 Silke Neumann
Silke Neumann Simon A. Shirley
Simon A. Shirley Roslyn A. Kemp
Roslyn A. Kemp Sarah M. Hook1
Sarah M. Hook1
- 1School of Pharmacy, University of Otago, Dunedin, New Zealand
- 2Department of Pathology, Dunedin School of Medicine, University of Otago, Dunedin, New Zealand
- 3Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand
Immune-suppressive cell populations impair antitumor immunity and can contribute to the failure of immune therapeutic approaches. We hypothesized that the non-steroidal anti-inflammatory drug licofelone, a dual cyclooxygenase-2/5-LO inhibitor, would improve therapeutic melanoma vaccination by reducing immune-suppressive cell populations. Therefore, licofelone was administered after tumor implantation, either alone or in combination with a peptide vaccine containing a long tyrosinase-related protein 2-peptide and the adjuvant α-galactosylceramide, all formulated into cationic liposomes. Mice immunized with the long-peptide vaccine and licofelone showed delayed tumor growth compared to mice given the vaccine alone. This protection was associated with a lower frequency of immature myeloid cells (IMCs) in the bone marrow (BM) and spleen of tumor-inoculated mice. When investigating the effect of licofelone on IMCs in vitro, we found that the prostaglandin E2-induced generation of IMCs was decreased in the presence of licofelone. Furthermore, pre-incubation of BM cells differentiated under IMC-inducing conditions with licofelone reduced the secretion of cytokines interleukin (IL)-10 and -6 upon lipopolysaccharides (LPS) stimulation as compared to untreated cells. Interestingly, licofelone increased IL-6 and IL-10 secretion when administered after the LPS stimulus, demonstrating an environment-dependent effect of licofelone. Our findings support the use of licofelone to reduce tumor-promoting cell populations.
Introduction
Successful cancer vaccines not only need to stimulate a robust antitumor immune response, but also need to alleviate the effects of immune-suppressive cell populations (1). Inflammatory factors secreted by tumor cells can polarize stromal and immune cells, including macrophages, immature myeloid cells (IMCs) [also known as myeloid-derived suppressor cells (MDSCs)], and T-cells, toward an immune-suppressive phenotype (2). Furthermore, the constant secretion of inflammatory molecules by tumor cells into the systemic circulation promotes IMCs to egress from the bone marrow (BM) and to infiltrate the tumor where they suppress antitumor immunity (3). IMCs have been identified as one of the major pro-tumor immune populations and are positively correlated with melanoma progression, which makes them an attractive target for immunotherapy (4, 5).
Immature myeloid cells are a heterogeneous population of myeloid precursor cells that are present in most cancers and which typically expand during disease progression (3). IMCs are induced by tumor-derived inflammatory factors [e.g., VEGF, granulocyte-macrophage colony-stimulating factor (GM-CSF), prostaglandins, interleukin (IL)-1β, IL-6, IL-10, TGF-β, S100A8, and S100A9] (2, 6, 7) that stimulate tumor growth and myelopoiesis and block the differentiation of IMCs into mature effector cells such as DCs, macrophages, and granulocytes (8, 9). In mouse models, IMCs can be divided into distinct subpopulations based on cell morphology and the expression of the cell surface markers Gr-1 and CD11b, as well as by their suppressive functions (10). The suppressive ability of monocytic IMCs (CD11b+ Gr-1intermediate) is mediated through an increased catabolism of the essential amino acid arginine, which results in decreased proliferation, anergy, and increased apoptosis of T-cells (2). Granulocytic IMCs (CD11b+ Gr-1high) produce large amounts of reactive oxygen species (ROS) that impede T-cell receptor signaling (10). Activated IMCs can themselves produce pro-inflammatory stimuli such as IL-6, prostaglandin E2 (PGE2), and VEGF, which provide a positive feedback loop for their recruitment and activation (8).
One of the key inflammatory factors closely linked to carcinogenesis as well as to the induction, accumulation, and activation of IMCs is PGE2 (11). PGE2 is a physiologically abundant eicosanoid, derived from arachidonic acid through the cyclooxygenase (COX)-1 and -2 enzymatic pathways (12). PGE2 synergizes with leukotriene B4 (LTB4) to induce local inflammation and to attract immune cells into tissue (13). Leukotrienes (LTs) are also produced from arachidonic acid but require the enzyme 5-lipoxygenase (5-LO) for the production of inflammatory mediators (14). 5-LO expression is detectable in granulocytes, monocytes, and macrophages, which produce the largest quantities of LTs, while expression in DCs, B-cells, and mast cells is lower (15). In contrast to COX-1, which is constitutively expressed in almost all tissues, the expression of COX-2 is tightly regulated. Apart from constitutive expression in the brain and kidneys, COX-2 expression in other tissues is low but can rapidly be induced by inflammatory factors (16). Furthermore, overexpression of COX-2 has been observed in many cancers (13).
Recently, blockade of PGE2 using the non-steroidal anti-inflammatory drug (NSAID), celecoxib (a specific COX-2 inhibitor), was shown to improve immunotherapy by inhibiting the generation of IMCs (17). Similarly, the blockage of LTB4-mediated effects improved antitumor immunity in a mouse model (18). Since COX-2 and 5-LO convert the same substrate, inhibition of either of these pathways frees up arachidonic acid that can be used by the other enzyme, therefore simultaneous inhibition of both enzymes could offer superior anti-inflammatory effects combined with a better safety profile (19). To date, several NSAIDs with dual COX-2/5-LO activity have undergone clinical testing for use in the treatment of inflammatory diseases, with licofelone (2-[2-(4-chlorophenyl)-6,6-dimethyl-1-phenyl-5,7-dihydropyrrolizin-3-yl]acetic acid) being the most advanced. In addition to demonstrating an excellent tolerability, licofelone has been shown to have a dose-dependent anticancer effect in cell culture and in animal models (20–22). So far studies have focused on the ability of licofelone to directly inhibit PG (21) and LT synthesis (23) and induce apoptosis in tumor cells; however, little is known about its direct effects on immune cell populations in the context of cancer.
We hypothesized that a combination therapy utilizing the NSAID licofelone along with a therapeutic cancer vaccine would decrease inflammatory signaling through COX-2/5-LO inhibition in immune cells and thus facilitate the development of antitumor immunity. To test this hypothesis, licofelone was administered to mice following implantation of tumor cells and was incorporated into a therapeutic liposomal cancer vaccine containing a long peptide and the adjuvant α-galactosylceramide (α-GalCer), previously shown to increase tumor survival in a melanoma model (24). Immunization with long peptides containing both a CD4 and CD8 T-cell epitope induce improved immune responses as compared to minimal T-cell epitopes, which do not require processing prior to presentation on MHC molecules and can induce tolerance (25, 26). The combination of a long peptide [tyrosinase-related protein (TRP) 2], the adjuvant α-GalCer, and licofelone prolonged the survival of tumor-bearing mice as compared to the TRP2–α-GalCer vaccine alone. Further, we show that the prolonged survival correlated with a decrease in the number of IMCs in tumor-bearing mice. Further investigation of the effects of licofelone on IMCs in vitro demonstrated changes in IMC phenotype including their ability to produce pro-inflammatory cytokines. These findings support the use of licofelone as an additional component in cancer vaccination regimens, enhancing their immunotherapeutic potential.
Materials and Methods
Materials
Lipopolysaccharides (LPS from Escherichia coli 055:B5), collagenase from Clostridium histolyticum Type IA, phosphatidylcholine (PC), 2′,7′-dichlorofluorescin diacetate (DCFDA), and PGE2 were purchased from Sigma Aldrich, USA. Murine IL-6 and -10 CBA flex set were purchased from BD Bioscience, USA. Murine GM-CSF was purchased from Biolegend, New Zealand. The lipids 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were bought from Avanti Polar Lipids, USA. α-GalCer, synthesized from galactose and phytosphingosine, TCI P1765, via α-specific glycosylation methodology (27), was kindly provided by Gavin Painter, Ferrier Research Institute, New Zealand. The long TRP2-peptide [TRP2(180–188),(88–102)-SVYDFFVWLKFFHRTCKCTGNFA] and the CD4 and CD8 OVA peptides (ISQAVHAAHAEINEAGR and SIINFEKL, respectively) were purchased from Mimotopes, Australia. B16F10luc2 melanoma cells were obtained from ATCC, New Zealand. Licofelone was from AdooQ Bioscience, CA, USA. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was purchased from Molecular Probes, USA. CD43 microbeads were from Miltenyi Biotec, Germany.
Preparation and Characterization of Cationic Liposomes
Cationic liposomes containing the long TRP2-peptide and αGalCer were prepared by hydrating thin lipid films as described previously (24). For some formulations, 4.43 mg licofelone was added to the lipid precursor prior to hydration. All liposomal formulations were diluted with sterile PBS in order to deliver 200 ng/mouse α-GalCer, 20 nmol/mouse long TRP2-peptide, and 5 mg/kg licofelone per injection. Cationic liposomes containing only 5 mg/kg licofelone were similarly prepared. Particle size (Z-average) and polydispersity (PDI) of the preparations were analyzed by photon correlation spectroscopy. Electrophoretic mobility was measured in order to determine the zeta potential of the formulations. Results are the average of three independent samples measured in triplicate at 25°C.
Mice
The 6- to 10-week-old C57BL/6J and OT-I mice were bred and maintained under specific pathogen-free conditions at the HTRU, University of Otago. The Animal Ethics Committee, University of Otago, approved all experiments (AEC 16/14).
Tumor Challenge
B16F10luc2 melanoma cells (1 × 105 cells) were injected subcutaneously into the right flank C57BL/6 mice and tumor growth and body weight were monitored every 2 days. Tumor size was assessed by measuring the length and the width of the tumor using digital calipers and mice were culled when tumors reached predetermined humane end points or at 20 days post-tumor injection.
Immunization Schedule
B16F10luc2 melanoma-bearing mice were randomly divided into five groups of seven mice each and were left untreated or were injected intravenously on day 6 with vaccine (TRP2-peptide and α-GalCer in cationic liposomes) or licofelone-vaccine (TRP2-peptide, α-GalCer, and licofelone in cationic liposomes) plus additional licofelone in cationic liposomes or with only the licofelone liposomes (no vaccine). The licofelone liposomes were given by subcutaneous injection in the neck once every second day, starting on the day of tumor grafting until day 14 of the study.
Phenotyping of Immune Subsets after Tumor Challenges
Mice were sacrificed 20 days after tumor cell injection and single cell suspensions were prepared from the tumor-draining lymph nodes (td-LNs), spleens, and one hind limb (for the recovery of BM cells). BM was extracted from the hind limbs as described previously (24). The cells were re-suspended at 1 × 106 cells/mL and incubated with the following antibodies after non-specific binding was blocked by incubation with Fc receptor block (F16/32): CD11b PE-Cy7, Gr-1 FITC, CD11c APC, F4/80 brilliant violet 421, and CD3 APC-Cy7 (all antibodies were purchased from Biolegend, New Zealand and were titrated on splenocytes and BM cells). Propidium iodide (PI) was added prior to acquiring data using a BD FACSCanto II and FlowJo software (version 9.5.2, TreeStar, Inc., USA) was used to analyze data.
Viability and ROS Production of Ex Vivo-Derived Melanoma Cells
Macerated tumors from C57BL/6 mice were incubated with collagenase from C. histolyticum Type IA (1 mg/mL) and calcium chloride (44.1 μg/mL) at 37°C for 30 min. Red blood cells were lysed and the tumor cells were re-suspended at 5 × 105 cells/mL in cIMDM and seeded into a 24 well plate. These cells were incubated with increasing doses of licofelone (1–20 μM) for 48 h and viability and ROS production were examined by flow cytometry after incubation with PI and 1 μL of freshly prepared DCFDA (10 μM stock solution).
Generation and Phenotyping of IMCs
Bone marrow cells were isolated from naïve C57BL/6 mice as described previously (28, 29) and seeded at 5 × 105 cells/mL in the presence of GM-CSF (20 ng/mL) and PGE2 (9.1 μg/mL). Increasing concentrations of licofelone (2.5–50 μM) were added to the culture and refreshed every other day (11) together with media and cytokines. Cells were harvested on day 5 of culture and the phenotype analyzed. In some experiments, BM cells harvested from tumor-bearing mice and were re-cultured for 24 h in the presence of either licofelone (5 μM), LPS (50 ng/mL), or a combination of both. Culture supernatants were stored at −20°C for subsequent cytokine analysis using BD Biosciences CBA Mouse Flex Sets. Samples were run on a BD FACSCanto II and the FCAP Array software (v1.0, Soft Flow) was used to calculate cytokine concentrations in the samples and the standards.
T-Cell Suppression Assay
The suppressive potential of IMCs was evaluated by their ability to inhibit antigen-specific T-cell proliferation. To prepare target T-cells, spleens were dissected from naive OT-I mice and CD43+ cells were isolated by positive selection on an AutoMACS Pro Separator (Miltenyi Biotec) to exclude B-cells. CFSE labeled (24) CD43+ splenocytes were seeded at 5 × 104 cells/well in a 96 well round-bottom plate and stimulated with the CD8 epitope of ovalbumin (SIINFEKL; 0.01 μg/mL) in the presence or absence of IMCs for 48 h. IMCs were generated in vitro as described above and incubated with licofelone (5 μM) for 20 h before LPS (50 ng/mL) was added for 4 h. Cells were washed, re-suspended, and added to the sorted splenocytes at the indicated ratios. After 48 h the proliferation of T-cells was assessed by flow cytometry.
Statistical Analysis
The statistical significance between values was compared using one-way ANOVA followed by post hoc Tukey’s pairwise comparison. All data are expressed as the mean + SD. Survival curves were compared by using the Log-rank test (Mantel–Cox). Statistical analysis was performed using GraphPad Prism version 6.00. All experiments were repeated at least twice.
Results
Incorporation of the NSAID Licofelone into Therapeutic Cancer Vaccine Regimens Improves Survival of Tumor-Bearing Mice
We have recently shown that a therapeutic vaccine, consisting of a long TRP2-peptide co-delivered with α-GalCer in cationic liposomes, increased cytotoxic T-cell responses, and improved tumor survival in melanoma-bearing mice (24). To investigate if the antitumor efficacy of the vaccine could be enhanced, the dual COX-2/5-LO inhibitor licofelone was added to the therapy. Licofelone was included as part of the vaccine and was also delivered by subcutaneous injection pre- and postimmunization, as we hypothesized that this would allow enhanced T-cell priming.
We characterized the cationic liposomes containing the long TRP2-peptide, α-GalCer ± licofelone regarding their size, PDI, and zeta potential as the size and charge of liposomes influences their immunogenicity (25). Cationic liposomes containing the long TRP2-peptide and α-GalCer displayed a size of 280 ± 87 nm and a positive surface charge of 43.6 ± 0.8 mV (Table 1). Incorporation of licofelone into cationic liposomes, together with the TRP2-peptide and α-GalCer, did not markedly alter the size (243 ± 43 nm) or surface charge of the cationic liposomes. However, liposomes containing only licofelone alone were significantly larger (802 ± 59 nm, p < 0.001).
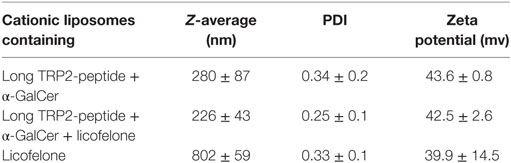
Table 1. Overview of average particle size, PDI index, and zeta potential of vaccine formulations.
Tumor-bearing mice were treated i.v. with the liposomal vaccine containing the long TRP2-peptide, α-GalCer ± licofelone, or with liposomal licofelone alone. In addition, groups immunized i.v. with the vaccine containing licofelone, or liposomal licofelone alone, received repeated s.c. injections of liposomal licofelone. Immunization with the long TRP2-peptide vaccine prolonged the survival of melanoma-bearing mice compared to untreated mice, with the best outcome being seen when mice were also given licofelone. Vaccination with the combination of long TRP2-peptide vaccine and licofelone resulted in 5/7 (71.4%) of the vaccinated mice remaining tumor free (p < 0.01) (Figure 1). Mice injected with licofelone had the same survival times as untreated mice. No adverse effects were observed with vaccine treatments, as indicated by steady body weight over the vaccine treatment period (data not shown).
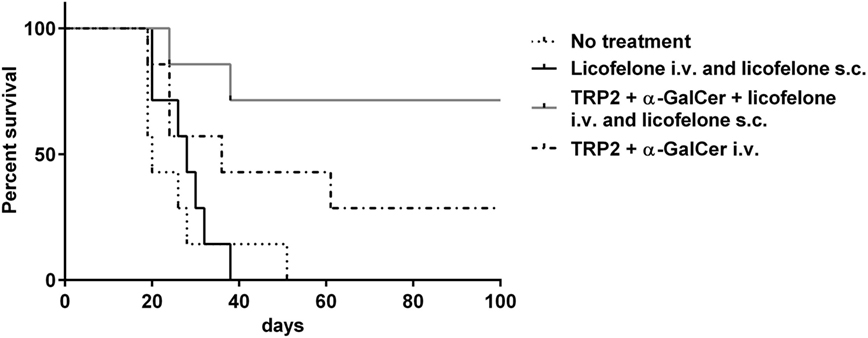
Figure 1. Addition of licofelone to a long-peptide cancer vaccine therapy prolongs survival of melanoma-bearing mice. Groups of mice (n = 7) were injected s.c. with 5 × 104 B16F10luc2 tumor cells and were subsequently left untreated or injected i.v. on day 6 with TRP2-peptide and α-GalCer in cationic liposomes or TRP2-peptide, α-GalCer, and licofelone in cationic liposomes. Mice that received a vaccine containing licofelone in cationic liposomes were also injected s.c. every second day with the licofelone formulated into cationic liposomes while a control group of mice received only s.c. injections of licofelone liposomes. The graph shows the Kaplan–Meier survival curve. Data are representative of two independent experiments with each n = 7 mice/group, ***p < 0.0001.
Treatment with Licofelone-Containing Vaccine Decreases Gr-1+ CD11b+ Cell Population in Tumor-Inoculated Mice
We hypothesized that the addition of licofelone prolonged the survival of tumor-bearing mice by reducing the frequency of immune-suppressive populations in lymphatic organs. Therefore, we examined the frequency of myeloid cell populations in the BM, spleen, and td-LNs 20 days after tumor inoculation. Tumor-inoculated mice treated with i.v. licofelone-vaccine plus additional s.c. licofelone showed a decrease in the frequency of Gr-1− F4/80+ macrophages and CD11c+ cells in spleen and, for Gr-1− F4/80+ macrophages, in BM (Figure 2). Examination of myeloid subsets in the td-LNs showed a trend toward a reduced frequency of Gr-1+ CD11b+ IMCs in mice treated with any of the three formulations; however, this was not significant.
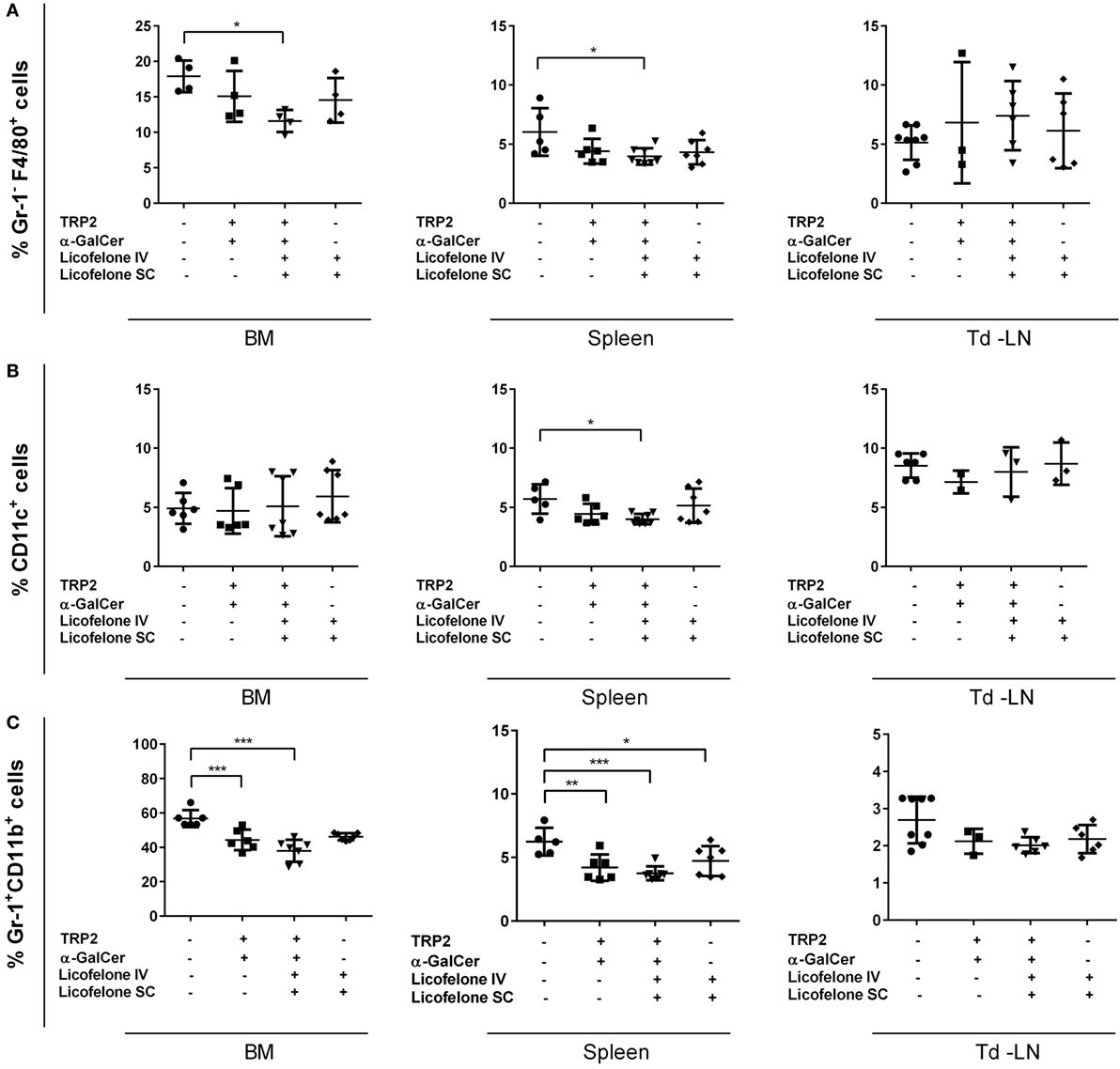
Figure 2. Addition of licofelone to the vaccine therapy reduces the frequency of myeloid cell populations in the BM and the spleens of tumor-bearing mice. Mice were manipulated as described in Figure 1 and the BM, spleens, and td-LNs were collected on day 20 and analyzed for the frequency of (A) Gr-1− F4/80+ macrophages, (B) Gr-1+ CD11b+ IMCs, and (C) CD11c+ DCs by flow cytometry. Frequencies are shown as a percentage of live cells (PIlow cells). Data shown are the mean ± SD of five to seven mice from two experiments. ***p < 0.001, **p < 0.05, *p < 0.01.
The frequency of Gr-1+ CD11b+ IMCs in the BM and the spleen was reduced in mice treated with either the i.v. vaccine or the i.v. licofelone-vaccine plus additional s.c. licofelone. In addition, the frequency of Gr-1+ CD11b+ IMCs in spleen was reduced after licofelone treatment alone (i.v. licofelone plus s.c. licofelone), without vaccination with the long TRP2-peptide and α-GalCer. No significant changes were observed in the frequency of CD4+ and CD8+ T-cell populations (data not shown).
Licofelone Does Not Inhibit Melanoma Cell Growth and ROS Production In Vitro
In order to examine if the antitumor effect of licofelone observed in vivo was attributable to a direct inhibition of tumor cell growth, we evaluated the viability of B16F10luc2 melanoma cells, derived ex vivo, after a 48-h incubation with licofelone. No significant reduction in viability was observed after incubation of B16F10luc2 melanoma cells harvested from tumor-bearing mice with licofelone at 1, 5, 10, or 20 μM (Figure 3A). Similarly, the production of ROS by melanoma cells, as measured by DCFDA staining, was not changed after treatment with licofelone (Figure 3B).
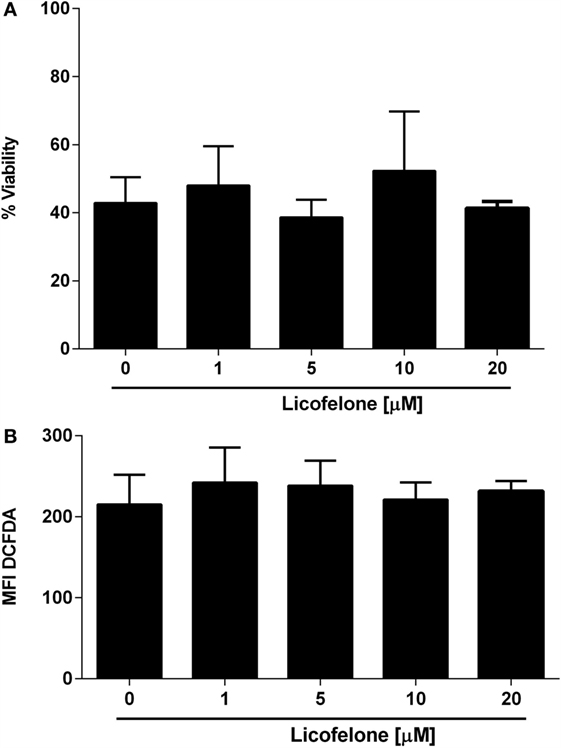
Figure 3. Viability (A) and ROS production (B) of melanoma cells derived ex vivo are not changed by licofelone treatment. B16F10luc2 melanoma cells were treated with licofelone at 1, 5, 10, or 20 μM for 48 h. Data shown are the mean + SD from three individual experiments.
Treatment of Gr-1+ CD11b+ IMCs with Licofelone Reduces the Production of IL-6 and -10 in a Time-Dependent Manner
Increased generation of IMCs in the BM and subsequent accumulation within the tumor has been associated with cancer progression (30). Since licofelone affected the phenotype of BM cells differentiated in the presence of inflammatory mediators associated with tumor progression, we examined if licofelone also influenced the secretion of pro-inflammatory cytokines by IMCs. IL-6 and IL-10 are important inflammatory mediators involved in Gr-1+ CD11b+ mediated immune suppression. The levels of these two cytokines were analyzed in PGE2 and GM-CSF differentiated BM cultures treated either with licofelone (5 μM) and/or LPS (50 ng/mL). LPS was added to activate the cells and induce cytokine production.
While concentrations of IL-6 increased when licofelone was added to the cultures for as little as 4 h after LPS activation (Figure 4A), the treatment of cells with licofelone and LPS only resulted in a significant increase in IL-10 when cultures were supplemented with licofelone for 20 h after LPS activation (Figure 4B). The reduced levels of IL-10 measured at 24 h may be due to autocrine IL-10 uptake as blocking uptake with anti-IL-10 receptor antibodies has shown to increase the amount of IL-10 detected in culture (31).
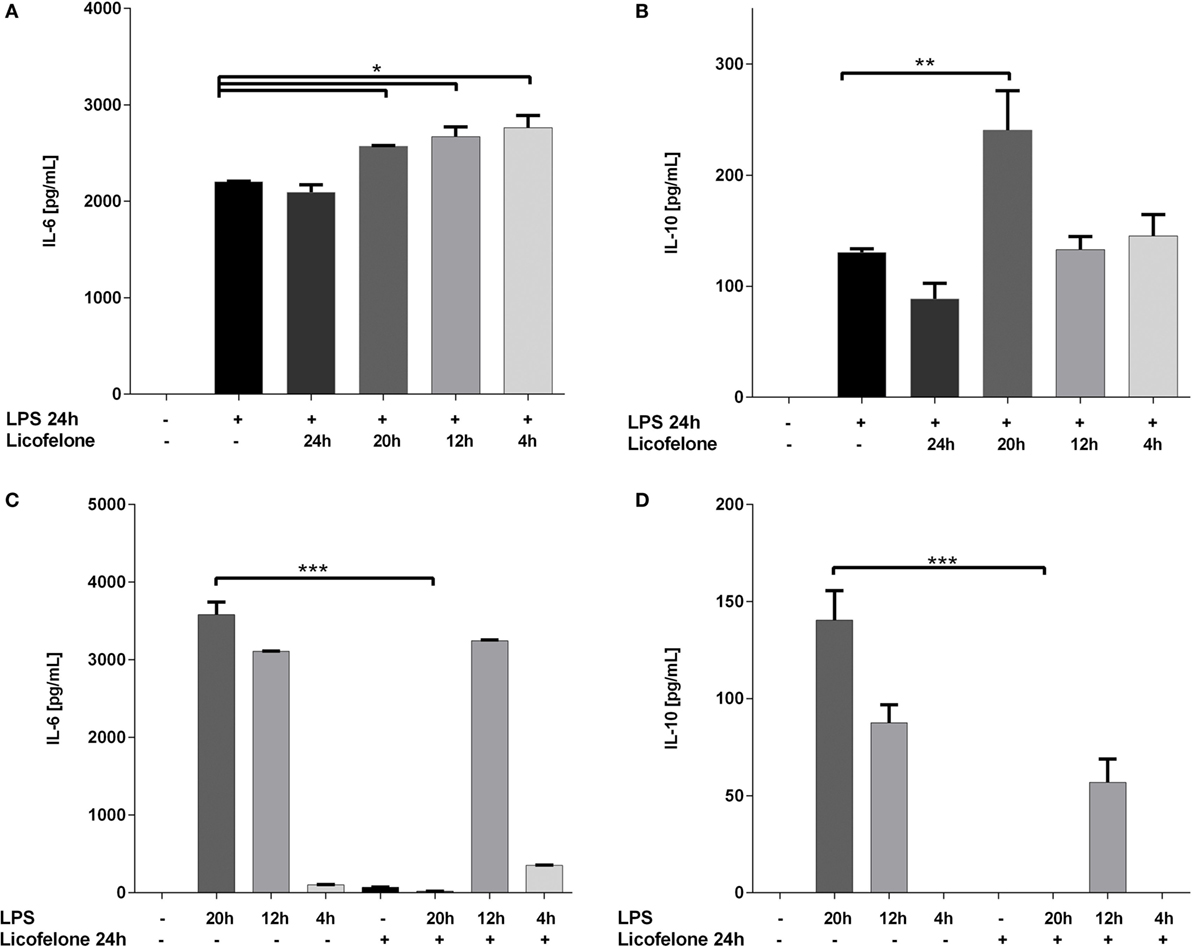
Figure 4. Licofelone increases IL-6 (A,C) and IL-10 (B,D) production when added to LPS-stimulated cells but decreases the production of the two cytokines when added prior to LPS stimulation. IMCs generated from the BM of tumor-bearing mice with GM-CSF (20 ng/mL) and PGE2 (9.1 μg/mL) for 5 days. Cells were treated either with licofelone (5 μM) and/or LPS (50 ng/mL) alone as described. Data shown are mean + SD of pooled groups from two independent experiments. ***p < 0.0001.
The reverse experiment was also carried out whereby the BM cells differentiated under IMC-inducing conditions were pre-incubated with licofelone before the addition of LPS for 4–20 h. Control levels of cytokine in LPS-stimulated cells were measured to assess the effect of licofelone pre-incubation. Treatment of cells with licofelone for 4 h prior to the addition of LPS (for the remaining 20 h) completely ablated the production of IL-6 and IL-10 (Figures 4C,D). Cells stimulated with LPS at later time points did not result in a decrease in amounts of both IL-6 and IL-10 produced.
Licofelone Suppresses the Generation of Gr-1+ CD11b+ IMCs and Gr-1− F4/80+ Macrophages In Vitro
The COX-2 inhibitor celecoxib (32) and several E-prostanoid receptor antagonists (11) have been shown to modulate the generation and the suppressive potential of IMCs. To investigate if the dual COX-2/5-LO inhibitor licofelone shapes myeloid cell populations, BM cells were cultured in the presence of inflammatory factors (PGE2 and GM-CSF) commonly secreted by tumor cells (11) and increasing doses of licofelone. The generation of Gr-1+ CD11b+ IMCs was significantly suppressed in the presence of 2.5 and 5 μM licofelone while the generation of Gr-1− F4/80+ macrophages was suppressed with all three concentrations of licofelone used (Figure 5). The addition of licofelone had no impact on the generation of CD11c+ cells.
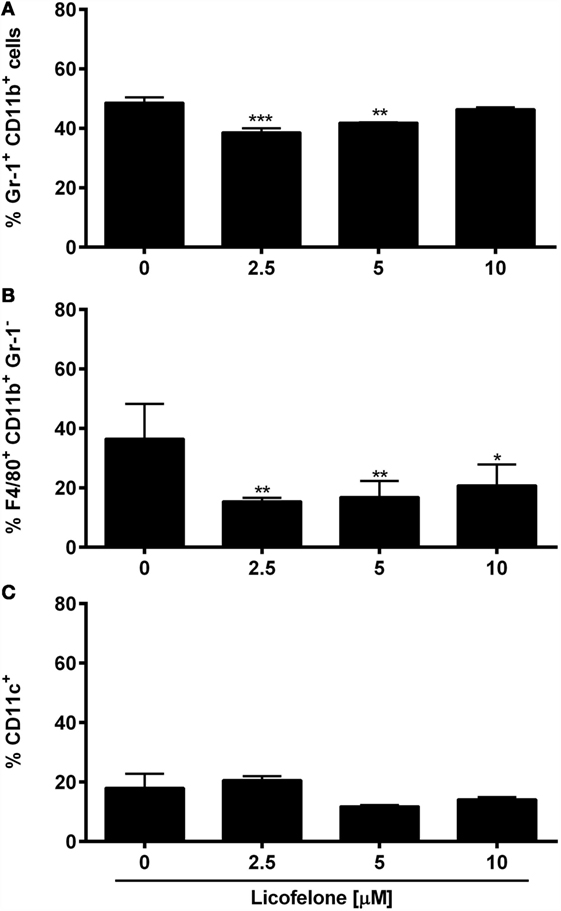
Figure 5. Licofelone affects the generation of IMCs and macrophages in vitro. BM cells were harvested from the femur of naive C57BL/6 mice and were incubated with media and GM-CSF (20 ng/mL), PGE2 (9.1 μg/mL), and licofelone (2.5, 5, and 10 μM) for 5 days. The frequency of (A) Gr-1+ CD11b+ IMCs, (B) Gr-1− F4/80+ macrophages, and (C) CD11c+ DCs was determined by flow cytometry. Frequencies are shown as a percentage of live cells (PIlow cells). The results are the mean + SD of three independent experiments (n = 2/experiment). ***p < 0.001, **p < 0.01, *p < 0.05.
Licofelone Attenuates the Suppressive Function of Gr-1+ CD11b+ IMCs
Next, we examined if licofelone was able to ameliorate or reverse the suppressive function of CD11b+ Gr-1+ IMCs in vitro. CD11b+ Gr-1+ IMCs were generated in vitro from BM precursor cells, pulsed with the SIINFEKL peptide, and activated with LPS prior to coculture with OT-I mouse splenocytes. The proliferation of T-cells was suppressed at all investigated ratios with licofelone being able to reverse the suppressive function of CD11b+ Gr-1+ IMCs partially (Figure 6).
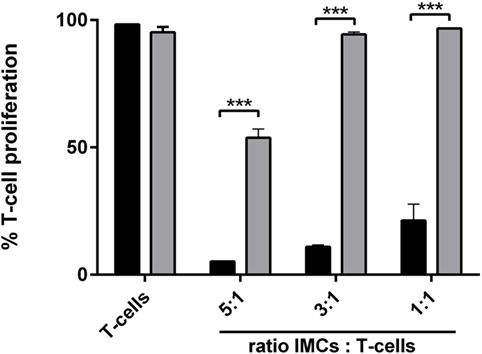
Figure 6. Incubation with licofelone reduces suppressive activity of Gr-1+ CD11b+ IMCs. BM cells differentiated in the presence of GM-CSF (20 ng/mL) and PGE2 (9.1 μg/mL) were incubated with licofelone (5 μM) for 20 h (gray bar) or media (black bar) and then pulsed with LPS and OVA257–264 for 4 h before they were cocultured with CFSE-stained splenocytes from OT-I mice in varying ratios. After 72 h, the proliferation of OVA stimulated T-cells was assessed by flow cytometry. Data are representative of three independent experiments with two replicates + SD each, ***p < 0.0001.
Discussion
One of the biggest obstacles preventing the success of cancer vaccines is the ability of tumor cells to evade killing by the immune system. This is facilitated by the development of immune-suppressive cell populations in the BM, which subsequently migrate to the tumor. Here we investigated the potential of the NSAID licofelone, a dual COX-2/5-LO inhibitor, to reduce the frequency of immune-suppressive cells in lymphatic organs and to stimulate antitumor immunity when used in combination with a cancer vaccine. Licofelone was administered in two ways, once i.v. on day 6 after tumor grafting with the other vaccine components (long TRP2-peptide and α-GalCer) in cationic liposomes, and also every second day s.c. over a period of 14 days beginning on the day of the tumor grafting, again formulated into cationic liposomes.
Previous reports have described a chemo-preventive effect of licofelone in colon cancer (20) and lung adenoma (22, 33) models when administered directly to the cancer site. While in vitro low concentrations (2.5–40 μM) of licofelone decreased proliferation of several prostate tumor cell lines by inducing apoptotic cell death (21), at higher concentrations (150 μM) licofelone induced mitochondrial dysfunction (34). However, we found no effect of systemic licofelone monotherapy on the development of solid tumors in the melanoma model and no direct effect of licofelone on the viability of B16F10luc2 tumor cells harvested from tumor-bearing mice in vitro. Similarly, licofelone did not influence the ROS production of B16 melanoma cells isolated from tumor-bearing mice. These results align with studies that demonstrate COX-2 independent inhibition of ROS production by chemo-preventive drugs, such as nitric oxide-donating aspirin, and suggest that these are off-target effects of the COX inhibitor (35). Together this indicates that in this model the NSAID needs to be given in combination with a vaccine in order to stimulate effective antitumor responses and improve survival. Experiments replacing licofelone with the COX-2 inhibitor celecoxib (data not shown) did not improve survival, suggesting that dual inhibition of both COX-2 and 5-LO is beneficial.
We chose to package the vaccine components and licofelone into cationic liposomes as they are an excellent delivery system for poorly water-soluble drugs, such as licofelone, and are inherently immune-stimulatory (36). The size and charge of the liposomes strongly affects their systemic distribution as well as the uptake and activation of APCs. The positive charge facilitates interactions with the negatively charged cell membranes and is thereby thought to enhance antigen delivery to DCs (37). Furthermore, cationic liposomes activate pro-inflammatory and pro-apoptotic pathways that lead to the generation of ROS (38), enhanced expression of costimulatory molecules such as CD80 and CD86, and secretion of chemokines and cytokines (39), which together should support the generation of an antitumor response. We have previously shown that immunization with a liposomal vaccine, with a similar size and charge as reported here, was superior in terms of eliciting CD8+ T-cell responses as compared to immunization with the vaccine components alone (24).
When we analyzed the composition of cells after immunization with the various vaccines, we detected significant differences in the frequency of myeloid cell populations in the spleens and BM of tumor-inoculated mice treated with a licofelone-containing vaccine with the levels of Gr-1+ CD11b+ IMCs and Gr-1− F4/80+ macrophages being decreased. A similar trend was observed in the td-LNs; however, these differences were not statistically significant which is likely due to variation in the size of tumors on the day they were harvested. The frequency of Gr-1+ CD11b+ IMCs was also decreased in mice treated with the vaccine alone (without licofelone), perhaps indicating that one of the other vaccine components was responsible for this change. For example, the glycolipid α-GalCer, used as an adjuvant in this study, activates iNKT cells, which are known to reduce the suppressive potential of IMCs (40). Moreover, α-GalCer has been shown to promote the maturation of DCs and to contribute to the induction of T-cell responses (41). Even though vaccines containing the iNKT adjuvant α-GalCer improved the survival of tumor-bearing mice, the antitumor effect was further improved by the addition of licofelone to the treatment regimen, significantly extending the median survival time.
The effect of licofelone on myeloid cells from immunized tumor-inoculated mice ex vivo and directly in vitro was examined. An important factor to consider when interpreting the results from the in vitro experiments is the phenotype of cells generated in the chosen culture conditions. Approximately 60–70% of BM-derived cells generated in the presence of GM-CSF express high levels of CD11c and MHCII and are able to stimulate T-cell proliferation in response to antigens (42). The addition of PGE2 and IL-4 has been shown to reduce the CD11c+ MHCII+ population and instead increases the frequency of Gr-1+ CD11b+ IMCs, which are able to suppress T-cell proliferation (11). In accordance with these studies, we found approximately half of our cells, cultured with GM-CSF and PGE2 for 5 days, expressed Gr-1 and CD11b and were able to suppress T-cell proliferation. The addition of licofelone at 2.5–5 μM was able to directly suppress the generation of Gr-1+ CD11b+ IMCs and Gr-1− F4/80+ macrophages. This supports the results of the ex vivo analysis where the frequency of Gr-1+ CD11b+ IMCs and Gr-1− F4/80+ macrophages was similarly reduced after licofelone treatment. While there is little other published information regarding dual COX-2/5-LO inhibitors; the COX-2 inhibitor celecoxib, when used in a mesothelioma mouse model, affected both the number and the function of IMCs by inhibiting PGE2 synthesis and decreasing ROS and NO production (43). The in vitro observed effect of licofelone on macrophages is intriguing as cytokines produced by macrophages upon exposure to PGE2, such as IL-6, are important modulators of IMC function (11). Since we observed an effect of licofelone on both, macrophages and IMCs, it remains to be determined if only one of these populations or both are responsible for the observed antitumor immunity. However, the expression of the same cell surface markers by both MDSCs and macrophages makes it difficult to distinguish between the cell populations and consequently the effects of licofelone. For example, an immune-suppressive subset of MDSCs with a low expression of Gr-1 has been described and found to also express F4/80 (44). Since both cell populations are able to suppress T-cell responses, the inhibition of their development by licofelone is advantageous.
Reduced COX-2 and 5-LO activity has also been associated with a decrease in pro-inflammatory cytokines detectable in the serum of tumor-bearing mice. This included IL-6, which is one of the major cytokines involved in the expansion of Gr-1+ CD11b+ IMCs (20, 45). Therefore the production of IL-6 and IL-10 [which activates the immune-suppressive and tumor-promoting effects of Gr-1+ CD11b+ IMCs via STAT3 and STAT6 signaling (46–48)] by licofelone-treated cells differentiated under IMC-inducing conditions was examined. We found that licofelone treatment of IMC-differentiated LPS-activated cells was unable to reduce production of both IL-6 and IL-10 and instead increased cytokine production in some conditions. However, when licofelone was given before LPS activation, both IL-6 and IL-10 production was almost completely suppressed. Interestingly, the effect of licofelone was transient with suppression only being apparent when licofelone was given around the time of LPS activation. Blidner et al. have shown a differential response of IMCs to the COX-1/2 inhibitor, indomethacin, depending on the IMC microenvironment (49). Under inflammatory conditions, associated with the tumor progression, indomethacin counteracted the suppressive actions of the IMCs and increased their expression of Gr-1. Interestingly, indomethacin treatment in a non-inflammatory environment led to an increase in NO and ROS production in IMCs, amplifying their immune suppression (49, 50). The changes in IL-6 and -10 production after licofelone treatment of LPS-stimulated and unstimulated of Gr-1+ CD11b+ IMCs complement these observations.
Immune suppression, the key feature of IMCs, is mediated through various metabolic and signaling pathways, as well as through direct cell-to-cell contact (8). Therefore, it was of interest to see that licofelone pre-treatment of BM cells differentiated under IMC-conditions could partially reverse CD8 T-cell suppression. This is in agreement with findings from studies that have found other anti-inflammatory agents, for example celecoxib, to reverse IMC induced T-cell suppression in vitro (32, 43, 51). It remains to be determined if the antitumor activity of licofelone solely depends on the inhibition of Gr-1+ CD11b+ IMCs numbers and function through effects on prostaglandin and LT synthesis; or if other factors, such as inhibition of VEGF production (21), are involved. However utilizing a safe, small molecule drug such as licofelone to reduce tumor-induced immune suppression and potentiate immune responses stimulated by therapeutic vaccines provides an attractive alternative to the use of checkpoint blockade inhibitors; which while proving to be very effective in some situations (52, 53) are expensive, cannot be delivered orally and have the potential for serious immune-related adverse events (53, 54).
Author Contributions
All the authors reviewed and approved the final version of the manuscript and agreed to be accountable for the content of the work. SN and SS designed the experiments; acquired, analyzed, and interpreted the data; prepared and critically revised the manuscript; and are accountable for all aspects of the work. RK and SH designed the experiments, interpreted the data, critically revised the manuscript, and are accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This publication was written during tenure of a New Zealand Lottery Health Postdoctoral Fellowship (Silke Neumann, Dunedin, New Zealand).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00537/full#supplementary-material.
Figure S1. Gating strategy for identification of IMCs harvested from tumor-bearing mice. All data collected were analyzed, and doublets were excluded by using forward scatter height and area (FSC-H/FSC-A). Live cells were detected by gating on PIlow cells based on the unstained control. The frequency of macrophages was determined by gating on Gr-1− and F4/80+ cells. Gating from PIlow cells, the gate for CD11c− cells was determined in comparison to an unstained sample. From there, IMCs were identified by the expression Gr-1 and CD11b. The gate for IMCs was chosen in comparison to the relevant FMO.
References
1. Umansky V. New strategies for melanoma immunotherapy: how to overcome immunosuppression in the tumor microenvironment. Oncoimmunology (2012) 1(5):765–7. doi: 10.4161/onci.19713
2. Gabrilovich D, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
3. Ueha S, Shand FH, Matsushima K. Myeloid cell population dynamics in healthy and tumor-bearing mice. Int Immunopharmacol (2011) 11(7):783–8. doi:10.1016/j.intimp.2011.03.003
4. Jordan KR, Amaria RN, Ramirez O, Callihan EB, Gao D, Borakove M, et al. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol Immunother (2013) 62(11):1711–22. doi:10.1007/s00262-013-1475-x
5. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res (2013) 20(6):1601–9. doi:10.1158/1078-0432.CCR-13-2508
6. Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol (2012) 13(4):343–51. doi:10.1038/ni.2224
7. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res (2007) 67(20):10019–26. doi:10.1158/0008-5472.CAN-07-2354
8. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol (2012) 22(4):275–81. doi:10.1016/j.semcancer.2012.01.011
9. Gabrilovich D, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
10. Youn J, Nagaraj S, Collazo M, Gabrilovich D. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181:5791–802. doi:10.4049/jimmunol.181.8.5791
11. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res (2007) 67(9):4507–13. doi:10.1158/0008-5472.CAN-06-4174
12. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol (2015) 15(8):511–23. doi:10.1038/nri3859
13. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol (2012) 188(1):21–8. doi:10.4049/jimmunol.1101029
14. Steinhilber D, Fischer AS, Metzner J, Steinbrink SD, Roos J, Ruthardt M, et al. 5-lipoxygenase: underappreciated role of a pro-inflammatory enzyme in tumorigenesis. Front Pharmacol (2010) 1:143. doi:10.3389/fphar.2010.00143
15. Goetzl EJ, An S, Smith WL. Specificity of expression and effects of eicosanoid mediators in normal physiology and human diseases. FASEB J (1995) 9(11):1051–8.
16. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LBA, et al. Cyclooxygenase in biology and disease. FASEB J (1998) 12(12):1063–73.
17. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res (2011) 71(24):7463–70. doi:10.1158/0008-5472.CAN-11-2449
18. Yokota Y, Inoue H, Matsumura Y, Nabeta H, Narusawa M, Watanabe A, et al. Absence of LTB4/BLT1 axis facilitates generation of mouse GM-CSF–induced long-lasting antitumor immunologic memory by enhancing innate and adaptive immune systems. Blood (2012) 120(17):3444–54. doi:10.1182/blood-2011-10-383240
19. Charlier C, Michaux C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur J Med Chem (2003) 38(7–8):645–59. doi:10.1016/S0223-5234(03)00115-6
20. Mohammed A, Janakiram NB, Li Q, Choi C-I, Zhang Y, Steele VE, et al. Chemoprevention of colon and small intestinal tumorigenesis in apcmin/+ mice by licofelone, a novel dual 5-lox/cox inhibitor: potential implications for human colon cancer prevention. Cancer Prev Res (2011) 4(12):2015–26. doi:10.1158/1940-6207.CAPR-11-0233
21. Narayanan NK, Nargi D, Attur M, Abramson SB, Narayanan BA. Anticancer effects of licofelone (ml-3000) in prostate cancer cells. Anticancer Res (2007) 27(4B):2393–402.
22. Sharma S, Lee J, Zhou J, Steele VE. Chemopreventive efficacy and mechanism of licofelone in a mouse lung tumor model via aspiration. Cancer Prev Res (2011) 4(8):1233–42. doi:10.1158/1940-6207.CAPR-10-0117
23. Fischer L, Hornig M, Pergola C, Meindl N, Franke L, Tanrikulu Y, et al. The molecular mechanism of the inhibition by licofelone of the biosynthesis of 5-lipoxygenase products. Br J Pharmacol (2007) 152(4):471–80. doi:10.1038/sj.bjp.0707416
24. Neumann S, Young K, Compton B, Anderson R, Painter G, Hook S. Synthetic TRP2 long-peptide and alpha-galactosylceramide formulated into cationic liposomes elicit CD8(+) T-cell responses and prevent tumour progression. Vaccine (2015) 33(43):5838–44. doi:10.1016/j.vaccine.2015.08.083
25. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol (2010) 10(11):787–96. doi:10.1038/nri2868
26. Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol (2008) 38(4):1033–42. doi:10.1002/eji.200737995
27. Du W, Gervay-Hague J. Efficient synthesis of alpha-galactosyl ceramide analogues using glycosyl iodide donors. Org Lett (2005) 7(10):2063–5. doi:10.1021/ol050659f
28. Scheicher C, Mehlig M, Zecher R, Reske K. Dendritic cells from mouse bone marrow: in vitro differentiation using low doses of recombinant granulocyte-macrophage colony-stimulating factor. J Immunol Methods (1992) 154(2):253–64. doi:10.1016/0022-1759(92)90199-4
29. Neumann S, Burkert K, Kemp R, Rades T, Rod Dunbar P, Hook S. Activation of the NLRP3 inflammasome is not a feature of all particulate vaccine adjuvants. Immunol Cell Biol (2014) 92(6):535–42. doi:10.1038/icb.2014.21
30. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13(10):739–52. doi:10.1038/nrc3581
31. Levings MK, Gregori S, Tresoldi E, Cazzaniga S, Bonini C, Roncarolo MG. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood (2005) 105(3):1162–9. doi:10.1182/blood-2004-03-1211
32. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood (2011) 118(20):5498–505. doi:10.1182/blood-2011-07-365825
33. Kumar G, Patlolla JM, Madka V, Mohammed A, Li Q, Zhang Y, et al. Simultaneous targeting of 5-LOX-COX and ODC block NNK-induced lung adenoma progression to adenocarcinoma in A/J mice. Am J Cancer Res (2016) 6(5):894–909.
34. Tavolari S, Bonafè M, Marini M, Ferreri C, Bartolini G, Brighenti E, et al. Licofelone, a dual COX/5-LOX inhibitor, induces apoptosis in HCA-7 colon cancer cells through the mitochondrial pathway independently from its ability to affect the arachidonic acid cascade. Carcinogenesis (2008) 29(2):371–80. doi:10.1093/carcin/bgm265
35. Sun Y, Chen J, Rigas B. Chemopreventive agents induce oxidative stress in cancer cells leading to COX-2 overexpression and COX-2-independent cell death. Carcinogenesis (2009) 30(1):93–100. doi:10.1093/carcin/bgn242
36. Yan W, Chen W, Huang L. Mechanism of adjuvant activity of cationic liposome: phosphorylation of a MAP kinase, ERK and induction of chemokines. Mol Immunol (2007) 44(15):3672–81. doi:10.1016/j.molimm.2007.04.009
37. Bramwell VW, Perrie Y. Particulate delivery systems for vaccines: what can we expect? J Pharm Pharmacol (2006) 58(6):717–28. doi:10.1211/jpp.58.6.0002
38. Yan W, Chen W, Huang L. Reactive oxygen species play a central role in the activity of cationic liposome based cancer vaccine. J Control Release (2008) 130(1):22–8. doi:10.1016/j.jconrel.2008.05.005
39. Vangasseri DP, Cui Z, Chen W, Hokey DA, Falo LD Jr, Huang L. Immunostimulation of dendritic cells by cationic liposomes. Mol Membr Biol (2006) 23(5):385–95. doi:10.1080/09687860600790537
40. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest (2008) 118(12):4036–48. doi:10.1172/JCI36264
41. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med (2003) 198(2):267–79. doi:10.1084/jem.20030324
42. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity (2015) 42(6):1197–211. doi:10.1016/j.immuni.2015.05.018
43. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. BMC Cancer (2010) 10:464. doi:10.1186/1471-2407-10-464
44. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol (2010) 40(1):22–35. doi:10.1002/eji.200939903
45. Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, et al. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol (2012) 42(8):2060–72. doi:10.1002/eji.201142335
46. Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol (2003) 4(12):1169–76. doi:10.1038/ni1012
47. Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immunol (2010) 199(3):273–81. doi:10.1007/s00430-010-0151-4
48. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol (2007) 7(1):41–51. doi:10.1038/nri1995
49. Blidner AG, Salatino M, Mascanfroni ID, Diament MJ, Bal de Kier Joffe E, Jasnis MA, et al. Differential response of myeloid-derived suppressor cells to the nonsteroidal anti-inflammatory agent indomethacin in tumor-associated and tumor-free microenvironments. J Immunol (2015) 194(7):3452–62. doi:10.4049/jimmunol.1401144
50. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity (2010) 32(6):790–802. doi:10.1016/j.immuni.2010.05.010
51. Nagaraj S, Schrum AG, Cho H-I, Celis E, Gabrilovich DI. Mechanism of T-cell tolerance induced by myeloid-derived suppressor cells. J Immunol (2010) 184(6):3106–16. doi:10.4049/jimmunol.0902661
52. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
53. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28(19):3167–75. doi:10.1200/JCO.2009.26.7609
Keywords: cancer vaccine, melanoma, NSAID, COX-2/5-LO, myeloid-derived suppressor cells, immature myeloid cells, liposomes, α-galactosylceramide
Citation: Neumann S, Shirley SA, Kemp RA and Hook SM (2016) Improved Antitumor Activity of a Therapeutic Melanoma Vaccine through the Use of the Dual COX-2/5-LO Inhibitor Licofelone. Front. Immunol. 7:537. doi: 10.3389/fimmu.2016.00537
Received: 14 September 2016; Accepted: 14 November 2016;
Published: 05 December 2016
Edited by:
Sherven Sharma, VA Greater Los Angeles Healthcare System (VHA), USAReviewed by:
Anna Kozlowska, University of California Los Angeles, USAThorbald Van Hall, Leiden University, Netherlands
Copyright: © 2016 Neumann, Shirley, Kemp and Hook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silke Neumann, c2lsa2UubmV1bWFubkBvdGFnby5hYy5ueg==
†Silke Neumann and Simon A. Shirley contributed equally to this work.