 Karolina A. Zielińska1
Karolina A. Zielińska1 Laura Van Moortel2
Laura Van Moortel2 Ghislain Opdenakker1
Ghislain Opdenakker1 Karolien De Bosscher2
Karolien De Bosscher2 Philippe E. Van den Steen1*
Philippe E. Van den Steen1*
- 1Laboratory of Immunobiology, Rega Institute for Medical Research, KU Leuven, Leuven, Belgium
- 2Receptor Research Laboratories, Nuclear Receptor Lab, VIB-UGent, VIB Medical Biotechnology Center, Ghent, Belgium
The endothelium plays a crucial role in inflammation. A balanced control of inflammation requires the action of glucocorticoids (GCs), steroidal hormones with potent cell-specific anti-inflammatory properties. Besides the classic anti-inflammatory effects of GCs on leukocytes, recent studies confirm that endothelial cells also represent an important target for GCs. GCs regulate different aspects of endothelial physiology including expression of adhesion molecules, production of pro-inflammatory cytokines and chemokines, and maintenance of endothelial barrier integrity. However, the regulation of endothelial GC sensitivity remains incompletely understood. In this review, we specifically examine the endothelial response to GCs in various inflammatory diseases ranging from multiple sclerosis, stroke, sepsis, and vasculitis to atherosclerosis. Shedding more light on the cross talk between GCs and endothelium will help to improve existing therapeutic strategies and develop new therapies better tailored to the needs of patients.
1. Introduction
Glucocorticoids (GCs) are endogenous stress hormones with strong anti-inflammatory properties. Therefore, GCs have been therapeutically exploited for more than six decades, often at dosages, which exceed the physiological levels (1). GCs influence the function of various subtypes of immune cells including T cells, dendritic cells, macrophages, and B cells (2). A growing amount of evidence indicates that GCs also regulate multiple aspects of endothelial physiology. In this review, we discuss the effects of GCs on endothelium and the mechanisms that control GC sensitivity in endothelial cells.
2. Endothelial Cell Biology in a Nutshell
2.1. Morphology and Heterogeneity of the Endothelium
The human body contains approximately 2.5 × 1012 endothelial cells that are typically flat with a thin basement membrane (also known as basal lamina) enriched in type IV collagen and laminin (3–5). However, in specific blood vessels used by lymphocytes to enter the lymph nodes (high endothelial venules) endothelial cells are plum and tall with thick basal lamina (6). The thickness of endothelium varies from 0.1 µm in capillaries and veins to 1 µm in the aorta (4).
Endothelial cell phenotypes vary between different organs, between different segments of the vascular bed within the same organ, and between neighboring endothelial cells of the same organ and blood vessel type (7). In arteries and veins, the endothelium forms a continuous layer of cells held together by tight junctions (TJs)—one of the two main types of intercellular junctions in endothelium (7, 8). Endothelial cells in arteries are long and narrow and are aligned in the direction of blood flow. In contrast, in veins endothelial cells are wider and shorter and lack the alignment in the direction of blood flow (9). Capillary endothelial cells are highly adapted to underlying tissues and present many phenotypic differences between different vascular beds (9). The endothelium of capillaries may be continuous, fenestrated, or discontinuous (7). The microvascular bed of each organ is composed of specialized endothelial cells endowed with a unique set of adhesion molecules, chemokines, transcription factors, and metabolic profiles (10).
2.2. Functions of the Endothelium
The endothelium participates not only in blood delivery but also in organ regeneration and maintenance of homeostasis (11). Endothelial cells are metabolically active and play an important role in physiological processes such as the control of vasomotor tone, the trafficking of leukocytes between blood and underlying tissue, angiogenesis, and both innate and adaptive immunity (7).
In the central nervous system (CNS), the endothelium forms the blood–brain barrier (BBB) that controls the exchange of immune cells and mediators between blood and CNS (12). The BBB consists of a thick glycocalyx, endothelial cells linked by tight junctions (TJs), a double basement membrane, and astrocytic end-feet (13). The double basement membrane enables precise localization of sites of immune cell extravasation (14). Endothelial TJs, which constitute a crucial element of the BBB, control paracellular movement of solutes through the BBB and maintain brain homeostasis. They consist of claudins, occludin, and junction adhesion molecules (JAMs) (15). TJs are intermingled with adherens junctions (AJs) that form a belt via interaction between cadherins (8, 15). Proper organization of TJs requires AJs assembly (16).
2.3. Endothelial Activation during Inflammation and Angiogenesis
Although at rest endothelial cells present a non-reactive surface at the interface between blood and tissue, upon activation the endothelium becomes a major player in the generation of the inflammatory response (3). Pro-inflammatory cytokines produced by immune cells activate the endothelium and mediate leukocyte recruitment. Endothelial selectins such as E- and P-selectin capture leukocytes from the blood flow and mediate rolling (17). This slows down the circulating leukocytes and enables chemokines, localized on the endothelial surface, to interact with their receptors on leukocytes leading to integrin activation (12). Pro-inflammatory mediators such as tumor necrosis factor (TNF), interleukin (IL)-1, -17A, -18, -33, -36γ, and interferon (IFN)-α, -β, -γ stimulate the endothelium to produce various chemokines including CC chemokine ligand 2 (CCL2, also known as monocyte chemoattractant protein-1 or MCP-1), CCL20 (also known as macrophage inflammatory protein-3α or MIP-3α), CXC chemokine ligand 1 (CXCL1, also known as growth-related oncogene-α or GRO-α), CXCL8 (also known as IL-8), CXCL9 (also known as monokine induced by IFN-γ or MIG), CXCL10 (also known as IFN-γ-induced protein-10 or IP-10), CXCL11 (also known as interferon-inducible T cell α chemoattractant or I-TAC), and also integrin ligands such as intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (3, 18–24). In contrast, IL-10 and IL-37 decrease CXCL8 (IL-8) and ICAM-1 expression in endothelial cells (25, 26). ICAM-1 and VCAM-1 control firm adhesion and leukocyte crawling on the endothelial surface until they transmigrate through the endothelial barrier (27). Platelet endothelial cell adhesion molecule-1 (PECAM-1) controls localization of junctional proteins such as VE-cadherin and β-catenin and mediates leukocyte extravasation to the sites of inflammation (28).
Angiogenesis—the growth of new blood vessels—plays an important role in the control of embryonic development and is involved in various human diseases. Increased angiogenesis is associated with neoplastic diseases, while insufficient blood vessel growth limits the repair process in ischemic cardiovascular diseases (29, 30). Vascular endothelial growth factor (VEGF) regulates vascular development during embryogenesis, blood vessel formation in adults, wound healing, and organ regeneration (31, 32). In mammals, five VEGF ligands (VEGF-A, -B, -C, -D, and placental growth factor—PLGF) can bind to three receptor tyrosine kinases—VEFG receptor-1, -2, and -3 (32). Binding of VEGF to its receptor also increases vascular permeability through activation of focal adhesion tyrosine kinase (FAK) that interacts with vascular endothelial cadherin (VE-cadherin) leading to VE-cadherin–β-catenin dissociation (33). The Tie receptors (Tie-1 and Tie-2) and their angiopoietin ligands (ANG1–ANG4) constitute the second receptor Tyr kinase system in the vasculature. ANG–Tie controls vessel quiescence and regulates later stages of angiogenesis (30). ANG1 exerts its anti-inflammatory effects through inhibition of VEGF-induced expression of ICAM-1, VCAM-1, and E-selectin (34). Moreover, ANG1 prevents VEGF and TNF-induced expression of the pro-coagulatory molecule tissue factor (TF) (35). Later, ANG2 was described as a ligand that antagonizes ANG1 activity on Tie-2 and acts as a switch between the quiescent and inflamed state of endothelium (36, 37). ANG2 also controls later steps of leukocyte adhesion. Furthermore, ANG2 sensitizes endothelial cells toward TNF and modulates TNF-induced expression of adhesion molecules (36).
Matrix metalloproteinases (MMPs) control turnover of the extracellular matrix and are crucial in the regulation of inflammation. MMPs regulate leukocyte recruitment and modulate cytokine and chemokine activity (38, 39). For example, MMP-9 induces endothelial barrier breakdown through degradation of endothelial junctions (40). MMP-9 also plays an important role in matrix remodeling during angiogenesis and mediates influx of proteins into angiogenic tissue (41, 42).
3. Glucocorticoids (GCs) and the Glucocorticoid Receptor (GR)
3.1. GCs as Inhibitors of Inflammation
Glucocorticoids (GCs) are steroidal hormones that are synthesized in the zona fasciculata of the adrenal cortex, starting from a cholesterol scaffold (43, 44). GCs are produced in a circadian pattern or in response to stress and regulate various physiological processes including metabolism of sugar, protein, fat, muscle and bone, cardiovascular function, reproduction, and cognition (45). The hypothalamo–pituitary axis controls the secretion of cortisol, which is the physiologically active human GC (46, 47). The periventricular nucleus of the hypothalamus produces corticotrophin-releasing hormone (CRH), which induces the synthesis of adrenocorticotropic hormone (ACTH, also called corticotrophin) in the anterior pituitary gland. ACTH subsequently induces the production of the active cortisol, which in turn inhibits the secretion of CRH in the hypothalamus via a negative feedback mechanism (45, 48, 49).
Importantly, GCs also act as strong anti-inflammatory mediators, providing a negative feedback on inflammation (50–52). The anti-inflammatory potential of GCs has led to the application of exogenous GCs for the treatment of a variety of inflammatory disorders such as asthma, rheumatoid arthritis, and inflammatory bowel diseases (53, 54). GCs are also prescribed to transplantation patients to prevent organ rejection and used for the treatment of lymphoid cancers including leukemia, lymphoma, and myeloma (44, 51, 55).
Due to their pleiotropic actions, the use of exogenous GCs has two major drawbacks. First, high therapeutic doses used to reach desired physiological effects also induce undesired side-effects including osteoporosis, diabetes, and hypertension (48, 54, 56). Second, prolonged and/or highly dosed GC use can lead to GC resistance. This phenomenon has been discussed extensively (48, 57, 58).
3.2. GR Structure and Isoforms
GCs exert their effects mainly by binding to the cytosolic glucocorticoid receptor (GR), a member of the nuclear receptor superfamily that is ubiquitously expressed (46, 51, 59). The human GR-coding nr3c1 gene is located on chromosome 5. Transcription generates multiple GR isoforms due to alternative splicing (45, 48). Each isoform is composed of three major functional domains: an N-terminal domain (NTD), a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) (Figure 1). The NTD mediates the recruitment of the basal transcriptional machinery and contains the transcriptional activation function 1 (AF-1) domain with residues that undergo posttranslational modifications (PTMs) and interact with cofactors in a ligand-independent way (45, 47). The DBD contains two zinc fingers that regulate GR dimerization and DNA binding (45, 56). The DBD is linked to the LBD via a hinge region. The LBD forms a hydrophobic ligand-binding pocket and contains the second transcriptional activation function domain (AF-2), which is responsible for ligand-dependent interaction with coregulators and recruitment of the basal transcriptional machinery (45–47). The receptor also contains two nuclear localization signals (NLS1 and NLS2, Figure 1) at the junction of the DBD and the hinge region, and within the LBD (45).
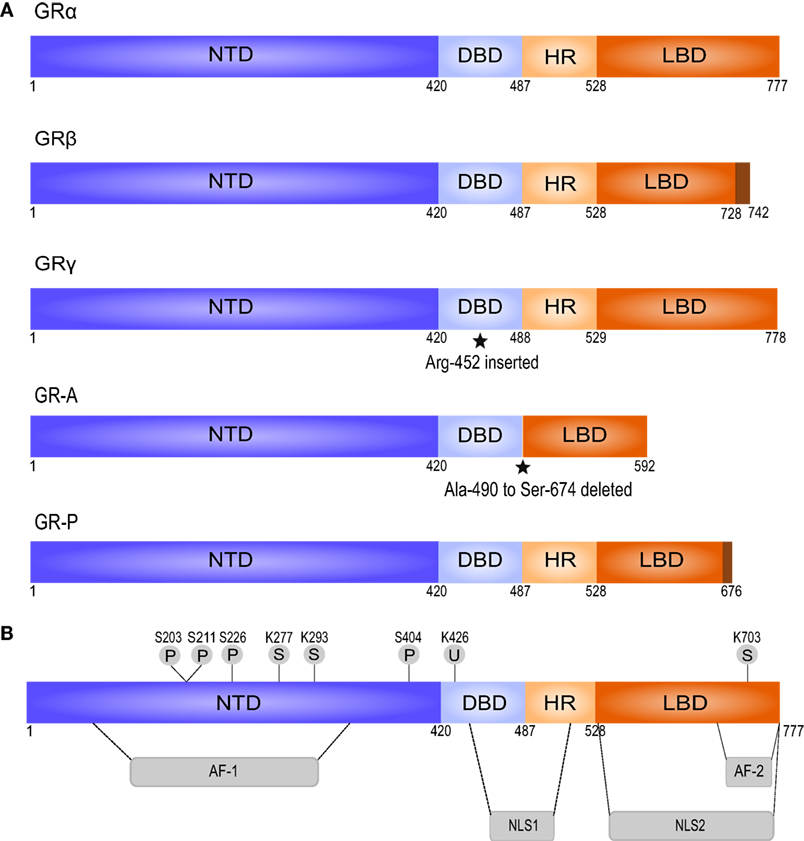
Figure 1. Splice variants and posttranslational modifications of the human glucocorticoid receptor (GR). (A) The splice variant GRβ differs from GRα in the C-terminal LBD, does not bind GCs, and acts as a dominant negative regulator of GRα. GRγ contains an insertion of an additional arginine residue in the DBD. This impairs its ability to regulate specific GC-responsive genes. GR-A and GR-P miss large regions in the LBD and fail to bind GCs, based on Oakley and Cidlowski (45). (B) GR contains several residues subjected to posttranslational modifications. AF, activation function; DBD, DNA-binding domain; HR, hinge region; LBD, ligand-binding domain; NLS, nuclear localization signal; NTD, N-terminal domain; P, phosphorylation; S, sumoylation; U, ubiquitination.
Alternative splicing of the LBD of the human GR gene generates GRα and GRβ—the best characterized isoforms of GR (45). GRα is the classic GR protein that mediates the majority of GCs actions. GRβ is considered a dominant inhibitor of GRα that resides constitutively in the nucleus but cannot bind to GCs and has no direct effect on GC-responsive genes. However, GRβ can antagonize the activity of GRα by competing for glucocorticoid response element (GRE) binding or for cofactors, or by formation of inactive GRα/GRβ heterodimers (45, 48). In general, the expression of GRβ is low but can be enhanced by pro-inflammatory cytokines (e.g., TNF and IL-1) and other immune activators (e.g., microbial superantigens) (60, 61). Increased GRβ expression has been involved in GC resistance in a variety of inflammatory diseases (45, 50). GRγ, GR-A, and GR-P are the three other transcriptional GR isoforms. They are less characterized than GRα and GRβ, but GRγ and GR-P are also known to be involved in GC resistance (62–64).
Alternative translation initiation results in further division of each isoform into subtypes. For instance, GRα has 8 subtypes, which are the result of 8 highly conserved start codons present in the GR mRNA. GRα-A is the full-length receptor and is the most abundant GR protein in many cell types, together with GRα-B (45). GR is also subject to various posttranslational modifications including phosphorylation, SUMOylation, and ubiquitination (43, 48, 65). Unless specified otherwise, it is always GRα that is referred to, since most known GC-mediated actions are exerted via this receptor isoform.
3.3. Molecular Mechanisms of GC Action
In the absence of ligand, GR mainly resides in the cytoplasm in association with chaperone proteins (e.g., hsp90, hsp70, and hsp90-binding protein p23) and immunophilins [e.g., FK506-binding protein (FKBP)-51 and FKBP-52] (45). Upon ligand binding, GR undergoes a conformational change that induces partial dissociation of the chaperone complex and exposes the nuclear localization sequences (43, 45). FKBP-51 is substituted by FKBP-52, and the transport protein dynein transports the GR complex along the cytoskeletal tracts to the nucleus (44, 65). This process also depends on importin-α and importin 13 (56, 57). In the nucleus, the complex dissociates and GR binds to DNA in homodimeric form (44). The cellular localization of GR is a dynamic process, since both active and inactive forms of the receptor have been demonstrated to shuttle back and forth between nucleus and cytoplasm (49, 65). Nevertheless, in most cases GR resides in the cytosol in the absence of GCs, while it translocates to the nucleus when bound to its ligand. The effect on transcription depends on binding to DNA and/or other transcription factors and on the recruitment of GR coregulators. The latter proteins include coactivators and corepressors that assist in GR transcriptional activity (66).
Binding of homodimeric GR to glucocorticoid response elements (GREs) results in transcriptional activation and rapid switches between the bound and the unbound state of GR (45, 49). This interaction induces transcription of various genes including sphingosine kinase 1 (Sphk1) and TGF-stimulated clone 22 domain protein-3 (Tsc22d3), the coding gene for GC-induced leucine zipper (GILZ) (56, 67). The latter protein is reported to bind to NF-κB and AP-1, and the protective effect of this interaction has been shown in several inflammatory models (56). The promotor region of MAPK phosphatase-1 gene mkp1 also contains a GRE site (56). The corresponding protein, MKP-1 or dual specificity phosphatase-1 (DUSP1) is a MAPK phosphatase that inactivates kinases involved in activation of AP-1—ERKs, p38, and JNK. MAPKs are also involved in the fine-tuning of NF-κB signaling (56, 68). Other anti-inflammatory mediators induced by GR bound to GREs include IL-10 and annexin-1 (AnxA1) (56, 69).
Moreover, GC-bound GR can also inhibit gene transcription via negative GREs (nGREs) with consensus sequence CTCC(n)0−2GGAGA. The nature of this sequence does not allow GR dimerization, thus these inhibitory actions are mediated by monomeric GR (70). GR via an nGRE reduces expression of thymic stromal lymphopoietin (TSLP). The GR-dependent repression through nGRE is probably mediated via the assembly of a corepressor complex and the recruitment of histone deacetylases (70).
DNA-bound GR can still interact with other transcription factors via a mechanism called composite regulation. GR and its interacting transcription factors both bind to their own response element and influence each others’ transcription in a DNA-dependent manner (65, 68). In the hypothalamus, GR bound to an nGRE interacts with DNA-bound AP-1 and negatively regulates CRH (71).
GR can also bind to DNA-bound transcription factors, without interacting with DNA itself (65, 72). This process—called tethering—occurs at lower cortisol levels than GRE-mediated GR actions (48). However, it remains unclear if GR tethers in a monomeric or dimeric form or works in yet another fashion (73, 74). Nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1) belong to the best characterized proteins that are tethered by GR (44). The GR-mediated inhibition of NF-κB and AP-1 represents the main mechanism for the anti-inflammatory actions of GCs, but GR can also interact with other transcription factors including interferon regulatory factor 3 (IRF3), signal transducer and activator of transcription (STAT) 3 and 5, and GATA3 (43, 65, 68, 75). GR-mediated tethering onto NF-κB and AP-1 implies direct binding of presumably monomeric GR to the DNA-bound Jun- and the p65-subunit (45, 65, 68, 76); however, the surface part of GR involved in these interactions differs for AP-1 and NF-κB (48). Tethering between GR and these transcription factors is reciprocal, which implies that AP-1 and NF-κB can inhibit DNA-bound GR (47, 51, 68). GR can also inhibit AP-1 and NF-κB through other mechanisms including the cofactor GRIP1-mediated inhibition of NF-κB/IRF3 dimer formation, via reduced histone acetyltransferase (HAT) activity of CBP or via recruitment of histone deacetylase 2 (HDAC2). The latter mechanism seems to be responsible for GR-mediated inhibition of CXCL8 (IL-8) expression (55, 65, 68). GCs also repress NF-κB-driven genes via upregulation of the cytoplasmic NF-κB inhibitor (IκB-α) (44).
Additionally, GR exerts some of its actions in a rapid non-genomic mode within minutes. Thereby GR influences signaling pathways, possibly via membrane-associated GR and a second messenger cascade (48, 51, 55, 77). Only recently it has been appreciated that GR elicits also pro-inflammatory pathways to prime cells for an adequate subsequent anti-inflammatory response (55).
4. Regulation of Endothelial Physiology by GCs
GCs exert various effects on immune cell function. GCs cause immunosuppression in pro-inflammatory T cells while increasing the activity of regulatory T cells. GCs seem to affect B cell proliferation. In macrophages, the effects of GCs are concentration-dependent and range from immunosuppression to immunostimulation. Furthermore, GCs mediate the induction of a tolerogenic phenotype in dendritic cells (2). GCs also induce apoptosis in various types of immune cells including T cells, B cells, and plasmacytoid dendritic cells (78–80). This process plays an important role in the development of the immune system and in fine-tuning of its function. Interestingly, GCs have opposite effects in macrophages and mediate their survival (81). GC-induced apoptosis also has been extensively reviewed in other cell types; however, the effects of GCs on endothelial apoptosis remain insufficiently studied (82).
It becomes more and more clear that GCs also regulate multiple aspects of endothelial physiology (Figure 2). GCs inhibit pro-inflammatory signaling pathways in endothelium and induce protective molecules that maintain endothelial function, especially upon inflammation. For instance, dexamethasone blocks nuclear translocation of NF-κB and reduces the binding of AP-1 and GATA to DNA in endothelial cells (83). GCs induce MKP-1 (also known as DUSP1), which inhibits MAPK signaling pathways, and tristetraprolin (TTP, also known as ZFP36), which destabilizes mRNAs of pro-inflammatory cytokines (84, 85). AnxA1, induced by GCs in endothelium, causes leukocyte detachment and regulates BBB integrity (86–88). Moreover, AnxA1 inhibits phospholipase A2, an enzyme that releases arachidonic acid from phospholipids to produce pro-inflammatory mediators such as prostaglandins and leukotrienes via cyclooxygenase (89).
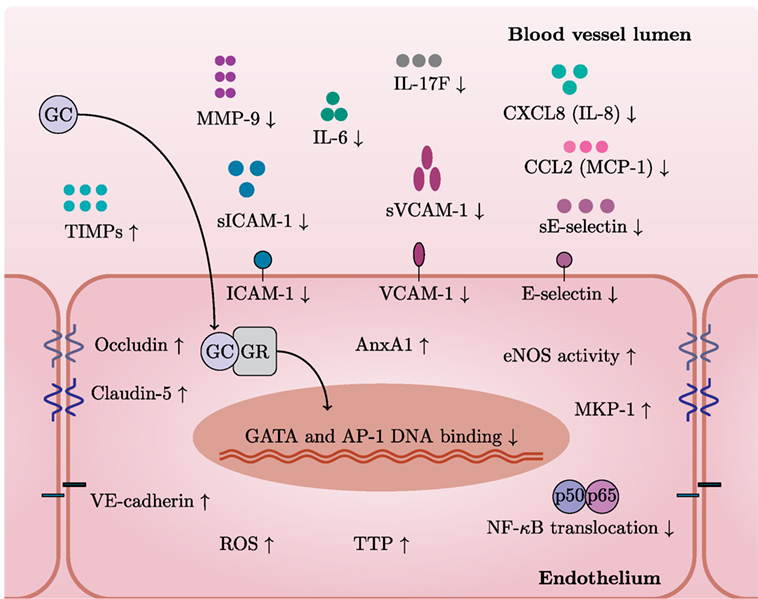
Figure 2. GCs exert specific actions in endothelial cells. In general, GCs have a variety of cell type-specific effects. This figure depicts GC actions that have been described in endothelium. After entering an endothelial cell, GCs bind to GC receptor (GR) and translocate to the nucleus. GR bound to GCs inhibits pro-inflammatory pathways by limiting GATA and AP-1 DNA binding and NF-κB translocation. GCs decrease levels of adhesion molecules (VCAM-1, ICAM-1, and E-selectin) and also their soluble forms and MMP-9 while increasing levels of junctional proteins—occludin, claudin-5, and VE-cadherin. GCs induce protective molecules such as AnxA1, TTP, MKP-1, and TIMPs. Furthermore, GCs reduce levels of IL-6, IL-17F, CXCL8 (IL-8), and CCL2 (MCP-1). Stimulation with GCs increases the activity of eNOS—a critical mediator of vascular integrity. Induction of ROS represents detrimental effects of GC excess on the vasculature.
GCs inhibit the endothelial production of several pro-inflammatory cytokines and chemokines including IL-6, IL-17F, CXCL8 (IL-8), and CCL2 (MCP-1) (84, 85, 90). GCs also downregulate ICAM-1, VCAM-1, and E-selectin (91–93). Moreover, GCs reduce levels of soluble forms of ICAM-1, VCAM-1, and E-selectin (sICAM-1, sVCAM-1, and sE-selectin) (94). Interestingly, GCs may also downregulate HLA-DR in IFN-γ-stimulated endothelial cells (92). Overall, GCs decrease leukocyte transmigration across the endothelium, thus limiting inflammation (95).
GCs also increase the activity of eNOS—a critical mediator of vascular integrity (96). Release of NO in the lumen inhibits platelet aggregation and leukocyte adhesion (97). Conversely, Iuchi et al. showed that GC excess induces reactive oxygen species and peroxynitrite formation, with possible detrimental effects on the vasculature (98).
Disruption of the endothelial barrier integrity is a common feature of various diseases including multiple sclerosis (MS) and stroke and leads to edema (99). GCs preserve endothelial barrier integrity through upregulation of junctional proteins such as occludin, claudin-5, and VE-cadherin (100–102) and down-regulation of MMP-9—an enzyme involved in junctional protein cleavage (103–105). GCs also induce endogenous MMP-9 inhibitors—TIMP-3 and TIMP-1 (103, 106). However, the induction of TIMP-1 seems controversial since contradictory results exist (106). Since MMP-9 is able to cleave CXCL8 (IL-8) and drastically potentiates its activities (107), it is tempting to speculate that downregulation of MMP-9 by GCs further affects neutrophil chemoattraction at the endothelial surface.
In summary, GCs influence endothelial barrier integrity, inhibit pro-inflammatory transcription factors, and induce protective molecules in endothelium (Figure 2). Investigation of molecular mechanisms of GC action in endothelium will enable comparison of these to the extensive data on regulation of inflammation by GC in other cell types.
5. Regulation of Endothelial GC Sensitivity
Although GCs have been the mainstay therapy for inflammatory diseases since 1950, GC treatment possesses significant challenges such as reduced efficacy and/or development of GC resistance (108). GC resistance has been reported in various diseases such as asthma and chronic obstructive pulmonary disease (COPD) (109) and in different cells types including peripheral blood mononuclear cells (PBMCs), B cells, and alveolar macrophages. A wide range of mechanisms causing GC resistance in these cell types have been described including increased GRβ expression, posttranslational GR modification, impaired nuclear translocation of GR, reduced MKP-1 expression, and decreased activity of HDAC2 (58, 110–113).
A limited number of studies addressed GC sensitivity of endothelium uncovering several mechanisms that regulate the response of endothelium to GCs (Figure 3). In dexamethasone-resistant HUVECs, GR interacted more strongly with BCL2-associated athanogene 1 (BAG1) protein than in dexamethasone-sensitive HUVECs (114). In general, BAG1 is a cytoplasmic protein, which can translocate to the nucleus and bind to DNA, decreasing GR transactivation (115). BAG1 also interferes with GR folding through interaction with HSP70 (116). Moreover, BAG1 may target interacting proteins for proteasomal degradation (117). In HUVECs, proteasome inhibition increased GR protein levels and abolished differences between GC-sensitive and GC-resistant cells, suggesting that BAG1-mediated proteasomal degradation of GR accounts in part for the human variability in endothelial sensitivity to GCs (114). In vivo, proteasome inhibition improved GC sensitivity of endothelium and protected against brain edema (118).
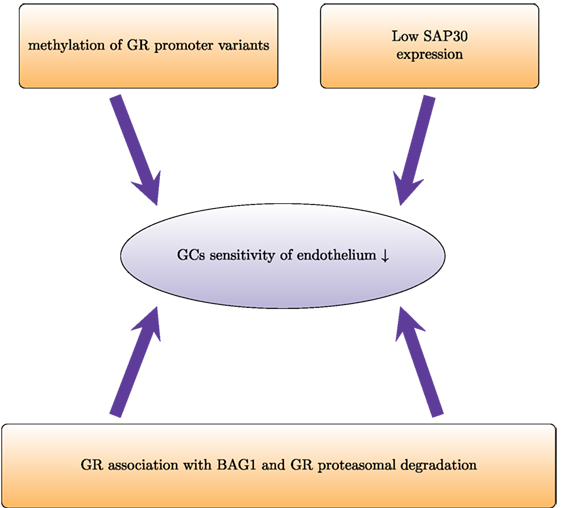
Figure 3. Proteasomal degradation and epigenetic modifications regulate glucocorticoid sensitivity in endothelial cells. Proteasomal degradation of the glucocorticoid receptor (GR) impairs GR activity and prevents physiological actions of GCs. In dexamethasone-resistant HUVECs, GR associates with the proteasomal recruiting protein BCL2-associated athanogene 1 (BAG1). This results in a shorter half-life of GR. In HUVECs, low induction of SAP30 (component of Sin3A-histone deacetylase complex) impairs GR-mediated transrepression and leads to lower GC sensitivity. Furthermore, higher methylation of the promoters of the different variants of the untranslated exon 1 of GR leads to lower expression and therefore decreased GC sensitivity.
Epigenetic mechanisms such as DNA methylation and histone modifications also regulate GC sensitivity of endothelial cells. Dexamethasone-sensitive and dexamethasone-resistant HUVECs have different GR promoter methylation patterns (119). The GR gene contains several variants of the untranslated exon 1:1A–1I (120). Each of these variants has its own promoter. Dexamethasone-sensitive cells show higher methylation levels of promoter 1D and lower methylation levels of promoter 1F. Pharmacological demethylation with 5-aza-2-deoxycytidine increased the mRNA expression of all isoforms (except 1D in resistant HUVECs) and enhanced the GC sensitivity (119). Another epigenetic mechanism that influences endothelial GC sensitivity involves Sin3A–HDAC. This multi-protein complex that regulates gene expression via histone deacetylation consists of SAP30, Sin3, histone deacetylases HDAC1 and HDAC2, histone-binding proteins RbAp46 and RbAp48, and other proteins. SAP30 represses gene transcription via tethering to gene promoters (121). Poor expression of Sap30 has been suggested as an explanation of impaired transrepression in HUVECs (122). Transgenic overexpression of SAP30 in HUVECs and analysis of its expression under inflammatory conditions would also improve the current understanding of how GC sensitivity is regulated in endothelium.
Variations in the expression of the GC-activating and -deactivating enzymes 11β-HSD1 and 11β-HSD2 have been described in endothelial cells (123). However, it remains unclear to what extent this affects the GC sensitivity.
In conclusion, the currently known mechanisms regulating GC sensitivity in endothelial cells summarized in Figure 3 include proteasomal degradation of GR and epigenetic modifications. However, the mechanisms that control GC sensitivity are complex and often cell type specific. The regulation of GC sensitivity in endothelial cells has not yet been sufficiently addressed, in particular under inflammatory conditions. Since endothelium plays a crucial role in inflammation, we believe that further studies on this subject will open up new perspectives for the development and improvement of the current treatment strategies.
6. Endothelial Response to GCs in Multiple Sclerosis
6.1. BBB Integrity in Multiple Sclerosis
Multiple sclerosis (MS) is mediated by autoreactive T cells that attack the myelin-sheath, but other inflammatory cells such as B cells, macrophages, and microglia may be as well involved in the pathogenesis. T cells infiltrate into the central nervous system (CNS) causing massive inflammation (124, 125). MS affects TJs and AJs protein expression in patients. Incubation of SVEC4–10 endothelial cells with serum from patients with MS in exacerbation phase reduced the levels of occludin and VE-cadherin (126). Claudin-5 expression was downregulated in immortalized human brain microvascular endothelial cells (TY09) after treatment with sera from the relapse phase of relapse-remitting MS (RRMS-R) or secondary progressive MS (SPMS) (127). Cytokines (TNF or IL-6) also reduced expression of TJ proteins in human brain microvascular endothelial cells (128).
6.2. Effects of GCs on BBB in Multiple Sclerosis
Episodes of neurological deficit (relapses) and periods of recovery (remission) characterize the most common form of MS. GCs remain standard therapy for MS (129). High-dose short-term GCs are prescribed to manage relapses although the opinions of neurologists and national guidelines on dose and duration of GC treatment vary (130, 131). In brain endothelial cells, GCs upregulate AnxA1 that regulates endothelial barrier integrity (86, 88). Indeed, AnxA1 KO mice show disrupted BBB as a result of actin cytoskeleton rearrangements (88). Junctional proteins also belong to GC target genes (132, 133). GCs induce occludin, claudin-5, and VE-cadherin in brain microvascular endothelial cells preventing endothelial barrier damage (100, 102). However, sera from patients with MS (with active disease and remission) downregulated expression of claudin-5 and occludin in cEND cells. Furthermore, dexamethasone failed to restore expression of these proteins (104). This suggests that treatment of cEND cells with sera from patients with MS impairs transactivation of junctional protein genes by GCs.
GCs downregulate expression of matrix metalloproteinase-9 (MMP-9, a key mediator of extracellular matrix remodeling and BBB disruption) in mouse brain endothelial cells incubated with sera from patients with MS (103, 104). Similarly, treatment with dexamethasone inhibits MMP-9 expression in primary rat brain endothelial cells stimulated with TNF or IL-1β (134). GCs also induce expression of the tissue inhibitor of metalloproteinases-1 (TIMP-1) (103).
High-dose methylprednisolone reduced expression of adhesion molecules (ICAM-1, VCAM-1) in human brain microvascular endothelial cells stimulated with TNF and in endothelial cells from patients with MS (91, 92). Methylprednisolone limited migration of PBMCs through endothelial barrier after 3 h, but this effect was less pronounced after 24 h (95). GCs also limit interactions of leukocytes with endothelium through downregulation of integrins (VLA-4, LFA-1) (135) and induction of AnxA1 that causes detachment of leukocytes from endothelial cells (87, 136).
Overall, in vitro studies indicate that GCs restore BBB integrity through induction of TJ proteins and protective molecules such as TIMPs. Inhibition of damaging molecules (MMP-9) and leukocyte transmigration constitute another important mode of GC action in MS.
7. Endothelial Response to GCs in Stroke
7.1. BBB Integrity in Ischemic Stroke
Ischemic stroke consists of two phases with pathological impact: ischemia and reperfusion. This induces oxidative stress in the brain and results in TJs damage and BBB disruption (15, 137). During the ischemic phase, loss of regional cerebral blood flow leads to deprivation of oxygen and nutrients in the surrounding tissue. Reperfusion reestablishes cerebral blood flow to the ischemic brain, but it also causes additional damage due to oxidative stress leading to increased blood–brain barrier permeability (15).
Oxygen and glucose deprivation and subsequent reperfusion disrupt TJs. In vitro experiments showed that hypoxia reduces claudin-5 levels and changes its localization in the plasma membrane in bEND.3 cells (138). MMP-2 and caveolin-1 (induced upon hypoxia) mediate occludin degradation and claudin-5 redistribution (139). Caspase-3, expressed by rat brain endothelial cells stimulated with TNF, is another enzyme involved in TJ disruption during ischemia (140, 141). Moreover, other molecules secreted in the brain such as VEGF and thrombin increase BBB permeability during stroke (142, 143).
7.2. Effects of GCs on BBB in Ischemic Stroke
Benefits from steroid use in stroke remain controversial according to clinical trials (144, 145). In vivo and in vitro experiments provide even more contradictory data. In one study, high-dose GCs administered within 2 h of transient cerebral ischemia, increase endothelial NOS activity and reduce infarct size. The underlying mechanism for activation of endothelial NOS by GCs involves the PI3K/Akt kinases (96). However, other studies reported lack of effect (145, 146) or even damaging effects (147) of GCs in stroke. GC signaling in myeloid and endothelial cells may aggravate ischemic infarcts (Figure 4) (147). Endothelial GR signaling reduces levels of junctional proteins in stroke since Tie-2-GR KO mice (with endothelium-specific GR deletion) produce more claudin-5, occluding, and caveolin-1 than WT mice after middle cerebral brain occlusion. This suggests that endogenous GCs, released post-injury, signal through endothelial GR and reduce levels of junctional proteins. Moreover, deletion of endothelial GR reduces infarct volume (147). These data are in contrast with the classic notion that synthetic GCs induce junctional proteins [see section 4 and Ref. (132)]. Thus, the classic inductive effects of GC on TJ proteins are overshadowed and even inverted during ischemic stroke. The reasons for this discrepancy remain unclear. GR deletion abolishes the physiological effects of endogenous and exogenous GCs and could possibly also affect GC-independent processes. Therefore, the consequences of GR deletion may not be in line with described effects of high doses of exogenous GCs.
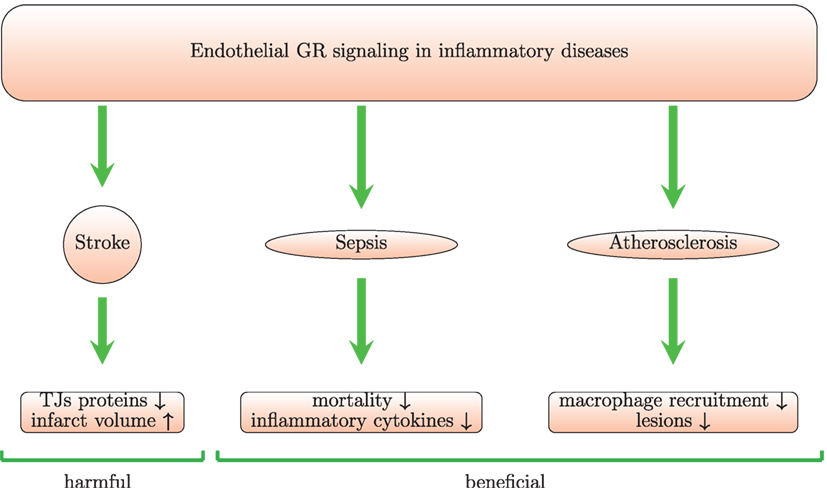
Figure 4. Endothelial GR signaling is beneficial in animal models of sepsis and atherosclerosis but harmful in stroke model. Mice with conditional GR deletion in endothelium provide a useful tool to study the role of endothelial GR signaling. In a model of stroke with these mice, it was shown that endothelial GR signaling increases infarct volume and reduces levels of junctional proteins (TJs) leading to increase in BBB permeability. In septic mice, the endothelial GR protects against LPS-induced inflammation and mortality. In an in vivo model of atherosclerosis, the endothelial GR reduces lesions and macrophage recruitment.
Kleinschnitz and colleagues showed occurrence of GC resistance at the hypoxic blood–brain barrier (118). Dexamethasone failed to induce junctional proteins in murine brain microvascular endothelial cells (cEND) following oxygen and glucose deprivation. Proteasome-dependent degradation of GR diminished transactivation of GC target genes. Bortezomib—a proteasome inhibitor—restored induction of junctional proteins by GCs after oxygen and glucose deprivation. Consequently, mice treated with bortezomib and GCs developed less edema (118).
In conclusion, the effects of GCs in stroke remain insufficiently understood. Beneficial, damaging effects and even GC resistance have been suggested.
8. Endothelial Response to GCs in Sepsis
8.1. Activation of Endothelium in Sepsis
Sepsis—a life-threatening organ dysfunction caused by a dysregulated host response to infection (148)—constitutes the most common cause of death in non-coronary intensive care units. The pathophysiology of sepsis remains poorly understood, and the host response mainly determines patient outcome. Alteration of endothelial cell function plays a crucial role in the pathophysiology of sepsis (149). Endothelial cells undergo excessive activation that involves release of pro-inflammatory mediators to recruit leukocytes and promote clotting to immobilize the pathogens upon tissue invasion. Functional changes observed after activation of endothelium include increased cell adhesion and leukocyte trafficking, loss of barrier function, and programed cell death (149). Cytokines such as VEGF-A, colony-stimulating factor 1 (CSF1), IL-1β, and TNF secreted by macrophages during tissue injury bind to receptors on endothelium resulting in endothelial activation and expression of adhesion molecules such as P-selectin, E-selectin, ICAM-1, and VCAM-1 (150, 151).
The NF-κB pathway plays a central role in the induction of cytokines and adhesion molecules mentioned above (152) and mediates multiple organ inflammation in sepsis (153). Endothelial ROS-mediated NF-κB and AP-1 activation as well as leukocyte recruitment in the lungs of septic mice require active MAPK kinase kinase 3 (154). Double transgenic mice that overexpress a degradation-resistant form of the NF-κB inhibitor IκBα in vascular endothelium showed endothelial selective NF-κB blockade, inhibition of adhesion molecules, reduced neutrophil infiltration into different organs, and decreased endothelial permeability after LPS challenge (153). In support, another study showed that inhibition of NF-κB may be beneficial to the microvasculature during endotoxemia. TNF treatment of mouse microvascular endothelial cells induced cytotoxicity and production of pro-inflammatory cytokines including IL-1β, IL-6, CCL5 (RANTES), and iNOS expression. NF-κB inhibition abrogated these changes (155).
Endothelial cells together with leukocytes recognize pathogens via pattern recognition receptors that interact with microbial products (156, 157). Bacterial products such as LPS from Gram-negative bacteria interact with toll-like receptors (TLRs) leading to the activation of inflammatory and coagulation pathways (158). In endothelial cells, TLR4 may be localized intracellularly and requires then LPS internalization for its activation. The uptake system also depends on LPS binding protein and soluble CD14 (159). LPS induces CCL2 (MCP-1) secretion in activated human endothelial cells (HUVECs) through non-receptor tyrosine kinase 2 (Pyk2) activation and p38 MAPK phosphorylation (160). LPS and non-LPS components of meningococci (human pathogens that cause fatal sepsis and meningitis) can upregulate E-selectin expression in human primary endothelial cells (HUVECs) leading to neutrophil recruitment. The LPS-independent mechanism involves activation of the activating transcription factor 2 (ATF2) via p38 MAPK (161).
Caveolin-1 (a signaling protein associated with caveolae—small invaginations of the plasma membrane in endothelial cells) regulates the inflammatory response to LPS mediated by TLR4. After LPS exposure, caveolin-1 undergoes phosphorylation and interacts with TLR4 resulting in NF-κB activation and pro-inflammatory cytokine expression in mouse lung microvascular endothelial cells. Mutation of the phosphorylation site in caveolin-1 protected mice from septic shock after LPS challenge (162). P120-catenin (p120)—a protein expressed in adherent cells—also regulates TLR4 signaling after LPS challenge. P120 blocks interaction of TLR4 with the adaptor molecule MyD88 thus inhibiting TLR4 signaling and NF-κB activation in endothelial cells. In mice injected with LPS, p120 levels were inversely correlated with inflammation severity. P120 suppression in lung endothelial cells in mice with siRNA resulted in greater susceptibility to LPS and higher mortality. P120 blockade also increased ICAM-1, TNF, and IL-6 expression after LPS stimulation (163).
8.2. Endothelial Barrier Breakdown in Sepsis
Hyperpermeability of endothelium is a hallmark of sepsis and causes tissue edema (149, 164). ANG1 and ANG2—important biomarkers of endothelial dysfunction—bind to Tie-2 receptor on endothelial cells. Under physiological conditions, ANG1 levels are higher and trigger pro-survival and anti-inflammatory pathways in endothelium. In sepsis, ANG2 is released from Weibel–Palade bodies. ANG2 Tie-2 interaction activates pro-inflammatory, pro-thrombic pathways and vascular leakage (150, 151). Septic patients show elevated serum levels of ANG2. Incubation of human microvascular endothelial cells with serum from septic patients disrupted endothelial barrier integrity. Moreover, ANG2 alone can induce such effects and excessive systemic ANG2 levels cause pulmonary leakage in healthy mice (165). Activated protein C (APC) upregulates Tie-2 and ANG1 expression and enhances endothelial barrier function. APC treatment of HUVECs reduces permeability and upregulates the tight junction-associated protein zona occludens (ZO-1) (166).
MMPs modulate endothelial barrier integrity through processing of adherens and tight junction proteins (167). MMP-8 inhibition protects mice from death in sepsis (168–170). Arpino and colleagues showed that inhibition of metalloproteinase-mediated adherens junctions’ disruption by tissue inhibitor of metalloproteinases-3 (TIMP-3) preserves normal endothelial barrier integrity. Pulmonary microvascular endothelial cells monolayers from TIMP-3 KO mice spontaneously displayed barrier dysfunction associated with disrupted surface VE-cadherin localization (167).
Bacterial products and host inflammatory mediators are also involved in increased endothelial permeability during sepsis. The neisserial antigen fragment C2 released after proteolysis of the surface-exposed protein neisserial heparin binding antigen (NHBA) from Neisseria meningitidis increases endothelial barrier permeability via internalization of VE-cadherin. ROS induction by C2 results in VE-cadherin phosphorylation and internalization contributing to severe vascular leakage observed in meningococcal sepsis (171). TNF or LPS treatment of human dermal microvascular endothelial cells also results in endothelial barrier disruption. Soluble VE-cadherin, released by a disintegrin and metalloproteinase 10 (ADAM10) in the culture supernatant, inhibits VE-cadherin binding thus contributing to breakdown of endothelial barrier during inflammation. Inhibition of ADAM10 blocks soluble VE-cadherin production and preserves endothelial barrier integrity upon TNF or LPS stimulation. In septic patients, plasma levels of soluble VE-cadherin correlate with disease severity (172).
Stimulation of human brain microvascular endothelial cells with LPS also reduces occludin and claudin-5 levels resulting in endothelial barrier breakdown. This is mediated by LPS-induced ROS. Indeed, adenoviral expression of superoxide dismutase or inhibition of NADPH oxidase by AMP-activated protein kinase (AMPK)—an enzyme that regulates redox homeostasis in endothelial cells—abolished these effects (173). Kang and colleagues showed that heat shock protein A12B (HSPA12B) also rescues endothelial barrier integrity upon LPS stimulation in HUVECs. HSPA12B induces VE-cadherin, myosin light chain, and CDC42. Moreover, HSPA12B silencing increased lung permeability in septic mice (174).
8.3. GC Therapy in Sepsis
Despite over 30 years of investigation, the use of GCs in septic patients remains controversial (175, 176). Prednisolone was beneficial in a model of Gram-negative sepsis in healthy volunteers. Treatment with prednisolone decreased plasma levels of TNF, IL-6, CXCL8 (IL-8), and CCL2 (MCP-1) and increased IL-10 levels. Prednisolone also blocked neutrophil activation (177). However, short treatment with high-dose GCs was ineffective in the majority of studies performed in sepsis. Long course treatment with a low dose seems a more promising strategy (178–180). The effects of GCs on coagulation, which is also involved in sepsis, constitute another controversial issue. GCs failed to attenuate the LPS-induced coagulation cascade in a human model of sepsis (177). In contrast, Bartko and coauthors showed that GCs locally reduce coagulation in a human model of lung inflammation with LPS (181). Different experimental settings used in these studies may in part explain contradictory results. Moreover, dexamethasone has been shown to enhance the stability of TF transcript in human monocytic cells stimulated with LPS (182), whereas in HUVECs, GR knockdown increased TF expression upon LPS stimulation (183). According to a systematic review, GCs appear to upregulate the activity of coagulation factors in healthy people, whereas during active inflammation GCs decrease levels of fibrinogen and von Willebrand factor and increase plasminogen activator inhibitor-1 (PAI-1) levels (184).
Contradictory effects of GCs in sepsis may be related to the promoter polymorphism of the NF-κB1 gene. Hydrocortisone failed to inhibit LPS-induced NF-κB translocation in monocytes from patients with a deletion in the promoter region of NF-κB1 gene (delATTG). As such, hydrocortisone treatment was also associated with increased 30-day mortality in these patients (185). This suggests that stratification of patients may help to predict the efficacy of GC treatment. This is also supported by an in vivo study with a sepsis model induced by cecal ligation and puncture. Stratification of mice according to the levels of circulating IL-6 not only predicted mortality but also efficacy of GC treatment, as only mice with high IL-6 responded to the GC treatment (186).
Nitric oxide (NO)-releasing derivatives of glucocorticoids show enhanced anti-inflammatory properties and may provide another pathway to improve GC therapy in sepsis (187). In particular, a NO-releasing derivative of dexamethasone, ND8008, was found to be more effective than dexamethasone in reducing the inflammatory response in LPS-stimulated mouse peritoneal macrophages and in an in vivo model of methicillin-resistant Staphylococcus aureus (MRSA) blood infection (188).
8.4. GR in Sepsis
Patients with septic shock exhibit high variability in GC responsiveness, and this may partially explain controversial effects of GC therapy in sepsis. Increased sickness was associated with lower GC sensitivity. However, GRβ and 11βHSD1 did not influence GC sensitivity in patients with sepsis (189). GR expression and translocation gradually decrease in experimental sepsis. This may explain why early dexamethasone treatment of septic mice improved clinical outcome compared to late treatment (190). Another study showed that GCs may worsen clinical outcome in septic patients with high GRβ levels through induction of miR-124 in T cells that downregulate GRα expression (191). However, other GC-regulated genes have been shown to have beneficial effects in sepsis. miR-511, induced by GCs via GR, confers protection in vivo against TNF-induced inflammation (192). In activated human CD4+ T cells, GCs induce miR-98 that inhibits the expression of pro-inflammatory mediators (193). Furthermore, GCs downregulate miR-155 resulting in reduction of inflammation in macrophages stimulated with LPS (194). GILZ protects SPRET/Ei mice against LPS-induced lethal inflammation (195). Pro-inflammatory cytokines and GCs, via GR, induce sphingosine kinase 1 (Sphk1) in a model of acute lung injury (ALI)—a complication of sepsis. This results in increase of sphingosine 1-phosphate levels (S1P), which bind to its receptor on the endothelium and triggers endothelial barrier enhancement (67). GR-mediated immunosuppression in macrophages improves survival during sepsis. Treatment of mice with a point mutation in the GR DNA-binding domain that impairs formation of transactivating GR dimers (GRdim) and mice lacking GR in macrophages with recombinant IL-1 receptor antagonist improves their survival after LPS challenge confirming that regulation of IL-1β in macrophages by GCs plays essential role in the control of sepsis (196). Conditional GR deletion in macrophages results in greater mortality and cytokine induction after LPS treatment (197).
8.5. Endothelial GR in Sepsis
Signaling through endothelial GR plays a beneficial role in models of sepsis (Figure 4). Mice with an endothelium-specific deletion of GR show increased mortality, higher levels of TNF, IL-6, and nitric oxide in comparison with control mice after challenge with LPS (183). GR deletion in HUVECs treated with LPS increases NF-κB levels and IL-6 levels suggesting that endothelial GR plays a crucial role in the regulation of the NF-κB pathway and nitric oxide synthesis. Moreover, mice lacking endothelial GR pretreated with dexamethasone show increased NF-κB activity after LPS injection (198). A possible limitation of this study is that GR deletion is driven by the Tie-1 promoter, which may be not fully endothelium-specific, as hematopoietic stem cells also express Tie-1 (199). Although GR-mediated induction of IκBα plays the main role in GC-induced suppression of NF-κB in monocytes and lymphocytes, in endothelial cells physical interaction between GR and NF-κB seems more important (200). Brostjan and colleagues also found that dexamethasone inhibits NF-κB-mediated transcription of E-selectin in porcine endothelial cells stimulated with LPS or TNF (93). Prednisolone reduced levels of soluble E-selectin and VCAM-1, respectively, in a human endotoxemia model (94). Maximal inhibition was achieved at 30 mg. However, levels of soluble ICAM-1 were not affected by the treatment (94). Interestingly, GCs also decreased LPS-induced expression of IL-17F in vitro in rat pulmonary endothelial cells and in vivo in a model of LPS-induced ALI. This was paralleled by inhibition of the lung injury (90).
In summary, endothelial GR signaling protects against LPS-induced sepsis (Figure 4). GCs also inhibit endothelial activation in in vitro and in vivo models of sepsis.
9. Endothelial Response to GCs in Vasculitis
9.1. Activation of Endothelium during Vasculitis
Vasculitis is a heterogeneous group of disorders characterized by self-sustaining inflammation of blood vessels. Small vessel vasculitis affects venules and capillaries and is a hallmark of immune-complex vasculitis or necrotizing vasculitis. Giant cell arteritis and Takayasu’s arteritis involve medium and large vessels (201). Diverse triggers such as bacterial and viral infections can cause vasculitis, but it can also occur as a primary disease of unknown origin (202, 203).
Activation of endothelium and leukocyte infiltration are hallmarks of vasculitis and are tightly interrelated. Anti-endothelial antibodies (AECA) constitute an important cause of endothelial activation. Stimulation of human kidney endothelial cells with AECA upregulated vascular adhesion protein-1 (VAP-1), CCL2 (MCP-1), MHC class I-related antigen A (MICA), and CXCL6 (GCP-2) expression. AECA also activated c-Jun, ATF2, and NF-κB (204). IgA anti-endothelial cell antibodies present in sera from children with active Henoch–Schönlein purpura induce ERK phosphorylation and CXCL8 (IL-8) expression in HUVECs (205).
Furthermore, several cytokines and enzymes in the plasma of vasculitis patients contribute to endothelial activation. Plasma from patients with Kawasaki disease [acute febrile childhood vasculitis characterized by the development of coronary artery abnormalities in 25–30% of untreated patients (206)] activates HUVECs and induces MMP-9 expression. This effect is counteracted by IFN-γ (207). Another study showed that sera from patients with acute Kawasaki disease induce ICAM-1 expression in HUVECs (208).
TNF-like weak inducer of apoptosis (TWEAK) serum levels of patients with acute Henoch–Schönlein purpura (the most common systemic vasculitis in children) are increased and correlate with severity of the disease. TWEAK induces CCL5 (RANTES) and CXCL8 (IL-8) expression in a human dermal endothelial cell line (209). TWEAK also upregulates E-selectin and ICAM-1 as shown in vitro and in an in vivo model of cutaneous vasculits (210).
Proteinase 3 (PR3), which is mainly produced by neutrophils and is the main autoantigen in granulomatosis with polyangiitis, binds also to endothelium and induces CXCL8 (IL-8) and CCL2 (MCP-1) that provide chemotactic signals for neutrophils and monocytes. PR3 also upregulates the expression of adhesion molecules such as ICAM-1 on endothelial cells (211, 212).
Among the signaling pathways induced in endothelial cells during vasculitis, NF-κB and endoplasmic reticulum stress response protein X-box binding protein-1 (XBP-1) appear important. In a local Shwartzman reaction model of TNF-induced vasculitis, XBP-1 contributes to the vascular damage as shown in mice with an endothelium-specific XBP-1 deletion (213). This transcription factor can be induced by TNF and upregulates the p65-subunit of NF-κB, resulting in sustained NF-κB-mediated transcription of pro-inflammatory molecules. XBP-1 also regulates leukocyte adhesion to endothelium in vitro and neutrophil infiltration in vivo (213).
9.2. Endothelial Injury in Vasculitis
Endothelial injury in vasculitis involves detachment of whole endothelial cells and apoptosis. These processes play an important role in the pathogenesis of vasculitis. Regulation of the balance between endothelial injury and endothelial repair is poorly studied. In children with active vasculitis the levels of VEGF, ANG2, circulating endothelial cells (CECs), and endothelial progenitor cells (EPCs) increase (214). EPCs migrate from bone marrow to the lesions. Chronic inflammation during vasculitis may impair EPC function and reduce endothelial repair capacity (215).
In a mouse model of Kawasaki disease, activation of Nlrp3 inflammasome results in endothelial injury (216). In small vessel vasculitis, neutrophil-derived myeloperoxidase contributes to endothelial injury. It causes loss of cell membrane integrity and morphological changes in endothelial cells (217). The transfer of myeloperoxidase from neutrophils to endothelial cells is cell contact dependent and is mediated by β2 integrin (218).
Bacterial products constitute another group of agents involved in endothelial injury during vasculitis. Haemophilus somnus infection often causes vasculitis and thrombosis in bovines (219). This pathogen and its lipooligosaccharide induce apoptosis in bovine pulmonary artery endothelial cells in vitro (220).
9.3. GC Therapy in Vasculitis
GC therapy remains the golden standard in vasculitis (221, 222). GCs are used to induce remission and as maintenance therapy (223). Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a group of unique diseases characterized by necrotizing inflammation of small blood vessels and presence of ANCA (224). GCs constitute the main therapy for AAV due to their rapid onset of action (225) and are an effective treatment for non-severe relapses in the majority of patients with AAV (226). However, there is no consensus among clinicians on induction doses and therapeutic schedules of GC therapy in AAV to induce and maintain remission (225). Patients with systemic necrotizing vasculitis who do not respond to a sole GC treatment are prescribed GC combined with cytotoxic agents (227).
In patients with giant cell arteritis, treatment with high-dose GCs reduced neutrophil adhesion to endothelium already after 48 h. However, 6 months after the GC dose the suppressor phenotype of neutrophils was less pronounced (228). Another study showed that GCs inhibit IL-17 release, but IFN-γ levels remained unaffected (222). A plethora of cytokines was identified in vasculitic lesions in giant cell arteritis with two dominant clusters: IL-6/IL-17 and IL-12/IFN-α. The IL-6/IL-17 cluster, active in early and untreated disease, is highly sensitive to GCs. Despite this inhibition, inflammation persists in patients with elevated levels of IL-12 and IFN-γ that are resistant to GCs (229). High doses of GCs inhibited IL-2 and IFN-γ from T cells, but IFN-γ mRNA levels were only slightly affected in severe combined immunodeficiency (SCID) mice engrafted with human temporal arteries. Moreover, activation of macrophages was only partially inhibited by GCs in this model, suggesting a strong GC-resistant component (230).
9.4. Suppression of Endothelial Response by GCs in Vasculitis
Endothelial activation clearly plays a prominent role in the vascular damage and overall pathophysiology of the various vasculitides. Although GCs have in general multiple beneficial effects on endothelial cells (see section 4), it remains unclear to what extent the therapeutic effects of GCs in vasculitis are mediated through the endothelium. For instance, one study indicated that GCs improve flow-mediated dilation, a marker of endothelial function in giant cell arteritis patients (231), but a later and larger study failed to obtain statistical significance of this parameter (232). In an in vitro model of vasculitis with HUVECs cocultured with neutrophils and stimulated with TNF or IL-1β, GCs inhibited E-selectin expression, which is involved in the damaging hyperadhesiveness of neutrophils to endothelium (233). Another study showed that dexamethasone inhibits IL-6 production and E-selectin expression in human coronary arterial endothelial cells stimulated with TNF (234).
Overall, more in vivo studies are needed to delineate the endothelial response and sensitivity to GCs in vasculitis. For instance, experiments with mice with endothelium-specific deletion of the GR may yield interesting insights.
10. Endothelial Response to GCs in Atherosclerosis
10.1. Activation of Endothelium during Atherosclerosis
Atherosclerosis affects large vessels and is the primary cause of heart disease and stroke. High-fat, high-cholesterol diet causes accumulation of lipoprotein particles and their aggregates in the intima at the lesion sites of predilection (235). Low density lipoproteins (LDL) passively diffuse through TJs and accumulate in the subendothelial matrix. Oxidized LDL stimulate the endothelium to produce adhesion molecules and reduce NO production, which is a critical mediator of vasorelaxation (235).
In animal models, endothelial cells in the arteries express VCAM-1 in response to cholesterol accumulation in the intima resulting in monocyte recruitment (236). Monocytes subsequently transmigrate into the intima, where they take up lipoproteins forming foam cells (235). Smooth muscle cells also express VCAM-1 that promotes recruitment and retention of mononuclear cells in the intima (237). Several pro-inflammatory mediators produced by endothelial cells play an important role in recruitment of immune cells (236). CCL2 (MCP-1) promotes monocyte recruitment and is the dominant mediator of macrophage accumulation in atherosclerotic lesions (238). CCR2 KO mice developed less atherosclerotic lesions (239). Macrophage migration inhibitory factor (MIF)—a key regulator in chronic and acute inflammation—also plays a role in the pathogenesis of atherosclerosis and is upregulated in monocytes and endothelial cells in human atherosclerotic lesions (240, 241). MIF blockade results in plaque regression and lower monocyte and T cell content in the lesions (242). Tissue factor (TF) triggers coagulation and its elevated levels are found in atherosclerotic plaques. IL-33 induces TF expression in HUVECs and coronary artery endothelial cells in a ST2 and NF-κB-dependent manner. In human carotid atherosclerotic plaques, TF levels positively correlate with IL-33 expression (243). Vascular endothelial cells and macrophages produce also T cell attractants CXCL9 (Mig), CXCL10 (IP-10), and CXCL11 (I-TAC). These chemokines play an important role in the recruitment of T cells within the lesions, since activated T cells express the corresponding receptor CXCR3 (244).
Bacterial infection-mediated inflammation has been shown to facilitate development of atherosclerosis via the following mechanism: flagellin (principal component of bacterial flagellum) induces interaction between NADPH oxidase-4 (Nox4) and TLR5 (245). Nox4 seems unusual because it releases H2O2 in contrast to other Nox family members that produce superoxide (246). Nox4 and TLR5 interaction results in H2O2 generation and induction of CXCL8 (IL-8) and ICAM-1 in human aortic endothelial cells. Nox4 deficiency results in resistance to flagellin-induced atherosclerosis in ApoE KO mice (245). Porphyromonas gingivalis, a bacterial species that causes periodontitis, impairs tube formation and induces adhesion molecules (ICAM-1, VCAM-1) in human coronary artery endothelial cells through TLR4 signaling (247).
10.2. The Role of Endogenous GCs in Atherosclerosis
The link between GCs and the cardiovascular system is complex. Epidemiological models showed correlation between endogenous plasma corticosteroid levels and severity of cardiovascular disease (248). The function of endogenous GCs in the development of atherosclerosis remains unclear. Adrenalectomy stimulates atherosclerosis in LDL receptor KO mice (249), but not in ApoE KO mice (250). Fine-tuning of GCs signaling by 11β-HSD1 (converts inactive GCs into active form) and 11β-HSD2 (converts active GCs into inactive form) also plays a role in atherosclerosis. Inhibition of 11β-HSD1 in atherosclerosis-prone apoE KO mice directly attenuates atherosclerotic plaques and decreases pro-inflammatory gene expression in the vasculature (251). 11β-HSD1 KO mice on apoE KO background challenged with western diet showed smaller plaques and lower macrophage content. Foam cell formation was decreased as well as expression of several TLRs (252). 11β-HSD1 inhibition reduces T cell infiltration, CCL2 (MCP-1) and VCAM-1 levels in atherosclerotic plaques (253).
10.3. Synthetic GCs in Atherosclerosis
The therapeutic effects of GCs in atherosclerosis development remain unclear. Mice with human-like lipoprotein metabolism treated with corticosterone showed a decrease in the total atherosclerotic lesion area and in macrophage content in the plaques. However, the treatment negatively affected body fat metabolism: it increased body weight, subcutaneous adipose tissue, and food intake (254). Therefore, a localized treatment would be beneficial, since this could help to avoid the systemic side-effects. This remains challenging because of the difficulty in implanting drug-eluting devices without subjecting vessels to mechanical damage. Kastrup and colleagues developed a drug-eluting bioadhesive gel that can be glued onto the inside surface of blood vessels (255). Atherosclerotic mice treated with this steroid-eluting adhesive gel show lower macrophage content, less plasma cytokines, and biomarkers of inflammation in the plaques.
Importantly, atherosclerosis may even occur as a side effect of GC therapy. For instance, in patients with rheumatoid arthritis GC therapy was associated with carotid plaque, arterial incompressibility, and dose-dependently also with all-cause and cardiovascular mortality (256, 257). As a possible explanation, GCs induce macrophage adipocyte fatty acid-binding protein (FABP4), a pro-atherogenic protein upregulated by long chain fatty acids and oxidized LDL. Pitavastatin is a HMG-CoA reductase inhibitor that lowers cholesterol levels and therefore also FABP4 levels. When combined with dexamethasone pitavastatin enhances in a synergistic manner FABP4 expression (258). Furthermore, co-treatment of apoE KO mice (challenged with high-fat diet) with dexamethasone and pitavastatin exacerbates diet-induced atherosclerotic lesions (259).
Although synthetic GCs reduce inflammation in animal models of atherosclerosis, metabolic side-effects may occur and overshadow beneficial effects. Localized GC treatment may therefore prove more effective and deserve further investigation.
10.4. Effects of GCs on Endothelium in Atherosclerosis
Endothelial GR suppresses atherogenesis in animals and plays an important role in the atheroprotective actions of endogenous GCs (Figure 4). ApoE KO mice lacking endothelial GR subjected to a high-fat diet developed more severe atherosclerotic lesions and showed increased macrophage recruitment (260).
GCs exert various anti-inflammatory actions such as inhibition of VCAM-1 and CCL2 (MCP-1) (see section 4) via endothelial GR. Two GC-inducible genes tristetraprolin (TTP, also known as ZFP36) and glucocorticoid-induced leucine zipper (GILZ, also known as TSC22 domain family protein-3) influence endothelial cell function during atherosclerosis (261, 262). GILZ is a key endogenous regulator of the immune response that interacts with signal transduction pathways (1). Degenerated aortocoronary saphenous vein bypass grafts (with inflammatory cell activation) showed lower protein and mRNA levels of GILZ in comparison with healthy controls. GILZ was also downregulated in HUVECs and macrophages upon TNF stimulation, and GILZ KO enhanced pro-inflammatory gene expression (261). TTP destabilizes pro-inflammatory cytokine mRNA through binding to AU-rich elements within their 3′-untranslated regions (263). LPS, GCs, and forskolin induce TTP expression in human aortic endothelial cells. Endothelial cells overlying atherosclerotic lesions in mice and humans express TTP that has been shown to reduce NF-κB activation during atherosclerosis (262).
In conclusion, the main protective effects of GCs on endothelium during inflammation include inhibition of pro-inflammatory molecules (e.g., VCAM-1 and MCP-1) and induction of protective molecules (such as GILZ and TTP). Moreover, endothelial GR protects against atherosclerosis in in vivo models. However, the overall effects of GCs in atherosclerosis are complex and also include deleterious metabolic effects.
11. Conclusion
A plethora of in vivo and in vitro studies have shown that the endothelium represents an important target for GCs. GCs induce junctional proteins and several protective molecules in endothelium. Furthermore, GCs inhibit endothelial expression of pro-inflammatory mediators such as cytokines, chemokines, and adhesion molecules. This greatly contributes to the beneficial effects of GCs in various inflammatory diseases, because the endothelium is a crucial player in inflammation. However, the beneficial actions of GCs may sometimes be overshadowed by detrimental side-effects and GC resistance, in particular on long-term treatment. GC resistance often arises as a consequence of disease in otherwise GC-sensitive individuals and tissues and is a major problem in clinical practice. Although various mechanisms, including proteasomal degradation and epigenetic modifications, have been reported to modulate GC sensitivity of endothelium, it appears that the current knowledge about the regulation of GC sensitivity in endothelial cells is still lagging behind our understanding of GC-mediated effects in other cell types such as macrophages and dendritic cells.
As future perspectives, we believe that further studies should be performed to investigate the precise interactions between GC signaling and pro- and anti-inflammatory pathways in endothelial cells. Such interactions are often cell type-specific and differ according to the cytokines and other stimuli used to activate the endothelial cells in vitro or according to the disease in vivo. Furthermore, exploring the regulation of GR activity through the variety of its isoforms and posttranslational modifications and by non-coding RNAs in endothelial cells is highly promising. These mechanisms fine-tune the interactions of GR with other signaling molecules, transcription factors, and cofactors and may dictate GC sensitivity and resistance. Finally, the variability of endothelial GC-mediated signaling between individuals and between endothelial cells within the body has to be addressed, since such information will help to define which diseases and which patient groups will benefit the most from GC therapy. Overall, this is a formidable task, but it will yield great benefits for fundamental science, pharmaceutical development, and precision medicine for patients.
Author Contributions
KZ: design and writing of all sections and figures except section 3 on GCs and GR. LVM: writing of section 3 on GCs and GR. KDB: design and writing of section 3 on GCs and GR and critical reading. GO: conception and design of the review and critical revision of text and figures. PVdS: conception, design, and participation in the writing of the review and critical revision of text and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work has been funded by the Fund for Scientific Research (F.W.O.-Vlaanderen), the Charcot Foundation, (Belgium) and the Geconcerteerde OnderzoeksActies (GOA 2013/014) of the Research Fund of the KU Leuven. LVM is a PhD student at UGent, KDB is a Research Professor at U Gent, and PVdS is a Research Professor at the KU Leuven.
References
1. Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheumatol (2011) 7(6):340–8. doi:10.1038/nrrheum.2011.59
2. Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, et al. The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev (2011) 10(6):305–10. doi:10.1016/j.autrev.2010.11.009
3. Pober JS, Sessa WC. Inflammation and the blood microvascular system. Cold Spring Harb Perspect Med (2015) 7(1):1–12. doi:10.1101/cshperspect.a016345
4. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res (2007) 100(2):158–73. doi:10.1161/01.RES.0000255691.76142.4a
5. Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, et al. An estimation of the number of cells in the human body. Ann Hum Biol (2013) 40:463–71. doi:10.3109/03014460.2013.807878
6. Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol (2004) 4(5):360–70. doi:10.1038/nri1354
7. Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med (2012) 2(1):a006429. doi:10.1101/cshperspect.a006429
8. Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol (2004) 5(4):261–70. doi:10.1038/nrm1357
9. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res (2007) 100(2):174–90. doi:10.1161/01.RES.0000255690.03436.ae
10. Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell (2013) 26(2):204–19. doi:10.1016/j.devcel.2013.06.017
11. Rafii S, Butler JM, Ding B-S. Angiocrine functions of organ-specific endothelial cells. Nature (2016) 529(7586):316–25. doi:10.1038/nature17040
12. Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol Rev (2012) 248(1):228–39. doi:10.1111/j.1600-065X.2012.01127.x
13. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun (2016). doi:10.1016/j.bbi.2016.03.010
14. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol (2010) 10(10):712–23. doi:10.1038/nri2852
15. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis (2008) 32(2):200–19. doi:10.1016/j.nbd.2008.08.005
16. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci (2008) 121(13):2115–22. doi:10.1242/jcs.017897
17. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res (2015) 107(3):331–9. doi:10.1093/cvr/cvv154
18. Pollheimer J, Bodin J, Sundnes O, Edelmann RJ, Skånland SS, Sponheim J, et al. Interleukin-33 drives a proinflammatory endothelial activation that selectively targets nonquiescent cells. Arterioscler Thromb Vasc Biol (2013) 33(2):47–55. doi:10.1161/ATVBAHA.112.253427
19. Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schönbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med (2002) 195(2):245–57. doi:10.1084/jem.20011022
20. Knipe L, Meli A, Hewlett L, Bierings R, Dempster J, Skehel P, et al. A revised model for the secretion of tPA and cytokines from cultured endothelial cells. Blood (2010) 116(12):2183–91. doi:10.1182/blood-2010-03-276170
21. Bridgewood C, Stacey M, Alase A, Lagos D, Graham A, Wittmann M. IL-36γ has proinflammatory effects on human endothelial cells. Exp Dermatol (2016) 1–20. doi:10.1111/exd.13228
22. Indraccolo S, Pfeffer U, Minuzzo S, Esposito G, Roni V, Mandruzzato S, et al. Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol (2007) 178(2):1122–35. doi:10.4049/jimmunol.178.2.1122
23. Loos T, Dekeyzer L, Struyf S, Schutyser E, Gijsbers K, Gouwy M, et al. TLR ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced CXCL9 in autoimmune arthritis. Lab Invest (2006) 86(9):902–16. doi:10.1038/labinvest.3700453
24. Mai J, Nanayakkara G, Lopez-Pastrana J, Li X, Li YF, Wang X, et al. Interleukin-17A promotes aortic endothelial cell activation via transcriptionally and post-translationally activating p38 mitogen-activated protein kinase (MAPK) pathway. J Biol Chem (2016) 291(10):4939–54. doi:10.1074/jbc.M115.690081
25. Lisinski TJ, Furie MB. Interleukin-10 inhibits proinflammatory activation of endothelium in response to Borrelia burgdorferi or lipopolysaccharide but not interleukin-1beta or tumor necrosis factor alpha. J Leukoc Biol (2002) 72(3):503–11.
26. Xie Y, Li Y, Cai X, Wang X, Li J. Interleukin-37 suppresses ICAM-1 expression in parallel with NF-κB down-regulation following TLR2 activation of human coronary artery endothelial cells. Int Immunopharmacol (2016) 38:26–30. doi:10.1016/j.intimp.2016.05.003
27. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15(11):692–704. doi:10.1038/nri3908
28. Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial PECAM-1 and its function in vascular physiology and atherogenic pathology. Exp Mol Pathol (2016) 100(3):409–15. doi:10.1016/j.yexmp.2016.03.012
29. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell (2011) 146(6):873–87. doi:10.1016/j.cell.2011.08.039
30. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi:10.1038/nrm2639
31. Hoeben ANN, Landuyt B, Highley MSM, Wildiers H, Oosterom ATVAN, Bruijn EADE, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev (2004) 56(4):549–80. doi:10.1124/pr.56.4.3.549
32. Olsson A-K, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol (2006) 7(May):359–71. doi:10.1038/nrm1911
33. Chen X, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell (2012) 22(1):146–57. doi:10.1016/j.devcel.2011.11.002
34. Kim I, Moon SO, Park SK, Chae SW, Koh GY. Angiopoietin-1 reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. Circ Res (2001) 89(6):477–89. doi:10.1097/INF.0b013e318211581e
35. Kim I, Oh JL, Ryu YS, So JN, Sessa WC, Walsh K, et al. Angiopoietin-1 negatively regulates expression and activity of tissue factor in endothelial cells. FASEB J (2002) 16(1):126–38. doi:10.1096/fj.01-0556fje
36. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med (2006) 12(2):235–9. doi:10.1038/nm1351
37. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science (1997) 277(5322):55–60. doi:10.1126/science.277.5322.55
38. Hu J, Van den Steen PE, Sang Q-XA, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov (2007) 6(6):480–98. doi:10.1038/nrd2308
39. Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol (2004) 4(8):617–29. doi:10.1038/nri1418
40. Rajashekhar G, Shivanna M, Kompella UB, Wang Y, Srinivas SP. Role of MMP-9 in the breakdown of barrier integrity of the corneal endothelium in response to TNF-α. Exp Eye Res (2014) 122:77–85. doi:10.1016/j.exer.2014.03.004
41. Genersch E, Hayess K, Neuenfeld Y, Haller H. Sustained ERK phosphorylation is necessary but not sufficient for MMP-9 regulation in endothelial cells: involvement of Ras-dependent and -independent pathways. J Cell Sci (2000) 113(Pt 23):4319–30.
42. Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol (2013) 48(3):222–72. doi:10.3109/10409238.2013.770819
43. Dejager L, Vandevyver S, Petta I, Libert C. Dominance of the strongest: inflammatory cytokines versus glucocorticoids. Cytokine Growth Factor Rev (2014) 25(1):21–33. doi:10.1016/j.cytogfr.2013.12.006
44. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev (2003) 24(4):488–522. doi:10.1210/er.2002-0006
45. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol (2013) 132(5):1033–44. doi:10.1016/j.jaci.2013.09.007
46. Lu NZ, Wardell SE, Burnstein KL, Defranco D, Fuller PJ, Giguere V, et al. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol Rev (2006) 58(4):782–97. doi:10.1124/pr.58.4.9
47. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol (2013) 380(1–2):41–54. doi:10.1016/j.mce.2012.12.014
48. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med (2005) 353:1711–23. doi:10.1056/NEJMra050541
49. De Bosscher K. Selective glucocorticoid receptor modulators. J Steroid Biochem Mol Biol (2010) 120(2–3):96–104. doi:10.1016/j.jsbmb.2010.02.027
50. Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (1998) 94(6):557–72. doi:10.1042/cs0940557
51. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev (2000) 21(1):55–89. doi:10.1210/edrv.21.1.0389
52. Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev (2004) 125(10–11 Spec. Iss):697–706. doi:10.1016/j.mad.2004.06.010
53. Joos GF, Brusselle GG, Van Hoecke H, Van Cauwenberge P, Bousquet J, Pauwels RA. Positioning of glucocorticosteroids in asthma and allergic rhinitis guidelines (versus other therapies). Immunol Allergy Clin North Am (2005) 25(3):597–612. doi:10.1016/j.iac.2005.06.001
54. Schacke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther (2002) 96(1):23–43. doi:10.1016/S0163-7258(02)00297-8
55. Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation (2014) 22:20–32. doi:10.1159/000362724
56. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology (2013) 154(3):993–1007. doi:10.1210/en.2012-2045
57. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet (2009) 373:1905–17. doi:10.1016/S0140-6736(09)60326-3
58. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol (2013) 131(3):636–45. doi:10.1016/j.jaci.2012.12.1564
59. De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A (2005) 102(44):15827–32. doi:10.1073/pnas.0505554102
60. Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A (2001) 98(12):6865–70. doi:10.1073/pnas.121455098
61. Haim YO, Unger ND, Souroujon MC, Mittelman M, Neumann D. Resistance of LPS-activated bone marrow derived macrophages to apoptosis mediated by dexamethasone. Sci Rep (2014) 4:1–10. doi:10.1038/srep04323
62. Haarman EG, Kaspers GJL, Pieters R, Rottier MMA, Veerman AJP. Glucocorticoid receptor alpha, beta and gamma expression vs in vitro glucocorticod resistance in childhood leukemia. Leukemia (2004) 18(3):530–7. doi:10.1038/sj.leu.2403225
63. Beger C, Gerdes K, Lauten M, Tissing WJE, Fernandez-Munoz I, Schrappe M, et al. Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br J Haematol (2003) 122(2):245–52. doi:10.1046/j.1365-2141.2003.04426.x
64. Sanchez-Vega B, Krett N, Rosen ST, Gandhi V, Sánchez-Vega B, Krett N, et al. Glucocorticoid receptor transcriptional isoforms and resistance in multiple myeloma cells. Mol Cancer Ther (2006) 5(12):3062–70. doi:10.1158/1535-7163.mct-06-0344
65. Beck IME, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev (2009) 30(7):830–82. doi:10.1210/er.2009-0013
66. Desmet SJ, Dejager L, Clarisse D, Thommis J, Melchers D, Bastiaensen N, et al. Cofactor profiling of the glucocorticoid receptor from a cellular environment. Methods Mol Biol (2014) 1204:83–94. doi:10.1007/978-1-4939-1346-6_8
67. Vettorazzi S, Bode C, Dejager L, Frappart L, Shelest E, Klaßen C, et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat Commun (2015) 6:1–12. doi:10.1038/ncomms8796
68. Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab (2013) 24(3):109–19. doi:10.1016/j.tem.2012.11.005
69. Mozo L, Suárez A, Gutiérrez C. Glucocorticoids up-regulate constitutive interleukin-10 production by human monocytes. Clin Exp Allergy (2004) 34(3):406–12. doi:10.1111/j.1365-2222.2004.01824.x
70. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell (2011) 145(2):224–41. doi:10.1016/j.cell.2011.03.027
71. Malkoski SP, Dorin RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol (1999) 13(10):1629–44. doi:10.1210/mend.13.10.0351
72. Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol (2007) 275(1–2):13–29. doi:10.1016/j.mce.2007.07.003
73. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol (2014) 12(3):1–14. doi:10.1371/journal.pbio.1001813
74. Presman DM, Ganguly S, Schiltz RL, Johnson TA, Karpova TS, Hager GL. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc Natl Acad Sci U S A (2016) 113(29):8236–41. doi:10.1073/pnas.1606774113
75. Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell (2012) 47(1):38–49. doi:10.1016/j.molcel.2012.04.021
76. De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF-kappa B-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci U S A (2000) 97(8):3919–24. doi:10.1073/pnas.97.8.3919
77. Löwenberg M, Stahn C, Hommes DW, Buttgereit F. Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids (2008) 73(9–10):1025–9. doi:10.1016/j.steroids.2007.12.002
78. Lepelletier Y, Zollinger R, Ghirelli C, Raynaud F, Hadj-Slimane R, Cappuccio A, et al. Toll-like receptor control of glucocorticoid-induced apoptosis in human plasmacytoid predendritic cells (pDCs). Blood (2010) 116(18):3389–97. doi:10.1182/blood-2010-05-282913
79. Brunetti M, Martelli N, Colasante A, Piantelli M, Musiani P, Aiello FB. Spontaneous and glucocorticoid-induced apoptosis in human mature T lymphocytes. Blood (1995) 86(11):4199–205.
80. Gruver-Yates AL, Quinn MA, Cidlowski JA. Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology (2014) 155(2):463–74. doi:10.1210/en.2013-1473
81. Saffar AS, Ashdown H, Gounni AS. The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr Drug Targets (2011) 12(4):556–62. doi:10.2174/138945011794751555
82. Gruver-Yates A, Cidlowski J. Tissue-specific actions of glucocorticoids on apoptosis: a double-edged sword. Cells (2013) 2(2):202–23. doi:10.3390/cells2020202
83. Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK, et al. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res (2000) 87(1):19–25. doi:10.1161/01.RES.87.1.19
84. Zakkar M, Luong LA, Chaudhury H, Ruud O, Punjabi PP, Anderson JR, et al. Dexamethasone arterializes venous endothelial cells by inducing mitogen-activated protein kinase phosphatase-1: a novel antiinflammatory treatment for vein grafts? Circulation (2011) 123(5):524–32. doi:10.1161/CIRCULATIONAHA.110.979542
85. Shi JX, Li JS, Hu R, Shi Y, Su X, Guo XJ, et al. Tristetraprolin is involved in the glucocorticoid-mediated interleukin 8 repression. Int Immunopharmacol (2014) 22(2):480–5. doi:10.1016/j.intimp.2014.07.031
86. Go KG, Ter Haar JG, de Ley L, Zuiderveen F, Parente L, Solito E, et al. The effect of steroid treatment on lipocortin immunoreactivity of rat brain. Mediators Inflamm (1994) 3(3):177–80. doi:10.1155/S0962935194000232
87. Gavins FNE, Hickey MJ. Annexin A1 and the regulation of innate and adaptive immunity. Front Immunol (2012) 3:1–11. doi:10.3389/fimmu.2012.00354
88. Cristante E, McArthur S, Mauro C, Maggioli E, Romero IA, Wylezinska-Arridge M, et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc Natl Acad Sci U S A (2013) 110(3):832–41. doi:10.1073/pnas.1209362110
89. Liu NK, Zhang YP, Han S, Pei J, Xu LY, Lu PH, et al. Annexin A1 reduces inflammatory reaction and tissue damage through inhibition of phospholipase A(2) activation in adult rats following spinal cord injury. J Neuropathol Exp Neurol (2007) 66(10):932–43. doi:10.1097/nen.0b013e3181567d59
90. You Q, Zhang D, Cheng Niu C, Ming Zhu Z, Wang N, Yue Y, et al. Expression of IL-17A and IL-17F in lipopolysaccharide-induced acute lung injury and the counteraction of anisodamine or methylprednisolone. Cytokine (2014) 66(1):78–86. doi:10.1016/j.cyto.2013.12.019
91. Dufour A, Corsini E, Gelati M, Ciusani E, Zaffaroni M, Giombini S, et al. Modulation of ICAM-1, VCAM-1 and HLA-DR by cytokines and steroids on HUVECs and human brain endothelial cells. J Neurol Sci (1998) 157(2):117–21. doi:10.1016/S0022-510X(98)00059-8
92. Gelati M, Corsini E, Dufour A, Massa G, Giombini S, Solero CL, et al. High-dose methylprednisolone reduces cytokine-induced adhesion molecules on human brain endothelium. Can J Neurol Sci (2000) 27(3):241–4. doi:10.1017/S0317167100000883
93. Brostjan C, Anrather J, Csizmadia V, Natarajan G, Winkler H. Glucocorticoids inhibit E-selectin expression by targeting NF-kappaB and not ATF/c-Jun. J Immunol (1997) 158:3836–44.
94. Lemaire LC, de Kruif MD, Giebelen IA, van Zoelen MAD, Veer CV, van der Poll T. Differential dose-dependent effects of prednisolone on shedding of endothelial adhesion molecules during human endotoxemia. Immunol Lett (2008) 121(2):93–6. doi:10.1016/j.imlet.2008.09.005
95. Gelati M, Corsini E, De Rossi M, Masini L, Bernardi G, Massa G, et al. Methylprednisolone acts on peripheral blood mononuclear cells and endothelium in inhibiting migration phenomena in patients with multiple sclerosis. Arch Neurol (2002) 59(5):774–80. doi:10.1001/archneur.59.5.774
96. Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, et al. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest (2002) 110(11):1729–38. doi:10.1172/JCI200215481
97. Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation (2006) 113(13):1708–14. doi:10.1161/CIRCULATIONAHA.105.602532
98. Iuchi T, Akaike M, Mitsui T, Ohshima Y, Shintani Y, Azuma H, et al. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res (2003) 92(1):81–7. doi:10.1161/01.RES.0000050588.35034.3C
99. Ishihara H, Kubota H, Lindberg RLP, Leppert D, Gloor SM, Errede M, et al. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor A 2 involves matrix metalloproteinases and tight junction proteins. J Neuropathol Exp Neurol (2008) 67(5):435–48. doi:10.1097/NEN.0b013e31816fd622
100. Förster C, Burek M, Romero IA, Weksler B, Couraud P-O, Drenckhahn D. Differential effects of hydrocortisone and TNF alpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol (2008) 586(7):1937–49. doi:10.1113/jphysiol.2007.146852
101. Kashiwamura Y, Sano Y, Abe M, Shimizu F, Haruki H, Maeda T, et al. Hydrocortisone enhances the function of the blood-nerve barrier through the up-regulation of claudin-5. Neurochem Res (2011) 36:849–55. doi:10.1007/s11064-011-0413-6
102. Blecharz KG, Drenckhahn D, Forster CY. Glucocorticoids increase VE-cadherin expression and cause cytoskeletal rearrangements in murine brain endothelial cEND cells. J Cereb Blood Flow Metab (2008) 28(6):1139–49. doi:10.1038/jcbfm.2008.2
103. Förster C, Kahles T, Kietz S, Drenckhahn D. Dexamethasone induces the expression of metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular endothelial cell line cEND. J Physiol (2007) 580(3):937–49. doi:10.1113/jphysiol.2007.129007
104. Blecharz KG, Haghikia A, Stasiolek M, Kruse N, Drenckhahn D, Gold R, et al. Glucocorticoid effects on endothelial barrier function in the murine brain endothelial cell line cEND incubated with sera from patients with multiple sclerosis. Mult Scler (2010) 16(3):293–302. doi:10.1177/1352458509358189
105. Cui N, Wang H, Long Y, Su L, Liu D. Dexamethasone suppressed LPS-induced matrix metalloproteinase and its effect on endothelial glycocalyx shedding. Mediators Inflamm (2015) 2015:1–8. doi:10.1155/2015/912726
106. Hartmann C, El-Gindi J, Lohmann C, Lischper M, Zeni P, Galla HJ. TIMP-3: a novel target for glucocorticoid signaling at the blood-brain barrier. Biochem Biophys Res Commun (2009) 390(2):182–6. doi:10.1016/j.bbrc.2009.08.158
107. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood (2000) 96(8):2673–81.
108. De Bosscher K, Beck IM, Ratman D, Berghe WV, Libert C. Activation of the glucocorticoid receptor in acute inflammation: the SEDIGRAM concept. Trends Pharmacol Sci (2016) 37(1):4–16. doi:10.1016/j.tips.2015.09.002
109. Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest (2008) 134(2):394–401. doi:10.1378/chest.08-0440
110. Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax (2008) 63(9):784–90. doi:10.1136/thx.2007.090027
111. Xu Q, Goleva E, Ou L-S, Li L-B, Leung DYM. CD56+ cells induce steroid resistance in B cells exposed to IL-15. J Immunol (2004) 172(11):7110–5. doi:10.4049/jimmunol.172.11.7110
112. Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, et al. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med (1997) 186(9):1567–74. doi:10.1084/jem.186.9.1567
113. Milara J, Lluch J, Almudever P, Freire J, Xiaozhong Q, Cortijo J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol (2014) 134(2):314.e–22.e. doi:10.1016/j.jaci.2014.02.001
114. Mata-Greenwood E, Stewart JM, Steinhorn RH, Pearce WJ. Role of BCL2-associated athanogene 1 in differential sensitivity of human endothelial cells to glucocorticoids. Arterioscler Thromb Vasc Biol (2013) 33(5):1046–55. doi:10.1161/ATVBAHA.113.301247
115. Schmidt U, Wochnik GM, Rosenhagen MC, Young JC, Hartl FU, Holsboer F, et al. Essential role of the unusual DNA-binding motif of BAG-1 for inhibition of the glucocorticoid receptor. J Biol Chem (2003) 278(7):4926–31. doi:10.1074/jbc.M212000200
116. Kanelakis KC, Morishima Y, Dittmar KD, Galigniana MD, Takayama S, Reed JC, et al. Differential effects of the hsp70-binding protein BAG-1 on glucocorticoid receptor folding by the hsp90-based chaperone machinery. J Biol Chem (1999) 274(48):34134–40. doi:10.1074/jbc.274.48.34134
117. Lüders J, Demand J, Höhfeld J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J Biol Chem (2000) 275(7):4613–7. doi:10.1074/jbc.275.7.4613
118. Kleinschnitz C, Blecharz K, Kahles T, Schwarz T, Kraft P, Göbel K, et al. Glucocorticoid insensitivity at the hypoxic blood-brain barrier can be reversed by inhibition of the proteasome. Stroke (2011) 42(4):1081–9. doi:10.1161/STROKEAHA.110.592238
119. Mata-Greenwood E, Jackson PN, Pearce WJ, Zhang L. Endothelial glucocorticoid receptor promoter methylation according to dexamethasone sensitivity. J Mol Endocrinol (2015) 55(2):133–46. doi:10.1530/JME-15-0124
120. Presul E, Schmidt S, Kofler R, Helmberg A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J Mol Endocrinol (2007) 38(1–2):79–90. doi:10.1677/jme.1.02183
121. Zhang Y, Sun Z-W, Iratni R, Erdjument-Bromage H, Tempst P, Hampsey M, et al. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol Cell (1998) 1(7):1021–31. doi:10.1016/S1097-2765(00)80102-1
122. Koenen P, Barczyk K, Wolf M, Roth J, Viemann D. Endothelial cells present an innate resistance to glucocorticoid treatment: implications for therapy of primary vasculitis. Ann Rheum Dis (2012) 71(5):729–36. doi:10.1136/annrheumdis-2011-200530
123. Gong R, Morris DJ, Brem AS. Variable expression of 11beta hydroxysteroid dehydrogenase (11beta-HSD) isoforms in vascular endothelial cells. Steroids (2008) 73(11):1187–96. doi:10.1016/j.steroids.2008.05.009
124. Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta (2011) 1812(2):220–30. doi:10.1016/j.bbadis.2010.07.019
125. Schweingruber N, Reichardt SD, Lühder F, Reichardt HM. Mechanisms of glucocorticoids in the control of neuroinflammation. J Neuroendocrinol (2011) 24(1):174–82. doi:10.1111/j.1365-2826.2011.02161.x
126. Minagar A, Ostanin D, Long AC, Jennings M, Kelley RE, Sasaki M, et al. Serum from patients with multiple sclerosis downregulates occludin and VE-cadherin expression in cultured endothelial cells. Mult Scler (2003) 9(3):235–8. doi:10.1191/1352458503ms916oa
127. Shimizu F, Tasaki A, Sano Y, Ju M, Nishihara H, Oishi M, et al. Sera from remitting and secondary progressive multiple sclerosis patients disrupt the blood-brain barrier. PLoS One (2014) 9(3):1–9. doi:10.1371/journal.pone.0092872
128. Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS One (2014) 9(7):1–13. doi:10.1371/journal.pone.0101815
129. Dietrich J-B. Endothelial cells of the blood-brain barrier: a target for glucocorticoids and estrogens? Front Biosci (2004) 9:684–93. doi:10.2741/1272
130. Galea I, Ward-Abel N, Heesen C. Relapse in multiple sclerosis. BMJ (2015) 350:1–8. doi:10.1136/bmj.h1765
131. Tremlett HL, Luscombe DK, Wiles CM. Use of corticosteroids in multiple sclerosis by consultant neurologists in the United Kingdom. J Neurol Neurosurg Psychiatry (1998) 65(3):362–5. doi:10.1136/jnnp.65.3.362
132. Salvador E, Shityakov S, Förster C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res (2014) 355(3):597–605. doi:10.1007/s00441-013-1762-z
133. Witt K, Sandoval K. Steroids and the blood-brain barrier: therapeutic implications. Adv Pharmacol (2014) 71:361–82. doi:10.1016/bs.apha.2014.06.018
134. Harkness KA, Adamson P, Sussman JD, Davies-Jones GA, Greenwood J, Woodroofe MN. Dexamethasone regulation of matrix metalloproteinase expression in CNS vascular endothelium. Brain (2000) 123(4):698–709. doi:10.1093/brain/123.4.698
135. Elovaara I, Lalla M, Spare E, Lehtimaki T, Dastidar P. Methylprednisolone reduces adhesion molecules in blood and cerebrospinal fluid in patients with MS. Neurology (1998) 51(6):1703–8. doi:10.1212/WNL.51.6.1703
136. Gavins FNE, Yona S, Kamal AM, Flower RJ, Perretti M. Leukocyte antiadhesive actions of annexin 1: aLXR- and FPR-related anti-inflammatory mechanisms. Blood (2003) 101(10):4140–7. doi:10.1182/blood-2002-11-3411
137. Shin JA, Yoon JC, Kim M-S, Park E-M. Activation of classical estrogen receptor subtypes reduces tight junction disruption of brain endothelial cells under ischemia/reperfusion injury. Free Radic Biol Med (2016) 92:78–89. doi:10.1016/j.freeradbiomed.2016.01.010
138. Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol (2007) 170(4):1389–97. doi:10.2353/ajpath.2007.060693
139. Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci (2012) 32(9):3044–57. doi:10.1523/JNEUROSCI.6409-11.2012
140. Wiggins-Dohlvik K, Merriman M, Shaji CA, Alluri H, Grimsley M, Davis ML, et al. Tumor necrosis factor-a disruption of brain endothelial cell barrier is mediated through matrix metalloproteinase-9. Am J Surg (2014) 208(6):954–60. doi:10.1016/j.amjsurg.2014.08.014
141. Zehendner CM, Librizzi L, de Curtis M, Kuhlmann CRW, Luhmann HJ. Caspase-3 contributes to ZO-1 and Cl-5 tight-junction disruption in rapid anoxic neurovascular unit damage. PLoS One (2011) 6(2):1–11. doi:10.1371/journal.pone.0016760
142. Bai Y, Zhu X, Chao J, Zhang Y, Qian C, Li P, et al. Pericytes contribute to the disruption of the cerebral endothelial barrier via increasing VEGF expression: implications for stroke. PLoS One (2015) 10(4):1–20. doi:10.1371/journal.pone.0124362
143. Hun Lee J, Won S, Stein DG. Progesterone attenuates thrombin-induced endothelial barrier disruption in the brain endothelial cell line bEnd.3: the role of tight junction proteins and the endothelial protein C receptor. Brain Res (2015) 1613:73–80. doi:10.1016/j.brainres.2015.04.002
144. Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev (2011) 9:CD000064. doi:10.1002/14651858.CD000064.pub2
145. Poungvarin N. Steroids have no role in stroke therapy. Stroke (2004) 35(1):229–30. doi:10.1161/01.STR.0000105931.81723.26
146. Norris JW. Steroids may have a role in stroke therapy. Stroke (2004) 35(1):228–9. doi:10.1161/01.STR.0000105930.29558.DB
147. Sorrells SF, Caso JR, Munhoz CD, Hu CK, Tran KV, Miguel ZD, et al. Glucocorticoid signaling in myeloid cells worsens acute CNS injury and inflammation. J Neurosci (2013) 33(18):7877–89. doi:10.1523/JNEUROSCI.4705-12.2013
148. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA (2016) 315(8):801–10. doi:10.1001/jama.2016.0287
149. Aird W. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Sepsis (2003) 101(10):3765–77. doi:10.1182/blood-2002-06-1887.Supported
150. Opal SM, van der Poll T. Endothelial barrier dysfunction in septic shock. J Intern Med (2015) 277(3):277–93. doi:10.1111/joim.12331
151. Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence (2013) 4:507–16. doi:10.4161/viru.24530
152. Fish JE, Cybulsky MI. Taming endothelial activation with a microRNA. J Clin Invest (2012) 122(6):1967–70. doi:10.1172/JCI63818
153. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med (2008) 205(6):1303–15. doi:10.1084/jem.20071393
154. Mannam P, Zhang X, Shan P, Zhang Y, Shinn AS, Zhang Y, et al. Endothelial MKK3 is a critical mediator of lethal murine endotoxemia and acute lung injury. J Immunol (2013) 190(3):1264–75. doi:10.4049/jimmunol.1202012
155. Zhou Z, Gengaro P, Wang W, Wang X-Q, Li C, Faubel S, et al. Role of NF-kappaB and PI 3-kinase/Akt in TNF-alpha-induced cytotoxicity in microvascular endothelial cells. Am J Physiol Renal Physiol (2008) 295(4):932–41. doi:10.1152/ajprenal.00066.2008
156. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol (2001) 1(2):135–45. doi:10.1038/35100529
157. Salvador B, Arranz A, Francisco S, Córdoba L, Punzón C, Llamas MÁ, et al. Modulation of endothelial function by toll like receptors. Pharmacol Res (2016) 108:46–56. doi:10.1016/j.phrs.2016.03.038
158. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell toll-like receptor pathways in sepsis. Innate Immun (2015) 21(8):827–46. doi:10.1177/1753425915606525
159. Dunzendorfer S, Lee H-K, Soldau K, Tobias PS. Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J (2004) 18(10):1117. doi:10.1096/fj.03-1263fje
160. Anand AR, Bradley R, Ganju RK. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-kappaB-dependent pathway. Mol Immunol (2009) 46(5):962–8. doi:10.1016/j.molimm.2008.09.022
161. Jacobsen MC, Dusart PJ, Kotowicz K, Bajaj-Elliott M, Hart SL, Klein NJ, et al. A critical role for ATF2 transcription factor in the regulation of E-selectin expression in response to non-endotoxin components of Neisseria meningitidis. Cell Microbiol (2016) 18(1):66–79. doi:10.1111/cmi.12483
162. Jiao H, Zhang Y, Yan Z, Wang Z-G, Liu G, Minshall RD, et al. Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signaling and sepsis-induced lung inflammation. J Immunol (2013) 191(12):6191–9. doi:10.4049/jimmunol.1300873
163. Wang Y-L, Malik AB, Sun Y, Hu S, Reynolds AB, Minshall RD, et al. Innate immune function of the adherens junction protein p120-catenin in endothelial response to endotoxin. J Immunol (2011) 186(5):3180–7. doi:10.4049/jimmunol.1001252
164. Li Y, Hadden C, Cooper A, Ahmed A, Wu H, Lupashin VV, et al. Sepsis-induced elevation in plasma serotonin facilitates endothelial hyperpermeability. Sci Rep (2016) 6:1–13. doi:10.1038/srep22747
165. Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med (2006) 3(3):356–70. doi:10.1371/journal.pmed.0030046
166. Minhas N, Xue M, Fukudome K, Jackson CJ. Activated protein C utilizes the angiopoietin/Tie2 axis to promote endothelial barrier function. FASEB J (2010) 24(3):873–81. doi:10.1096/fj.09-134445
167. Arpino V, Mehta S, Wang L, Bird R, Rohan M, Pape C, et al. Tissue inhibitor of metalloproteinases 3-dependent microvascular endothelial cell barrier function is disrupted under septic conditions. Am J Physiol Heart Circ Physiol (2016) 10:1455–67. doi:10.1152/ajpheart.00796.2015
168. Hu J, Van Den Steen PE, Dillen C, Opdenakker G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochem Pharmacol (2005) 70(4):535–44. doi:10.1016/j.bcp.2005.04.047
169. Qiu Z, Chen J, Xu H, Van den Steen PE, Opdenakker G, Wang M, et al. Inhibition of neutrophil collagenase/MMP-8 and gelatinase B/MMP-9 and protection against endotoxin shock. J Immunol Res (2014) 2014:1–10. doi:10.1155/2014/747426
170. Vandenbroucke RE, Dejonckheere E, Van Lint P, Demeestere D, Van Wonterghem E, Vanlaere I, et al. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci (2012) 32(29):9805–16. doi:10.1523/JNEUROSCI.0967-12.2012
171. Casellato A, Rossi Paccani S, Barrile R, Bossi F, Ciucchi L, Codolo G, et al. The C2 fragment from Neisseria meningitidis antigen NHBA increases endothelial permeability by destabilizing adherens junctions. Cell Microbiol (2014) 16(6):925–37. doi:10.1111/cmi.12250
172. Flemming S, Burkard N, Renschler M, Vielmuth F, Meir M, Schick MA, et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc Res (2015) 107(1):32–44. doi:10.1093/cvr/cvv144
173. Zhao Z, Hu J, Gao X, Liang H, Liu Z. Activation of AMPK attenuates lipopolysaccharide-impaired integrity and function of blood-brain barrier in human brain microvascular endothelial cells. Exp Mol Pathol (2014) 97(3):386–92. doi:10.1016/j.yexmp.2014.09.006
174. Kang Q, Chen Y, Zhang X, Yu G, Wan X, Wang J, et al. Heat shock protein A12B protects against sepsis-induced impairment in vascular endothelial permeability. J Surg Res (2016) 202(1):87–94. doi:10.1016/j.jss.2015.12.034
175. Marik PE. Glucocorticoids in sepsis: dissecting facts from fiction. Crit Care (2011) 15(3):1–3. doi:10.1186/cc10428
176. Annane D. The role of ACTH and corticosteroids for sepsis and septic shock: an update. Front Endocrinol (2016) 7:1–7. doi:10.3389/fendo.2016.00070
177. de Kruif MD, Lemaire LC, Giebelen IA, van Zoelen MAD, Pater JM, van den Pangaart PS, et al. Prednisolone dose-dependently influences inflammation and coagulation during human endotoxemia. J Immunol (2007) 178(3):1845–51. doi:10.4049/jimmunol.178.3.1845
178. Annane D. Glucocorticoids in the treatment of severe sepsis and septic shock. Curr Opin Crit Care (2005) 11(5):449–53. doi:10.1097/01.ccx.0000176691.95562.43
179. Annane D, Bellissant E, Bollaert PE, Briegel J, Keh D, Kupfer Y. Corticosteroids for severe sepsis and septic shock: a systematic review and meta-analysis. BMJ (2004) 329(7464):2–9. doi:10.1136/bmj.38181.482222.55
180. Patel GP, Balk RA. Systemic steroids in severe sepsis and septic shock. Am J Respir Crit Care Med (2012) 185(2):133–9. doi:10.1164/rccm.201011-1897CI
181. Bartko J, Schoergenhofer C, Schwameis M, Buchtele N, Wojta J, Schabbauer G, et al. Dexamethasone inhibits endotoxin-induced coagulopathy in human lungs. J Thromb Haemost (2016) 14:1–7. doi:10.1111/jth.13504
182. Reddy KV, Bhattacharjee G, Schabbauer G, Hollis A, Kempf K, Tencati M, et al. Dexamethasone enhances LPS induction of tissue factor expression in human monocytic cells by increasing tissue factor mRNA stability. J Leukoc Biol (2004) 76(1):145–51. doi:10.1189/jlb.0204068
183. Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A (2013) 110(1):306–11. doi:10.1073/pnas.1210200110
184. van Zaane B, Nur E, Squizzato A, Gerdes VEA, BÜLler HR, Dekkers OM, et al. Systematic review on the effect of glucocorticoid use on procoagulant, anti-coagulant and fibrinolytic factors. J Thromb Haemost (2010) 8(11):2483–93. doi:10.1111/j.1538-7836.2010.04034.x
185. Schäfer ST, Gessner S, Scherag A, Rump K, Frey UH, Siffert W, et al. Hydrocortisone fails to abolish NF-κB1 protein nuclear translocation in deletion allele carriers of the NFKB1 promoter polymorphism (-94ins/delATTG) and is associated with increased 30-day mortality in septic shock. PLoS One (2014) 9(8):1–9. doi:10.1371/journal.pone.0104953
186. Osuchowski MF, Connett J, Welch K, Granger J, Remick DG. Stratification is the key: inflammatory biomarkers accurately direct immunomodulatory therapy in experimental sepsis. Crit Care Med (2009) 37(5):1567–73. doi:10.1097/CCM.0b013e31819df06b
187. Paul-Clark MJ, Mancini L, Del Soldato P, Flower RJ, Perretti M. Potent antiarthritic properties of a glucocorticoid derivative, NCX-1015, in an experimental model of arthritis. Proc Natl Acad Sci U S A (2002) 99(3):1677–82. doi:10.1073/pnas.022641099
188. Yang Y, Li H, Sun H, Gong L, Guo L, Shi Y, et al. A novel nitro-dexamethasone inhibits agr system activity and improves therapeutic effects in MRSA sepsis models without antibiotics. Sci Rep (2016) 6:1–13. doi:10.1038/srep20307
189. Cohen J, Pretorius CJ, Ungerer JPJ, Cardinal J, Blumenthal A, Presneill J, et al. Glucocorticoid sensitivity is highly variable in critically ill patients with septic shock and is associated with disease severity. Crit Care Med (2016) 44:1034–41. doi:10.1097/CCM.0000000000001633
190. Bergquist M, Nurkkala M, Rylander C, Kristiansson E, Hedenstierna G, Lindholm C. Expression of the glucocorticoid receptor is decreased in experimental Staphylococcus aureus sepsis. J Infect (2013) 67(6):574–83. doi:10.1016/j.jinf.2013.07.028
191. Ledderose C, Möhnle P, Limbeck E, Schütz S, Weis F, Rink J, et al. Corticosteroid resistance in sepsis is influenced by microRNA-124–induced downregulation of glucocorticoid receptor-α. Crit Care Med (2012) 40(10):2745–53. doi:10.1097/CCM.0b013e31825b8ebc
192. Puimège L, Van Hauwermeiren F, Steeland S, Van Ryckeghem S, Vandewalle J, Lodens S, et al. Glucocorticoid-induced microRNA-511 protects against TNF by down-regulating TNFR1. EMBO Mol Med (2015) 7(8):1004–17. doi:10.15252/emmm.201405010
193. Davis TE, Kis-Toth K, Szanto A, Tsokos GC. Glucocorticoids suppress T cell function by up-regulating microRNA-98. Arthritis Rheum (2013) 65(7):1882–90. doi:10.1002/art.37966
194. Zheng Y, Xiong S, Jiang P, Liu R, Liu X, Qian J, et al. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: a novel anti-inflammation mechanism. Free Radic Biol Med (2012) 52(8):1307–17. doi:10.1016/j.freeradbiomed.2012.01.031
195. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimège L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med (2013) 5(3):456–70. doi:10.1002/emmm.201201683
196. Kleiman A, Hubner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J (2012) 26(2):722–9. doi:10.1096/fj.11-192112
197. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood (2007) 109(10):4313–9. doi:10.1182/blood-2006-10-048215
198. Goodwin JE, Feng Y, Velazquez H, Zhou H, Sessa WC. Loss of the endothelial glucocorticoid receptor prevents the therapeutic protection afforded by dexamethasone after LPS. PLoS One (2014) 9(10):1–7. doi:10.1371/journal.pone.0108126
199. Batard P, Sansilvestri P, Scheinecker C, Knapp W, Debili N, Vainchenker W, et al. The Tie receptor tyrosine kinase is expressed by human hematopoietic progenitor cells and by a subset of megakaryocytic cells. Blood (1996) 87(6):2212–20.
200. Brostjan C, Anrather J, Csizmadia V, Stroka D, Soares M, Bach FH, et al. Glucocorticoid-mediated repression of NFkappaB activity in endothelial cells does not involve induction of IkappaBalpha synthesis. J Biol Chem (1996) 271(32):19612–6. doi:10.1074/jbc.271.32.19612
201. Maugeri N, Rovere-Querini P, Baldini M, Sabbadini MG, Manfredi AA. Translational mini-review series on immunology of vascular disease: mechanisms of vascular inflammation and remodelling in systemic vasculitis. Clin Exp Immunol (2009) 156(3):395–404. doi:10.1111/j.1365-2249.2009.03921.x
202. Keller TT, Mairuhu ATA, de Kruif MD, Klein SK, Gerdes VEA, ten Cate H, et al. Infections and endothelial cells. Cardiovasc Res (2003) 60(1):40–8. doi:10.1016/S0008-6363(03)00354-7
203. Khasnis A, Langford CA. Update on vasculitis. J Allergy Clin Immunol (2009) 123(6):1226–36. doi:10.1016/j.jaci.2009.04.024
204. Holmén C, Elsheikh E, Christensson M, Liu J, Johansson A-S, Qureshi AR, et al. Anti endothelial cell autoantibodies selectively activate SAPK/JNK signalling in Wegener’s granulomatosis. J Am Soc Nephrol (2007) 18(9):2497–508. doi:10.1681/ASN.2006111286
205. Yang YH, Huang YH, Lin YL, Wang LC, Chuang YH, Yu HH, et al. Circulating IgA from acute stage of childhood Henoch-Schönlein purpura can enhance endothelial interleukin (IL)-8 production through MEK/ERK signalling pathway. Clin Exp Immunol (2006) 144(2):247–53. doi:10.1111/j.1365-2249.2006.03076.x
206. Galeotti C, Bayry J, Kone-Paut I, Kaveri SV. Kawasaki disease: aetiopathogenesis and therapeutic utility of intravenous immunoglobulin. Autoimmun Rev (2010) 9(6):441–8. doi:10.1016/j.autrev.2009.12.004
207. Sakata K, Hamaoka K, Ozawa S, Niboshi A, Yahata T, Fujii M, et al. Matrix metalloproteinase-9 in vascular lesions and endothelial regulation in Kawasaki disease. Circ J (2010) 74(8):1670–5. doi:10.1253/circj.CJ-09-0980
208. Inoue H, Kato M, Okada Y, Morikawa A, Kimura Y. Sera from patients with Kawasaki disease induce intercellular adhesion molecule-1 but not Fas in human endothelial cells. Int Arch Allergy Immunol (2001) 125:250–5. doi:10.1159/000053823
209. Chen T, Guo ZP, Li MM, Li JY, Jiao XY, Zhang YH, et al. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK), an important mediator of endothelial inflammation, is associated with the pathogenesis of Henoch-Schonlein purpura. Clin Exp Immunol (2011) 166(1):64–71. doi:10.1111/j.1365-2249.2011.04442.x
210. Chen T, Guo ZP, Li L, Li MM, Wang TT, Jia RZ, et al. TWEAK enhances E-selectin and ICAM-1 expression, and May contribute to the development of cutaneous vasculitis. PLoS One (2013) 8(2):1–11. doi:10.1371/journal.pone.0056830
211. Berger SP, Seelen MA, Hiemstra PS, Gerritsma JS, Heemskerk E, van der Woude FJ, et al. Proteinase 3, the major autoantigen of Wegener’s granulomatosis, enhances IL-8 production by endothelial cells in vitro. J Am Soc Nephrol (1996) 7(5):694–701.
212. Taekema-Roelvink ME, Kooten C, Kooij SV, Heemskerk E, Daha MR. Proteinase 3 enhances endothelial monocyte chemoattractant protein-1 production and induces increased adhesion of neutrophils to endothelial cells by upregulating intercellular cell adhesion molecule-1. J Am Soc Nephrol (2001) 12(5):932–40.
213. Ziogas A, Muders MH, Economopoulou M, Sprott D, Grossklaus S, Siegert G, et al. Brief report: endothelial-specific X-box binding protein 1 deficiency limits tumor necrosis factor-induced leukocyte recruitment and vasculitis. Arthritis Rheumatol (2015) 67(12):3279–85. doi:10.1002/art.39309
214. Clarke LA, Hong Y, Eleftheriou D, Shah V, Arrigoni F, Klein NJ, et al. Endothelial injury and repair in systemic vasculitis of the young. Arthritis Rheum (2010) 62(6):1770–80. doi:10.1002/art.27418
215. Hong Y, Eleftheriou D, Klein NJ, Brogan PA. Impaired function of endothelial progenitor cells in children with primary systemic vasculitis. Arthritis Res Ther (2015) 17(1):1–10. doi:10.1186/s13075-015-0810-3
216. Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta (2015) 1853(2):396–408. doi:10.1016/j.bbamcr.2014.11.012
217. Pitanga TN, de Aragão França L, Rocha VCJ, Meirelles T, Borges VM, Gonçalves MS, et al. Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol (2014) 15:1–10. doi:10.1186/1471-2121-15-21
218. Jerke U, Rolle S, Purfürst B, Luft FC, Nauseef WM, Kettritz R. B2 integrin-mediated cell-cell contact transfers active myeloperoxidase from neutrophils to endothelial cells. J Biol Chem (2013) 288(18):12910–9. doi:10.1074/jbc.M112.434613
219. Gogolewski RP, Leathers CW, Liggitt HD, Corbeil LB. Experimental Haemophilus somnus pneumonia in calves and immunoperoxidase localization of bacteria. Vet Pathol (1987) 24(3):250–6.
220. Sylte MJ, Leite FP, Kuckleburg CJ, Inzana TJ, Czuprynski CJ. Caspase activation during Haemophilus somnus lipooligosaccharide-mediated apoptosis of bovine endothelial cells. Microb Pathog (2003) 35(6):285–91. doi:10.1016/j.micpath.2003.08.002
221. Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med (2007) 146(9):621–30. doi:10.7326/0003-4819-146-9-200705010-00004
222. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation (2010) 121(7):906–15. doi:10.1161/CIRCULATIONAHA.109.872903
223. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis (2009) 68(3):310–7. doi:10.1136/ard.2008.088351
224. Kallenberg CGM. Pathogenesis of ANCA-associated vasculitis, an update. Clin Rev Allergy Immunol (2011) 41(2):224–31. doi:10.1007/s12016-011-8258-y
225. Keller SF, Miloslavsky EM. Corticosteroids in antineutrophil cytoplasmic antibody-associated vasculitis. Rheum Dis Clin North Am (2016) 42(1):91–101. doi:10.1016/j.rdc.2015.08.010
226. Miloslavsky EM, Specks U, Merkel PA, Seo P, Spiera R, Langford CA, et al. Outcomes of nonsevere relapses in antineutrophil cytoplasmic antibody-associated vasculitis treated with glucocorticoids. Arthritis Rheumatol (2015) 67(6):1629–36. doi:10.1002/art.39104
227. Guillevin L. Treatment of severe and/or refractory ANCA-associated vasculitis. Curr Rheumatol Rep (2014) 16(8):1–6. doi:10.1007/s11926-014-0430-y
228. Nadkarni S, Dalli J, Hollywood J, Mason JC, Dasgupta B, Perretti M. Investigational analysis reveals a potential role for neutrophils in giant-cell arteritis disease progression. Circ Res (2014) 114(2):242–8. doi:10.1161/CIRCRESAHA.114.301374
229. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol (2013) 9(12):731–40. doi:10.1038/nrrheum.2013.161
230. Brack A, Rittner HL, Younge BR, Kaltschmidt C, Weyand CM, Goronzy JJ. Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras. J Clin Invest (1997) 99(12):2842–50. doi:10.1172/JCI119477
231. Gonzalez-Juanatey C, Llorca J, Garcia-Porrua C, Sanchez-Andrade A, Martín J, Gonzalez-Gay MA. Steroid therapy improves endothelial function in patients with biopsy-proven giant cell arteritis. J Rheumatol (2006) 33(1):74–8.
232. Hafner F, Haas E, Belaj K, Froehlich H, Gary T, Eller P, et al. Endothelial function and carotid intima-media thickness in giant-cell arteritis. Eur J Clin Invest (2014) 44(3):249–56. doi:10.1111/eci.12227
233. Heimbürger M, Lärfars G, Bratt J. Prednisolone inhibits cytokine-induced adhesive and cytotoxic interactions between endothelial cells and neutrophils in vitro. Clin Exp Immunol (2000) 119(3):441–8. doi:10.1046/j.1365-2249.2000.01165.x
234. Makata H, Ichiyama T, Uchi R, Takekawa T, Matsubara T, Furukawa S. Anti-inflammatory effect of intravenous immunoglobulin in comparison with dexamethasone in vitro: implication for treatment of Kawasaki disease. Naunyn Schmiedebergs Arch Pharmacol (2006) 373(5):325–32. doi:10.1007/s00210-006-0084-z
236. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol (2006) 6(7):508–19. doi:10.1038/nri1882
237. Li H, Cybulsky MI, Gimbrone MA, Libby P. Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am J Pathol (1993) 143(6):1551–9.
238. Dhawan L, Liu B, Blaxall BC, Taubman MB. A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein-1 mRNA stability. J Biol Chem (2007) 282(14):10146–52. doi:10.1074/jbc.M605925200
239. Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature (1998) 394(6696):894–7. doi:10.1038/29788
240. Santos LL, Morand EF. Macrophage migration inhibitory factor: a key cytokine in RA, SLE and atherosclerosis. Clin Chim Acta (2009) 399(1–2):1–7. doi:10.1016/j.cca.2008.09.014
241. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med (2011) 17(11):1410–22. doi:10.1038/nm.2538
242. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13(5):587–96. doi:10.1038/nm1567
243. Stojkovic S, Kaun C, Basilio J, Rauscher S, Hell L, Krychtiuk KA, et al. Tissue factor is induced by interleukin-33 in human endothelial cells: a new link between coagulation and inflammation. Sci Rep (2016) 6:1–11. doi:10.1038/srep25171
244. Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, et al. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest (1999) 104(8):1041–50. doi:10.1172/JCI6993
245. Kim J, Seo M, Kim SK, Bae YS. Flagellin-induced NADPH oxidase 4 activation is involved in atherosclerosis. Sci Rep (2016) 6(April):1–16. doi:10.1038/srep25437
246. Nisimoto Y, Diebold BA, Constentino-Gomes D, Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry (2014) 53(31):5111–20. doi:10.1021/bi500331y
247. Huang C-Y, Shih C-M, Tsao N-W, Lin Y-W, Shih C-C, Chiang K-H, et al. The GroEL protein of Porphyromonas gingivalis regulates atherogenic phenomena in endothelial cells mediated by upregulating toll-like receptor 4 expression. Am J Transl Res (2016) 8(2):384–404.
248. Hadoke PWF, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol (2009) 156(5):689–712. doi:10.1111/j.1476-5381.2008.00047.x
249. van der Sluis RJ, van Puijvelde GH, Van Berkel TJC, Hoekstra M. Adrenalectomy stimulates the formation of initial atherosclerotic lesions: reversal by adrenal transplantation. Atherosclerosis (2012) 221(1):76–83. doi:10.1016/j.atherosclerosis.2011.12.022
250. Hoekstra M, Frodermann V, Van Den Aardweg T, Van Der Sluis RJ, Kuiper J. Leukocytosis and enhanced susceptibility to endotoxemia but not atherosclerosis in adrenalectomized APOE knockout mice. PLoS One (2013) 8(11):1–12. doi:10.1371/journal.pone.0080441
251. Luo MJ, Thieringer R, Springer MS, Wright SD, Hermanowski-Vosatka A, Plump A, et al. 11β-HSD1 inhibition reduces atherosclerosis in mice by altering proinflammatory gene expression in the vasculature. Physiol Genomics (2013) 45(1):47–57. doi:10.1152/physiolgenomics.00109.2012
252. García RA, Search DJ, Lupisella JA, Ostrowski J, Guan B, Chen J, et al. 11β-hydroxysteroid dehydrogenase type 1 gene knockout attenuates atherosclerosis and in vivo foam cell formation in hyperlipidemic apoE/ mice. PLoS One (2013) 8(2):e53192. doi:10.1371/journal.pone.0053192
253. Kipari T, Hadoke PWF, Iqbal J, Man TY, Miller E, Coutinho AE, et al. 11 Beta-hydroxysteroid dehydrogenase type 1 deficiency in bone marrow-derived cells reduces atherosclerosis. FASEB J (2013) 27(4):1519–31. doi:10.1096/fj.12-219105
254. Auvinen HE, Wang Y, Princen H, Romijn JA, Havekes LM, Smit JWA, et al. Both transient and continuous corticosterone excess inhibit atherosclerotic plaque formation in APOE*3-Leiden. CETP mice. PLoS One (2013) 8(5):e63882. doi:10.1371/journal.pone.0063882
255. Kastrup CJ, Nahrendorf M, Figueiredo JL, Lee H, Kambhampati S, Lee T, et al. Painting blood vessels and atherosclerotic plaques with an adhesive drug depot. Proc Natl Acad Sci U S A (2012) 109(52):21444–9. doi:10.1073/pnas.1217972110
256. del Rincón I, Battafarano DF, Restrepo JF, Erikson JM, Escalante A. Glucocorticoid dose thresholds associated with all-cause and cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatol (2014) 66(2):264–672. doi:10.1002/art.38210
257. del Rincón I, O’Leary DH, Haas RW, Escalante A. Effect of glucocorticoids on the arteries in rheumatoid arthritis. Arthritis Rheum (2004) 50(12):3813–22. doi:10.1002/art.20661
258. Hu W, Zhou X, Jiang M, Duan Y, Chen Y, Li X, et al. Statins synergize dexamethasone-induced adipocyte fatty acid binding protein expression in macrophages. Atherosclerosis (2012) 222(2):434–43. doi:10.1016/j.atherosclerosis.2012.03.007
259. Zhang W, Wang X, Hu W, Liu L, Li X, Han J, et al. Co-treatment of pitavastatin and dexamethasone exacerbates the high-fat diet-induced atherosclerosis in apoE-deficient mice. J Cardiovasc Pharmacol (2015) 66(2):189–95. doi:10.1097/FJC.0000000000000264
260. Goodwin JE, Zhang X, Rotllan N, Feng Y, Zhou H, Fernández-Hernando C, et al. Endothelial glucocorticoid receptor suppresses atherogenesis–brief report. Arterioscler Thromb Vasc Biol (2015) 35(4):779–82. doi:10.1161/ATVBAHA.114.304525
261. Hahn RT, Hoppstadter J, Hirschfelder K, Hachenthal N, Diesel B, Kessler SM, et al. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis (2014) 234(2):391–400. doi:10.1016/j.atherosclerosis.2014.03.028
262. Zhang H, Taylor WR, Joseph G, Caracciolo V, Gonzales DM, Sidell N, et al. mRNA-binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells. Arterioscler Thromb Vasc Biol (2013) 33(6):1212–20. doi:10.1161/ATVBAHA.113.301496
Keywords: endothelium, glucocorticoids, glucocorticoid resistance, inflammation, cytokines, tight junctions, adhesion molecules
Citation: Zielińska KA, Van Moortel L, Opdenakker G, De Bosscher K and Van den Steen PE (2016) Endothelial Response to Glucocorticoids in Inflammatory Diseases. Front. Immunol. 7:592. doi: 10.3389/fimmu.2016.00592
Received: 07 October 2016; Accepted: 29 November 2016;
Published: 14 December 2016
Edited by:
Rudolf Lucas, Augusta University, USAReviewed by:
Jean-Marc Cavaillon, Institut Pasteur, FranceAndrea Huwiler, University of Bern, Switzerland
Takato Takenouchi, National Agriculture and Food Research Organization, Japan
Copyright: © 2016 Zielińska, Van Moortel, Opdenakker, De Bosscher and Van den Steen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe E. Van den Steen, cGhpbGlwcGUudmFuZGVuc3RlZW5Aa3VsZXV2ZW4uYmU=