 Feliciano Chanana Paquissi
Feliciano Chanana Paquissi- Department of Medicine, Clínica Girassol, Luanda, Angola
Non-alcoholic fatty liver disease (NAFLD) is an increasing problem worldwide and is associated with negative outcomes such as cirrhosis, hepatocellular carcinoma, insulin resistance, diabetes, and cardiovascular events. Current evidence shows that the immune response has an important participation driving the initiation, maintenance, and progression of the disease. So, various immune imbalances, from cellular to cytokines levels, have been studied, either for better compression of the disease pathophysiology or as biomarkers for severity assessment and outcome prediction. In this article, we performed a thorough review of studies that evaluated the role of inflammatory/immune imbalances in the NAFLD. At the cellular level, we gave special focus on the imbalance between neutrophils and lymphocytes counts (the neutrophil-to-lymphocyte ratio), and that which occurs between T helper 17 (Th17) and regulatory T cells as emerging biomarkers. By extension, we reviewed the reflection of these imbalances at the molecular level through pro-inflammatory cytokines including those involved in Th17 differentiation (IL-6, IL-21, IL-23, and transforming growth factor-beta), and those released by Th17 cells (IL-17A, IL-17F, IL-21, and IL-22). We gave particular attention to the role of IL-17, either produced by Th17 cells or neutrophils, in fibrogenesis and steatohepatitis. Finally, we reviewed the potential of these pathways as new therapeutic targets in NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a health problem with increasing prevalence worldwide (1, 2), mainly due to the increased rates of obesity and type 2 diabetes (3, 4), with increasingly installation at an early age (4, 5). The prevalence of NAFLD in the general adult population is around 25.24% (6), reaching 67.5 and 74% among those with obesity or diabetes, respectively (7), with 10–12.2% of them having subclinical hepatic inflammation, known as non-alcoholic steatohepatitis (NASH), and/or fibrosis (8). NAFLD is associated with significant morbidity and mortality, increasing the risk of cirrhosis, hepatocellular carcinoma (HCC), insulin resistance, metabolic syndrome, diabetes, cardiovascular events, and cardiovascular and liver disease mortality (2, 9–13), being, therefore, recognized as a multisystem disease (14).
Recent investigations have highlighted the function of the immune response as a driver in the initiation, maintenance, and progression of NAFLD (15–17). General inflammatory/immunity biomarkers, as C-reactive protein (CRP), interleukins, have been associated with the occurrence and prognosis of NAFLD, including the link to vascular events (15, 18–21). In the cellular level, various immune imbalances have also emerged as biomarkers in NAFLD, from those in general white blood cells such as neutrophil-to-lymphocyte ratio (NLR), to specific lymphocytes subsets (22–24). The NLR expresses an imbalance in leukocytes with the dominance of neutrophils over lymphocytes and has been increasingly recognized as a predictor of outcomes in NAFLD (25), a role also shown in other chronic liver diseases such as viral hepatitis, liver cirrhosis, and HCC (26–28). Another described cellular imbalance is what occurs at the CD4+ cells level with the dominance of T helper 17 (Th17) subset over the regulatory T (Treg) cells, which results from the polarization of the differentiation of T helper cells, also present in NAFLD and other chronic liver diseases (24, 29–32). The functional equilibrium between Th17 and Treg in peripheral blood is an important element to ensure the equilibrium between the defense and the autoimmunity; and there is an interplay and plasticity between these cells and their subsets, with cellular polarizations and different cytokine profiles in the presence of different stimuli (33–36). The cellular imbalances reflect at the molecular level through pro-inflammatory cytokines including those involved in Th17 differentiation [IL-6, IL-21, IL-23, and transforming growth factor-beta (TGF-β)] (33, 37–39), and those released by Th17 cells (IL-17A, IL-17F, IL-21, and IL-22) (29, 40–42).
In this article, we performed a thorough review of studies that evaluated the role of inflammatory/immune imbalances in the pathophysiology and outcome prediction in NAFLD. We focused the neutrophil/lymphocyte and Th17/Treg imbalances as emerging biomarkers at the cellular level; its reflection at the molecular level through pro-inflammatory cytokines with particular attention to the role of IL-17, either produced by Th17 cells or neutrophils, in fibrogenesis and steatohepatitis. Finally, we reviewed the potential of these pathways as new therapeutic targets in NAFLD.
An Overview of the Predictive Role of General Inflammatory Biomarkers in NAFLD
Several inflammatory markers such as CRP and cytokines have been associated with NAFLD (15, 16, 20, 43). In a study with individuals without obesity, the presence of hs-CRP, with or without insulin resistance, was associated with higher prevalence of NAFLD, with a significant risk increase as increased the value of hs-CRP [odds ratio (OR) de 1.37; 95% confidence interval (CI): 1.06–1.77] for each 1 SD above (1.48 mg/L) (16). In another study, a positive association between the degree of steatosis and hs-CRP was observed (P < 0.05) after adjusting for BMI (44). Additionally, an elevated CRP is a predictor of disease progression and severity, as shown that hs-CRP was significantly higher in cases of NASH than in simple steatosis (19, 45); furthermore, among patients with NASH, hs-CRP was significantly elevated in those with advanced fibrosis compared with those with mild, even after adjustment for confounders (19).
Other inflammatory biomarkers associated with NAFLD and its progression are cytokines. The expression of interleukin-6 (IL-6) is markedly increased in the liver cells of patients with simple steatosis (P < 0.005) or NASH (P < 0.010) compared to normal subjects (46). IL-6 expressed in hepatocytes, and its level in the blood, correlates positively with the degree of liver inflammation, and fibrosis (46). Another key cytokine in NAFLD is interleukin-17 (IL-17) (41). The activation of the IL-17 axis has shown to have a central role in the progression from NAFLD to NASH in experimental studies (15, 24, 41, 42). The role of IL-17 family cytokines will be discussed further elsewhere in this paper. Similarly, TGF-β, a cytokine known by its fibrotic effect in many organs is involved in the progression of NAFLD (47, 48). In a study that evaluated the gene expression of this cytokine in NAFLD, there was a marked increase in TGF-β1 gene expression in patients with NASH compared with simple steatosis (P = 0.0002) (49). In another study with 1,322 healthy subjects without other risk factors, serum TGF-β3 levels was significantly higher in those who developed NAFLD than in those who did not (mean 554 vs. 285 pg/ml; P = 0.002) after 4 years of follow-up; and as the TGF-β3 tertiles increased, there was a significant increase in NAFLD incidence (6.3, 38.0, and 55.7% for the first, second, and third tertiles, respectively; P < 0.05) (50).
The inflammatory response seems to be an important mediator of many NAFLD-associated outcomes such as the HCC by creating an inflammatory microenvironment (28, 51), cardiovascular disease by promoting atherogenesis (52, 53). Table 1 summarizes the clinical studies that have assessed the role of general inflammatory biomarkers and cytokines to predict outcomes in NAFLD.
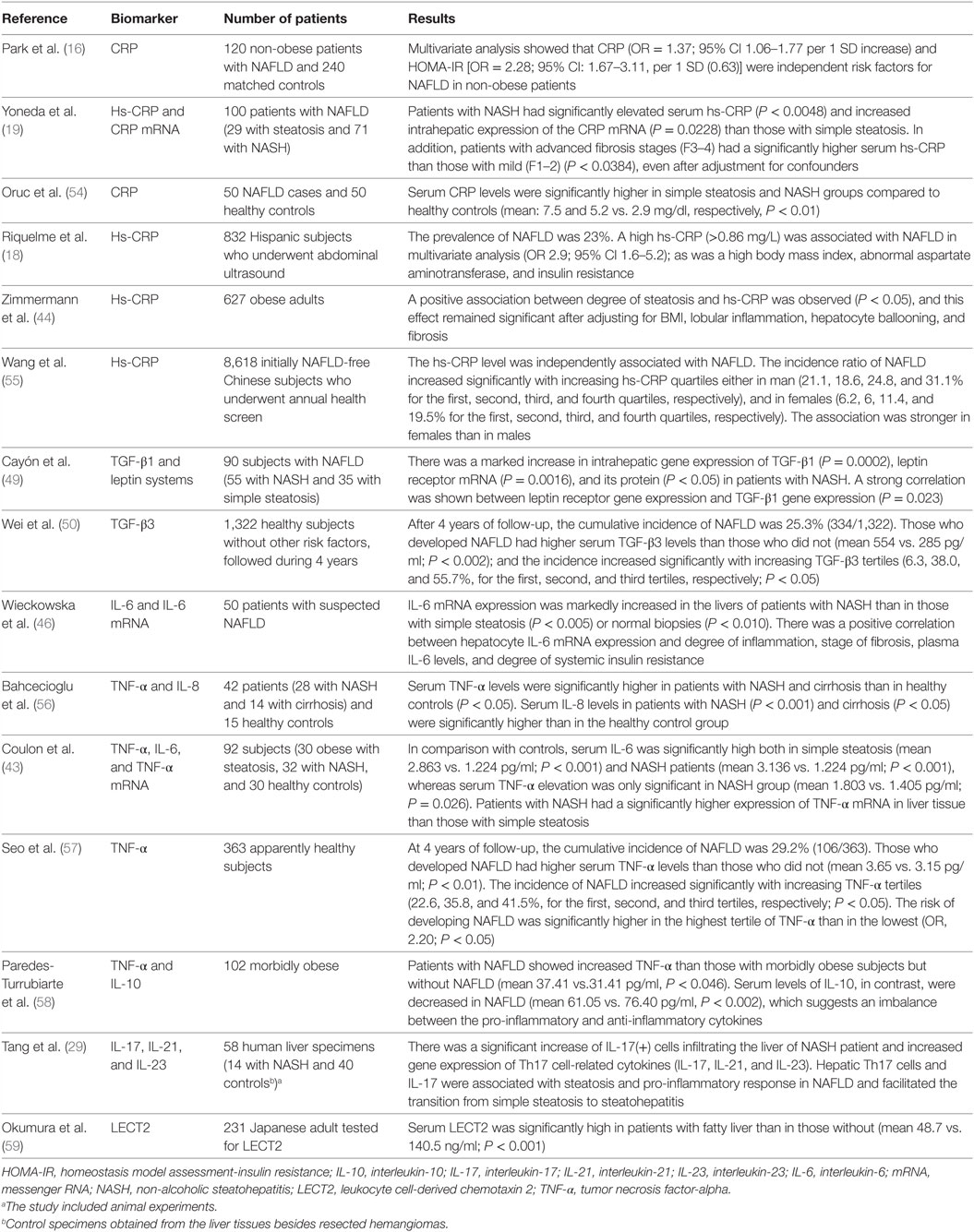
Table 1. Clinical studies that have assessed the role of general inflammatory biomarkers and cytokines to predict outcomes in NAFLD.
The Role of Cellular Immune Imbalances in NAFLD
In the last 5 years, many cellular imbalances in the immune response have been associated with NAFLD and its prognosis (23, 25), which have brought a cellular background to what was observed through pro-inflammatory cytokines. These cellular imbalances range from the simple increase in the total count of leukocytes (23) to specific lymphocytes subpopulations (25). This was remarkable in a study with 3,681 healthy subjects in which, as increased the WBC count quartile above the lowest, there was a significant increase in NAFLD risk in both men [OR (95% CIs): 1.48 (1.10–1.98), 1.59 (1.18–2.14), and 1.84 (1.35–2.51) for the second, third, and fourth quartiles, respectively]; and women [OR (95% CIs): 1.15 (0.67–1.96) 1.88 (1.13–3.11), and 2.74 (1.68–4.46) for the second, third, and fourth quartiles, respectively] (23). Among these imbalances, we highlight that which occurs at the level of T helper cellular subsets (Th17/Treg imbalance), and that between neutrophils and lymphocytes counts (expressed by NLR).
The Th17/Treg Imbalance
An important cellular imbalance that has emerged as a cellular background in the role of the inflammatory response in NAFLD is the Th17/Treg imbalance. In a study with 94 subjects (30 with NASH, 31 with NAFLD, and 43 healthy controls), patients with NASH (and in less degree with steatosis) had a lower frequency of T regulatory cells in peripheral blood, in comparison with controls (30). In addition, the progression from steatosis to NASH was marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting T regulatory cell ratio in the liver and in peripheral blood (30).
In experimental models, liver infiltration by Th17 cells showed to be a critical element for NASH initiation and development of fibrosis in wild-type mice, and this infiltration was accompanied by an increase in the production of pro-inflammatory cytokines (IL-6, TNF-α, and TGF-β) (42). In another study using mice fed with high-fat diet (HFD), there was a significant increase of Th17 cells in the liver (P < 0.02) and the abdominal adipose tissue (AAT) (P < 0.01), without a concurrent increase of Treg (60). NASH and metabolic alterations occurred in mice-fed HFD, and Th17 cells (either AAT or liver-derived) positively correlated with NASH (60). Other studies have shown in parallel that the reduction, dysfunction, or disproportionate number of Treg cells contributes to the progression to NASH because Treg cells play a critical role in regulating the inflammatory processes in the liver (24, 29, 30). This cellular imbalance is accompanied by the activation of the IL-17 axis, and an increase of other pro-inflammatory cytokines such as IL-6, and TNF-α (42, 58); and its value has been highlighted by the demonstration that therapies targeted to reverse this imbalance have shown the potential to alleviate steatosis and the progression to NASH (32, 61, 62).
Lymphocytes Site-Specific Source
An important element to consider is the site specificity of lymphocytes in NAFLD, as shown in an experimental study where it was observed that the cells infiltrating the liver were labeled lymphocytes that migrated predominantly from mesenteric lymph nodes (MLN) than from spleen, bone marrow, or thymus (63), suggesting that the gut is the primary source of cellular elements involved in NAFLD pathogenesis, which is in turn affected by the microbiota (64, 65).
Neutrophil-to-Lymphocyte Ratio as a Cellular Biomarker in NAFLD
Neutrophil-to-lymphocyte ratio is a derivative biomarker obtained from the absolute counts of neutrophils and lymphocytes. NLR is a cellular imbalance (with the dominance of neutrophils over the lymphocytes) that has been found to be related to a lot of diseases that share the chronic inflammatory response as critical in pathogenesis, such as cancer and cardiovascular diseases (66, 67). In a nationally representative American sample, including 9,427 subjects, the average NLR was 2.15 in the general population, and values above these were found in those with chronic inflammatory states including cardiovascular diseases and diabetes (68). The potential of NLR as a biomarker has also been shown in liver diseases, which reflects the pathologic effects of the dominance of activated neutrophils, an important effector cell of the innate immunity, in diseases of this organ (69, 70).
NLR and NAFLD Severity
In NAFLD, the NLR is associated with high disease severity, as found in a study with 101 patients where NASH patients had higher NLR compared with those without (mean 2.5 vs. 1.6, P < 0.001) (25). NLR showed a good correlation with the NAFLD activity score and its individual components (steatosis, inflammation, and ballooning P < 0.001), and advanced fibrosis stages (F3–4) compared with lower stages (F1–2) (median 2.9 vs. 1.8, respectively, P < 0.001) (25). In another study including 873 patients with biopsy-proven NAFLD (and 150 healthy controls), NLR was higher in NASH patients than in non-NASH cases (mean 2.6 vs. 1.9, respectively, P < 0.001); and similarly, patients with advanced fibrosis stages (F3–4) had a higher NLR compared with those in early (F1–2) (mean 2.5 vs.1.8, respectively, P < 0.001) (71). In study comparing the role of this biomarker in three liver diseases (NASH, HBV, and HCV hepatitis), NLR was significantly higher in NASH patients compared to HBV, and HCV, or controls (P < 0.001, P < 0.001, and P < 0.001, respectively) (22), suggesting a higher contribution of this imbalance in NASH than in other chronic liver diseases.
NLR and NAFLD Prognostisis
Besides being a marker of disease severity, NLR is also a predictor of mortality (72). In a study including 570 patients with end-stage cirrhosis (including 54 due to NAFLD) listed for liver transplantation, NLR ≥ 5 was associated with higher 3-month mortality (OR 6.02, P < 0.043); and as increased NLR, there was a significant increase in proportion of patients who died within 3 months of listing (3, 13.8, and 37.3%, for NLR < 2, 2–4.9, and ≥5, respectively, P < 0.001) (72). NLR is also a predictor of higher NAFLD score, advanced fibrosis, and severe ascites (25, 71, 72).
NLR in NAFLD-Associated Conditions
The role of NLR seems to begin long before and extend beyond the NAFLD. This is evident because subjects with obesity and diabetes, which are the main risk factors for NAFLD, show higher average NLR in relation to controls (68, 73), and among patients with morbid obesity, the mean NLR values were significantly higher in those who developed type 2 diabetes (T2DM) than those who did not (mean 4.11 vs. 3.46, P < 0.001) (73), showing that a higher inflammatory states precedes both (NAFLD and T2DM) obesity-related outcomes.
A high NLR remains an independent predictor of poorer outcome even in those that evolved to terminal stages of chronic liver disease – cirrhosis and/or HCC (27, 74, 75). In the evolution of patients with HCC undergoing radiofrequency ablation, or surgical resection, a high NLR predicted poor outcomes with higher recurrence and mortality rates (74–77) and poor overall survival even after a curative liver resection (76). And even in those undergoing liver transplantation, a high NLR was associated with poorer overall and recurrence-free survival (78).
Neutrophils as Important Source of IL-17 in Liver Diseases
Recent investigations have found that neutrophils are themselves an important source of IL-17 in the human liver, especially in late fibrosis stages (56, 70). Therefore, these researches come to bring a pathophysiological background to the predictive role of elevated NLR in this disease, while it occurs with an important IL-17 axis activation, besides other possible mechanisms beyond our understanding. This role of neutrophils as a source of IL-17 has just been found in other organic diseases such as the kidney (79) and airways (80).
Other Cellular Imbalances in NAFLD
Other immune system cells that have been found imbalanced in frequency and that appear to be involved in the cross talk with hepatocytes, hepatocellular damage, and in the transition from NASH to HCC are natural killer T cells and CD8+ lymphocytes (81, 82). Table 2 summarizes the clinical studies that evaluated the role of cellular imbalances as drivers and predictors of outcomes in NAFLD.
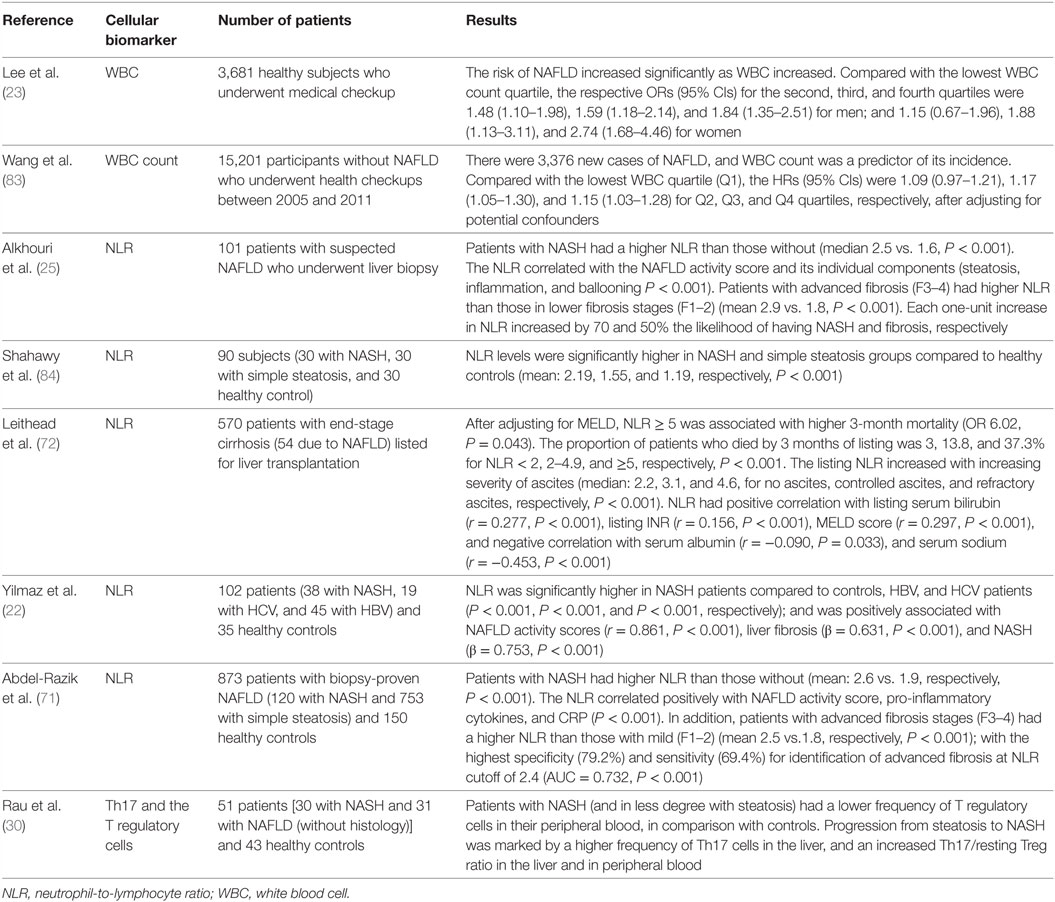
Table 2. Clinical studies on the value of cellular immune imbalances as drivers and predictors of outcomes in NAFLD.
Underlying Mechanisms, Pathways, and Relationship Between Cells and Cytokines in NAFLD
The understanding of the pathophysiological mechanisms linking the cellular and cytokines immune imbalances to NAFLD is still limited, and a subject of ongoing studies, as is the clarification between initiators and perpetuator imbalances. However, recent studies have been consistent in pointing out the hyperactivation of the interleukin-17 axis and TGF-β signaling pathway as the central elements in the pathogenesis of NAFLD, as well as other chronic liver diseases independently of underlying cause (15, 24, 29, 31, 48). So, the sum of the various imbalances in the immune system results in the dominance of activated pro-inflammatory pathways over the regulatory, as shown by a concurrent imbalance in Th17/Treg cells (30), culminating in the activation of the IL-17 axis.
Neutrophils and Interleukin-6
As shown in Table 2, NLR is one of the most studied cell imbalances in recent times on outcome prediction in NAFLD. Neutrophils are the main cells of innate immunity, and its dominance is associated with the polarization to a more pro-inflammatory response, including its participation in the IL-17 activation chain, a key cytokine in organic fibrosis (85, 86). This occurs because neutrophils (and macrophages) produces the IL-6, as the main cytokine, which is in turn important in the differentiation of Th17 cells from naive T helper cells (37, 38, 87). On the other hand, a recent study showed that neutrophils are themselves an important source of IL-17 in the human liver, especially in late fibrosis stages (70). In addition to the IL-17 axis activation, the predominance of neutrophils may be associated with other mechanisms such as oxidative stress and activation/release of matrix metalloproteinases (MMPs) (88, 89).
The Th17/Treg Differentiation and the IL-17 Axis Activation
The Th17 differentiate from the naive T helper cells in the presence of IL-6, TGF-β, IL-21, and IL-23 (37, 38, 90). Th17 cells secrete the IL-17, IL-21, and e IL-22, being important in immunity against extracellular infectious agents such as bacteria and fungi but also contribute in the immunopathogenesis of many diseases such as psoriasis and tumors (51, 91–93). IL-17 is the most studied Th17-secreted cytokine in liver disease (15, 41, 42). The differentiation of Treg, the functional counterpart of Th17 cells, has the TGF-β as a pivotal factor in the presence of retinoic acid (94–97). The main function of the Treg cells is to regulate different aspects of the immune response in order to ensure the immunologic tolerance (98, 99). The dynamic Th17/Treg balance in peripheral blood is an important element to ensure the equilibrium between the defense and the autoimmunity and is regulated by various factors, such as IL-6, IL-10, TGF-β, and the microbiome (33, 34, 90, 100). So, these cells and their precursors are interconnected and have plasticity, which causes to direct their response, in the presence of different stimuli, to different cellular type and/or cytokine profile (34–36). For example, in the presence of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α, the normal TGF-β-driven Treg differentiation is shifted to Th17 differentiation (33, 90). So, neutrophils (and macrophages), through the production of the IL-6, participates in the IL-17 activation chain; and IL-17, in turn, is an important in granulopoiesis (101) and participates in neutrophil recruitment and organs infiltration after initial injury, and induces neutrophils cytokines and chemokines production, promoting further injury (24, 79, 102–104).
The Role of IL-17 Axis Activation and Associated Signaling Pathways in Fibrogenesis and Steatohepatitis
Interleukin-17 is a pro-inflammatory cytokine that is known to be produced mainly by T helper lymphocytes sub type 17 (Th17) and neutrophils, as discovered more recently, which is associated with the progression of NAFLD (15, 24, 41, 70). In the liver, the IL-17 exacerbates the liver tissue inflammation (29, 105, 106), enhancing tissue leukocytes infiltration (107), is a mediator of the cross talk between the immune system and liver cells (85, 108–111), has a profibrotic effect as noted in liver biopsies (70, 85, 86), among several others effects. In addition, is a potent stimulator of production of other inflammatory mediators, such as tumor necrosis factor (TNF-α), interleukin-1 (IL-1), and IL-6 (85, 111, 112). And by induction of IL-6 production in the hepatic cells and serum, it mediates the cross talk between liver cells, the innate, and adaptative immune responses (85, 109, 113) and has a feedback on its axis at both local and systemic level (40).
In experimental studies, the activation of the IL-17 axis showed to be central to the development of NAFLD and progression to NASH and fibrosis (15, 41, 85, 110). And the neutralization or the lack of this axis caused significant attenuation of obesity, methionine choline-deficient diet (MCDD), or schistosoma-induced liver inflammation and fibrosis (15, 41, 42, 86, 104, 114). In addition, livers of IL-17(−/−) mice were protected from NASH development (42).
One of the mechanisms by which the IL-17A exerts its profibrotic effect is using the TGF-β signaling pathways, promoting an upregulation of its receptor on hepatic stellate cells (48, 109). In addition, IL-17 inhibits the natural TGF-β-driven Treg differentiation by the pro-inflammatory environment it promotes, and by stimulating the IL-6 production, the most potent inductor of Th17 cells differentiation (38, 87, 90, 111), thereby enhancing further Th17/Treg imbalance (29, 33, 39, 115).
The relationship of the IL-17, NLR, and fibrosis has been found in other liver diseases such as viral hepatitis (116, 117), cirrhosis, and HCC (27, 28), which suggests the involvement of common points in pathogenic pathways (22, 31, 116–118). The maintenance of these imbalances seems to favor the inflammatory microenvironment, which would explain their prognostic implications, and therapeutics potentials, from NAFLD, to cirrhosis, and HCC (28, 51). In addition, the role of the IL-17 axis in fibrogenesis has been shown in organs other than the liver, including the heart (119, 120), lung (121), and kidney (122). This model has been reasonably proven by evidence of elevation of neutrophils, Th17, and related cytokines, both in the systemic circulation and in the liver (28, 29, 70). So, is this inflammatory arsenal that would act in both hepatic inflammatory infiltration and in fibrogenesis.
Deficient Synthesis or Release of Anti-inflammatory and Antifibrotic Cytokines
The NAFLD immune imbalances, in addition to the above, appears to be also associated with deficient synthesis or release of anti-inflammatory and antifibrotic cytokines as IL-10 (58, 61), IL-4 (61, 123), IL-22 (42), and interferon gamma (IFN-γ) (124) that have a protective effect by suppressing the maturation of Th17 cells or counterbalancing the IL-17 effects (42, 61, 125–127). For example, Treg requires IL-10 signaling to suppress the Th17 cell-mediated inflammation (100), and this anti-inflammatory cytokine was decreased in morbidly obese patients with NAFLD (58). Figure 1 shows a simply proposed model connecting the cellular to cytokines imbalances, including the activation of the IL-17 and the progression of NAFLD.
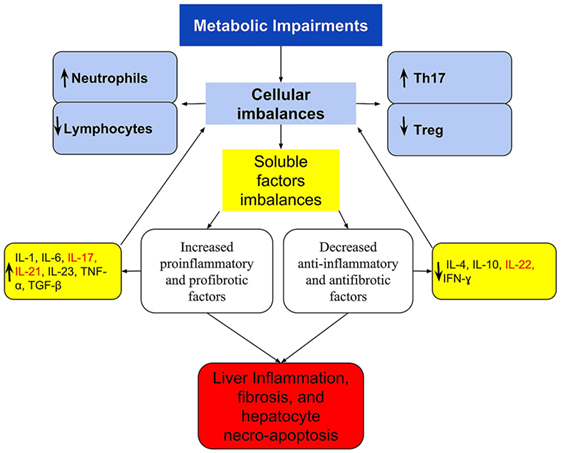
Figure 1. A simplified scheme of imbalanced immune response, connecting the cellular and cytokines imbalances with the activation of IL-17 axis and the progression of NAFLD. Metabolic disorders lead to immune imbalances in the peripheral blood and/or in the liver that are expressed at the cellular level by Th17/Treg imbalance and by the dominance of neutrophil over lymphocytes. These are reflected in imbalanced soluble factors with the dominance of pro-inflammatory and profibrotic over the anti-inflammatory and antifibrotic, which culminates in the Th17/IL-17 axis hyperactivation. In the liver, these imbalances are responsible for the recruitment and organ infiltration by neutrophil (102, 103), for increased hepatic gene expression of Th17-related cytokines (IL-17, IL-21, and IL-23) (29), resulting in steatohepatitis and fibrosis. The extra-hepatic effect, is increased production and release of neutrophils (101), and greater polarization to Th17 response, increasing further cellular imbalances, and acting as a feedback loop. Th17-secreted cytokines are listed in red.
Emerging and Potentials Therapeutics Agents Targeting the Immune Response in NAFLD
The immune imbalances underlying the evolution of NAFLD has been explored as therapeutic targets for new drugs (or pleiotropic effects of old drugs) with the potential to slow the disease progression (114, 128).
Targeting the IL-17 Axis and Related Signaling Pathway
Statins
Some of the drugs that have emerged with therapeutic potential in NAFLD are statins (129–131). Recent studies have increasingly highlighted the immunomodulatory role of statins (132–134); and shown in some diseases (other than NAFLD), its potential to interfere in the IL-17 axis, both by inhibiting the differentiation of Th17 cells, decreasing the production of IL-17 (62, 133, 135, 136), and inducing upregulation and recruitment of Treg (133, 137–139), shifting thus T cells response to Treg dominance (134, 140), which has an anti-inflammatory role and is a coordinator of immunologic tolerance (98, 99). This effect was also evident in atherosclerosis where the administration of statins was associated with the accumulation of T regulatory cells in atherosclerotic plaque (141, 142). Among the mechanisms underlying this effect on Treg induction is by modulating the TGF-β1 signal transduction (139). It is very likely that other pleiotropic effects, often unpredictable on statins, may participate in the mediation of this benefit, such as those related to the antioxidant effect (143).
In NAFLD, the use statins reduced the risk of both NASH (OR 0.57, P = 0.055) and fibrosis (OR 0.47, P = 0.011) (144). In a study involving 42 patients with dyslipidemia and biopsy-proven NASH who underwent treatment with atorvastatin (10 mg/day) for 12 months, atorvastatin improved NASH activity score and increased liver to spleen density ratio, and this improvement was accompanied by a significant reduction of inflammation markers (145). In parallel, atorvastatin significantly decreased liver transaminase, γ-glutamyl transpeptidase, low-density lipoprotein cholesterol, and triglycerides (145). In another study with 20 patients with HIV and biopsy-proven NASH, the use of rosuvastatin ameliorated NASH in 19 of 20 patients within 12 months (130). Given this known plausibility of immune imbalances in NAFLD pathophysiology and the effect of statins in reestablishing the balance, studies with appropriate design are needed to confirm or refute this effect.
Vitamin D
Another agent that has shown important participation on Th17/Treg lymphocytes differentiation and IL-17 axis modulation is vitamin D and its isoforms (146–148). In the liver, studies have shown a significant association between vitamin D deficiency or insufficiency with NAFLD, principally in men and diabetics patients (149–152), and increased significantly the risk of NASH, fibrosis, and NASH severity in both adults and children with NAFLD (5, 153). Vitamin D supplementation in subjects with NAFLD reduced liver fibrosis through counteracting TGF-β-induced fibrogenesis (154, 155) and reduced the inflammatory response and insulin resistance as surrogate outcomes (155, 156). In one of these studies, patients with NASH had higher levels of TGF-β1 than those with simple steatosis, and the improvement of the inflammation and fibrosis after treatment was accompanied by a reduction in TGF-β1 levels (155). In the experimental model, the supplementation slowed the development and progression of NASH (157). So, available vitamin D compounds or vitamin D receptor agonists can bring another target to treat NAFLD, either as by modulating the IL-17 axis or other mechanisms.
Monoclonal Antibodies
It is worth remembering that there are already monoclonal antibodies against IL-17 (secukinumab and ixekizumab), already released for the treatment of rheumatic diseases that have activation of IL-17-axis as a crucial point in its pathogenesis (91), and the newest and attractive tregalizumab a monoclonal antibody that binds to CD4 T cells and induces Treg activation (158). Considering the key role of IL-17 in NAFLD, these agents may have a protective effect on the progression of NAFLD. In fact, in experimental studies, anti-IL-17 antibody improved hepatic steatosis by suppressing interleukin-17-related fatty acid metabolism (159). However, there are no clinical studies that have tested the use of this agent in NAFLD.
Probiotics and Retinoic Acid
Another aspect that opens new therapeutic potentials is the consideration of the intestine as a primary source of lymphocytes in NAFLD, and the modulator role of microbiota in this cellular population (64, 65). In fact, the administration of lactobacillus and other probiotics shows to decrease Th17 cell population and IL-17 secretion, while increasing Treg cell population (160–162). In addition, it has been shown that retinoic acid has an important role as a modulator of the cell response differentiation in gut, favoring to the Treg cells (94, 95, 163, 164). So studies with retinoic acid or its receptor agonists may bring the next NAFLD treatments targeting the IL-17 axis. It wins a particular interest by the observation that low levels of retinoic acid in serum or its receptor in hepatic tissue are associated with higher severity of NAFLD (165); and the demonstration that the administration of all-trans retinoic acid ameliorates hepatic steatosis in experimental research (166). Like statins, retinoic acid modulates the TGF-β1 signal transduction inducing to Treg response, and this may be one of the mechanisms underlying its beneficial effect (95, 97).
Other Potential Agents
Several other agents have the potential to act on the IL-17 axis (shifting the Th17/Treg balance in favor of Treg dominance), including agents such as rennin–angiotensin system blockers and digoxin (51, 142, 167). In a study including 159 hypertensive patients, the association of telmisartan with rosuvastatin showed a synergistic effect on ameliorating Th17/Treg functional imbalance, with a significant decrease in Th17 cells frequency, IL-17, IL-6, TNF-α, IL-1β, IL-2, IFN-γ, hs-CRP, and MCP-1, TGF-β3 (142). In another study, animals treated with digoxin, which also inhibits Th17 differentiation, presented with reduced levels of circulating Th17 cells and serum IL-17A, associated with reduced liver steatosis, liver immune cell infiltration, and liver injury; and increased glucose tolerance and insulin sensitivity than non-treated mice (51). Other substances able to shift the imbalance of Treg/Th17 cells to Treg dominance and that have shown to relieve NAFLD are 3, 3′-diindolylmethane and flavonoids (32, 61).
Conclusion and Future Directions
The available studies point to an important value of immunes imbalances, either at cellular or cytokines levels, in the pathogenesis of NAFLD. Particularly, the imbalances between neutrophils and lymphocytes counts (NLR) and at T helper cellular subsets (expressed by Th17/Treg imbalance). The reflection of this at the molecular level is a pro-inflammatory environment that includes IL-6, TNF-α, and TGF-β, and culminates in the hyperactivation of the IL-17 axis. The knowledge of this participation can help better understand the pathogenesis, offer non-invasive tools to evaluate the disease, and support the development of new therapeutic targets. Among these targets, the IL-17 axis and related signaling pathway is a potential; and agents such as statins, vitamin D, retinoic acid, probiotics, and monoclonal antibodies against IL-17 have a promissory perspective.
New studies should be designed for clarification between initiators, and perpetuator imbalances, and to prove the overall clinical utility of these imbalances as potential disease biomarkers. The effectiveness of therapies targeting the various mediators and pathways involved in disease pathogenesis should be evaluated by well-designed clinical randomized trials with adequate sample size and with histological assessment of the disease.
Author Contributions
FP: prepared the manuscript text and figures.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JW and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
1. Kanwal F, Kramer JR, Duan Z, Yu X, White D, El-Serag HB. Trends in the burden of nonalcoholic fatty liver disease in a United States cohort of veterans. Clin Gastroenterol Hepatol (2016) 14:301–8.e1–e2. doi:10.1016/j.cgh.2015.08.010
2. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med (2010) 363:1341–50. doi:10.1056/NEJMra0912063
3. Targher G, Bertolini L, Padovani R, Rodella S, Tessari R, Zenari L, et al. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care (2007) 30:1212–8. doi:10.2337/dc06-2247
4. Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr (2013) 162:496–500.e1. doi:10.1016/j.jpeds.2012.08.043
5. Nobili V, Giorgio V, Liccardo D, Bedogni G, Morino G, Alisi A, et al. Vitamin D levels and liver histological alterations in children with nonalcoholic fatty liver disease. Eur J Endocrinol (2014) 170:547–53. doi:10.1530/EJE-13-0609
6. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (2016) 64:73–84. doi:10.1002/hep.28431
7. Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology (2011) 140:124–31. doi:10.1053/j.gastro.2010.09.038
8. Koehler EM, Schouten JNL, Hansen BE, van Rooij FJA, Hofman A, Stricker BH, et al. Prevalence and risk factors of non-alcoholic fatty liver disease in the elderly: results from the Rotterdam study. J Hepatol (2012) 57:1305–11. doi:10.1016/j.jhep.2012.07.028
9. Dunn W, Xu R, Wingard DL, Rogers C, Angulo P, Younossi ZM, et al. Suspected nonalcoholic fatty liver disease and mortality risk in a population-based cohort study. Am J Gastroenterol (2008) 103:2263–71. doi:10.1111/j.1572-0241.2008.02034.x
10. Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology (2005) 129:113–21. doi:10.1053/j.gastro.2005.04.014
11. Targher G, Bertolini L, Padovani R, Rodella S, Zoppini G, Pichiri I, et al. Prevalence of non-alcoholic fatty liver disease and its association with cardiovascular disease in patients with type 1 diabetes. J Hepatol (2010) 53:713–8. doi:10.1016/j.jhep.2010.04.030
12. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology (2010) 51:1820–32. doi:10.1002/hep.23594
13. Cheung C-L, Lam KSL, Wong ICK, Cheung BMY. Non-invasive score identifies ultrasonography-diagnosed non-alcoholic fatty liver disease and predicts mortality in the USA. BMC Med (2014) 12:154. doi:10.1186/s12916-014-0154-x
14. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol (2015) 62:S47–64. doi:10.1016/j.jhep.2014.12.012
15. Harley ITW, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology (2014) 59:1830–9. doi:10.1002/hep.26746
16. Park SH, Kim BI, Yun JW, Kim JW, Park DI, Cho YK, et al. Insulin resistance and C-reactive protein as independent risk factors for non-alcoholic fatty liver disease in non-obese Asian men. J Gastroenterol Hepatol (2004) 19:694–8. doi:10.1111/j.1440-1746.2004.03362.x
17. Farrell GC, Rooyen van D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver (2012) 6:149–71. doi:10.5009/gnl.2012.6.2.149
18. Riquelme A, Arrese M, Soza A, Morales A, Baudrand R, Pérez-Ayuso RM, et al. Non-alcoholic fatty liver disease and its association with obesity, insulin resistance and increased serum levels of C-reactive protein in Hispanics. Liver Int (2009) 29:82–8. doi:10.1111/j.1478-3231.2008.01823.x
19. Yoneda M, Mawatari H, Fujita K, Iida H, Yonemitsu K, Kato S, et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J Gastroenterol (2007) 42:573–82. doi:10.1007/s00535-007-2060-x
20. Lizardi-Cervera J, Chavez-Tapia NC, Pérez-Bautista O, Ramos MH, Uribe M. Association among C-reactive protein, fatty liver disease, and cardiovascular risk. Dig Dis Sci (2007) 52:2375–9. doi:10.1007/s10620-006-9262-6
21. Chiang C-H, Huang C-C, Chan W-L, Chen J-W, Leu H-B. The severity of non-alcoholic fatty liver disease correlates with high sensitivity C-reactive protein value and is independently associated with increased cardiovascular risk in healthy population. Clin Biochem (2010) 43:1399–404. doi:10.1016/j.clinbiochem.2010.09.003
22. Yilmaz H, Yalcin KS, Namuslu M, Celik HT, Sozen M, Inan O, et al. Neutrophil-lymphocyte ratio (NLR) could be better predictor than C-reactive protein (CRP) for liver fibrosis in non-alcoholic steatohepatitis(NASH). Ann Clin Lab Sci (2015) 45:278–86.
23. Lee Y-J, Lee H-R, Shim J-Y, Moon B-S, Lee J-H, Kim J-K. Relationship between white blood cell count and nonalcoholic fatty liver disease. Dig Liver Dis (2010) 42:888–94. doi:10.1016/j.dld.2010.04.005
24. Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol (2011) 2011:345803. doi:10.1155/2011/345803
25. Alkhouri N, Morris-Stiff G, Campbell C, Lopez R, Tamimi TA-R, Yerian L, et al. Neutrophil to lymphocyte ratio: a new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int (2012) 32:297–302. doi:10.1111/j.1478-3231.2011.02639.x
26. Liu H, Zhang H, Wan G, Sang Y, Chang Y, Wang X, et al. Neutrophil-lymphocyte ratio: a novel predictor for short-term prognosis in acute-on-chronic hepatitis B liver failure. J Viral Hepat (2014) 21:499–507. doi:10.1111/jvh.12160
27. Biyik M, Ucar R, Solak Y, Gungor G, Polat I, Gaipov A, et al. Blood neutrophil-to-lymphocyte ratio independently predicts survival in patients with liver cirrhosis. Eur J Gastroenterol Hepatol (2013) 25:435–41. doi:10.1097/MEG.0b013e32835c2af3
28. Motomura T, Shirabe K, Mano Y, Muto J, Toshima T, Umemoto Y, et al. Neutrophil-lymphocyte ratio reflects hepatocellular carcinoma recurrence after liver transplantation via inflammatory microenvironment. J Hepatol (2013) 58:58–64. doi:10.1016/j.jhep.2012.08.017
29. Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol (2011) 166:281–90. doi:10.1111/j.1365-2249.2011.04471.x
30. Rau M, Schilling A-K, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol (2016) 196:97–105. doi:10.4049/jimmunol.1501175
31. Li J, Qiu S-J, She W-M, Wang F-P, Gao H, Li L, et al. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PLoS One (2012) 7:e39307. doi:10.1371/journal.pone.0039307
32. Liu Y, She W, Wang F, Li J, Wang J, Jiang W. 3, 3’-diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol (2014) 23:489–98. doi:10.1016/j.intimp.2014.09.024
33. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
34. Geginat J, Paroni M, Maglie S, Alfen JS, Kastirr I, Gruarin P, et al. Plasticity of human CD4 T cell subsets. Front Immunol (2014) 5:630. doi:10.3389/fimmu.2014.00630
35. Mai J, Wang H, Yang X-F. Th 17 cells interplay with Foxp3+ Tregs in regulation of inflammation and autoimmunity. Front Biosci (Landmark Ed) (2010) 15:986–1006. doi:10.2741/3657
36. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13:668–77. doi:10.1016/j.autrev.2013.12.004
37. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nat Immunol (2007) 8:1390–7. doi:10.1038/ni1539
38. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8:967–74. doi:10.1038/ni1488
39. Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol (2009) 130:27–33. doi:10.1016/j.clim.2008.08.018
40. Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity (2008) 29:628–36. doi:10.1016/j.immuni.2008.07.018
41. Giles DA, Moreno-Fernandez MEM, Stankiewicz TTE, Cappelletti M, Huppert SS, Iwakura Y, et al. Regulation of inflammation by IL-17A and IL-17F modulates non-alcoholic fatty liver disease pathogenesis. PLoS One (2016) 11:e0149783. doi:10.1371/journal.pone.0149783
42. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (Lond) (2016) 130:193–203. doi:10.1042/CS20150405
43. Coulon S, Francque S, Colle I, Verrijken A, Blomme B, Heindryckx F, et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine (2012) 59:442–9. doi:10.1016/j.cyto.2012.05.001
44. Zimmermann E, Anty R, Tordjman J, Verrijken A, Gual P, Tran A, et al. C-reactive protein levels in relation to various features of non-alcoholic fatty liver disease among obese patients. J Hepatol (2011) 55:660–5. doi:10.1016/j.jhep.2010.12.017
45. Uchihara M, Izumi N. [High-sensitivity C-reactive protein (hs-CRP): a promising biomarker for the screening of non-alcoholic steatohepatitis (NASH)]. Nihon Rinsho (2006) 64:1133–8.
46. Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol (2008) 103:1372–9. doi:10.1111/j.1572-0241.2007.01774.x
47. Stärkel P, Sempoux C, Leclercq I, Herin M, Deby C, Desager J-P, et al. Oxidative stress, KLF6 and transforming growth factor-β up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J Hepatol (2003) 39:538–46. doi:10.1016/S0168-8278(03)00360-X
48. Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology (2014) 59:483–95. doi:10.1002/hep.26698
49. Cayón A, Crespo J, Mayorga M, Guerra A, Pons-Romero F. Increased expression of Ob-Rb and its relationship with the overexpression of TGF-beta1 and the stage of fibrosis in patients with nonalcoholic steatohepatitis. Liver Int (2006) 26:1065–71. doi:10.1111/j.1478-3231.2006.01337.x
50. Wei Y, Tian Q, Zhao X, Wang X. Serum transforming growth factor beta 3 predicts future development of nonalcoholic fatty liver disease. Int J Clin Exp Med (2015) 8:4545–50.
51. Gomes AL, Teijeiro A, Burén S, Tummala KS, Yilmaz M, Ari W, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell (2016) 30:161–75. doi:10.1016/j.ccell.2016.05.020
52. Kim EJ, Kim B, Seo HS, Lee YJ, Kim HH, Son H-H, et al. Cholesterol-induced non-alcoholic fatty liver disease and atherosclerosis aggravated by systemic inflammation. PLoS One (2014) 9:e97841. doi:10.1371/journal.pone.0097841
53. Moon SH, Noh TS, Cho YS, Hong SP, Hyun SH, Choi JY, et al. Association between nonalcoholic fatty liver disease and carotid artery inflammation evaluated by 18F-fluorodeoxyglucose positron emission tomography. Angiology (2015) 66:472–80. doi:10.1177/0003319714537872
54. Oruc N, Ozutemiz O, Yuce G, Akarca US, Ersoz G, Gunsar F, et al. Serum procalcitonin and CRP levels in non-alcoholic fatty liver disease: a case control study. BMC Gastroenterol (2009) 9:16. doi:10.1186/1471-230X-9-16
55. Wang L-R, Liu W-Y, Wu S-J, Zhu G-Q, Lin Y-Q, Braddock M, et al. Parabolic relationship between sex-specific serum high sensitive C reactive protein and non-alcoholic fatty liver disease in Chinese adults: a large population-based study. Oncotarget (2016) 7:14241–50. doi:10.18632/oncotarget.7401
56. Bahcecioglu IH, Yalniz M, Ataseven H, Ilhan N, Ozercan IH, Seckin D, et al. Levels of serum hyaluronic acid, TNF-alpha and IL-8 in patients with nonalcoholic steatohepatitis. Hepatogastroenterology (2005) 52:1549–53.
57. Seo YY, Cho YK, Bae J-C, Seo MH, Park SE, Rhee E-J, et al. Tumor necrosis factor-α as a predictor for the development of nonalcoholic fatty liver disease: a 4-year follow-up study. Endocrinol Metab (Seoul) (2013) 28:41. doi:10.3803/EnM.2013.28.1.41
58. Paredes-Turrubiarte G, González-Chávez A, Pérez-Tamayo R, Salazar-Vázquez BY, Hernández VS, Garibay-Nieto N, et al. Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin Exp Med (2016) 16:193–202. doi:10.1007/s10238-015-0347-4
59. Okumura A, Unoki-Kubota H, Matsushita Y, Shiga T, Moriyoshi Y, Yamagoe S, et al. Increased serum leukocyte cell-derived chemotaxin 2 (LECT2) levels in obesity and fatty liver. Biosci Trends (2013) 7(6):276–83. doi:10.5582/bst.2013.v7.6.276
60. Vonghia L, Ruyssers N, Schrijvers D, Pelckmans P, Michielsen P, De Clerck L, et al. CD4+ROR γ t++ and Tregs in a mouse model of diet-induced nonalcoholic steatohepatitis. Mediators Inflamm (2015) 2015:239623. doi:10.1155/2015/239623
61. Li J, Chen Z, Jiang J, He B. Regulation of pure total flavonoids from Citrus on TH17/Treg balance in mice with NASH. Zhongguo Zhong Yao Za Zhi (2015) 40:2644–8.
62. Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol (2008) 180:6988–96. doi:10.4049/jimmunol.180.10.6988
63. Hu Y, Zhang H, Li J, Cong X, Chen Y, He G, et al. Gut-derived lymphocyte recruitment to liver and induce liver injury in non-alcoholic fatty liver disease mouse model. J Gastroenterol Hepatol (2016) 31:676–84. doi:10.1111/jgh.13183
64. Omenetti S, Pizarro TT. The Treg/Th17 axis: a dynamic balance regulated by the gut microbiome. Front Immunol (2015) 6:639. doi:10.3389/fimmu.2015.00639
65. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep (2015) 5:8096. doi:10.1038/srep08096
66. Proctor MJ, McMillan DC, Morrison DS, Fletcher CD, Horgan PG, Clarke SJ. A derived neutrophil to lymphocyte ratio predicts survival in patients with cancer. Br J Cancer (2012) 107:695–9. doi:10.1038/bjc.2012.292
67. Paquissi FC. The role of inflammation in cardiovascular diseases: the predictive value of neutrophil-lymphocyte ratio as a marker in peripheral arterial disease. Ther Clin Risk Manag (2016) 12:851–60. doi:10.2147/TCRM.S107635
68. Azab B, Camacho-Rivera M, Taioli E. Average values and racial differences of neutrophil lymphocyte ratio among a nationally representative sample of United States subjects. PLoS One (2014) 9:e112361. doi:10.1371/journal.pone.0112361
69. Xu R, Huang H, Zhang Z, Wang F-S. The role of neutrophils in the development of liver diseases. Cell Mol Immunol (2014) 11:224–31. doi:10.1038/cmi.2014.2
70. Macek Jilkova Z, Afzal S, Marche H, Decaens T, Sturm N, Jouvin-Marche E, et al. Progression of fibrosis in patients with chronic viral hepatitis is associated with IL-17(+) neutrophils. Liver Int (2016) 36:1116–24. doi:10.1111/liv.13060
71. Abdel-Razik A, Mousa N, Shabana W, Refaey M, ElMahdy Y, Elhelaly R, et al. A novel model using mean platelet volume and neutrophil to lymphocyte ratio as a marker of nonalcoholic steatohepatitis in NAFLD patients. Eur J Gastroenterol Hepatol (2016) 28:e1–9. doi:10.1097/MEG.0000000000000486
72. Leithead JA, Rajoriya N, Gunson BK, Ferguson JW. Neutrophil-to-lymphocyte ratio predicts mortality in patients listed for liver transplantation. Liver Int (2015) 35:502–9. doi:10.1111/liv.12688
73. Yilmaz H, Ucan B, Sayki M, Unsal I, Sahin M, Ozbek M, et al. Usefulness of the neutrophil-to-lymphocyte ratio to prediction of type 2 diabetes mellitus in morbid obesity. Diabetes Metab Syndr (2015) 9:299–304. doi:10.1016/j.dsx.2014.04.009
74. Chen T-M, Lin C-C, Huang P-T, Wen C-F. Neutrophil-to-lymphocyte ratio associated with mortality in early hepatocellular carcinoma patients after radiofrequency ablation. J Gastroenterol Hepatol (2012) 27:553–61. doi:10.1111/j.1440-1746.2011.06910.x
75. Ji F, Liang Y, Fu S-J, Guo Z-Y, Shu M, Shen S-L, et al. A novel and accurate predictor of survival for patients with hepatocellular carcinoma after surgical resection: the neutrophil to lymphocyte ratio (NLR) combined with the aspartate aminotransferase/platelet count ratio index (APRI). BMC Cancer (2016) 16:137. doi:10.1186/s12885-016-2189-1
76. Huang G-Q, Zhu G-Q, Liu Y-L, Wang L-R, Braddock M, Zheng M-H, et al. Stratified neutrophil-to-lymphocyte ratio accurately predict mortality risk in hepatocellular carcinoma patients following curative liver resection. Oncotarget (2015) 7:5429–39. doi:10.18632/oncotarget.6707
77. Liao R, Tang Z-W, Li D-W, Luo S-Q, Huang P, Du C-Y. Preoperative neutrophil-to-lymphocyte ratio predicts recurrence of patients with single-nodule small hepatocellular carcinoma following curative resection: a retrospective report. World J Surg Oncol (2015) 13:265. doi:10.1186/s12957-015-0670-y
78. Limaye AR, Clark V, Soldevila-Pico C, Morelli G, Suman A, Firpi R, et al. Neutrophil-lymphocyte ratio predicts overall and recurrence-free survival after liver transplantation for hepatocellular carcinoma. Hepatol Res (2013) 43:757–64. doi:10.1111/hepr.12019
79. Li L, Huang L, Vergis AL, Ye H, Bajwa A, Narayan V, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest (2010) 120:331–42. doi:10.1172/JCI38702
80. Taylor PR, Bonfield TL, Chmiel JF, Pearlman E. Neutrophils from F508del cystic fibrosis patients produce IL-17A and express IL-23 – dependent IL-17RC. Clin Immunol (2016) 170:53–60. doi:10.1016/j.clim.2016.03.016
81. Syn W-K, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology (2010) 51:1998–2007. doi:10.1002/hep.23599
82. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell (2014) 26:549–64. doi:10.1016/j.ccell.2014.09.003
83. Wang S, Zhang C, Zhang G, Yuan Z, Liu Y, Ding L, et al. Association between white blood cell count and non-alcoholic fatty liver disease in urban Han Chinese: a prospective cohort study. BMJ Open (2016) 6:e010342. doi:10.1136/bmjopen-2015-010342
84. El Shahawy MS. Neutrophil to lymphocyte ratio as a new marker for predicting steatohepatitis in patients with nonalcoholic fatty liver disease. Int J Adv Res (2015) 3:688–97.
85. Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology (2012) 143:765–76.e1–3. doi:10.1053/j.gastro.2012.05.049
86. Zhang Y, Huang D, Gao W, Yan J, Zhou W, Hou X, et al. Lack of IL-17 signaling decreases liver fibrosis in murine schistosomiasis japonica. Int Immunol (2015) 27:317–25. doi:10.1093/intimm/dxv017
87. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol (2010) 40:1830–5. doi:10.1002/eji.201040391
88. Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med (2012) 52:59–69. doi:10.1016/j.freeradbiomed.2011.10.003
89. Okazaki I, Noro T, Tsutsui N, Yamanouchi E, Kuroda H, Nakano M, et al. Fibrogenesis and carcinogenesis in nonalcoholic steatohepatitis (NASH): involvement of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinase (TIMPs). Cancers (Basel) (2014) 6:1220–55. doi:10.3390/cancers6031220
90. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
91. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti–interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med (2012) 366:1190–9. doi:10.1056/NEJMoa1109997
92. Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol (2010) 130:1373–83. doi:10.1038/jid.2009.399
93. Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol (2010) 10:248–56. doi:10.1038/nri2742
94. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317:256–60. doi:10.1126/science.1145697
95. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun C-M, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204:1757–64. doi:10.1084/jem.20070590
96. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
97. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol (2008) 181:2277–84. doi:10.4049/jimmunol.181.4.2277
98. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
99. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol (2012) 30:733–58. doi:10.1146/annurev-immunol-020711-075043
100. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich J-M, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34:566–78. doi:10.1016/j.immuni.2011.03.018
101. McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol (2005) 175:404–12. doi:10.4049/jimmunol.175.1.404
102. Griffin GK, Newton G, Tarrio ML, Bu D, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-α sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol (2012) 188:6287–99. doi:10.4049/jimmunol.1200385
103. Roos AB, Sethi S, Nikota J, Wrona CT, Dorrington MG, Sandén C, et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2015) 192:428–37. doi:10.1164/rccm.201409-1689OC
104. Zhang Y, Chen L, Gao W, Hou X, Gu Y, Gui L, et al. IL-17 neutralization significantly ameliorates hepatic granulomatous inflammation and liver damage in Schistosoma japonicum infected mice. Eur J Immunol (2012) 42:1523–35. doi:10.1002/eji.201141933
105. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi:10.1038/ni1261
106. Giles D, Moreno-Fernandez M, Divanovic S. IL-17 axis driven inflammation in non-alcoholic fatty liver disease progression. Curr Drug Targets (2015) 16:1315–23. doi:10.2174/1389450116666150531153627
107. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28:454–67. doi:10.1016/j.immuni.2008.03.004
108. Vonghia L, Francque S. Cross talk of the immune system in the adipose tissue and the liver in non-alcoholic steatohepatitis: pathology and beyond. World J Hepatol (2015) 7:1905–12. doi:10.4254/wjh.v7.i15.1905
109. Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol (2014) 193:3925–33. doi:10.4049/jimmunol.1400861
110. Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol (2013) 191:1835–44. doi:10.4049/jimmunol.1203013
111. Chen J, Liao M, Gao X, Zhong Q, Tang T, Yu X, et al. IL-17A induces pro-inflammatory cytokines production in macrophages via MAPKinases, NF-κB and AP-1. Cell Physiol Biochem (2013) 32:1265–74. doi:10.1159/000354525
112. Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One (2011) 6:e18909. doi:10.1371/journal.pone.0018909
113. Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol (2007) 19:353–61. doi:10.1016/j.smim.2007.10.008
114. Xu R, Tao A, Zhang S, Zhang M. Neutralization of interleukin-17 attenuates high fat diet-induced non-alcoholic fatty liver disease in mice. Acta Biochim Biophys Sin (Shanghai) (2013) 45:726–33. doi:10.1093/abbs/gmt065
115. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat Immunol (2008) 9:650–7. doi:10.1038/ni.1613
116. Abdel-Razik A, Mousa N, Besheer TA, Eissa M, Elhelaly R, Arafa M, et al. Neutrophil to lymphocyte ratio as a reliable marker to predict insulin resistance and fibrosis stage in chronic hepatitis C virus infection. Acta Gastroenterol Belg (2015) 78:386–92.
117. Du W-J, Zhen J-H, Zeng Z-Q, Zheng Z-M, Xu Y, Qin L-Y, et al. Expression of Interleukin-17 associated with disease progression and liver fibrosis with hepatitis B virus infection: IL-17 in HBV infection. Diagn Pathol (2013) 8:40. doi:10.1186/1746-1596-8-40
118. Wang L, Chen S, Xu K. IL-17 expression is correlated with hepatitis B-related liver diseases and fibrosis. Int J Mol Med (2011) 27:385–92. doi:10.3892/ijmm.2011.594
119. Liu Y, Zhu H, Su Z, Sun C, Yin J, Yuan H, et al. IL-17 contributes to cardiac fibrosis following experimental autoimmune myocarditis by a PKCβ/Erk1/2/NF-κB-dependent signaling pathway. Int Immunol (2012) 24:605–12. doi:10.1093/intimm/dxs056
120. Feng W, Li W, Liu W, Wang F, Li Y, Yan W. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp Mol Pathol (2009) 87:212–8. doi:10.1016/j.yexmp.2009.06.001
121. Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J Exp Med (2010) 207:535–52. doi:10.1084/jem.20092121
122. Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int (2015) 88:776–84. doi:10.1038/ki.2015.200
123. Cooney LA, Fox DA. Regulation of Th17 maturation by interleukin 4. Crit Rev Immunol (2013) 33:379–87. doi:10.1615/CritRevImmunol.2013007096
124. Woo KS, Kim BG, Choi JL, Kim BR, Kim KH. Neutrophil-to-lymphocyte ratio is associated with impaired interferon-gamma release to phytohemagglutinin. PLoS One (2015) 10:e0125794. doi:10.1371/journal.pone.0125794
125. Lee J, Lee J, Park MK, Lim MA, Park EM, Kim EK, et al. Interferon gamma suppresses collagen-induced arthritis by regulation of Th17 through the induction of indoleamine-2,3-deoxygenase. PLoS One (2013) 8:e60900. doi:10.1371/journal.pone.0060900
126. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity (2007) 27:647–59. doi:10.1016/j.immuni.2007.07.023
127. Lu D-H, Guo X-Y, Qin S-Y, Luo W, Huang X-L, Chen M, et al. Interleukin-22 ameliorates liver fibrogenesis by attenuating hepatic stellate cell activation and downregulating the levels of inflammatory cytokines. World J Gastroenterol (2015) 21:1531–45. doi:10.3748/wjg.v21.i5.1531
128. Yamaguchi K, Nishimura T, Ishiba H, Seko Y, Okajima A, Fujii H, et al. Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int (2015) 35:550–61. doi:10.1111/liv.12645
129. Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek atorvastatin and coronary heart disease evaluation (GREACE) Study: a post-hoc analysis. Lancet (2010) 376:1916–22. doi:10.1016/S0140-6736(10)61272-X
130. Kargiotis K, Athyros VG, Giouleme O, Katsiki N, Katsiki E, Anagnostis P, et al. Resolution of non-alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World J Gastroenterol (2015) 21:7860. doi:10.3748/wjg.v21.i25.7860
131. Gómez-Domínguez E, Gisbert JP, Moreno-Monteagudo JA, García-Buey L, Moreno-Otero R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol Ther (2006) 23:1643–7. doi:10.1111/j.1365-2036.2006.02926.x
132. Zhang X, Markovic-Plese S. Statins’ immunomodulatory potential against Th17 cell-mediated autoimmune response. Immunol Res (2008) 41:165–74. doi:10.1007/s12026-008-8019-z
133. Li XL, Dou YC, Liu Y, Shi CW, Cao LL, Zhang XQ, et al. Atorvastatin ameliorates experimental autoimmune neuritis by decreased Th1/Th17 cytokines and up-regulated T regulatory cells. Cell Immunol (2011) 271:455–61. doi:10.1016/j.cellimm.2011.08.015
134. Forero-Peña DA, Gutierrez FRS. Statins as modulators of regulatory T-cell biology. Mediators Inflamm (2013) 2013:167086. doi:10.1155/2013/167086
135. Zhang X, Tao Y, Troiani L, Markovic-Plese S. Simvastatin inhibits IFN regulatory factor 4 expression and Th17 Cell differentiation in CD4+ T cells derived from patients with multiple sclerosis. J Immunol (2011) 187:3431–7. doi:10.4049/jimmunol.1100580
136. Zhang X, Tao Y, Wang J, Garcia-Mata R, Markovic-Plese S. Simvastatin inhibits secretion of Th17-polarizing cytokines and antigen presentation by DCs in patients with relapsing remitting multiple sclerosis. Eur J Immunol (2013) 43:281–9. doi:10.1002/eji.201242566
137. Tang T-T, Song Y, Ding Y-J, Liao Y-H, Yu X, Du R, et al. Atorvastatin upregulates regulatory T cells and reduces clinical disease activity in patients with rheumatoid arthritis. J Lipid Res (2011) 52:1023–32. doi:10.1194/jlr.M010876
138. Mira E, Leon B, Barber DF, Jimenez-Baranda S, Goya I, Almonacid L, et al. Statins induce regulatory T cell recruitment via a CCL1 dependent pathway. J Immunol (2008) 181:3524–34. doi:10.4049/jimmunol.181.5.3524
139. Kim YC, Kim KK, Shevach EM. Simvastatin induces Foxp3+ T regulatory cells by modulation of transforming growth factor-β signal transduction. Immunology (2010) 130:484–93. doi:10.1111/j.1365-2567.2010.03269.x
140. Maneechotesuwan K, Kasetsinsombat K, Wamanuttajinda V, Wongkajornsilp A, Barnes PJ. Statins enhance the effects of corticosteroids on the balance between regulatory T cells and Th17 cells. Clin Exp Allergy (2013) 43:212–22. doi:10.1111/cea.12067
141. Meng X, Zhang K, Li J, Dong M, Yang J, An G, et al. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol Med (2012) 18:598–605. doi:10.2119/molmed.2011.00471
142. Liu Z, Zhao Y, Wei F, Ye L, Lu F, Zhang H, et al. Treatment with telmisartan/rosuvastatin combination has a beneficial synergistic effect on ameliorating Th17/Treg functional imbalance in hypertensive patients with carotid atherosclerosis. Atherosclerosis (2014) 233:291–9. doi:10.1016/j.atherosclerosis.2013.12.004
143. Foster T, Budoff MJ, Saab S, Ahmadi N, Gordon C, Guerci AD. Atorvastatin and antioxidants for the treatment of nonalcoholic fatty liver disease: the St Francis heart study randomized clinical trial. Am J Gastroenterol (2011) 106:71–7. doi:10.1038/ajg.2010.299
144. Nascimbeni F, Aron-Wisnewsky J, Pais R, Tordjman J, Poitou C, Charlotte F, et al. Statins, antidiabetic medications and liver histology in patients with diabetes with non-alcoholic fatty liver disease. BMJ Open Gastroenterol (2016) 3:e000075. doi:10.1136/bmjgast-2015-000075
145. Hyogo H, Yamagishi S, Maeda S, Kimura Y, Ishitobi T, Chayama K, et al. Atorvastatin improves disease activity of nonalcoholic steatohepatitis partly through its tumour necrosis factor-α-lowering property. Dig Liver Dis (2012) 44:492–6. doi:10.1016/j.dld.2011.12.013
146. Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther (2007) 324:23–33. doi:10.1124/jpet.107.127209
147. Joshi S, Pantalena L-C, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol (2011) 31:3653–69. doi:10.1128/MCB.05020-11
148. Terrier B, Derian N, Schoindre Y, Chaara W, Geri G, Zahr N, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther (2012) 14:R221. doi:10.1186/ar4060
149. Wang D, Lin H, Xia M, Aleteng Q, Li X, Ma H, et al. Vitamin D levels are inversely associated with liver fat content and risk of non-alcoholic fatty liver disease in a Chinese middle-aged and elderly population: the Shanghai Changfeng Study. PLoS One (2016) 11:e0157515. doi:10.1371/journal.pone.0157515
150. Rhee E-J, Kim MK, Park SE, Park C-Y, Baek KH, Lee W-Y, et al. High serum vitamin D levels reduce the risk for nonalcoholic fatty liver disease in healthy men independent of metabolic syndrome. Endocr J (2013) 60:743–52. doi:10.1507/endocrj.EJ12-0387
151. Barchetta I, Angelico F, Del Ben M, Baroni MG, Pozzilli P, Morini S, et al. Strong association between non alcoholic fatty liver disease (NAFLD) and low 25(OH) vitamin D levels in an adult population with normal serum liver enzymes. BMC Med (2011) 9:85. doi:10.1186/1741-7015-9-85
152. Seo JA, Eun CR, Cho H, Lee SK, Yoo HJ, Kim SG, et al. Low vitamin D status is associated with nonalcoholic fatty liver disease independent of visceral obesity in Korean adults. PLoS One (2013) 8:e75197. doi:10.1371/journal.pone.0075197
153. Nelson JE, Roth CL, Wilson LA, Yates KP, Aouizerat B, Morgan-Stevenson V, et al. Vitamin D deficiency is associated with increased risk of non-alcoholic steatohepatitis in adults with non-alcoholic fatty liver disease: possible role for MAPK and NF-κB? Am J Gastroenterol (2016) 111:852–63. doi:10.1038/ajg.2016.51
154. Beilfuss A, Sowa J-P, Sydor S, Beste M, Bechmann LP, Schlattjan M, et al. Vitamin D counteracts fibrogenic TGF-β signalling in human hepatic stellate cells both receptor-dependently and independently. Gut (2015) 64:791–9. doi:10.1136/gutjnl-2014-307024
155. Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther (2001) 15:1667–72. doi:10.1046/j.1365-2036.2001.01083.x
156. Foroughi M, Maghsoudi Z, Askari G. The effect of vitamin D supplementation on blood sugar and different indices of insulin resistance in patients with non-alcoholic fatty liver disease (NAFLD). Iran J Nurs Midwifery Res (2016) 21:100–4. doi:10.4103/1735-9066.174759
157. Han H, Cui M, You X, Chen M, Piao X, Jin G. A role of 1,25(OH)2D3 supplementation in rats with nonalcoholic steatohepatitis induced by choline-deficient diet. Nutr Metab Cardiovasc Dis (2015) 25:556–61. doi:10.1016/j.numecd.2015.02.011
158. König M, Rharbaoui F, Aigner S, Dälken B, Schüttrumpf J. Tregalizumab – a monoclonal antibody to target regulatory T cells. Front Immunol (2016) 7:11. doi:10.3389/fimmu.2016.00011
159. Shi W, Zhu Q, Gu J, Liu X, Lu L, Qian X, et al. Anti-IL-17 antibody improves hepatic steatosis by suppressing interleukin-17-related fatty acid synthesis and metabolism. Clin Dev Immunol (2013) 2013:253046. doi:10.1155/2013/253046
160. Jan R-L, Yeh K-C, Hsieh M-H, Lin Y-L, Kao H-F, Li P-H, et al. Lactobacillus gasseri suppresses Th17 pro-inflammatory response and attenuates allergen-induced airway inflammation in a mouse model of allergic asthma. Br J Nutr (2012) 108:130–9. doi:10.1017/S0007114511005265
161. Li J, Sung CYJ, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci U S A (2016) 113:E1306–15. doi:10.1073/pnas.1518189113
162. Chen R-C, Xu L-M, Du S-J, Huang S-S, Wu H, Dong J-J, et al. Lactobacillus rhamnosus GG supernatant promotes intestinal barrier function, balances Treg and TH17 cells and ameliorates hepatic injury in a mouse model of chronic-binge alcohol feeding. Toxicol Lett (2016) 241:103–10. doi:10.1016/j.toxlet.2015.11.019
163. Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol (2009) 86:959–69. doi:10.1189/jlb.0109006
164. Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol (2015) 8:265–78. doi:10.1038/mi.2014.64
165. Liu Y, Chen H, Wang J, Zhou W, Sun R, Xia M. Association of serum retinoic acid with hepatic steatosis and liver injury in nonalcoholic fatty liver disease. Am J Clin Nutr (2015) 102:130–7. doi:10.3945/ajcn.114.105155
166. Kim SC, Kim C-K, Axe D, Cook A, Lee M, Li T, et al. All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology (2014) 59:1750–60. doi:10.1002/hep.26699
Keywords: non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, neutrophil-to-lymphocyte ratio, Th17 cells, Treg cells, interleukin-17
Citation: Paquissi FC (2016) Immune Imbalances in Non-Alcoholic Fatty Liver Disease: From General Biomarkers and Neutrophils to Interleukin-17 Axis Activation and New Therapeutic Targets. Front. Immunol. 7:490. doi: 10.3389/fimmu.2016.00490
Received: 09 August 2016; Accepted: 25 October 2016;
Published: 11 November 2016
Edited by:
Fulvio D’Acquisto, Queen Mary University of London, UKReviewed by:
Philippe Saas, EFS Bourgogne Franche-Comté, FranceJames R. Whiteford, Queen Mary University of London, UK
Copyright: © 2016 Paquissi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feliciano Chanana Paquissi, ZmVwYXF1aXNzaUBnbWFpbC5jb20=