 Celia A. McKee
Celia A. McKee John R. Lukens
John R. Lukens- Department of Neuroscience, Center for Brain Immunology and Glia, School of Medicine, University of Virginia, Charlottesville, VA, USA
Traumatic brain injury (TBI) affects an ever-growing population of all ages with long-term consequences on health and cognition. Many of the issues that TBI patients face are thought to be mediated by the immune system. Primary brain damage that occurs at the time of injury can be exacerbated and prolonged for months or even years by chronic inflammatory processes, which can ultimately lead to secondary cell death, neurodegeneration, and long-lasting neurological impairment. Researchers have turned to rodent models of TBI in order to understand how inflammatory cells and immunological signaling regulate the post-injury response and recovery mechanisms. In addition, the development of numerous methods to manipulate genes involved in inflammation has recently expanded the possibilities of investigating the immune response in TBI models. As results from these studies accumulate, scientists have started to link cells and signaling pathways to pro- and anti-inflammatory processes that may contribute beneficial or detrimental effects to the injured brain. Moreover, emerging data suggest that targeting aspects of the immune response may offer promising strategies to treat TBI. This review will cover insights gained from studies that approach TBI research from an immunological perspective and will summarize our current understanding of the involvement of specific immune cell types and cytokines in TBI pathogenesis.
Introduction
Traumatic brain injury (TBI) affects millions of people worldwide every year, and current estimates from the World Health Organization (WHO) suggest that TBI will be the third leading cause of death and disability by the year 2020 (1). In the US alone, upwards of 1.7 million Americans seek medical treatment for some form of brain trauma each year (2, 3), and nearly 2% of the American population, or approximately 5–6 million people, currently suffer from TBI-related disabilities (4). TBI is a particularly serious threat to health in newborns, children, the elderly, military service personnel, and athletes involved in contact sports. Trauma to the brain can result in persistent and debilitating impairments in cognition, sensory function, mental health, and motor function. Furthermore, TBI-induced inflammation and pathology have been strongly linked to increased risks of developing numerous neurological disorders including anxiety, depression, PTSD, Alzheimer’s disease (AD), chronic traumatic encephalopathy (CTE), Parkinson’s disease, and amyotrophic lateral sclerosis (ALS) (5–10).
Despite being a prevalent and pressing global medical issue, there are currently no FDA-approved therapeutics to treat TBI. In recent years, mounting evidence from both TBI patients and animal models of brain injury implicate dysregulated immune responses in the potentiation of TBI-induced neurological dysfunction and brain pathology (11–16). For instance, elevated cytokine production is one of the strongest prognostic indicators of poor clinical outcomes in TBI (17–21), and brain trauma has been shown to induce immune-mediated inflammatory responses that can last for years post-injury (22, 23).
In addition to providing vital protective measures against pathogens and tumors, the immune system is also centrally involved in the restoration of tissue homeostasis following injury. Critical functions that are carried out by the immune system in response to injury include the sequestration of tissue-damaging irritants, engulfment and disposal of cellular debris, and the promotion of the wound-healing response. Tissue damage that results from trauma, ischemia-reperfusion injury, metabolic distress, and environmental irritants provokes the release of damage-associated molecular patterns (DAMPs) [e.g., ATP, reactive oxygen species (ROS), damaged mitochondria, and necrotic cells] and alarmins [e.g., interleukin (IL)-1α, IL-33, HMGB1]. The recognition of DAMPs and alarmins by immune receptors then stimulates the local production of cytokines and chemokines at the site of injury, which subsequently coordinates the activation, expansion, and recruitment of immune cells to areas of tissue damage.
Brain trauma results in two phases of tissue injury. The first round of injury is a direct result of exorbitant mechanical impact to the brain tissue. The aftermath of a severe blow to the head results in immediate neuronal and glial cell death, axonal injury, disruption of the blood–brain barrier (BBB), edema, and the release of DAMPs and excitotoxic agents (24). The immune response to TBI is intended to promote neuroprotection and repair, but can become maladaptive if dysregulation occurs. Whether the immune response contributes to repair or further destruction ultimately depends on the nature, duration, and magnitude of the immunological events that develop in response to brain injury. If not properly controlled, the immune system can provoke a secondary phase of tissue damage and neuroinflammation. In contrast to the acute nature of the primary brain injury, the secondary tissue damage generally results in a diffuse, long-lasting injury. The fundamental role that the immune system plays in driving the secondary phase of tissue damage following brain trauma has led many to believe that immunomodulatory approaches may offer a much-needed strategy to treat TBI. In this review, we discuss how aspects of the immune response can influence clinical outcomes following TBI. In particular, we highlight recent findings from experimental models of TBI that define central roles for individual immune cell types and cytokines in TBI pathogenesis (Figure 1; Table 1).
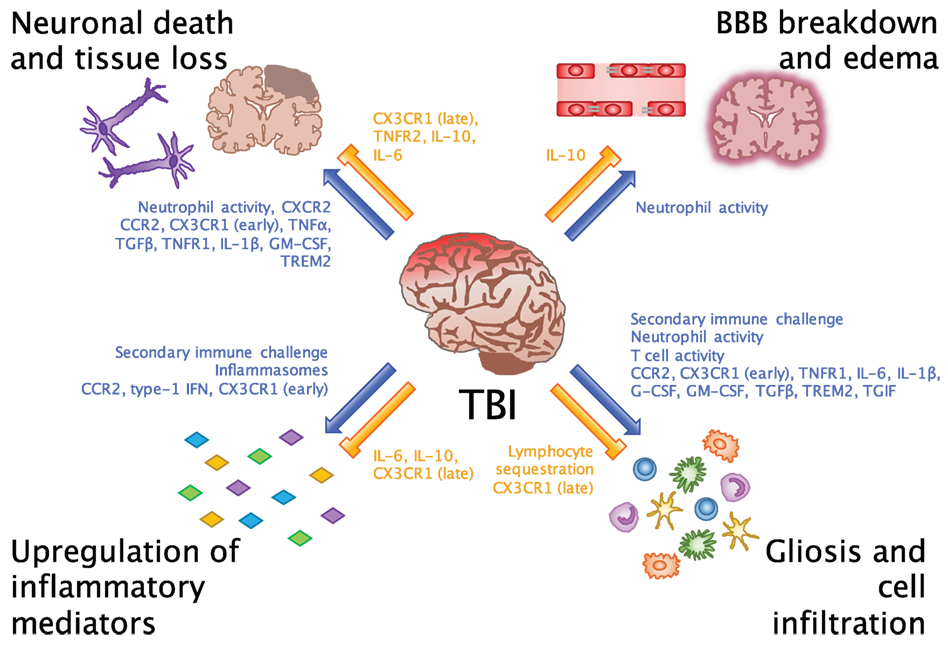
Figure 1. Beneficial and detrimental roles for the immune system in TBI. Common consequences of neuroinflammation after TBI include neuronal death and tissue loss, BBB breakdown and edema, upregulation of inflammatory mediators, and gliosis and cell infiltration. Researchers have evaluated these processes in order to understand which inflammatory cells and molecules potentiate (blue arrows) and inhibit (orange bars) the inflammatory environment of the brain. While we are beginning to link certain cells and molecules to their beneficial and detrimental effects in CNS injury, an important takeaway from these findings is that facilitators of inflammation may be involved in multiple processes at different points in time after injury.
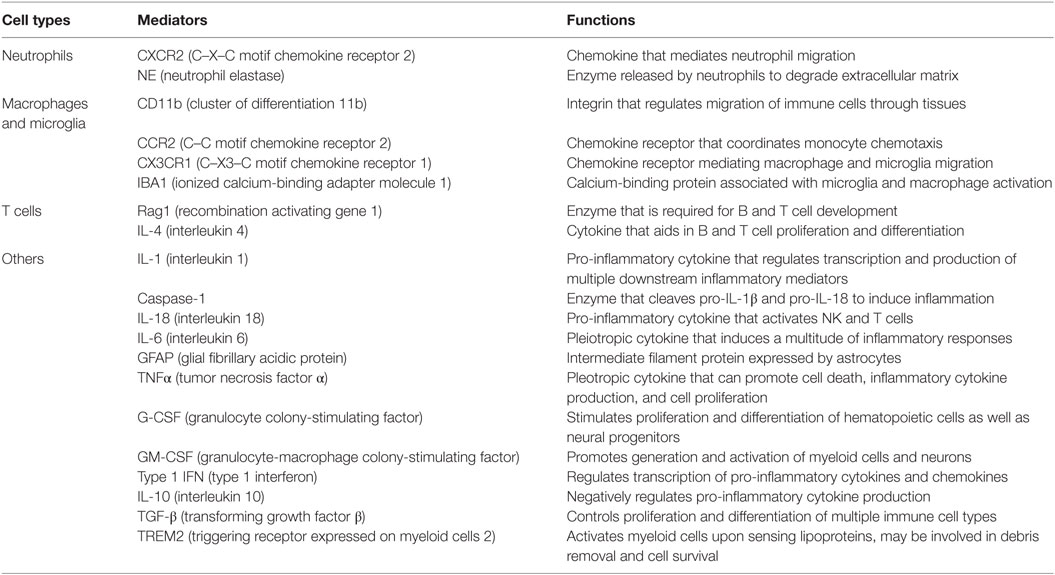
Table 1. Key immune mediators involved in TBI pathogenesis.
The Kinetics of the Immune Response to Brain Injury
Upon brain injury, DAMPs and alarmins are released into the extracellular space where they can then signal through pattern-recognition receptors (PRRs) and cytokine receptors on CNS resident cells. This promotes the production of cytokines and chemokines that are involved in coordinating the recruitment of immune cells to sites of tissue damage (Figure 2). Neutrophils are the first immune cells that are recruited to the brain in response to trauma (25, 26). They first appear in the sub-arachnoid and vascular space surrounding the site of tissue damage within hours of injury. Neutrophils then begin to infiltrate into the brain parenchyma at 24 h post-injury (27). As the first responders, they play critical roles in the containment of the injury lesion and in the removal of cellular debris and damaged cells. Neutrophils predominate during the first days following injury; however, their numbers diminish greatly between days 3 and 5 post-injury. This time point coincides with the recruitment of other peripheral immune cells and the local activation of microglia and astrocytes. CCR2-expressing monocytes are the major immune cell population that infiltrates into the damaged tissue at days 3–5 post-injury, although T cells, natural killer (NK) cells, and dendritic cells (DCs) can also be detected around the injury site (15, 28–30). The coordinated production of chemokines following trauma orchestrates the recruitment of immune cells to areas of brain injury. The major chemokine pathways that are involved in mobilizing immune cells to brain damage have been comprehensively reviewed recently (11, 31–33) and will not be covered in great detail in this review. By 2 weeks post-injury, the brain is largely devoid of any infiltrating immune cells. However, activated microglia and astrocytes and elevated levels of inflammatory cytokines can be detected for months to years following brain injury (22, 23, 34, 35). This is unlike what is seen following tissue damage in other peripheral organs, where tissue resident macrophages and stromal cells typically return to a resting or immunologically quiescent state within weeks post-injury. The existence of activated glial cells and aberrant regulation of cytokine expression for months to years post-TBI suggests that the immune response to TBI can persist for long periods beyond the initial trauma.
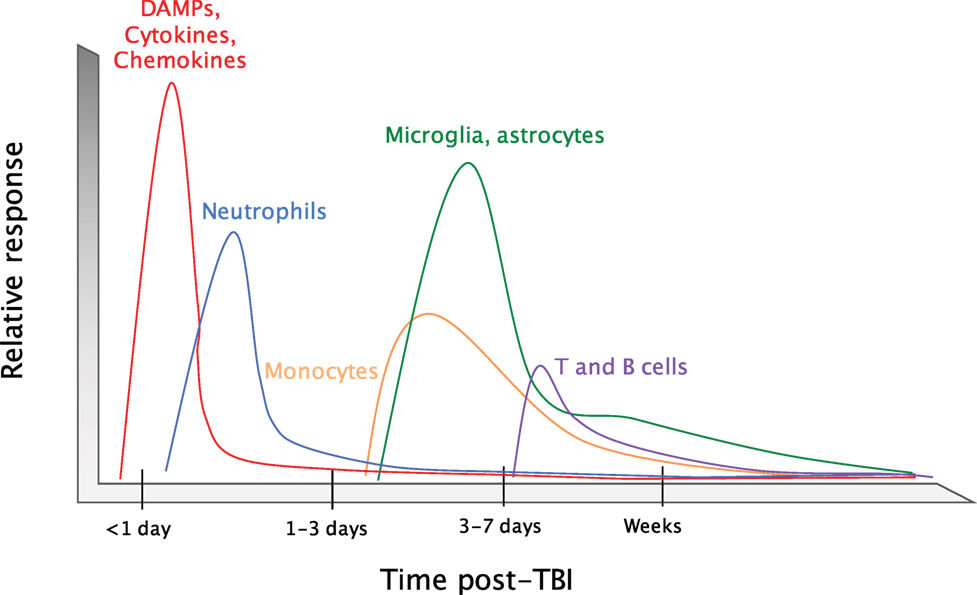
Figure 2. Timeline of the immune response to TBI. Upon an impact to the head, cellular damage results in the rapid release of damage-associated molecular patterns (DAMPs) that prompt resident cells to release cytokines and chemokines. These signals quickly call in neutrophils, which aid in the containment of the injury site and promote the removal of debris and damaged cells. As neutrophil numbers begin to decline after a period of days, infiltrating monocytes and activated glia begin to accumulate around the site of injury to perform reparative functions. Depending on the severity of the brain injury, T and B cells can also be recruited to sites of brain pathology at later time points in the response (3–7 days post-injury).
Involvement of Immune Cell Types in TBI
Neutrophils
Considering their role in vascular permeability and edema in peripheral tissues, neutrophils have largely been implicated in BBB breakdown and edema in TBI. However, it is still unclear how their activity is related to these processes. Early TBI papers agree that neutrophils can accumulate at sites of injury within hours post-injury (26, 36) and that the number of neutrophils that are recruited to sites of brain trauma typically correlates with the severity of the injury (37). However, studies on the role of neutrophils in mediating BBB breakdown, edema, and neurodegeneration have been inconclusive. Although neutrophilia has been reported to coincide with BBB breakdown and neurodegeneration, these processes seem to be disconnected spatially and temporally from the invasion of neutrophils (26). Furthermore, efforts to deplete neutrophils have been unsuccessful in linking them to loss in BBB integrity (36), which was thought to be the event responsible for subsequent edema and neuronal death.
Due to these early findings, researchers began to think of neutrophil activity and tissue edema as having important consequences independent of BBB breakdown. Kenne et al. used an anti-Gr-1 antibody to deplete neutrophils in a cortical controlled impact (CCI) model and found that neutrophil depletion led to decreased edema for at least 48 h after injury, but did so without ameliorating BBB permeability (38). Neutrophil depletion was associated with decreased numbers of apoptotic cells, reduced macrophage/microglia activation in the cortex, and mitigated tissue loss. These data are similar to results from CXCR2 knockout mice, which were used by Semple et al. to reduce CXCR2-mediated infiltration of neutrophils after TBI (39). These mice did show reduced neutrophil infiltration into the brain, but BBB breakdown appeared similar to wild-type mice. While they also showed significantly less cell death within the lesion, this did not have an impact on functional outcome. Collectively, these two studies suggest that neutrophil depletion may have neuroprotective effects in TBI without necessarily being linked to BBB breakdown.
Further research into the importance of neutrophil activity post-TBI has begun to characterize the mechanisms involved in neutrophil-mediated neurodegeneration at early time points. For instance, Semple et al. used neutrophil elastase (NE) knockout mice in a CCI model to investigate how neutrophil effector functions contribute to secondary tissue damage and neurological dysfunction following brain trauma (40). They found that NE-deficient mice exhibit significantly diminished edema at 24 h after injury. However, this was not associated with reductions in neutrophil numbers or decreased production of matrix metalloproteinase-9 (MMP-9), which is known to regulate neutrophil migration by promoting extracellular matrix breakdown and/or through the modulation of chemokine activity. NE knockout mice also had reduced numbers of apoptotic neurons as well as lower heme-oxygenase levels in the hippocampus at 24 h after injury, indicating attenuated cell death and a less severe oxidative state. However, these early neuroprotective effects did not prevent cortical or hippocampal volume loss in the long-term, which may explain why NE deficiency was not found to improve behavioral performance at 2 months post-injury. These findings suggest that NE activity contributes to injury-induced edema and early neurodegeneration.
Thus, it is becoming clearer that neutrophils are linked to cerebral edema and neuronal death in TBI, but the relationship between neutrophil activity and BBB breakdown is not as clear as previously thought. It is likely that the distinct differences between the BBB and other vascular barriers outside the brain mean that the BBB structure has a different relationship with neutrophils that remains to be elucidated. In future studies, it will be important to investigate whether vascular edema in TBI is directly responsible for releasing cytotoxic substances that cause neuronal death following TBI or whether neutrophils and other inflammatory cells within the parenchyma are the primary source of neurotoxic factors that promote cytotoxic edema and early neurodegeneration in TBI.
Macrophages and Microglia
There has been tremendous interest in defining the discrete roles of macrophages and microglia in TBI. Activated microglia and macrophages release pro- and anti-inflammatory factors that can signal to resident and peripheral cells to promote or resolve the inflammatory response to trauma. Chronically activated microglia and macrophages have been found in rodent models and humans after TBI (22, 23, 41, 42) and are considered to be one of the hallmarks of unresolved inflammation that may have long-term consequences (43, 44).
Groups have utilized different methods to deplete microglia and macrophages in vivo in order to characterize their roles in TBI-induced neuroinflammation, tissue damage, and neurological dysfunction. Two of these methods use targeted depletion of CD11b-expressing cells with transgenic CD11b-TK (thymidine kinase) and CD11b-DTR (diphtheria toxin receptor) mice (45, 46). While both methods were effective in reducing their target cell types post-TBI, neither attenuated signs of tissue damage such as axonal injury and lesion size. However, it should be noted that both of the treatment approaches that were used to deplete CD11b-expressing cells in these studies were found to cause inflammation even in uninjured mice. Therefore, it is likely that triggering inflammation before injury had an effect on the outcomes that were observed in these studies.
The chemokine receptor CCR2 plays critical roles in the recruitment of monocytes/macrophages to the brain, and, as a result, suppression of CCR2 signaling is often exploited to reduce the effects of infiltrating monocytes/macrophages in TBI studies. Numerous recent reports have shown that abrogating CCR2-mediated events can markedly limit both TBI-induced neuroinflammation and cognitive decline. For instance, Morganti et al. found that the CCR2 antagonist CCX872 reduces accumulation of peripheral macrophages in the brain and alters the regulation of several pro- and anti-inflammatory cytokines as well as NADPH oxidase (NOX2) production after CCI (47). These effects were associated with less severe hippocampal-dependent cognitive dysfunction. Similarly, CCR2 deficiency in another CCI study reduced numbers of infiltrating monocytes and rescued long-term spatial learning and memory deficits in the Morris water maze (MWM) test (48). Another group disrupted CCR2 activity by knocking in a red fluorescent protein (RFP) protein at the Ccr2 gene locus in mice. In their studies, they found that impaired CCR2 signaling prevents monocyte recruitment into the brain and reduces cavity volume and axonal pathology following fluid percussion injury (FPI) (49). Taken together, these studies indicate that inhibition of CCR2-mediated cell infiltration limits neurodegeneration and neurological decline following brain trauma.
A recent study by Zanier et al. used CX3CR1 knockout mice to disrupt CX3CL1 chemokine signaling in order to understand its importance in controlling myeloid cell activity in TBI (50). After receiving a CCI injury, CX3CR1 knockout animals showed neurological protection 4 days following TBI. However, while wild-type mice returned to pre-injury levels of neuroscore performance by 5 weeks post-injury, CX3CR1-deficient mice still exhibited appreciable impairments in neuroscore performance at this time point. This decline in neuroscore performance at later time points in Cx3cr1−/− mice was associated with persistent neuronal death and an overall decrease in neuronal numbers. Further investigation into the effects of disrupted CX3CR1 signaling on macrophages and microglia showed that these cell types exhibit a more protective, anti-inflammatory phenotype in injured CX3CR1-null mice than seen in injured controls at early time points. However, at 5 weeks post-TBI, CX3CR1-deficient mice showed signs of elevated myeloid cell activation as compared to wild-type animals. Taken together, these results indicate that while early CX3CR1 signaling may have detrimental effects, this signaling is necessary at later time points post-brain injury to prevent long-term inflammation and cognitive impairment.
Another issue facing the TBI field is how to best define inflammatory cell types. Using principal component analysis (PCA) and microarray analysis of brain macrophages, Hsieh et al. found that the subset of macrophages expressing the M2 (alternatively activated macrophages)-associated marker arginase-1 (Arg1) had a distinctly different transcriptional profile from arginase-1-negative cells, but that the genes they expressed after TBI did not match traditional M2 markers (51). They found that while Arg1+ and Arg1− macrophages expressed a variety of M1 and M2 markers, they differed distinctly in their chemokine profiles and several genes involved in injury protection and wound healing. These data indicate that delineating macrophages by an M1 (classically activated macrophages) or M2 phenotype in TBI obscures other macrophage subsets that may have distinct roles in the injury response.
Emerging data also suggest that macrophage phenotypes may be more flexible than once thought. Wang et al. set out to characterize the timeline of M1 and M2 macrophage/microglia activity after CCI. By tracking M1 macrophages/microglia with the marker CD16/32 and M2 macrophages/microglia with CD206, they found that at 3 and 7 days after injury the majority of Iba1+ cells assumed an M1 phenotype, yet at day 5 there was a rise in M2 macrophage/microglial cell numbers (52). This shift from an M1 state to an M2 phenotype and back may provide protection from possible detrimental effects of a prolonged state of either phenotype. The authors also found that white matter injury correlated with M1 cells, peaking at 3 and 7 days.
Clarifying the activation timeline and phenotypes of macrophages and microglia will likely be important in understanding how unresolved inflammation can lead to long-term detrimental consequences. An emerging body of literature is beginning to define how microglia and macrophages can be primed by TBI and generate exaggerated immune response and functional deficits upon secondary immune challenge. For example, injection of LPS at 30 days after injury in an FPI model induced more robust inflammatory cytokine production by CD11b-expressing cells in TBI animals than in controls (53). This was associated with decreased social exploratory behavior at 24 h after LPS injection as well as depressive behaviors. This same group found that secondary immune challenge also caused learning and memory deficits that could be linked to TBI-mediated microglia priming (54). These data indicate that at long-term time points, when behavioral deficits appear to have normalized following brain injury, a second immune challenge can produce further cognitive decline.
Taken together, these studies provide examples of how the TBI field is beginning to characterize macrophage and microglia migration, activation, and priming in relation to functional deficits after TBI. An important consideration for this field is that many authors choose to study both macrophages and microglia as a combined population, acknowledging that it is difficult to distinguish them within an inflammatory context using current markers, such as CD11b, CD45, CX3CR1, and IBA1. However, considering the importance of these cells in both the short and long-term inflammatory states, more specifically targeted techniques would help to define their discrete roles. In addition, considering the timeline of their activation, adapting methods to study macrophage and microglia signaling over the acute and chronic phases of TBI will be necessary in order to uncover time-dependent beneficial and detrimental effects as well as identifying effective therapeutic windows.
T Cells
The kinetics of T cell infiltration have been described in TBI patients and animal models (55–57), but it still remains unclear what role(s) they play in brain trauma-associated wound-healing responses. In a study by Weckbach et al., Rag1−/− mice were used to investigate how the absence of B and T cells influences brain pathology and neurological impairment following weight drop-induced TBI (58). Surprisingly, lacking the adaptive arm of the immune system did not appreciably affect neurological outcome, BBB integrity, pro- or anti-apoptotic mediators, hippocampal architecture, or astroglial activation in these studies.
In a separate study, Mencl et al. used the sphingosine-1-phosphate receptor agonist and lymphocyte sequesterer FTY720 to inhibit T cell migration to the brain following TBI (59). While FTY720 did decrease the numbers of circulating lymphocytes, it did not provide any protection to TBI animals in terms of lesion volume, neuroscore, apoptotic neurons, BBB maintenance, or edema. However, FTY720 was found to reduce the numbers of neutrophils and macrophages/microglia in the ipsilateral hemisphere at 1 day after injury. Thus, future investigations should evaluate the ability of T cells to regulate the infiltration of other immune cells into sites of brain injury.
In future studies, it will be important to move away from methods that promote global defects in T cell responses and consider more specific effects of T cell subsets on TBI progression. In other models of CNS injury, T cells have been found to confer neuroprotection (60–62). For example, Walsh et al. recently reported that protection after spinal cord injury (SCI) is guided by specific T cell-derived cytokines, particularly IL-4 (63). Their interest in IL-4 stemmed from the observation that T cells within the site of injury were the major producers of IL-4 in their model and that functional recovery was markedly delayed following SCI in IL-4 knockout mice. They found that reconstituting Rag1−/− mice with IL-4-deficient T cells prior to injury did not lead to functional recovery, but transfer of wild-type T cells did. In addition, a MyD88-dependent Th2 skew of T cells was necessary to produce IL-4 and induce elevated neurotrophin signaling and axonal outgrowth both in vitro and in vivo. This insight into T cell subsets in injury recovery may apply to the TBI field, and thus warrants more specific investigations.
Inflammatory Mediators in TBI
Interleukin-1
Interleukin-1 is a potent pro-inflammatory cytokine that has been implicated in numerous inflammatory and neurological disorders. Secretion of IL-1 must be tightly regulated in the brain, as unchecked IL-1 production has been shown to provoke neuroinflammation and neurodegeneration. There are two distinct forms of IL-1 – IL-1α and IL-1β – both of which can induce similar levels of inflammatory signaling following engagement of IL-1 receptor (IL-1R). Although IL-1α and IL-1β evoke almost identical downstream inflammatory responses, their expression patterns and requirements for activation differ greatly. IL-1α is constitutively expressed by all nucleated cells, and secreted full-length IL-1α can transmit inflammatory signaling without the need for further modification or processing. In contrast, IL-1β is generated as a biologically inactive pro-form protein that requires cleavage to elicit its inflammatory properties and secretion (64). Caspase-1 activation in inflammasome complexes has emerged as a major mechanism for both IL-1β cleavage and IL-1α release (14, 65–69), although recent studies have also begun to identify additional inflammasome-independent pathways that promote IL-1 production (64, 70–72).
Interleukin-1β is one of the most frequently measured cytokines in the TBI literature, and it has been shown to be increased after TBI in humans and mouse models (46, 47, 53, 73–83). During neuroinflammation, IL-1β is known to have profound effects on BBB permeability, glial activation, immune cell recruitment, and neurodegeneration (84–86) and is likely one of the first immune mediators as it peaks early after injury. IL-1β is known to strongly enhance inflammatory responses following TBI, and this has led many to postulate that IL-1 production may negatively impact clinical outcomes following brain trauma (75).
Recently, progress has been made using methods to neutralize or antagonize the activity of IL-1β in TBI. In two different studies, Clausen et al. administered an anti-IL-1β neutralizing antibody to CCI-injured animals through 14 days after TBI (87, 88). In these studies, IL-1β neutralization led to a decrease in the numbers of microglia/macrophages, neutrophils, and T cells in the brain, especially at 7 days after injury. Although they did not report appreciable differences in motor coordination performance during the rotarod test, they did observe better performance during learning trials in the MWM, as well as decreased tissue loss at experimental endpoints for anti-IL-1β-treated animals.
In other models of CNS injury, IL-1α upregulation precedes that of IL-1β and IL-1α deletion limits neuronal damage and promotes accelerated functional recovery (89). IL-1α has also been suggested to jump start an inflammatory loop that is sustained and enhanced upon the upregulation of IL-1β, accounting for the excess of IL-1 signaling commonly seen in inflammatory states (90). Thus, future studies should investigate the effects of IL-1α separately from IL-1β, as their baseline expression, regulation, and secretion differ significantly and may thus control the kinetics of inflammation in different ways.
In humans, the recombinant IL-1 receptor antagonist anakinra is currently being tested to treat severe TBI, as it has shown some promise in the treatment of stroke (91). Helmy et al. used anakinra with PCA analysis to demonstrate that IL-1 signaling is a pivotal upstream regulator of TBI-induced cytokine production (92), which, they showed in a later trial, may lead to a shift in macrophages to express higher levels of pro-inflammatory cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-1β (93). This result is somewhat surprising considering the antagonistic effect on IL-1 signaling anakinra would be expected to have, but it suggests that further exploration into the mechanisms involved as well as delineation of the distinct functions of IL-1α and IL-1β during neuroinflammation will likely yield critical insights into the regulation of TBI pathogenesis by IL-1 signaling.
As described briefly above, inflammasome signaling has emerged as a major mechanism involved in IL-1 production. Inflammasomes are multiprotein complexes that coordinate caspase-1-mediated inflammatory cytokine production and cell death. Recent studies have shown that aberrant regulation of inflammasome signaling is a major driver of inflammation and pathology in multiple models of tissue damage, including stroke, macular degeneration, and renal ischemia (16, 66, 73, 94, 95). Inflammasomes consist of a sensor molecule such as a Nod-like receptor (NLR) or a pyrin/HIN domain-containing protein (PYHIN) family receptor, the adaptor protein ASC (apoptosis associated speck-like protein containing a CARD), and caspase-1. To date, five receptors – NLRP1, NLRP3, NLRC4, AIM2, and PYRIN – have been discovered to promote inflammasome signaling. Following the detection of their cognate danger- or pathogen-associated triggers, inflammasome-associated NLRs and PYHIN family receptors promote rapid inflammasome complex formation. The coordinated assembly of this multiprotein inflammasome platform promotes activation-inducing auto-cleavage of caspase-1. Activated caspase-1 can then cleave both pro-IL-1β and pro-IL-18, which is required to elicit their inflammatory properties and for their secretion. Bioactive caspase-1 also provokes pyroptosis, which is a gasdermin D-mediated inflammatory form of cell death that is associated with the release of the pro-inflammatory alarmins IL-1α and HMGB-1 (96, 97).
Since the finding that inflammasome proteins are upregulated after TBI in human patients (98), significant attention has been paid to identify the inflammasome-associated signaling events that are engaged in response to brain trauma (14, 99, 100). Inflammasome literature has identified the expression of NLRP1, NLRP2, and NLRP3 as well as AIM2 inflammasomes in microglia, neurons, and astrocytes in the CNS (100–104). Furthermore, recent studies in CNS injury models have uncovered critical roles for inflammasome signaling in driving inflammatory responses following tissue damage in the CNS. For instance, SCI leads to the upregulation and assembly of NLRP1 inflammasome components in spinal cord neurons (105). Moreover, neutralizing anti-ASC antibody treatment was also found to improve histopathological and functional outcome following SCI in these studies. In stroke models, methods to reduce inflammasome signaling, such as anti-NLRP1 neutralizing antibodies and caspase-1 inhibitors, as well as NLRP3, ASC, NLRC4, and AIM2 knockout mice, have all shown signs of improved functional recovery and reductions in inflammasome signaling (73, 103, 106, 107). Similarly, in an intracerebral hemorrhage model, both small interfering RNA and a selective inhibitor of the purinergic receptor P2X7R, which has been shown to promote NLRP3 activation in some experimental settings (108), limited inflammasome activation and led to neuroprotection (109). Considering the consistent benefits of inhibiting inflammasome components across these models, the inflammasome provides a tempting target for alleviating CNS injury.
Additional insights into the timing and importance of inflammasomes in CNS injury have been gained from recent TBI studies. In an FPI model, inflammasome components, such as ASC and caspase-1, were shown to be upregulated in cortical neurons for up to 24 h post-injury (110). Co-immunoprecipitation of inflammasome proteins also demonstrated that NLRP1 and ASC could be detected in multiprotein complexes in the brain. Treatment with an ASC-neutralizing antibody reduced caspase-1 activation and IL-1β production while also decreasing lesion volume, suggesting beneficial effects of targeting inflammasome activity. Liu et al. also recently showed that TBI results in upregulated expression of NLRP3, ASC, and caspase-1. Moreover, they report that the expression of these inflammasome-associated proteins can remain elevated out to 7 days post-injury (111). Importantly, inflammasome components in this model localized not only to neurons, but also to astrocytes and microglia, suggesting a wide range of inflammasome activation across cell types. Measurements of IL-1β and IL-18 protein levels also demonstrated that while IL-1β peaks around 6 h after injury and subsequently decreases over time, IL-18 expression remains elevated through 7 days after injury. In agreement with these findings, a separate study also reported elevated IL-18 production for at least a week post-TBI in both humans and experimental animals (112). These data suggest that early inflammasome production of IL-1β may be involved in acute inflammation and tissue damage, while inflammasome-driven IL-18 may contribute to the perpetuation of TBI-induced inflammation. It should be noted, however, that in a more recent study neither NLRP1 nor ASC knockout mice exhibited any improvements in lesion volume, histopathology, cell death, or motor function following CCI injury (81). It is possible that differences in the extent of caspase-1 abrogation and/or the timing of inflammasome inhibition or differences in injury models may help explain the disparate results that were reported in these studies.
Although key roles for inflammasomes have been clearly identified in other models of sterile inflammation and trauma, the specific contributions of inflammasome activation to TBI pathogenesis have only recently been investigated and multiple questions remain. For instance, although the formation of inflammasome complexes has been reported following TBI, the roles that specific inflammasomes play in driving TBI-associated pathology and neurological dysfunction have not been studied in great detail in animal models. In addition, the individual contributions of inflammasome-derived cytokines (i.e., IL-1α, IL-1β, and IL-18) and caspase-1-mediated cell death in TBI pathogenesis still remain poorly characterized. The major cell types in which inflammasomes operate to promote TBI progression have also not been formally defined to date. The genetic targeting of inflammasome signaling components in mice has aided in the discovery of critical roles for inflammasomes in other models of sterile inflammation. Future in vivo TBI studies that utilize these genetic tools should help to more fully characterize the contributions of specific aspects of inflammasome signaling in brain trauma.
Interleukin-6
Interleukin-6 has frequently been associated with TBI outcome in humans, but it is unclear whether its role is primarily beneficial or detrimental. Microdialysis fluid detection of parenchymal IL-6 production has been associated with improved survival in TBI patients (21). However, more recent evidence points to a detrimental role for IL-6 in TBI (113). In these studies, plasma levels of IL-6 were shown to be significantly higher in severe TBI patients over moderate TBI patients. Both subacute and chronic serum levels of IL-6 have been associated with unfavorable short and long-term outcomes (75). In separating human patients by high or low cerebrospinal fluid (CSF) IL-6 trajectory, high trajectory patients are much more likely to have unfavorable clinical outcomes (76). Thus, while the role of IL-6 in TBI is still somewhat unclear, data from TBI patients indicate that IL-6 is consistently upregulated after TBI and can remain elevated in chronic stages, making it a potentially important mediator of long-term outcome.
Early animal studies verified that IL-6 is elevated in CSF and serum after TBI (114). Evidence from IL-6 knockout mice has also confirmed it as a pro-inflammatory cytokine that recruits activated glia and immune cells to sites of injury. Indeed, genetic ablation of IL-6 in cryolesioned mice resulted in fewer reactive astrocytes and macrophages and increased neuronal death (115). Conversely, overexpression of IL-6 in astrocytes enhanced recruitment of glia and immune cells to the lesion site and decreased both oxidative stress and neuronal death (115, 116). These studies suggest that IL-6’s role in inducing inflammation and glial scar formation is important in reducing prolonged cell death. A later CCI study also pointed to beneficial effects of IL-6 by showing that its deficiency leads to significantly poorer performance on behavioral tests as well as higher IL-1β protein levels in the cortex, suggesting that IL-6 may be an important regulator of IL-1β in TBI (117). However, a more recent study using a weight drop model showed that systemic neutralization of IL-6 mitigates some of the inflammatory and behavioral effects of hypoxia on exacerbating post-injury responses, implying that reducing the inflammatory response induced by IL-6 can indeed provide neuroprotection and lead to better outcome (118). When considering these types of studies, it is important to keep in mind the difference between complete or partial removal of a gene and/or its product. It is likely that some level of IL-6 is necessary to produce an inflammatory state that positively affects outcome such that either complete elimination or overexpression of IL-6 can be detrimental.
Tumor Necrosis Factor α
Early work on the role of tumor necrosis factor (TNFα) in TBI mouse models suggested that it has early deleterious effects after TBI while exhibiting more protective effects in chronic stages (119). However, other work suggested that TNFα is necessary to protect from early mortality within a week of injury (120). Regardless of these contradictions, literature on TNFα in TBI consistently shows an upregulation of this cytokine after injury (74, 75, 113, 121), suggesting an important role for TNFα in both the acute and chronic phases.
The importance of TNFα early after injury was recently confirmed in a weight drop model. In this study, mice receiving a TNFα inhibitor at 1 and 12 h after injury showed improved cognitive performance 1 week post-injury, but mice administered the inhibitor at 18 h post-injury did not, implying a very short window for TNFα-targeting therapeutics after TBI (122). Further investigation of the mice given the inhibitor at 1 h showed fewer apoptotic neurons and less astrogliosis at 72 h after injury in both the cortex and dentate gyrus. This study outlines a 12-h window after injury during which the detrimental effects of TNFα may be attenuated, and points toward a tentative link between TNFα and prolonged astrogliosis and neuronal death.
Aside from defining the timing of TNFα activity in TBI, it will be important to elucidate the pro-apoptotic and pro-survival pathways in which it participates following brain trauma. In conjunction with its role as a major inflammatory switch, TNFα is known to induce both cell proliferation and apoptosis through several signaling pathways. While its activation of transcription factors, such as NF-κB and AP-1, can lead to transcription of inflammatory and apoptotic mediators, signaling through death receptors to activate caspases can also play an important part in determining cell death or survival. A recent article by Longhi et al. showed that separate deletion of either TNF receptor 1 (TNFR1) or 2 (TNFR2) can have opposite effects on cell survival and behavioral deficits (123). Using p55 (TNFR1) and p75 (TNFR2) knockout mice in a CCI model of TBI, they showed that TNFR1 deletion attenuated neuroscore deficits through 4 weeks post-injury and led to a shift to pro-survival signaling along with attenuated neuronal death and smaller lesion volume. TNFR2 knockout had the opposite effect in worsening neuroscore with no signs of pro-survival signaling or protection from cell and tissue loss. The TNFR1 knockout mice also showed a smaller area stained for CD11b as well as a higher area stained for Ym1, a marker for anti-inflammatory macrophage phenotypes, compared to the TNFR2 knockout group, suggesting opposite roles for the two receptors in recruiting inflammatory macrophages and microglia to the site of injury. These data agree with a paper by Yang et al. in which TNFR2/Fas knockout mice showed worse motor and cognitive performance after CCI TBI, although in that study neither TNFR1 or TNFR2 knockout alone had an effect on lesion volume or the number of dead cells in the cortex (124). Together, these studies indicate that the TNF receptors may play different roles post-injury, with TNFR2 providing a neuroprotective role and TNFR1 playing a detrimental one.
An important consideration about TNFα signaling is that due to the much wider expression of TNFR1 across cell types as well as its ability to respond to both forms of TNF (both soluble and transmembrane), this receptor could have more potent inflammatory consequences than its counterpart TNFR2. In addition, it has been shown that TNFR1 can signal through NF-κB, JNK, and caspase-mediated apoptotic pathways, while it is more common for TNFR2 to engage NF-κB and PI3K to induce pro-inflammatory and pro-survival signaling (125, 126). Thus, consolidating seemingly contradictory evidence for the role of TNFα in TBI with regards to cell death and clinical outcome will likely involve understanding the conditions under which its various forms and receptors participate in different survival or death pathways and the timeline on which this signaling can occur.
Granulocyte Colony-Stimulating Factor/Granulocyte Macrophage Colony-Stimulating Factor
Both granulocyte colony-stimulating factor (G-CSF) and GM-CSF are involved in the expansion and mobilization of immune cells from the bone marrow and act as key cytokines in the inflammatory response. Interestingly, some recent evidence suggests that both G-CSF and GM-CSF may play a protective role in TBI. A recent paper using G-CSF administration after a CCI injury showed that G-CSF injection improves cognitive recovery and increases neurogenesis in the hippocampus (127). This was accompanied by higher activation of astrocytes and microglia as well as higher levels of brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF), indicating that G-CSF may regulate production of neurotrophic factors by activated glia post-TBI to promote neurogenesis. Similarly, a study using GM-CSF knockout mice showed that GM-CSF deficiency in TBI results in more cognitive deficits with higher tissue and neuronal loss after FPI (128). GM-CSF knockout mice also showed reductions in astrogliosis, which may suggest that GM-CSF plays a role in activating astrocytes to protect cells and boost tissue repair. Understanding how both of these molecules interact with glia to promote neuronal protection and regeneration may elucidate how other neuroprotective processes may involve glial functions.
Type 1 Interferon
Increasing evidence points toward central roles of type 1 interferon (IFN) signaling in inflammatory CNS disorders and age-related cognitive decline (129–132). A recent study by Karve et al. is one of the first to investigate the involvement of type 1 IFN signaling in TBI pathogenesis (82). They found that deficiency in type 1 IFN signaling produced by either type 1 IFN receptor (IFNAR) knockout or an IFNAR blocking antibody reduces lesion volume. This neuroprotection was associated with a shift toward more anti-inflammatory cytokine signaling; however, this also coincided with increased GFAP and IBA1 staining. In addition, using a bone marrow chimera, they found that IFNAR deficiency in hematopoietic cells alone was sufficient to confer lesion volume protection and elevated GFAP and IBA1 staining. Importantly, they also showed that brain trauma in humans promotes enhanced expression of type-1 IFN, which suggests that type-1 IFN signaling may potentially influence clinical outcome in TBI patients.
Interleukin-10
Interleukin-10 has been shown to be elevated in TBI patients (78, 133–135) and has been associated with unfavorable outcome and mortality (75, 134, 135). Despite these associations, Chen et al. found a role for IL-10 in conferring neuroprotection with hyperbaric oxygen (HBO) treatment (80). They found that the protective effects of HBO in TBI included reduced lesion volume and edema, improvements in cognitive performance, and the dampening of pro-inflammatory cytokine production in the cortex. It also led to a shift from apoptotic to cell survival pathways and greater BBB integrity. This wide range of positive effects was diminished in IL-10-knockout mice, and IL-10 injection by itself improved lesion volume, edema, and cognitive outcome in both wild-type and IL-10-knockout animals. This indicates an important protective role for IL-10 in TBI as well as a way to induce its production through HBO treatment. It is possible that the association between IL-10 (and likely many other cytokines) and poor outcome is primarily due to a widespread upregulation of cytokines after TBI, and that a more informative approach to understanding the role of IL-10 after brain injury involves dissecting its specific roles in damage responses.
Transforming Growth Factor β
Transforming growth factor β (TGFβ) increases acutely in the serum and CSF of TBI patients (136). Several mediators of TGFβ signaling have been shown to be upregulated in TBI models (79, 137, 138). For example, transforming growth factor beta-activated kinase 1 (TAK1) was shown to increase in expression and is detected in cortical neurons and astrocytes after weight drop TBI (79). Inhibition of TAK1 signaling improved neuronal survival and motor function and also decreased NF-κB activity and inflammatory cytokine release. Transforming growth-interacting factor (TGIF), a transcriptional co-repressor of TGFβ that can inhibit transcriptional activation of TGFβ, was demonstrated to be upregulated in TBI animals and localized to both neurons and microglia (138). Using small hairpin RNA to knockdown TGIF levels in the brain, the authors found that lower TGIF levels led to a decrease in infarct volume and microglia number around the lesion, as well as a change in microglia morphology. Knockdown of TGIF also improved motor function through 2 weeks after injury. These data indicate that mediators of TGFβ signaling can have important inflammatory consequences.
Future Perspectives
As highlighted in this review, increasing experimental evidence indicates that the immune system can profoundly influence clinical outcomes following TBI. Importantly, various recent studies have shown that targeting immune signaling with genetic and pharmacological approaches can lead to significant improvements in neurological function and tissue repair post-TBI. Both neuroprotective and detrimental roles have been assigned to the immune system in TBI. Whether the immune response contributes to beneficial tissue repair or further brain damage largely depends on the nature, kinetics, and magnitude of the inflammatory response. Although targeting the immune system has emerged as an exciting potential strategy to treat TBI, there are numerous outstanding questions that need to be addressed to better characterize the involvement of immune signaling in TBI etiology and to realize the full potential of immune-based therapeutics.
For one, there is still an overall lack of consensus on the overarching roles that discrete immune cell types and pathways play in TBI. Future efforts are needed to help reconcile the biological reasons that account for the disparate results that have been reported on immune mediators in TBI by different labs. Much of the inconsistency in the literature can be attributed to the utilization of different approaches to induce brain trauma and modulate immune signaling between labs (Table 2). Pinpointing what is mechanistically responsible for the conflicting findings in the TBI literature will help to uncover the important nuances of the immune response to brain trauma and will aid in the identification of optimal therapeutic regimens to treat discrete types of CNS injury.
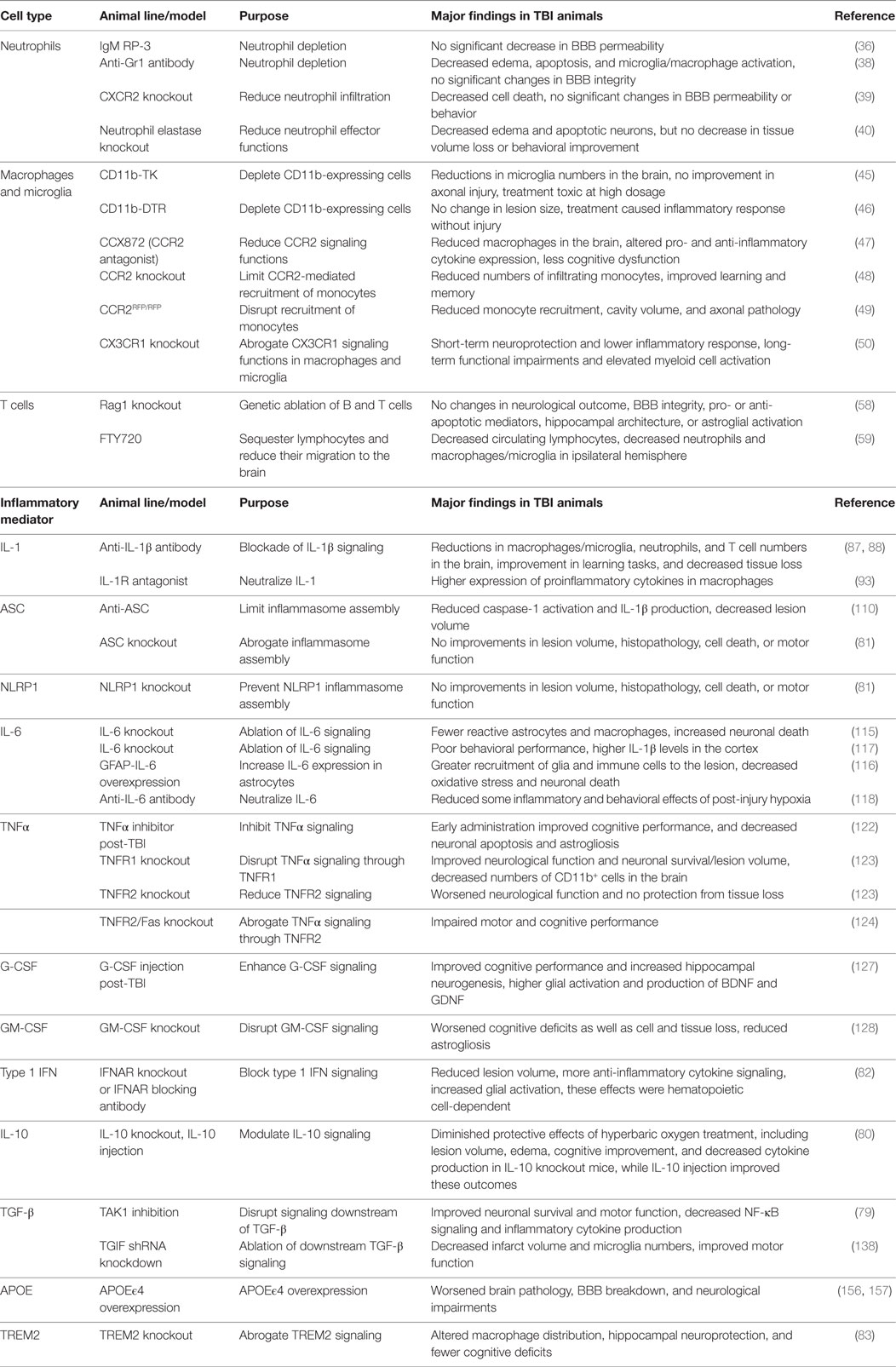
Table 2. Genetic models used to characterize the role of immune cell types and signaling pathways in TBI.
One thing that is largely agreed upon in the literature is that no two brain injuries are alike and that seemingly similar types of trauma can result in diverse clinical outcomes. Unfortunately, the reasons for the heterogeneity in disease sequelae and recovery time associated with comparable forms of trauma remain poorly understood. Heterogeneity in recovery time and extent of neurological dysfunction in the TBI patient population can only be partially explained by differences in brain injury severity and location. This has led to greater appreciation for the roles of host genetics, environmental factors, lifestyle choices, and previous TBI history in overall clinical outcome. It is currently difficult to predict how interplay between these diverse non-injury factors affect TBI pathogenesis, and future studies are greatly needed to uncover their influence on TBI. Unfortunately, current TBI treatment approaches do not fully take into consideration many of the non-injury factors that are described above. The utilization of immune-based biomarkers in the future may offer a strategy to improve the stratification and treatment of TBI patient groups. Recent advancements from experimental TBI models indicate that the nature and kinetics of the immune responses can vary depending on brain injury location and severity. Furthermore, immune responses are also significantly affected by environmental and lifestyle factors (e.g., diet, antibiotics usage, prescription, or recreational drug use), microbiome composition, individual genetic factors, and previous TBI history. Therefore, it is feasible that immune cell frequencies and cytokine production in patients may serve as valuable biomarkers to predict clinical outcomes, stratify patient groups, and to maximize therapeutic approaches to treat TBI.
Mounting epidemiological evidence indicates that TBI is a major risk factor for developing numerous neurological disorders including AD, ALS, CTE, and posttraumatic stress disorder (PTSD) (5–10, 139, 140) and also possibly MS (141–143). Although it is widely appreciated that TBI predisposes individuals to other neurological disorders, how TBI mechanistically contributes to CNS disease later in life still remains poorly understood. Dysregulated immune responses have been identified to play key roles in the pathogenesis of the majority of neurological disorders and mental illnesses that have been linked to TBI. As a result, it is tempting to speculate that the aberrant inflammatory conditions that are generated in response to brain trauma may set in motion a series of events that can contribute to the development of other neurological disorders over time. Indeed, recent advances have been made in characterizing how TBI can contribute to AD pathogenesis. These studies have shown that brain trauma can spur the aberrant release and deposition of both amyloid beta (Aβ) and tau (144). The accumulation of neurotoxic forms of Aβ and tau are believed to be major drivers of AD pathogenesis (145), and studies of postmortem brains following brain trauma indicate that Aβ and tau deposition are also hallmarks of TBI (144, 146–152). The mechanism(s) by which Aβ and tau promote AD is still a matter of great debate; however, emerging data clearly point to roles for Aβ- and tau-induced neuroinflammation in this process (153). Therefore, it is conceivable that neuroinflammation and neuronal damage that is incited by the deposition of Aβ and tau following TBI may instigate a pathological cycle of continued Aβ and tau release and inflammation that initiates early AD progression. Furthermore, targeting the hyperinflammatory responses that are generated in response to Aβ and tau accumulation post-brain trauma could help to limit the risk of AD development in TBI patients (154, 155).
Interest in the link between TBI and AD has also extended to some of the major genetic susceptibility factors that are associated with AD. Genome-wide association studies (GWAS) have determined that carrying an allele of APOEϵ4 (apolipoprotein E ϵ4) or a mutation in TREM2 (triggering receptor expressed on myeloid cells 2) is associated with significantly higher rates of AD in humans (156). Interestingly, recent reports suggest that manipulation of either TREM2 or APOE biology can also affect the severity of brain pathology and neurological dysfunction following TBI (83, 157). For instance, it was shown that TBI results in more severe memory and functional impairments in individuals who carry the APOEϵ4 allele than in people who possess other APOE alleles (154, 158). Moreover, transgenic overexpression of APOEϵ4 in mice was also found to cause worsened brain pathology, BBB breakdown, and neurological impairments following brain injury (159, 160).
TREM2 is an immunoglobulin-superfamily receptor that is predominantly expressed on myeloid cells including microglia, macrophages, and osteoblasts. Recent studies have shown that TREM2 is involved in the removal of debris, misfolded proteins, and phospholipids from the CNS (161). An important feature of both AD and TBI is that Aβ can accumulate into plaques (157), which are thought to lead to detrimental effects on neurological function if not cleared by phagocytes. TREM2 can help microglia and infiltrating macrophages detect lipoprotein-bound Aβ in the brain and trigger phagocytosis (162) and may also participate in maintaining the survival of these cells (163) (Figure 3). These activated cells can then recruit more phagocytes to the sites of Aβ accumulation. Downstream effects of this phagocytic response remain unclear, but multiple studies have agreed that TREM2 deficiency in various CNS disorders does lead to a decreased number of phagocytes, which likely impairs debris clearance (83, 164–166). In such cases of TREM2 mutation or dysfunction, it is possible that Aβ and other misfolded proteins may not only be insufficiently cleared, but may also accumulate faster. A recent article by Saber et al. used an FPI injury model to explore the effect of TREM2 on TBI-induced neuroinflammation, tissue loss, and neurological function (83). They found that TREM2-knockout mice did indeed show fewer macrophages throughout the brain, but more were present close to the site of injury. This was associated with hippocampal neuroprotection and fewer cognitive deficits in the TREM2 knockouts. Interestingly, sham TREM2-knockout mice appeared to have some differences from sham wild-type mice in certain behaviors. This paper shows that TREM2 is also likely to be important in TBI recovery; thus, it will be interesting going forward to elucidate the mechanisms by which it mediates debris clearance and neuroinflammation.
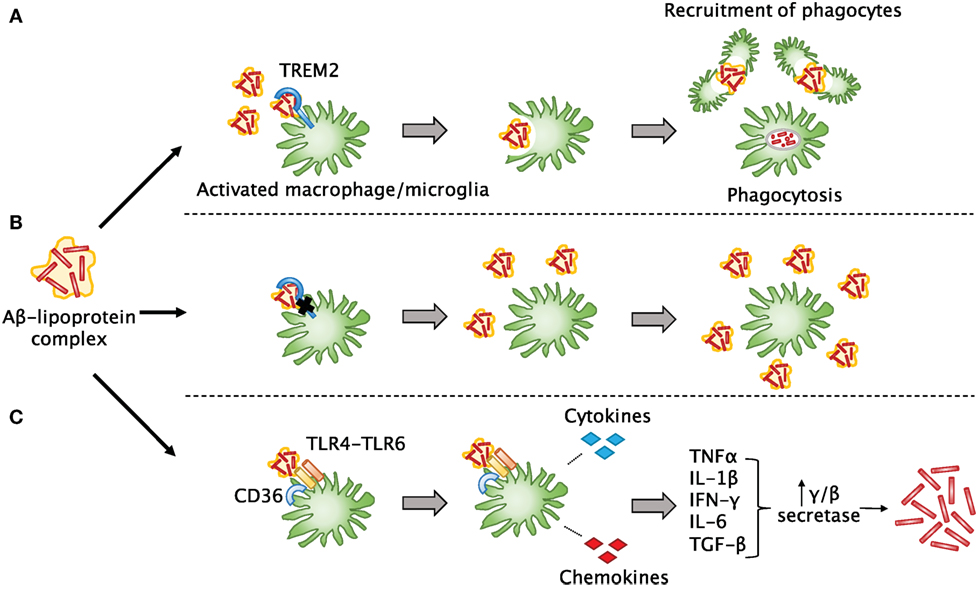
Figure 3. The involvement of TREM2 in post-TBI amyloid beta clearance. (A) Aβ released after TBI quickly forms into plaques and may bind to lipoproteins. TREM2 assists surrounding myeloid cells in sensing Aβ-lipoprotein complexes, engulfing and breaking down the Aβ, and recruiting other phagocytes to the site of injury. (B) In the case of TREM2 mutations or dysfunction, these cells may not be able to properly sense and clear Aβ or recruit other cells, so that more Aβ builds up over time and leads to plaque formation as seen in Alzheimer’s disease. (C) Another possibility is that in the absence of TREM2 signaling, other signaling pathways may predominate in myeloid cells. For example, when a complex of CD36 with a TLR4-TLR6 heterodimer senses Aβ, instead of phagocytosis, it leads to release of pro-inflammatory cytokines and chemokines that can induce upregulation of secretases in other cells, which are known to lead to increased production of Aβ.
Despite the recent progress that has been made in characterizing how having a history of TBI can predispose individuals to AD, little is currently known about what biologically accounts for the increased risk of developing other neurological conditions in TBI patients. Immune dysfunction has been implicated in the pathogenesis of many of these CNS disorders that are more prevalent following brain trauma, including CTE, mental illness, and PTSD (167–169). Therefore, targeting the immune system following TBI may help to lower the risk of developing other neurological diseases later in life.
Conclusion
Once considered a silent epidemic, TBI is now recognized as a serious threat to global human health. In recent years, considerable efforts and resources have been paid to reduce the rates and severity of TBI. Unfortunately, these preventative measures have been largely unsuccessful, and the number of individuals who sustain debilitating brain trauma each year continues to rise. As highlighted in this review, recent advancements in animal models of TBI clearly indicate that immune responses are centrally involved in the development of brain pathology and neurological dysfunction following TBI. Importantly, these emerging studies suggest that targeting the immune system could offer a much-needed therapeutic approach to treat TBI. Future studies that are geared toward further defining the major immunological pathways that influence TBI pathogenesis will lead to an improved understanding of brain injury etiology and will aid in the identification of novel immune-based TBI treatment strategies.
Author Contributions
CM wrote the sections of the manuscript related to the involvement of immune cell types and inflammatory mediators in TBI; designed the manuscript; and created the figures. JL wrote the introduction, future perspectives, conclusion, and sections of the manuscript related to inflammasomes and the kinetics of the immune response to brain injury; designed the manuscript; and oversaw the entire process of manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to authors whose work could not be referenced in this review due to space limitations. The authors also thank members of the Lukens Lab for their valuable comments on the manuscript. This work was supported by the Owens Family Foundation (JL) and the University of Virginia Research and Development Committee Award (JL).
References
1. The changing landscape of traumatic brain injury research. Lancet Neurol (2012) 11(8):651. doi: 10.1016/S1474-4422(12)70166-7
2. Faul M, Coronado V. Epidemiology of traumatic brain injury. Handb Clin Neurol (2015) 127:3–13. doi:10.1016/B978-0-444-52892-6.00001-5
3. Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, et al. Surveillance for traumatic brain injury-related deaths – United States, 1997-2007. MMWR Surveill Summ (2011) 60(5):1–32.
4. Selassie AW, Zaloshnja E, Langlois JA, Miller T, Jones P, Steiner C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. J Head Trauma Rehabil (2008) 23(2):123–31. doi:10.1097/01.HTR.0000314531.30401.39
5. Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol (1991) 20(Suppl 2):S28–35. doi:10.1093/ije/20.Supplement_2.S28
6. Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol (2007) 166(7):810–6. doi:10.1093/aje/kwm153
7. Holsinger T, Steffens DC, Phillips C, Helms MJ, Havlik RJ, Breitner JC, et al. Head injury in early adulthood and the lifetime risk of depression. Arch Gen Psychiatry (2002) 59(1):17–22. doi:10.1001/archpsyc.59.1.17
8. Goldman SM, Tanner CM, Oakes D, Bhudhikanok GS, Gupta A, Langston JW. Head injury and Parkinson’s disease risk in twins. Ann Neurol (2006) 60(1):65–72. doi:10.1002/ana.20882
9. Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N Engl J Med (2008) 358(5):453–63. doi:10.1056/NEJMoa072972
10. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol (2009) 68:709–35. doi:10.1097/NEN.0b013e3181a9d503
11. Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation (2012) 9:236. doi:10.1186/1742-2094-9-236
12. Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer’s disease prevention. J Neuroinflammation (2012) 9:185. doi:10.1186/1742-2094-9-185
13. Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol (2013) 4:18. doi:10.3389/fneur.2013.00018
14. Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci (2014) 15(2):84–97. doi:10.1038/nrn3638
15. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol (2015) 72(3):355–62. doi:10.1001/jamaneurol.2014.3558
16. Gadani Sachin P, Walsh James T, Lukens John R, Kipnis J. Dealing with danger in the CNS: the response of the immune system to injury. Neuron (2015) 87(1):47–62. doi:10.1016/j.neuron.2015.05.019
17. Woiciechowsky C, Schöning B, Cobanov J, Lanksch WR, Volk HD, Döcke WD. Early IL-6 plasma concentrations correlate with severity of brain injury and pneumonia in brain-injured patients. J Trauma (2002) 52(2):339–45.
18. Arand M, Melzner H, Kinzl L, Bruckner UB, Gebhard F. Early inflammatory mediator response following isolated traumatic brain injury and other major trauma in humans. Langenbecks Arch Surg (2001) 386(4):241–8. doi:10.1007/s004230100204
19. Shiozaki T, Hayakata T, Tasaki O, Hosotubo H, Fuijita K, Mouri T, et al. Cerebrospinal fluid concentrations of anti-inflammatory mediators in early-phase severe traumatic brain injury. Shock (2005) 23(5):406–10. doi:10.1097/01.shk.0000161385.62758.24
20. Chiaretti A, Genovese O, Aloe L, Antonelli A, Piastra M, Polidori G, et al. Interleukin 1beta and interleukin 6 relationship with paediatric head trauma severity and outcome. Childs Nerv Syst (2005) 21(3):185–93; discussion 94. doi:10.1007/s00381-004-1032-1
21. Winter CD, Pringle AK, Clough GF, Church MK. Raised parenchymal interleukin-6 levels correlate with improved outcome after traumatic brain injury. Brain (2004) 127(2):315–20. doi:10.1093/brain/awh039
22. Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain (2013) 136(Pt 1):28–42. doi:10.1093/brain/aws322
23. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol (2011) 70(3):374–83. doi:10.1002/ana.22455
24. Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron (2012) 76(5):886–99. doi:10.1016/j.neuron.2012.11.021
25. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi:10.1038/nri3399
26. Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci (1995) 15(12):8223–33.
27. Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol (1997) 61(3):279–85.
28. Szmydynger-Chodobska J, Strazielle N, Gandy JR, Keefe TH, Zink BJ, Ghersi-Egea JF, et al. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab (2012) 32(1):93–104. doi:10.1038/jcbfm.2011.111
29. Schwarzmaier SM, Plesnila N. Contributions of the immune system to the pathophysiology of traumatic brain injury – evidence by intravital microscopy. Front Cell Neurosci (2014) 8:358. doi:10.3389/fncel.2014.00358
30. Russo MV, McGavern DB. Immune surveillance of the CNS following infection and injury. Trends Immunol (2015) 36(10):637–50. doi:10.1016/j.it.2015.08.002
31. Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care (2002) 8(2):101–5. doi:10.1097/00075198-200204000-00002
32. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics (2010) 7(1):22–30. doi:10.1016/j.nurt.2009.10.016
33. Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci (2015) 36(7):471–80. doi:10.1016/j.tips.2015.04.003
34. Villapol S, Byrnes KR, Symes AJ. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front Neurol (2014) 5:82. doi:10.3389/fneur.2014.00082
35. Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, et al. “Hit & Run” model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab (2013) 33(6):834–45. doi:10.1038/jcbfm.2013.30
36. Whalen MJ, Carlos TM, Kochanek PM, Clark RSB, Heineman S, Schiding JK, et al. Neutrophils do not mediate blood-brain barrier permeability early after controlled cortical impact in rats. J Neurotrauma (1999) 16(7):583–94. doi:10.1089/neu.1999.16.583
37. Clark RSB, Schiding JK, Kaczorowski SL, Marion DW, Kochanek PM. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma (1994) 11(5):499–506. doi:10.1089/neu.1994.11.499
38. Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation (2012) 9:17. doi:10.1186/1742-2094-9-17
39. Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis (2010) 40(2):394–403. doi:10.1016/j.nbd.2010.06.015
40. Semple BD, Trivedi A, Gimlin K, Noble-Haeusslein LJ. Neutrophil elastase mediates acute pathogenesis and is a determinant of long-term behavioral recovery after traumatic injury to the immature brain. Neurobiol Dis (2015) 74:263–80. doi:10.1016/j.nbd.2014.12.003
41. Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WST, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int (2004) 146(2–3):97–104. doi:10.1016/j.forsciint.2004.06.027
42. Beschorner R, Nguyen TD, Gozalan F, Pedal I, Mattern R, Schluesener HJ, et al. CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol (2002) 103(6):541–9. doi:10.1007/s00401-001-0503-7
43. Block ML, Hong JS. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans (2007) 35(Pt 5):1127–32. doi:10.1042/bst0351127
44. Gao H-M, Hong J-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol (2008) 29(8):357–65. doi:10.1016/j.it.2008.05.002
45. Bennett RE, Brody DL. Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury. J Neurotrauma (2014) 31(19):1647–63. doi:10.1089/neu.2013.3320
46. Frieler RA, Nadimpalli S, Boland LK, Xie A, Kooistra LJ, Song J, et al. Depletion of macrophages in CD11b diphtheria toxin receptor mice induces brain inflammation and enhances inflammatory signaling during traumatic brain injury. Brain Res (2015) 1624:103–12. doi:10.1016/j.brainres.2015.07.011
47. Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci (2015) 35(2):748–60. doi:10.1523/jneurosci.2405-14.2015
48. Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma (2014) 31(20):1677–88. doi:10.1089/neu.2013.3252
49. Gyoneva S, Kim D, Katsumoto A, Kokiko-Cochran ON, Lamb BT, Ransohoff RM. Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J Neuroinflammation (2015) 12(1):1–12. doi:10.1186/s12974-015-0443-0
50. Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T, et al. Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J Neurotrauma (2015) 33(11):1060–72. doi:10.1089/neu.2015.4041
51. Hsieh CL, Kim CC, Ryba BE, Niemi EC, Bando JK, Locksley RM, et al. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol (2013) 43(8):2010–22. doi:10.1002/eji.201243084
52. Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab (2013) 33(12):1864–74. doi:10.1038/jcbfm.2013.146
53. Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP. Immune activation promotes depression one month after diffuse brain injury: a role for primed microglia. Biol Psychiatry (2014) 76(7):575–84. doi:10.1016/j.biopsych.2013.10.014
54. Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, et al. Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun (2016) 54:95–109. doi:10.1016/j.bbi.2016.01.009
55. Czigner A, Mihály A, Farkas O, Büki A, Krisztin-Péva B, Dobó E, et al. Kinetics of the cellular immune response following closed head injury. Acta Neurochir (2007) 149(3):281–9. doi:10.1007/s00701-006-1095-8
56. Clausen F, Lorant T, Lewén A, Hillered LT. Lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma (2007) 24(8):1295–307. doi:10.1089/neu.2006.0258
57. Hausmann R, Kaiser A, Lang C, Bohnert M, Betz P. A quantitative immunohistochemical study on the time-dependent course of acute inflammatory cellular response to human brain injury. Int J Legal Med (1999) 112(4):227–32. doi:10.1007/s004140050241
58. Weckbach S, Neher M, Losacco JT, Bolden AL, Kulik L, Flierl MA, et al. Challenging the role of adaptive immunity in neurotrauma: Rag1(-/-) mice lacking mature B and T cells do not show neuroprotection after closed head injury. J Neurotrauma (2012) 29(6):1233–42. doi:10.1089/neu.2011.2169
59. Mencl S, Hennig N, Hopp S, Schuhmann MK, Albert-Weissenberger C, Siren AL, et al. FTY720 does not protect from traumatic brain injury in mice despite reducing posttraumatic inflammation. J Neuroimmunol (2014) 274(1–2):125–31. doi:10.1016/j.jneuroim.2014.07.010
60. Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med (1999) 5(1):49–55. doi:10.1038/4734
61. Ellwardt E, Walsh JT, Kipnis J, Zipp F. Understanding the role of T cells in CNS homeostasis. Trends Immunol (2016) 37(2):154–65. doi:10.1016/j.it.2015.12.008
62. Jones KJ, Lovett-Racke AE, Walker CL, Sanders VM. CD4+ T cells and neuroprotection: relevance to motoneuron injury and disease. J Neuroimmune Pharmacol (2015) 10(4):587–94. doi:10.1007/s11481-015-9625-x
63. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest (2015) 125(2):699–714. doi:10.1172/jci76210
64. Lukens JR, Kanneganti TD. Beyond canonical inflammasomes: emerging pathways in IL-1-mediated autoinflammatory disease. Semin Immunopathol (2014) 36(5):595–609. doi:10.1007/s00281-014-0434-8
65. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi:10.1038/nri3452
66. Lukens JR, Gross JM, Kanneganti TD. IL-1 family cytokines trigger sterile inflammatory disease. Front Immunol (2012) 3:315. doi:10.3389/fimmu.2012.00315
67. Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol (2012) 13:333–42. doi:10.1038/ni.2237
68. Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci (2014) 8:315. doi:10.3389/fnins.2014.00315
69. de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab (2014) 34(3):369–75. doi:10.1038/jcbfm.2013.227
70. Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, Vogel P, et al. Critical role for inflammasome-independent IL-1beta production in osteomyelitis. Proc Natl Acad Sci U S A (2014) 111(3):1066–71. doi:10.1073/pnas.1318688111
71. Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, et al. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature (2013) 498(7453):224–7. doi:10.1038/nature12174
72. Lukens JR, Kanneganti TD. SHP-1 and IL-1alpha conspire to provoke neutrophilic dermatoses. Rare Dis (2014) 2:e27742. doi:10.4161/rdis.27742
73. Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J, et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci U S A (2015) 112(13):4050–5. doi:10.1073/pnas.1419090112
74. Juengst SB, Kumar RG, Failla MD, Goyal A, Wagner AK. Acute inflammatory biomarker profiles predict depression risk following moderate to severe traumatic brain injury. J Head Trauma Rehabil (2015) 30(3):207–18. doi:10.1097/htr.0000000000000031
75. Kumar RG, Boles JA, Wagner AK. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil (2015) 30(6):369–81. doi:10.1097/htr.0000000000000067
76. Kumar RG, Diamond ML, Boles JA, Berger RP, Tisherman SA, Kochanek PM, et al. Acute CSF interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain Behav Immun (2015) 45:253–62. doi:10.1016/j.bbi.2014.12.021
77. Hutchinson PJ, O’Connell MT, Rothwell NJ, Hopkins SJ, Nortje J, Carpenter KLH, et al. Inflammation in human brain injury: intracerebral concentrations of IL-1α, IL-1β, and their endogenous inhibitor IL-1ra. J Neurotrauma (2007) 24(10):1545–57. doi:10.1089/neu.2007.0295
78. Helmy A, Carpenter KLH, Menon DK, Pickard JD, Hutchinson PJA. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab (2011) 31(2):658–70. doi:10.1038/jcbfm.2010.142
79. Zhang D, Hu Y, Sun Q, Zhao J, Cong Z, Liu H, et al. Inhibition of transforming growth factor beta-activated kinase 1 confers neuroprotection after traumatic brain injury in rats. Neuroscience (2013) 238:209–17. doi:10.1016/j.neuroscience.2013.02.022
80. Chen X, Duan XS, Xu LJ, Zhao JJ, She ZF, Chen WW, et al. Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience (2014) 266:235–43. doi:10.1016/j.neuroscience.2013.11.036
81. Brickler T, Gresham K, Meza A, Coutermarsh-Ott S, Williams TM, Rothschild DE, et al. Nonessential role for the NLRP1 inflammasome complex in a murine model of traumatic brain injury. Mediators Inflamm (2016) 2016:6373506. doi:10.1155/2016/6373506
82. Karve IP, Zhang M, Habgood M, Frugier T, Brody KM, Sashindranath M, et al. Ablation of Type-1 IFN signaling in hematopoietic cells confers protection following traumatic brain injury. eNeuro (2016) 3(1). doi:10.1523/eneuro.0128-15.2016
83. Saber M, Kokiko-Cochran ON, Puntambekar S, Lathia J, Lamb BT. TREM2 deficiency alters acute macrophage distribution and improves recovery after TBI. J Neurotrauma (2016). doi:10.1089/neu.2016.4401
84. Shaftel SS, Griffin WST, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation (2008) 5(1):1–12. doi:10.1186/1742-2094-5-7
85. Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol (2005) 5(8):629–40. doi:10.1038/nri1664
86. Wang W-Y, Tan M-S, Yu J-T, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Annals of Translational Medicine (2015) 3(10):136. doi:10.3978/j.issn.2305-5839.2015.03.49
87. Clausen F, Hånell A, Björk M, Hillered L, Mir AK, Gram H, et al. Neutralization of interleukin-1β modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci (2009) 30(3):385–96. doi:10.1111/j.1460-9568.2009.06820.x
88. Clausen F, Hånell A, Israelsson C, Hedin J, Ebendal T, Mir AK, et al. Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci (2011) 34(1):110–23. doi:10.1111/j.1460-9568.2011.07723.x
89. Luheshi NM, Kovács KJ, Lopez-Castejon G, Brough D, Denes A. Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J Neuroinflammation (2011) 8(1):1–5. doi:10.1186/1742-2094-8-186
90. Di Paolo NC, Shayakhmetov DM. Interleukin 1[alpha] and the inflammatory process. Nat Immunol (2016) 17(8):906–13. doi:10.1038/ni.3503
91. Emsley HCA, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry (2005) 76(10):1366–72. doi:10.1136/jnnp.2004.054882
92. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab (2014) 34(5):845–51. doi:10.1038/jcbfm.2014.23
93. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J Cereb Blood Flow Metab (2015) 36(8):1434–48. doi:10.1177/0271678x15620204
94. Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell (2012) 149(4):847–59. doi:10.1016/j.cell.2012.03.036
95. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A (2009) 106(48):20388–93. doi:10.1073/pnas.0908698106
96. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi:10.1038/nature15541
97. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi:10.1038/nature15514
98. Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg (2012) 117(6):1119–25. doi:10.3171/2012.9.jns12815
99. Hanamsagar R, Aldrich A, Kielian T. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J Neurochem (2014) 129(4):704–11. doi:10.1111/jnc.12669
100. Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ III, Nonner D, Bullock MR, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab (2014) 34(4):621–9. doi:10.1038/jcbfm.2013.236
101. Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One (2015) 10(6):e0130624. doi:10.1371/journal.pone.0130624
102. Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem (2009) 284(27):18143–51. doi:10.1074/jbc.M109.004804
103. Fann DY, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis (2013) 4:e790. doi:10.1038/cddis.2013.326
104. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia (2013) 61(7):1113–21. doi:10.1002/glia.22499
105. de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci (2008) 28(13):3404–14. doi:10.1523/jneurosci.0157-08.2008
106. Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab (2009) 29(3):534–44. doi:10.1038/jcbfm.2008.143
107. Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab (2014) 34(4):660–7. doi:10.1038/jcbfm.2013.242
108. Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol (2012) 3:414. doi:10.3389/fimmu.2012.00414
109. Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation (2015) 12(1):1–17. doi:10.1186/s12974-015-0409-2
110. de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab (2009) 29(7):1251–61. doi:10.1038/jcbfm.2009.46
111. Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res (2013) 38(10):2072–83. doi:10.1007/s11064-013-1115-z
112. Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab (2002) 22(8):971–8. doi:10.1097/00004647-200208000-00008
113. Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One (2013) 8(7):e68963. doi:10.1371/journal.pone.0068963
114. Hans VH, Kossmann T, Lenzlinger PM, Probstmeier R, Imhof HG, Trentz O, et al. Experimental axonal injury triggers interleukin-6 mRNA, protein synthesis and release into cerebrospinal fluid. J Cereb Blood Flow Metab (1999) 19(2):184–94. doi:10.1097/00004647-199902000-00010
115. Penkowa M, Giralt M, Carrasco J, Hadberg H, Hidalgo J. Impaired inflammatory response and increased oxidative stress and neurodegeneration after brain injury in interleukin-6-deficient mice. Glia (2000) 32(3):271–85. doi:10.1002/1098-1136(200012)32:3<271:AID-GLIA70>3.0.CO;2-5
116. Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernández J, et al. Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Exp Neurol (2003) 181(2):130–48. doi:10.1016/S0014-4886(02)00051-1
117. Ley EJ, Clond MA, Singer MB, Shouhed D, Salim A. IL6 deficiency affects function after traumatic brain injury. J Surg Res (2011) 170(2):253–6. doi:10.1016/j.jss.2011.03.006
118. Yang SH, Gangidine M, Pritts TA, Goodman MD, Lentsch AB. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock (2013) 40(6):471–5. doi:10.1097/shk.0000000000000037
119. Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A (1999) 96(15):8721–6. doi:10.1073/pnas.96.15.8721
120. Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, et al. Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab (2000) 20(2):369–80. doi:10.1097/00004647-200002000-00019
121. Yan EB, Satgunaseelan L, Paul E, Bye N, Nguyen P, Agyapomaa D, et al. Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J Neurotrauma (2014) 31(7):618–29. doi:10.1089/neu.2013.3087
122. Baratz R, Tweedie D, Wang J-Y, Rubovitch V, Luo W, Hoffer BJ, et al. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J Neuroinflammation (2015) 12:45. doi:10.1186/s12974-015-0237-4
123. Longhi L, Perego C, Ortolano F, Aresi S, Fumagalli S, Zanier ER, et al. Tumor necrosis factor in traumatic brain injury: effects of genetic deletion of p55 or p75 receptor. J Cereb Blood Flow Metab (2013) 33(8):1182–9. doi:10.1038/jcbfm.2013.65
124. Yang J, You Z, Kim H-H, Hwang S-K, Khuman J, Guo S, et al. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma (2010) 27(6):1037–46. doi:10.1089/neu.2009.1229
125. McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation (2008) 5:45. doi:10.1186/1742-2094-5-45
126. Probert L. TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience (2015) 302:2–22. doi:10.1016/j.neuroscience.2015.06.038
127. Song S, Kong X, Acosta S, Sava V, Borlongan C, Sanchez-Ramos J. Granulocyte colony-stimulating factor promotes behavioral recovery in a mouse model of traumatic brain injury. J Neurosci Res (2016) 94(5):409–23. doi:10.1002/jnr.23714
128. Shultz SR, Tan XL, Wright DK, Liu SJ, Semple BD, Johnston L, et al. Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma (2014) 31(10):976–83. doi:10.1089/neu.2013.3106
129. Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging (2014) 35(5):1012–23. doi:10.1016/j.neurobiolaging.2013.10.089
130. Wang R, Yang B, Zhang D. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia (2011) 59(6):946–58. doi:10.1002/glia.21167
131. Khorooshi R, Owens T. Injury-induced type I IFN signaling regulates inflammatory responses in the central nervous system. J Immunol (2010) 185(2):1258–64. doi:10.4049/jimmunol.0901753
132. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (2014) 346(6205):89–93. doi:10.1126/science.1252945
133. Yan EB, Hellewell SC, Bellander BM, Agyapomaa DA, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. J Neuroinflammation (2011) 8:147. doi:10.1186/1742-2094-8-147
134. Di Battista AP, Rhind SG, Hutchison MG, Hassan S, Shiu MY, Inaba K, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflammation (2016) 13(1):40. doi:10.1186/s12974-016-0500-3
135. Schneider Soares FM, Menezes de Souza N, Liborio Schwarzbold M, Paim Diaz A, Costa Nunes J, Hohl A, et al. Interleukin-10 is an independent biomarker of severe traumatic brain injury prognosis. Neuroimmunomodulation (2012) 19(6):377–85. doi:10.1159/000342141
136. Morganti-Kossmann MC, Hans VHJ, Lenzlinger PM, Dubs R, Ludwig E, Trentz O, et al. TGF-β is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J Neurotrauma (1999) 16(7):617–28. doi:10.1089/neu.1999.16.617
137. Liu Y, Liu Z, Li X, Luo B, Xiong J, Gan W, et al. Accumulation of connective tissue growth factor+ cells during the early phase of rat traumatic brain injury. Diagn Pathol (2014) 9:141. doi:10.1186/1746-1596-9-141
138. Chio CC, Chang CP, Lin MT, Su FC, Yang CZ, Tseng HY, et al. Involvement of TG-interacting factor in microglial activation during experimental traumatic brain injury. J Neurochem (2014) 131(6):816–24. doi:10.1111/jnc.12971
139. Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology (1985) 35(2):264–7. doi:10.1212/WNL.35.2.264
140. Guo Z, Cupples L, Kurz A, Auerbach S, Volicer L, Chui H, et al. Head injury and the risk of AD in the MIRAGE study. Neurology (2000) 54:1316–23. doi:10.1212/wnl.54.6.1316
141. Warren SA, Olivo SA, Contreras JF, Turpin KVL, Gross DP, Carroll LJ, et al. Traumatic injury and multiple sclerosis: a systematic review and meta-analysis. Can J Neurol Sci (2013) 40(2):168–76. doi:10.1017/S0317167100013688
142. Lunny CA, Fraser SN, Knopp-Sihota JA. Physical trauma and risk of multiple sclerosis: a systematic review and meta-analysis of observational studies. J Neurol Sci (2014) 336(1–2):13–23. doi:10.1016/j.jns.2013.08.011
143. Kang JH, Lin HC. Increased risk of multiple sclerosis after traumatic brain injury: a nationwide population-based study. J Neurotrauma (2012) 29(1):90–5. doi:10.1089/neu.2011.1936
144. Washington PM, Morffy N, Parsadanian M, Zapple DN, Burns MP. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J Neurotrauma (2014) 31(1):125–34. doi:10.1089/neu.2013.3017
145. Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol (2014) 71(4):505–8. doi:10.1001/jamaneurol.2013.5847
146. Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, Sengupta U, Prough DS, Jackson GR, et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. J Biol Chem (2013) 288(23):17042–50. doi:10.1074/jbc.M113.472746
147. Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet (1991) 338(8780):1422–3. doi:10.1016/0140-6736(91)92724-G
148. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry (1994) 57(4):419–25. doi:10.1136/jnnp.57.4.419
149. Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, et al. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology (2003) 60(9):1457–61. doi:10.1212/01.WNL.0000063313.57292.00
150. Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, et al. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science (2008) 321(5893):1221–4. doi:10.1126/science.1161591
151. Washington PM, Villapol S, Burns MP. Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol (2016) 275(Pt 3):381–8. doi:10.1016/j.expneurol.2015.06.015
152. Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med (2009) 15(4):377–9. doi:10.1038/nm.1940
153. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol (2015) 16(3):229–36. doi:10.1038/ni.3102
154. Crawford FC, Vanderploeg RD, Freeman MJ, Singh S, Waisman M, Michaels L, et al. APOE genotype influences acquisition and recall following traumatic brain injury. Neurology (2002) 58(7):1115–8. doi:10.1212/WNL.58.7.1115
155. Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology (1999) 52(2):244–8. doi:10.1212/WNL.52.2.244
156. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med (2016) 18(5):421–30. doi:10.1038/gim.2015.117
157. Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci (2010) 11(5):361–70. doi:10.1038/nrn2808
158. Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA (1997) 278(2):136–40. doi:10.1001/jama.1997.03550020068040
159. Sabo T, Lomnitski L, Nyska A, Beni S, Maronpot RR, Shohami E, et al. Susceptibility of transgenic mice expressing human apolipoprotein E to closed head injury: the allele E3 is neuroprotective whereas E4 increases fatalities. Neuroscience (2000) 101(4):879–84. doi:10.1016/S0306-4522(00)00438-3
160. Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature (2012) 485(7399):512–6. doi:10.1038/nature11087
161. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med (2005) 201:647–57. doi:10.1084/jem.20041611
162. Yeh Felix L, Wang Y, Tom I, Gonzalez Lino C, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron (2016) 91(2):328–40. doi:10.1016/j.neuron.2016.06.015
163. Wang Y, Cella M, Mallinson K, Ulrich Jason D, Young Katherine L, Robinette Michelle L, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell (2015) 160(6):1061–71. doi:10.1016/j.cell.2015.01.049
164. Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med (2015) 212:287–95. doi:10.1084/jem.20142322
165. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci (2015) 35(8):3384–96. doi:10.1523/jneurosci.2620-14.2015
166. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell (2015) 160:1061–1071. doi:10.1016/j.cell.2015.01.049
167. Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-a unifying hypothesis. Surg Neurol Int (2011) 2:107. doi:10.4103/2152-7806.83391
168. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9(1):46–56. doi:10.1038/nrn2297
169. Ramirez K, Fornaguera-Trias J, Sheridan JF. Stress-induced microglia activation and monocyte trafficking to the brain underlie the development of anxiety and depression. In: Geyer MA, Ellenbroek BA, Marsden CA, Barnes Th. RE, editors. Current Topics in Behavioral Neurosciences. Cham, Switzerland: Springer International Publishing (2016). p. 1–18.
Keywords: traumatic brain injury, neuroinflammation, cytokine, inflammasome, innate immunology, neuroprotection, microglia, neurodegeneration
Citation: McKee CA and Lukens JR (2016) Emerging Roles for the Immune System in Traumatic Brain Injury. Front. Immunol. 7:556. doi: 10.3389/fimmu.2016.00556
Received: 16 September 2016; Accepted: 18 November 2016;
Published: 05 December 2016
Edited by:
Ihssane Zouikr, RIKEN Brain Science Institute, JapanCopyright: © 2016 McKee and Lukens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John R. Lukens, anJsN25AdmlyZ2luaWEuZWR1