 Catharina C. Gross1
Catharina C. Gross1 Andreas Schulte-Mecklenbeck1
Andreas Schulte-Mecklenbeck1 Heinz Wiendl1
Heinz Wiendl1 Emanuela Marcenaro2,3
Emanuela Marcenaro2,3 Nicole Kerlero de Rosbo4
Nicole Kerlero de Rosbo4 Antonio Uccelli2,4,5
Antonio Uccelli2,4,5 Alice Laroni2,4,5*
Alice Laroni2,4,5*
- 1Department of Neurology, University Hospital Münster, Münster, Germany
- 2Centre of Excellence for Biomedical Research, University of Genova, Genova, Italy
- 3Department of Experimental Medicine, University of Genova, Genova, Italy
- 4Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genova, Genova, Italy
- 5IRCCS San Martino-IST, Genova, Italy
There is increasing evidence that natural killer (NK) cells exhibit regulatory features. Among them, CD56bright NK cells have been suggested to play a major role in controlling T cell responses and maintaining homeostasis. Dysfunction in NK cell-mediated regulatory features has been recently described in untreated multiple sclerosis (MS), suggesting a contribution to MS pathogenesis. Moreover, biological disease-modifying treatments effective in MS apparently enhance the frequencies and/or regulatory function of NK cells, further pointing toward an immunoprotective role of NK cells in MS. Here, we summarize the current knowledge on the regulatory functions of NK cells, based on their interactions with other cells belonging to the innate compartment, as well as with adaptive effector cells. We review the more recent data reporting disruption of NK cell/T cell interactions in MS and discuss how disease-modifying treatments for MS affect NK cells.
Natural Killer (NK) Cells as Controllers of (Auto)Immune Responses
Regulatory Features of NK Cells
Autoimmune reactivity occurs in every subject, but only 5–10% of humans develop an autoimmune disease (1). Keeping autoreactive cells under control and, thus, preventing them to cause disease is the task of specialized immune cell subsets, called “regulatory” cells. The best characterized regulatory immune cell populations belong to the adaptive immune system (IS) and include regulatory T cells (Tregs), type-1 regulatory T cells, and regulatory B cells. However, there is increasing evidence that the innate IS also plays an important role in controlling autoreactive cells.
An important population of regulatory immune cells belongs to the natural killer (NK) cells. These, so-called CD56bright NK cells, owe their name to high surface expression of CD56 (also known as neural cell adhesion molecule), are CD16−/dim, express the inhibitory receptor NKG2A, and do not express killer cell immunoglobulin-like receptors (KIR). CD56bright NK cells were first considered “immunoregulatory” by Cooper et al., due to increased production of cytokines and reduced cytotoxicity compared to CD56dim NK cells (2).
It is now established that CD56bright NK cells regulate other immune cells belonging to both the innate and adaptive IS. Although most studies on CD56bright NK cell function have been conducted ex vivo with cells purified from peripheral blood, lymph nodes (LNs) are likely a key place where CD56bright NK cells exert their regulatory function (3), since they preferentially home to parafollicular T cell areas (4) where immune responses develop. In addition to CD56bright NK cells, the major NK cell subset in peripheral blood, CD56dim NK cells, which derive from CD56bright NK cells and are more differentiated, also exert regulatory functions as discussed below.
Interactions between Regulatory NK Cells and Innate Immune Cells
CD56bright NK cells express receptors for cytokines such as interleukin (IL)-12, IL-15, and IL-18 (5–7), which are produced by activated antigen-presenting cells (APCs). These cytokines can trigger proliferation of CD56bright NK cells and their production of molecules such as IFN-γ, IL-10 and IL-13, TNF-β, and GM-CSF (2). In this context, Ferlazzo et al. demonstrated that dendritic cells (DCs) are a key source of IL-12 and IL-15 for activation of CD56bright NK cells (8), and we have shown that DC-derived IL-27 can modulate proliferation and function of these cells (9). Thus, APCs modulate NK cell functions and phenotype (10–13). Infections most likely modulate the function of CD56bright NK cells indirectly through APCs, because co-culturing CD56bright with APCs activated via TLR4 (macrophages, DC) or TLR9 (plasmacytoid DCs) stimulates their proliferation and cytokine production (2, 8, 14, 15). Conversely, activated NK cells modulate the function of APCs: they stimulate monocytes to produce TNF-α (16) and kill immature DCs in a process called DC editing (17, 18).
Interactions between Regulatory NK Cells and Adaptive Immune Cells
Natural killer cells also interact with adaptive effector cells. IFN-γ secreted by CD56bright NK cells in response to T cell-derived IL-2 has been demonstrated to stimulate T cells in LNs (4). Along this line, increased local bioavailability of IL-2 by blocking the IL-2Rα chain (CD25) on recently activated T cells upon treatment with daclizumab is associated with expansion and activation of CD56bright NK cells in multiple sclerosis (MS) patients (19–21). Indeed, while T cells express the high-affinity form of the IL-2 receptor, which comprises CD25, CD56bright NK cells express both high-affinity and intermediate-affinity (not comprising CD25) forms of the IL-2 receptor (20, 22). Thus, upon daclizumab treatment, NK cells are stimulated through binding of IL-2 to their intermediate-affinity receptor. This results in control of T cell activation through direct killing (19, 21), which, for the CD56bright subset, involves release of cytotoxic granzyme K (23). Furthermore, IL-27-stimulated CD56bright NK cells have been shown to suppress the proliferation of autologous CD4+ T cells in a contact-dependent manner associated with increased perforin content (9). CD56bright NK cells, stimulated with the pro-inflammatory cytokines IL-12 and IL-15, prevent autologous CD4+ T cell proliferation through a cytotoxic mechanism involving the engagement of the natural cytotoxicity receptors (NCRs), such as NKp30 and NKp46 (24), on NK cells and the release of granzyme B (25). CD56bright NK cells were also shown to inhibit proliferation of autologous CD4+ T cells by secreting the immunosuppressive molecule adenosine. Inhibition of CD38 (“ADP ribosyl-cyclase”), an enzyme involved in the production of adenosine, restored proliferation of T cells in the presence of CD56bright NK cells (26). While these studies described the effects of CD56bright NK cells on T cells undergoing activation, others reported direct cytotoxicity of CD56bright NK cells on previously activated T cells. Nielsen and coauthors found that killing of pre-activated T cells by CD56bright NK cells involves the activating receptors NKG2D, LFA-1, and TRAIL and is enhanced when blocking NKG2A (27). Another study demonstrated that both CD56bright and CD56dim NK cells kill autologous antigen-activated CD4+ T cells through engagement of DNAM-1 and 2B4 and their cognate receptors CD155 and CD48, respectively (21). These and other studies reveal that different stimuli activate NK cells toward cytotoxicity and/or suppression of T cell proliferation (Figure 1).
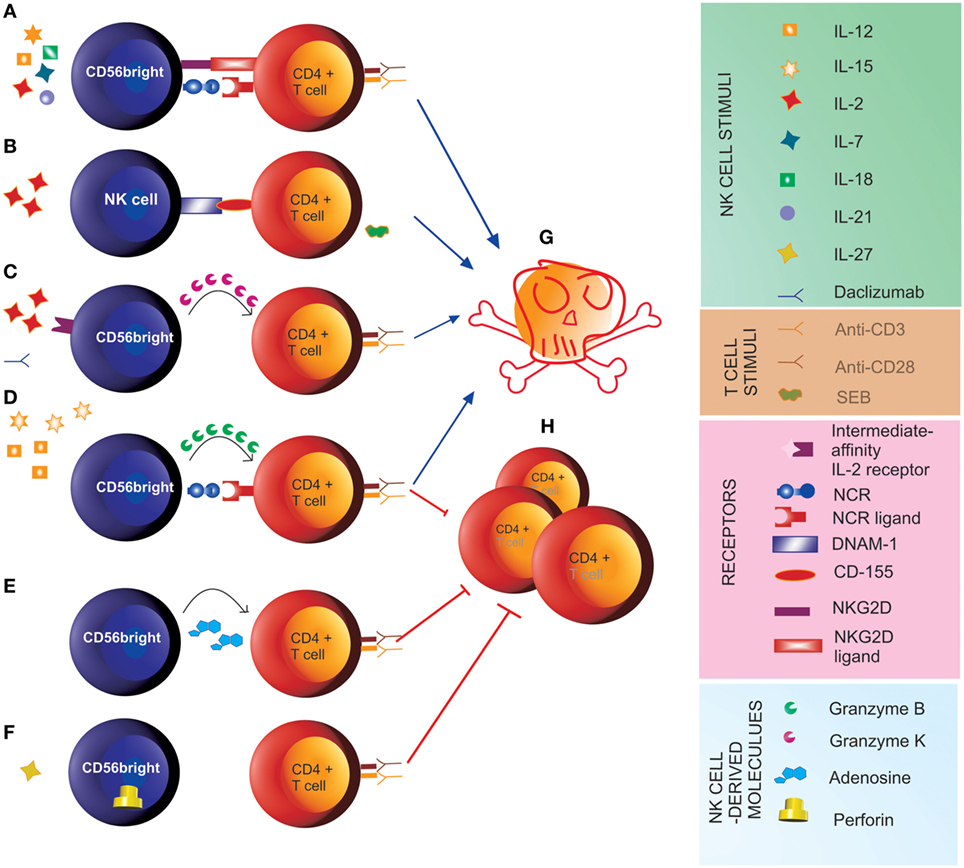
Figure 1. Natural killer (NK) cell-mediated control of T cell responses. (A) Stimulus of CD56bright with IL-2, IL-7, IL-12, IL-15, IL-18, and IL-21 induces cytotoxicity (G) toward previously activated autologous CD4+ T cells through the engagement of NKG2D and the natural cytotoxicity receptors (NCRs) (27). (B) Pre-activation of NK cells with IL-2 and of autologous CD4+ T cells with staphylococcal enterotoxin B (SEB) induces cytotoxicity of NK cells toward autologous T cells (G) through engagement of the activating receptor DNAM-1 on NK cells and its ligand CD155 on T cells (21). (C) In the presence of the anti-IL-2Rα monoclonal antibody daclizumab, IL-2 signal through the intermediate-affinity receptor induces cytotoxicity of CD56bright NK cells toward autologous activated CD4+ T cells (G) involving the transfer of granzyme K to target cells (23). (D) The pro-inflammatory cytokines IL-12 and IL-15 induce anti-proliferative (H) and cytotoxic function of CD56bright NK cells toward CD4+ T cells undergoing activation through engagement of the NCRs NKp30 and NKp46 and release of granzyme B (25). (E) CD56bright suppress proliferation of autologous CD4+ T cells (H) by releasing adenosine (26). (F) IL-27 induces suppressor function of CD56bright NK cells toward autologous CD4+ T cells (H), which is associated with increased perforin content (9).
Possible Role of NK Cells in MS and Its Animal Model
Multiple sclerosis is an autoimmune disease of the central nervous system (CNS) characterized by an attack of the myelin sheath that surrounds and protects CNS axons by autoreactive T cells. Its murine model, experimental autoimmune encephalomyelitis (EAE), is triggered by active immunization with myelin antigens or transfer of activated autoreactive myelin-specific T cells to naïve recipients. Until recently, defects in regulatory mechanisms had only been described in MS in cells of the adaptive compartment (28).
Conflicting data on a beneficial vs. detrimental role of NK cells in EAE have been published (29–31), but studies on regulatory NK cells in mice are difficult to translate into humans, because murine NK cells do not express CD56 and the murine counterparts of CD56bright and CD56dim subsets have not been identified with certainty.
Enhancing regulatory features of NK cells ameliorates the disease course of EAE. In particular, NK cells expressing NKG2A (which is expressed by all CD56bright NK cells in humans) were shown to decrease CNS inflammation by killing T cells and microglial cells, when the interaction between NKG2A and its ligand Qa-1 (the murine equivalent to the human HLA-E) expressed on the target cells was blocked by antibodies specific for either antigen (32, 33). Decreased expression of Qa-1 on microglial cells upon CNS inflammation rendered them more sensitive to NKG2A+ NK cell-mediated lysis. Importantly, enrichment of NK cells through treatment with IL-2 coupled with IL-2 mAb (“IL-2 complexes”) (34) ameliorated EAE (35). A recent work from the same group has shed light on the differential effects of NK cells in early vs. late stages of EAE and possibly MS (36), which may depend on their interactions with neural stem cells (NSCs). Indeed, Liu et al. found that, in MS and EAE brains, NK cells are in contact with NSCs and that, in EAE NSCs released IL-15 upon contact with NK cells, thereby supporting NK cell proliferation and survival; in turn, NK cells killed NSCs, particularly during the late stages of EAE, as a result of reduced expression of Qa-1 on NSCs. Accordingly, removal of NK cells during the late phase of EAE reduced disease severity (36). These observations suggest that cytotoxicity of NK cells may be a double-edged sword in EAE, with NK cells attenuating inflammation in the acute phase of disease by killing immune cells (T cells, microglial cells), but impairing potential repair during the late stage by killing NSCs.
While Liu et al. did not ascertain whether NK cells in contact with NSCs belonged to the CD56bright or CD56dim NK subset (36), others analyzed the phenotype of NK cells in the cerebrospinal fluid (CSF). The majority of intrathecal NK cells in healthy individuals, MS patients and patients with other neurological diseases are CD56bright NK cells (21, 37, 38), suggesting that CSF enrichment in CD56bright NK cells is not MS specific, but rather CNS specific (37). This may reflect organ-specific, rather than blood-specific, tropism of CD56bright NK cells (39). The recently discovered lymphatic vessels in the brain (40, 41) may be the route of entry for CD56bright NK cells, which were shown to circulate in the lymph (42). Of note, a higher migratory capacity of CD56bright compared to CD56dim NK cells was observed in a model of the human blood–brain barrier (BBB) (21).
Given the evidence that CD56bright NK cells are a regulatory population of the IS in healthy individuals, we have explored their function in untreated MS patients or patients with clinically isolated syndrome suggestive of MS (25). The number of CD56bright NK cells was similar in MS patients and healthy subjects (HS). However, upon stimulus with pro-inflammatory cytokines, CD56bright NK cells from MS patients suppressed much less efficiently the proliferation of autologous CD4+ T cells compared to those from HS. This was associated with an increased expression of HLA-E on CD4+ T cells in MS and was reverted by blocking HLA class I on T cells, suggesting that the cytotoxic function of CD56bright NK cells on their targets is inhibited through binding of HLA-E on T cells to the NK cell inhibitory ligand NKG2A. HLA-E is a non-classic major HLA class I molecule expressed by immune cells and, outside the immune compartment, by endothelial cells, which release its soluble form upon inflammation (43). HLA-E upregulation was found in MS CNS within white matter lesions, in endothelial cells and astrocytes (44, 45). Similarly, Morandi et al. detected increased levels of soluble HLA-E in the CSF and expression of HLA-E within immune cells and neural cells in MS plaques, which correlated with decreased NK cell cytotoxicity (46). The causes of such upregulation are, as yet, unknown. Thus, an impairment of CD56bright NK cell immunoregulatory function in MS may occur not only in the periphery but also within the CNS. Recently, we also described decreased cytolytic activity of NK cells in MS as a consequence of reduced upregulation of CD155 on T cells after activation, concomitantly with a reduced NK cell surface expression of DNAM-1 (21). These studies point toward a resistance of T cells to NK cell suppressive functions rather than an intrinsic defect in the NK cells in MS.
The relevance of the immunoregulatory function of NK cells in MS is emphasized by studies from the group of Takashi Yamamura, who described a particular peripheral NK phenotype to be characteristic of MS patients in remission, with increased production of the anti-inflammatory cytokine, IL-5 (“NK2” cells), and high expression of CD95 (47), which inhibited the production of IFN-γ by Th1 clones (48). Among these patients, a further division of NK2 cells as CD11c-high (not producing IL-5) and CD11c-low (producing IL-5) subsets identified CD11c-high patients at risk of relapse (49), suggesting that CD11c + NK cells are pro-inflammatory. In another study, a subpopulation of NK cells characterized by low expression of CD8, a phenotype associated with CD56bright NK cells, was observed to be reduced in untreated patients with relapsing-remitting MS (50).
Previous infections may influence the development of MS (51). Interestingly, infections not only activate NK cells but also shape NK cell functions (52–54). Thus, cytomegalovirus (CMV) induces expansion of NK cells that produce IL-10 in mice, to prevent excess of activation of CD8 T cells (55, 56). In humans, CMV infection is associated with expansion of terminally differentiated NK cells bearing the NKG2C receptor and has been implicated both in MS etiology and/or “protection” (57). In this context, Martinez-Rodriguez et al. explored the expression of NKG2C on NK cells from MS patients and controls, in relation to their CMV+ serostatus and to the NKG2C genotype, finding that the expansion of NKG2C+ NK cells in CMV+ patients was associated to lower risk of disease progression, suggesting that CMV may exert a beneficial influence on MS, either through expansion of NKG2C+ NK cells or through other mechanisms (58). Differently to CMV, infection with Epstein–Barr virus, which has also been associated with an increased risk of MS, expands early differentiated NKG2A + CD56dimNK cells (59, 60), but whether such cells have any role in the pathogenesis of MS is unknown.
The Impact of MS Therapies on NK Cells
In addition to first-line MS therapies, interferon beta (IFN-β) and glatiramer acetate, novel immune-modulating therapies such as the anti-inflammatory dimethyl fumarate, the T cell proliferation inhibitor teriflunomide, the migration inhibitors natalizumab and fingolimod (FTY720), the IL-2 receptor-modulating daclizumab, and the immune cell-depleting alemtuzumab are now available for treatment of MS (61, 62). Many of these immune-modulating biologicals alter the NK cell compartment (Figures 2A,B) by increasing NK cell frequencies (Figure 2A) and/or NK-mediated immune regulatory functions (Figure 2B) (61, 63), which points to an immune-protective role of NK cells in MS. Furthermore, antibody-dependent cell-mediated cytotoxicity (ADCC) by CD56dim NK cells has been proposed as an essential therapeutic mechanism in alemtuzumab-mediated T and B cell depletion as well as rituximab-mediated B cell depletion (Figure 2C) (64, 65).
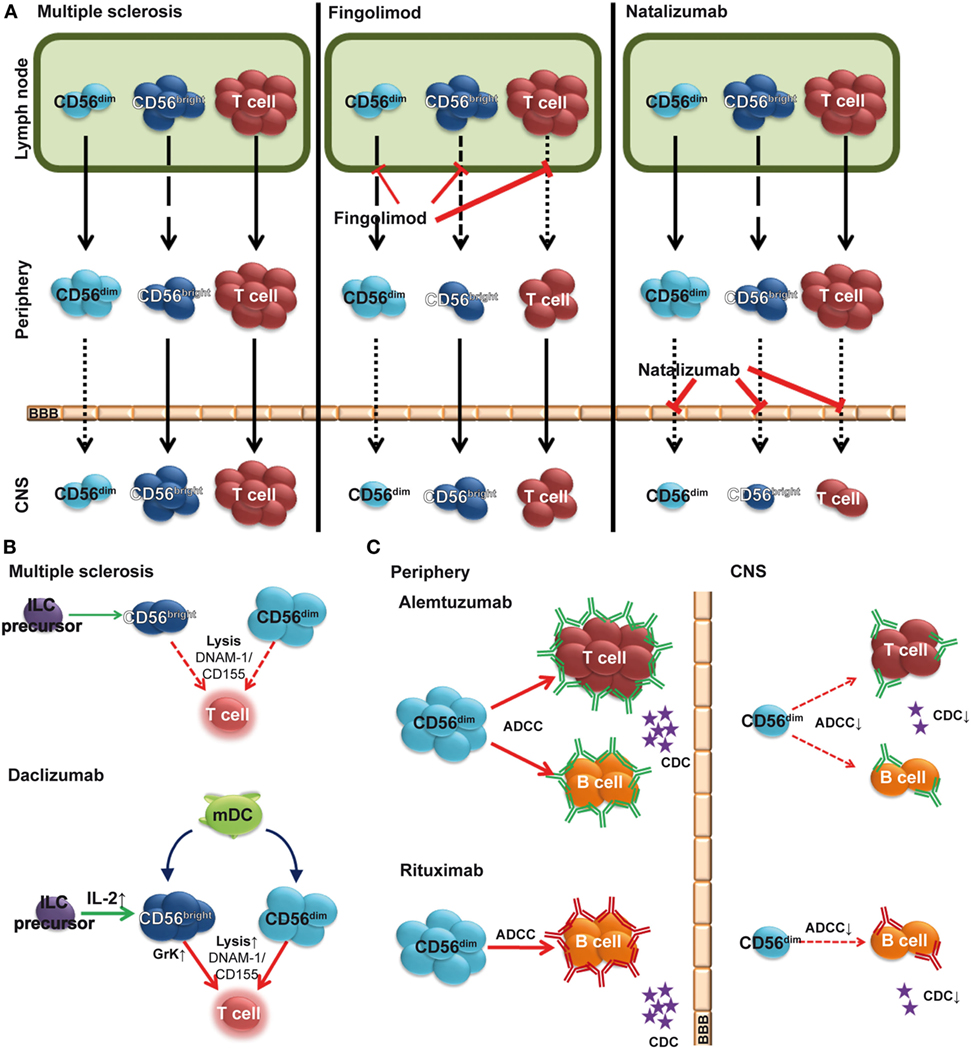
Figure 2. Impact of multiple sclerosis (MS) therapies on the natural killer (NK) cell compartment. (A) Fingolimod (FTY720) inhibits egress of CD56bright, and to a lower degree, CD56dim NK cells from the lymph node (LN), resulting in a relative increase of the latter subset in the periphery (middle). Natalizumab inhibits transmigration of lymphocytes including NK cells across the blood–brain barrier (BBB). (B) Elevated levels of IL-2 in daclizumab-treated patients promote differentiation and expansion of CD56bright NK cells. Daclizumab both boosts NK cell cytolytic function in a DC-dependent manner and renders antigen-activated T cells more sensitive toward NK-mediated lysis, thus restoring defective NK-mediated control of T cell activity in MS. (C) In addition to complement-dependent cytotoxicity (CDC), alemtuzumab (top) and rituximab (bottom) use CD56dim NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC) to deplete T and/or B cells in peripheral blood. However, in the CSF, sparseness of CD56dim NK cells, reduced levels of complement proteins, and lack of antibodies crossing the BBB limit local immune-modulating efficacy [(C) adapted from Ref. (66)].
One therapeutic approach in MS is the reduction of inflammatory lesions by inhibiting infiltration of autoreactive lymphocytes into the CNS. While the humanized monoclonal antibody (hMA) anti-CD49d (alpha 4 integrin) natalizumab prevents transmigration of circulating lymphocytes across the BBB (67), the sphingosine 1-phosphate receptor (S1P) modulator FTY720 reduces CNS inflammation in MS (68) indirectly by preventing lymphocyte egress from LNs (69). A natalizumab-induced increase of total NK cells and CD56bright NK cells in blood concomitantly with reduced NK cell numbers in CSF (Figure 2A) (21, 70, 71) suggests CD49d-dependent transmigration of NK cells into the CNS. Trafficking of NK cells in steady state and inflammatory conditions requires S1P (72) and decreased numbers of circulating CD56bright NK cells have been observed 6 h after FTY720 treatment (73) (Figure 2A). Long-term treatment also resulted in reduced numbers of both NK cell subsets in the CSF (own observations). Despite reduced NK cell numbers, a relative increase in peripheral and intrathecal NK cell subsets within the lymphocyte compartment (74) indicates that FTY720 inhibits NK cell emigration less than that of other lymphocytes (Figure 2A). This might be due to the fact that egress of NK cells from LNs is regulated through both S1P1 and S1P5 (72, 75), whereas other lymphocytes use only S1P1 (76). Since S1P1 and S1P5 seem to trigger the activation of distinct intracellular signal transduction pathways, it has been suggested that S1P5 might be less susceptible to FTY720 than S1P1 (72). Along this line, a higher expression of S1P5 on CD56dim NK cells than on CD56bright ones (72) might explain the relative increase of the CD56dim NK cell subset (77). While treatment of MS patients with FTY720 alters NK cell trafficking, it has no impact on cytokine secretion or cytolytic function of NK cells (77).
In contrast, treatment with daclizumab affects both peripheral (20, 78) and intrathecal CD56bright NK cell numbers (79), as well as their immunoregulatory function (19, 21) (Figures 1 and 2B). Daclizumab is a recently approved hMA directed against IL-2Rα, which showed enhanced efficacy in MS compared to IFN-β [DECIDE trial (80)]. Daclizumab enhances endogenous mechanisms of immune tolerance by reducing early T cell activation (81), expanding CD56bright NK cells (20, 78), while reducing lymphoid tissue-inducer cells (82), and restoring defective NK cell-mediated control of T cell activity in MS (19, 21). The mechanism of the effect of daclizumab on CD56bright NK cells has been discussed in chapter 1.3 (Figure 2C). Daclizumab both boosts NK cell cytolytic function in a DC-dependent manner and renders antigen-activated T cells more sensitive toward NK-mediated lysis, thus restoring defective NK cell-mediated control of T cell activity in MS (19, 21) (Figure 2B).
Alemtuzumab is a hMA directed against the cell surface molecule CD52, which demonstrated a high clinical efficacy in MS (83, 84). CD52 is highly expressed on T and B cells and to a lower degree on NK cell subsets (85, 86). Accordingly, a relative increase of circulating NK cells with increased numbers of CD56bright NK cells was observed 6 months after alemtuzumab therapy, whereas the CD56dim subset remained unaltered. However, there was no change in NK cell cytolytic function (87). ADCC is mediated via the FcγRIII (CD16)-expressing CD56dim NK cell subset (88). Since intrathecal CD56dim NK cells are sparse (21, 89), the therapeutic efficacy of alemtuzumab within the CNS might be limited (Figure 2C). Along this line, insufficient disease inhibition in progressive MS by intrathecal application of rituximab was proposed to be due to low numbers of CD56dim NK cells and reduced levels of complement proteins within the CNS (66). Further studies are required to shed more light on NK cell-mediated ADCC as a mechanism of action of human monoclonal antibody-mediated depleting therapies in MS.
Summary/Outlook
NK cells are important players in controlling T cell activation in CNS autoimmunity, and impaired immune regulatory function of NK cells might be one of the driving forces in the pathogenesis of MS. Thus, a better understanding of the underlying mechanisms of NK cell-mediated regulation of T cell activation might help to improve treatment strategies in MS.
Author Contributions
CG wrote chapter 3, conceptualized Figure 2, revised the manuscript, and approved the final version. AS-M drew Figure 2, revised the manuscript, and approved the final version. HW wrote chapter 3, revised the manuscript, and approved the final version. EM critically reviewed the manuscript for important intellectual content and approved the final version. NKdeR conceptualized Figure 1, critically reviewed the manuscript for important intellectual content, and approved the final version. AU critically reviewed the manuscript for important intellectual content and approved the final version. AL organized the sections of the manuscript, wrote chapters 1 and 2, conceptualized and drew Figure 1, revised the manuscript, and approved the final version.
Conflict of Interest Statement
CG received speaker honoraria and travel expenses for attending meetings from Genzyme, Novartis Pharma GmbH, and Bayer Health Care. AS-M has no financial disclosures. HW received compensation for serving on Scientific Advisory Boards/Steering Committees for Bayer Healthcare, Biogen, Merck Serono, Novartis, and Sanofi-Genzyme. He also received speaker honoraria and travel support from Bayer vital GmbH, Bayer Schering AG, Biogen, CSL Behring, Fresenius Medical Care, Glaxo Smith Kline, GW Pharmaceuticals, Lundbeck, Merck Serono, Omniamed, Novartis, and Sanofi-Genzyme. He received compensation as a consultant from Biogen, Merck Serono, Novartis, and Sanofi-Aventis. He received research support from Bayer Vital, Biogen, Genzyme, Merck Serono, Novartis, Sanofi-Genzyme Germany, and Sanofi US. EM has no financial disclosures. NKdeR has no financial disclosures. AU received grants and contracts from Fondazione Italiana Sclerosi Multipla (FISM), Novartis, Fondazione Cariplo, and Italian Ministry of Health; received honoraria or consultation fees from Biogen, Roche, Teva, Merck Serono, Genzyme, and Novartis. He is a member of a company advisory board, board of directors, or other similar groups for Roche and Genzyme. AL received honoraria for speaking by Biogen, Novartis, and Teva, consulting fees by Merck Serono, Sanofi-Genzyme, and Novartis, and funding for travel from Teva, Merck Serono, Biogen, and Novartis.
Funding
The work of CG and HW has been supported by the Collaborative Research Centre CRC TR128 “Initiating/Effector vs. Regulatory Mechanisms in Multiple Sclerosis – Progress towards Tackling the Disease”; Project A09 “Analysis and therapeutic modification of innate immune regulatory networks controlling T-cell responses in MS” and the Disease Related Competence Network for Multiple Sclerosis (Krankheitsbezogenes Kompetenznetzwerk MS, KKNMS); Subproject ImmuneMS (FKZ 01FI1603A) funded by the Federal Ministry of Education and Research. AL was supported by a fellowship from Fondazione Italiana Sclerosi Multipla entitled “Dissection of the function of CD56bright NK cells in multiple sclerosis” (Prot. N. 297/09/F14). Some of the results discussed in this review were generated through the grant “Personalizing health care in multiple sclerosis using systems medicine tools (Sys4MS)” supported by the Italian Ministry of Health (ERA-NET Cofund grant supported by the European Commission, call ERACoSysMed) to the IRCCS San Martino-IST (grant number “PER-2012-2361136”) and through the grant “Studio dell’effetto immunomodulante e neuroriparativo delle cellule staminali mesenchimali in pazienti con sclerosi multipla” supported by Compagnia di San Paolo to AU (grant number 2015.AAI4109.U4916).
References
1. Cooper GS, Bynum MLK, Somers EC. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun (2009) 33:197–207. doi:10.1016/j.jaut.2009.09.008
2. Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56bright subset. Blood (2001) 97:3146–51. doi:10.1182/blood.V97.10.3146
3. Ferlazzo G, Morandi B. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front Immunol (2014) 5:159. doi:10.3389/fimmu.2014.00159
4. Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, et al. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood (2003) 101:3052–7. doi:10.1182/blood-2002-09-2876
5. Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, et al. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med (1994) 180:1395–403. doi:10.1084/jem.180.4.1395
6. Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56(bright) natural killer (NK) cells: an important NK cell subset. Immunology (2009) 126:458–65. doi:10.1111/j.1365-2567.2008.03027.x
7. Cichocki F, Schlums H, Theorell J, Tesi B, Miller JS, Ljunggren HG, et al. Diversification and functional specialization of human NK cell subsets. Curr Top Microbiol Immunol (2016) 395:63–94. doi:10.1007/82_2015_487
8. Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci U S A (2004) 101:16606–11. doi:10.1073/pnas.0407522101
9. Laroni A, Gandhi R, Beynon V, Weiner HL. IL-27 imparts immunoregulatory function to human NK cell subsets. PLoS One (2011) 6:e26173. doi:10.1371/journal.pone.0026173
10. Piccioli D, Sbrana S, Melandri E, Valiante NM. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J Exp Med (2002) 195:335–41. doi:10.1084/jem.20010934
11. Mailliard RB, Alber SM, Shen H, Watkins SC, Kirkwood JM, Herberman RB, et al. IL-18-induced CD83+CCR7+ NK helper cells. J Exp Med (2005) 202:941–53. doi:10.1084/jem.20050128
12. Marcenaro E, Ferranti B, Moretta A. NK-DC interaction: on the usefulness of auto-aggression. Autoimmun Rev (2005) 4:520–5. doi:10.1016/j.autrev.2005.04.015
13. Marcenaro E, Cantoni C, Pesce S, Prato C, Pende D, Agaugue S, et al. Uptake of CCR7 and acquisition of migratory properties by human KIR+ NK cells interacting with monocyte-derived DC or EBV cell lines: regulation by KIR/HLA-class I interaction. Blood (2009) 114:4108–16. doi:10.1182/blood-2009-05-222265
14. Vitale M, Chiesa MD, Carlomagno S, Romagnani C, Thiel A, Moretta L, et al. The small subset of CD56brightCD16 – natural killer cells is selectively responsible for both cell proliferation and interferon-γ production upon interaction with dendritic cells. Eur J Immunol (2004) 34:1715–22. doi:10.1002/eji.200425100
15. Romagnani C, Della Chiesa M, Kohler S, Moewes B, Radbruch A, Moretta L, et al. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. Eur J Immunol (2005) 35:2452–8. doi:10.1002/eji.200526069
16. Dalbeth N, Gundle R, Davies RJ, Lee YC, Mcmichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol (2004) 173:6418–26. doi:10.4049/jimmunol.173.10.6418
17. Della Chiesa M, Vitale M, Carlomagno S, Ferlazzo G, Moretta L, Moretta A. The natural killer cell-mediated killing of autologous dendritic cells is confined to a cell subset expressing CD94/NKG2A, but lacking inhibitory killer Ig-like receptors. Eur J Immunol (2003) 33:1657–66. doi:10.1002/eji.200323986
18. Morandi B, Mortara L, Chiossone L, Accolla RS, Mingari MC, Moretta L, et al. Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PLoS One (2012) 7:e39170. doi:10.1371/journal.pone.0039170
19. Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103:5941–6. doi:10.1073/pnas.0601335103
20. Martin JF, Perry JS, Jakhete NR, Wang X, Bielekova B. An IL-2 paradox: blocking CD25 on T cells induces IL-2-driven activation of CD56(bright) NK cells. J Immunol (2010) 185:1311–20. doi:10.4049/jimmunol.0902238
21. Gross CC, Schulte-Mecklenbeck A, Runzi A, Kuhlmann T, Posevitz-Fejfar A, Schwab N, et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc Natl Acad Sci U S A (2016) 113:E2973–82. doi:10.1073/pnas.1524924113
22. Caligiuri MA, Zmuidzinas A, Manley TJ, Levine H, Smith KA, Ritz J. Functional consequences of interleukin 2 receptor expression on resting human lymphocytes. Identification of a novel natural killer cell subset with high affinity receptors. J Exp Med (1990) 171:1509–26. doi:10.1084/jem.171.5.1509
23. Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol (2011) 187:781–90. doi:10.4049/jimmunol.1100789
24. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223. doi:10.1146/annurev.immunol.19.1.197
25. Laroni A, Armentani E, Kerlero De Rosbo N, Ivaldi F, Marcenaro E, Sivori S, et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J Autoimmun (2016) 72:8–18. doi:10.1016/j.jaut.2016.04.003
26. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16- NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J Immunol (2015) 195(3):965–72. doi:10.4049/jimmunol.1500591
27. Nielsen N, Odum N, Urso B, Lanier LL, Spee P. Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS One (2012) 7:e31959. doi:10.1371/journal.pone.0031959
28. Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev (2014) 259:231–44. doi:10.1111/imr.12169
29. Gandhi R, Laroni A, Weiner HL. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol (2010) 221:7–14. doi:10.1016/j.jneuroim.2009.10.015
30. Lunemann JD, Munz C. Do natural killer cells accelerate or prevent autoimmunity in multiple sclerosis? Brain (2008) 131:1681–3. doi:10.1093/brain/awn132
31. Morandi B, Bramanti P, Bonaccorsi I, Montalto E, Oliveri D, Pezzino G, et al. Role of natural killer cells in the pathogenesis and progression of multiple sclerosis. Pharmacol Res (2008) 57:1–5. doi:10.1016/j.phrs.2007.11.003
32. Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity (2007) 26:593–604. doi:10.1016/j.immuni.2007.03.017
33. Leavenworth JW, Schellack C, Kim HJ, Lu L, Spee P, Cantor H. Analysis of the cellular mechanism underlying inhibition of EAE after treatment with anti-NKG2A F(ab’)2. Proc Natl Acad Sci U S A (2010) 107:2562–7. doi:10.1073/pnas.0914732107
34. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311:1924–7. doi:10.1126/science.1122927
35. Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med (2010) 207:1907–21. doi:10.1084/jem.20092749
36. Liu Q, Sanai N, Jin WN, La Cava A, Van Kaer L, Shi FD. Neural stem cells sustain natural killer cells that dictate recovery from brain inflammation. Nat Neurosci (2016) 19:243–52. doi:10.1038/nn.4211
37. Han S, Lin YC, Wu T, Salgado AD, Mexhitaj I, Wuest SC, et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J Immunol (2014) 192:2551–63. doi:10.4049/jimmunol.1302884
38. Rodriguez-Martin E, Picon C, Costa-Frossard L, Alenda R, Sainz De La Maza S, Roldan E, et al. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol (2015) 180:243–9. doi:10.1111/cei.12580
39. Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol (2014) 192:3805–15. doi:10.4049/jimmunol.1301889
40. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi:10.1038/nature14432
41. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med (2015) 212:991–9. doi:10.1084/jem.20142290
42. Montalto E, Mangraviti S, Costa G, Carrega P, Morandi B, Pezzino G, et al. Seroma fluid subsequent to axillary lymph node dissection for breast cancer derives from an accumulation of afferent lymph. Immunol Lett (2010) 131:67–72. doi:10.1016/j.imlet.2010.03.002
43. Coupel S, Moreau A, Hamidou M, Horejsi V, Soulillou JP, Charreau B. Expression and release of soluble HLA-E is an immunoregulatory feature of endothelial cell activation. Blood (2007) 109:2806–14. doi:10.1182/blood-2006-06-030213
44. Durrenberger PF, Webb LV, Sim MJ, Nicholas RS, Altmann DM, Boyton RJ. Increased HLA-E expression in white matter lesions in multiple sclerosis. Immunology (2012) 137:317–25. doi:10.1111/imm.12012
45. Pannemans K, Broux B, Goris A, Dubois B, Broekmans T, Van Wijmeersch B, et al. HLA-E restricted CD8+ T cell subsets are phenotypically altered in multiple sclerosis patients. Mult Scler (2014) 20:790–801. doi:10.1177/1352458513509703
46. Morandi F, Venturi C, Rizzo R, Castellazzi M, Baldi E, Caniatti ML, et al. Intrathecal soluble HLA-E correlates with disease activity in patients with multiple sclerosis and may cooperate with soluble HLA-G in the resolution of neuroinflammation. J Neuroimmune Pharmacol (2013) 8:944–55. doi:10.1007/s11481-013-9459-3
47. Takahashi K, Miyake S, Kondo T, Terao K, Hatakenaka M, Hashimoto S, et al. Natural killer type 2 bias in remission of multiple sclerosis. J Clin Invest (2001) 107:R23–9. doi:10.1172/JCI11819
48. Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T. The regulatory role of natural killer cells in multiple sclerosis. Brain (2004) 127:1917–27. doi:10.1093/brain/awh219
49. Aranami T, Miyake S, Yamamura T. Differential expression of CD11c by peripheral blood NK cells reflects temporal activity of multiple sclerosis. J Immunol (2006) 177:5659–67. doi:10.4049/jimmunol.177.8.5659
50. De Jager PL, Rossin E, Pyne S, Tamayo P, Ottoboni L, Viglietta V, et al. Cytometric profiling in multiple sclerosis uncovers patient population structure and a reduction of CD8low cells. Brain (2008) 131:1701–11. doi:10.1093/brain/awn118
51. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23:683–747. doi:10.1146/annurev.immunol.23.021704.115707
52. Saghafian-Hedengren S, Sohlberg E, Theorell J, Carvalho-Queiroz C, Nagy N, Persson JO, et al. Epstein-Barr virus coinfection in children boosts cytomegalovirus-induced differentiation of natural killer cells. J Virol (2013) 87:13446–55. doi:10.1128/JVI.02382-13
53. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity (2015) 42:443–56. doi:10.1016/j.immuni.2015.02.008
54. Muntasell A, Pupuleku A, Cisneros E, Vera A, Moraru M, Vilches C, et al. Relationship of NKG2C copy number with the distribution of distinct cytomegalovirus-induced adaptive NK cell subsets. J Immunol (2016) 196:3818–27. doi:10.4049/jimmunol.1502438
55. Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med (2009) 206:2235–51. doi:10.1084/jem.20082387
56. Tarrio ML, Lee SH, Fragoso MF, Sun HW, Kanno Y, O’shea JJ, et al. Proliferation conditions promote intrinsic changes in NK cells for an IL-10 response. J Immunol (2014) 193:354–63. doi:10.4049/jimmunol.1302999
57. Vanheusden M, Stinissen P, ‘t Hart BA, Hellings N. Cytomegalovirus: a culprit or protector in multiple sclerosis? Trends Mol Med (2015) 21:16–23. doi:10.1016/j.molmed.2014.11.002
58. Martinez-Rodriguez JE, Cobo-Calvo A, Villar LM, Munteis E, Blanco Y, Rasal R, et al. Adaptive natural killer cell response to cytomegalovirus and disability progression in multiple sclerosis. Mult Scler (2016) 22:741–52. doi:10.1177/1352458515601215
59. Azzi T, Lünemann A, Murer A, Ueda S, Béziat V, Malmberg K-J, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood (2014) 124:2533. doi:10.1182/blood-2014-01-553024
60. Chijioke O, Landtwing V, Munz C. NK cell influence on the outcome of primary Epstein-Barr virus infection. Front Immunol (2016) 7:323. doi:10.3389/fimmu.2016.00323
61. Wiendl H, Gross CC. Modulation of IL-2Ralpha with daclizumab for treatment of multiple sclerosis. Nat Rev Neurol (2013) 9:394–404. doi:10.1038/nrneurol.2013.95
62. Ziemssen T, De Stefano N, Pia Sormani M, Van Wijmeersch B, Wiendl H, Kieseier BC. Optimizing therapy early in multiple sclerosis: an evidence-based view. Mult Scler Relat Disord (2015) 4:460–9. doi:10.1016/j.msard.2015.07.007
63. Chanvillard C, Jacolik RF, Infante-Duarte C, Nayak RC. The role of natural killer cells in multiple sclerosis and their therapeutic implications. Front Immunol (2013) 4:63. doi:10.3389/fimmu.2013.00063
64. Anolik JH, Campbell D, Felgar RE, Young F, Sanz I, Rosenblatt J, et al. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum (2003) 48:455–9. doi:10.1002/art.10764
65. Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology (2009) 128:260–70. doi:10.1111/j.1365-2567.2009.03115.x
66. Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol (2016) 3:166–79. doi:10.1002/acn3.293
67. Coyle PK. The role of natalizumab in the treatment of multiple sclerosis. Am J Manag Care (2010) 16:S164–70.
68. Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman CH, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med (2006) 355:1124–40. doi:10.1056/NEJMoa052643
69. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol (2010) 33:91–101. doi:10.1097/WNF.0b013e3181cbf825
70. Putzki N, Baranwal MK, Tettenborn B, Limmroth V, Kreuzfelder E. Effects of natalizumab on circulating B cells, T regulatory cells and natural killer cells. Eur Neurol (2010) 63:311–7. doi:10.1159/000302687
71. Skarica M, Eckstein C, Whartenby KA, Calabresi PA. Novel mechanisms of immune modulation of natalizumab in multiple sclerosis patients. J Neuroimmunol (2011) 235:70–6. doi:10.1016/j.jneuroim.2011.02.010
72. Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol (2007) 8:1337–44. doi:10.1038/ni1523
73. Mehling M, Burgener AV, Brinkmann V, Bantug GR, Dimeloe S, Hoenger G, et al. Tissue distribution dynamics of human NK cells inferred from peripheral blood depletion kinetics after sphingosine-1-phosphate receptor blockade. Scand J Immunol (2015) 82:460–6. doi:10.1111/sji.12347
74. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kumpfel T, Buck D, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology (2011) 76:1214–21. doi:10.1212/WNL.0b013e3182143564
75. Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med (2009) 206:2469–81. doi:10.1084/jem.20090525
76. Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol (2007) 8:1295–301. doi:10.1038/ni1545
77. Johnson TA, Evans BL, Durafourt BA, Blain M, Lapierre Y, Bar-Or A, et al. Reduction of the peripheral blood CD56(bright) NK lymphocyte subset in FTY720-treated multiple sclerosis patients. J Immunol (2011) 187:570–9. doi:10.4049/jimmunol.1003823
78. Bielekova B, Howard T, Packer AN, Richert N, Blevins G, Ohayon J, et al. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol (2009) 66:483–9. doi:10.1001/archneurol.2009.50
79. Bielekova B, Richert N, Herman ML, Ohayon J, Waldmann TA, Mcfarland H, et al. Intrathecal effects of daclizumab treatment of multiple sclerosis. Neurology (2011) 77:1877–86. doi:10.1212/WNL.0b013e318239f7ef
80. Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A, et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med (2015) 373:1418–28. doi:10.1056/NEJMoa1501481
81. Wuest SC, Edwan JH, Martin JF, Han S, Perry JS, Cartagena CM, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med (2011) 17:604–9. doi:10.1038/nm.2365
82. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med (2012) 4:145ra106. doi:10.1126/scitranslmed.3004140
83. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet (2012) 380:1819–28. doi:10.1016/S0140-6736(12)61769-3
84. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet (2012) 380:1829–39. doi:10.1016/S0140-6736(12)61768-1
85. Freedman MS, Kaplan JM, Markovic-Plese S. Insights into the mechanisms of the therapeutic efficacy of alemtuzumab in multiple sclerosis. J Clin Cell Immunol (2013) 4:1000152. doi:10.4172/2155-9899.1000152
86. Ruck T, Bittner S, Wiendl H, Meuth SG. Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int J Mol Sci (2015) 16:16414–39. doi:10.3390/ijms160716414
87. Gross CC, Ahmetspahic D, Ruck T, Schulte-Mecklenbeck A, Schwarte K, Jörgens S, et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3:e289. doi:10.1212/NXI.0000000000000183
88. Perussia B, Trinchieri G, Jackson A, Warner NL, Faust J, Rumpold H, et al. The Fc receptor for IgG on human natural killer cells: phenotypic, functional, and comparative studies with monoclonal antibodies. J Immunol (1984) 133:180–9.
Keywords: natural killer cells, CD56bright NK cells, CD56dim NK cells, regulatory immune cells, innate immune system, multiple sclerosis
Citation: Gross CC, Schulte-Mecklenbeck A, Wiendl H, Marcenaro E, Kerlero de Rosbo N, Uccelli A and Laroni A (2016) Regulatory Functions of Natural Killer Cells in Multiple Sclerosis. Front. Immunol. 7:606. doi: 10.3389/fimmu.2016.00606
Received: 20 October 2016; Accepted: 01 December 2016;
Published: 19 December 2016
Edited by:
Miguel López-Botet, IMIM (Hospital del Mar Research Institute), SpainReviewed by:
Debby Burshtyn, University of Alberta, CanadaJacques Zimmer, Centre de Recherche Public de la Santé, Luxembourg
Copyright: © 2016 Gross, Schulte-Mecklenbeck, Wiendl, Marcenaro, Kerlero de Rosbo, Uccelli and Laroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alice Laroni, YWxpY2UubGFyb25pQHVuaWdlLml0