 Anders J. Svendsen1*
Anders J. Svendsen1* Kristina Gervin2Robert Lyle2Lene Christiansen1Kirsten Kyvik3Peter Junker4Christian Nielsen5
Kristina Gervin2Robert Lyle2Lene Christiansen1Kirsten Kyvik3Peter Junker4Christian Nielsen5 Gunnar Houen6
Gunnar Houen6 Qihua Tan1,7
Qihua Tan1,7
- 1The Danish Twin Registry, Epidemiology, Institute of Public Health, University of Southern Denmark, Odense, Denmark
- 2Department of Medical Genetics, Oslo University Hospital, University of Oslo, Oslo, Norway
- 3Denmark and Odense Patient data Explorative Network (OPEN), Institute of Clinical Research, Odense University Hospital, University of Southern Denmark, Odense, Denmark
- 4Department of Rheumatology, Odense University Hospital, University of Southern Denmark, Odense, Denmark
- 5Department of Clinical Immunology, Odense University Hospital, Odense, Denmark
- 6Department of Clinical Biochemistry and Immunology, Statens Serum Institute, Copenhagen, Denmark
- 7Unit of Human Genetics, Department of Clinical Research, University of Southern Denmark, Odense, Denmark
Objectives: In an explorative epigenome-wide association study (EWAS) to search for gene independent, differentially methylated DNA positions and regions (DMRs) associated with rheumatoid arthritis (RA) by studying monozygotic (MZ) twin pairs discordant for RA.
Methods: Genomic DNA was isolated from whole blood samples from 28 MZ twin pairs discordant for RA. DNA methylation was measured using the HumanMethylation450 BeadChips. Smoking, anti-cyclic citrullinated peptide antibodies, and immunosuppressive treatment were included as covariates. Pathway analysis was performed using GREAT.
Results: Smoking was significantly associated with hypomethylation of a DMR overlapping the promoter region of the RNF5 and the AGPAT1, which are implicated in inflammation and autoimmunity, whereas DMARD treatment induced hypermethylation of the same region. Additionally, the promotor region of both S100A6 and EFCAB4B were hypomethylated, and both genes have previously been associated with RA. We replicated several candidate genes identified in a previous EWAS in treatment-naïve RA singletons. Gene-set analysis indicated the involvement of immunologic signatures and cancer-related pathways in RA.
Conclusion: We identified several differentially methylated regions associated with RA, which may represent environmental effects or consequences of the disease and plausible biological pathways pertinent to the pathogenesis of RA.
Introduction
Rheumatoid arthritis (RA) belongs to the group of complex autoimmune diseases mediated by interactions between genetic and environmental exposures. Several genome-wide association studies (GWAS) have been undertaken. Around 60 risk alleles for RA have been identified, and it is anticipated that currently known genetic risk factors only account for 16% of the total susceptibility (1, 2). However, heritability estimates from RA twin studies vary considerably from 12 to 60% (3–5). In addition, there is evidence that only comparisons between dizygotic (DZ) and monochorionic monozygotic (MZ) twins are valid for inference of genetic heritability in classical twin studies, because dichorionic MZ twins are not only identical with regard to DNA sequence but also have a higher intraclass correlation of DNA methylation (DNAm) than both monochorionic MZ twins and DZ twins (6). Therefore, higher concordance in MZ twins may not only be due to a higher degree of DNA sequence similarity since molecular mechanisms of heritability may not be limited to the DNA sequence. In addition, the diverse clinical manifestations of RA within MZ pairs concordant for RA indicate the importance of non-genetic factors on the expression of the disease (7). Thus, there is mounting evidence that environmental factors, or stochasticity, play a strong role in the etiology and expression of RA and that the effect may be mediated through epigenetic mechanisms (6, 8). Thus, to identify environmentally induced DNAm changes associated with RA, we have therefore taken advantage of the disease-discordant identical twin design to adjust for most genetic effects and many non-genetic effects such as early environment, maternal-, age-, sex-, and cohort effects. Smoking is the hitherto strongest environmental risk factor associated with RA, and in particular in the subset of RA patients possessing anti-citrullinated protein antibodies (ACPA) (9). In addition, there is strong evidence to suggest that smoking is associated with changes in DNAm (10), and genome-wide methylation data obtained on DNA from peripheral blood leukocytes suggest the existence of dynamic, site-specific methylation changes in response to smoking, which may contribute to the extended risks associated with cigarette smoking that may persist many years after cessation (11). For these reasons we have included both smoking and the presence of ACPA as covariates in the data analysis.
Inflammatory arthritis may be associated with global genomic DNA hypomethylation and with specificity for some blood cell subpopulations that is reversed with methotrexate (MTX) treatment. These changes are accompanied by parallel changes in the levels of enzymes involved in methylation, suggesting the possibility of regulation at this level (12, 13). Treatment of RA patients with MTX has also been shown to regulate defective Treg cell function through demethylation of specific genetic loci (13). As we have investigated twins with established disease, who are currently or previously treated with disease-modifying anti-rheumatic drugs (DMARDs), we have included current DMARD treatment as a covariate in the data analysis. To our knowledge, the effect of other conventional DMARDs on DNAm has not been investigated. A recent study has suggested that DNA methylation profiling may provide a new biomarker of response to biologics (14), but so far, there is no evidence to suggest that biologics themselves have a direct effect on DNAm.
Epigenetics comprise a wide range of regulatory mechanisms including histone modification, miRNA expression, and DNAm. DNAm is under constant environmental influence, highly dynamic, and differs between cell-types (15). In RA, global DNA hypomethylation has been observed in both synovial fibroblasts (RASF) (12, 16–18), peripheral blood mononuclear cells (PBMCs), and in specific subsets of T- and B-lymphocytes that may be reversed by treatment (19). Several studies have focused on DNAm of candidate loci in PBMC from RA patients, while only few studies have included multiple loci or at the epigenome-wide level (20). We therefore performed an explorative epigenome-wide association study (EWAS) characterizing DNAm differences in PMBCs from RA discordant MZ twin pairs in order to identify potential genetically independent DNAm marks associated with RA.
Materials and Methods
Twins
Recruitment of 28 MZ twin pairs discordant for RA was done as previously described (4, 21). The median discordance time was 18 years (interquartile range 11–30 years). RA was classified according to the ACR 1987 criteria (22). Absence of RA was verified in the co-twins based on clinical examination. Zygosity was confirmed by genetic markers (23). DNA was extracted from EDTA blood and kept at −80°C until use. RA characteristics: females 78% mean age at disease onset 38 years, anti-CCP antibody positive 61%, ever smokers 69% (smoking information missing in 14%). Sixty-eight percent were currently treated with DMARD of which 80% were treated with MTX, and none were treated with biologics or steroids. Most of the twins were in clinical remission, and the average CRP value was 3.9 mg/ml, range 0–29.
DNAm Measurements
Genomic DNA from peripheral blood was bisulfite converted using the EZ DNA methylation Kit (ZYMO research), and DNAm status was assessed using the Infinium 450 K HumanMethylation BeadChip (Illumina) according to the manufacturer’s instructions at the Norwegian Microarray Consortium in Oslo. In order to minimize the batch effect on intra-pair DNAm differences, co-twins were processed together on the same chip. Data normalization was done using the free R package minfi, which employs subset quantile within-array normalization (24). The level of DNAm was summarized by calculating the “beta” value defined by the Illumina’s formula as β = M/(M + U + 100). We also performed QC using minfi to calculate the detection p-value defined as the proportion of control probes, which have intensities greater than that probe on the same array. A β value with its assigned detection p-value >0.01 was treated as missing. CpGs with more than 5% missing data were dropped from the subsequent analysis.
To adjust for differences in cell type composition between co-twins, we applied a statistical algorithm integrated in minfi (25, 26). All downstream analyses were based on this cell type adjusted dataset.
Statistical Analysis
Identification of Differentially Methylated Positions
To identify the differentially methylated positions (DMPs) associated with RA, we fitted a linear regression model (27) to predict the mean fold change in DNAm between co-twins discordant for RA at each CpG site with adjustment for age, sex, smoking, anti-CCP antibody, and current DMARD treatment. In this model, association with RA is indicated by an intercept α that is statistically different from 0 with α > 0 for increased and α < 0 for decreased methylation levels in diseased versus healthy co-twin. A slope parameter for smoking, anti-CCP antibody, or DMARD that is statistically above or below 0 indicates exposure associated hypermethylation (β > 0) or hypomethylation (β < 0). Genome-wide significance of DMPs was determined after correcting for multiple testing by calculating the false discovery rate (FDR) with a threshold of 0.05. DMPs reaching an uncorrected p-value <5 × 10−5 were defined as suggestive.
Identification of Differentially Methylated Regions
Differentially methylated regions (DMRs) were identified by the free R package bumphunter (28). First, we calculated the 99th percentile of the smoothed βs to obtain upper and lower thresholds. These thresholds were then used to define hypermethylated or hypomethylated DMRs with smoothed peaks above or below the thresholds defined as putative DMRs. For each putative DMR identified, bumphunter calculates a sum statistic by taking the sum of the absolute values of all the smoothed βs within that region. The sum statistic was then used to rank all DMRs with the top-most important DMRs having the highest sum statistic value. To determine the statistical significance of each putative DMR, we performed 1000 permutations of case–control status and estimated random DMRs for each permutation. Empirical genome-wide p-values were calculated based on family-wise error rate (FWER) that computed, for each observed DMR-area, the proportion of maximum area values per permutation that are larger than the observed area. A DMR reaching an empirical p-value <0.05 was defined as significant. We defined an observed DMR as suggestive if its area was larger than the smallest area in the 1000 maximum areas from each permutation. In addition to the empirical genome-wide p-value, we also estimated the empirical uncorrected p-value for a single DMR as the proportion of all random DMRs from 1000 permutations that are larger than the area of the observed DMR. Multiple testing was corrected for by calculating FDR to obtain genome-wide significance defined as FDR < 0.05. The results from this latter method were consistent with the results based on FWER. We present the mean fold change in RA twins compared to co-twins adjusted for age, sex, smoking, anti-CCP antibody, and DMARD treatment as well as the fold change in RA twins predicted by each of the covariates smoking, anti-CCP antibody, and DMARD treatment.
Gene-Set Analysis
We applied the DMRs as input genomic regions to Genomic Regions Enrichment of Annotations Tool [(GREAT) – version 2.0] to analyze the functional significance of cis-regulatory regions (29). Genome Reference Consortium Human Build 37 (GRCh37) was used as RefSeq database. GREAT was run against a whole genome background, and it performed both the binomial test over genomic regions and the hypergeometric test over genes to provide an accurate picture of annotation enrichments for genomic regions. We only present pathways with a fold enrichment of at least two by either test that are also significant at an FDR of 0.05 by both tests.
Results
DNAm in MZ Twins Discordant for RA
Since smoking is associated with both extensive changes in DNAm (10) and with anti-cyclic citrullinated peptide antibodies (anti-CCP antibodies) (9), smoking and anti-CCP antibodies were included as covariates. Further, MTX may elicit profound effects on DNAm (12). Since 68% of the patients were treated with DMARDs (80% with MTX), current treatment with DMARD was also included as a covariate. Analysis of these data revealed no genome-wide significant DMPs associated with RA or any of the covariates between co-twins (Figure 1).
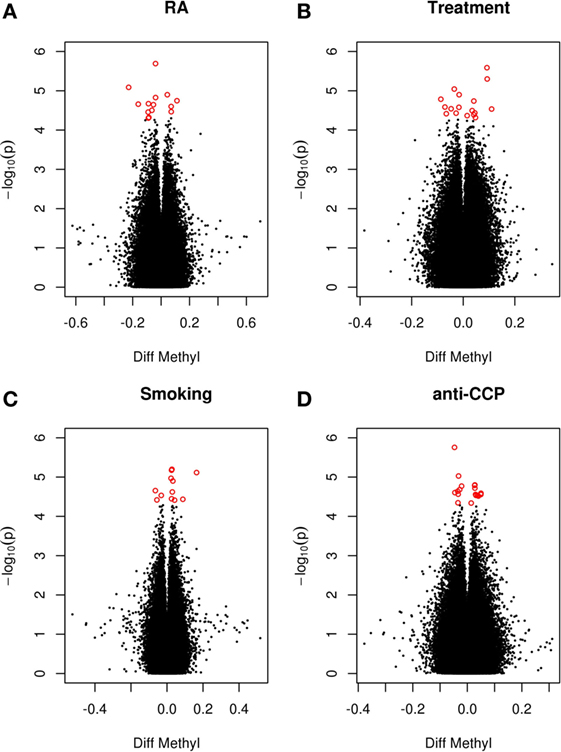
Figure 1. RA-associated DMPs. Volcano plot of detectable genome-wide differentially methylated CpG sites in 28 monozygotic RA discordant twin pairs adjusted for cell type. The −log values represent the logarithm with base 10 of the p-value against the methylation difference (beta values) between the RA twin and the non-RA co-twin. Positive values indicate hypermethylation in the RA twin compared to the co-twin. The red dots represent suggestive DMPs with a nominal p-value <5 × 10−5. (A) RA twin versus co-twin adjusting for smoking, anti-CCP antibody, and treatment. (B) RA twin versus co-twin predicted by smoking adjusting for anti-CCP antibody and treatment. (C) RA twin versus co-twin predicted by treatment adjusting for smoking and anti-CCP antibody. (D) RA twin versus co-twin predicted by anti-CCP antibody adjusting for smoking and treatment.
Regional Analyses Identify Differentially Methylated Regions
It has been reported that DNAm levels are strongly correlated across the genome and functionally relevant findings have in general been associated with genomic regions rather than single CpGs (30). The hitherto largest EWAS in RA singletons reported clustering of the most significant CpGs in regions and supports the use of region-based statistical approaches (31). Regional analysis is also less prone to be affected by the technical artifacts associated with individual probes. We therefore also performed regional analyses (i.e., “bump hunting”) (28), which allow effective modeling of measurement error and biological variability. We identified 603 putative DMRs (“bumps”) associated with RA adjusted for the other covariates. The unadjusted raw beta values of the six top ranked DMRs are presented in Figure S1 in Supplementary Material. In addition, we investigated the interaction between RA and each of the covariates smoking, anti-CCP antibodies, and treatment and identified 702, 570, and 906 putative DMRs, respectively. The top ranked DMR (364 bps) associated with RA reached borderline genome-wide significance after permutation test (p < 0.07). This DMR was hypomethylated with a mean fold change of 0.33 and located in the promoter region of the S100A (S100 calcium-binding protein A6) (Table 1; Table S1 in Supplementary Material).
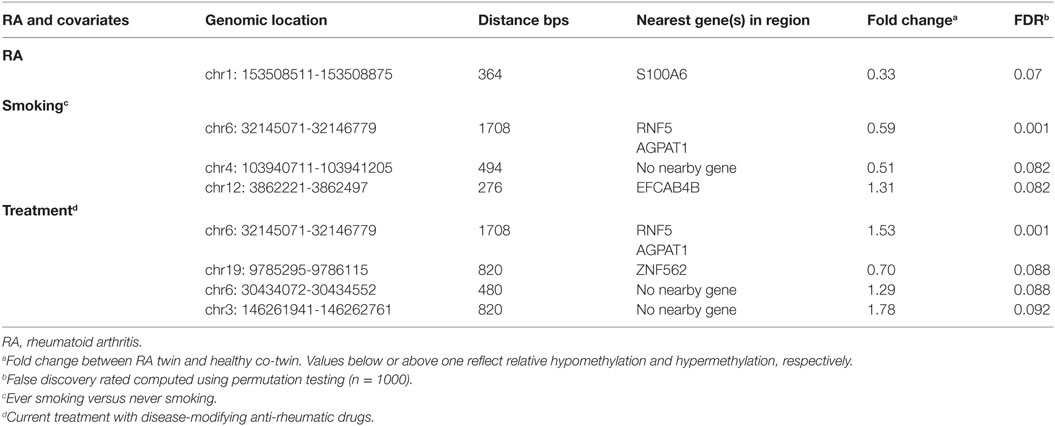
Table 1. The top-ranked DMRs associated with RA.
The region comprises a CpG island, and data from the Encode Project point to the presence of both multiple transcription factor binding sites, and a large DNAse hypersensitivity region. Furthermore, two nearby regions showed enrichment of histone marks, which indicate a regulatory role for this region. S100A6 belongs to a cluster of genes on chromosome 1q21 encoding S100 proteins localized in the cytoplasm and/or nucleus of a wide range of cells.
Subsequently, we searched for DMRs associated with RA and predicted by any of the three covariates. The top ranked DMR (1708 bps) associated with RA and predicted by smoking reached genome-wide significance (p < 0.001). This region overlaps with the promoters of both RNF5 (ring finger protein 5) and AGPAT1 (1-acylglycerol-3-phosphate O-acyltransferase 1 located in the class III region of the human major histocompatibility complex) (Figure 2; Table 1; Table S1 in Supplementary Material). This region also contains several transcription factor binding sites, DNAse hypersensitivity sites, and enrichment of histone marks suggesting a regulatory role for this region. Notably, this region also reached genome-wide significance (FDR adjusted p < 0.001) with treatment as predictor, but with reversed DNAm pattern to hypermethylation (Figure 2; Table 1; Table S1 in Supplementary Material). This suggests that smoking, which is the strongest known environmental risk factor for RA, induces hypomethylation in this promoter region and that DMARD treatment may reverse this. It is notable that 35 of 36 CpGs were hypomethylated in RA twin modulated by smoking, and 36 of 36 CpGs were hypermethylated in RA modulated by treatment (Table S2 in Supplementary Material).
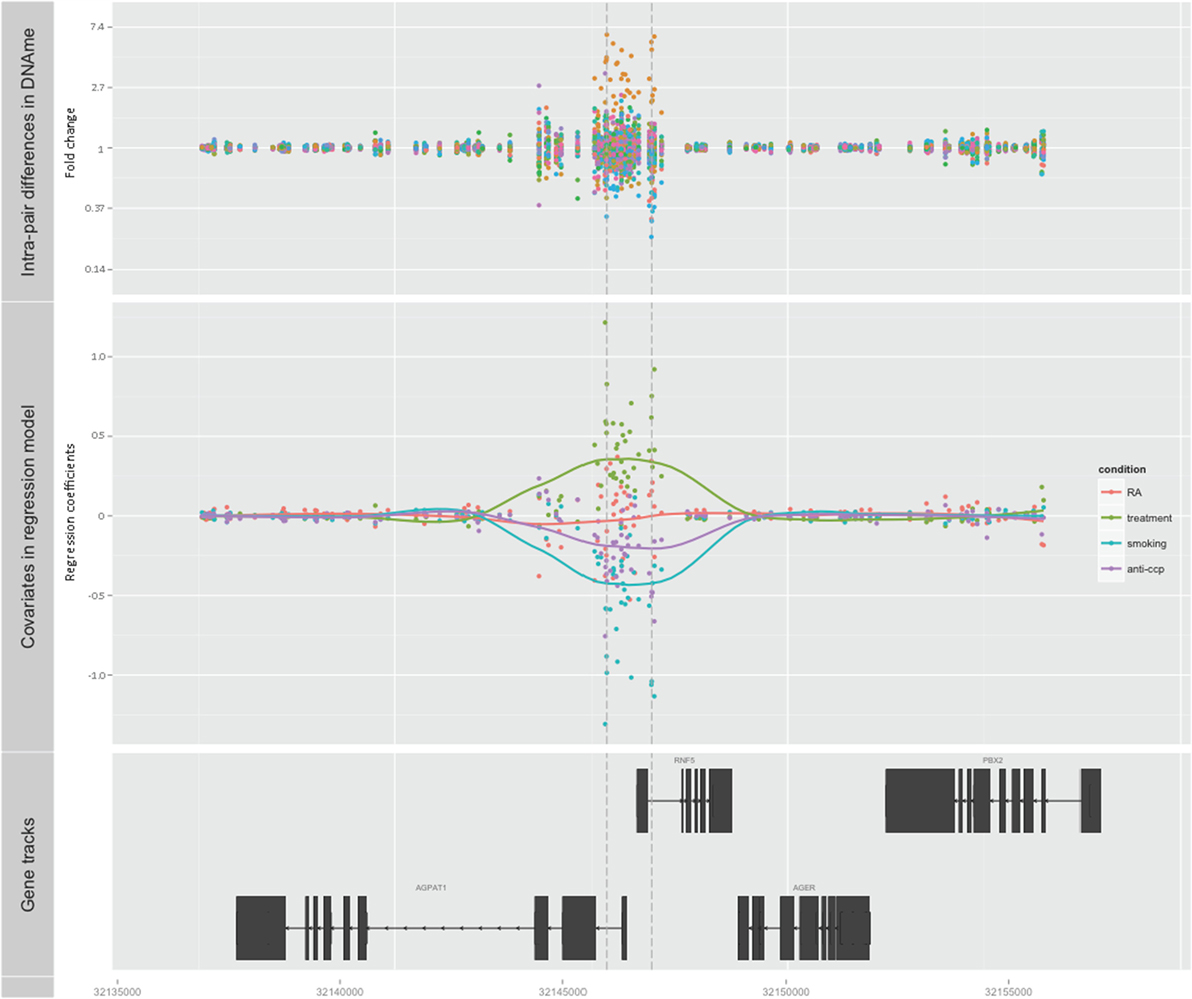
Figure 2. Top-ranked DMR associated with RA. The genome-wide significant differentially methylated region is located at the promoter region of AGPAT1 and RNF5. The top panel shows the unadjusted fold change on the y-axis (ratio of beta values, RA twin numerator, and co-twin denominator) of the 28 twin pairs. The middle panel displays the regression coefficients for RA (orange) and each of the covariates treatment (green), smoking (blue), and anti-CCP antibody (purple). The corresponding smoothed lines denote mean methylation level. The bottom panel depicts the genes and their genomic location.
The second and third ranked DMRs predicted by smoking reached borderline significance, p < 0.08 (Table 1; Table S1 in Supplementary Material). There are no nearby genes within the second DMR, but the third co-localizes with a CpG island covering parts of 5′-UTR and the first exon of EFCAB4B (EF-hand calcium binding domain 4B).
The second ranked DMR (p < 0.09) predicted by treatment included the upstream region and the first part of ZNF562 (zinc finger protein 562) and also harbors a CpG island and other elements suggestive of regulatory functions.
The top ranked DMR predicted by anti-CCP antibodies overlaps with the promotor region of two genes; CRYZ (Quinone oxidoreductase) and TYW3 (tRNA-yW synthesizing protein 3 homolog), but this region did not reach genome-wide significance (p < 0.2).
No independent population of RA discordant twin pairs was available for replication. We therefore compared our results with the hitherto largest EWAS based on peripheral blood from anti-CCP antibody positive and treatment naïve RA singletons by Liu et al. (31). This study also corrected for cell type heterogeneity, age, gender, and smoking but did not investigate the individual effect of the covariates. These authors searched for DNAm associated with RA at the single-CpG-site level and not at the regional level, and they reduced the number of interrogated CpGs from 450 to 300 K. Consequently, a comparison at the CpG level was not optimal, and we therefore searched for replication at the gene-level among the genes associated with DMRs predicted by RA. Among our nine top ranked DMRs, we identified six nearby genes suggestively associated with RA. These genes were also reported by Liu et al. (Table 2; Table S1 in Supplementary Material), but only S100A6, C13orf38, and SDCCAG1 exhibited on average the same direction of methylation.
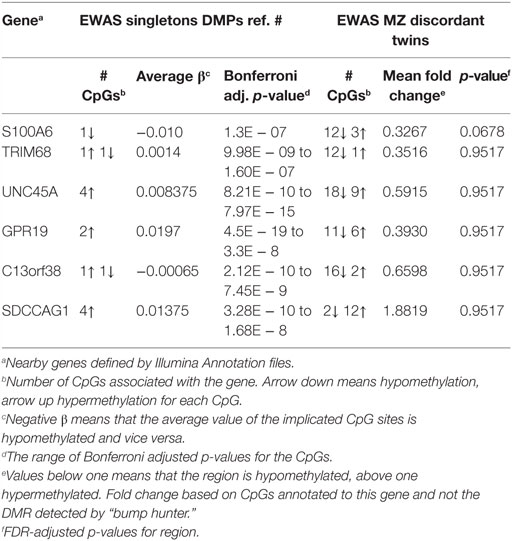
Table 2. Validation of genes nearby differentially methylated CpG sites identified in EWAS of case–control study in singletons.
By contrast, the two CpGs linked to TRIM68 and C13orf38 showed opposite directions of methylation in the study by Liu et al., whereas 12 of 13 and 16 of 18 CpGs in our study had the same direction of methylation (Table 2). Clearly, this illustrates the strength of the regional approach and indicates that these genes on average are associated with hypomethylation in RA. In total, 36 genes were overlapping the DMRs suggestively associated with RA in our data set. According to the study by Liu et al., all 36 of these genes were covered by from 1 to 7 CpGs reaching genome-wide significance in their study, but we cannot compare the direction of association beyond the 6 genes mentioned above because Liu et al. did not investigate the effect of the covariates. Interestingly, the promoters of the RNF5 and AGPAT1 genes were within 100-kb distance of the identified DMPs in the study by Liu et al.
Gene-Set Analysis
We then performed GSAs to explore the potential of shared biological functions and pathways among the identified DMRs. The 603, 702, 570, and 906 putative DMRs predicted by RA, smoking, anti-CCP antibody, and treatment, respectively, comprised the input genomic regions applied to GREAT (29) to compute ontology term enrichment and identify processes or pathways that are perturbed in established RA. In Table S3 in Supplementary Material, we present the entire list of significant ontology terms and pathways. Genes with promoter regions containing the binding site for ELK1 were enriched in RA (binomial FDR 1.2 × 10−11) as well as in RA predicted by treatment (binomial FDR 2.7 × 10−15). ELK1 is a member of the E-twenty-six (ETS) oncogene family (32) and is an intracellular transcription factor of the p38MAPK signaling cascade involved in inflammation and tissue destruction in RA (33). It binds to three sites in the promoter region of tumor necrosis factor alpha (TNF-α) (34), a key player in the inflammation of RA. Genes upregulated in cervical cancer, thyroid carcinoma, and breast tumor were enriched using the RA dataset, and genes upregulated in breast and ovarian cancer were enriched in RA predicted by smoking. Gene sets that represent cell states and perturbations within the immune system were also enriched.
A key assumption of GSA requires that all genes, a priori, have the same probability of appearing. In cases, where some genes are tested many more times than others, genes with more associated probes are more likely to fulfill whatever ad hoc criteria to define differentially methylated genes. This may cause a strong bias and as the Illumina 450 K BeadChip contains from 1 to 1288 probes per gene, this type of bias should not be neglected. However, the GSA presented in this study is based on regions, which may average out the number of probes per region and thereby mitigating this bias and reduce the number of spurious findings. Thus, we did not find any correlation between the significance level of the pathways and the number of probes per gene and the most significant pathways clustered around the mean number of CpGs per gene for the whole microarray (Figure S2 in Supplementary Material).
Discussion
This is the first comprehensive EWAS in MZ twins discordant for RA. We did not disclose DMPs associated with RA but identified one genome-wide significant DMR and several candidate DMRs. EWAS in MZ discordant twin pairs are particularly useful to detect DMRs caused by environmental or stochastic effects as compared with findings from case–control studies in singletons, which are also influenced by genetic variation. Some of our top ranking DMRs located in the promoter region of genes have previously been reported in a large EWAS on RA singletons (31). Thus, replication of these findings in a different setting of MZ twin pairs discordant for RA significantly adds to the evidence that DNAm changes in these genes may mediate the effect of environmental exposures, including drug treatment.
The strongest signal was observed in the promoter region of RNF5 and AGPAT1. The protein encoded by RFN5 is an 18-kDa RING finger membrane-bound ubiquitin E3 ligase, which has not previously been associated with RA (35). It has been shown to decrease the level of autophagy. It has been reported previously that macrophages from RNF5 knock-out mice contain a greater number of autophagosomes surrounding bacterial pathogens than wild-type mice, and this RNF5 is also implicated in antiviral innate immune signaling (36). RNF5 has been shown to be downregulated in patients with established Crohn’s disease and ulcerative colitis (37) as well as in patients with spondyloarthritis and chronic gut inflammation (35). Increased expression of this gene has been reported in PBMCs from RA patients with active disease (38), and SNP genotypes of both RNF5 and AGPAT1 have been associated with susceptibility to type 1 diabetes (39). Thus, both RNF5 and AGPAT1 have been associated with inflammation and autoimmunity.
The S100A6 has been shown to be overexpressed in peripheral blood from RA patients and the expression level correlated to MMP3 levels (40), which accords with our finding of hypomethylation in the promoter region. The active gene may therefore be a predictor of cartilage and bone destruction. The S100A6 is also overexpressed in salivary glandular epithelial cells in patients with Sjögren’s syndrome (41).
EFCAB4B is a Ca(2+)-binding protein that plays a key role in store-operated calcium entry (SOCE) in T-cells (42), and five different single nucleotide variations in EFCAB4B have previously been associated with RA (43). ZNF562 may be involved in transcriptional regulation and has to our knowledge not previously been associated with RA.
Although the DMR nearby CRYZ and TYWS did not reach genome-wide significance in our study, DMPs in the promotor regions of these genes did reach genome-wide significance in the study by Liu et al. (31) DMRs overlapping these genes have previously been related to response to biologics (44), and the genes have been associated with both inflammation and Type 2 diabetes (45).
None of the genes nearby our top ranked DMRs were among the five different genes that according to Liu. et al. (31) may mediate their effect through changes in DNAm. If the DNAm differences predominantly mediate genetic effects, we would not expect to find these DNAm differences within our RA discordant MZ twin pairs. Hence, this discordance with Liu et al. indirectly supports that the effect of these polymorphisms are mediated via DNAm variation.
Previous studies by Nile et al. and Ishida et al. reported hypomethylation of single, but distinctive, CpG motifs in the promoter region of interleukin-6 in genomic DNA from PBMCs, which were associated with RA (46, 47). However, both the number and position of CpG motifs in the IL-6 promoter region differed between the two populations and neither of the two distinct motifs were among the interrogated sites on the 450 K Illumina assay. Among the other five interrogated CpG motifs in the promoter region of IL-6 on the 450 K assay, none were associated with RA in our study in accordance with the findings in the UK (46) and the Japanese studies (47).
In a study by Liao et al., 10 CpG motifs in the promoter region of CD40L on the X chromosome from CD4+ T cells were found to be hypomethylated in female RA patients and correlated with mRNA expression (48). We found no aberrant methylation pattern when analyzing the same promoter region separately for male and female twin pairs. This discrepancy may be explained by genetic variation between cases and controls in the Chinese study as well as differences in ethnicity, interrogated motifs, and cell type specificity. Furthermore, differences in treatment may also contribute to divergence from their results.
Genes upregulated in poorly differentiated thyroid carcinoma compared to normal thyroid tissue were enriched using the RA dataset. Autoimmune thyroid diseases (AITD) are associated with RA, and several studies have shown an association of AITD and papillary thyroid cancer (49). A higher prevalence of papillary thyroid cancers has been reported in RA patients (50) and other systemic autoimmune disorders including Sjøgrens syndrome (51, 52). Although the increased malignancy risk in RA is primarily associated with lymphomas and lung cancer (53–55), our GSA also revealed enrichment of genes upregulated in breast cancer and ovarian cancer. Even though there in no evidence of a direct association of RA with these cancers, it has previously been emphasized that the destructive process of RA may share features with neoplastic tissue. Thus, RA synoviocytes can grow under anchorage-independent conditions with defective contact inhibition, and synovial dedifferentiation and angiogenesis and mutations in the p53 tumor suppressor gene have been described in synovial tissue (56). From an epigenetic viewpoint, cancer and RA are characterized by genome-wide hypomethylation (12, 57, 58), and the demonstration of pathways shared by cancer and RA may provide new insights into disease-overlapping aspects of these two disease categories and might help to explain the invasive aspects of the diseases.
Although our study has taken advantage of the efficient MZ co-twin control method with adjustment for important confounders, some limitations should be considered. Thus, mosaicism for de novo mutations, retrotranspositions, indels, duplications, and chromosomal rearrangements may play a role in MZ twin discordance. In addition, copy number variants (CNVs) may manifest as DNAm changes, as suggested in one study on autism (59), although CNVs in general seem to have little impact on bead-array-based measures of DNAm (60). Also, we can not exclude the possibility of residual confounding due to cell type heterogeneity.
The inflammatory process in RA is systemic and may beside joints target a variety of extra-articular sites. While the etiology of RA remains elusive, there is robust evidence that T cells, B cells, and proinflammatory cytokine and chemokine networks are core pathogenetic mediators in RA. However, additional peripheral blood cells are implicated as well including innate effector cells like macrophages, mast cells, dendritic cells, and natural killer cells (61). Synovitis is caused by activation of these subpopulation of mononuclear cells in conjunction with granulocytes and angiogenesis (62). Although epigenetic studies at the level of specific cellular types seem attractive, it is worth considering that in a cross-sectional design like the present, there is an inherent risk that demethylation changes co-occurring in different cell subpopulations may go unrecognized by focusing on one particular cell type or that the significance of single cell type methylation changes are overestimated. Accordingly, in this exploratory study, which relies on a strong experimental design including MZ twins who are discordant for RA, we found it appropriate to study epigenetic marks in whole PBMCs. Despite the risk of a “dilution effect” by normally methylated subsets of cells, demethylation alterations were actually detected in several candidate regions reaching genome-wide significance at p levels between 0.001 and 0.1. These effect sizes accord well with the so far largest EWAS on singletons, which also relied on DNA from PMBC (31).
We have not done technical verification, but investigator-initiated studies on the validity of the 450 K Illumina assay have shown robust results when compared to DNAm measurement based on sequencing (63, 64). Furthermore, we have minimized batch effects because samples from cases and controls were processed simultaneously and have been placed on the same plates. In order to reduce biological variation, blood samples were drawn simultaneously from either twin in each. Last but not least, since we are dealing with MZ discordant twin pairs, the effects of genetic variants affecting probe binding or read-out are indistinguishable between cases and controls.
Although, we did not collect samples for quantification of mRNA in whole blood, our results accord with previously published gene expression studies on the RNF5 and the S100A6 in peripheral blood from RA patients and patients with Sjögren’s syndrome (38, 40, 41).
We also considered treatment as a source of epigenetic modification. However, our study is based on a population of RA twins with different disease profiles, which may influence our findings (65). Notably, all our RA discordant twin pairs underwent standardized clinical examination, and we therefore have optimally validated information of treatment at the time of blood sampling. Thus, this is in fact the first study to address the effect of treatment in a EWAS. It may be argued that the study is underpowered because only 68% of the cases were currently treated with a DMARD. Nevertheless, we observed a significant genome-wide effect of treatment on DMAm, and interestingly that the effect of smoking on DNAm in RA was partially antagonized by DMARD treatment. Concerning power, it has previously been estimated that 15 MZ disease-discordant twin pairs have reasonable power to detect disease-associated DMRs (66). Our disease-discordant MZ twin design provides a perfect match for genetics, maternal, and cohort effects as well as age and sex. But in addition, we have also been able to adjust for important covariates known to influence DNAm, e.g., smoking and treatment, which further increases the power of our study.
By focusing on MZ twins, the case co-twin design is especially useful in epigenetic studies as one of the main tasks in these studies is to find environmental exposures that are associated with the observed epigenetic changes linked to disease status. In our model, we have measured the intra-pair differential DNAm as a function of environmental exposure, providing adjustment for confounding factors as fixed effects and taking into account random effects such as batch effect, arrangement of samples on the array, etc. The effects of age and sex are matched out in the co-twin design; however in epigenetic studies, this argument does not hold because within-twin pair difference in epigenetic measurement can be higher in old than in young twin pairs, and for some specific sites within-twin pairs, epigenetic difference can differ according to sex. Therefore, age and sex were included as pair-specific covariates with fixed effects and their effects adjusted (27).
This is a cross-sectional study including cases with already established disease implying that the epigenetic changes may reflect both cause and effect of chronic inflammation. Yet, secondary effects may help to further elucidate the RA pathogenesis and lead to discovery of early diagnostic and prognostic markers. Furthermore, individual epigenetic signatures that remain stable over time have been described (67) and hold promise for use at the level of the individual by analogy with genotypes. Such regions can be considered as candidates for assessment of DNAm associations with disease, whereas those that are particularly labile may be relevant when assessing epigenetic marks of, e.g., treatment effects. Further epigenetic studies replicating our findings in samples collected in early life and before clinical disease onset may help to resolve these issues.
In conclusion, this exploratory EWAS on a well-characterized sample of MZ twin pairs discordant for RA has identified candidate regions and plausible biological pathways pertinent to the pathogenesis of RA, which are not explained by genetic variation. Our study also strongly suggests that there is an interaction between important covariates, DNAm, and RA. The present data are available for application in other levels of, e.g., genomics, transcriptomic, and proteomics to enhance meta-dimensional analyses to achieve a more detailed comprehension of the RA disease pathways (68).
Ethics
The study was approved by all the regional scientific ethics committees in Denmark (Projekt ID: S-20070088) and the Danish Data Protection board (J. nr. 2007-41-0747). We obtained informed written consent from all participants in the study.
Author Contributions
AS conceived the study. QT, KG, RL, LC, and AS performed the analysis and interpretation of the data. GH, CN, LC, RL, KG, and KK contributed reagents/materials/analysis tools. AS, QT, KG, RL, LC, and PJ prepared the manuscript. All the authors read and approved the manuscript.
Conflict of Interest Statement
This is an investigator initiated study, and the funding source had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. The authors have declared that no competing interests exist.
Acknowledgments
We thank Henriette Cederholm for help with the logistics and interviews, Susanne Knudsen for the establishment of a blood and DNA repository, and Lars Hvidberg for data management.
Funding
This work was supported by The Danish Rheumatism Association and The KID foundation.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00510/full#supplementary-material.
References
1. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet (2010) 42(6):508–14. doi: 10.1038/ng.582
2. Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol (2013) 9(3):141–53. doi:10.1038/nrrheum.2012.237
3. MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum (2000) 43(1):30–7. doi:10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B
4. Svendsen AJ, Kyvik KO, Houen G, Junker P, Christensen K, Christiansen L, et al. On the origin of rheumatoid arthritis: the impact of environment and genes – a population based twin study. PLoS One (2013) 8(2):e57304. doi:10.1371/journal.pone.0057304
5. Haj Hensvold A, Magnusson PKE, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis (2015) 74(2):375–80. doi:10.1136/annrheumdis-2013-203947
6. Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet (2009) 41(2):240–5. doi:10.1038/ng.286
7. MacGregor AJ, Bamber S, Carthy D, Vencovsky J, Mageed RA, Ollier WE, et al. Heterogeneity of disease phenotype in monozygotic twins concordant for rheumatoid arthritis. Br J Rheumatol (1995) 34:215–20. doi:10.1093/rheumatology/34.3.215
8. Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature (2010) 465(7299):721–7. doi:10.1038/nature09230
9. Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet (2009) 373(9664):659–72. doi:10.1016/S0140-6736(09)60008-8
10. Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One (2013) 8(5):e63812. doi:10.1371/journal.pone.0063812
11. Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet (2012) 21(13):3073–82. doi:10.1093/hmg/dds135
12. Kim YI, Logan JW, Mason JB, Roubenoff R. DNA hypomethylation in inflammatory arthritis: reversal with methotrexate. J Lab Clin Med (1996) 128(2):165–72. doi:10.1016/S0022-2143(96)90008-6
13. Cribbs AP, Kennedy A, Penn H, Amjadi P, Green P, Read JE, et al. Methotrexate restores regulatory T cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol (2015) 67(5):1182–92. doi:10.1002/art.39031
14. Plant D, Webster A, Nair N, Oliver J, Smith SL, Eyre S, et al. Differential methylation as a biomarker of response to etanercept in patients with rheumatoid arthritis. Arthritis Rheumatol (2016) 68(6):1353–60. doi:10.1002/art.39590
15. Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LTY, Kohlbacher O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature (2013) 500(7463):477–81. doi:10.1038/nature12433
16. Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum (2009) 60(12):3613–22. doi:10.1002/art.25018
17. Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis (2013) 72(1):110–7. doi:10.1136/annrheumdis-2012-201526
18. Liu C-C, Fang T-J, Ou T-T, Wu C-C, Li R-N, Lin Y-C, et al. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol Lett (2011) 135(1–2):96–9. doi:10.1016/j.imlet.2010.10.003
19. de Andres MC, Perez-Pampin E, Calaza M, Santaclara FJ, Ortea I, Gomez-Reino JJ, et al. Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res Ther (2015) 17(1):1–9. doi:10.1186/s13075-015-0748-5
20. Bottini N, Firestein GS. Epigenetics in rheumatoid arthritis: a primer for rheumatologists. Curr Rheumatol Rep (2013) 15(11):372. doi:10.1007/s11926-013-0372-9
21. Svendsen AJ, Holm NV, Kyvik K, Petersen PH, Junker P. Relative importance of genetic effects in rheumatoid arthritis: historical cohort study of Danish nationwide twin population. BMJ (2002) 324(7332):264–6. doi:10.1136/bmj.324.7332.264
22. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum (1988) 31:315–24. doi:10.1002/art.1780310302
23. Christiansen L, Frederiksen H, Schousboe K, Skytthe A, von Wurmb-Schwark N, Christensen K, et al. Age- and sex-differences in the validity of questionnaire-based zygosity in twins. Twin Res (2003) 6(4):275–8. doi:10.1375/136905203322296610
24. Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol (2012) 13(6):R44. doi:10.1186/gb-2012-13-6-r44
25. Houseman EA, Christensen BC, Yeh RF, Marsit CJ, Karagas MR, Wrensch M, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics (2008) 9:365. doi:10.1186/1471-2105-9-365
26. Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics (2014) 30(10):1431–9. doi:10.1093/bioinformatics/btu029
27. Tan Q, Christiansen L, von Bornemann Hjelmborg J, Christensen K. Twin methodology in epigenetic studies. J Exp Biol (2015) 218(Pt 1):134–9. doi:10.1242/jeb.107151
28. Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol (2012) 41(1):200–9. doi:10.1093/ije/dyr238
29. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol (2010) 28(5):495–501. doi:10.1038/nbt.1630
30. Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet (2006) 38(12):1378–85. doi:10.1038/ng1909
31. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol (2013) 31(2):142–7. doi:10.1038/nbt.2487
32. Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol (2001) 2(11):827–37. doi:10.1038/35099076
33. Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis (2008) 67(7):909–16. doi:10.1136/ard.2007.074278
34. Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, et al. A lipopolysaccharide-specific enhancer complex involving ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo. Mol Cell Biol (2000) 20(16):6084–94. doi:10.1128/MCB.20.16.6084-6094.2000
35. Laukens D, Peeters H, Cruyssen BV, Boonefaes T, Elewaut D, De Keyser F, et al. Altered gut transcriptome in spondyloarthropathy. Ann Rheum Dis (2006) 65(10):1293–300. doi:10.1136/ard.2005.047738
36. Kuang E, Okumura CYM, Sheffy-Levin S, Varsano T, Shu VC-W, Qi J, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet (2012) 8(10):e1003007. doi:10.1371/journal.pgen.1003007
37. Dooley TP, Curto EV, Reddy SP, Davis RL, Lambert GW, Wilborn TW, et al. Regulation of gene expression in inflammatory bowel disease and correlation with IBD drugs. Screening by DNA microarrays. Inflamm Bowel Dis (2004) 10(1):1–14. doi:10.1097/00054725-200401000-00001
38. Edwards CJ, Feldman JL, Beech J, Shields KM, Stover JA, Trepicchio WL, et al. Molecular profile of peripheral blood mononuclear cells from patients with rheumatoid arthritis. Mol Med (2007) 13(1–2):40–58. doi:10.2119/2006-000056.Edwards
39. McKinnon E, Morahan G, Nolan D, James I; Diabetes Genetics Consortium. Association of MHC SNP genotype with susceptibility to type 1 diabetes: a modified survival approach. Diabetes Obes Metab (2009) 11(Suppl 1):92–100. doi:10.1111/j.1463-1326.2008.01009.x
40. Lee HM, Sugino H, Aoki C, Murakami M, Matsutani T, Ochi T, et al. Correlations between S100 gene expression levels and the local and systemic inflammatory markers (matrix metalloproteinase-3, MMP3; erythrocyte sedimentation rate, ESR) in rheumatoid arthritis patients [abstract]. Arthritis Rheum (2011) 63(Suppl 10):1925.
41. Steinfeld S, Rommes S, Francois C, Decaestecker C, Maho A, Appelboom T, et al. Big prolactin 60 kDa is overexpressed in salivary glandular epithelial cells from patients with Sjogren’s syndrome. Lab Invest (2000) 80(2):239–47. doi:10.1038/labinvest.3780027
42. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol (2007) 7(9):690–702. doi:10.1038/nri2152
43. Mitsunaga S, Hosomichi K, Okudaira Y, Nakaoka H, Kunii N, Suzuki Y, et al. Exome sequencing identifies novel rheumatoid arthritis-susceptible variants in the BTNL2. J Hum Genet (2013) 58(4):210–5. doi:10.1038/jhg.2013.2
44. Webster A, Plant D, Eyre S, Wilson G, Morgan A, Isaacs J, et al. OP0257 differential DNA methylation related to response to adalimumab and etanercept in patients with rheumatoid arthritis. Ann Rheum Dis (2014) 73(Suppl 2):158–9. doi:10.1136/annrheumdis-2014-eular.4104
45. Qi Q, Menzaghi C, Smith S, Liang L, de Rekeneire N, Garcia ME, et al. Genome-wide association analysis identifies TYW3/CRYZ and NDST4 loci associated with circulating resistin levels. Hum Mol Genet (2012) 21(21):4774–80. doi:10.1093/hmg/dds300
46. Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum (2008) 58(9):2686–93. doi:10.1002/art.23758
47. Ishida K, Kobayashi T, Ito S, Komatsu Y, Yokoyama T, Okada M, et al. Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J Periodontol (2012) 83(7):917–25. doi:10.1902/jop.2011.110356
48. Liao J, Liang G, Xie S, Zhao H, Zuo X, Li F, et al. CD40L demethylation in CD4+ T cells from women with rheumatoid arthritis. Clin Immunol (2012) 145(1):13–8. doi:10.1016/j.clim.2012.07.006
49. Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmun Rev (2015) 14(2):174–80. doi:10.1016/j.autrev.2014.10.016
50. Lee Y-A, Lee S-H, Song R, Yang H-I, Hong S-J, Min K. Higher prevalence of coexisting papillary thyroid cancer as well as autoimmune thyroid diseases in patients with rheumatoid arthritis [abstract]. Arthritis Rheum (2010) 62(Suppl 10):1038.
51. Weng M-Y, Huang Y-T, Liu M-F, Lu T-H. Incidence of cancer in a nationwide population cohort of 7852 patients with primary Sjögren’s syndrome in Taiwan. Ann Rheum Dis (2012) 71(4):524–7. doi:10.1136/annrheumdis-2011-200402
52. Liang Y, Yang Z, Qin B, Zhong R. Primary Sjögren’s syndrome and malignancy risk: a systematic review and meta-analysis. Ann Rheum Dis (2014) 73(6):1151–6. doi:10.1136/annrheumdis-2013-203305
53. Smitten A, Simon T, Hochberg M, Suissa S. A meta-analysis of the incidence of malignancy in adult patients with rheumatoid arthritis. Arthritis Res Ther (2008) 10(2):R45. doi:10.1186/ar2404
54. Mercer LK, Davies R, Galloway JB, Low A, Lunt M, Dixon WG, et al. Risk of cancer in patients receiving non-biologic disease-modifying therapy for rheumatoid arthritis compared with the UK general population. Rheumatology (2013) 52(1):91–8. doi:10.1093/rheumatology/kes350
55. Mellemkjaer L, Linet MS, Gridley G, Frisch M, Møller H, Olsen JH. Rheumatoid arthritis and cancer risk. Eur J Cancer (1996) 32(10):1753–7. doi:10.1016/0959-8049(96)00210-9
56. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature (2003) 423(6937):356–61. doi:10.1038/nature01661
57. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics (2009) 1(2):239–59. doi:10.2217/epi.09.33
58. Karouzakis E, Gay RE, Gay S, Neidhart M. Epigenetic control in rheumatoid arthritis synovial fibroblasts. Nat Rev Rheumatol (2009) 5(5):266–72. doi:10.1038/nrrheum.2009.55
59. Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry (2014) 19:862–71. doi:10.1038/mp.2013.114
60. Houseman EA, Christensen BC, Karagas MR, Wrensch MR, Nelson HH, Wiemels JL, et al. Copy number variation has little impact on bead-array-based measures of DNA methylation. Bioinformatics (2009) 25(16):1999–2005. doi:10.1093/bioinformatics/btp364
61. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365(23):2205–19. doi:10.1056/NEJMra1004965
62. Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov (2003) 2(6):473–88. doi:10.1038/nrd1109
63. Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar S, Kreipe H, et al. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes (2012) 5(1):210. doi:10.1186/1756-0500-5-210
64. Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the infinium methylation 450K technology. Epigenomics (2011) 3(6):771–84. doi:10.2217/epi.11.105
65. Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol (2013) 190(3):1297–303. doi:10.4049/jimmunol.1202572
66. Kaminsky Z, Petronis A, Wang SC, Levine B, Ghaffar O, Floden D, et al. Epigenetics of personality traits: an illustrative study of identical twins discordant for risk-taking behavior. Twin Res Hum Genet (2008) 11(1):1–11. doi:10.1375/twin.11.1.1
67. Feinberg AP, Irizarry RA, Fradin D, Aryee MJ, Murakami P, Aspelund T, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med (2010) 2(49):49ra67. doi:10.1126/scitranslmed.3001262
Keywords: rheumatoid arthritis, DNA methylation, monozygotic twins, epigenome-wide association study, pathway analysis
Citation: Svendsen AJ, Gervin K, Lyle R, Christiansen L, Kyvik K, Junker P, Nielsen C, Houen G and Tan Q (2016) Differentially Methylated DNA Regions in Monozygotic Twin Pairs Discordant for Rheumatoid Arthritis: An Epigenome-Wide Study. Front. Immunol. 7:510. doi: 10.3389/fimmu.2016.00510
Received: 25 August 2016; Accepted: 02 November 2016;
Published: 17 November 2016
Edited by:
Masaaki Murakami, Hokkaido University, JapanReviewed by:
Florence Burté, Newcastle University, UKKa Man Law, University of California Los Angeles, USA
Copyright: © 2016 Svendsen, Gervin, Lyle, Christiansen, Kyvik, Junker, Nielsen, Houen and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anders J. Svendsen, YXN2ZW5kc2VuQGhlYWx0aC5zZHUuZGs=