 Diego Sánchez-Martínez1
Diego Sánchez-Martínez1 Pilar M. Lanuza1Natalia Gómez2
Pilar M. Lanuza1Natalia Gómez2 Aura Muntasell3
Aura Muntasell3 Elisa Cisneros2
Elisa Cisneros2 Manuela Moraru2Gemma Azaceta4
Manuela Moraru2Gemma Azaceta4 Alberto Anel5
Alberto Anel5 Luis Martínez-Lostao4,6Martin Villalba7,8Luis Palomera4
Luis Martínez-Lostao4,6Martin Villalba7,8Luis Palomera4 Carlos Vilches3José A. García Marco9
Carlos Vilches3José A. García Marco9 Julián Pardo1,6,10,11*
Julián Pardo1,6,10,11*
- 1Biomedical Research Center of Aragón (CIBA), Aragón Health Research Institute (IIS Aragón), University of Zaragoza, Zaragoza, Spain
- 2Immunogenetics and HLA, Instituto de Investigación Sanitaria Puerta de Hierro, Majadahonda, Spain
- 3Immunity and infection Lab, IMIM (Hospital del Mar Medical Research Institute), Barcelona, Spain
- 4Hospital Clínico Universitario Lozano Blesa, Instituto Aragonés de Ciencias de la Salud (IACS)/Aragón Health Research Institute (IIS Aragón), Zaragoza, Spain
- 5Department of Biochemistry and Molecular and Cellular Biology, Aragón Health Research Institute (IIS Aragón), University of Zaragoza, Zaragoza, Spain
- 6Nanoscience Institute of Aragon (INA), University of Zaragoza, Zaragoza, Spain
- 7INSERM U1183, Université de Montpellier 1, UFR Médecine, Montpellier, France
- 8Institute for Regenerative Medicine and Biotherapy (IRMB), CHU Montpellier, Montpellier, France
- 9Unidad de Citogenética Molecular/Servicio de Hematología, Hospital Universitario Puerta de Hierro-Majadahonda, Madrid, Spain
- 10Aragón I+D Foundation (ARAID), Government of Aragon, Zaragoza, Spain
- 11Department of Microbiology, Preventive Medicine and Public Health, University of Zaragoza, Zaragoza, Spain
Mutational status of TP53 together with expression of wild-type (wt) IGHV represents the most widely accepted biomarkers, establishing a very poor prognosis in B-cell chronic lymphocytic leukemia (B-CLL) patients. Adoptive cell therapy using allogeneic HLA-mismatched Natural killer (NK) cells has emerged as an effective and safe alternative in the treatment of acute myeloid and lymphoid leukemias that do not respond to traditional therapies. We have described that allogeneic activated NK cells eliminate hematological cancer cell lines with multidrug resistance acquired by mutations in the apoptotic machinery. This effect depends on the activation protocol, being B-lymphoblastoid cell lines (LCLs) the most effective stimulus to activate NK cells. Here, we have further analyzed the molecular determinants involved in allogeneic NK cell recognition and elimination of B-CLL cells, including the expression of ligands of the main NK cell-activating receptors (NKG2D and NCRs) and HLA mismatch. We present preliminary data suggesting that B-CLL susceptibility significantly correlates with HLA mismatch between NK cell donor and B-CLL patient. Moreover, we show that the sensitivity of B-CLL cells to NK cells depends on the prognosis based on TP53 and IGHV mutational status. Cells from patients with worse prognosis (mutated TP53 and wt IGHV) are the most susceptible to activated NK cells. Hence, B-CLL prognosis may predict the efficacy of allogenic activated NK cells, and, thus, NK cell transfer represents a good alternative to treat poor prognosis B-CLL patients who present a very short life expectancy due to lack of effective treatments.
Introduction
B-cell chronic lymphocytic leukemia (B-CLL), a heterogeneous disease with variable clinic presentation and evolution, is the most common leukemia in adults in Western World (1, 2). It is characterized by the accumulation of CD5+ B cells in peripheral blood and lymphoid organs (3). This leukemia is usually treated with chemotherapy and anti-CD20 antibodies (i.e., Rituximab) (4), but remains incurable largely due to development of refractory disease. This is frequently associated with the expression of molecular markers that confer bad prognosis (2).
Several prognostic and predictive markers have been described for B-CLL, including cytogenetic abnormalities like 17p and 11q deletions, associated in some instances with rapid clinical progression, chemotherapy resistance, and inferior survival (5). Expression of membrane-proteins such as CD38 (6) and CD49d (7) or intracellular ZAP70 (8) have also been described as adverse prognostic factors. Among them, a recent study indicates that CD49d expression is the best immunophenotypic predictor of the overall patient survival (9). Some single mutations like NOTCH1 (10, 11), the splicing factor 3b (SF3B1) (12, 13), BIRC3 (14), exportin 1 (XPO1), MYD88, and KLHL6 (15) have also been related to poor prognosis, although their utility as prognostic markers is still under clinical evaluation. Among all of them, TP53 mutation/deletion and expression of unmutated IGHV are widely accepted as indicators of poor prognosis at the time of diagnosis (16–19).
Unmutated IGHV is associated with higher aggressiveness of B-CLL cells since proliferating signals through B cell receptor are unaffected. In contrast, mutated IGHV produces unresponsive B cell receptors. TP53 is a tumor suppressor that plays a key role in DNA repair as well as apoptosis trigger in response to DNA damage. Thus, inactivation of TP53 favors malignant cell transformation and confers resistance to chemo and radiotherapy (20).
Natural killer (NK) cells belong to the innate immune system and were originally identified as lymphocytes capable of killing cells that have downregulated MHC-I expression due to pathogen infection or transformation (21–26). They constitute a heterogeneous cell population with distinct phenotypic and functional characteristics, including, but not limited to, their ability to mediate cytolytic activity (27, 28). NK cell activity is regulated by the equilibrium between signals transduced by inhibitory and activating receptors, which dictates target cell elimination and pro-inflammatory cytokine production (29, 30). The main inhibitory receptors, NKG2A killer-cell immunoglobulin-like receptors (KIRs) family, bind to MHC-I molecules on target cells. The main activating receptors, NKG2D and NCRs (NKp30, NKp44, and NKp46) recognize stress ligands on target cells (31, 32). The balance between inhibitory and activating signals dictates if NK cells will recognize and destroy target cells.
During allogeneic hematopoietic stem cell transplantation, in a context of KIR–MHC mismatch, HLA alleles expressed on target cells may not inhibit NK cells. Accordingly, allogeneic NK cells have been proposed to kill hematological cancer cells and improve prognosis, mainly in the context of mismatched hematopoietic stem cell transplantation (33–37). Clinical protocols based on these concepts have been designed to treat some hematological malignancies, including lymphoma, acute myeloid and lymphoid leukemia, and multiple myeloma (34, 37–42). Regarding B-CLL, at present, it is unclear whether KIR–HLA mismatch may also regulate B-CLL allogeneic NK cell recognition. NK cells activated with high concentrations of IL-2, known as lymphokine-activated killer (LAK) cells, were shown to kill B-CLL cells (43–45). In contrast, other authors reported that autologous and allogeneic LAK cells were unable to kill B-CLL cells (46–48). More recently, it was shown that unstimulated NK cells did not kill B-CLL cells, but cytotoxicity was recovered using IL-15-activated NK cells in combination with rituximab (49). Clinical trials based on autologous NK cells have not shown benefits (50).
We have previously shown that the selection of a proper activating stimulus is critical to generate activated NK cells able to kill chemoresistant hematological cancer cell lines as well as cells from B-CLL patients (51, 52). Allogeneic NK cells activated in the presence of EBV-transformed B-cell lymphoblastoid cell lines (LCL) presented significantly higher cytotoxicity than those generated with K562 cells and IL-2/IL-15. This activation protocol has been now employed to (i) analyze the molecular determinants that drive allogeneic NK cell recognition of B-CLL cells and (ii) to test the susceptibility of adverse prognosis B-CLL cells, defined according to IGHV mutational status and TP53 deletion/mutation, to allogeneic activated NK cells.
Materials and Methods
Isolation and Activation of Human NK Cells
Human ex vivo NK cells were enriched by using anti-CD56 MicroBeads with a MultiStand MACS (MACS, Miltenyi Biotec) from freshly isolated peripheral blood mononuclear cells (PBMCs).
Activation of human primary NK cells was pursued by culturing PBMCs for 5 days with Mitomycin C-treated R69- or 721.221-LCL at 10:1 PBMC:stimulator ratio. Subsequently, NK cells were enriched using anti-CD56 MicroBeads with a MultiStand MACS (MACS, Miltenyi Biotec).
PBMCs were obtained from blood from healthy donors by Ficoll gradient centrifugation (Blood and Tissue Bank of Aragon; approved by the CEICA, number: C.I.PI11/006). NK cell purity (CD56+/CD3−) was higher than 90% in all cases. Contamination with CD8+ CD3+ cells was less than 2%.
B-CLL Patients
Blood samples from patients with B-CLL were obtained from Hospital Clinico Lozano Blesa (Zaragoza) and Hospital Puerta de Hierro-Majadahonda (Madrid). They were processed by Ficoll gradient centrifugation to obtain PBMCs and stored frozen in liquid nitrogen until their use. In all cases, the percentage of CD19+CD5+ cells was higher than 80% and no differences were observed between frozen and fresh B-CLL cells regarding their susceptibility to NK cells. This study was approved by Ethics Committee for Clinical Research of Aragon (CEICA), number: C.I.PI11/006; and by Ethics Committee for Clinical Research of Hospital Puerta de Hierro-Majadahonda, number: PI31_13.
Analyses of TP53 and IGHV Mutational Status
IGHV gene mutational status was analyzed and classified according to ERIC recommendations (53). TP53 genetic abnormalities were detected by conventional cytogenetics, FISH, and Sanger sequencing analysis.
NK Cell-Mediated Cytotoxicity
NK cells were labeled with 1 μM of CellTracker™ Green CMFDA (Life Technologies) and incubated with target cells at 9:1 e:t ratio for 4 h. Subsequently, phosphatidylserine (PS) translocation and membrane damage were analyzed in the green or violet fluorescence negative target cell population by flow cytometry using annexin V and 7AAD as previously described (51).
Analyses of HLA Genotype and Prediction of KIR–HLA Mismatch
Killer-cell immunoglobulin-like receptor ligands were deduced from the cell HLA types, determined by standard methods approved by the European Federation for Immunogenetics, as in Ref. (54) or directly assessed by PCR with sequence-specific primers targeted to the critical polymorphic positions (oligonucleotide sequences and PCR conditions available upon request). Mismatch was defined, according to the missing-self model, as the absence in the target cell of a KIR ligand present in the effector cells.
Flow Cytometry
The antibodies against HLA-E-PE (clone 3D12), HLA-ABC/E/F/G-FITC (clone W6/32), DR4-PE (clone DJR1), and DR5-PE (clone MD5-1) were from eBioscience. The antibody against ICAM-1-APC (clone HA58) was from BD. The antiCD19-APC (clone LT19) and antiCD5-FITC (clone UCHT2) were from Miltenyi Biotec. The NKR-Fc quimeras were from R&D Systems (NKG2D-Fc) or were provided by Miguel López-Botet (NKp30-Fc and NKp46-Fc). The anti-human IgG secondary antibody labeled with PE was from Jackson Immunoresearch. All antibodies were diluted in PBS with 5% FCS and 0.1% sodium azide for cell staining. To stain NKR ligands using NKR-Fc quimeras, cells were blocked with rabbit serum before staining with the respective chimeras.
Statistical Analyses
Statistical analyses were performed using the GraphPad Prism v 4.0 software by one-way analysis of variance (ANOVA) as indicated in the figure/table legends. Regression-tree analyses (Decision tree learning) were used to classify B-CLL samples according to its susceptibility to NK cells. This is a predictive model that maps the values of cell death (percentage) to conclusions about categorical predictor variables.
Results
NK Cells Require Activation to Kill CLL Cells
As mentioned above, it is unclear whether NK cells activated with exogenous cytokines may kill B-CLL cells. Our group has previously described that NK cells activated with EBV-transformed R69 or 721.211 LCLs are able to eliminate cells from B-CLL patients without requiring supplementation with exogenous cytokines (51). Importantly, activated NK cells did not kill healthy non-transformed PBMCs. Here, we confirmed that the cytotoxic potential of NK cells activated with R69 LCLs was increased in comparison with freshly isolated NK cells from the same donors. As shown in Figure 1A, freshly isolated allogeneic NK cells from healthy donors did not kill B-CLL cells. In contrast, activated NK cells presented a significantly increased cytotoxic potential against B-CLL cells, even in cells from a patient (CLL2) with chemotherapy and Rituximab resistance (Table 1). Killing of Jurkat cells and R69 LCL (that expresses all known HLA ligands for inhibitory KIRs) was also significantly enhanced after NK cell activation. Cell death was almost completely inhibited by EGTA indicating the granule exocytosis was the main mechanism employed by NK cells to kill B-CLL cells (data not shown). As previously found in NK cells activated with R69 LCLs (51), the NK cell receptors NKG2A, NKp30, NKp44, and DNAM and the cytotoxic protease granzyme B were upregulated in activated NK cells (data not shown).
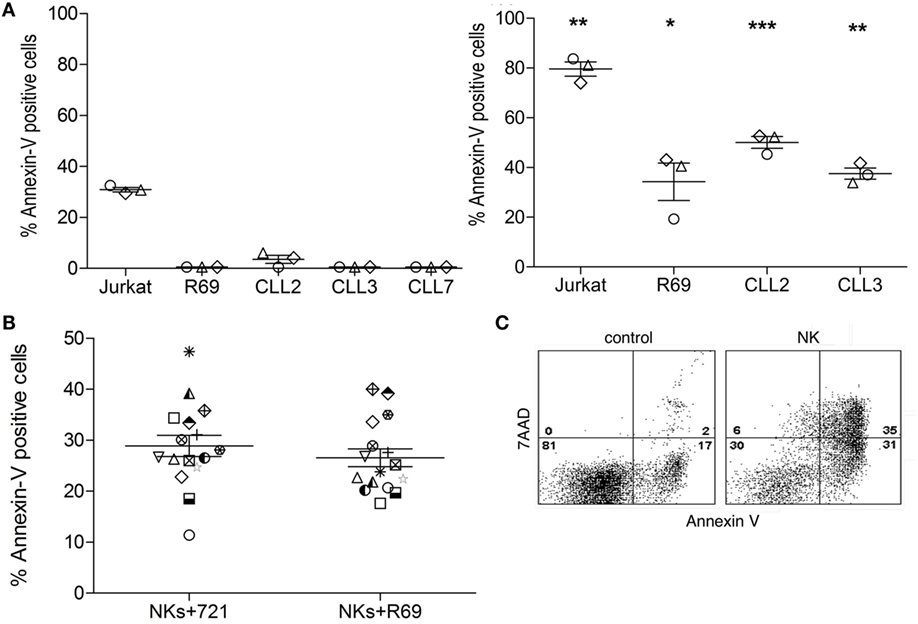
Figure 1. Allogeneic NK cells require activation to kill B-CLL cells. (A) Jurkat and R69 cells or cells from B-CLL patients were incubated with naïve (left) or R69-LCL activated (right) NK cells (MACS enriched, >90% CD56+CD3− cells; CTGreen labeled) for 4 h at 9:1 effector:target ratio as described in Section “Materials and Methods.” Subsequently, PS traslocation (annexin V DY634) and membrane permeabilization (7AAD uptake) were analyzed by flow cytometry in the cell population negative for CTGreen. (B) Cells from B-CLL were incubated with R69- or 721-LCL-activated NK cells (MACS enriched, >90% CD56+CD3− cells; CTGreen labeled) for 4 h at 9:1 effector:target ratio as described in Section “Materials and Methods.” Subsequently, PS traslocation (annexin V DY634) and membrane permeabilization (7AAD uptake) were analyzed by flow cytometry in the cell population negative for CTGreen. Data in the graphics are represented as the mean ± SEM from three independent NK cell donors (A) or from 14 independent B-CLL patients (B). Annexin V+ cells represent the % of AnnexinV+7AAD− plus AnnexinV+7AAD+ cells. Statistical analysis was performed by comparing the means of naive versus activated NK cells within each group using 2-way ANOVA with Tukey HSD post hoc test; ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001. (C) Representative dot plot of Annexin V/7AAD staining in a B-CLL sample incubated in medium alone (control) or together with NK cells (NK). Numbers correspond to the % of cells in each quadrant.
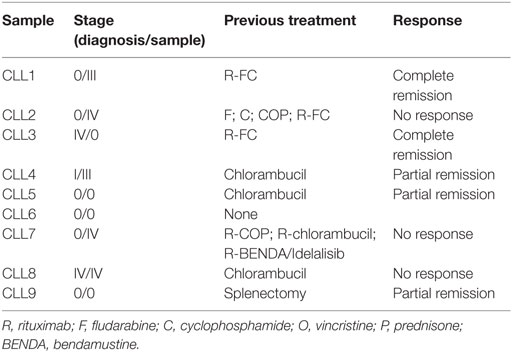
Table 1. Characteristics of CLL patients used in the study of compatibility.
Next, we analyzed whether HLA-I expression in the LCLs used to activate NK cells might influence their anti-leukemic potential. To this aim we compared the cytotoxic activity of NK cells from 4 independent donors, activated either with HLA-I+ R69 LCLs or with HLA-I− 721.221 cells, against B-CLL cells from four different patients. As shown in Figure 1, although with some individual variations, the cytotoxic potential of activated NK cell did not depend on HLA-I expression in stimulating LCLs (Figure 1B). Thus, we employed NK cells isolated from PBMC cultures activated for 5 days with R69 LCLs in the next experiments.
Expression of NK Cell Ligands by B-CLL Cells
Our data confirm that LCL-activated NK cells are able to kill B-CLL cells. However, we have previously found that the cytotoxic potential of allogeneic NK cells against B-CLL cells varies depending on the NK cell donor/B-CLL patient pair (51).
Thus, experiments were set up to address the basis for the variable susceptibility of B-CLL cells to activated NK cells. Seven NK cell donors and six B-CLL patients were selected, and the level of cell death was evaluated in all combinations (42). Four and three different NK cells donors were blindly incubated with the six B-CLL patients on two different days. As shown in Figure 2A, the level of cell death varied depending on the pair of NK cell donors and B-CLL patients incubated in agreement with our previous results (51). Some donors like the one represented with circles presented a similar profile of killing against all CLL samples. However, this was not a general trend since others like the ones represented with triangles, diamonds, or inverted triangles presented a high variability depending on the CLL sample. Indeed, the profile of killing between the different NK cell donors was statistically similar (Supplementary figure). These killing profiles were not dependent on the level of NK cell activation since all of them similarly killed Jurkat cells and expressed comparable levels of granzyme B, NKG2D, and NCRs (NKp30, Nkp44, and Nkp46) (data not shown). Thus, a priori, this variability could be partly explained by (i) the degree of HLA–KIR mismatch between donor and patient as found in AML and ALL or (ii) the intrinsic resistance of cells from some B-CLL patients to activated NK cells.
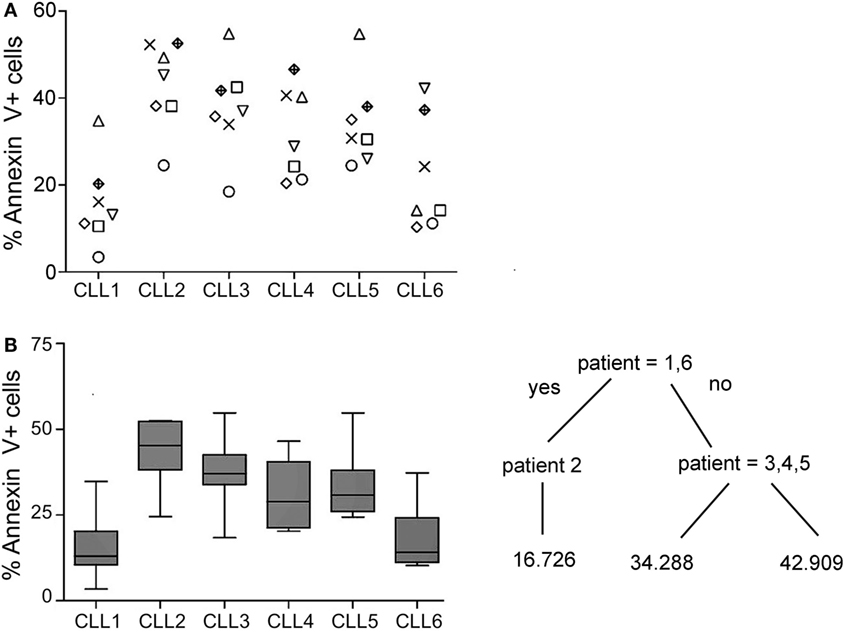
Figure 2. Analyses of the cytotoxic potential of activated allogeneic NK cells employing six CLL patients and seven NK cell donors. Cells from B-CLL patients were incubated with R69-activated NK cells (MACS enriched, >90% CD56+CD3− cells; CTGreen labeled) from seven independent donors for 4 h at 9:1 effector:target ratio in two independent experiments as described in Section “Materials and Methods.” Subsequently, PS traslocation (annexin V DY634) and membrane permeabilization (7AAD uptake) were analyzed by flow cytometry in the cell population negative for CTGreen. (A) The graph represents the % of annexin V positive cells in the 42 combinations (six B-CLL × seven NK). Annexin V+ cells represent the % of Annexin V+7AAD− plus AnnexinV+7AAD+ cells. Each symbol represents a NK cell donor. All NK cell donors were incubated with every B-CLL sample. (B) Cell death (Annexin V+) in every B-CLL sample was represented in a boxplot where median ± SD is indicated. Statistical analysis was performed employing a regression tree in which patient samples are clustered according to their similarity of sensitivity to NK cell cytotoxicity.
As shown in the regression-tree analysis in Figure 2B, some B-CLL cells like CLL1 and CLL6 are more resistant than others to activated NK cells, irrespectively of the NK cell donor, supporting our second hypothesis. Thus, we decided to analyze the reason for the increased resistance of CLL1 and CLL6 to activated NK cells. First, we analyzed whether this could be related to the level of expression of ligands recognized by the NK cell receptors NKG2D, NKp30, NKp46, KIRs, and LFA-1. As shown in Figure 3A, the expression of NKG2D and NKp30 ligands were very low in most CLL samples, and only some CLLs expressed higher levels of NKp46 ligands. Jurkat cells expressed ligands for all these receptors, validating our approach. Next, we analyzed the expression of the adhesion molecule ICAM-1 and of HLA-I using both an antibody (clone W6/32) that recognizes all HLA-I molecules and another one specific for HLA-E. As shown in Figure 3B, all cells expressed HLA-I (ABC/E/F/G and E) and ICAM1, although the level of expression was quite variable. Of note, the level of expression of HLA-ABC and HLA-E in CLL1 and CLL6 was higher in comparison with the other B-CLL samples, suggesting that this expression could be related to the increased resistance of these samples. However, blocking of CD94 in NK cells, a protein required for the inhibitory signaling of the HLA-E ligand NKG2A, by using specific antibodies did not significantly enhance NK cell-mediated cytotoxicity (data not shown) suggesting that HLA-E expression in CLL1 and CLL6 was not the main responsible for NK cell resistance.
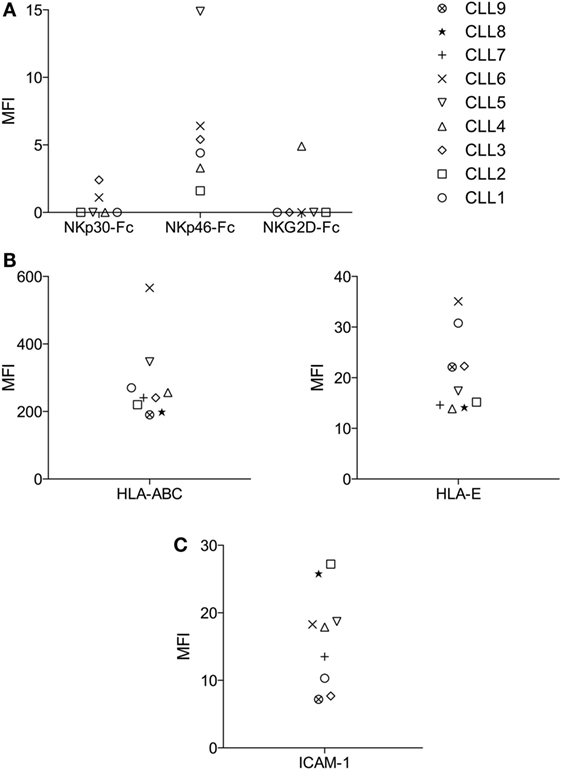
Figure 3. Analyses of the expression of activating and inhibitory ligands of NK cell receptors in B-CLL cells. Expression of NKp30, NKp46, and NKG2D activating ligands using Fc quimeras (A), HLA-ABC and HLA-E inhibitory ligands (B), and the adhesion molecule ICAM-1 (C) using specific antibodies were analyzed in B-CLL cells by flow cytometry as described in Section “Materials and Methods.” The Mean fluorescence intensity (MFI) of every B-CLL sample is represented in the graphs. MFI = (MFI specific Ab) − (MFI isotype control).
The adhesion molecule ICAM-1 was expressed by all B-CLL samples (Figure 3C), and the level of expression did not correlate with NK cell susceptibility. Although we did not test other adhesion molecules involved in NK cell recognition like LFA-3, we have previously shown that blocking ICAM-1/LFA-1 interaction almost completely eliminate NK cell cytotoxicity in hematological cancer cells (including B-CLL) [(55) and data not shown] suggesting a minor role for other adhesion molecules.
KIR–HLA Receptor–Ligand Mismatch
As mentioned above KIR–HLA mismatch has been shown to promote NK cell-mediated elimination of some hematological neoplasias, including acute monocytic and lymphocytic leukemias, and to a lesser extent multiple myeloma (56). However, this effect is not clear for B-CLL, and it could help to explain the different susceptibility of some B-CLL cells. Thus, we analyzed whether mismatches between NK cell donor KIRs and patient HLA ligands (HLA-A3/A11, -Bw4, -C1/2) would explain the differences observed. To this end, we determined the HLA epitopes for the seven NK cell donors and the six B-CLL patients. The results of these analyses are shown in Table 2 where the expression of known ligands for KIR3DL2 (A3/A11), KIR3DL1 (Bw4), KIR2DL1 (C2), and KIR2DL2/2DL3 (C1) are indicated.
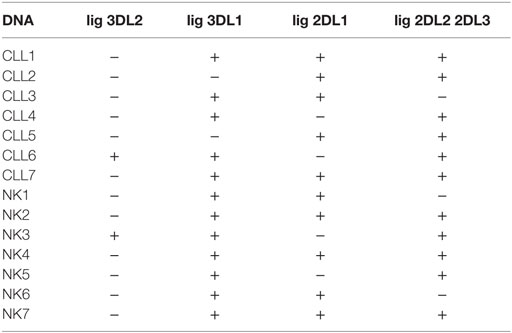
Table 2. KIR ligands expressed in CLL patients and NK cell donors used in the study of compatibility.
Based on the degree of KIR ligand compatibility in donor to patient direction, we predicted matched (compatible) and mismatched (incompatible) combinations in the 42 NK-CLL pairs and analyzed whether the degree of mismatch significantly correlated with the susceptibility observed in the B-CLL samples. A summary of the percentage of matched (0) and mismatched (1) combinations is shown in Table 3. As expected, since the experimental design followed a double blind protocol, in which HLA genotype and mismatch was analyzed after testing NK cell-mediated cytotoxicity, a higher number of unmatched than matched combinations were analyzed. However, as shown in Figure 3, matched and unmatched NK-CLL combinations were tested at the same time, which partially compensate this potential limitation. As shown in Figure 4, except for one NK cell donor in CLL1, matched NK cell donors induced the lowest level of cell death in every B-CLL case, although in some instances differences were low. The median cell death in compatible B-CLL/NK cell combinations was 18%, and this value increased to 34% in mismatched combinations (Table 4). These differences became statistically significant using a one mixed-model analysis of variance (ANOVA). The model controlled for the within-subject nature of the seven NK cell donors by including random effects for B-CLL patient and B-CLL patient × NK cell interaction. Thus, although the statistical significance was close to 0.05, it should be noted that the model used for these analyses is very conservative taking into account that there is a non-controlled intrinsic variability in the components within each group (NK cell donors and B-CLL patients). Thus there was a good correlation between mismatching and NK cell cytotoxicity supporting that NK cell alloreactivity explains the variability observed in each B-CLL sample.
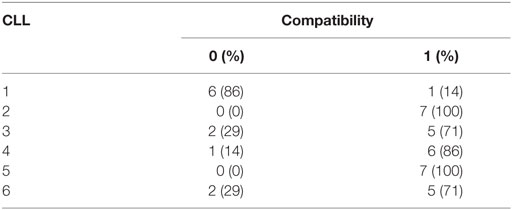
Table 3. Distribution of matched (0)/mismatched (1) combinations for each CLL patient.
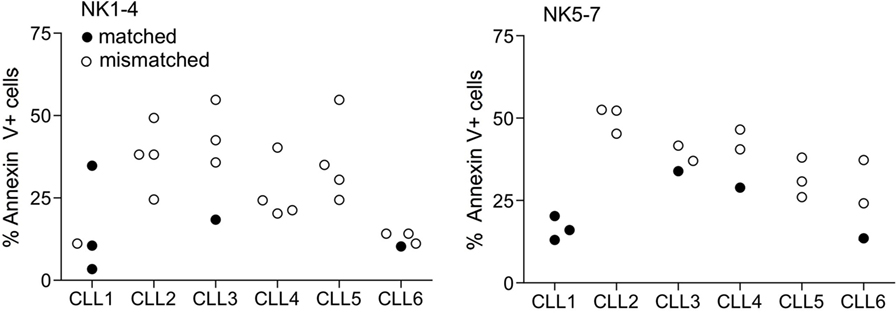
Figure 4. Correlation between matched B-CLL/NK cell combinations and cell death. The data in Figure 2 were now represented as matched and mismatched combinations. The graph represents the % of annexin V+ cells in the 42 combinations (six B-CLL × seven NK) as described in legend to Figure 2 separated in two independent experiments (NK1-4 and NK5-7). White and solid symbols correspond to mismatched and matched combinations, respectively. Statistical analyses and results are described in Tables 4 and 5.
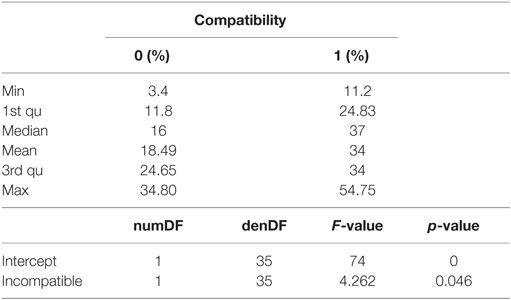
Table 4. Statistical analyses of the difference in cell death between matched (0) and mismatched (1) groups.
Poor Prognosis B-CLL Cells Are More Susceptible to Activated NK Cells than Good Prognosis B-CLL Cells
Although mismatch partly explained the variability observed in the different B-CLL/NK cell combinations, our results also suggest that in each B-CLL case there is/are intrinsic characteristic/s that regulate/s the susceptibility of B-CLL cells to NK cells which is independent on the expression of NK cell receptor ligands.
When we analyzed retrospectively the clinical evolution (response to treatment) of these patients, we realized that except for CLL3, paradoxically, samples from patients with a better response (CLL1) or in which no treatment was required (CLL6) were those more resistant to NK cell cytotoxicity (Table 1; Figure 2B). In contrast, the patient that did not respond to treatments (CLL2) was the most susceptible to NK cells. Based on this observation, we established a new experiment to analyze in more detail whether the prognosis of CLL patients correlated with the susceptibility of B-CLL cells to NK cells. Prognosis was established by determination of TP53 and IGHV mutational status as previously shown (16–19). Three different groups of B-CLL patients were established retrospectively according to the expression of wild-type (wt) or mutated/deleted (mut) TP53 and wt or mut IGHV: TP53wt IGHVmut (nine patients), TP53wt IGHVwt (eight patients), and TP53mut IGHVwt (five patients) as good, intermediate, and poor prognostic groups (Table 5). The samples of these groups were blindly incubated with NK cells from several donors randomly selected and the level of cell death was determined.
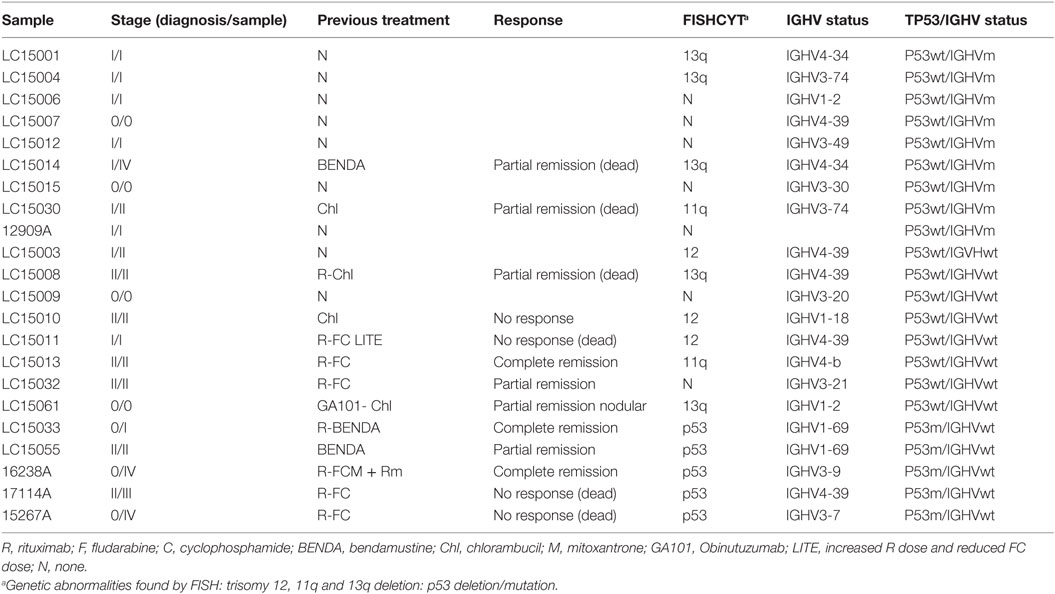
Table 5. Characteristics of CLL patients used in the study of susceptibility in relation to prognosis.
As shown in Figure 5, the susceptibility of B-CLL cells to NK cells was clearly enhanced in the worse prognostic group in comparison with good and intermediate prognosis. Expression of wt IGHV in the context of wt TP53 also increased the susceptibility of B-CLL cells to NK cells although did not reach statistic significance.
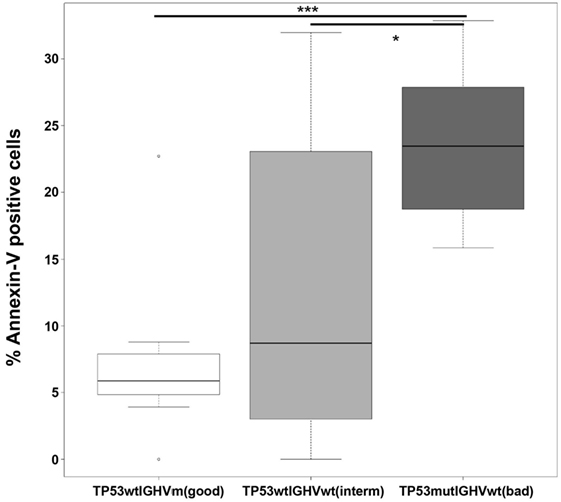
Figure 5. Poor prognosis B-CLL cells are more susceptible to allogeneic R69-LCL-activated NK cells than good prognosis samples. R69-LCL-activated allogeneic NK cells (MACS enriched, >90% CD56+CD3− cells; CTGreen labeled) were incubated for 4 h at 9:1 effector:target ratio with B-CLL cells from three groups of patients classified according to prognosis based on the mutational status of TP53 and IGHV (good: TP53wt IGHVmut, n = 9; intermediate: TP53wt IGHVwt, n = 8; bad: TP53mut IGHVwt, n = 5). Subsequently, PS traslocation (annexin V DY634) and membrane permeabilization (7-AAD uptake) were analyzed by flow cytometry in the cell population negative for CTGreen. Annexin V+ cells represent the % of AnnexinV+7AAD− plus AnnexinV+7AAD+ cells. Every B-CLL sample was incubated with NK cells from the several healthy donors and the % of Annexin V+ cells for every B-CLL sample were represented in a boxplot where median ± SD is indicated. Statistical analysis was performed by comparing the means of every group using 1-way ANOVA with Tukey HSD post hoc test; ns, not significant, *p < 0.05, ***p < 0.001.
Discussion
Allogeneic NK cells have been shown to be effective in the treatment of poor prognosis acute myeloid and lymphoid leukemia (AML and ALL) (34, 37–42). Here, we show that allogeneic activated NK cells also recognize B-CLL cells and that, as shown for AML and ALL, our results suggest that recognition might be partially dependent on HLA mismatch. In addition, we show that allogeneic NK cells are able to efficiently kill leukemic cells from B-CLL patients with very poor prognosis categorized according to expression of mutated TP53 and wt IGHV. Notably, a worse prognosis was associated with an increased susceptibility of B-CLL cells to activated NK cells suggesting that those tumors expressing more aggressive phenotypes due to mutations may be good targets for NK cell immunotherapy. Indeed, it has recently been shown that B-CLL cells expressing wt IGHV present a higher number and a different pattern of somatic mutations, conferring those associated with IGHV a more aggressive and drug-resistant phenotype (15, 57, 58). Although we do not have yet a molecular explanation for this finding, it suggests that activated allogeneic NK cells could be a good alternative to treat them. Indeed, these results are somehow in agreement with previous findings indicating that cells with a higher resistance like undifferentiated/stem like tumors are more susceptible to NK cells (59–63). Since two different cohorts of patients were used for these studies, the formal proof that a combination of mismatch and prognosis predicts the sensitivity of B-CLL cells to allogeneic NK cells is still lacking. However, our data suggest that both factors should be taken into account when selecting therapies against bad prognosis B-CLL.
Controversial findings have been reported on the susceptibility of B-CLL cells to NK cells. Some studies found that activation of NK cells with IL2 increased its cytotoxic potential against B-CLL cells (43–45), by contrast others reported opposite results (46–48). However, these studies did not analyze the degree of HLA compatibility between donor NK cells and B-CLL cells, which could explain these a priori contradictory results. A general explanation for the relative resistance of B-CLL cells to syngeneic NK cells is the low expression of NKR ligands in B-CLL cells together with a high expression of classical HLA-I genes (64–66). Although we have not specifically identified which NKRs are involved in the elimination of B-CLL cells, our data suggest that allogeneic activated NK cells recognize and kill B-CLL cells even expressing low levels of activating NKR ligands. An enhanced NKR expression after activation as previously found (51) and a reduction of inhibitory signals due to HLA mismatch would promote the allogeneic NK cell response against B-CLL cells. This strategy is being currently used to develop therapeutic alternatives like blocking KIR with the antibody, Lirilumab (67). In this case the effect maybe more limited due the inability of patient NK cells to be reactivated against leukemic cells. Our results suggest that combining activated NK cells with blocking KIR antibodies might enhance the therapeutic potential of these therapies.
Another critical factor that may explain the differences regarding the susceptibility of B-CLL cells to NK cells is the use of NK cells activated under different protocols. We have previously found the stimulus employed to activate NK cells is critical in predicting the effectiveness of NK cells against resistant hematological cancer cells (51, 52). Thus, in contrast to studies mainly employing cytokines (IL-2 and/or IL-15) to activate NK cells, we have used here an EBV-transformed B-LCL as feeder cells. This protocol has been shown to enhance NK-mediated cytotoxicity against hematological neoplasia more efficiently than cytokines even in combination with K562 feeder cells (51, 52).
Following this protocol, we have analyzed the molecular determinants that might explain the relative resistance of B-CLL to NK cells. First, we have confirmed that activation is a prerequisite to generate allogeneic NK cells able to kill B-CLL cells. We ruled out that the activation level of NK cells from the different donors was responsible for the different susceptibility of B-CLL [Data not shown and (51)]. In addition, we did not find a clear correlation between expression of ligands for the major activating NK cell receptors (e.g., NKG2D, NKp30, and NKp46) or the ICAM-1 adhesion molecule and B-CLL susceptibility to NK cells. Concerning the major ligands for NK inhibitory receptors, a good correlation between HLA-ABC and HLA-E expression and B-CLL susceptibility to NK cells was observed. However, a more rigorous analysis of HLA-ABC expression should be performed in a higher number of samples to reach a clear conclusion.
Employing a total of 42 NK donor-B-CLL patient combinations, we obtained data suggesting that HLA mismatching was associated with the level of susceptibility of every B-CLL case. However to definitively confirm it, the expression of KIRs at the protein level should be also analyzed in a higher number of combinations, as a receptor–ligand model could improve the prediction of NK cell effectivity (68). Despite these potential limitations, our results underline the importance of selecting a suitable NK cell donor to treat a given B-CLL patient. In particular the analysis of HLA mismatch would be useful to readily exclude NK cell donors with low effectivity (compatible). Yet, testing susceptibility to NK cells from mismatched donors in vitro is mandatory to select a good NK cell donor, since B-CLL from some patients are inherently more resistant than others, irrespectively of HLA mismatch.
The general resistance of some B-CLL patients could be due to previously reported immune evasion mechanisms, i.e., shedding of NK cell receptor-soluble ligands (66). Although these authors did not test the role of HLA mismatch in B-CLL resistance, and we have not formally tested this potential evasion mechanism, our data comparing the susceptibility of B-CLL cells from patients with good, intermediate, and bad prognosis suggest that the low susceptibility of some B-CLL patients is paradoxically related with a less aggressive cancer cell phenotype.
Irrespective of which signals regulate NK cell recognition of B-CLL cells, it seems clear that expression of poor prognosis markers like mutated or deleted TP53 and wt IGHV, related to low life expectancy, may be converted into a chance to employ activated allogeneic NK cells to treat aggressive B-CLL phenotypes. Studies employing larger cohort or patients and clinical trials will be required to confirm the therapeutic value of our findings. Encouraging the development of these trials our conclusions are supported by recent findings indicating that allogeneic stem cell transplantation is a good option for poor prognosis B-CLL if the balance between GVL effect and GVHD is evaluated properly (69).
Author Contributions
DS-M performed most experimental work, analyzed results, and wrote the first draft of the manuscript. PL performed experimental work and analyzed results. NG, GA, LP, and JM selected and provided LLC samples, analyzed clinical data, and wrote the manuscript. AM provided NKR-chimeras. EC, MM, and CV performed immunophenotyping of NK cell donors and patient samples and the mismatch study. AA, LM-L, and MV discussed the experimental data and wrote the manuscript. JP designed, supervised, and evaluated the experiments and wrote the final version of the manuscript. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the use of Servicios Cientifico-Tecnicos (SCT) del CIBA (Instituto Aragones de Ciencias de la Salud, Universidad de Zaragoza, Fundacion IIS Aragón) and Servicio de Citómica del SAI (Universidad de Zaragoza). The authors also would like to acknowledge Alberto Cebollada for providing assistance to perform the statistical analysis. All our funders are public or charitable organizations. A grant from Fondation de France (0057921) to MV and DS-M. SUDOE to AA, JP, MV and CV. SAF2011-25390 and SAF2014-54763-C2-1-R to JP. Fondo Social Europeo (FSE) to JP and AA. JP is supported by Fundacion ARAID.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00454/full#supplementary-material
References
1. Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med (1995) 333:1052–7. doi:10.1056/NEJM199510193331606
2. Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer (2010) 10:37–50. doi:10.1038/nrc2764
3. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med (2005) 352:804–15. doi:10.1056/NEJMra041720
4. Keating MJ, O’Brien S, Albitar M, Lerner S, Plunkett W, Giles F, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol (2005) 23:4079–88. doi:10.1200/JCO.2005.12.051
5. Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med (2000) 343:1910–6. doi:10.1056/NEJM200012283432602
6. Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood (2011) 118:3470–8. doi:10.1182/blood-2011-06-275610
7. Zucchetto A, Bomben R, Dal Bo M, Bulian P, Benedetti D, Nanni P, et al. CD49d in B-cell chronic lymphocytic leukemia: correlated expression with CD38 and prognostic relevance. Leukemia (2006) 20:523–5; author reply 8–9. doi:10.1038/sj.leu.2404087
8. Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med (2003) 348:1764–75. doi:10.1056/NEJMoa023143
9. Bulian P, Shanafelt TD, Fegan C, Zucchetto A, Cro L, Nuckel H, et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J Clin Oncol (2014) 32:897–904. doi:10.1200/JCO.2013.50.8515
10. Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med (2011) 208:1389–401. doi:10.1084/jem.20110921
11. Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood (2011) 119:521–9. doi:10.1182/blood-2011-09-379966
12. Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood (2011) 118:6904–8. doi:10.1182/blood-2011-08-373159
13. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet (2012) 44:47–52. doi:10.1038/ng.1032
14. Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood (2012) 119:2854–62. doi:10.1182/blood-2011-12-395673
15. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475:101–5. doi:10.1038/nature10113
16. Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood (2008) 112:3322–9. doi:10.1182/blood-2008-04-154070
17. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood (1999) 94:1840–7.
18. Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol (2010) 29:2223–9. doi:10.1200/JCO.2010.32.0838
19. Oscier DG, Gardiner AC, Mould SJ, Glide S, Davis ZA, Ibbotson RE, et al. Multivariate analysis of prognostic factors in CLL: clinical stage, IGVH gene mutational status, and loss or mutation of the p53 gene are independent prognostic factors. Blood (2002) 100:1177–84.
20. Wickremasinghe RG, Prentice AG, Steele AJ. p53 and Notch signaling in chronic lymphocytic leukemia: clues to identifying novel therapeutic strategies. Leukemia (2011) 25:1400–7. doi:10.1038/leu.2011.103
21. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5:117–21. doi:10.1002/eji.1830050208
22. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–7. doi:10.1002/eji.1830050208
23. Karre K. Natural killer cell recognition of missing self. Nat Immunol (2008) 9:477–80. doi:10.1038/ni0508-477
24. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today (1990) 11:237–44. doi:10.1016/0167-5699(90)90097-S
25. Herberman RB, Holden HT. Natural cell-mediated immunity. Adv Cancer Res (1978) 27:305–77. doi:10.1016/S0065-230X(08)60936-7
27. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22:633–40. doi:10.1016/S1471-4906(01)02060-9
28. Krzywinska E, Allende-Vega N, Cornillon A, Vo DN, Cayrefourcq L, Panabieres C, et al. Identification of anti-tumor cells carrying natural killer (NK) cell antigens in patients with hematological cancers. EBioMedicine (2015) 2:1364–76. doi:10.1016/j.ebiom.2015.08.021
29. Anel A, Aguilo JI, Catalan E, Garaude J, Rathore MG, Pardo J, et al. Protein kinase C-theta (PKC-theta) in natural killer cell function and anti-tumor immunity. Front Immunol (2012) 3:187. doi:10.3389/fimmu.2012.00187
30. Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science (2004) 306:1517–9. doi:10.1126/science.1103478
31. Bottino C, Moretta L, Moretta A. NK cell activating receptors and tumor recognition in humans. Curr Top Microbiol Immunol (2006) 298:175–82. doi:10.1007/3-540-27743-9_9
32. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi:10.1038/ni1581
33. Chan CJ, Andrews DM, Smyth MJ. Can NK cells be a therapeutic target in human cancer? Eur J Immunol (2008) 38:2964–8. doi:10.1002/eji.200838764
34. Fuchs EJ, Huang XJ, Miller JS. HLA-haploidentical stem cell transplantation for hematologic malignancies. Biol Blood Marrow Transplant (2009) 16:S57–63. doi:10.1016/j.bbmt.2009.10.032
35. Locatelli F, Pende D, Mingari MC, Bertaina A, Falco M, Moretta A, et al. Cellular and molecular basis of haploidentical hematopoietic stem cell transplantation in the successful treatment of high-risk leukemias: role of alloreactive NK cells. Front Immunol (2013) 4:15. doi:10.3389/fimmu.2013.00015
36. Ruggeri L, Mancusi A, Burchielli E, Perruccio K, Aversa F, Martelli MF, et al. Natural killer cell recognition of missing self and haploidentical hematopoietic transplantation. Semin Cancer Biol (2006) 16:404–11. doi:10.1016/j.semcancer.2006.07.007
37. Velardi A. Natural killer cell alloreactivity 10 years later. Curr Opin Hematol (2012) 19:421–6. doi:10.1097/MOH.0b013e3283590395
38. Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother (2010) 59:1739–44. doi:10.1007/s00262-010-0896-z
39. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118:3273–9. doi:10.1182/blood-2011-01-329508
40. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28:955–9. doi:10.1200/JCO.2009.24.4590
41. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood (2007) 110:433–40. doi:10.1182/blood-2006-07-038687
42. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol (2008) 143:641–53. doi:10.1111/j.1365-2141.2008.07340.x
43. Santiago-Schwarz F, Panagiotopoulos C, Sawitsky A, Rai KR. Distinct characteristics of lymphokine-activated killer (LAK) cells derived from patients with B-cell chronic lymphocytic leukemia (B-CLL). A factor in B-CLL serum promotes natural killer cell-like LAK cell growth. Blood (1990) 76:1355–60.
44. van der Harst D, Brand A, van Luxemburg-Heys SA, Kooy-Winkelaar EM, van Rood JJ. Lymphokine-activated killer cell functions in patients with leukemic B-lymphoproliferative diseases. Blood (1989) 74:2464–70.
45. Kay NE, Zarling J. Restoration of impaired natural killer cell activity of B-chronic lymphocytic leukemia patients by recombinant interleukin-2. Am J Hematol (1987) 24:161–7. doi:10.1002/ajh.2830240207
46. Kay NE, Zarling JM. Impaired natural killer activity in patients with chronic lymphocytic leukemia is associated with a deficiency of azurophilic cytoplasmic granules in putative NK cells. Blood (1984) 63:305–9.
47. Jewell AP, Worman CP, Giles FJ, Goldstone AH, Lydyard PM. Resistance of chronic lymphocytic leukaemia cells to interferon-alpha generated lymphokine activated killer cells. Leuk Lymphoma (1992) 7:473–80. doi:10.3109/10428199209049804
48. Foa R, Fierro MT, Raspadori D, Bonferroni M, Cardona S, Guarini A, et al. Lymphokine-activated killer (LAK) cell activity in B and T chronic lymphoid leukemia: defective LAK generation and reduced susceptibility of the leukemic cells to allogeneic and autologous LAK effectors. Blood (1990) 76:1349–54.
49. Veuillen C, Aurran-Schleinitz T, Castellano R, Rey J, Mallet F, Orlanducci F, et al. Primary B-CLL resistance to NK cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J Clin Immunol (2011) 32:632–46. doi:10.1007/s10875-011-9624-5
50. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32:177–86. doi:10.1038/sj.bmt.1704086
51. Sanchez-Martinez D, Azaceta G, Muntasell A, Aguilo N, Nunez D, Galvez EM, et al. Human NK cells activated by EBV lymphoblastoid cells overcome anti-apoptotic mechanisms of drug resistance in haematological cancer cells. Oncoimmunology (2015) 4:e991613. doi:10.4161/2162402X.2014.991613
52. Sanchez-Martinez D, Krzywinska E, Rathore MG, Saumet A, Cornillon A, Lopez-Royuela N, et al. All-trans retinoic acid (ATRA) induces miR-23a expression, decreases CTSC expression and granzyme B activity leading to impaired NK cell cytotoxicity. Int J Biochem Cell Biol (2014) 49:42–52. doi:10.1016/j.biocel.2014.01.003
53. Ghia P, Stamatopoulos K, Belessi C, Moreno C, Stilgenbauer S, Stevenson F, et al. ERIC recommendations on IGHV gene mutational status analysis in chronic lymphocytic leukemia. Leukemia (2007) 21(1):1–3. doi:10.1038/sj.leu.2404457
54. Moraru M, Cisneros E, Gomez-Lozano N, de Pablo R, Portero F, Canizares M, et al. Host genetic factors in susceptibility to herpes simplex type 1 virus infection: contribution of polymorphic genes at the interface of innate and adaptive immunity. J Immunol (2012) 188:4412–20. doi:10.4049/jimmunol.1103434
55. Núñez D, Domingo MP, Sánchez-Martínez D, Cebolla V, Chiou A, Velázquez-Campoy A, et al. Recombinant production of human ICAM-1 chimeras by single step on column refolding and purification. Process Biochem (2013) 48:708–15. doi:10.1016/j.procbio.2013.03.006
56. Velardi A, Ruggeri L, Mancusi A, Aversa F, Christiansen FT. Natural killer cell allorecognition of missing self in allogeneic hematopoietic transplantation: a tool for immunotherapy of leukemia. Curr Opin Immunol (2009) 21:525–30. doi:10.1016/j.coi.2009.07.015
57. Martinez-Trillos A, Quesada V, Villamor N, Puente XS, Lopez-Otin C, Campo E. Recurrent gene mutations in CLL. Adv Exp Med Biol (2013) 792:87–107. doi:10.1007/978-1-4614-8051-8_4
58. Delgado J, Salaverria I, Baumann T, Martinez-Trillos A, Lee E, Jimenez L, et al. Genomic complexity and IGHV mutational status are key predictors of outcome of chronic lymphocytic leukemia patients with TP53 disruption. Haematologica (2014) 99:e231–4. doi:10.3324/haematol.2014.108365
59. Jewett A, Tseng HC, Arasteh A, Saadat S, Christensen RE, Cacalano NA. Natural killer cells preferentially target cancer stem cells; role of monocytes in protection against NK cell mediated lysis of cancer stem cells. Curr Drug Deliv (2012) 9(1):5–16. doi:10.2174/156720112798375989
60. Tseng HC, Arasteh A, Kaur K, Kozlowska A, Topchyan P, Jewett A. Differential cytotoxicity but augmented IFN-γ secretion by NK cells after interaction with monocytes from humans, and those from wild type and myeloid-specific COX-2 knockout mice. Front Immunol (2015) 6:259. doi:10.3389/fimmu.2015.00259
61. Tseng HC, Inagaki A, Bui VT, Cacalano N, Kasahara N, Man YG, et al. Differential targeting of stem cells and differentiated glioblastomas by NK cells. J Cancer. (2015) 6(9):866–76. doi:10.7150/jca.11527
62. Bui VT, Tseng HC, Kozlowska A, Maung PO, Kaur K, Topchyan P, et al. Augmented IFN-γ and TNF-α induced by probiotic bacteria in NK cells mediate differentiation of stem-like tumors leading to inhibition of tumor growth and reduction in inflammatory cytokine release; regulation by IL-10. Front Immunol (2015) 6:576. doi:10.3389/fimmu.2015.00576
63. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol (2009) 182(6):3530–9. doi:10.4049/jimmunol.0802845
64. Maki G, Hayes GM, Naji A, Tyler T, Carosella ED, Rouas-Freiss N, et al. NK resistance of tumor cells from multiple myeloma and chronic lymphocytic leukemia patients: implication of HLA-G. Leukemia (2008) 22:998–1006. doi:10.1038/leu.2008.15
65. Nuckel H, Switala M, Sellmann L, Horn PA, Durig J, Duhrsen U, et al. The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia (2010) 24:1152–9. doi:10.1038/leu.2010.74
66. Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M, et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood (2013) 121:3658–65. doi:10.1182/blood-2013-01-476606
67. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123:678–86. doi:10.1182/blood-2013-08-519199
68. Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol (2004) 172:644–50. doi:10.4049/jimmunol.172.1.644
Keywords: allogeneic NK cells, bad prognosis leukemia, mismatch, chronic lymphocytic leukemia, leukemia resistance
Citation: Sánchez-Martínez D, Lanuza PM, Gómez N, Muntasell A, Cisneros E, Moraru M, Azaceta G, Anel A, Martínez-Lostao L, Villalba M, Palomera L, Vilches C, García Marco JA and Pardo J (2016) Activated Allogeneic NK Cells Preferentially Kill Poor Prognosis B-Cell Chronic Lymphocytic Leukemia Cells. Front. Immunol. 7:454. doi: 10.3389/fimmu.2016.00454
Received: 26 July 2016; Accepted: 11 October 2016;
Published: 27 October 2016
Edited by:
Anahid Jewett, University of California, Los Angeles, USAReviewed by:
Junko Matsuzaki, Roswell Park Cancer Institute, USACarlos Alfaro, University of Navarra, Spain
Copyright: © 2016 Sánchez-Martínez, Lanuza, Gómez, Muntasell, Cisneros, Moraru, Azaceta, Anel, Martínez-Lostao, Villalba, Palomera, Vilches, García Marco and Pardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julián Pardo, cGFyZG9qaW1AdW5pemFyLmVz