 Barbara Rossi
Barbara Rossi Gabriela Constantin
Gabriela Constantin- Section of General Pathology, Department of Medicine, University of Verona, Verona, Italy
Experimental autoimmune encephalomyelitis (EAE) is the most common animal model of multiple sclerosis (MS), a chronic inflammatory autoimmune disease of the central nervous system (CNS) characterized by multifocal perivascular infiltrates that predominantly comprise lymphocytes and macrophages. During EAE, autoreactive T cells first become active in the secondary lymphoid organs upon contact with antigen-presenting cells (APCs), and then gain access to CNS parenchyma, through a compromised blood–brain barrier, subsequently inducing inflammation and demyelination. Two-photon laser scanning microscopy (TPLSM) is an ideal tool for intravital imaging because of its low phototoxicity, deep tissue penetration, and high resolution. In the last decade, TPLSM has been used to visualize the behavior of T cells and their contact with APCs in the lymph nodes (LNs) and target tissues in several models of autoimmune diseases. The leptomeninges and cerebrospinal fluid represent particularly important points for T cell entry into the CNS and reactivation following contact with local APCs during the preclinical phase of EAE. In this review, we highlight recent findings concerning the pathogenesis of EAE and MS, emphasizing the use of TPLSM to characterize T cell activation in the LNs and CNS, as well as the mechanisms of tolerance induction. Furthermore, we discuss how advanced imaging unveils disease mechanisms and helps to identify novel therapeutic strategies to treat CNS autoimmunity and inflammation.
Introduction
Immune responses directed against self-antigens of the central nervous system (CNS) underlie several diseases, including multiple sclerosis (MS), neuromyelitis optica, and acute disseminated encephalomyelitis. MS is a chronic inflammatory demyelinating disease of the CNS affecting approximately 2–2.5 million people worldwide and leading to chronic progressive disability in the majority of cases (1). MS is heterogeneous both clinically and histopathologically, suggesting that different effector cells and molecular mechanisms are involved in the induction of tissue destruction (2). The most common form of MS, known as relapsing–remitting MS (RRMS), is associated with acute inflammatory episodes that reduce neurological function. RRMS patients may experience some recovery between relapses, but in 80% of cases, the disease evolves to a more progressive form, termed secondary progressive MS (SPMS). The latter is associated with a gradual loss of neurological function and ascending paralysis, both of which are believed to be less dependent on inflammation (3). Epidemiological and identical twin studies suggest that both genetics and environmental factors may play a role in disease pathogenesis (4, 5). Although the cause of MS is unknown, the presence of perivascular mononuclear cell infiltrates and demyelination suggest the disease is induced by an autoimmune response (4, 6, 7). Autoreactive T cell activation in MS is mainly associated with infection and is probably mediated by molecular mimicry, novel antigen presentation, bystander activation, or the coexpression of T cell receptors (TCRs) with different specificities (8–12). Antigen-presenting cells (APCs) containing myelin antigens were identified in the cervical lymph nodes (LNs) from MS patients, suggesting that the induction and propagation of autoimmune responses in MS starts at the periphery, within CNS draining LNs (13–15).
Several animal models have been developed to simulate clinical and histopathological patterns of demyelinating CNS inflammation, and these have been studied to determine the key steps in MS pathogenesis. The preferred animal model of MS is experimental autoimmune encephalomyelitis (EAE), which was introduced in 1925 by Koritschoner and Schweinburg, who induced spinal cord (SC) inflammation in rabbits by inoculation with human SC material (16). Since then, EAE has been induced by the immunization of susceptible animal strains, including primates and rodents, and has been shown to mimic several aspects of human MS (17). EAE can also be induced by the adoptive transfer of myelin-specific CD4+ T cell lines produced in vitro, proving that EAE is induced by an autoimmune response to myelin antigens (18). The shared clinical and histopathological features of EAE and MS suggest that both are autoimmune diseases induced by CNS-specific CD4+ autoreactive major histocompatibility complex class II (MHC-II) restricted T cells, which trigger a cascade of pathogenic events resulting in inflammation, demyelination, and neurodegeneration (19). Indeed, DR15 and DQw6 are the most important genetic susceptibility factors for MS, and myelin antigen-specific CD4+ MHC-II restricted T cells are more abundant in the blood and cerebrospinal fluid (CSF) of MS patients (20–23). During EAE, CNS-specific T cells are activated in the peripheral lymphoid organs and then migrate into the CNS (24). Inside the CNS, the T cells are reactivated by resident or infiltrating activated APCs, which present MHC-II associated peptides, resulting in inflammation, demyelination, and axonal damage.
Experimental autoimmune encephalomyelitis is considered an invaluable tool to study the activation of autoreactive T cells in the peripheral immune compartment, their migration into and reactivation within the CNS, and the subsequent induction of CNS inflammation. The activation and trafficking of immune system cells in EAE/MS is not fully understood. Recently, in vivo imaging techniques, such as two-photon laser scanning microscopy (TPLSM), have provided insights into the underlying disease mechanisms, leading to the development of novel therapeutic strategies to delay the progression of the disease. In this review, we discuss recent work on immune responses during EAE, highlighting the use of in vivo imaging to investigate T cell activation in lymphoid organs and the CNS and to study the basis of novel disease mechanisms.
Immune Responses and Their Regulation During EAE
The most widely used protocol for EAE induction is currently based on the subcutaneous (sc) injection of an encephalitogenic peptide, which is emulsified in complete Freund’s adjuvant (CFA) containing mineral oil and Mycobacterium tubercolosis strain H37Ra, followed by intravenous (iv) administration of pertussis toxin as adjuvant. In the Swiss Jim Lambert (SJL) mouse (H-2s), EAE can be actively induced by immunization with CNS homogenate, proteolipid protein (PLP), myelin basic protein (MBP), or encephalitogenic epitopes of PLP (PLP139–151, PLP178–191), myelin oligodendrocyte protein (MOG92–106), or MBP (MBP84–104) in an emulsion with CFA (25). The disease follows a predictable clinical course, characterized by a prodromal period of 10–15 days followed by ascending paralysis beginning in the tail and hind limbs and progressing to the forelimbs concurrent with weight loss. In SJL mice, the disease involves a relapsing–remitting course of paralysis, allowing for mechanistic studies or immunomodulatory strategies in a relapsing autoimmune disease setting. MOG35–55 is a potent encephalitogenic peptide in C57BL/6 (H-2b) mice, and immunization with this peptide leads to chronic progressive disease. Generally, the resulting clinical EAE phenotype depends mainly on the antigen source and the genetic background of the animal species and strain.
Experimental autoimmune encephalomyelitis is a useful model for the investigation of immunological mechanisms responsible for the inflammatory autoimmune process in MS. During EAE, naïve autoreactive CD4+ T cells are activated in the secondary lymphoid organs and reach the CNS through the blood by extravasation across the blood–brain barrier (BBB) (26). Inside the CNS, the autoreactive CD4+ T cells are reactivated by resident or migrating APCs displaying CNS self-antigens, which are necessary for T-cell reactivation. This process is required for the pathogenesis of MS and EAE because it induces the production of soluble pro-inflammatory mediators (26). These molecules may trigger the recruitment of other inflammatory cells, including innate immune system cells, which are key contributors to demyelination and axonal damage (26).
Autoimmune diseases also reflect a failure to sustain immune tolerance to self and/or cross-reactive molecules. EAE models have contributed to the understanding of immunoregulatory processes during the pathogenesis of MS, and CD4+CD25+FoxP3+ regulatory T (Treg) cells represent the most efficient immunoregulatory cellular mechanism (27–30). Abnormalities in Treg generation and function are considered a primary cause of autoimmune disease and other immunological disorders (31). These cells represent 5–10% of the CD4+ T lymphocytes in healthy adult mice and humans, and they have a specialized role in controlling both the innate and adaptive immune systems (32, 33). Treg cells have been shown to modulate neuroinflammatory processes in several EAE studies. For example, Rag−/− MBP-TCR transgenic mice develop spontaneous EAE and the depletion or inactivation of Treg cells by the injection of an anti-CD25 monoclonal antibody results in a massive activation of autoreactive T cells, leading to more severe EAE and a delayed or abrogated recovery phase (34–36). In EAE induced by MOG35–55, both antigen-specific T-effector and Treg cells differentiate and proliferate in the periphery before migrating to the CNS, with Treg cells necessary for natural recovery after the disease peak observed in immunized EAE mice (36, 37). In both actively induced and passively induced EAE models, the accumulation and expansion of Treg cells in the CNS correlates with recovery (36, 38). Dendritic cells (DCs) may be a major target of Treg-dependent immunoregulation in lymphoid organs during EAE and other animal models of autoimmunity (39–41).
Visualization of T Cell Dynamics In Vivo by TPLSM – An Introduction
Two-photon laser scanning microscopy is advantageous because it achieves deep tissue penetration and high resolution with low phototoxicity, making it ideal to visualize immune system cells in living animals and to provide dynamic views of leukocytes (42, 43). TPLSM movies are acquired by rendering a raw time series of three-dimensional images with one or more fluorescent channels. Cell tracking requires the location of each cell within three-dimensional space at successive time points, and repeated imaging at defined time intervals can be used to determine a cell’s migratory path, velocity, motility, chemotactic index, and physical interactions with tissue components (43). The behavior of different cell types and the molecular mechanisms controlling their migration can be studied by using TPLSM to compare two or more experimental conditions (e.g., wild-type versus knockout animals, or animals with and without defined cell populations).
T cells are constantly in motion under physiological and pathological conditions, traveling over long distances in the blood, migrating into lymphoid or non-lymphoid organs through the endothelial wall, and then actively moving inside the tissues. TPLSM has been used extensively to study the motility behavior of these cells inside lymphoid and non-lymphoid tissues during autoimmune responses. For example, TPLSM has shown that following extravasation in the LNs, T cells initially move at high velocity, displaying a random walk or directed migration pattern to facilitate antigen surveillance (44–46). If migrating T cells come into direct contact with APCs in the vicinity of high endothelial venules (HEVs), their dynamic behavior undergoes changes, such as reduced motility or organization into clusters, leading to cell activation and proliferation (47, 48). T cell crawling on DCs has also been visualized in the LNs in the T cell area (49, 50). During viral infections, central memory CD8+ T cells may encounter antigen also in the peripheral areas in LNs presented by subcapsular sinus macrophages (51). Tolerance induction is accompanied by less stable DC/T cell interactions with shorter T cell/APC contact times, smaller DC/T cell clusters, and a rapid restoration of effector T cell motility (39–41, 52–54). Therefore, the detection of antigens and subsequent immune responses requires the long-range migration of cells, short-range communication, and direct cell–cell contact with APCs.
The reactivation of primed T cells in the CNS is necessary for the initiation of inflammatory responses and requires physical contact between T cells and local APCs. In the last decade, TPLSM has revealed the motility behavior of migrating T cells in the CNS and their reactivation inside the SC parenchyma, which is the main site of CNS inflammation in most EAE models (55–58). Importantly, T cells interact with APCs in the subarachnoid space during the early stages of EAE, and these contacts seem critical for T cell reactivation and expansion in the CNS (57, 59). TPLSM studies have been pivotal to the investigation of immune system cell behavior in the lymphoid tissue and CNS during autoimmune diseases, and the dynamic behavior of effector T cells and Treg cells during the course of EAE is discussed in detail below.
T Cell Dynamics in the LNs During EAE
In the absence of immunization, naïve T cells continuously migrate and scan antigen-presenting DC populations within the T cell zones of LNs (46, 49). Their fast and stochastic motility behavior facilitates a series of random encounters between antigen-specific T cells and DCs, thus favoring the initiation of potential adaptive immune responses (48, 60, 61). However, this apparently non-directed motility in the LNs is guided on rails, namely the fibroblastic reticular cells (FRCs), which are covered in chemokines and form tight networks in secondary lymphoid organs (62).
Dendritic cells in LNs upregulate their surface-processed antigen about 3 h after immunization, whereas migratory DCs appear after ~24 h (63, 64). Following immunization, interactions between antigen-specific T cells and antigen-loaded DCs in the LNs are prolonged compared to the steady state (65). They also undergo changes in motility involving initial transient serial encounters, followed by a phase of slowing and stable contacts, and then by a return to high motility (38, 48, 49, 66, 67). During antigen recognition, TCR ligation reduces or arrests T cell motility by sending stop signals through LFA-1 integrin, allowing the formation of stable T cell/APC conjugates and efficient activation (67–69). In agreement with TPLSM studies in other experimental models of autoimmunity, the induction of active EAE is followed within 20–24 h by a substantial reduction in the speed and motility coefficients of antigen-specific naïve T cells in response to antigen challenge in the draining LNs. Simultaneously, the arrest coefficient of these cells increases significantly, suggesting there is direct physical contact with DCs as confirmed by measuring individual T cell/DC interactions frame by frame (40, 41).
Dendritic cells are essential not only for the induction of antigen-specific immune responses but also for the maintenance of peripheral tolerance (70). In vivo studies have shown that immunity and tolerance both require the activation and clonal expansion of T cells following antigen-specific interactions between naïve T cells and APCs (71, 72). For example, an elegant TPLSM study using a tolerogenic immunoglobulin carrying the MOG35–55 peptide (Ig-MOG) has revealed T cell motility and T cell/DC interactions leading to T cell tolerance during EAE induced in 2D2 TCR transgenic mice (73, 74). This study showed that tolerized T cells had an activated T-helper (Th)2 phenotype resulting in the secretion of IL-4 and IL-5 cytokines, and their motility was reduced following tolerogen exposure similar to Th1 cells after immunization (74). However, the Th2 cells had higher migration velocities and took longer to exhibit changes in motility than Th1 cells, suggesting that both Th1 immunity and Th2 tolerance alter T cell migration following antigen recognition (74).
Although T cell activation during the initial phase of the immune response in the LNs has been studied in detail, less is known about the later stages of the disease. We used TPLSM to show that efficient antigen-dependent priming is also maintained during the later stages of active EAE induced by MOG35–55 (41). Our results showed that 7 days after immunization, during the preclinical phase of the disease, ovalbumin (OVA)-specific T cells moved faster, had a higher motility coefficient and a lower arrest coefficient than MOG35–55-specific T cells. MOG-specific effector T cells were more motile, had a lower arrest coefficient and a shorter contact time with DCs 7 days after immunization compared to MOG-specific T cells after 1 day, suggesting antigen-dependent activation is less effective. However, the number of stable contacts between T cells and DCs was higher after 7 days, suggesting that efficient antigen-dependent priming is also maintained during later phases of the immune response. Interestingly, the percentage of intermediate contacts was strongly reduced after 7 days, and the immune response was more polarized, with most T cells establishing transient or prolonged contacts with DCs. Adoptively transferred T cells appeared to lack contact with B cells and macrophages, suggesting that DCs provide the main mechanism for ongoing T cell activation during the late phase of the immune response during EAE. Activated MOG35–55-specific (but not OVA-specific) T cells tended to cluster, keeping close to each other without physical contact and swarming into organized spatial patterns, suggesting that proliferation and therefore clustering are necessary for the clonal expansion and maintenance of an immune response in the LNs. Sustained T cell activation and proliferation requires persistent antigen availability and the presentation of antigen by migratory DCs (75, 76). Subcutaneous immunization with an emulsion containing CFA in the EAE model allows the slow release and persistent presentation of antigen, favoring prolonged antigen-dependent T cell activation (77). In addition, some T cells may be primed in the LNs and may therefore be activated more efficiently due to the presence of chemokines and cytokines released during the 7 days of continuous antigen recognition. The presence of cytokines and chemokines is supported by the clustering of T cells, which suggests potential clonal expansion. Taken together, these observations suggest that antigen-dependent T cell activation is maintained during the late phase of the immune response, highlighting the importance of prolonged and sustained immune responses for the successful induction of autoimmunity.
Treg Cell-Dependent Dynamics in the LNs During EAE
Regulatory T cells are a subset of suppressor T cells that contribute to the maintenance of immunological homeostasis and self-tolerance (78). The mechanisms by which Treg cells regulate immune responses are complex and incompletely understood, but there is a consensus that DCs are the major target of Treg cells in lymphoid organs (39, 40, 79, 80). The contacts between T cells and DCs are highly regulated events influenced by the timing of activation, the signal strength and the inflammatory environment, so the regulation of these contact dynamics by Treg cells is an essential component of the T cell activation process (60, 81, 82).
Two-photon laser scanning microscopy studies during preclinical EAE have shown that the presence of Treg cells increases the velocity and motility of T cells in draining LNs (as shown in Figure 1) (40). In addition, Treg cells reduce the arrest coefficient of T cells in draining LNs by limiting the contact time between T cells and antigen-loaded DCs (40, 41). It is unclear how Treg cells regulate T cell/DC interactions during EAE, but previous studies suggest that CTLA-4 may be necessary for the suppressive function of Treg cells in the draining LNs after immunization (83). Supporting this hypothesis, previous TPLSM studies have shown that the presence of CTLA-4 on Treg cells increases OVA-specific T cell motility and modulates the threshold for T cell activation (83). These studies of T cells specific for non-myelin antigens agree with TPLSM studies in EAE showing that Treg cells control the priming of autoreactive T cells at very early stages of CNS disease by preventing persistent T cell interactions with DCs in the LNs (40, 41). Further support comes from previous studies showing that the presence of CTLA-4 on Treg cells modulates CD80 and CD86 expression on DCs following antigen stimulation and suppresses the production of IL-6 and TNF-α by DCs (84–86). Finally, the analysis of explanted pancreatic LNs from non-obese diabetic mice has shown that persistent Treg/DC contacts prevent autoreactive T cell activation by DCs (39).
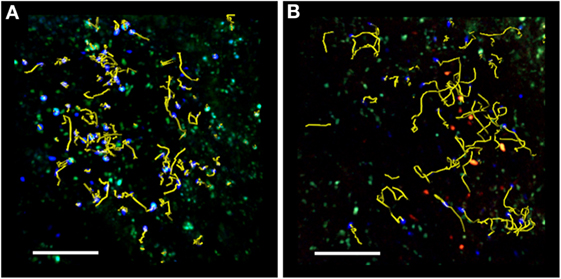
Figure 1. TPLSM imaging of an exposed lymph node after MOG35–55 immunization. Representative image of autoreactive T cells (blue) showing motility in the absence (A) or presence (B) of exogenous transplanted Treg cells (red). TPLSM was performed using MHC-II-GFP transgenic mice to visualize APCs (green). For an intuitive assessment of cell motility, yellow cell tracks are displayed graphically to indicate the progression of naïve T cell movement during the imaging period. The resulting positional information in four dimensions (xyz coordinates and time) is the basis for all subsequent computational analysis. Two-photon multidimensional data analysis parameters revealed that during MOG35–55 immunization, Treg cells modify the behavior of autoreactive T cells, increasing their motility and reducing their contact time with APCs in the peripheral LNs. Both figures are original. Scale bar = 100 micron.
We recently used TPLSM imaging to investigate the negative modulation exerted by Treg cells during the late phase of the immune response in the draining LNs in the EAE mouse. Imaging was carried out 7 days after immunization, when efficient antigen-dependent priming is maintained and the antigen-dependent clustering of effector T cells is associated with their high capacity for proliferation. We found that the presence of exogenous adoptively transferred Treg cells increased the velocity and motility of antigen-specific T cells and reduced the contact time between T cells and DCs (41). Furthermore, Treg cells inhibited the clustering of T cells and suppressed T cell proliferation in vivo, preferentially affecting Th1 rather than Th17 cell expansion during the late phase of the immune response (41). Previous studies have shown that the modulation of T cell proliferation by Treg cells depends on the strength of the antigenic stimulus, whereas the modulation of chemokine production in DCs by Treg cells does not (87). In this scenario, our data suggest that Treg cells may inhibit pro-inflammatory cytokine/chemokine production and secretion by DCs, which is responsible for T cell clustering and may efficiently inhibit weaker antigen-specific T cell activation 7 days after immunization during the late phase of the immune response in EAE (41).
The role of P-selectin glycoprotein ligand 1 (PSGL-1), a trafficking receptor for leukocytes during inflammation, is not restricted to its function as an adhesion molecule. Indeed, its role extends to the regulation of immune responses (41, 88). PSGL-1 is associated with a DC tolerogenic phenotype in mice, and PSGL-1 deficiency has been correlated with increased T-cell proliferation and autoimmunity in several disease models including EAE, suggesting a role for PSGL-1 in tolerogenic mechanisms (41, 89–92). To investigate the function of PSGL-1 in the amelioration of EAE by Treg cells, we recently used TPLSM to compare T cell dynamics in the presence of wild-type and PSGL-1 deficient Treg cells in the LNs of CD11c-YFP-immunized mice. We found that the transfer of exogenous wild-type Treg cells restored the motility of effector T cells by reducing the cell-cell contact time between effector T cells and DCs in immunological synapses during early-phase EAE (1 day after immunization) induced by MOG35–55. In this context, the presence of Treg cells lacking PSGL-1 increased the velocity of T cells to the same extent as wild-type Treg cells, confirming that PSGL-1 does not play any role during strong antigenic stimulation. At the later, preclinical phase of the disease (7 days after immunization) when the antigen-dependent activation of T cells declines but is still maintained in the LNs, Treg cells reduced the duration of contact between MOG35–55 T cells and DCs, as well as their spatial clustering. However, the presence of exogenous transplanted PSGL-1 deficient Treg cells was unable to inhibit the characteristic swarming into organized clusters and the resulting proliferation of MOG35–55 T effector cells (41). These data suggest that PSGL-1 is a key mediator of Treg-dependent suppression during the later stages of preclinical EAE, when antigenic stimulation in the LNs appears to be less efficient (41). The molecular mechanisms used by Treg cells to modulate later-phase immune responses are still unknown, but we propose that PSGL-1 expression on the surface of Treg cells could regulate DC activity by controlling contacts between the two cell types, as previously shown for LFA-1 (85). The molecular basis of such interactions may involve PSGL-1 on the surface of Treg cells binding to L-selectin on the surface of DCs, although mature DCs express such low levels of this molecule that alternative ligands are more likely (93). PSGL-1 may also inhibit antigen-dependent chemokine secretion by DCs (80, 94). Therefore, the recruitment of inflammatory cells by LNs and the co-localization of antigen-bearing DCs with antigen-specific T cells suggests that the regulation of interactions between Treg cells and DCs by PSGL-1 could modulate the local cytokine/chemokine profile in the LNs by reducing the clustering and activation of antigen-specific T cells (80, 94). In addition, PSGL-1 expressed on the surface of Treg cells may sequester chemokines, such as CCL19/CCL21, and thus prevent interactions between newly arrived CCR7+ T cells and APCs during EAE (95, 96).
Another aspect of tolerance induction during EAE that remains unclear is the requirement for antigen-specific Treg cells. The transfer of polyclonal Treg cells ameliorated EAE in one study (97), but conflicting results were produced in another (98). However, two independent TPLSM studies have demonstrated that polyclonal naïve Treg cells rapidly affect the motility of antigen-specific T cells, suggesting that antigenic specificity is not required by Treg cells to modulate the development of EAE in the secondary lymphoid organs (40, 41). These results are supported by studies suggesting that Treg cells may produce suppressive cytokines, thus overriding their antigen specificity (99, 100). Even so, studies in other experimental models support the need for antigen specificity, so further investigation is required to clarify whether antigen-specific suppression by Treg cells is required in the LNs during EAE (39, 101, 102).
The Dynamics of T Cell Activation in the Spinal Cord During EAE
In organ-specific autoimmune disease models, pathogenic autoreactive T cells are activated in the periphery of the lymphoid organs before entering the target organ. For example, in an intravenous injection model of transfer EAE, encephalitogenic T cells activated in vitro migrate into lymphoid organs before they cross the BBB and enter the CNS (103). The reactivation of primed CD4+ T cells in the CNS requires interactions with local APCs during the initiation of an inflammatory response (Figure 2). The behavior of autoreactive T cells was investigated in vitro for the first time using a combination of TPLSM and fluorescence video microscopy to study SC slices obtained at the onset of EAE in a rat model (55). The results showed that MBP-specific effector T cells move through the CNS with two distinct migratory patterns: ~65% of the cells moved rapidly and randomly through the compact white and gray matter, whereas the remaining ~35% appeared tethered to a fixed point, suggesting the formation of immune synapses (55). Importantly, pretreatment of the SC tissue with neutralizing anti-MHC-II monoclonal antibodies significantly reduced the number of arrested autoreactive T cells (55). In contrast to the MBP-specific T cells, OVA-specific T cells did not form synapse-like contacts in the SC in this model but invaded the CNS and moved rapidly through the tissue (55). More recent in vivo TPLS studies based on laminectomy in the lumbar SC of a rat EAE model were used to visualize T cells within the meningeal areas and the adjacent white matter. These studies revealed that an intravenous infusion of soluble MBP during the acute EAE phase caused rapid antigen uptake by APCs in the CNS, resulting in the deceleration of MBP-specific T cells, a higher frequency of T cell immobilization, a stronger activating state, and the enhanced production of inflammatory cytokines (56). In this EAE model, BBB leakage allowed the uptake of circulating antigen that was quickly processed by CNS cells expressing MHC-II, which were preferentially located in meningeal areas and in the vicinity of vessels, i.e., the regions mainly infiltrated by T cells. However, effector and resting memory T cells specific for the non-CNS antigen OVA were also recruited to EAE lesions and moved there without contacting APCs. As shown for the MPB-specific T cells, the OVA-specific T cells were activated and arrested following the intravenous infusion of OVA, which increased the severity of disease symptoms. This suggested that effector T cells home toward acute EAE lesions and can be reactivated locally by exogenous antigen regardless of the specificity of the intruding T cell population (56). Thus, non-CNS antigens leaking into the chronically inflamed CNS through the bloodstream may trigger relapses in MS (56).
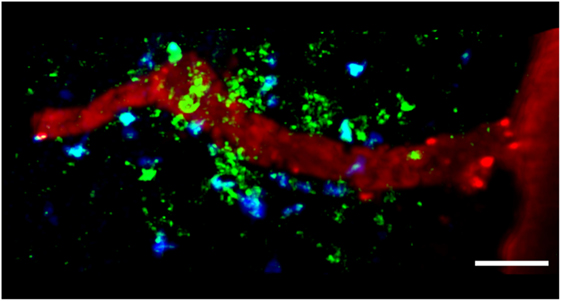
Figure 2. TPLSM imaging of autoreactive T cells interacting with perivascular MHC-II+ APCs in the exposed spinal cord during EAE. TPLSM was performed in MHC-II-GFP transgenic mice to visualize perivascular APCs (green) in contact with autoreactive T cells (blue). In order to investigate the types of APCs that can establish contacts with T cells, perivascular phagocytes were identified by intrathecally infused Texas Red-tagged dextran in MHC-II-GFP transgenic mice. These cells were located strategically around vessels, monitoring the environment with their cellular processes and sharing some morphological features with DCs, and others with macrophages. The merge of Texas Red and GFP positive cells provided evidence that perivascular phagocytes are functional APCs, expressing MHC-II determinants. The figure is original. Scale bar = 50 micron.
The MHC-II+ APCs located in the CNS during EAE mainly comprise perivascular macrophages and DCs, which induce the local reactivation of primed myelin-reactive T cells resulting in CNS inflammation and EAE progression (Figure 2) (55, 58). Flow cytometry showed that the most rapidly labeled phagocytes (30 min after the administration of intravenous antigen during the acute phase of EAE) share characteristics with macrophages, which express high levels of CD45, suggesting that these cells originate from the blood and are not resident cells (56). Later, the proteolytic digestion of the fluorescent antigen was observed in monocytes/macrophages, microglial cells, and B cells, suggesting these cells are involved in antigen presentation during EAE (56). The quantification of cell types in SC homogenates from mouse EAE models has confirmed the presence of CD11c+ DCs in the CNS, which are similar in phenotype and genotype to splenic DCs (104, 105). CNS-derived DCs can induce the proliferation of MOG-specific T cells, suggesting their importance in antigen presentation during EAE (105). Near-infrared imaging of DC transmigration has confirmed that these APCs can also migrate from the periphery into the CNS during EAE, and that their migration correlates with the severity of inflammation (106).
Recently, TPLSM imaging allowed the visualization of dynamic contacts between T cells and perivascular phagocytes (57, 107, 108). These results demonstrated that the leptomeninges and CSF are major routes for the migration of encephalitogenic T cells into the CNS during early phases of EAE in rats, and a site for T cell activation before the invasion of CNS parenchymal vessels (57, 107, 108). These studies showed that intravenously administrated MBP-specific T cells arrested on the leptomeningeal vessels and crawled intravascularly, preferentially against the blood flow (57). Following extravasation, T cells continue to scan the abluminal vascular surface and the underlying pial membrane, encountering phagocytes that effectively present antigens. These contacts induce the effector T cells to produce pro-inflammatory mediators, trigger tissue invasion and promote the formation of inflammatory infiltrates (57). Other reports confirm that T cells become activated in the perivascular space, but not within the vascular lumen, although previous in vitro studies have shown that endothelial cells can present antigen to adherent myelin-specific T cells (107, 109). In a MBP transfer EAE rat model, the comparison of highly immunogenic MBP-specific and modestly immunogenic MOG-specific T cells revealed that regardless of antigen specificity, the two cell types crawled with similar velocities in blood vessels (107). Once inside the CNS, the MBP-specific T cells moved within the leptomeningeal space with a lower velocity and mean square displacement than the MOG-specific T cells and had longer lasting contacts, whereas the MOG-specific T cells moved continuously in a straight line. However, activation signaling was not sufficient to completely arrest the highly immunogenic myelin-specific T cells in the leptomeningeal space, suggesting that contacts with APCs in the leptomeningeal area guide immigrant autoimmune T cells into the CNS parenchyma, rather than arresting them for extended periods of time (107). Before entering the CNS parenchyma, effector T cells traffic between the leptomeninges and CSF, with less adherent and activated T cells being flushed from the leptomeningeal network of collagen fibers by the CSF flow (108). TPLSM studies have shown that T cell adhesiveness to leptomeningeal structures is enforced by VLA-4 and LFA-1 integrins, CCR5/CXCR3-dependent signaling, and the antigenic stimulation of T cells in contact with leptomeningeal macrophages (108). However, T cells from the CSF fully retained their encephalitogenic potential and were able to reattach to the leptomeninges and invade the parenchyma (108). Together, these studies show that leptomeninges represent a key checkpoint for T-cell infiltration into the CNS during autoimmune inflammation or immune surveillance (57, 58, 108, 110).
It is unclear whether FRC structures like those in the LNs are present in the CNS parenchyma, but activated T cells can move in a similar manner in the CNS and LNs, suggesting analogous stromal structures are present in the CNS (111). Accordingly, TPLSM analysis in an experimental model of toxoplasmic encephalitis indicated the presence of an analogous reticular system associated with areas of parasite replication and local CNS inflammation, but not in normal brain tissues (111). CD4+ T-cell motility is actively promoted in the SC during EAE (112), and CD4+ T cells can gain access to CNS parenchyma and partially migrate along inflammation-induced extracellular matrix (ECM) structures, which are similar to those seen in LNs (112). These observations suggest that preexisting scaffolds guide lymphocyte migration in lymphoid tissues, but specialized structures are induced during CNS inflammation and guide T cell migration, potentially facilitating the screening of APCs and integrating relevant stimulatory, costimulatory, and regulatory signals (111–114).
The disease phase of effector T cell activation is crucial for the subsequent inflammatory process and the clinical disease (115). Using TPLSM, it was previously shown that the migration of myelin-specific T cells was mostly restricted to leptomeninges and perivascular areas during the preclinical phase and disease onset. However, during chronic phase of disease, T cells were spread throughout the white and gray matter (115). These studies have also shown that leptomeningeal phagocytes are responsible for the initial activation of encephalitogenic T cells in the CNS by mediating short cell–cell contacts with T cells (115). T cell activation process continues during disease progression and extends deeper in the CNS parenchyma through contacts with microglia and recruited phagocytes. However the initial activation during the preclinical phase of EAE seems to determine the clinical outcome of the autoimmune process (115). The visualization of effector T cells in their natural environment could also represent a useful tool to investigate the functional mechanisms behind therapeutic approaches during EAE. Using TPSLM, it was previously shown that administration of soluble myelin peptides or soluble intact MBP protein during the preclinical phase of diseases leads to a drastic reduction of antigen-specific T cell velocity in lymphoid peripheral organs (116). This efficiently prevented T cell migration into the CNS and abrogated CNS inflammation and disease development (116). In contrast, soluble antigen administration during established disease phase led to a strong aggravation of the clinical symptoms, increasing T cell activation and trafficking in the CNS (56).
Tolerogenic Dynamics in the Spinal Cord During EAE
The presence of Treg cells during early-phase autoimmune diseases increases the motility of antigen-specific T cells in the LNs and reduces their contact time with DCs in lymphoid organs, contributing to the maintenance of self-tolerance (39–41). This view of preventive immune suppression in the LNs during EAE may be too simplistic because Treg cells have also been detected in the inflamed CNS, where they can modify the motility behavior of antigen-specific T cells and their reactivation inside the CNS (Figure 3) (36, 37). The trafficking of Treg cells to peripheral inflammation sites is necessary for their suppression of inflammation (117). In actively and passively induced EAE models, the accumulation of Treg cells in the CNS correlates with restrained inflammation and recovery (36, 38). Also, Treg cells are enriched in the CSF of relapsing–remitting MS patients (118) suggesting they migrate to the inflamed CNS and are potentially responsible for immunosuppression also during human disease.
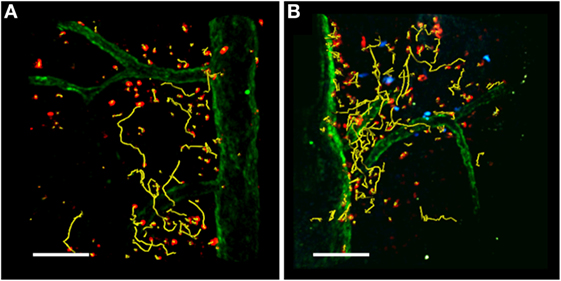
Figure 3. TPLSM imaging of the exposed spinal cord after MOG35–55 immunization. TPLSM representative dynamics (yellow tracks) of autoreactive Th1 cells (red) injected into C57Bl/6 EAE mice in the absence (A) or presence (B) of exogenous transplanted Treg cells (blue). Spinal cord vessels (green) were labeled by the systemic injection of 525 nm non-targeted quantum dots. All figures are original. Scale bar = 100 micron.
Two-photon laser scanning microscopy was recently combined with the in vivo depletion of Treg cells in a mouse EAE model to investigate the function of these cells in the CNS during EAE (119). Effector T cells were shown to move in confined trajectories in the meningeal SC of Treg cell-depleted mice, with a reduced mean velocity and linearity index compared to the control non-depleted animals. In parallel, the stationary phase of T effector cells was increased in the absence of Treg cells, indicating enhanced interactions with potential APCs within the inflamed SC during EAE associated with the increased capacity of T effector cells to proliferate in in the CNS. The suppression of T cell proliferation in the CNS of control mice with the normal complement of Treg cells was associated with lower levels of IFNγ but similar levels of IL-17 compared to Treg cell-depleted mice (119). These studies agree with previous data showing that CNS-derived Treg cells obtained during recovery suppress the in vitro proliferation of CNS-derived T effector cells in response to antigen by limiting the activation of cells producing IFNγ but not those producing IL-17 (38). Furthermore, CNS-derived Treg cells fail to control pathogenic T cells at peak EAE due to the local presence of IL-6 and TNF-α, potentially secreted by Th17 cells (37). Finally, the rapid progression of CNS autoimmunity can be prevented by Treg cells via their ability to suppress IFNγ-mediated immune responses (120). Collectively, these results suggest that Treg cells have a selective role at the site of CNS inflammation, depending on the local inflammatory setting, and the composition of the effector T cell population.
Conclusion
TPLSM allows the visualization of cell motility and interaction dynamics in the immune system of living animals, revealing the behavior of effector T-cells in LNs and peripheral tissue sites during inflammation and autoimmunity. Efficient T cell motility in the SC parenchyma is regulated by CNS resident cells and the ECM. Therefore, inhibiting the movement of effector T cells or their interactions with local APCs, or potentiating the activity of Treg cells in the CNS, may affect the development and progression of EAE/MS. The molecular mechanisms controlling the intra-tissue motility of activated T cell subsets, such as Th17 and Th1 cells in the CNS, and their interactions with local APCs could help to identify new treatments, ultimately for intrathecal administration, to complement existing systemic therapies for neurodegenerative autoimmune diseases. TPLSM could also be used to determine the molecular mechanisms by which Treg cells regulate the immune response during EAE/MS, which is necessary to fully understand their role. Advanced imaging techniques will also help in the future to provide insights into the mechanisms controlling T cell activation during different disease phases, such as the chronic phase, and may lead to the development of novel therapeutic strategies based on the inhibition of stage-specific mechanisms to delay the progression of the disease.
Author Contributions
BR and GC equally contributed to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by Fondazione Italiana Sclerosi Multipla (FISM) Cod. 2013/R/21 (to BR), and by the National Multiple Sclerosis Society (NMSS) (RG-1501-02926), New York, NY, USA, European Research Council grants (261079 NEUROTRAFFICKING, 693606 IMPEDE, and 695714 IMMUNOALZHEIMER), Italian Ministry of Health grant GR2009, Italian Ministry of Education and Research (MIUR), Alzheimer’s Drug Discovery Foundation (ADDF) USA (to GC).
References
1. Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev (2010) 9:387–94. doi:10.1016/j.autrev.2009.11.010
2. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol (2000) 47:707–17. doi:10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
3. Compston A, Coles A. Multiple sclerosis. Lancet (2008) 372:1502–17. doi:10.1016/S0140-6736(08)61620-7
4. Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol (1992) 10:153–87. doi:10.1146/annurev.iy.10.040192.001101
5. Sadovnick AD, Ebers GC. Epidemiology of multiple sclerosis: a critical overview. Can J Neurol Sci (1993) 20:17–29. doi:10.1017/S0317167100047351
6. Arnason BG. Relevance of experimental allergic encephalomyelitis to multiple sclerosis. Neurol Clin (1983) 1:765–82.
7. Raine CS. Biology of disease. Analysis of autoimmune demyelination: its impact upon multiple sclerosis. Lab Invest (1984) 50:608–35.
8. Delogu LG, Deidda S, Delitala G, Manetti R. Infectious diseases and autoimmunity. J Infect Dev Ctries (2011) 5:679–87. doi:10.3855/jidc.2061
9. Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity (2009) 30:348–57. doi:10.1016/j.immuni.2009.01.009
10. Muenz C, Luenemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol (2009) 9:246–58. doi:10.1038/nri2527
11. Olson JK, Croxford JL, Calenoff MA, Dal Canto MC, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. J Clin Invest (2001) 108:311–8. doi:10.1172/JCI13032
12. Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol (2010) 11:628–34. doi:10.1038/ni.1888
13. Fabriek BO, Zwemmer JN, Teunissen CE, Dijkstra CD, Polman CH, Laman JD, et al. In vivo detection of myelin proteins in cervical lymph nodes of MS patients using ultrasound-guided fine-needle aspiration cytology. J Neuroimmunol (2005) 161:190–4. doi:10.1016/j.jneuroim.2004.12.018
14. de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol (2002) 169:5415–23. doi:10.4049/jimmunol.169.10.5415
15. Weller RO, Galea I, Carare RO, Minagar A. Pathophysiology of the lymphatic drainage of the central nervous system: implications for pathogenesis and therapy of multiple sclerosis. Pathophysiology (2010) 17:295–306. doi:10.1016/j.pathophys.2009.10.007
16. Koritschoner RS, Schweinburg F. Induktion von Paralyse und Rückenmarksentzündung durch Immunisierung von Kaninchen mit menschlichem Rückenmarksgewebe. Z Immunitätsf Exp Therapie (1925) 42:217–83.
17. Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain (2006) 129:1953–71. doi:10.1093/brain/awl075
18. Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol (1981) 11:195–9. doi:10.1002/eji.1830110307
19. Martin R, McFarland HF. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit Rev Clin Lab Sci (1995) 32:121–82. doi:10.3109/10408369509084683
20. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23:683–747. doi:10.1146/annurev.immunol.23.021704.115707
21. Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science (1990) 247:718–21. doi:10.1126/science.1689076
22. Burns J, Bartholomew B, Lobo S. Isolation of myelin basic protein-specific T cells predominantly from the memory T cell compartment in multiple sclerosis. Ann Neurol (1999) 45:33–9. doi:10.1002/1531-8249(199901)45:1<33::AID-ART7>3.0.CO;2-G
23. Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med (1994) 179:973–84. doi:10.1084/jem.179.3.973
24. Furtado GC, Marcondes MC, Latkowski JA, Tsai J, Wensky A, Lafaille JJ. Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol (2008) 181:4648–55. doi:10.4049/jimmunol.181.7.4648
25. Mix E, Meyer-Rienecker H, Hartung HP, Zettl UK. Animal models of multiple sclerosis – potentials and limitations. Prog Neurobiol (2010) 92:386–404. doi:10.1016/j.pneurobio.2010.06.005
26. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol (2009) 9:393–407. doi:10.1038/nri2550
27. Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev (2014) 259:231–44. doi:10.1111/imr.12169
28. Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood (2008) 111:715–22. doi:10.1182/blood-2007-03-079947
29. Paintlia AS, Paintlia MK, Singh I, Singh AK. Combined medication of lovastatin with rolipram suppresses severity of experimental autoimmune encephalomyelitis. Exp Neurol (2008) 214:168–80. doi:10.1016/j.expneurol.2008.07.024
30. Tischner D, Weishaupt A, van den Brandt J, Müller N, Beyersdorf N, Ip CW, et al. Polyclonal expansion of regulatory T cells interferes with effector cell migration in a model of multiple sclerosis. Brain (2006) 129:2635–47. doi:10.1093/brain/awl213
31. Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi:10.1146/annurev.immunol.21.120601.141122
32. Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol (2002) 2:389–400. doi:10.1038/nri821
33. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
34. Olivares-Villagomez D, Wang Y, Lafaille JJ. Regulatory CD4+ T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med (1998) 188:1883–94. doi:10.1084/jem.188.10.1883
35. Cassan C, Piaggio E, Zappulla JP, Mars L, Couturier N, Bucciarelli F, et al. Pertussis toxin reduces the number of splenic Foxp3+ regulatory T cells. J Immunol (2006) 177:1552–60. doi:10.4049/jimmunol.177.3.1552
36. McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol (2005) 175:3025–32. doi:10.4049/jimmunol.175.5.3025
37. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med (2007) 13:423–31. doi:10.1038/nm1564
38. O’Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol (2007) 179:958–66. doi:10.4049/jimmunol.179.2.958
39. Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol (2006) 7:83–92. doi:10.1038/ni1289
40. Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, et al. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med (2006) 203:505–11. doi:10.1084/jem.20050783
41. Angiari S, Rossi B, Piccio L, Zinselmeyer BH, Budui S, Zenaro E, et al. Regulatory T cells suppress the late phase of the immune response in lymph nodes through P-selectin glycoprotein ligand-1. J Immunol (2013) 191:5489–500. doi:10.4049/jimmunol.1301235
42. Ustione A, Piston DW. A simple introduction to multiphoton microscopy. J Microsc (2011) 243:221–6. doi:10.1111/j.1365-2818.2011.03532.x
43. Zenaro E, Rossi B, Angiari S, Constantin G. Use of imaging to study leukocyte trafficking in the central nervous system. Immunol Cell Biol (2013) 91:271–80. doi:10.1038/icb.2012.81
44. Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol (2005) 23:127–59. doi:10.1146/annurev.immunol.23.021704.115628
45. Cahalan MD, Parker I. Choreography of cell motility and interaction dynamics imaged by two-photon microscopy in lymphoid organs. Annu Rev Immunol (2008) 26:585–626. doi:10.1146/annurev.immunol.24.021605.090620
46. Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc Natl Acad Sci U S A (2003) 100:2604–9. doi:10.1073/pnas.2628040100
47. Bajenoff M, Granjeaud S, Guerder S. The strategy of T cell antigen presenting cell encounter in antigen-draining lymph nodes revealed by imaging of initial T cell activation. J Exp Med (2003) 198:715–24. doi:10.1084/jem.20030167
48. Miller MJ, Safrina O, Parker I, Cahalan MD. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med (2004) 200:847–56. doi:10.1084/jem.20030167
49. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427:154–9. doi:10.1038/nature02238
50. Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol (2003) 4:579–85. doi:10.1038/ni928
51. Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, et al. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell (2012) 150:1249–63. doi:10.1016/j.cell.2012.08.015
52. Hugues S, Fetler L, Bonifaz L, Helft J, Amblard F, Amigorena S. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat Immunol (2004) 5:1235–42. doi:10.1038/ni1134
53. Shakhar G, Lindquist RL, Skokos D, Dudziak D, Huang JH, Nussenzweig MC, et al. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat Immunol (2005) 6:707–14. doi:10.1038/ni1210
54. Zinselmeyer BH, Dempster J, Gurney AM, Wokosin D, Miller M, Ho H, et al. In situ characterization of CD4+ T cell behavior in mucosal and systemic lymphoid tissues during the induction of oral priming and tolerance. J Exp Med (2005) 201:1815–23. doi:10.1038/ni1134
55. Kawakami N, Nägerl UV, Odoardi F, Bonhoeffer T, Wekerle H, Flügel A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J Exp Med (2005) 201:1805–14. doi:10.1084/jem.20050011
56. Odoardi F, Kawakami N, Klinkert WE, Wekerle H, Flügel A. Blood-borne soluble protein antigen intensifies T cell activation in autoimmune CNS lesions and exacerbates clinical disease. Proc Natl Acad Sci U S A (2007) 104:18625–30. doi:10.1073/pnas.0705033104
57. Bartholomäus I, Kawakami N, Odoardi F, Schläger C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature (2009) 462:94–8. doi:10.1038/nature08478
58. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med (2005) 11:328–34. doi:10.1038/nm1197
59. Kivisäkk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, et al. Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol (2009) 65:457–69. doi:10.1002/ana.21379
60. Bousso P. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat Rev Immunol (2008) 8:675–84. doi:10.1038/nri2379
61. Miller MJ, Hejazi AS, Wei SH, Cahalan MD, Parker I. T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc Natl Acad Sci U S A (2004) 101:998–1003. doi:10.1073/pnas.0306407101
62. Bajénoff M, Glaichenhaus N, Germain RN. Fibroblastic reticular cells guide T lymphocyte entry into and migration within the splenic T cell zone. J Immunol (2008) 181:3947–54. doi:10.4049/jimmunol.181.6.3947
63. Itano AA, McSorley SJ, Reinhardt RL, Ehstm BD, Ingulli E, Rudensky AY, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity (2003) 19:47–57. doi:10.1016/S1074-7613(03)00175-4
64. Manickasingham S, Reis e Sousa C. Microbial and T cell-derived stimuli regulate antigen presentation by dendritic cells in vivo. J Immunol (2000) 165:5027–34. doi:10.4049/jimmunol.165.9.5027
65. Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science (2002) 96:1873–6. doi:10.1126/science.1071065
66. Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science (2002) 296:1869–73. doi:10.1126/science.1070051
67. Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, et al. In vivo imaging of T cell priming. Sci Signal (2008) 1:t2. doi:10.1126/stke.112pt2
68. Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity (1996) 4:421–30. doi:10.1016/S1074-7613(00)80409-4
69. Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci U S A (1997) 94:3909–13. doi:10.1073/pnas.94.8.3909
70. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21:685–711. doi:10.1146/annurev.immunol.21.120601.141040
71. Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualisation of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity (1994) 1:327–39. doi:10.1016/1074-7613(94)90084-1
72. Smith KM, Davidson JM, Garside P. T-cell activation occurs simultaneously in local and peripheral lymphoid tissue following oral administration of a range of doses of immunogenic or tolerogenic antigen although tolerized T cells display a defect in cell division. Immunology (2002) 106:144–58. doi:10.1046/j.1365-2567.2002.01427.x
73. Legge KL, Gregg RK, Maldonado-Lopez R, Li L, Caprio JC, Moser M, et al. On the role of dendritic cells in peripheral T cell tolerance and modulation of autoimmunity. J Exp Med (2002) 196:217–27. doi:10.1084/jem.20011061
74. Divekar RD, Haymaker CL, Cascio JA, Guloglu BF, Ellis JS, Tartar DM, et al. T cell dynamics during induction of tolerance and suppression of experimental allergic encephalomyelitis. J Immunol (2011) 187:3979–86. doi:10.4049/jimmunol.1100531
75. Obst R, van Santen HM, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med (2005) 201:1555–65. doi:10.1084/jem.20042521
76. Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity (2008) 29:795–806. doi:10.1016/j.immuni.2008.08.013
77. Billiau A, Matthys P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J Leukoc Biol (2001) 70:849–60.
78. Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev (2001) 182:18–32. doi:10.1034/j.1600-065X.2001.1820102.x
79. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. doi:10.1038/nri2343
80. Dal Secco V, Soldani C, Debrat C, Asperti-Boursin F, Donnadieu E, Viola A, et al. Tunable chemokine production by antigen presenting dendritic cells in response to changes in regulatory T cell frequency in mouse reactive lymph nodes. PLoS One (2009) 4:e7696. doi:10.1371/journal.pone.0007696
81. Zinselmeyer BH, Dempster J, Wokosin DL, Cannon JJ, Pless R, Parker I, et al. Chapter 16. Two-photon microscopy and multidimensional analysis of cell dynamics. Methods Enzymol (2009) 461:349–78. doi:10.1016/S0076-6879(09)05416-0
82. Henrickson SE, von Andrian UH. Single-cell dynamics of T-cell priming. Curr Opin Immunol (2007) 19:249–58. doi:10.1016/j.coi.2007.04.013
83. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi:10.1126/science.1131078
84. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:27. doi:10.1126/science.1160062
85. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A (2008) 2105:10113–8. doi:10.1073/pnas.0711106105
86. Dejean AS, Beisner DR, Ch’en IL, Kerdiles YM, Babour A, Arden KC, et al. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat Immunol (2009) 10:504–13. doi:10.1038/ni.1729
87. Morlacchi S, Dal Secco V, Soldani C, Glaichenhaus N, Viola A, Sarukhan A. Regulatory T cells target chemokine secretion by dendritic cells independently of their capacity to regulate T cell proliferation. J Immunol (2011) 186:6807–14. doi:10.4049/jimmunol.1003265
88. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi:10.1038/nri2156
89. Urzainqui A, Martínez del Hoyo G, Lamana A, de la Fuente H, Barreiro O, Olazabal IM, et al. Functional role of P-selectin glycoprotein ligand 1/P-selectin interaction in the generation of tolerogenic dendritic cells. J Immunol (2007) 179:7457–65. doi:10.4049/jimmunol.179.11.7457
90. Matsumoto M, Miyasaka M, Hirata T. P-selectin glycoprotein ligand-1 negatively regulates T-cell immune responses. J Immunol (2009) 183:7204–11. doi:10.4049/jimmunol.0902173
91. He X, Schoeb TR, Panoskaltsis-Mortari A, Zinn KR, Kesterson RA, Zhang J, et al. Deficiency of P-selectin or P-selectin glycoprotein ligand-1 leads to accelerated development of glomerulonephritis and increased expression of CC chemokine ligand 2 in lupus-prone mice. J Immunol (2006) 177:8748–56. doi:10.4049/jimmunol.177.12.8748
92. Kerfoot SM, Norman MU, Lapointe BM, Bonder CS, Zbytnuik L, Kubes P. Reevaluation of P-selectin and alpha 4 integrin as targets for the treatment of experimental autoimmune encephalomyelitis. J Immunol (2006) 176:6225–34. doi:10.4049/jimmunol.176.10.6225
93. von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol (2003) 3:867–78. doi:10.1038/nri1222
94. Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol (2000) 18:593–620. doi:10.1146/annurev.immunol.18.1.593
95. Veerman KM, Williams MJ, Uchimura K, Singer MS, Merzaban JS, Naus S, et al. Interaction of the selectin ligand PSGL-1 with chemokines CCL21 and CCL19 facilitates efficient homing of T cells to secondary lymphoid organs. Nat Immunol (2007) 8:532–9. doi:10.1038/ni1456
96. Friedman RS, Jacobelli J, Krummel MF. Surface-bound chemokines capture and prime T cells for synapse formation. Nat Immunol (2006) 7:1101–8. doi:10.1038/ni1384
97. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol (2002) 169:4712–6. doi:10.4049/jimmunol.169.9.4712
98. Mekala DJ, Geiger TL. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Blood (2005) 105:2090–2. doi:10.1182/blood-2004-09-3579
99. Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-β-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med (2003) 9:1202–8. doi:10.1038/nm924
100. Homann D, Holz A, Bot A, Coon B, Wolfe T, Petersen J, et al. Autoreactive CD4+ T cells protect from autoimmune diabetes via bystander suppression using the IL-4/Stat6 pathway. Immunity (1999) 11:463–72. doi:10.1016/S1074-7613(00)80121-1
101. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med (2004) 199:1467–77. doi:10.1084/jem.20040180
102. Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol (2005) 174:6772–80. doi:10.4049/jimmunol.174.11.6772
103. Flügel A, Berkowicz T, Ritter T, Labeur M, Jenne D, Li Z, et al. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity (2001) 14:547–60. doi:10.1016/S1074-7613(01)00143-1
104. Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol (2008) 180:7376–84. doi:10.4049/jimmunol.180.11.7376
105. Anandasabapathy N, Victora GD, Meredith M, Feder R, Dong B, Kluger C, et al. Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J Exp Med (2011) 208:1695–705. doi:10.1084/jem.20102657
106. Sagar D, Lamontagne A, Foss CA, Khan ZK, Pomper MG, Jain P. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood-brain barrier through paracellular transmigration and ERK activation. J Neuroinflammation (2012) 9:245–60. doi:10.1186/1742-2094-9-245
107. Pesic M, Bartholomäus I, Kyratsous NI, Heissmeyer V, Wekerle H, Kawakami N. 2-photon imaging of phagocyte-mediated T cell activation in the CNS. J Clin Invest (2013) 123:1192–201. doi:10.1172/JCI67233
108. Schläger C, Körner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature (2016) 530:349–53. doi:10.1038/nature16939
109. Lopes Pinheiro MA, Kamermans A, Garcia-Vallejo JJ, van Het Hof B, Wierts L, O’Toole T, et al. Internalization and presentation of myelin antigens by the brain endothelium guides antigen-specific T cell migration. Elife (2016) 5:e13149. doi:10.7554/eLife.13149
110. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med (2011) 365:2188–97. doi:10.1056/NEJMoa1100648
111. Wilson EH, Harris TH, Mrass P, John B, Tait ED, Wu GF, et al. Behavior of parasite-specific effector CD8+ T cells in the brain and visualization of a kinesis-associated system of reticular fibers. Immunity (2009) 30:300–11. doi:10.1016/j.immuni.2008.12.013
112. Herz J, Paterka M, Niesner RA, Brandt AU, Siffrin V, Leuenberger T, et al. In vivo imaging of lymphocytes in the CNS reveals different behaviour of naïve T cells in health and autoimmunity. J Neuroinflammation (2011) 8:131. doi:10.1186/1742-2094-8-131
113. Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol (2000) 6:1991–2002. doi:10.1016/S0002-9440(10)64838-9
114. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol (2006) 65:124–41. doi:10.1097/65.2.124
115. Lodygin D, Odoardi F, Schläger C, Körner H, Kitz A, Nosov M, et al. A combination of fluorescent NFAT and H2B sensors uncovers dynamics of T cell activation in real time during CNS autoimmunity. Nat Med (2013) 9:784–90. doi:10.1038/nm.3182
116. Odoardi F, Kawakami N, Li Z, Cordiglieri C, Streyl K, Nosov M, et al. Instant effect of soluble antigen on effector T cells in peripheral immune organs during immunotherapy of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2007) 104:920–5. doi:10.1073/pnas.0608383104
117. Siegmund K, Feuerer M, Siewert C, Ghani S, Haubold U, Dankof A, et al. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood (2005) 106:3097–104. doi:10.1182/blood-2005-05-1864
118. Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol (2007) 147:412–8. doi:10.1111/j.1365-2249.2006.03271.x
119. Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol Commun (2014) 2:163. doi:10.1186/s40478-014-0163-1
Keywords: experimental autoimmune encephalomyelitis, T cell activation, regulatory T cells, two-photon microscopy
Citation: Rossi B and Constantin G (2016) Live Imaging of Immune Responses in Experimental Models of Multiple Sclerosis. Front. Immunol. 7:506. doi: 10.3389/fimmu.2016.00506
Received: 18 August 2016; Accepted: 01 November 2016;
Published: 21 November 2016
Edited by:
Melanie P. Matheu, Prellis Biologics, USAReviewed by:
Egle Solito, Queen Mary University of London, UKLara Campana, University of Edinburgh, UK
Copyright: © 2016 Rossi and Constantin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Rossi, YmFyYmFyYS5yb3NzaUB1bml2ci5pdA==;
Gabriela Constantin, Z2FicmllbGEuY29uc3RhbnRpbkB1bml2ci5pdA==