 Florian Wirsdörfer
Florian Wirsdörfer Verena Jendrossek
Verena Jendrossek- Institute of Cell Biology (Cancer Research), University Hospital Essen, Essen, Germany
Radiation-induced pneumonitis and fibrosis are dose-limiting side effects of thoracic irradiation. Thoracic irradiation triggers acute and chronic environmental lung changes that are shaped by the damage response of resident cells, by the resulting reaction of the immune system, and by repair processes. Although considerable progress has been made during the last decade in defining involved effector cells and soluble mediators, the network of pathophysiological events and the cellular cross talk linking acute tissue damage to chronic inflammation and fibrosis still require further definition. Infiltration of cells from the innate and adaptive immune systems is a common response of normal tissues to ionizing radiation. Herein, lymphocytes represent a versatile and wide-ranged group of cells of the immune system that can react under specific conditions in various ways and participate in modulating the lung environment by adopting pro-inflammatory, anti-inflammatory, or even pro- or anti-fibrotic phenotypes. The present review provides an overview on published data about the role of lymphocytes in radiation-induced lung disease and related damage-associated pulmonary diseases with a focus on T lymphocytes and B lymphocytes. We also discuss the suspected dual role of specific lymphocyte subsets during the pneumonitic phase and fibrotic phase that is shaped by the environmental conditions as well as the interaction and the intercellular cross talk between cells from the innate and adaptive immune systems and (damaged) resident epithelial cells and stromal cells (e.g., endothelial cells, mesenchymal stem cells, and fibroblasts). Finally, we highlight potential therapeutic targets suited to counteract pathological lymphocyte responses to prevent or treat radiation-induced lung disease.
Introduction
About 60% of all cancer patients receive radiotherapy (RT) at some point during the course of their disease, and good results in terms of long-term survival and tumor cure are achieved in a variety of tumors by multimodal combinations of surgery, RT, and chemotherapy. Concurrent radiochemotherapy could improve the prognosis of glioma, lung, head and neck, esophageal, cervical, anal, and rectal cancer (1–8) and is part of standard therapy for locally advanced tumors of these entities. Yet, treatment outcome is still unsatisfactory for common forms of cancer with high loco-regional failure rates or frequent development of metastases. Although patient-specific clinical factors may explain some of these failures, it is commonly assumed that biological factors adversely affecting the response of tumor cells to treatment, such as intrinsic radioresistance, tumor promoting mutations, unfavorable gene expression profiles, heterogeneity in radiation responses, or a resistance-promoting microenvironment, significantly contribute to treatment failures (9–14). Acute and late toxicity to normal tissues also limits the radiation dose that can be applied to the tumor, and tolerable doses are often linked to suboptimal tumor control—even accepting side effects that lead to decreased quality of life (15). Normal tissue toxicity also precludes therapy intensification efforts for many locally advanced tumors by the combination with cytotoxic chemotherapy (16–18). As a consequence, there is high interest in improving the therapeutic ratio either by technical and physical innovations in treatment delivery, e.g., intensity-modulated radiation therapy or particle therapy, or by developing effective strategies to prevent or treat the toxic effects of ionizing radiation (IR) in normal tissues without protecting the tumor cells, or to increase intrinsic radiosensitivity of cancer cells without increasing sensitivity of normal tissue cells, respectively.
Dose-limiting side effects in the lung tissue after RT of the thoracic region or total body irradiation in conditioning regimens for hematopoietic stem cell transplantation include inflammatory (pneumonitis) and fibrotic changes (pulmonary fibrosis) (19–21). Radiation-induced damage to the lung tissue leads, like infectious, thermal, or physical damage, to the activation of the immune system. This inflammatory response is needed to orchestrate tissue repair and regeneration in order to restore tissue homeostasis. Depending on the degree of the resulting aseptic inflammation, patients can present with pneumonitis. Radiation-induced pneumonitis can develop at 4–12 weeks after RT with symptoms like fever, chest pain, dry cough, and dyspnea or even respiratory failure in severe cases and occurs in 5–20% of patients with lung or breast cancer (22–24). The pneumonitic phase is characterized by the recruitment of diverse immune cells of myeloid and lymphoid origin and a perpetual cascade of cytokines/chemokines resulting in various degrees of lung inflammation and the described symptoms (Figure 1).
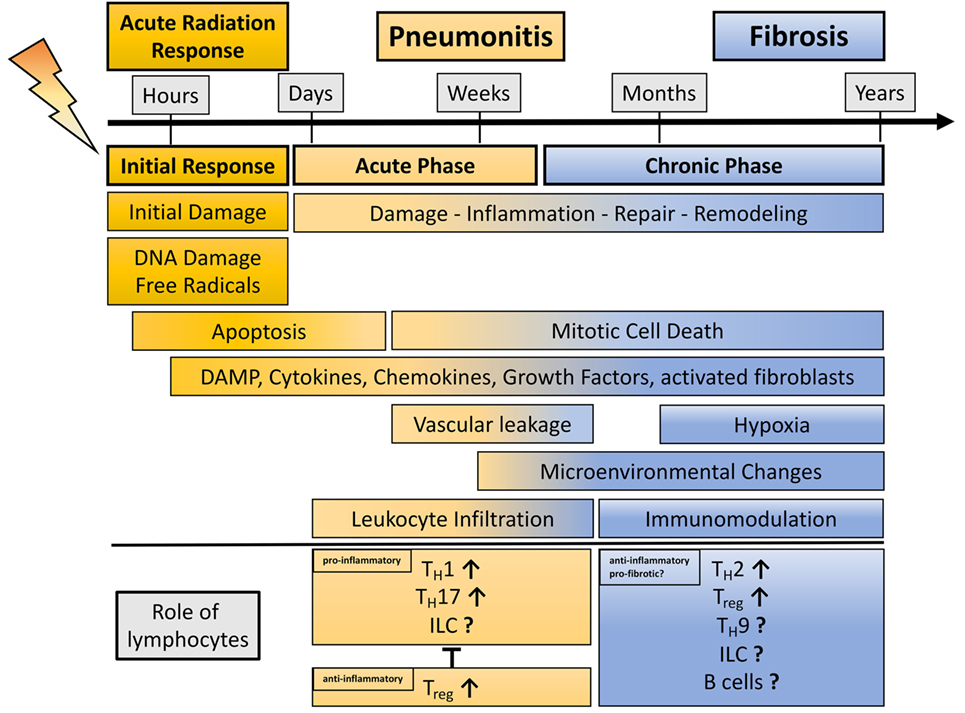
Figure 1. Schematic representation showing the phases of radiation-induced lung injury over time with a view on the dual role of lymphocytes during radiation-induced pneumopathy. Damage to the lung results in an initial response (acute radiation response) due to DNA damage, ROS induction, and apoptosis. Release of damage-associated molecular patterns (DAMPs) and secretion of cytokines and chemokines activate the immune system. This phase passes over into an acute inflammatory phase (pneumonitis) that is characterized by an enhanced pro-inflammatory response and vascular leakage. In this phase, diverse lymphocyte subpopulations like TH1, TH17, and potentially innate lymphoid cells (ILC) can contribute to inflammation, whereas it is believed that the lymphocyte subpopulations Treg are needed to control harmful, excessive pro-inflammatory responses. Resolution of inflammation and repair induction is paralleled by late mitotic cell death subsequent, hypoxia, release of DAMP, cytokines, and growth factors. These alterations in the lung micromilieu are described for the chronic phase of radiation-induced pneumopathy. These environmental changes can contribute to immunomodulation; here, it is believed that lymphocytes (TH2, TH9, Treg, and potentially ILC) show an anti-inflammatory or even pro-fibrotic phenotype, thereby having the potential to further alter the environment in the lung toward the induction of disease-promoting myofibroblasts and fibrosis development.
Development of radiation-induced lung fibrosis is mostly observed 6–24 months after RT and may become chronic in patients with a large irradiated lung volume (24). Major symptoms of lung fibrosis are breathing difficulties and subsequent volume loss of the lung (25). Studies from various groups using rodent models and patient samples helped to reveal a complex response of the lung tissue toward irradiation with multiple interactions between resident lung cells including lung-resident mesenchymal stem cells (MSC), locally generated or recruited fibroblasts, and infiltrating immune cells, respectively (26–37). We speculate that a sophisticated network between damaged resident cells (epithelial cells, endothelial cells, and lung-resident MSC), recruited immune cells, and soluble mediators (cytokines, chemokines, growth factors, and proteases) and the resulting environmental changes participate in shaping the observed inflammatory and fibrotic alterations of the lung tissue (Figures 1 and 2).
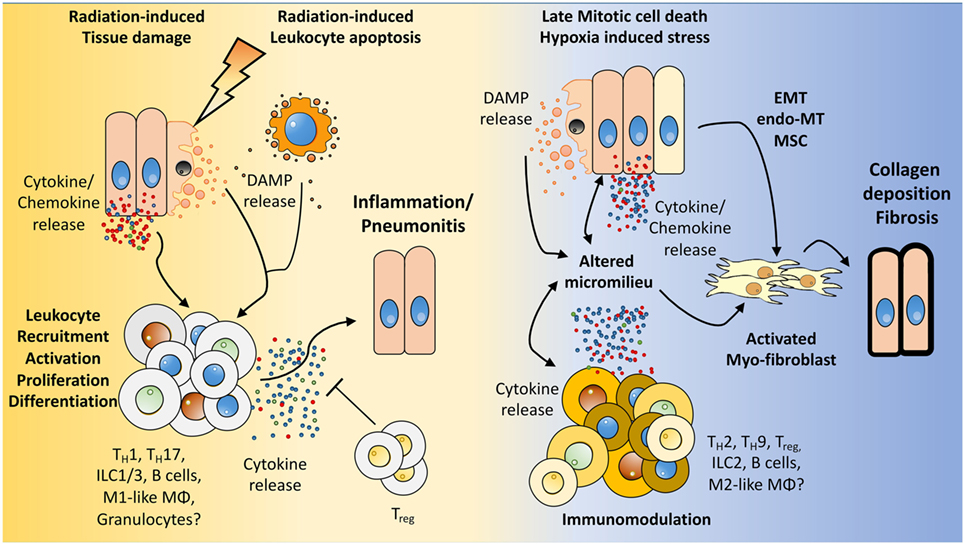
Figure 2. How the microenvironment shapes the immune response and vice versa. We hypothesize that radiation induces damage to tissue resident cells, e.g., endothelial and epithelial lung cells, mesenchymal stem cells (MSC) as well as in resident immune cells. The resulting tissue damage can initiate stress responses or cell death with subsequent release of cytokines/chemokines and damage-associated molecular patterns (DAMPs). This initial damage response leads to the recruitment and activation of diverse immune cells to the lung, among them lymphocytes. Further activation, proliferation of these cells, and secretion of cytokines shape the pulmonary micromilieu toward inflammation and—if this response is too excessive—to the development of severe pneumonitis. Late chronic mitotic cell death and subsequent tissue hypoxia lead to the release of DAMPs and cytokines/chemokines from resident cells thereby altering the micromilieu in the lung. These environmental changes impact on the immune cells present in the lung tissue and promote an altered cytokine release of immune cells. Finally, epithelial-mesenchymal-transition, endothelial-mesenchymal-transition, mesenchymal stem cell differentiation, and the altered environment contribute to the induction of activated myofibroblasts, collagen deposition, and fibrosis.
Among other molecular markers there is evidence from preclinical and clinical studies that T lymphocytes infiltrate the lung to a considerable extent, particularly during the pneumonitic phase at 3–12 weeks post-irradiation, although lymphocyte infiltration was also observed at later time points during the chronic inflammatory/fibrotic phase at 16–30 weeks post-irradiation (30, 35, 38–40). Interestingly, earlier studies described a correlation between the presence of CD4+ T cells in the bronchoalveolar lavage of irradiated breast or lung cancer patients and the development of pneumonitis (39, 41, 42). Furthermore, depletion of CD4+ T cells during the pneumonitic phase attenuated the development of lung fibrosis upon thoracic irradiation in a murine model (43). These findings suggest a complex role of CD4+ T cells in the pathogenesis of radiation-induced lung disease. Antibody-mediated inhibition of the accumulation of CD3+ lymphocytes or depletion of CD4 and CD8 T lymphocytes also reduced fibrosis levels in the murine model of pulmonary fibrosis induced by the radiomimetic and DNA-damaging drug bleomycin (BLM) (44, 45). In contrast, the lack of mature T and B lymphocytes in recombination-activating gene 2 (RAG2)-deficient mice exacerbated radiation-induced fibrosis (46). Altogether, these findings highlight that lymphocytes play a complex role in DNA damage-induced lung disease and suggest that depending on the disease stage and the environmental conditions, shaped by the tissue response to the damage, specific lymphocyte subpopulations exert either beneficial or adverse effects (Figure 1). We propose that a disturbed balance between tissue inflammation and repair processes participates in the development of radiation-induced pulmonary fibrosis as it has been described for other fibrotic diseases and that lymphocytes are involved in these processes (47). Nevertheless, it remains to be demonstrated whether lymphocytes directly contribute to radiation-induced lung disease or only modulate disease progression. Furthermore, it remains to be explored whether, besides the myeloid compartment, innate lymphoid cells (ILC) might contribute to radiation-induced fibrosis. Finally, the mechanisms driving radiation-induced lymphocyte deviation remain to be defined.
Lymphocytes: Effector Cells of the Immune System
Lymphocytes are characterized as white blood cells that are homogeneous in appearance but that have various functions. They include T cells, B cells, and ILC among them conventional natural killer (NK) cells. T cells, ILC, and B cells are responsible for the production of cytokines and antibodies (B cells), whereas NK cells can induce direct cell-mediated killing of virus-infected cells and tumor cells. Here, we will focus on a potential role of B and T lymphocytes as well as ILC.
The different major subpopulations of T lymphocytes include CD8+, CD4+ T cells, NK T cells, and γδT cells. CD8+ T cells comprise cytotoxic T cells or cytolytic T cells. They control and eliminate intracellular pathogens and tumor cells and can further differentiate into CD8+ memory cells (48). γδT cells express a T cell receptor differing from the conventional αβT cells. The function of γδT cells is poorly understood, but current knowledge implies a role in immunoregulation in pathogen and allergen responses (49). NK T cells are a unique subpopulation of lymphocytes that are mainly involved in innate immunity and will not be further discussed in the present review.
CD4+ T cells comprise TH1 and TH2 subpopulations. Furthermore, advances in immunology have led to the characterization of newly appreciated CD4+ T cell effector populations that regulate the immune response such as interleukin (IL)-17-producing T cells (TH17 cells), T cells with regulatory function [regulatory T cells (Treg)], IL-9-secreting TH9 cells, IL-22-dominant TH22 cells, and B cell-interacting follicular helper T cells (TFH), thus revising established paradigms (50–58).
The secretion of interferon (IFN)-γ and the directed elimination of intracellular pathogens characterize a TH1 response. In contrast, TH2 responses are shaped by the cytokines IL-4 and IL-13, supporting the defense against parasites, and moreover contribute to the generation of antibodies (59).
TH17 cells preferentially produce IL-17A-F and play a role in inflammatory processes such as autoimmune diseases and the defense against extracellular pathogens. TH17 cells further produce the cytokines IL-21, IL-22, and IL-23, which exert strong pro-inflammatory effects (60). TH17 cells are induced by IL-6, IL-21, and transforming growth factor beta (TGF-β), a potent regulator of lung homeostasis as well as in pathologies (61, 62).
Another important subpopulation of T cells are Treg. Treg show a suppressive capacity, control immune reactions, and inhibit exaggerated inflammation (60, 63). Treg exist as natural occurring Treg (nTreg) and induced Treg (iTreg). Murine thymus-derived nTreg are CD4+/CD25+ and express the transcription factor FoxP3 (murine cell marker), whereas in humans not all Treg express FoxP3. Therefore, Treg in humans are mainly characterized via the marker profile CD4+/CD25hi/CD127low (64). Treg show their suppressive capacity by secreting anti-inflammatory cytokines like IL-10 and TGF-β, which can induce cell cycle arrest or apoptosis in T effector cells.
Recent studies also revealed a small population of CD8+ Treg at steady state; these CD8+ Treg are present in the human and the murine system, and they express the marker CD25 as well as FoxP3. Churlaud et al. showed that CD8+ Treg are highly suppressive and responsive to IL-2 (65), but further studies are needed to uncover the origin and the role of CD8+ Treg in health and pathologies.
B lymphocytes represent the second heterogeneous group of lymphocytic cells. B cells originate from the bone marrow, mature in the spleen, and differentiate in the lymph nodes into germinal center cells after contact with antigens and T cells (66, 67). Besides their capacity for antibody secretion, they have functions in antigen presentation and secretion of diverse cytokines (68, 69).
The newly identified group of the lymphoid cells, namely ILC, plays an important junction between innate and adaptive immunity. Like other lymphoid cells, they originate from a common lymphoid progenitor in the bone marrow. But in contrast to their relatives, they lack (RAG)-dependent rearrangement of antigen receptors as well as phenotypical markers of myeloid and dendritic cells (70, 71). ILC have been characterized by their expression pattern of the master transcription factors (T-bet, GATA3, and RORγt) and specific cytokines that usually define T cell subpopulations. Based on this categorization, three different subpopulations (ILC1, ILC2, and ILC3) have been defined as follows: (i) ILC1 cells include conventional NK cells and T-bet+/IFN-γ-producing cells; (ii) ILC2 cells show GATA3 expression, and they secrete TH2 cytokines, IL-4, IL-5, IL-9, and IL-13 in response to IL-25 and IL-33; and (iii) ILC3 cells express the transcription factor RORγt, and they release the cytokines IL-17, IL-22, granulocyte–macrophage colony-stimulating factor (GM-CSF) as well as lymphotoxins. ILC act on tissue homeostasis and tissue remodeling; moreover, they participate in regulating immune responses to inflammation and infection in tissues like liver, lymph nodes, and mucosal barriers like gut and lung (72, 73). These characteristics make it highly likely that ILC also modulate the progression of radiation-induced lung inflammation and fibrosis (see Role of Lymphocytes in the Defense of the Lung Tissue).
Role of Lymphocytes in the Defense of the Lung Tissue
In the lung, T cells are found in relatively high numbers in the mucosa, in the intraepithelial part, in the underlying lamina propria, and also in the lung parenchyma. T cells of the intraepithelial region express CD8, while CD4 is the dominant surface marker of T cells in the lamina propria. Furthermore, it has been described that CD45RO is expressed on both, CD4 and CD8 T cell subsets, indicating their role as effector and/or memory cells (74). For CD8+ T cells it is known that they protect the lung against influenza infection. Nevertheless, there it is believed that CD8+ T cells also contribute to lung injury, e.g., during influenza infection due to their cytotoxic effects and the massive production of the pro-inflammatory cytokines IFN-γ and tumor necrose factor (TNF)-α (75), respectively.
Among the CD4+ T cells, lung TH17 cells may not only play a role in neutrophil recruitment and pathogen clearance but also be highly relevant in respiratory inflammatory diseases (76). Moreover, a recent study demonstrated that TH17 cells also play a role in lung cancer progression: here, the authors analyzed blood samples from patients and described that TH17 as well as Treg subsets are involved in the immunopathology of NSCLC (77). In other studies, Treg were found to counteract the inflammation-induced injury to the airways associated with lung infections as well as the development of atopic diseases. Treg also play a role in mediating inhalation tolerance and in controlling allergen-specific T cells from activation (74, 78). Interestingly, there is evidence from some studies that Treg contribute to fibrotic diseases in the lung by promoting a pro-fibrotic microenvironment (79–81).
Also, γδ T cells show functions in the airways. For the lungs it is described that γδ T cells reside in the subepithelium of alveolar and non-alveolar regions (82). Here, they modulate immune responses against allergens and pathogens (49, 83). A study from Simonian et al. also revealed a role in inflammation (hypersensitivity pneumonitis)-induced lung fibrosis. Here, γδ T cells diminished CD4+ cell recruitment by the secretion of regulatory IL-22 thereby protecting the lung from fibrosis (84).
However, the role of TH9, TH22, and TFH cells in lung pathologies is still underexplored. So far, current studies hint to a role for TH22 and TFH cells in host defense against viruses and bacteria in the lung, whereas TH9 cells seem to play an important role in asthma (85–87).
Similar to T cell populations, B cells are also present in the parenchymal lung as well as in the conducting airways. In the lamina propria, they act as antibody-secreting plasma cells producing immunoglobulin (Ig)A but may also contribute to local antigen presentation (74, 88). In the lung, B cells can act, for example, as memory B cells producing IgA and IgG neutralizing antibodies that have the ability to protect against pulmonary viral reinfections (89).
The group of ILC is in the focus of current research. ILC have been implicated in immunity of the mucosal barrier in the lung, e.g., in allergic asthma, hyper responsiveness, and viral infection (90–93). The present knowledge on ILC has nicely been summarized in two recent reviews with a focus on their roles in the lung tissue (94, 95). Though these reviews emphasize that the role of ILC in the lung is still poorly characterized, we will highlight some important observations at this point. Among the three ILC subsets defined so far, ILC2 present the main ILC in the murine lung, but with 2–3 × 104 cells per lung and thus 0.4–1% of total lung cells, these ILC seem to be a relatively rare population, at least under physiological conditions (91). But, pathological conditions in the lung are associated with changes in the ILC population (96). In this context, ILC2 and ILC3 seem to play more imported roles than ILC1 during chronic lung disease in both, mice and men (94, 97, 98). For example, in patients with chronic obstructive pulmonary disease (COPD) ILC3 constitute the major population with 60% of all ILC, whereas ILC2 amount to 30% and ILC1 to 10% of the ILC (98). Of note, depletion of ILC2 reduced epithelial integrity, induced epithelial degeneration, and impaired lung functions during influenza virus infection highlighting a protective role of ILC2 for these processes (91).
First reports suggest that ILC may also play an important role during acute pulmonary inflammation. In this context, ILC3 have recently been identified as a major source of IL-17A thereby inducing neutrophil recruitment in a murine model of LPS-induced acute respiratory distress syndrome (99). Furthermore, in eosinophilic crystalline pneumonia, a murine idiopathic type 2 lung inflammation, IL-2 was shown to function as an important activator of ILC2 functions; the authors further highlight a potential cross talk between ILC2 and ILC3 as well as T cells and Treg during disease pathogenesis (100). Even more important, in a murine model for allergic asthma, pulmonary epithelial cell-derived TGF-β1 and IL-33 contributed to ILC2-mediated responses (101).
Besides their effects on acute pulmonary responses, ILC also impact the development of pulmonary fibrosis [for a recent review, refer to Ref. (102)]. In this context, an interplay between ILC, macrophages, and cells of the adaptive immune system was shown to participate—together with IL-13, IL-25, and IL-33—in the pathogenesis of BLM-induced pulmonary fibrosis in mice (103–105). Moreover, Hams et al. also found elevated levels of IL-25 and ILC2 in the lungs of patients with idiopathic pulmonary fibrosis (IPF) (105, 106), whereas others described a role for ILC2 and ILC3 in fibroblast activation providing a mechanistic link between ILC and fibrotic diseases (102).
Since work from our own laboratory and others implicate TH17 cells and Treg in the pathogenesis of radiation-induced lung disease, these observations strongly suggest that ILC, particularly ILC2 and ILC3, might also participate in the cross talk between damaged resident cells, recruited immune cells, and activated fibroblasts and thus modulate the extent of lung inflammation and progression of radiation-induced fibrosis. But, an involvement of ILC in radiation-induced pneumopathy remains to be demonstrated, and potential beneficial or disease-promoting effects during the different disease stages (inflammation, fibrosis) have to be explored.
Impact of Ionizing Radiation on Lymphocytes
Radiation therapy is an essential and common approach in cancer treatment. So far, the use of IR in cancer treatment is based on its high potential to induce tumor cell death and to abrogate survival of clonogenic tumor cells. The toxic effects of IR result from the deposition of energy from IR in tumor and normal tissue cells including immune cells. Energy deposition results in damage to cellular macromolecules, particularly cellular DNA, e.g., by direct breakage of chemical bonds within the DNA as well as by the generation of free radicals (107, 108). Among the diverse damaging effects of IR, the induction of DNA double strand breaks is considered as the most toxic lesion in cells.
The hematopoietic compartment is particularly sensitive to IR, for example, blood sample analysis revealed the rapid development of a hematopoietic syndrome in patients exposed to a total body irradiation of 1–2 Gray (Gy), which was characterized by a decline of the hematopoietic compartment (109). Similar to other hematopoietic cells, lymphocytes are particularly sensitive to radiation-induced cell death. Nevertheless, the various lymphocyte subtypes differ in their radiosensitivity: up to now it has been demonstrated that B cells, naive T cells, and NK cells are highly radiosensitive, whereas T memory cells, NK T cells, and Treg cells are more resistant to the toxic effects of IR (110–114). Moreover, a study from 1995 revealed a higher radiosensitivity of IL-4-producing TH2 cells compared to TH1 cells (115). Based on their high radiosensitivity and easy accessibility, blood lymphocytes are frequently used in biodosimetry (116–119).
Further direct effects described in irradiated lymphocytes concentrate on the transcriptional response of these cells to IR, e.g., by gene expression profiling. These studies revealed that a majority of the strongly activated genes are p53 targets, like DNA damage-binding protein 2, the BCL-2-associated gene BAX, and tumor necrosis factor receptor superfamily, member 10b (TNFRSF10B) that are involved in DNA repair and apoptosis regulation (120–122).
However, in addition to these direct or “targeted” effects of IR on lymphocytes such as the induction of cellular stress responses and cell death in lymphocytes within the radiation field, lymphocytes can also mount an indirect response to radiation-induced tissue damage. In this context, “danger signals” released from damaged or dying cells in irradiated tissues result in lymphocyte activation, infiltration into the damaged tissue, and the release of inflammatory mediators (Figure 2).
Of note, high-dose irradiation also efficiently triggers direct tumor cell death and augments innate immune responses and tumor-specific immunity thereby enhancing the local and distant antitumor effects of RT. This was nicely summarized by one of the pioneers in the filed of radioimmunotherpy, Silvia Formenti (123). Accordingly, high numbers of tumor-infiltrating cytotoxic lymphocytes may predict the response of certain tumors to treatments involving RT (105, 124). This is a hot topic in the field of radiation biology and oncology highlighted in other reviews; here, we will concentrate on the contribution of radiation-induced modulation of the lymphocyte compartment to the adverse late effects of IR in the lung.
Role of Lymphocytes in the Irradiated Lung
As nicely highlighted in a recent review on the general effects of IR on T lymphocytes and normal tissue responses, our understanding of the interaction between lymphocytes and radiation-induced tissue damage is still rudimentary (125). Because of the complexity of the involved cellular systems and soluble factors, investigations about the mechanisms underlying radiation-induced adverse late effects, for example, in the lung can only be performed in patients as well as animal models in vivo, particularly rodent models.
Importance of the Experimental Model
Experimental models that use a single high-dose whole thorax or hemithorax irradiation of fibrosis-sensitive mice (C57BL/6) mimic human disease with respect to the time course and major symptoms of the disease (pneumonitis, fibrosis) and are therefore frequently used to study the underlying mechanisms, to define disease biomarkers and novel therapeutic targets, and to explore potential toxic effects of new combination strategies using IR in combination with molecularly targeted drugs (26, 126–130).
In this context, it is important to consider that mice with different backgrounds differ in their sensitivity to acute and chronic responses (pneumonitis, fibrosis), and these differences seem to be associated with differences in immune response in fibrosis-sensitive (C57BL/6, C57BL/6J) and fibrosis-resistant mice (e.g., BALB/c, C3Hf, and A/J) (131–133). In support of this assumption, a comparison of the transcriptome of lung tissue of fibrosis-prone C57BL/6J or fibrosis-resistant C3Hf/KAM mice upon BLM treatment revealed that the differences between the mouse strains included genes important for apoptosis, oxidative stress, and immune regulation (134).
On the other hand, due to the long latency of radiation-induced adverse side effects, researchers frequently use local or systemic administration of the DNA-damaging drug BLM as a radiomimetic drug that rapidly provokes a pronounced lung fibrosis in rodent models (135). However, work from our own group and from others suggests that the impact of the immune system on disease outcome may play different roles in the BLM and RT model, particularly when the acute model of intratracheal application of BLM is used (35, 136, 137). Since the C57BL/6 model is suited for the analysis of both, pneumonitis and fibrosis, we focus particularly on this model in the subsequent paragraphs. But, we are aware of the fact that future studies are needed in a more clinically relevant setting with fractionated irradiation.
Investigations in rodent models using experimental whole thorax or hemithorax irradiation are still underrepresented. Thus, preclinical studies about the contribution of RT-induced immunomodulation in normal tissues to radiation-induced lung disease are rare particularly with respect to lymphocyte responses. We therefore included data about lymphocytes responses from studies in other models of chronic respiratory disease or pulmonary fibrosis, where appropriate. We are aware that the described models are different with respect to the initial injury, the time course, and some of the involved mediators. However, from an immunological point of view, the different models of (chronic) inflammation/fibrosis share a sterile inflammation/repair/remodeling response to an initial damage/trauma and will therefore help to understand lymphocyte responses in the lung in response to radiation-induced tissue damage.
Early Effects of Thoracic Irradiation on Lymphocytes in the Lung
Early immune suppression with subsequent lymphocyte infiltration are common responses of irradiated tissues during the acute and chronic phase after irradiation, including the lung tissue (40, 42, 138, 139). Preclinical studies in mice corroborated the infiltration and reconstitution of lymphocytes observed in patients (for more details, see Table 1). For example, Paun et al. analyzed the primary radiation injury response of the lung at 6 h, 1, and 7 days after 18 Gy whole thorax irradiation in different mouse strains and characterized infiltrating T cell populations and their cytokine profile in the lung tissue and in the bronchoalveolar lavage fluid (BALF). The authors reveal lower T cell levels in the BALF and increased numbers of infiltrating TH1 and TH2 cells in the lung at day 1 and 7 (140). Another murine study from Zheng et al. uncovered a more delayed reconstitution of CD4+ T cells compared to that of CD8+ T cells upon low dose total body irradiation (2.5 Gy); furthermore, TH1 reconstitution was also impaired, whereas TH17 and Treg cells were elevated (141). In an own study, we showed that a 15 Gy whole thorax irradiation in mice led to a slight decrease in the percentage of CD3+ T cells in the lung at day 10 and 21 post-irradiation. In line with these findings, we found significant decreased levels of CD3+ T cells, including CD4+ and CD8+ T cells, at these time points in peripheral lymphoid organs like cervical lymph nodes and the spleen (142).
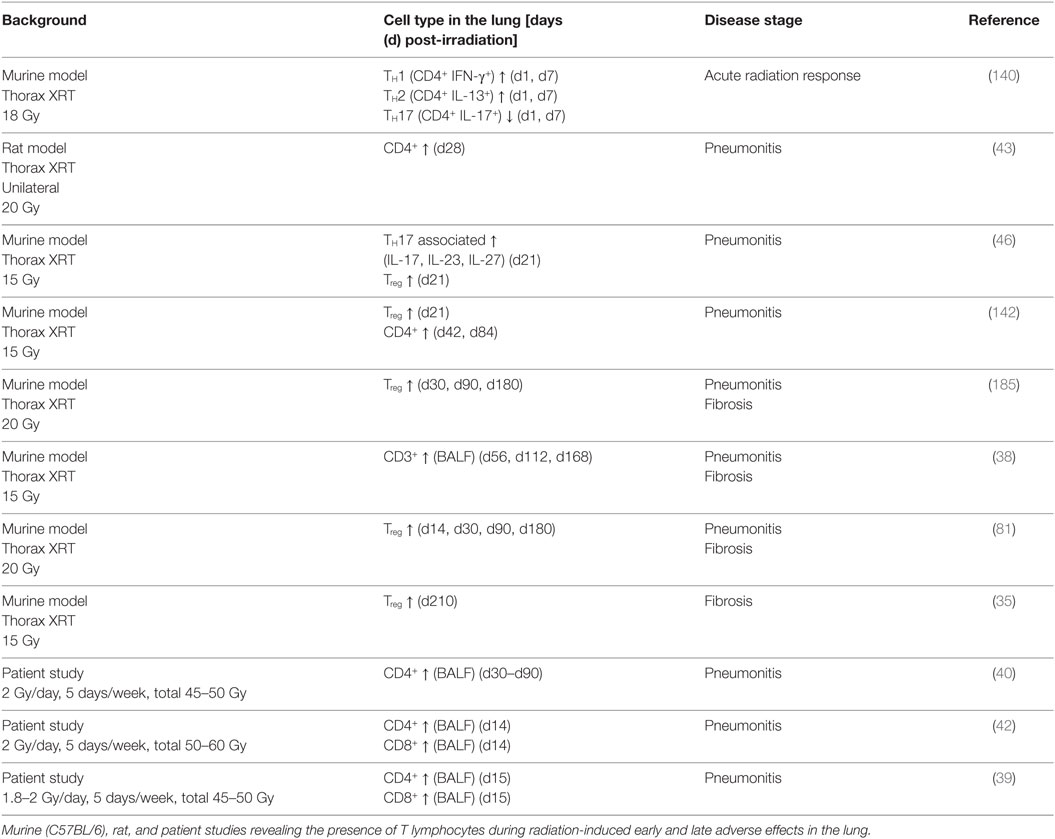
Table 1. Lymphocytes in the irradiated lung.
Altogether, these studies highlight that lymphocyte subsets differ in their rates of radiosensitivity, recovery, and infiltration during different disease stages suggesting that they may have a distinct contribution to the dynamic changes in the environment in the irradiated lung tissue.
Lymphocyte Responses to Signals from the Irradiated Lung
Damage-Associated Molecular Patterns (DAMPs)
In the past decade, several studies revealed how the immune system recognizes sterile tissue damage. Bianchi demonstrated how sterile tissue stress and damage in general led to the release of DAMPs (143). These endogenous “danger signals” induce and dictate an immune response to orchestrate repair, growth, and tissue homeostasis after damage (144, 145).
Besides this direct effect of IR, the response of the damaged resident tissue cells toward irradiation, e.g., epithelial cells, endothelial cells, or smooth muscle cells, involves a systemic “danger signal” that orchestrates immune cell recruitment and tissue repair (34). The radiation-induced oxidative injury in the lung and the release of DAMPs induce resident cells to secrete inflammatory and chemotactic cytokines. Human and murine studies revealed that DAMPs in the injured lung include among others extracellular heat shock proteins, S100 proteins, defensins, high-mobility group box-1 (146), extracellular nucleotides and nucleosides (35), as well as extracellular matrix (ECM) components like fibronectin, hyaluronan, uric acid, and surfactant proteins (37, 147). This is in line with findings from other studies dealing with lung injury, induced, for example, by smoke (148) or mechanical ventilation (149). Furthermore, animal studies with BLM-induced alveolitis revealed elevated levels of hyaluronan in the BALF and lung tissue on day 5 and was paralleled by an increased influx of polymorphonuclear leukocytes in the BALF and an interstitial–alveolar edema (150, 151).
For radiation-induced lung injury it has been described that the released DAMPs act through signaling via P2X, P2Y receptors, toll-like receptors (TLR-2 and TLR-4), receptor for advanced glycation end-products, and NOD-like receptors (NLRP3), respectively (152). Consistent with a role of TLR signaling in radiation-induced lung disease, Myd88 knockout mice displayed increased as well decreased fibrosis development depending on the type of injury induced. In a BLM-induced lung injury model, Myd88 knockout mice showed attenuated fibrosis and reduced cell infiltration in contrast to WT mice (136). In contrast, a study from Brickey et al. revealed that Myd88 was protective in irradiated lungs and that irradiated Myd88−/− mice had increased pro-fibrogenic factors and TH2 cytokines and displayed enhanced fibrosis levels at 24–27 weeks post-irradiation compared to WT mice (137).
Thus, the release of DAMPs, activators of an inflammatory cascade, leads to recruitment of inflammatory cells, leading to tissue inflammation, the so-called radiation-induced acute phase.
Cytokines/Chemokines with Impact on Lymphocyte Recruitment or Function
Moreover, thoracic irradiation triggers a rapid upregulation of the transcriptional regulator NFκB resulting in the production of pro-inflammatory cytokines (e.g., IL-6, IL-1α, IL-1β, TNF-α, and IFN-γ) within minutes to hours post-irradiation, at least at the mRNA level (34, 133, 153–155). However, as highlighted above, mouse strains differ in their cytokine profile after lung irradiation. Radiation induced distinct temporal changes in diverse cytokines in the pulmonary fibrosis-sensitive C57BL/6 mice compared to C3H mice (133). Rübe and colleagues demonstrated in a murine study that cells from the bronchiolar epithelium are a source for IL-6, TNF-α, and IL-1α in irradiated lungs (31). In line with these observations, Ao et al. described elevated IL-6 levels in the lungs of C57BL/6 model at 6 h post thorax irradiation with 12 Gy. In contrast, Paun et al. did not detect an increase in IL-6, IL-1β, IL-13, IL-17, and IFN-γ after 6 h post thorax irradiation with 18 Gy. These conflicting data highlight that the early stress response of the irradiated lung tissue requires further definition.
It is assumed that secreted mediators recruit immune cells, including neutrophils, granulocytes, macrophages, and lymphocytes, into the damaged tissue. In this context, a recent murine study described that the radiation-induced early lung inflammation was accelerated by induction of the inflammasome (Nlrp3, caspase 1, IL-1α, and IL-1β), highlighting the contribution of an early innate response (156). Exposure of lung tissue to IR triggered an increased influx of lymphocytes (30, 39, 140, 157). One of the driving forces of lymphocyte infiltration into the lung is the chemokine (C-C motif) ligand 18 (CCL18). Patients suffering from different lung diseases displayed elevated levels of CCL18, and these were associated with T cell recruitment (158–160). CCL18 is also known as pulmonary and activation-regulated chemokine; overexpression of CCL18 by intratracheal instillation of adenoviral vector AdV–CCL18 and subsequent overexpression of CCL18 in a murine BLM-induced injury model uncovered that CCL18 is highly selective for T cells and attracts these cells into the injured lung (161). Other potent chemoattractants that have been described to induce lymphocyte recruitment to a damaged lung tissue include the monocyte chemoattractant protein 1, IL-16, thymus and activation-regulated chemokine, macrophage-derived chemokine, CCL1 (I-309), CCL5 (RANTES), and stromal cell-derived factor 1 (46, 154, 162–164). In the injured tissue, the infiltrating lymphocytes get activated, start secreting diverse mediators, and finally contribute to a complex inflammatory milieu characteristic for pneumonitis.
As described above, various factors in the changing microenvironment of the irradiated lung have an impact on the response of recruited T lymphocytes that can act either in a pro- or in an anti-inflammatory way. For example, Paun et al. investigated pulmonary T helper cell populations during the acute radiation response of the lung after 18 Gy whole thorax irradiation in C57BL/6 mice and uncovered that TH1 (CD4+ IFN-γ+) and TH2 (CD4+ IL-13+) cells were increased at 1 and 7 days, but not at 6 h post-irradiation. Furthermore, they found a decrease in TH17 (CD4+ IL-17+) cells at 6 h, 1, and 7 day post-irradiation (140). Own investigations revealed the appearance of IL-17-expressing CD4+ T cells and CD4+FoxP3+ T-lymphocytes in the lung tissue of C57BL/6 mice at 21 days after whole thorax irradiation with a single high dose of 15 Gy (46).
During the early pneumonitic phase between 3 and 12 weeks post-irradiation (127) recruited T lymphocytes secrete TH1-like, pro-inflammatory TNF-α, IFN-γ, IL-2, and lymphotactin to attract and activate more immune cells to the site of damage (165). IFN-γ, for example, activates “classically activated” macrophages (M1) with high nitric-oxide synthase 2 expression but, on the other hand, shows suppressive effects on myofibroblasts and inhibits the production of ECM proteins (166, 167). Besides this classical TH1 response, a pro-inflammatory TH17-dominant response was observed after thoracic irradiation in mice. Cytokine levels of IL-16, IL-17, IL-23, and IL-27 were elevated 3 weeks post-irradiation where they might promote chronic inflammation and tissue damage (46).
The observed findings reveal that besides the early induction of cytokines like IL-1, IL-6, and TNF-α primarily lymphoid TH1 and TH17 responses contribute to the pro-inflammatory, pneumonitic phase.
Reconstitution of Lymphocytes in the Irradiated Lung
Of course, the imbalance in the hematopoietic compartment after exposure to IR needs to be restored. Interestingly, the overall recovery rate of different lymphocyte subsets varies in different organs and is chemokine dependent (168). For example, Santin et al. analyzed blood samples from irradiated patients with squamous cervical cancer: in this study CD8+ cells recovered in the blood faster than CD4+ cells over a time period of 40 days after a 5-week radiation treatment (169). This is consistent with earlier studies suggesting a prolonged reduction in lymphocyte proliferation, a persistent reduction in cell counts, or even an inversion of the CD4+/CD8+ ratio after whole body irradiation and thorax irradiation in breast cancer patients (170–173). Furthermore, in a recent study with 1,423 lung cancer patients, Yan et al. found significantly increased levels in blood CD3+ T-cells, especially CD8+ T cells, compared to CD4+ T cells in the patients 3 months after RT of the lung (174). Analysis of total lymphocytes in limited-stage small cell lung cancer (LS-SCLC) patients revealed a decrease in total lymphocytes during RT followed by recovery after the end of treatment; interestingly, the authors correlated the severity of radiation-related lymphopenia to treatment outcome revealing a potential use of radiation-related lymphopenia for prediction of poor survival in LS-SCLC (175).
Chronic Effects of Thoracic Irradiation on Lymphocytes in the Lung
TH Subsets
Substantial changes in the lung environment are observed during the chronic inflammatory and fibrotic phase in the irradiated lung. It is thought that in this context the shift from a TH1 cytokine profile toward a TH2 cytokine profile could be a key event. The signature of TH2 cytokines IL-4, IL-5, IL-10, and IL-13 is known to convey strong anti-inflammatory and pro-fibrotic effects, to mediate fibroblast activation, and activate “alternatively activated” macrophages (M2) with high arginase-1 expression (43, 166, 176, 177). Of further interest is a recent study from 2016 showing that radiation-induced lung fibrosis in a tumor-bearing mouse model was associated with enhanced type 2 immunity (178). In this study, the authors revealed that GATA3 expression in tumors appeared to affect the response to the normal lung tissue to radiation-induced damage. Lung fibrosis was more severe in tumor-bearing mice than in normal mice post-irradiation, highlighting that the enhanced type 2 immunity in tumors appeared to influence the outcome of radiation damage.
Despite the known pro-fibrotic actions of the IL-4/IL-13 axis, a recent study suggested a potential protective role for the IL-13Rα1 in a murine model of BLM-induced fibrosis; here, the authors speculated that IL-4 and/or IL-13 may act through the type 2 IL-4 receptor to regulate epithelial cell healing and immune responses to lung damage and further protection against pulmonary fibrosis (179). In line with this study, Han et al. also identified increased expression of the type 2 key transcription factor GATA3 in C57BL/6 mice that had received 12 Gy thorax irradiation (176).
Interestingly, first data about the role of the subset of TH9 cells have been obtained in a model of pneumonitis and fibrosis induced by silica particles; these suggest that IL-9 expression may reduce lung fibrosis and type 2 immune polarization (180). Taken together, until now the impact of the cytokine production of TH1, TH2, TH9, and TH17 subsets in radiation-induced pneumopathy has not been fully described and understood, but it is highly likely that TH2-driven responses are a key event of radiation-induced fibrosis development.
Up to now, there is only limited information available about the impact of the B cell compartment on the outcome of radiation-induced pneumopathy and other models of fibrotic lung disease. A recent transcriptome analysis of irradiated mouse lungs at 24 weeks after exposure to IR revealed that genes associated with B cell proliferation and activation were significantly induced in irradiated lungs of fibrosis-prone C57BL/6 mice suggesting a possible role of B cells in radiation-induced pneumopathy (181). Interestingly, deficiency in the B cell surface molecule CD19 in mice reduced the susceptibility to BLM-induced fibrosis, whereas CD19 overexpression in mice aggravated BLM-induced fibrosis (182). In contrast, B cells under the influence of IL-9 participated in the protection against lung fibrosis in a murine model of silica particle-induced lung fibrosis, and their protective effect was associated with the overexpression of prostaglandin-E2 (PGE2) in macrophages (183). Interestingly, PGE2 itself is thought to be anti-fibrotic due to its suppressing effects on fibroblast proliferation and its ability to reduce the expression of collagen mRNA (184). These controversial findings highlight the need for further studies to clarify the protective or destructive role of B cells in radiation-induced pneumopathy and the involved mediators.
TGF-β and Treg
Besides the influx of T helper lymphocytes, the Treg subset also infiltrates the irradiated lung tissue, where it is thought to suppress an exaggerated inflammation (80, 81, 142, 185). Treg can be generally induced by TGF-β (186). This cytokine is released early after tissue injury by type II pneumocytes, fibroblasts, and immune cells (187, 188), but the latent form can also be activated by radiation in vitro (189). During this early phase TGF-β is thought to function mainly as a pro-inflammatory mediator to attract neutrophils but may also provide signals for limitation of tissue inflammation, e.g., by inducing Treg (190, 191). During the later remodeling phase (24–30 weeks), macrophages can be a source for TGF-β; during this phase TGF-β is known to promote repair processes and to favor fibrosis. This might explain the observed biphasic appearance of Treg in the acute and in the chronic phase of radiation-induced lung injury described recently (35, 142, 185). Vice versa, Treg can also produce TGF-β as well as IL-10, revealing their suppressive capacity and suggesting an additional pro-fibrotic action (79, 192). In this context, it is discussed that besides TGF-β epithelial cell-derived IL-18 and IL-33—released after tissue damage—might be also important for the induction and maintenance of Treg. The activation of Treg via IL-33 and its receptor suppression of tumorigenicity 2 (ST2, also known as IL33R, IL-1RL1) lead to a TH2-like character, expressing GATA3 and secreting TH2 cytokines IL-5 and IL-13. Furthermore, IL-18- and IL-33-activated Treg showed higher suppressive capacity by enhanced activation and secretion of the anti-inflammatory and pro-fibrotic cytokines IL-10 and TGF-β as well as amphiregulin (AREG) (193–195). Thus, we speculate that IL-18/IL-33-driven Treg-activation contributes to tissue repair and a pro-fibrotic actions in the lung. Further studies are needed to confirm this in a radiation-induced lung injury model.
It has been shown that Treg contribute to fibrotic diseases in the lung such as radiation-induced, BLM-induced lung injury and IPF by modulating the microenvironment through mechanisms involving among others induction of Th17 responses, shifting the IFN-gamma, IL-12/IL-4, IL-5 balance, and promoting endothelial to mesenchymal transition (79–81, 196, 197).
Nevertheless, the role of Treg in pneumopathy remains to be further elucidated as these cells play distinct roles in different disease stages and disease models (79, 80, 192, 196, 198, 199). While depletion of CD4+CD25+ T cells with an anti-CD25 antibody during an early stage of BLM-induced lung disease reduced the levels of inflammatory cells, collagen deposition, TGF-β, and lung fibrosis in mice, Treg depletion during later stages in this model led to a more pronounced infiltration of inflammatory cells and increased fibrosis scores (196). This highlights a disease-promoting effect of CD4+CD25+ T cells during the acute phase of BLM-induced lung disease and a protective role of these cells during the fibrotic phase. By contrast, abrogation of the long-lasting (6 months) increase in CD4+CD25+ Treg observed in irradiated lungs of C57BL/6 mice by long-term CD4+CD25+ T cell depletion with an anti-CD25 antibody reduced the increase in fibrocytes and attenuated radiation-induced lung fibrosis (80). In this model, depletion of CD4+CD25+ T cells covered both, the pneumonitic and the fibrotic phase so that the two publications are not directly comparable. Nevertheless, the latter report implicates that a disease-promoting effect of CD4+CD25+ T cells is predominant in radiation-induced lung disease. To our present view, the suppressive properties of Treg are needed during the pneumonitic phase to dampen an overwhelming and excessive pro-inflammatory response that is however initially needed to induce repair and regeneration. Yet, during the chronic disease stage under the influence of a changing environment, Treg seem to adopt a pathologic character, secreting mediators like IL-10, TGF-β, and AREG, thereby contributing to a fibrosis-promoting intercellular cross talk. Our current hypothesis of the underling mechanisms is summarized in Figure 2; depending on the cell type the persistent damage caused by irradiation of resident cells will result either in a delayed partial cell loss (e.g., endothelial cells, alveolar epithelial cells) or in a chronic cell activation (e.g., MSC, fibroblasts) thereby driving chronic environmental changes (e.g., chronic increase in tissue hypoxia, adenosine, hyaluronan, macrophage-derived IL-10 and TGF-β, and epithelial-derived IL-18/IL-33) that promote among others the generation and a phenotypic adaptation of Treg. Under such conditions, the phenotype of ILC—like that of Treg cells and myeloid cells—may shift toward a disease-promoting phenotype supporting chronic lung inflammation and pulmonary fibrosis in irradiated lung tissue. It is therefore highly likely that the distinct roles of lymphocytes and Treg in BLM-induced versus radiation-induced pulmonary fibrosis may be due to differences in impact of an acute but reversible damage by the drug BLM and a chronic, persistent impact of IR on the environmental changes and associated immune changes.
The findings reported so far highlight the need for more detailed mechanistic analyses about the role of Treg for adverse late effects of IR in the lung.
Therapeutic Approaches for Radiation-Induced Pneumopathy
There is increasing evidence that lymphocytes play a role in RT-induced adverse late effects in the lung. Thus, these cells or the mediators associated with their pro- or anti-fibrotic function may constitute valuable targets for the prevention or treatment of radiation-induced lung disease. However, so far there is only little knowledge about the use of specific lymphocyte subpopulations or their mediators as potential diagnostic or predictive biomarkers for early or late adverse effects of IR in the lung. Consequently, so far treatment strategies targeting immune cells or associated mediators suspected to participate in disease pathogenesis are only tested in preclinical investigations in mice.
Instead, current treatment options for patients suffering from radiation-induced pneumonitis are limited to the symptomatic administration of anti-inflammatory drugs such as glucocorticoids thought to limit the toxic effects of the overwhelming inflammation by reducing the levels of pro-inflammatory cytokines, chemokines, and growth factors. But, these anti-inflammatory therapies may also indirectly impact on lymphocyte responses, their activation state, or both.
Current Treatment with an Impact on Lymphocyte Responses
Experimental studies in patients mostly aim or address either the impact of the treatment on radiation-induced pneumonitis or on radiation-induced lung fibrosis, respectively. However, immunomodulatory strategies will mostly influence both, early and late disease stages. For example, treatment of patients developing pneumonitis with anti-inflammatory drugs such as glucocorticoids and pentoxifylline (PTX) will also impact on the chronic radiation-induced immune changes thereby potentially influencing progression to lung fibrosis.
Due to their anti-inflammatory and immunosuppressive properties, glucocorticoids such as dexamethasone and prednisone are widely used after lung irradiation in symptomatic patients. Both drugs affect the expression of inflammation-associated genes by interaction with the steroid receptor. For example, glucocorticoid treatment involves the inhibition of the NF-κB pathway (200) as well as inhibition of the expression of IL-17A, TGF-β, IL-6, and TNF-α, thereby reducing radiation-related inflammation (201). Furthermore, dexamethasone also reduced the deposition of collagen in the lung tissue of irradiated mice (201, 202).
Pentoxifylline and alpha-tocopherol (vitamin E; Vit E) also exert anti-inflammatory actions and have already been used in patients. PTX is a xanthine derivative that acts by inhibiting TNF-α, IL-1, fibroblast growth factor, TGF-β as well as the SMAD pathway, whereas Vit E is known to counteract TGF-β and the SMAD signaling (203). In a study with 40 patients PTX was shown to provide significant protection against the early and late adverse late effects of RT in the lung (204). Furthermore, in another randomized trial study radiation-induced adverse effects were more frequent for all disease stages in the untreated control group of patients receiving irradiation alone compared to the groups where irradiation was combined with PTX and Vit E (205).
Another interesting approach that is already being explored since 1990 is the use of angiotensin-converting enzyme (ACE) inhibitors to treat radiation-induced adverse late effects in the lung (206). Interestingly, these inhibitors also interfere with immune responses, e.g., in T lymphocytes (207, 208). The membrane-bound ACE hydrolyzes a spectrum of substrates with physiologic relevance like angiotensin, bradykinin, or neurotensin (209) and is expressed in human CD4+ and CD8+ T-lymphocytes but not in B cells (210). Investigations with ACE inhibitors like captopril and ramipril in patients revealed a potential benefit in decreasing the incidence of radiation-induced pneumonitis (211–214). Unfortunately, a recent clinical study from 2016 validating the protective effect of the ACE inhibitor captopril in radiation-induced lung toxicity failed due to low accrual and a high number of patients who had to be excluded from the analysis. Nevertheless, the study confirmed safety of the ACE inhibitor treatment; the authors suggest that the use of newer ACE inhibitors (e.g., enalapril or lisinopril) during RT may be suited to solve the problems identified in their trial (215).
Other immunosuppressive agents that have been tested earlier preferentially in single case reports are cyclosporin and azathioprine. Cyclosporin is a common immunosuppressive agent that acts on CD4+ T lymphocytes by inhibiting the transcription of the interleukin-2 gene (216). Cyclosporin has already been described in 1991 as a treatment option in interstitial lung disease (217). Moreover, treatment with cyclosporine successfully reduced the symptoms of radiation-induced pneumonitis in a case study (218). Instead, case studies testing the use of azathioprine, a drug with suspected inhibitory effects on T lymphocyte formation and B lymphocyte proliferation (219, 220), revealed either a beneficial effect (221) or no effect in radiation-induced pneumonitis (222).
So far, current treatment options are limited and focus on the control of overwhelming pro-inflammatory responses during the pneumonitic phase. Furthermore, risk factors are not well understood, and predictive biomarkers are lacking. This highlights the urgent need for further preclinical and clinical studies to gain a more comprehensive understanding of the underlying mechanisms as well as the complex cellular cross talk and the mediators driving disease pathogenesis, if we aim to define predictive biomarkers and novel therapeutic approaches.
Current Research and Future Perspectives
Anti-inflammatory drugs are effectively reducing the symptoms of radiation-induced pneumonitis, and most patients completely recover from pneumonitis. However, many patients suffering from thorax-associated neoplasms, particularly lung cancer, or patients with chronic respiratory disease have an increased risk to develop lethal pneumonitis. Therefore, it is important to uncover diagnostic and prognostic biomarkers for radiation-induced lung disease, particularly lethal pneumonitis. Several patient-associated factors have been described to be associated with an increased risk to develop (severe) radiation pneumonitis such as the patient age and fitness, additional disease (e.g., tumor, COPD), as well as treatment (e.g., XRT fractions, drug administration) (223–225). Furthermore, several potential biomarkers such as pulmonary function, dose–volume histogram, end/pre-RT plasma levels of TGF-β1, IL1α, and IL6 have been described that may be suited to predict a higher risk for radiation-induced lung toxicity (226, 227). A promising novel approach to predict and understand genetic risk factors for radiation-induced toxicity may be the use of radiogenomics (228).
Despite the high efficacy of anti-inflammatory drugs in reducing the symptoms of radiation-induced pneumonitis, patients recovering from pneumonitis still have the risk of developing subsequent pulmonary complications. However, effective mechanism-based options for the treatment of pulmonary fibrosis are still limited (229). Therefore, another important topic of current research is to develop effective therapeutic strategies to prevent or treat radiation-induced lung fibrosis (223, 230); experimental approaches mostly target radiation-induced formation of free radicals, cell death, or specific cytokines or growth factors, respectively.
Importantly, a review from 2010 highlighting patient data as well as experimental studies mentioned already that “One last target that may need further investigation is that of the immune system, more specifically the alteration in immune responses…” (231). As shown in Table 1, the majority of studies in public databases identified CD4+ T cells and Treg as key populations among lymphocytes infiltrating lungs during radiation-induced pneumonitis and fibrosis. Though molecular approaches to reduce radiation-induced lung fibrosis by inhibiting pro-fibrotic mediators (e.g., TGF-β) are known to modulate lymphocyte induction/activation (232), 6 years later, our knowledge about the role of diverse lymphocytes during fibrosis is still limited. Targeted approaches to modulate lymphocyte recruitment, activation, or signaling are rare and still limited to preclinical studies in various murine models of chronic respiratory disease or fibrosis-associated disease, respectively.
One promising approach is to target the cytokine IL-17A as it has been suggested that the protective effects of anti-inflammatory drugs such as dexamethasone involve the reduction of IL-17A (233). In this context, treatment with an antibody against IL-17A reduced IL-17A, TGF-β, and IL-6 concentrations and alleviated radiation-induced pneumonitis and subsequent fibrosis in mice (233). Another interesting approach to target IL-17 might be the use of phosphodiesterase-4 inhibitors such as roflumilast that prevent the breakdown of cAMP thereby inhibiting fibroblast activation and TGF-β induction (234). In a murine model of chronic asthma, treatment with roflumilast reduced the expression of IL-17A, TNF-α, GM-CSF, and IL-6 to a similar extent as dexamethasone implying a potential use of roflumilast for the treatment of adverse events in the irradiated lung (235).
Another potential target currently in the focus of research of other diseases including cancer are Treg. As mentioned above, Treg can be induced by TGF-β and also secrete or bind TGF-β (236, 237). This suggests that targeting Treg might be suited to counteract radiation-induced adverse late effects in the lung and other diseases rich in tissue TGF-β-levels such as, skin, liver, and kidney (238–240). In support of this assumption, abrogation of the long-lasting (6 months) increase in Treg by depletion with an anti-CD25 antibody counteracted the development of radiation-induced fibrosis in mice (80) thereby corroborating findings in other fibrotic diseases (79). Still, further studies are needed to strengthen these results with respect to radiation-induced (pulmonary) fibrosis. The above findings of Xiong et al. are of particular interest because several studies highlight the potential importance of Treg depletion in enhancing antitumor immunity during RT (241, 242). One major challenge in targeting Treg will be defining the optimal treatment schedule, since Treg might also be beneficial in counteracting exaggerated inflammation during the pneumonitic phase (142).
In this context, we recently showed that the CD73/adenosine axis is a potential target in radiation-induced lung fibrosis. Lymphocytes, especially Treg, showed high CD73 expression after irradiation, and a CD73 deficiency in mice led to reduced expression of pro-fibrotic mediators like TGF-β and osteopontin during the fibrotic phase (35). Further unpublished data reveal that genetic deficiency of CD73 also precludes the accumulation of alternatively activated macrophages in prefibrotic macrophage clusters in the irradiated lung tissue (deLeve and Wirsdörfer, unpublished observations). The contribution of the CD73/adenosine pathway in fibrosis has already been described for BLM-induced pneumopathy and in other fibrotic diseases (243–245). Adenosine can bind to four different adenosine receptors to induce anti-inflammatory and pro-fibrotic actions. It is known that it inhibits lymphocyte proliferation, activation, and cytokine secretion. Furthermore, it promotes the induction and activation of Treg, highlighting its role in immunomodulation (246). In our hands, therapeutic targeting of the CD73/adenosine pathway by either enzymatic inhibition of adenosine accumulation or antibody blockade of adenosine-converting CD73 attenuated fibrosis development upon a single high-dose (15 Gy) whole thorax irradiation of C57BL/6 mice (35). The complex mechanism of the adenosine action in the pathogenesis of fibrotic diseases is not fully understood and is under current investigation. Importantly, the CD73/adenosine pathway has recently emerged as a novel immune checkpoint that tumor cells use to dampen intratumoral immune responses (247). Therefore, pharmacologic strategies for modulating CD73 or adenosine may limit radiation-induced adverse late effects presumably without increasing or even decreasing radiation resistance of tumor cells (35).
A novel potential candidate for future treatment options might be the group of ILC. ILC seem to play a critical role in lung inflammation, tissue remodeling, and fibrosis development thereby revealing a therapeutic potential of modulating ILC responses in the lung. Although it is highly likely that ILC may also impact radiation-induced lung disease, their role has not yet been investigated. Further studies are needed to clarify their contribution to acute and chronic disease stages after irradiation and to uncover a therapeutic potential in the context of ILC signaling, e.g., by targeting IL-33 or ST2. The cytokine IL-33 is important in innate and adaptive immunity and contributes to tissue homeostasis and is induced under environmental stress. IL-33 can be released by epithelial cells after tissue damage and is a trigger of tissue repair induced by ILC2 and Treg (193). Regarding lung fibrosis, it was revealed that IL-33 enhanced BLM-induced fibrosis by increasing the levels of the TH2 cytokines IL-4, IL-5, or IL-13 (103), leading to the assumption that ILC2 or TH2 cells are induced. Moreover, it was shown that treatment with an anti-IL-33 antibody attenuated BLM-induced lung inflammation and fibrosis (104). A current review nicely summarizes the role of IL-33 and ST2 in health and disease highlighting its potential for therapeutic intervention also in fibrotic lung disease (193). The observations on IL-33 activity in the lung with an impact on TH2 responses and tissue repair make it highly likely that IL-33/ST2 signaling may also impact on radiation-induced lung fibrosis. Further studies are needed to clarify the role of this signaling axis in radiation-induced pneumopathy.
Another interesting and novel therapeutic option to treat radiation-induced adverse effects in the lung with a potential interaction with lymphocytes is the therapeutic application of MSC or of microvesicles/exosomes secreted by MSC (36, 248–252). This approach is based on the initial observation that healthy resident MSC are important to lung homeostasis and protect the lungs after injury among others by immunomodulation mostly through paracrine mechanisms (253–256). However, when resident-specific endogenous MSC are damaged or lost, e.g., by differentiation into myofibroblasts, this cell population contributes to TGF-β production, tissue remodeling, and fibrosis in various models, including thoracic irradiation, exposure to BLM, and IPF (37, 257–262). In this context, impaired regulation of effector T cell proliferation upon loss of resident pulmonary MSC has been implicated in the development of BLM-induced fibrosis (257).
Instead exogenously applied MSC may exert tissue protective effects by differentiating into an epithelium-like phenotype and replacing damaged cells, although our own data hint to a minor contribution of this effect to their protective effects (36, 263). Interestingly, recent findings highlight the ability of MSC to transfer (healthy) organelles or molecules by direct cell-to-cell contact through tunneling nanotubes or by the release of exosomes or microvesicles, respectively (255).
Several reports including own studies revealed that MSC show anti-fibrotic and protective effects in the irradiated lung, and current reviews highlight a potential therapeutic benefit of MSC therapy for the treatment of radiation-induced and BLM-induced tissue damage (36, 37, 248, 263). In our hands, adoptively transferred MSC normalized certain aspects of radiation-induced immune deviation in the lung tissue, normalized vascular function, and attenuated radiation-induced pulmonary fibrosis (36, 37). We and others showed that the anti-inflammatory and anti-fibrotic action of MSC is mediated by the inhibition of TNF-a, IL-1alpha, and interleukin 1 receptor antagonist (249, 251), stimulating the secretion of hepatocyte growth factor (HGF) and PGE2 (250, 252) and restoration of the superoxide dismutase 1 expression (37).
Generally, MSC might also exert protective effects during pneumonitis and fibrosis development due to their antiproliferative effects (253) and their suppressive capacity on innate and adaptive immune responses (254). The suppressive capacity on lymphocytes is mediated by the secretion of soluble factors like HGF, PGE2, truncated CCL-2, IL-10, and PD-1 ligation thereby inhibiting CD4+ T cell proliferation and the polarization toward a TH1 and TH17 phenotype (254). Due to a potential inhibition of TH1 and TH17 cells during radiation-induced pneumonitis, MSC might dampen an excessive immune response. This effect may be complemented by the ability of MSC to favor the development of a TH2 phenotype and Treg (254, 264). This effect could be beneficial during the pneumonitic phase but may be disadvantageous during the fibrotic phase. We speculate that by dampening tissue inflammation and remodeling and restoration of resident cell function, MSC limit resident cell loss/dysfunction, chronic inflammation, and fibrosis-promoting environmental changes. In this context, others and we described that MSC treatment has protective effects in the lung due to a transdifferentiation into an epithelium-like phenotype (263) and by the protection from endothelial cell loss (37), respectively.
Finally, another experimental approach to reduce DNA damage-induced late effects in the lung with an impact on lymphocytes is the use of the lysophosphatidic acid receptor (LPA1) antagonist AM966: treatment with AM966 revealed reduced lung injury, vascular leakage, lymphocyte recruitment, and fibrosis development at 14 days after BLM treatment (265). However, this drug has not yet been tested in radiation-induced lung disease.
Nevertheless, current studies investigating the role of lymphocytes and their inducers and mediators as therapeutic targets in radiation-induced adverse effects in the lung are still rare. This might be due to insufficient knowledge on the beneficial and adverse role of specific lymphocytes subsets in different stages of disease pathogenesis.
Final Remarks
Up to now, mechanistic knowledge about the role of lymphocytes in radiation-induced pneumopathy is still limited, and no reliable diagnostic or predictive biomarkers for beneficial and adverse effects of specific lymphocyte subsets are available to date. Nevertheless, preclinical and clinical investigations indicate that radiation-induced immune changes are important to the outcome of RT and that lymphocytes contribute to the beneficial and adverse effects of IR in tumors and normal tissues, including the lung.
We hypothesize that radiation-induced acute damage to resident cells including progenitor cells of mesenchymal origin and a perpetual cascade of cytokines/chemokines triggers immune cell recruitment and activation to promote tissue repair. However, in cases where the immune response cannot be controlled by anti-inflammatory cells such as Treg or M2-like macrophages, patients may develop pneumonitis. If this initial damage response is not sufficient to repair the radiation-induced damage, the persistent damage results in chronic inflammation and delayed changes of resident cells, such as epithelial-mesenchymal-transition, endothelial-mesenchymal-transition, activation of MSC, or even chronic (mitotic) cell death of endothelial cells and alveolar epithelial cells. The resulting delayed environmental changes involve among others tissue hypoxia (by chronic endothelial cell loss), chronic inflammation, and chronic increase in fibroblasts and fibrosis-promoting mediators such as adenosine, hyaluronic acid, and TGFβ. These changes act together in the generation/activation of disease-promoting cell phenotypes such as activated myofibroblasts, pathologic Treg, or M2-like macrophages thereby promoting exaggerated ECM deposition and fibrosis development (Figure 2).
Here, we want to stress that radiation-induced immune changes can be either pro-inflammatory (acute phase) or anti-inflammatory/pro-fibrotic (chronic phase), and that lymphocytes exert distinct functions during radiation-induced pneumopathy (see Figure 1). We speculate that Treg might be beneficial during radiation-induced pneumonitis due to their suppressive action on pro-inflammatory cells as seen in hypersensitivity pneumonitis (266). In contrast, as outlined above current preclinical studies in diverse murine fibrosis models highlight a potential contribution of Treg in fibrosis development, and own work supports such a disease-promoting role also for radiation-induced pulmonary fibrosis (35). We therefore assume that these immunosuppressive cells or the environmental factors promoting their recruitment/expansion may constitute promising therapeutic targets to prevent or treat radiation-induced fibrosis. We are aware that the use of specific lymphocyte populations or associated signaling molecules as therapeutic targets is complicated by the fact these cells exert either beneficial or harmful roles. This depends on the disease stage, and potentially patient-specific genetic factors. Consequently this requires the careful definition of optimal treatment schedules to avoid immune cell-associated complications during both, pneumonitis and fibrosis. We also want to point out that recently discovered lymphocyte subsets like ILC as well as signaling pathways like IL-33/ST2 are of interest due to their potential contribution to RT-induced pneumopathy highlighting the need of related studies.
Further important issues that also need to be addressed are (i) the potential high intrinsic radioresistance of thorax-associated solid tumors treated by thoracic RT and (ii) a potential tumor immune escape. Therefore, it is important to consider that any inflammation-modulating or immune cell-targeting strategy for the treatment of radiation-induced pneumopathy may alter the antitumor effect of RT or combined treatment strategies involving immunomodulation or immunoboost; unfortunately, this issue is mostly not addressed by preclinical studies investigating the mechanisms of radiation-induced normal tissue toxicity. Vice versa, a potential increased normal tissue toxicity of such antitumor treatments is not analyzed when studying new combination treatments in preclinical models. Therefore, there is a high need to develop appropriate preclinical models if we want to identify treatment strategies balancing radiation-induced tumor cell clearance and normal tissue protection. Nevertheless, we are convinced that a better understanding of radiation-induced immunomodulation in tumors and normal tissues will offer novel opportunities for widening the therapeutic window by targeting immune cells or immune-associated mediators that promote both, tumor growth/resistance and normal tissue toxicity—of these, TGF-β and adenosine constitute perfect examples.
Taken together, it is important to deepen our knowledge about radiation-induced immune changes, including the modulation of lymphocyte recruitment, proliferation, and/or function of specific lymphocyte subsets during the different stages of radiation-induced lung disease. Further studies are needed to optimize therapeutic strategies for the prevention or treatment of adverse late effects of IR to normal tissues that also take into account the immune repertoire of the respective malignant disease before and after RT, if we aim at protecting the normal tissue without promoting tumor growth and vice versa.
Author Contributions
FW and VJ equally contributed to the writing of this review article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank S. de Leve for the careful revision of the manuscript.
Funding
The work was supported by grants of the DFG (GRK1739/1; JE275/1) and the BMBF (ZISS 02NUK024-D).
References
1. Bartelink H, Rubens RD, van der Schueren E, Sylvester R. Hormonal therapy prolongs survival in irradiated locally advanced breast cancer: a European organization for research and treatment of cancer randomized phase III trial. J Clin Oncol (1997) 15(1):207–15.
2. Bartelink H, Roelofsen F, Eschwege F, Rougier P, Bosset JF, Gonzalez DG, et al. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: results of a phase III randomized trial of the European organization for research and treatment of cancer radiotherapy and gastrointestinal cooperative groups. J Clin Oncol (1997) 15(5):2040–9.
3. Blanchard P, Baujat B, Holostenco V, Bourredjem A, Baey C, Bourhis J, et al. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): a comprehensive analysis by tumour site. Radiother Oncol (2011) 100(1):33–40. doi:10.1016/j.radonc.2011.05.036
4. Bosset JF, Collette L, Calais G, Mineur L, Maingon P, Radosevic-Jelic L, et al. Chemotherapy with preoperative radiotherapy in rectal cancer. N Engl J Med (2006) 355(11):1114–23. doi:10.1056/NEJMoa060829
5. Green JA, Kirwan JM, Tierney JF, Symonds P, Fresco L, Collingwood M, et al. Survival and recurrence after concomitant chemotherapy and radiotherapy for cancer of the uterine cervix: a systematic review and meta-analysis. Lancet (2001) 358(9284):781–6. doi:10.1016/S0140-6736(01)05965-7
6. Herskovic A, Martz K, al-Sarraf M, Leichman L, Brindle J, Vaitkevicius V, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med (1992) 326(24):1593–8. doi:10.1056/NEJM199206113262403
7. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi:10.1056/NEJMoa043330
8. Auperin A, Le Pechoux C, Rolland E, Curran WJ, Furuse K, Fournel P, et al. Meta-analysis of concomitant versus sequential radiochemotherapy in locally advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(13):2181–90. doi:10.1200/JCO.2009.26.2543
9. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer (2008) 8(7):545–54. doi:10.1038/nrc2419
10. Yu VY, Nguyen D, Pajonk F, Kupelian P, Kaprealian T, Selch M, et al. Incorporating cancer stem cells in radiation therapy treatment response modeling and the implication in glioblastoma multiforme treatment resistance. Int J Radiat Oncol Biol Phys (2015) 91(4):866–75. doi:10.1016/j.ijrobp.2014.12.004
11. Leder K, Pitter K, Laplant Q, Hambardzumyan D, Ross BD, Chan TA, et al. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell (2014) 156(3):603–16. doi:10.1016/j.cell.2013.12.029
12. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366(10):883–92. doi:10.1056/NEJMoa1113205
13. Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer (2013) 13(5):365–76. doi:10.1038/nrc3498
14. Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer (2005) 5(7):516–25. doi:10.1038/nrc1650
15. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer (2011) 11(4):239–53. doi:10.1038/nrc3007
16. Herman JM, Narang AK, Griffith KA, Zalupski MM, Reese JB, Gearhart SL, et al. The quality-of-life effects of neoadjuvant chemoradiation in locally advanced rectal cancer. Int J Radiat Oncol Biol Phys (2013) 85(1):e15–9. doi:10.1016/j.ijrobp.2012.09.006
17. Budach W, Hehr T, Budach V, Belka C, Dietz K. A meta-analysis of hyperfractionated and accelerated radiotherapy and combined chemotherapy and radiotherapy regimens in unresected locally advanced squamous cell carcinoma of the head and neck. BMC Cancer (2006) 6:28. doi:10.1186/1471-2407-6-28
18. Morgan MA, Parsels LA, Maybaum J, Lawrence TS. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov (2014) 4(3):280–91. doi:10.1158/2159-8290.CD-13-0337
19. Tsoutsou PG, Koukourakis MI. Radiation pneumonitis and fibrosis: mechanisms underlying its pathogenesis and implications for future research. Int J Radiat Oncol Biol Phys (2006) 66(5):1281–93. doi:10.1016/j.ijrobp.2006.08.058
20. Kong FM, Ten Haken R, Eisbruch A, Lawrence TS. Non-small cell lung cancer therapy-related pulmonary toxicity: an update on radiation pneumonitis and fibrosis. Semin Oncol (2005) 32(2 Suppl 3):S42–54. doi:10.1053/j.seminoncol.2005.03.009
21. Kelsey CR, Horwitz ME, Chino JP, Craciunescu O, Steffey B, Folz RJ, et al. Severe pulmonary toxicity after myeloablative conditioning using total body irradiation: an assessment of risk factors. Int J Radiat Oncol Biol Phys (2011) 81(3):812–8. doi:10.1016/j.ijrobp.2010.06.058
22. McDonald S, Rubin P, Phillips TL, Marks LB. Injury to the lung from cancer therapy: clinical syndromes, measurable endpoints, and potential scoring systems. Int J Radiat Oncol Biol Phys (1995) 31(5):1187–203. doi:10.1016/0360-3016(94)00429-O
23. Provatopoulou X, Athanasiou E, Gounaris A. Predictive markers of radiation pneumonitis. Anticancer Res (2008) 28(4C):2421–32.
24. Graves PR, Siddiqui F, Anscher MS, Movsas B. Radiation pulmonary toxicity: from mechanisms to management. Semin Radiat Oncol (2010) 20(3):201–7. doi:10.1016/j.semradonc.2010.01.010
25. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med (2012) 18(7):1028–40. doi:10.1038/nm.2807
26. Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med (2005) 201(6):925–35. doi:10.1084/jem.20041393
27. Arpin D, Perol D, Blay JY, Falchero L, Claude L, Vuillermoz-Blas S, et al. Early variations of circulating interleukin-6 and interleukin-10 levels during thoracic radiotherapy are predictive for radiation pneumonitis. J Clin Oncol (2005) 23(34):8748–56. doi:10.1200/JCO.2005.01.7145
28. Kasper M, Haroske G. Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis. Histol Histopathol (1996) 11(2):463–83.
29. Penney DP, Siemann DW, Rubin P, Maltby K. Morphological correlates of fractionated radiation of the mouse lung: early and late effects. Int J Radiat Oncol Biol Phys (1994) 29(4):789–804. doi:10.1016/0360-3016(94)90568-1
30. Roberts CM, Foulcher E, Zaunders JJ, Bryant DH, Freund J, Cairns D, et al. Radiation pneumonitis: a possible lymphocyte-mediated hypersensitivity reaction. Ann Intern Med (1993) 118(9):696–700. doi:10.7326/0003-4819-118-9-199305010-00006
31. Rübe CE, Uthe D, Wilfert F, Ludwig D, Yang K, Konig J, et al. The bronchiolar epithelium as a prominent source of pro-inflammatory cytokines after lung irradiation. Int J Radiat Oncol Biol Phys (2005) 61(5):1482–92. doi:10.1016/j.ijrobp.2004.12.072
32. Trott KR, Herrmann T, Kasper M. Target cells in radiation pneumopathy. Int J Radiat Oncol Biol Phys (2004) 58(2):463–9. doi:10.1016/j.ijrobp.2003.09.045
33. Rubin P, Johnston CJ, Williams JP, McDonald S, Finkelstein JN. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys (1995) 33(1):99–109. doi:10.1016/0360-3016(95)00095-G
34. McBride WH, Chiang CS, Olson JL, Wang CC, Hong JH, Pajonk F, et al. A sense of danger from radiation. Radiat Res (2004) 162(1):1–19. doi:10.1667/RR3196
35. Wirsdörfer F, de Leve S, Cappuccini F, Eldh T, Meyer AV, Gau E, et al. Extracellular adenosine production by ecto-5′-nucleotidase (CD73) enhances radiation-induced lung fibrosis. Cancer Res (2016) 76(10):3045–56. doi:10.1158/0008-5472.CAN-15-2310
36. Klein D, Schmetter A, Imsak R, Wirsdorfer F, Unger K, Jastrow H, et al. Therapy with multipotent mesenchymal stromal cells protects lungs from radiation-induced injury and reduces the risk of lung metastasis. Antioxid Redox Signal (2016) 24(2):53–69. doi:10.1089/ars.2014.6183
37. Klein D, Steens J, Wiesemann A, Schulz F, Kaschani F, Rock K, et al. Mesenchymal stem cell therapy protects lungs from radiation-induced endothelial cell loss by restoring superoxide dismutase 1 expression. Antioxid Redox Signal (2016). doi:10.1089/ars.2016.6748
38. Johnston CJ, Williams JP, Elder A, Hernady E, Finkelstein JN. Inflammatory cell recruitment following thoracic irradiation. Exp Lung Res (2004) 30(5):369–82. doi:10.1080/01902140490438915
39. Martin C, Romero S, Sanchez-Paya J, Massuti B, Arriero JM, Hernandez L. Bilateral lymphocytic alveolitis: a common reaction after unilateral thoracic irradiation. Eur Respir J (1999) 13(4):727–32. doi:10.1034/j.1399-3003.1999.13d05.x
40. Toma CL, Serbescu A, Alexe M, Cervis L, Ionita D, Bogdan MA. The bronchoalveolar lavage pattern in radiation pneumonitis secondary to radiotherapy for breast cancer. Maedica (Buchar) (2010) 5(4):250–7.
41. Morgan GW, Breit SN. Radiation and the lung: a reevaluation of the mechanisms mediating pulmonary injury. Int J Radiat Oncol Biol Phys (1995) 31(2):361–9. doi:10.1016/0360-3016(94)00477-3
42. Nakayama Y, Makino S, Fukuda Y, Min KY, Shimizu A, Ohsawa N. Activation of lavage lymphocytes in lung injuries caused by radiotherapy for lung cancer. Int J Radiat Oncol Biol Phys (1996) 34(2):459–67. doi:10.1016/0360-3016(95)02101-9
43. Westermann W, Schobl R, Rieber EP, Frank KH. Th2 cells as effectors in postirradiation pulmonary damage preceding fibrosis in the rat. Int J Radiat Biol (1999) 75(5):629–38. doi:10.1080/095530099140276
44. Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med (1989) 170(3):655–63. doi:10.1084/jem.170.3.655
45. Sharma SK, MacLean JA, Pinto C, Kradin RL. The effect of an anti-CD3 monoclonal antibody on bleomycin-induced lymphokine production and lung injury. Am J Respir Crit Care Med (1996) 154(1):193–200. doi:10.1164/ajrccm.154.1.8680680
46. Cappuccini F, Eldh T, Bruder D, Gereke M, Jastrow H, Schulze-Osthoff K, et al. New insights into the molecular pathology of radiation-induced pneumopathy. Radiother Oncol (2011) 101(1):86–92. doi:10.1016/j.radonc.2011.05.064
47. Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair (2012) 5(1):11. doi:10.1186/1755-1536-5-11
48. Youngblood B, Davis CW, Ahmed R. Making memories that last a lifetime: heritable functions of self-renewing memory CD8 T cells. Int Immunol (2010) 22(10):797–803. doi:10.1093/intimm/dxq437
49. Born WK, Jin N, Aydintug MK, Wands JM, French JD, Roark CL, et al. Gammadelta T lymphocytes-selectable cells within the innate system? J Clin Immunol (2007) 27(2):133–44. doi:10.1007/s10875-007-9077-z
50. Larosa DF, Orange JS. 1. Lymphocytes. J Allergy Clin Immunol (2008) 121(2 Suppl):S364–9. doi:10.1016/j.jaci.2007.06.016
51. Holtmeier W, Kabelitz D. Gammadelta T cells link innate and adaptive immune responses. Chem Immunol Allergy (2005) 86:151–83. doi:10.1159/000086659
52. Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol (2012) 24(3):297–302. doi:10.1016/j.coi.2012.01.014
53. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi:10.1038/ni1254
54. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
55. Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol (2004) 4(3):231–7. doi:10.1038/nri1309
56. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med (2000) 192(11):1553–62. doi:10.1084/jem.192.11.1553
57. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest (2009) 119(12):3573–85. doi:10.1172/JCI40202
58. Stassen M, Schmitt E, Bopp T. From interleukin-9 to T helper 9 cells. Ann N Y Acad Sci (2012) 1247:56–68. doi:10.1111/j.1749-6632.2011.06351.x
60. Homey B. [After TH1/TH2 now comes Treg/TH17: significance of T helper cells in immune response organization]. Hautarzt (2006) 57(8):730–2. doi:10.1007/s00105-006-1199-3
61. Chen Z, O’Shea JJ. Regulation of IL-17 production in human lymphocytes. Cytokine (2008) 41(2):71–8. doi:10.1016/j.cyto.2007.09.009
62. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity (2006) 24(6):677–88. doi:10.1016/j.immuni.2006.06.002
63. Noma T. [Helper T cell paradigm: Th17 and regulatory T cells involved in autoimmune inflammatory disorders, pathogen defense and allergic diseases]. Nihon Rinsho Men’eki Gakkai kaishi (2010) 33(5):262–71. doi:10.2177/jsci.33.262
64. Hartigan-O’Connor DJ, Poon C, Sinclair E, McCune JM. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods (2007) 319(1–2):41–52. doi:10.1016/j.jim.2006.10.008
65. Churlaud G, Pitoiset F, Jebbawi F, Lorenzon R, Bellier B, Rosenzwajg M, et al. Human and mouse CD8(+)CD25(+)FOXP3(+) regulatory T cells at steady state and during interleukin-2 therapy. Front Immunol (2015) 6:171. doi:10.3389/fimmu.2015.00171
66. Drolet JP, Frangie H, Guay J, Hajoui O, Hamid Q, Mazer BD. B lymphocytes in inflammatory airway diseases. Clin Exp Allergy (2010) 40(6):841–9. doi:10.1111/j.1365-2222.2010.03512.x
67. Tussiwand R, Bosco N, Ceredig R, Rolink AG. Tolerance checkpoints in B-cell development: Johnny B good. Eur J Immunol (2009) 39(9):2317–24. doi:10.1002/eji.200939633
68. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi:10.1146/annurev-immunol-020711-074934
69. Shimabukuro-Vornhagen A, Draube A, Liebig T, Popov A, Rothe A, von Bergwelt-Baildon M. The properties of human CD40-activated B cells as antigen-presenting cells are not affected by PGE2. Oncol Rep (2013) 29(3):1061–5. doi:10.3892/or.2012.2215
70. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells – how did we miss them? Nat Rev Immunol (2013) 13(2):75–87. doi:10.1038/nri3349
71. Gasteiger G, Rudensky AY. Interactions between innate and adaptive lymphocytes. Nat Rev Immunol (2014) 14(9):631–9. doi:10.1038/nri3726
72. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science (2015) 348(6237):aaa6566. doi:10.1126/science.aaa6566
73. Ishizuka IE, Constantinides MG, Gudjonson H, Bendelac A. The innate lymphoid cell precursor. Annu Rev Immunol (2016) 34:299–316. doi:10.1146/annurev-immunol-041015-055549
74. Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol (2008) 8(2):142–52. doi:10.1038/nri2236
75. Hamada H, Bassity E, Flies A, Strutt TM, Garcia-Hernandez Mde L, McKinstry KK, et al. Multiple redundant effector mechanisms of CD8+ T cells protect against influenza infection. J Immunol (2013) 190(1):296–306. doi:10.4049/jimmunol.1200571
76. Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med (2007) 13(2):139–45. doi:10.1038/nm1551
77. Duan MC, Han W, Jin PW, Wei YP, Wei Q, Zhang LM, et al. Disturbed Th17/treg balance in patients with non-small cell lung cancer. Inflammation (2015) 38(6):2156–65. doi:10.1007/s10753-015-0198-x
78. Umetsu DT, DeKruyff RH. The regulation of allergy and asthma. Immunol Rev (2006) 212:238–55. doi:10.1111/j.0105-2896.2006.00413.x
79. Liu F, Liu J, Weng D, Chen Y, Song L, He Q, et al. CD4+CD25+Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS One (2010) 5(11):e15404. doi:10.1371/journal.pone.0015404
80. Xiong S, Guo R, Yang Z, Xu L, Du L, Li R, et al. Treg depletion attenuates irradiation-induced pulmonary fibrosis by reducing fibrocyte accumulation, inducing Th17 response, and shifting IFN-gamma, IL-12/IL-4, IL-5 balance. Immunobiology (2015) 220(11):1284–91. doi:10.1016/j.imbio.2015.07.001
81. Xiong S, Pan X, Xu L, Yang Z, Guo R, Gu Y, et al. Regulatory T cells promote beta-catenin – mediated epithelium-to-mesenchyme transition during radiation-induced pulmonary fibrosis. Int J Radiat Oncol Biol Phys (2015) 93(2):425–35. doi:10.1016/j.ijrobp.2015.05.043
82. Wands JM, Roark CL, Aydintug MK, Jin N, Hahn YS, Cook L, et al. Distribution and leukocyte contacts of gammadelta T cells in the lung. J Leukoc Biol (2005) 78(5):1086–96. doi:10.1189/jlb.0505244
83. Nanno M, Shiohara T, Yamamoto H, Kawakami K, Ishikawa H. Gammadelta T cells: firefighters or fire boosters in the front lines of inflammatory responses. Immunol Rev (2007) 215:103–13. doi:10.1111/j.1600-065X.2006.00474.x
84. Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP. Gammadelta T cells protect against lung fibrosis via IL-22. J Exp Med (2010) 207(10):2239–53. doi:10.1084/jem.20100061
85. Boyden AW, Legge KL, Waldschmidt TJ. Pulmonary infection with influenza A virus induces site-specific germinal center and T follicular helper cell responses. PLoS One (2012) 7(7):e40733. doi:10.1371/journal.pone.0040733
86. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14(3):275–81. doi:10.1038/nm1710
87. Ying X, Su Z, Bie Q, Zhang P, Yang H, Wu Y, et al. Synergistically increased ILC2 and Th9 cells in lung tissue jointly promote the pathological process of asthma in mice. Mol Med Rep (2016) 13(6):5230–40. doi:10.3892/mmr.2016.5174
88. Lund FE, Hollifield M, Schuer K, Lines JL, Randall TD, Garvy BA. B cells are required for generation of protective effector and memory CD4 cells in response to pneumocystis lung infection. J Immunol (2006) 176(10):6147–54. doi:10.4049/jimmunol.176.10.6147
89. Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, et al. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc Natl Acad Sci U S A (2012) 109(7):2485–90. doi:10.1073/pnas.1115369109
90. Monticelli LA, Sonnenberg GF, Artis D. Innate lymphoid cells: critical regulators of allergic inflammation and tissue repair in the lung. Curr Opin Immunol (2012) 24(3):284–9. doi:10.1016/j.coi.2012.03.012
91. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi:10.1031/ni.2131
92. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol (2012) 129(1):e1–6. doi:10.1016/j.jaci.2011.10.036
93. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol (2011) 12(7):631–8. doi:10.1038/ni.2045
94. Marashian SM, Mortaz E, Jamaati HR, Alavi-Moghaddam M, Kiani A, Abedini A, et al. Role of innate lymphoid cells in lung disease. Iran J Allergy Asthma Immunol (2015) 14(4):346–60.
95. Lai DM, Shu Q, Fan J. The origin and role of innate lymphoid cells in the lung. Mil Med Res (2016) 3:25. doi:10.1186/s40779-016-0093-2
96. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350(6263):981–5. doi:10.1126/science.aac9593
97. Li BW, Hendriks RW. Group 2 innate lymphoid cells in lung inflammation. Immunology (2013) 140(3):281–7. doi:10.1111/imm.12153
98. De Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS One (2016) 11(1):e0145961. doi:10.1371/journal.pone.0145961
99. Muir R, Osbourn M, Dubois AV, Doran E, Small DM, Monahan A, et al. Innate lymphoid cells are the predominant source of IL-17A during the early pathogenesis of acute respiratory distress syndrome. Am J Respir Crit Care Med (2016) 193(4):407–16. doi:10.1164/rccm.201410-1782OC
100. Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol (2015) 136(6):1653–63.e1–7. doi:10.1016/j.jaci.2015.03.043
101. Denney L, Byrne AJ, Shea TJ, Buckley JS, Pease JE, Herledan GM, et al. Pulmonary epithelial cell-derived cytokine TGF-beta1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity (2015) 43(5):945–58. doi:10.1016/j.immuni.2015.10.012
102. Zhang Y, Tang J, Tian Z, van Velkinburgh JC, Song J, Wu Y, et al. Innate lymphoid cells: a promising new regulator in fibrotic diseases. Int Rev Immunol (2015) 19:1–16. doi:10.3109/08830185.2015.1068304
103. Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP, et al. Interleukin-33 potentiates bleomycin-induced lung injury. Am J Respir Cell Mol Biol (2013) 49(6):999–1008. doi:10.1165/rcmb.2013-0093OC
104. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol (2014) 134(6):1422.e–32.e. doi:10.1016/j.jaci.2014.05.011
105. Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A (2014) 111(1):367–72. doi:10.1073/pnas.1315854111
106. Hams E, Bermingham R, Fallon PG. Macrophage and innate lymphoid cell interplay in the genesis of fibrosis. Front Immunol (2015) 6:597. doi:10.3389/fimmu.2015.00597
107. Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol (1994) 65(1):7–17. doi:10.1080/09553009414550021
108. Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol (1994) 65(1):27–33. doi:10.1080/09553009414550041
109. Dainiak N. Hematologic consequences of exposure to ionizing radiation. Exp Hematol (2002) 30(6):513–28. doi:10.1016/S0301-472X(02)00802-0
110. Belka C, Ottinger H, Kreuzfelder E, Weinmann M, Lindemann M, Lepple-Wienhues A, et al. Impact of localized radiotherapy on blood immune cells counts and function in humans. Radiother Oncol (1999) 50(2):199–204. doi:10.1016/S0167-8140(98)00130-3
111. Yao Z, Jones J, Kohrt H, Strober S. Selective resistance of CD44hi T cells to p53-dependent cell death results in persistence of immunologic memory after total body irradiation. J Immunol (2011) 187(8):4100–8. doi:10.4049/jimmunol.1101141
112. Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys (2011) 81(4):1128–35. doi:10.1016/j.ijrobp.2010.09.034
113. Qu Y, Jin S, Zhang A, Zhang B, Shi X, Wang J, et al. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat Res (2010) 173(2):148–57. doi:10.1667/RR0978.1
114. Bogdandi EN, Balogh A, Felgyinszki N, Szatmari T, Persa E, Hildebrandt G, et al. Effects of low-dose radiation on the immune system of mice after total-body irradiation. Radiat Res (2010) 174(4):480–9. doi:10.1667/RR2160.1
115. DeKruyff RH, Fang Y, Umetsu DT. IL-4-based helper activity of CD4+ T cells is radiation sensitive. Cell Immunol (1995) 160(2):248–56. doi:10.1016/0008-8749(95)80035-H
116. Chua ML, Rothkamm K. Biomarkers of radiation exposure: can they predict normal tissue radiosensitivity? Clin Oncol (R Coll Radiol) (2013) 25(10):610–6. doi:10.1016/j.clon.2013.06.010
117. Vandevoorde C, Depuydt J, Veldeman L, De Neve W, Sebastia N, Wieme G, et al. In vitro cellular radiosensitivity in relationship to late normal tissue reactions in breast cancer patients: a multi-endpoint case-control study. Int J Radiat Biol (2016) 92(12):823–36. doi:10.1080/09553002.2016.1230238
118. Geara FB, Peters LJ, Ang KK, Wike JL, Brock WA. Prospective comparison of in vitro normal cell radiosensitivity and normal tissue reactions in radiotherapy patients. Int J Radiat Oncol Biol Phys (1993) 27(5):1173–9. doi:10.1016/0360-3016(93)90540-C
119. Ozsahin M, Crompton NE, Gourgou S, Kramar A, Li L, Shi Y, et al. CD4 and CD8 T-lymphocyte apoptosis can predict radiation-induced late toxicity: a prospective study in 399 patients. Clin Cancer Res (2005) 11(20):7426–33. doi:10.1158/1078-0432.CCR-04-2634
120. Mori M, Benotmane MA, Tirone I, Hooghe-Peters EL, Desaintes C. Transcriptional response to ionizing radiation in lymphocyte subsets. Cell Mol Life Sci (2005) 62(13):1489–501. doi:10.1007/s00018-005-5086-3
121. Gruel G, Voisin P, Vaurijoux A, Roch-Lefevre S, Gregoire E, Maltere P, et al. Broad modulation of gene expression in CD4+ lymphocyte subpopulations in response to low doses of ionizing radiation. Radiat Res (2008) 170(3):335–44. doi:10.1667/RR1147.1
122. Gridley DS, Rizvi A, Luo-Owen X, Makinde AY, Pecaut MJ. Low dose, low dose rate photon radiation modifies leukocyte distribution and gene expression in CD4(+) T cells. J Radiat Res (2009) 50(2):139–50. doi:10.1269/jrr.08095
123. Vatner RE, Cooper BT, Vanpouille-Box C, Demaria S, Formenti SC. Combinations of immunotherapy and radiation in cancer therapy. Front Oncol (2014) 4:325. doi:10.3389/fonc.2014.00325
124. Balermpas P, Michel Y, Wagenblast J, Seitz O, Weiss C, Rodel F, et al. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br J Cancer (2014) 110(2):501–9. doi:10.1038/bjc.2013.640
125. Schaue D, McBride WH. T lymphocytes and normal tissue responses to radiation. Front Oncol (2012) 2:119. doi:10.3389/fonc.2012.00119
126. Travis EL, Vojnovic B, Davies EE, Hirst DG. A plethysmographic method for measuring function in locally irradiated mouse lung. Br J Radiol (1979) 52(613):67–74. doi:10.1259/0007-1285-52-613-67
127. Eldh T, Heinzelmann F, Velalakan A, Budach W, Belka C, Jendrossek V. Radiation-induced changes in breathing frequency and lung histology of C57BL/6J mice are time- and dose-dependent. Strahlenther Onkol (2012) 188(3):274–81. doi:10.1007/s00066-011-0046-3
128. Chiang CS, Liu WC, Jung SM, Chen FH, Wu CR, McBride WH, et al. Compartmental responses after thoracic irradiation of mice: strain differences. Int J Radiat Oncol Biol Phys (2005) 62(3):862–71. doi:10.1016/j.ijrobp.2005.02.037
129. Flechsig P, Dadrich M, Bickelhaupt S, Jenne J, Hauser K, Timke C, et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-beta and BMP-associated proinflammatory and proangiogenic signals. Clin Cancer Res (2012) 18(13):3616–27. doi:10.1158/1078-0432.CCR-11-2855
130. Mangoni M, Vozenin MC, Biti G, Deutsch E. Normal tissues toxicities triggered by combined anti-angiogenic and radiation therapies: hurdles might be ahead. Br J Cancer (2012) 107(2):308–14. doi:10.1038/bjc.2012.236
131. Jackson IL, Vujaskovic Z, Down JD. A further comparison of pathologies after thoracic irradiation among different mouse strains: finding the best preclinical model for evaluating therapies directed against radiation-induced lung damage. Radiat Res (2011) 175(4):510–8. doi:10.1667/RR2421.1
132. Walkin L, Herrick SE, Summers A, Brenchley PE, Hoff CM, Korstanje R, et al. The role of mouse strain differences in the susceptibility to fibrosis: a systematic review. Fibrogenesis Tissue Repair (2013) 6(1):18. doi:10.1186/1755-1536-6-18
133. Ao X, Zhao L, Davis MA, Lubman DM, Lawrence TS, Kong FM. Radiation produces differential changes in cytokine profiles in radiation lung fibrosis sensitive and resistant mice. J Hematol Oncol (2009) 2:6. doi:10.1186/1756-8722-2-6
134. Haston CK, Tomko TG, Godin N, Kerckhoff L, Hallett MT. Murine candidate bleomycin induced pulmonary fibrosis susceptibility genes identified by gene expression and sequence analysis of linkage regions. J Med Genet (2005) 42(6):464–73. doi:10.1136/jmg.2004.027938
135. Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol (2008) 40(3):362–82. doi:10.1016/j.biocel.2007.08.011
136. Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest (2007) 117(12):3786–99.
137. Brickey WJ, Neuringer IP, Walton W, Hua X, Wang EY, Jha S, et al. MyD88 provides a protective role in long-term radiation-induced lung injury. Int J Radiat Biol (2012) 88(4):335–47. doi:10.3109/09553002.2012.652723
138. Yoshii Y. Pathological review of late cerebral radionecrosis. Brain Tumor Pathol (2008) 25(2):51–8. doi:10.1007/s10014-008-0233-9
139. Moravan MJ, Olschowka JA, Williams JP, O’Banion MK. Cranial irradiation leads to acute and persistent neuroinflammation with delayed increases in T-cell infiltration and CD11c expression in C57BL/6 mouse brain. Radiat Res (2011) 176(4):459–73. doi:10.1667/RR2587.1
140. Paun A, Kunwar A, Haston CK. Acute adaptive immune response correlates with late radiation-induced pulmonary fibrosis in mice. Radiat Oncol (2015) 10:45. doi:10.1186/s13014-015-0359-y
141. Zheng X, Guo Y, Wang L, Zhang H, Wang S, Wang L, et al. Recovery profiles of T-cell subsets following low-dose total body irradiation and improvement with cinnamon. Int J Radiat Oncol Biol Phys (2015) 93(5):1118–26. doi:10.1016/j.ijrobp.2015.08.034
142. Wirsdorfer F, Cappuccini F, Niazman M, de Leve S, Westendorf AM, Ludemann L, et al. Thorax irradiation triggers a local and systemic accumulation of immunosuppressive CD4+ FoxP3+ regulatory T cells. Radiat Oncol (2014) 9:98. doi:10.1186/1748-717X-9-98
143. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81(1):1–5. doi:10.1189/jlb.0306164
144. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol (2015) 6:422. doi:10.3389/fimmu.2015.00422
145. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296(5566):301–5. doi:10.1126/science.1071059
146. Wang L, Zhang J, Wang B, Wang G, Xu J. Blocking HMGB1 signal pathway protects early radiation-induced lung injury. Int J Clin Exp Pathol (2015) 8(5):4815–22.
147. Tolle LB, Standiford TJ. Danger-associated molecular patterns (DAMPs) in acute lung injury. J Pathol (2013) 229(2):145–56. doi:10.1002/path.4124
148. Pouwels SD, Heijink IH, van Oosterhout AJ, Nawijn MC. A specific DAMP profile identifies susceptibility to smoke-induced airway inflammation. Eur Respir J (2014) 43(4):1183–6. doi:10.1183/09031936.00127813
149. Kuipers MT, van der Poll T, Schultz MJ, Wieland CW. Bench-to-bedside review: damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care (2011) 15(6):235. doi:10.1186/cc10437
150. Nettelbladt O, Tengblad A, Hallgren R. Lung accumulation of hyaluronan parallels pulmonary edema in experimental alveolitis. Am J Physiol (1989) 257(6 Pt 1):L379–84.
151. Nettelbladt O, Hallgren R. Hyaluronan (hyaluronic acid) in bronchoalveolar lavage fluid during the development of bleomycin-induced alveolitis in the rat. Am Rev Respir Dis (1989) 140(4):1028–32. doi:10.1164/ajrccm/140.4.1028
152. Ratikan JA, Micewicz ED, Xie MW, Schaue D. Radiation takes its toll. Cancer Lett (2015) 368(2):238–45. doi:10.1016/j.canlet.2015.03.031
153. Schaue D, Kachikwu EL, McBride WH. Cytokines in radiobiological responses: a review. Radiat Res (2012) 178(6):505–23. doi:10.1667/RR3031.1
154. Johnston CJ, Williams JP, Okunieff P, Finkelstein JN. Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat Res (2002) 157(3):256–65. doi:10.1667/0033-7587(2002)157[0256:RIPFEO]2.0.CO;2
155. Hong JH, Chiang CS, Tsao CY, Lin PY, McBride WH, Wu CJ. Rapid induction of cytokine gene expression in the lung after single and fractionated doses of radiation. Int J Radiat Biol (1999) 75(11):1421–7. doi:10.1080/095530099139287
156. Sohn SH, Lee JM, Park S, Yoo H, Kang JW, Shin D, et al. The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice. Environ Toxicol Pharmacol (2015) 39(2):917–26. doi:10.1016/j.etap.2015.02.019
157. Hong JH, Jung SM, Tsao TC, Wu CJ, Lee CY, Chen FH, et al. Bronchoalveolar lavage and interstitial cells have different roles in radiation-induced lung injury. Int J Radiat Biol (2003) 79(3):159–67. doi:10.1080/0955300031000076894
158. Pardo A, Smith KM, Abrams J, Coffman R, Bustos M, McClanahan TK, et al. CCL18/DC-CK-1/PARC up-regulation in hypersensitivity pneumonitis. J Leukoc Biol (2001) 70(4):610–6.
159. Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med (2006) 173(7):781–92. doi:10.1164/rccm.200509-1518OC
160. Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol (2002) 26(5):549–57. doi:10.1165/ajrcmb.26.5.4683
161. Pochetuhen K, Luzina IG, Lockatell V, Choi J, Todd NW, Atamas SP. Complex regulation of pulmonary inflammation and fibrosis by CCL18. Am J Pathol (2007) 171(2):428–37. doi:10.2353/ajpath.2007.061167
162. Inoue T, Fujishima S, Ikeda E, Yoshie O, Tsukamoto N, Aiso S, et al. CCL22 and CCL17 in rat radiation pneumonitis and in human idiopathic pulmonary fibrosis. Eur Respir J (2004) 24(1):49–56. doi:10.1183/09031936.04.00110203
163. Yogo Y, Fujishima S, Inoue T, Saito F, Shiomi T, Yamaguchi K, et al. Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis. Respir Res (2009) 10:80. doi:10.1186/1465-9921-10-80
164. Xu J, Mora A, Shim H, Stecenko A, Brigham KL, Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol (2007) 37(3):291–9. doi:10.1165/rcmb.2006-0187OC
165. Choi YW, Munden RF, Erasmus JJ, Park KJ, Chung WK, Jeon SC, et al. Effects of radiation therapy on the lung: radiologic appearances and differential diagnosis. Radiographics (2004) 24(4):985–97; discussion 98. doi:10.1148/rg.244035160
166. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi:10.1038/nri978
167. Oldroyd SD, Thomas GL, Gabbiani G, El Nahas AM. Interferon-gamma inhibits experimental renal fibrosis. Kidney Int (1999) 56(6):2116–27. doi:10.1046/j.1523-1755.1999.00775.x
168. Pelus LM, Fukuda S. Chemokine-mobilized adult stem cells; defining a better hematopoietic graft. Leukemia (2008) 22(3):466–73. doi:10.1038/sj.leu.2405021
169. Santin AD, Hermonat PL, Ravaggi A, Bellone S, Roman J, Pecorelli S, et al. Effects of concurrent cisplatinum administration during radiotherapy vs. radiotherapy alone on the immune function of patients with cancer of the uterine cervix. Int J Radiat Oncol Biol Phys (2000) 48(4):997–1006. doi:10.1016/S0360-3016(00)00769-0
170. Rotstein S, Blomgren H, Petrini B, Wasserman J, Baral E. Long term effects on the immune system following local radiation therapy for breast cancer. I. Cellular composition of the peripheral blood lymphocyte population. Int J Radiat Oncol Biol Phys (1985) 11(5):921–5. doi:10.1016/0360-3016(85)90114-2
171. Newman GH, Rees GJ, Jones RS, Grove EA, Preece AW. Changes in helper and suppressor T lymphocytes following radiotherapy for breast cancer. Clin Radiol (1987) 38(2):191–3. doi:10.1016/S0009-9260(87)80032-6
172. Maity A, McKenna WG, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother Oncol (1994) 31(1):1–13. doi:10.1016/0167-8140(94)90408-1
173. Goans RE, Waselenko JK. Medical management of radiological casualties. Health Phys (2005) 89(5):505–12. doi:10.1097/01.HP.0000172144.94491.84
174. Yan L, Wu M, Ba N, Shi G, Wang L, Zhang H. Changes in T-lymphocytes in lung cancer patients after hyperthermic intraperitoneal chemotherapy or radiotherapy. Genet Mol Res (2016) 15(2). doi:10.4238/gmr.15027865
175. Cho O, Oh YT, Chun M, Noh OK, Lee HW. Radiation-related lymphopenia as a new prognostic factor in limited-stage small cell lung cancer. Tumour Biol (2016) 37(1):971–8. doi:10.1007/s13277-015-3888-y
176. Han G, Zhang H, Xie CH, Zhou YF. Th2-like immune response in radiation-induced lung fibrosis. Oncol Rep (2011) 26(2):383–8.
177. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med (2001) 194(6):809–21. doi:10.1084/jem.194.6.809
178. Chen J, Wang Y, Mei Z, Zhang S, Yang J, Li X, et al. Radiation-induced lung fibrosis in a tumor-bearing mouse model is associated with enhanced type-2 immunity. J Radiat Res (2016) 57(2):133–41. doi:10.1093/jrr/rrv077
179. Karo-Atar D, Bordowitz A, Wand O, Pasmanik-Chor M, Fernandez IE, Itan M, et al. A protective role for IL-13 receptor alpha 1 in bleomycin-induced pulmonary injury and repair. Mucosal Immunol (2016) 9(1):240–53. doi:10.1038/mi.2015.56
180. Arras M, Huaux F, Vink A, Delos M, Coutelier JP, Many MC, et al. Interleukin-9 reduces lung fibrosis and type 2 immune polarization induced by silica particles in a murine model. Am J Respir Cell Mol Biol (2001) 24(4):368–75. doi:10.1165/ajrcmb.24.4.4249
181. Paun A, Lemay AM, Haston CK. Gene expression profiling distinguishes radiation-induced fibrosing alveolitis from alveolitis in mice. Radiat Res (2010) 173(4):512–21. doi:10.1667/RR1798.1
182. Komura K, Yanaba K, Horikawa M, Ogawa F, Fujimoto M, Tedder TF, et al. CD19 regulates the development of bleomycin-induced pulmonary fibrosis in a mouse model. Arthritis Rheum (2008) 58(11):3574–84. doi:10.1002/art.23995
183. Arras M, Louahed J, Simoen V, Barbarin V, Misson P, van den Brule S, et al. B lymphocytes are critical for lung fibrosis control and prostaglandin E2 regulation in IL-9 transgenic mice. Am J Respir Cell Mol Biol (2006) 34(5):573–80. doi:10.1165/rcmb.2004-0383OC
184. Clark JG, Kostal KM, Marino BA. Modulation of collagen production following bleomycin-induced pulmonary fibrosis in hamsters. Presence of a factor in lung that increases fibroblast prostaglandin E2 and cAMP and suppresses fibroblast proliferation and collagen production. J Biol Chem (1982) 257(14):8098–105.
185. Xu L, Xiong S, Guo R, Yang Z, Wang Q, Xiao F, et al. Transforming growth factor beta3 attenuates the development of radiation-induced pulmonary fibrosis in mice by decreasing fibrocyte recruitment and regulating IFN-gamma/IL-4 balance. Immunol Lett (2014) 162(1 Pt A):27–33. doi:10.1016/j.imlet.2014.06.010
186. Prochazkova J, Fric J, Pokorna K, Neuwirth A, Krulova M, Zajicova A, et al. Distinct regulatory roles of transforming growth factor-beta and interleukin-4 in the development and maintenance of natural and induced CD4+ CD25+ Foxp3+ regulatory T cells. Immunology (2009) 128(1 Suppl):e670–8. doi:10.1111/j.1365-2567.2009.03060.x
187. Kumar RK, O’Grady R, Maronese SE, Wilson MR. Epithelial cell-derived transforming growth factor-beta in bleomycin-induced pulmonary injury. Int J Exp Pathol (1996) 77(3):99–107. doi:10.1046/j.1365-2613.1996.586969.x
188. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med (1989) 170(3):727–37. doi:10.1084/jem.170.3.727
189. Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int (1997) 51(5):1376–82. doi:10.1038/ki.1997.188
190. Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys (2000) 47(2):277–90. doi:10.1016/S0360-3016(00)00435-1
191. Rube CE, Uthe D, Schmid KW, Richter KD, Wessel J, Schuck A, et al. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys (2000) 47(4):1033–42. doi:10.1016/S0360-3016(00)00482-X
192. Lo Re S, Lecocq M, Uwambayinema F, Yakoub Y, Delos M, Demoulin JB, et al. Platelet-derived growth factor-producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am J Respir Crit Care Med (2011) 184(11):1270–81. doi:10.1164/rccm.201103-0516OC
193. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol (2016) 16(11):676–89. doi:10.1038/nri.2016.95
194. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory T cells are highly activated, Th2 biased and suppress CD4 T cell proliferation through IL-10 and TGFbeta release. PLoS One (2016) 11(8):e0161507. doi:10.1371/journal.pone.0161507
195. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell (2015) 162(5):1078–89. doi:10.1016/j.cell.2015.08.021
196. Boveda-Ruiz D, D’Alessandro-Gabazza CN, Toda M, Takagi T, Naito M, Matsushima Y, et al. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology (2013) 218(2):245–54. doi:10.1016/j.imbio.2012.05.020
197. Kotsianidis I, Nakou E, Bouchliou I, Tzouvelekis A, Spanoudakis E, Steiropoulos P, et al. Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2009) 179(12):1121–30. doi:10.1164/rccm.200812-1936OC
198. Song L, Weng D, Liu F, Chen Y, Li C, Dong L, et al. Tregs promote the differentiation of Th17 cells in silica-induced lung fibrosis in mice. PLoS One (2012) 7(5):e37286. doi:10.1371/journal.pone.0037286
199. Trujillo G, Hartigan AJ, Hogaboam CM. T regulatory cells and attenuated bleomycin-induced fibrosis in lungs of CCR7-/- mice. Fibrogenesis Tissue Repair (2010) 3:18. doi:10.1186/1755-1536-3-18
200. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science (1995) 270(5234):286–90. doi:10.1126/science.270.5234.286
201. Wang LP, Wang YW, Wang BZ, Sun GM, Wang XY, Xu JL. Expression of interleukin-17A in lung tissues of irradiated mice and the influence of dexamethasone. ScientificWorldJournal (2014) 2014:251067. doi:10.1155/2014/251067
202. Hong JH, Chiang CS, Tsao CY, Lin PY, Wu CJ, McBride WH. Can short-term administration of dexamethasone abrogate radiation-induced acute cytokine gene response in lung and modify subsequent molecular responses? Int J Radiat Oncol Biol Phys (2001) 51(2):296–303. doi:10.1016/S0360-3016(01)01702-3
203. Hamama S, Gilbert-Sirieix M, Vozenin MC, Delanian S. Radiation-induced enteropathy: molecular basis of pentoxifylline-vitamin E anti-fibrotic effect involved TGF-beta1 cascade inhibition. Radiother Oncol (2012) 105(3):305–12. doi:10.1016/j.radonc.2012.08.023
204. Ozturk B, Egehan I, Atavci S, Kitapci M. Pentoxifylline in prevention of radiation-induced lung toxicity in patients with breast and lung cancer: a double-blind randomized trial. Int J Radiat Oncol Biol Phys (2004) 58(1):213–9. doi:10.1016/S0360-3016(03)01444-5
205. Misirlioglu CH, Demirkasimoglu T, Kucukplakci B, Sanri E, Altundag K. Pentoxifylline and alpha-tocopherol in prevention of radiation-induced lung toxicity in patients with lung cancer. Med Oncol (2007) 24(3):308–11. doi:10.1007/s12032-007-0006-z
206. Ward WF, Lin PJ, Wong PS, Behnia R, Jalali N. Radiation pneumonitis in rats and its modification by the angiotensin-converting enzyme inhibitor captopril evaluated by high-resolution computed tomography. Radiat Res (1993) 135(1):81–7. doi:10.2307/3578400
207. Delfraissy JF, Galanaud P, Balavoine JF, Wallon C, Dormont J. Captopril and immune regulation. Kidney Int (1984) 25(6):925–9. doi:10.1038/ki.1984.111
208. Smart YC, Gillies AH, Waga SW, Carney SL, Smith AJ, Burton RC. Effects of captopril on circulating T lymphocyte subsets. Int J Clin Pharmacol Ther Toxicol (1987) 25(7):389–91.
209. Ehlers MR, Riordan JF. Angiotensin-converting enzyme: new concepts concerning its biological role. Biochemistry (1989) 28(13):5311–8. doi:10.1021/bi00439a001
210. Costerousse O, Allegrini J, Lopez M, Alhenc-Gelas F. Angiotensin I-converting enzyme in human circulating mononuclear cells: genetic polymorphism of expression in T-lymphocytes. Biochem J (1993) 290(Pt 1):33–40. doi:10.1042/bj2900033
211. Kharofa J, Cohen EP, Tomic R, Xiang Q, Gore E. Decreased risk of radiation pneumonitis with incidental concurrent use of angiotensin-converting enzyme inhibitors and thoracic radiation therapy. Int J Radiat Oncol Biol Phys (2012) 84(1):238–43. doi:10.1016/j.ijrobp.2011.11.013
212. Jenkins P, Watts J. An improved model for predicting radiation pneumonitis incorporating clinical and dosimetric variables. Int J Radiat Oncol Biol Phys (2011) 80(4):1023–9. doi:10.1016/j.ijrobp.2010.03.058
213. Kohl RR, Kolozsvary A, Brown SL, Zhu G, Kim JH. Differential radiation effect in tumor and normal tissue after treatment with ramipril, an angiotensin-converting enzyme inhibitor. Radiat Res (2007) 168(4):440–5. doi:10.1667/RR0707.1
214. Wang H, Liao Z, Zhuang Y, Xu T, Nguyen QN, Levy LB, et al. Do angiotensin-converting enzyme inhibitors reduce the risk of symptomatic radiation pneumonitis in patients with non-small cell lung cancer after definitive radiation therapy? Analysis of a single-institution database. Int J Radiat Oncol Biol Phys (2013) 87(5):1071–7. doi:10.1016/j.ijrobp.2013.08.033
215. Small W Jr, James JL, Moore TD, Fintel DJ, Lutz ST, Movsas B, et al. Utility of the ACE inhibitor captopril in mitigating radiation-associated pulmonary toxicity in lung cancer: results from NRG oncology RTOG 0123. Am J Clin Oncol (2016). doi:10.1097/COC.0000000000000289
216. Randak C, Brabletz T, Hergenrother M, Sobotta I, Serfling E. Cyclosporin A suppresses the expression of the interleukin 2 gene by inhibiting the binding of lymphocyte-specific factors to the IL-2 enhancer. EMBO J (1990) 9(8):2529–36.
217. Moolman JA, Bardin PG, Rossouw DJ, Joubert JR. Cyclosporin as a treatment for interstitial lung disease of unknown aetiology. Thorax (1991) 46(8):592–5. doi:10.1136/thx.46.8.592
218. Muraoka T, Bandoh S, Fujita J, Horiike A, Ishii T, Tojo Y, et al. Corticosteroid refractory radiation pneumonitis that remarkably responded to cyclosporin A. Intern Med (2002) 41(9):730–3. doi:10.2169/internalmedicine.41.730
219. Abdou NI, Zweiman B, Casella SR. Effects of azathioprine therapy on bone marrow-dependent and thymus-dependent cells in man. Clin Exp Immunol (1973) 13(1):55–64.
220. Dimitriu A, Fauci AS. Activation of human B lymphocytes. XI. Differential effects of azathioprine on B lymphocytes and lymphocyte subpopulations regulating B cell function. J Immunol (1978) 121(6):2335–9.
221. McCarty MJ, Lillis P, Vukelja SJ. Azathioprine as a steroid-sparing agent in radiation pneumonitis. Chest (1996) 109(5):1397–400. doi:10.1378/chest.109.5.1397
222. Kwok E, Chan CK. Corticosteroids and azathioprine do not prevent radiation-induced lung injury. Can Respir J (1998) 5(3):211–4. doi:10.1155/1998/896131
223. Giridhar P, Mallick S, Rath GK, Julka PK. Radiation induced lung injury: prediction, assessment and management. Asian Pac J Cancer Prev (2015) 16(7):2613–7. doi:10.7314/APJCP.2015.16.7.2613
224. Vogelius IR, Bentzen SM. A literature-based meta-analysis of clinical risk factors for development of radiation induced pneumonitis. Acta Oncol (2012) 51(8):975–83. doi:10.3109/0284186X.2012.718093
225. Takeda A, Kunieda E, Ohashi T, Aoki Y, Oku Y, Enomoto T, et al. Severe COPD is correlated with mild radiation pneumonitis following stereotactic body radiotherapy. Chest (2012) 141(4):858–66. doi:10.1378/chest.11-1193
226. Zhang XJ, Sun JG, Sun J, Ming H, Wang XX, Wu L, et al. Prediction of radiation pneumonitis in lung cancer patients: a systematic review. J Cancer Res Clin Oncol (2012) 138(12):2103–16. doi:10.1007/s00432-012-1284-1
227. Chen Y, Hyrien O, Williams J, Okunieff P, Smudzin T, Rubin P. Interleukin (IL)-1A and IL-6: applications to the predictive diagnostic testing of radiation pneumonitis. Int J Radiat Oncol Biol Phys (2005) 62(1):260–6. doi:10.1016/j.ijrobp.2005.01.041
228. Herskind C, Talbot CJ, Kerns SL, Veldwijk MR, Rosenstein BS, West CM. Radiogenomics: a systems biology approach to understanding genetic risk factors for radiotherapy toxicity? Cancer Lett (2016) 382(1):95–109. doi:10.1016/j.canlet.2016.02.035
229. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med (2013) 5(167):167sr1. doi:10.1126/scitranslmed.3004700
230. Straub JM, New J, Hamilton CD, Lominska C, Shnayder Y, Thomas SM. Radiation-induced fibrosis: mechanisms and implications for therapy. J Cancer Res Clin Oncol (2015) 141(11):1985–94. doi:10.1007/s00432-015-1974-6
231. Williams JP, Johnston CJ, Finkelstein JN. Treatment for radiation-induced pulmonary late effects: spoiled for choice or looking in the wrong direction? Curr Drug Targets (2010) 11(11):1386–94. doi:10.2174/1389450111009011386
232. Anscher MS. Targeting the TGF-beta1 pathway to prevent normal tissue injury after cancer therapy. Oncologist (2010) 15(4):350–9. doi:10.1634/theoncologist.2009-S101
233. Wang BZ, Wang LP, Han H, Cao FL, Li GY, Xu JL, et al. Interleukin-17A antagonist attenuates radiation-induced lung injuries in mice. Exp Lung Res (2014) 40(2):77–85. doi:10.3109/01902148.2013.872210
234. Togo S, Liu X, Wang X, Sugiura H, Kamio K, Kawasaki S, et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am J Physiol Lung Cell Mol Physiol (2009) 296(6):L959–69. doi:10.1152/ajplung.00508.2007
235. Herbert C, Hettiaratchi A, Webb DC, Thomas PS, Foster PS, Kumar RK. Suppression of cytokine expression by roflumilast and dexamethasone in a model of chronic asthma. Clin Exp Allergy (2008) 38(5):847–56. doi:10.1111/j.1365-2222.2008.02950.x
236. Wan YY, Flavell RA. TGF-beta and regulatory T cell in immunity and autoimmunity. J Clin Immunol (2008) 28(6):647–59. doi:10.1007/s10875-008-9251-y
237. Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol (2012) 4(1):29–37. doi:10.1093/jmcb/mjr033
238. Varga J, Whitfield ML. Transforming growth factor-beta in systemic sclerosis (scleroderma). Front Biosci (Schol Ed) (2009) 1:226–35. doi:10.2741/s22
239. Meng XM, Chung AC, Lan HY. Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond) (2013) 124(4):243–54. doi:10.1042/CS20120252
240. Dooley S, ten Dijke P. TGF-beta in progression of liver disease. Cell Tissue Res (2012) 347(1):245–56. doi:10.1007/s00441-011-1246-y
241. Smilowitz HM, Sasso D, Lee EW, Goh G, Micca PL, Dilmanian FA. Therapy model for advanced intracerebral B16 mouse melanoma using radiation therapy combined with immunotherapy. Cancer Immunol Immunother (2013) 62(7):1187–97. doi:10.1007/s00262-013-1423-9
242. Byrne WL, Mills KH, Lederer JA, O’Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res (2011) 71(22):6915–20. doi:10.1158/0008-5472.CAN-11-1156
243. Fernandez P, Perez-Aso M, Smith G, Wilder T, Trzaska S, Chiriboga L, et al. Extracellular generation of adenosine by the ectonucleotidases CD39 and CD73 promotes dermal fibrosis. Am J Pathol (2013) 183(6):1740–6. doi:10.1016/j.ajpath.2013.08.024
244. Peng Z, Fernandez P, Wilder T, Yee H, Chiriboga L, Chan ES, et al. Ecto-5’-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. Nucleosides Nucleotides Nucleic Acids (2008) 27(6):821–4. doi:10.1080/15257770802146403
245. Zhou Y, Schneider DJ, Morschl E, Song L, Pedroza M, Karmouty-Quintana H, et al. Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J Immunol (2011) 186(2):1097–106. doi:10.4049/jimmunol.1002907
246. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16(3):177–92. doi:10.1038/nri.2016.4
247. Allard D, Allard B, Gaudreau PO, Chrobak P, Stagg J. CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy (2016) 8(2):145–63. doi:10.2217/imt.15.106
248. Nicolay NH, Lopez Perez R, Debus J, Huber PE. Mesenchymal stem cells – a new hope for radiotherapy-induced tissue damage? Cancer Lett (2015) 366(2):133–40. doi:10.1016/j.canlet.2015.06.012
249. Martire A, Bedada FB, Uchida S, Poling J, Kruger M, Warnecke H, et al. Mesenchymal stem cells attenuate inflammatory processes in the heart and lung via inhibition of TNF signaling. Basic Res Cardiol (2016) 111(5):54. doi:10.1007/s00395-016-0573-2
250. Dong LH, Jiang YY, Liu YJ, Cui S, Xia CC, Qu C, et al. The anti-fibrotic effects of mesenchymal stem cells on irradiated lungs via stimulating endogenous secretion of HGF and PGE2. Sci Rep (2015) 5:8713. doi:10.1038/srep08713
251. Ortiz LA, Dutreil M, Fattman C, Pandey AC, Torres G, Go K, et al. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci U S A (2007) 104(26):11002–7. doi:10.1073/pnas.0704421104
252. Lan YW, Choo KB, Chen CM, Hung TH, Chen YB, Hsieh CH, et al. Hypoxia-preconditioned mesenchymal stem cells attenuate bleomycin-induced pulmonary fibrosis. Stem Cell Res Ther (2015) 6:97. doi:10.1186/s13287-015-0081-6
253. Jones S, Horwood N, Cope A, Dazzi F. The antiproliferative effect of mesenchymal stem cells is a fundamental property shared by all stromal cells. J Immunol (2007) 179(5):2824–31. doi:10.4049/jimmunol.179.5.2824
254. Glenn JD, Whartenby KA. Mesenchymal stem cells: emerging mechanisms of immunomodulation and therapy. World J Stem Cells (2014) 6(5):526–39. doi:10.4252/wjsc.v6.i5.526
255. Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther (2016) 7(1):125. doi:10.1186/s13287-016-0363-7
256. Alkhouri H, Poppinga WJ, Tania NP, Ammit A, Schuliga M. Regulation of pulmonary inflammation by mesenchymal cells. Pulm Pharmacol Ther (2014) 29(2):156–65. doi:10.1016/j.pupt.2014.03.001
257. Jun D, Garat C, West J, Thorn N, Chow K, Cleaver T, et al. The pathology of bleomycin-induced fibrosis is associated with loss of resident lung mesenchymal stem cells that regulate effector T-cell proliferation. Stem Cells (2011) 29(4):725–35. doi:10.1002/stem.604
258. Chow K, Fessel JP, Kaoriihida S, Schmidt EP, Gaskill C, Alvarez D, et al. Dysfunctional resident lung mesenchymal stem cells contribute to pulmonary microvascular remodeling. Pulm Circ (2013) 3(1):31–49. doi:10.4103/2045-8932.109912
259. Marriott S, Baskir RS, Gaskill C, Menon S, Carrier EJ, Williams J, et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Physiol Cell Physiol (2014) 307(8):C684–98. doi:10.1152/ajpcell.00114.2014
260. Luo Y, Xu W, Chen H, Warburton D, Dong R, Qian B, et al. A novel profibrotic mechanism mediated by TGFbeta-stimulated collagen prolyl hydroxylase expression in fibrotic lung mesenchymal cells. J Pathol (2015) 236(3):384–94. doi:10.1002/path.4530
261. Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell (2015) 16(1):51–66. doi:10.1016/j.stem.2014.11.004
262. Xie T, Liang J, Liu N, Huan C, Zhang Y, Liu W, et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest (2016) 126(8):3063–79. doi:10.1172/JCI85328
263. Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, et al. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci U S A (2003) 100(14):8407–11. doi:10.1073/pnas.1432929100
264. Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther (2011) 2(4):34. doi:10.1186/scrt75
265. Swaney JS, Chapman C, Correa LD, Stebbins KJ, Bundey RA, Prodanovich PC, et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br J Pharmacol (2010) 160(7):1699–713. doi:10.1111/j.1476-5381.2010.00828.x
Keywords: lymphocytes, radiotherapy, lung, pneumonitis, fibrosis
Citation: Wirsdörfer F and Jendrossek V (2016) The Role of Lymphocytes in Radiotherapy-Induced Adverse Late Effects in the Lung. Front. Immunol. 7:591. doi: 10.3389/fimmu.2016.00591
Received: 29 September 2016; Accepted: 29 November 2016;
Published: 14 December 2016
Edited by:
Udo S. Gaipl, University Hosptial Erlangen, GermanyReviewed by:
Kelly M. Mcnagny, University of British Columbia, CanadaFranz Rödel, University Hospital Frankfurt, Germany
Copyright: © 2016 Wirsdörfer and Jendrossek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Verena Jendrossek, dmVyZW5hLmplbmRyb3NzZWtAdW5pLWR1ZS5kZQ==