Matthaei M, et al. Hantaviral mechanisms driving HLA class I antigen presentation require both RIG-I and TRIF. Eur J Immunol (2013) 43:2566–76. doi:10.1002/eji.201243066
97. Outinen TK, Kuparinen T, Jylhava J, Leppanen S, Mustonen J, Makela S, et al. Plasma cell-free DNA levels are elevated in acute Puumala hantavirus infection. PLoS One (2012) 7:e31455. doi:10.1371/journal.pone.0031455
98. Vaheri A, Strandin T, Jaaskelainen AJ, Vapalahti O, Jarva H, Lokki ML, et al. Pathophysiology of a severe case of Puumala hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Antiviral Res (2014) 111:23–5. doi:10.1016/j.antiviral.2014.08.007
99. Massberg S, Grahl L, Von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16:887–96. doi:10.1038/nm.2184
100. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi:10.1182/blood-2011-03-343061
101. Gillrie MR, Lee K, Gowda DC, Davis SP, Monestier M, Cui L, et al. Plasmodium falciparum histones induce endothelial proinflammatory response and barrier dysfunction. Am J Pathol (2012) 180:1028–39. doi:10.1016/j.ajpath.2011.11.037
102. Simmons CP, Farrar JJ, Nguyen VV, Wills B. Dengue. N Engl J Med (2012) 366:1423–32. doi:10.1056/NEJMra1110265
103. Beatty PR, Puerta-Guardo H, Killingbeck SS, Glasner DR, Hopkins K, Harris E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci Transl Med (2015) 7:304ra141. doi:10.1126/scitranslmed.aaa3787
104. Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, et al. Dengue virus NS1 protein activates cells via toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med (2015) 7:304ra142. doi:10.1126/scitranslmed.aaa3863
105. Jenne CN, Kubes P. Platelets in inflammation and infection. Platelets (2015) 26:286–92. doi:10.3109/09537104.2015.1010441
Keywords: neutrophil extracellular traps, immunopathogenesis, neutrophils, viruses, viral immune evasion
Citation: Schönrich G and Raftery MJ (2016) Neutrophil Extracellular Traps Go Viral. Front. Immunol. 7:366. doi: 10.3389/fimmu.2016.00366
Received: 22 July 2016; Accepted: 05 September 2016;
Published: 19 September 2016
Edited by:
Marko Radic, University of Tennessee, USAReviewed by:
Winfried Barchet, University of Bonn, GermanyShahram Salek-Ardakani, University of Florida, USA
Paul Hasler, Kantonsspital Aarau, Switzerland
Copyright: © 2016 Schönrich and Raftery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günther Schönrich, Z3VlbnRoZXIuc2Nob2VucmljaEBjaGFyaXRlLmRl