 Kevin A. Robertson
Kevin A. Robertson Peter Ghazal
Peter Ghazal- Division of Infection and Pathway Medicine, University of Edinburgh, Edinburgh, UK
The sterol metabolic network is emerging center stage in inflammation and immunity. Historically, observational clinical studies show that hypocholesterolemia is a common side effect of interferon (IFN) treatment. More recently, comprehensive systems-wide investigations of the macrophage IFN response reveal a direct molecular link between cholesterol metabolism and infection. Upon infection, flux through the sterol metabolic network is acutely moderated by the IFN response at multiple regulatory levels. The precise mechanisms by which IFN regulates the mevalonate-sterol pathway—the spine of the network—are beginning to be unraveled. In this review, we discuss our current understanding of the multifactorial mechanisms by which IFN regulates the sterol pathway. We also consider bidirectional communications resulting in sterol metabolism regulation of immunity. Finally, we deliberate on how this fundamental interaction functions as an integral element of host protective responses to infection and harmful inflammation.
Introduction
Immunity depends on and employs metabolic pathways for its function. Our knowledge of the molecular and functional mechanisms for this coupling has grown dramatically in recent years and it is now accepted that a remodeling of glycololytic, lipid biosynthetic, and associated homeostatic molecular “circuitry” is an integral component of innate and adaptive immune responses (1–3). In particular, multiple immune-mediated mechanisms for the transcriptional, posttranscriptional, translational, and posttranslational regulation of lipid biosynthesis, storage, influx, and efflux in immune cells have been described (4–7). Broadly, with some notable exceptions, these mechanisms have been defined in vitro in specific cell types (e.g., macrophages) and their general significance and relative importance in vivo have yet to be fully characterized.
Immediately after infection, the ligation of cellular pattern-recognition receptors by, for example, dsRNA leads to an induction of NFkB, ATF2/c-jun, and interferon regulatory factor 3 (IRF3), a rapid upregulation of IFNα/β gene expression and secretion of type I IFNs by cells. The autocrine/paracrine binding of IFNα/β or IFN-γ (from activated NK and T cells) to type I or type II IFN receptors, respectively, leads to the activation of JAK/STAT signaling pathways and rapid alterations in the abundance of hundreds of transcripts in the cell. These IFN-stimulated changes reflect an acute re-programing of the cell to resist infection and limit cellular damage. Figure 1 shows a high-resolution temporal (every 30 min for the first 12 h) analysis of genome-wide alterations in gene expression upon IFN-γ activation of bone marrow-derived macrophages. Importantly, alongside many IFN-stimulated genes, this data reveal an equivalent number of transcripts are significantly suppressed by IFN.
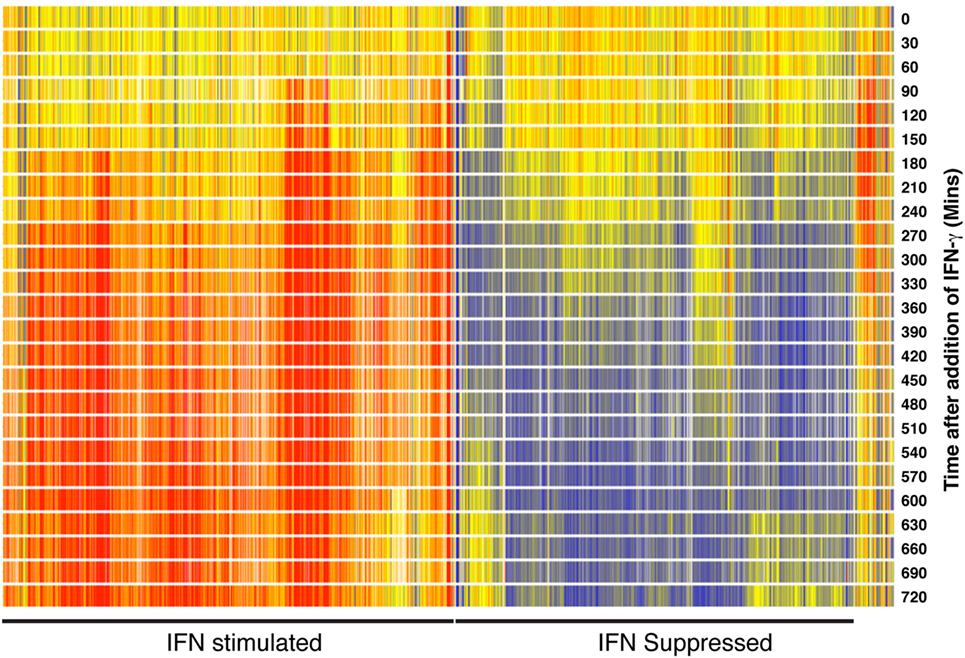
Figure 1. Heat map showing 1,048 genes significantly increased or decreased in expression after interferon (IFN) simulation of macrophages. Bone marrow-derived macrophages were mock treated or treated with 10 U/ml IFN and then sampled at 30-min intervals for a period of 12 h. Total RNA was then labeled and hybridized to Mouse Agilent V2 (G4121A) microarrays. Gene expression is shown as a pseudo-color—blue, decrease; red, increase. Explorative and statistical analyses were undertaken as previously described (4). Data are available for download from the NCBI gene expression omnibus (https://www.ncbi.nlm.nih.gov/geo/) (series GSE42504).
While interferon (IFN)-stimulated genes (ISG) such as NOS2, OAS2, MX2, and IFITM3 have intensively investigated antiviral or antibacterial effects, IFN downregulated transcripts have received relatively little attention (8–11). Notably, a statistical over-representation analysis of the IFN suppressed genes presented in Figure 1 identified the sterol metabolic network as a significantly over-represented component of this dataset. Importantly, consequent mechanistic studies demonstrated that a suppression of sterol biosynthesis is an integral component of the innate immune response to infection (4). This work raised several significant questions about the coupling of sterol metabolism to immunity. In particular, what are the molecular mechanisms by which IFN mediates a downregulation of the sterol biosynthesis pathway and how does the suppression of sterol biosynthesis benefit the infected host? Recent studies are beginning to answer some of these questions.
Here, we first discuss early clinical work showing iatrogenic effects of IFN on sterol metabolism. Next, with an emphasis on molecular oxysterol and miRNA-mediated mechanisms, we consider what is known about how IFN regulates sterol metabolism. Overall, we advance the notion that the mevalonate–sterol pathway is an effector arm of immunity and highlight how this response helps the host limit excessive inflammation and resist infection.
Hypocholesterolemic Effects of IFN Treatment in Humans
Although interest in IFN-mediated regulation of the sterol pathway has increased dramatically in recent years, IFN-induced alterations in cholesterol in humans have been reported for several decades (Table 1). In 1979, Baillie and Orr reported that acute viral infections are regularly associated with reductions in systemic cholesterol in patients (12). Subsequently, Cantell et al. (13) showed that the administration of partly purified human leukocyte IFN to volunteers led to a 20% drop in high-density lipoprotein (HDL), a transient declining trend in total cholesterol and put forward the first proposal that viral infections elicit a drop in cholesterol via the induction of IFN (13). Table 1 presents a chronological summary of wide-ranging studies in which natural and recombinant type I and type II IFNs have been administered to volunteers or patients with cancer, multiple sclerosis, human papilloma, or hepatitis C virus (HCV) infections. In all studies, despite differences in the preparation of IFN used, a drop in circulating total cholesterol and/or HDL was observed. Arguably, the strongest clinical evidence comes from prospective double blind studies such as those reported by Rosenzweig et al. (14) and Borden et al. (15). The former utilized a double blind analysis to demonstrate a dose-dependent effect of IFN administration on plasma cholesterol (14). The latter employed a prospective double blind placebo-controlled analysis of IFN treatment in renal carcinoma patients, demonstrating a significant decrease in mean plasma total cholesterol (15). It is worth noting that Rosenzweig et al. (14) also showed that the effects of IFN were not permanent and that after cessation of treatment circulating cholesterol levels returned to normal in patients (14). In subsequent metabolic tracer experiments, the primary effect of IFN was shown to occur via a modulation of cholesterol synthesis (16).
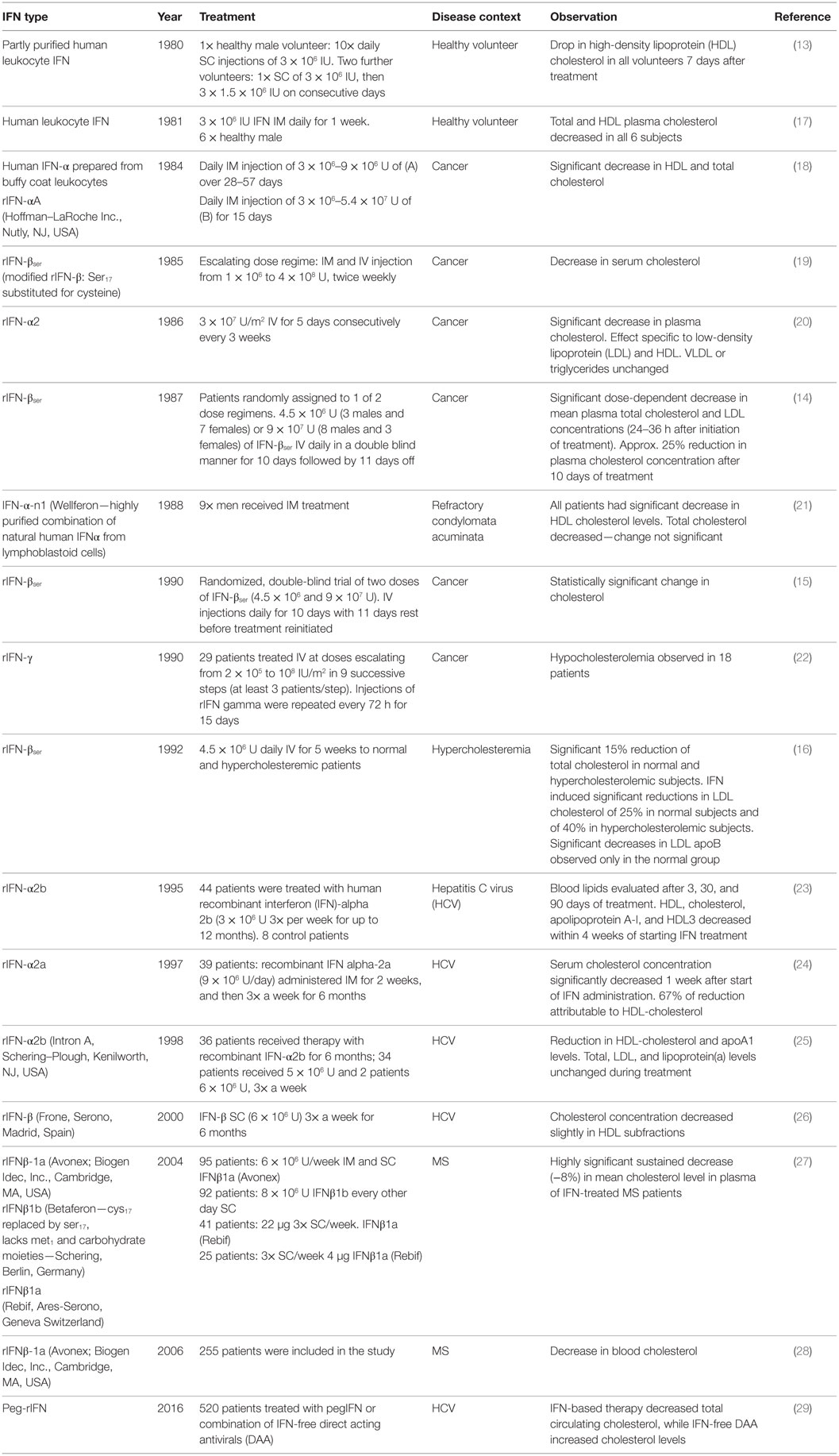
Table 1. Representative clinical studies reporting decreases in cholesterol following treatment with type 1 or 2 IFN.
In summary, the induction of hypocholesteremia by IFN has been recorded clinically for many years via an analysis of total cholesterol, HDL, or LDL in the circulation. Despite this recognition, physiological roles related to human health and underpinning this observation have not been further investigated.
The Mevalonate-Sterol Pathway is an Intrinsic Component of the IFN Response to Infection
Alongside, clinical studies demonstrating exogenously administered IFN can regulate sterol metabolism, a number of groups have also associated cholesterol regulation with IFN responses in experimental animal studies. In 1984, Kuo et al. showed that IFN-inducing agents significantly reduced cholesterol deposits in the aortas of rabbits fed a pro-atherogenic diet (30). Further, in 1987, Pereira et al. showed that a hypercholesteremic diet resulted in an increased susceptibility to murine hepatitis virus in A/J mice—a result in part due to a decreased response to IFN and reduced antiviral state (31). A key question in this context is: what benefit to the host is conferred by the IFN regulation of sterol metabolism? While studies prior to 2011 showed that toll-like receptor 3 (TLR3) or TLR4 ligation results in an IFN-independent inhibition of cholesterol efflux from the cell, little was known at this point about how IFN signaling directly influences cholesterol homeostasis and the physiological purpose this could serve (32). In 2011, Blanc et al. demonstrated that viral infection or treatment of macrophages with type I or II IFN results in a coordinate, negative regulation of the entire sterol biosynthesis pathway and that inflammatory cytokines such as TNF, IL-6, and IL1β are incapable of eliciting a similar effect. This study further showed that the regulation of the sterol pathway by IFN is, at least partly, due to a reduction in SREBF2 transcription and SREBP2 abundance and that this event is an integral component of the cell-intrinsic antiviral response (4). Notably, a recent study highlighted the interdependent reciprocal nature of the molecular circuitry coupling IFN and sterol metabolism. In 2015, York et al. described a STING-dependent recognition of decreased flux through the sterol biosynthetic pathway leading to positive feedback that enhances the type I IFN response and antiviral gene expression in the context of gammaherpesvirus infection (33). The implications of this data are discussed later in this review.
Functional Roles for the Mevalonate-Sterol Pathway in Governing Adaptive Immune Responses
Beyond the intracellular and/or cell-intrinsic environment, IFN-mediated regulation of sterol metabolism has the potential to influence many aspects of immunity. The functions that the sterol metabolic network plays in a wide range of adaptive immune responses have recently been reviewed (3). These include: an absolute requirement for SREBP2 functionality during activated T lymphocyte clonal expansion, a requirement for flux through the sterol biosynthesis pathway during the activation of T regulatory cell function, the observation that a hypercholesteremia can alter the balance of the Treg and T effector cells, and the induction of lymphocyte hyper-proliferation due to impaired cholesterol efflux (34–38). Cholesterol is also indispensable in the formation of lipid raft microdomains—crucial to the assembly of cell surface signaling molecules such as the T and B cell receptors—and has recently been identified as a critical allosteric regulator of TCR priming (39, 40). Notably, cholesterol is not the only output of the sterol biosynthesis pathway on which cells depend. For example, prenylation of the Ras family of GTPases by the side branch of the mevalonate pathway is integral to the control of T cell differentiation, proliferation, and cytokine production [reviewed in Ref. (41)]. Further, sterol pathway intermediates have also been identified as endogenous ligands for the transcription factor RORγt. RORγt is required for the differentiation of naïve CD4+ T lymphocytes into TH17 cells, a subset of lymphocytes associated with a range of autoimmune diseases and mediating protective immune responses to pathogens such as Klebsiella pneumoniae, Bordetella pertussis, Mycobacterium tuberculosis, and Candida albicans (42–45). Santori and colleagues identified sterol pathway intermediates generated downstream of lanosterol and above zymosterol as natural ligands of RORγt, while Hu et al. suggested that desmosterol (downstream of zymosterol and recently shown to be negatively regulated by IFN) is a potential endogenous ligand for this transcription factor (7, 46). Through the utilization of chemical library screening studies, the oxysterol 7β,26-dihydroxycholesterol (synthesized from 7β-hydroxycholesterol, a metabolite immediately downstream of cholesterol) has also been identified as a potent ligand of RORγt (47). In summary, the sterol metabolic network is increasingly viewed as integral to the activation and differentiation of T lymphocytes. More work, however, is required to better understand the precise mechanisms by which the network and/or specific metabolites function in these processes.
In B lymphocytes, IFN regulation of sterol metabolism may lead to alterations in lipid raft cholesterol composition and, in doing so, affect antigen processing/presentation (48) and B-cell receptor signaling (48–50). Notably, significant roles for the inflammatory sterol pathway product 25-hydroxycholesterol and 7α,25-dihydroxycholesterol in class-switching and the chemoattraction of B lymphocytes to germinal centers have recently been described and these will be discussed in detail later.
Benefits of IFN-Mediated Sterol Regulation During Infection
An IFN-mediated suppression of sterol metabolism has the potential to directly curtail the replication of microorganisms in the host. Pathogens with a dependency on the host sterol metabolic network include HCV, human immunodeficiency virus (HIV), Ebola, the Herpesvirus family [HCMV, murine cytomegalovirus (MCMV), herpes simplex virus type (HSV1) MHV-68, and varicella zoster virus (VZV)], Influenza A virus (IAV), Listeria monocytogenes, and M. tuberculosis (51–57). Importantly, the requirements of these organisms on the system vary dramatically. For example, lipid rafts play an integral role in the entry, assembly, and release of a wide range of unrelated viruses (enveloped and non-enveloped) such as HIV1, Ebola, Influenza A, and Echovirus 1 [reviewed here in Ref. (58)]. In contrast, HCMV uses cholesterol for envelopment and limiting the availability of intracellular cholesterol levels has been shown to restrict infectivity of this virus (59). The replication of several viruses also requires prenylation of host and/or virus proteins. For example, hepatitis D virus requires prenylation of its large delta antigen for optimal virion morphogenesis, HCV requires the geranylgeranylated host protein FBL2 for replication, and respiratory syncytial virus (RSV) F glycoprotein binds to the prenylated host protein RHOA enabling membrane fusion (60–62). In the context of bacterial infection, Listeriolysin O, the major virulence factor of the intracellular bacteria L. monocytogenes, is a cholesterol-dependent cytolysin (CDC) responsible for a wide array of functions including disruption of the internalization vacuole (54). Further, M. tuberculosis has cholesterol uptake machinery, an enzyme system capable of catabolizing sterols for growth and potentially utilizes sterols as a carbon and energy source (55, 63).
It is perhaps unsurprising, given the essential role sterol metabolism plays in their replication, that examples are appearing of pathogens subverting or co-opting the regulation of this system for their own benefit. In 2007, Mackenzie et al. showed that a West Nile virus (WNV) infection of Vero cells induced an upregulation of cholesterol biosynthesis and redistribution of cholesterol resulting in defective IFN-stimulated JAK/STAT signaling. This result was attributed to a disrupted recruitment and activation of the type 1 IFN receptor and IFN signaling proteins and emphasizes the tight integration of IFN and cholesterol regulation in the cell (64).
In summary, evidence increasingly reveals an intimate molecular coupling between IFN signaling and the sterol metabolic network. This underscores the importance of immune-mediated regulation of sterol metabolism as an integral component of the host response to infection.
Cellular Mechanisms for IFN Regulation of the Sterol Metabolic Network
A prototypic outcome of IFN signaling in the cell is the transcriptional activation or suppression of hundreds of genes. Over the past 5 years, significant progress in characterizing which of these genes contribute to regulation of cholesterol in the cell has been made. In this regard, oxysterol and miRNA-mediated mechanisms have risen to prominence and, in the following sections, we will review what is known about the functions of IFN-elicited CH25H/25-HC and miRNA-mediated sterol regulation. We will then conclude by considering mechanisms of cholesterol regulation by the “conventional” ISG Viperin and the IFITM protein family.
IFN-Induced 25-Hydroxycholesterol in Infection and Immunity
While cholesterol is a critical component of cell membranes and a precursor of bile acids and steroid hormones, at high concentrations, it may be toxic to the cell. Intracellular cholesterol homeostasis is, therefore, stringently controlled by tightly coupled regulatory mechanisms including influx and efflux, esterification, and storage and biosynthesis (65). Oxysterols are oxygenated forms of cholesterol formed directly from cholesterol (or oxysterols derived from cholesterol) by enzymatic and non-enzymatic mechanisms (55). Functionally, oxysterols such as 22(R)-hydroxycholesterol and 24(S)-hydroxycholesterol potently bind ligand-activated transcription factors liver X receptor (LXR)-α and/or LXR-β and induce the upregulation of cholesterol homeostasis-related proteins such as ABCA1—responsible for cholesterol efflux from the cell (66, 67). Notably, however, despite its identification over 50 years ago and an early demonstration of potent sterol biosynthesis regulatory feedback functionality, until recently, physiological roles for 25-hydroxycholesterol (25-HC) have proven elusive (68, 69).
25-HC binds the INSIG protein in the ER and, in doing so, prevents SREBP2 transport to the golgi/nucleus and cholesterol biosynthesis [reviewed in Ref. (70)]. It is not, however, a strong activator of the LXRs nor does it play a significant role in systemic cholesterol homeostasis in vivo (71). In 2009, independent studies showed that CH25H, the enzyme responsible for 25-HC synthesis, is transcriptionally upregulated in macrophages following treatment with a TLR agonist (72, 73). Park and Scott (74) then showed that type I IFNs are also capable of upregulating CH25H (74). While this evidence supported the notion that 25-HC may play a role in immunity, in fact, studies had been emerging for decades implicating 25-HC in the immune response to infection. In 1986, Kournikakis et al. demonstrated that 25-HC can suppress antibody-dependent cell cytotoxicity, and in 1998, Moog et al. showed 25-HC (but not cholesterol) can inhibit HIV in vitro (75, 76). Over the next decade, several groups independently investigated the effects of 25-HC on HCV subgenomic replicon replication and found the oxysterol inhibited this process (77–80). Notably, the effects of 25-HC are not restricted to viruses, and in 2006, Howe and Heinzen described a partial inhibition of the bacteria Coxiella burnetii following treatment of Vero cells with this oxysterol (81).
Broad Antiviral Functionality of 25-HC
Interest in the regulation and functions of 25-HC (and its derivatives) has increased dramatically in recent years [reviewed in Ref. (82)]. In 2012, Gold et al. demonstrated that ATF3 directly suppresses the transcription of CH25H and the production of 25-HC. They further showed that a deletion of ATF3 in APOE−/− mice results in enhanced aortic 25-HC expression and foam cell development (83). In 2013, Blanc et al. showed that 25-HC is the only oxysterol synthesized (and secreted) in significant quantities by murine macrophages after IFN activation and demonstrated that the transcription of CH25H is directly regulated by IFN through the binding of STAT1 to its promoter. These studies also showed that physiological levels of 25-HC have a broad antiviral functionality mediated, in the case of cytomegalovirus (CMV), post-entry via regulation of the sterol biosynthesis pathway. Data presented by Blanc et al. supported an important role for the prenylation side-branch of the sterol biosynthesis pathway, rather than cholesterol, in mediating antiviral effects against CMV (52). At the same time, Liu et al. (84), using a molecular screening approach, also identified CH25H as an important IFN-stimulated gene and demonstrated a broad antiviral functionality for 25-HC (84). Contrary to the CMV-related work of Blanc et al., however, Liu et al. found this effect was mediated via an inhibition of pathogen [vesicular stomatitis virus (VSV) and (HIV)] entry to the cell. The distinct modes of 25-HC action described likely reflect differences between the cell/virus systems examined. Liu et al. further showed that CH25H−/− mice are more susceptible to MHV-68 infection and the therapeutic administration of 25-HC to humanized mice suppressed HIV-induced T cell depletion (84). Together, these studies identified a significant new role for 25-HC as an effector in the immune response to infection and, since 2013, several independent studies have described further roles for 25-HC in this context. In 2014, Roulin et al. showed that 25-HC suppresses picornavirus infections by displacing cholesterol binding to the oxysterol sterol-binding protein (OSBP1). In doing so, 25-HC disrupts a cholesterol-phosphatidylinositol 4-phosphate counter-current essential for formation of the replication organelle at ER–Golgi membrane contact sites (85). Building on early studies investigating sterol pathway regulation, in 2015, Lu et al. showed that, alongside its ability to inhibit SREBP2 migration to the nucleus, IFN-elicited 25-HC induces a rapid proteosomal degradation of HMGCR in macrophages (6, 86). Work has also shown that 25-HC can inhibit a wide range of unrelated enveloped and non-enveloped viruses including poliovirus, Hepatitis B and C viruses, human papillomavirus (HPV-16), human rotavirus, encephalomyocarditis virus, and SFTS virus (87–92). Recent studies have also revealed more detail regarding the regulation of CH25H. Mboko et al. (93) showed that CH25H expression in mice is, at least partly, dependent on IRF1 and Xiang et al. (94) demonstrated an IFN-independent induction of CH25H in hepatocytes in response to viral infection (93, 94). In this regard, evidence to-date suggests that CH25H gene expression is regulated in a cell-specific manner and it cannot be considered a prototypic ISG. While the majority of studies have broadly focused on the ability of 25-HC to suppress infection via regulation of lipid metabolism, data from Shibata et al. (5) suggest that it may also achieve this via a specific activation of the GCN2/eIF2α/ATF4 branch of the integrated stress response (ISR) (5).
Notably, recent studies describe direct interactions of both CH25H and 25-HC with gene products of the microorganism. Chen et al. (95) describe a direct interaction between CH25H and NS5A of HCV leading to an inhibition of NS5A dimerization and inhibition of HCV replication (95). More recently, Ren et al. (96) describe an INSIG homolog with predicted 25-HC-binding capacity in the bacterium Mycobacterium vanbaalenii (96). What physiological role this would play, however, remains unclear.
In summary, it is now accepted that 25-HC is an important component of the IFN-induced response to infection and a range of studies have identified divergent mechanisms for the inhibition of entry, replication, and exit from the cell.
25-HC as an Inflammatory Mediator
Recent evidence has emerged supporting a role for 25-HC as a pro- and/or anti-inflammatory mediator. In 2010, it was demonstrated that 25-HC has the capacity to suppress CCR7 expression and thus impair DC migration (97). Wang et al. subsequently described a 25-HC-elicited RIG-I-dependent induction of IL-8 and Raccosta et al. showed that 25-HC can bind CXCR2 (98, 99). In 2014, Data from Reboldi et al. showed that in macrophages, through its ability to antagonize SREBP, 25-HC can reduce IL-1β expression and inflammasome activation. They further demonstrated that CH25H−/− mice are more sensitive to septic shock and have an enhanced ability to suppress L. monocytogenes infection (100). In contrast, Gold et al. (101) describe 25-HC as an amplifier of inflammation, showing a reduction in pro-inflammatory gene expression in poly I:C treated CH25H−/− macrophages and decreased inflammatory pathology in the lungs of Influenza virus-infected mice (101). Further evidence of a pro-inflammatory role for 25-HC has very recently emerged from Jang et al. (102) who describe a role for the oxysterol as an endogenous signal for NLRP3/inflammasome activation during cerebral inflammation (102). At present, therefore, evidence would appear to support multiple roles for 25-HC in the regulation of inflammation.
An important consideration in the analysis and interpretation of 25-HC-related data is the concentration of exogenous oxysterol utilized in vitro. Others, and ourselves, have demonstrated that nanomolar concentrations of 25-HC elicit profound effects in primary macrophages, e.g., Ref. (52, 100). In the literature, however, functional roles for 25-HC have been defined after treatment of cells with considerably higher concentrations (e.g., 10–100µM)—for instance (99, 102). Caution should be exercised when interpreting data from experiments utilizing arguably supraphysiological concentrations of the oxysterol. In this regard, more work—in particular in vivo—is required to characterize the specific concentrations, circumstances, locations, and times at which pro- or anti-inflammatory effects are observed during infection.
CH25H and Acquired Immune Responses
In 2009, Bauman et al. described a role for 25-HC in the direct repression of B cell proliferation and immunoglobulin class switching (72). Oxysterols are often subject to consecutive modifications in order that a functional effector molecule can be synthesized, and in 2011, two groups identified 7α,25-HC—generated via the hydroxylation of 25-HC by CYP7B1—as a ligand for the receptor EBI2 (71, 82, 103, 104). While studies of 25-HC have broadly focused on its production and function in macrophages, 7α,25-HC is primarily synthesized in radiation resistant stromal cells (105). The 7α,25-HC receptor EBI2 is expressed throughout the immune system and, to-date, has been shown to play a critical role in B lymphocyte and dendritic cell biology. In B lymphocytes, EBI2 binding of 7α,25-HC, induces a series of temporally regulated migratory events. B cells first move to the outer follicles of lymphoid tissues, then migrate to the T cell margin, and finally move to interfollicular regions before EBI2 is downregulated and germinal centers form (82, 106). Ultimately, binding of 7α,25-HC to EBI2 and subsequent B cell repositioning events are crucial to antibody responses and CH25H−/− mice have reduced IgG and IgM responses to T cell-dependent antigens (107, 108). In dendritic cells, EBI2 and 7α,25-HC are also crucial and determine cellular migration/location and ability of these cells to support CD4 and B cells responses to blood borne antigens (82, 109).
Importantly, roles for 25-HC and downstream metabolites in the regulation of T lymphocyte responses are emerging. In 2014, data from Chalmin et al. suggested that 7α,25-HC may direct the migration of activated T cells into the CNS in a model of autoimmune encephalomyelitis. Further, a very recent study from Li et al. (110) has described a role for 25-HC in T helper cell development. Specifically, through an interaction with T lymphocyte EBI2, 7α,25-HC functions to orientate T cells at the interface of the follicle and the T cell zone. In doing so, it promotes TFH cell differentiation by facilitating interactions first between the lymphocytes and ICOSLHI CD25+ dendritic cells and subsequently between lymphocytes and B cells (110).
Evidence is rapidly accumulating that multiple complementary mechanisms are responsible for the molecular coupling of IFN to sterol metabolism. In this context, Singaravelu et al. (111) recently described an ability of 25-HC to induce the expression miR-185 and, in doing so, regulate host lipid metabolism pathways critical to HCV replication (111). This finding will be discussed in more detail later.
How Does 25-HC Suppress Infection?
For almost four decades, a physiological role for 25-HC remained elusive, however, multiple lines of evidence now show that the direct induction of CH25H expression and 25-HC synthesis by IFN is a fundamentally important feature of immune responses to infection. A key unanswered question is: what are the mechanisms by which 25-HC can suppress infection?
Studies to-date have primarily utilized 25-HC as a research tool to study the functional role of cholesterol homeostasis and its effects on membrane composition, vesicular trafficking, and isoprenylation. The addition of exogenous side-chain oxysterols such as 25-HC to cells is known to elicit trafficking of cholesterol from the membrane to the ER—an event that may perturb membrane architecture and the orientation and composition of, for example, lipid rafts (112). Via an interaction with OSBP1, 25-HC is also known to disrupt a cholesterol-phosphatidylinositol 4-phosphate “counter-current” required for ER to golgi cholesterol transport and Rhinovirus replication (85). In 2013, Liu et al. found 25-HC-inhibited membrane fusion and cellular infections by HIV, Ebola, and HCV (84). In contrast, Blanc et al. (52) found that 25-HC had a minimal effect on MCMV entry and, by utilizing a mathematical model, estimated that entry-related effects of this oxysterol account for only 10% of its overall antiviral activity for this virus. Data from Blanc et al. (52) support the view that 25-HC primarily exerts its effects by limiting mevalonate–sterol biosynthesis pathway flux. In particular, flux associated with the isoprenoid branch is responsible for protein prenylation (52). Prenylation refers to the posttranslational modification of proteins by the covalent addition of farnesyl (C15) or geranylgeranyl (C20) to conserved amino acid motifs and is key to protein–membrane interactions/intracellular localization of, for example, the Rab proteins. The Rab GTPase superfamily has more than 20 members playing essential roles in vesicle trafficking and protein localization in the cell. Prenylation is the key to this function as it allows attachment of the protein to the lipid bilayer. An ability to inhibit the prenylation and, therefore, the function of Rab GTPases may allow 25-HC to influence a wide range of pathogens that depend on or utilize this family of proteins. For example, Rab11 is key to the recycling endosome pathway in cells and plays a critical role in the assembly of multiple negative strand RNA viruses such as respiratory syncitial virus, Influenza A, and Sendai virus (113–115). Notably, the direct prenylation of pathogen proteins is also an important event in some bacterial infections. For example, the PelH and AnkB proteins of Legionella pneumophila are known to require farnesylation, while SifA of Salmonella typhimurium requires geranylgeranylation for membrane association (116–118). The effects of 25-HC on these bacteria have yet to be characterized. Given the complexity of the Rab superfamily and the differential dependency of a range of pathogens on its functions, more work is required to determine whether 25-HC effects are mediated via this route.
An intriguing possibility is that microorganisms may exploit the disruption of Rab prenylation by 25-HC. Rab5a contributes to lysosomal degradation of L. monocytogenes in macrophages and a disruption of its geranylgeranylation in this context may prove advantageous to the bacterium. In this regard, data show that the growth of L. monocytogenes is greater in WT macrophages when compared to their CH25H−/− counterparts (100).
While several studies have identified an important role for 25-HC in vivo, questions remain regarding: where and when 25-HC is synthesized after infection, how 25-HC synthesis is regulated in different anatomical locations, functional in vivo intra- and extracellular concentrations, half-life in the tissues and circulation and therapeutic potential (84, 108). In 2014, Ikegami et al. analyzed oxysterol concentrations in serum of patients with chronic HCV infection and found 25-HC levels 44% greater than those in the controls. Notably, 25-HC levels decreased significantly in these HCV-infected patients after they had received PEGylated IFN and Ribavirin therapy for a period of 3 months (119).
The presence of INSIG homologs with potentially conserved 25-HC-binding capacity in bacteria and yeast raises the intriguing possibility that the oxysterol can directly influence these organisms. An incomplete understanding of sterol metabolism in these organisms and, in some cases, an absence of SREBP, SCAP, or HMGCR homologs makes progress in this field challenging at present (96).
IFN-Induced miRNA Regulation of Sterol Metabolism in Infection and Immunity
miRNA are small (20–25 nt) RNA encoded in introns, exons, and intergenic regions of the mammalian genome and are typically co-expressed with a protein-coding or non-coding primary transcript. miRNA function to regulate gene expression via imperfect base-pairing to the 3′UTR of mRNA which results in the targeting of the transcript for degradation and/or an inhibition of translation. A key functional characteristic of miRNA is that they can target and regulate the expression of multiple transcripts in the cell. Since the discovery that miRNA, in particular miR-33, function to regulate cholesterol homeostasis, interest in this area has grown dramatically and more than 20 miRNAs are now known to directly target the sterol metabolic network [reviewed in Ref. (120)]. Notably, a small number of sterol-associated miRNA have been shown to be IFN regulated and a subset of these also contribute to the immune response to infection. Here, we will review what is known about these IFN-regulated sterol regulatory miRNA and discuss the mechanisms employed to inhibit pathogens.
Sterol Pathway Targeting by miR-342-5P Generates Broad Antiviral Immunity
miR-342 is encoded in an intron of the Ena-vasodilator-stimulated phosphoprotein gene (EVL) in the mouse or Ena-Vasp-Like (EVL) gene in the human and co-transcribed with this transcript. Transcription of miR-342 can be induced by all-trans retinoic acid or IFN and suppressed by CpG island methylation upstream of EVL. Processing of the primary transcript results in the production of a pre-miRNA hairpin encoding two functional miRNA—miR-342-3p and miR-342-5p (7, 121–124). In vivo, the EVL transcript is primarily expressed in cells of the immune and nervous systems, however, a systematic tissue and cell-type analysis of miR-342 expression has yet to be undertaken (125). In macrophages, miR-342 has been identified as a PU.1-regulated miRNA contributing to myeloid differentiation and miR-342-5p shown to be a pro-inflammatory mediator capable of enhancing miR-155 expression (123, 124). miR-342-5p has recently been implicated in the regulation of SREBP1 and SREBP2 in a cancer cell line; however, biological roles and precise mechanisms for the miRNA in relation to sterol biosynthesis and the immune response were not addressed (126). In this regard, we recently demonstrated that, in BMDM, miR-342-5p is directly regulated by IFN via IRF1 (7). We further showed that miR-342-5p directly targets the master transcriptional regulator of the pathway SREBP2, multiple members of the sterol biosynthesis pathway including (IDI1 and SC4MOL) and can reduce miR-33 abundance in the cell (7). In doing so, miR-342-5p contributes to IFN-induced suppression of the sterol metabolic network—reducing the abundance of both sterol pathway metabolic intermediates and total cholesterol in macrophages. Notably, IFN-induced miR-342-5p suppression of the sterol metabolic network enables this miRNA to inhibit the replication of unrelated viruses including Influenza A and HSV1 (7). This study, in conjunction with our previous analysis of the antiviral effects 25-HC, highlights the complex, temporally coordinated, and redundant molecular circuitry utilized by the cell to regulate the sterol metabolic network during infection.
A summary of the molecular circuitry underlying the regulation of sterol metabolism by IFN is presented in Figure 2. In murine BMDM, CH25H mRNA expression is regulated by STAT1 and increases dramatically in the first half hour after activation of cells by IFN (52). In contrast, miR-342-5p expression increases from 2 to 3 h after IFN-γ activation of BMDM and is regulated by IRF1. Data from others and ourselves suggest, therefore, that a sequential IFN-elicited regulation of the sterol metabolic network exists in which 25-HC provides a rapid mechanism for decreasing sterol biosynthesis. It does this via an immediate proteosomal degradation of HMGCR and subsequent inhibition of SREBP2 nuclear translocation. This leads to a suppression of viral entry and/or replication via an inhibition of sterol pathway flux—an outcome that will also activate STING to further stimulate type I IFN production. Since IFN-stimulated ATF3 swiftly inhibits CH25H transcription and 25-HC is rapidly catabolized, the CH25H response is primarily effective in limiting sterol synthesis for the first few hours of IFN induction. Importantly, however, miR-342-5p then further promotes a longer, sustained fine-tuning of sterol metabolism, and antiviral effects in the cell by targeting SREBF2 RNA and transcripts encoding select enzymes of the sterol biosynthesis pathway (e.g., IDI1 and SC4MOL). In this role, miR-342-5p complements and reinforces the antiviral functions of the rapidly induced oxysterol 25-HC on sterol biosynthesis.
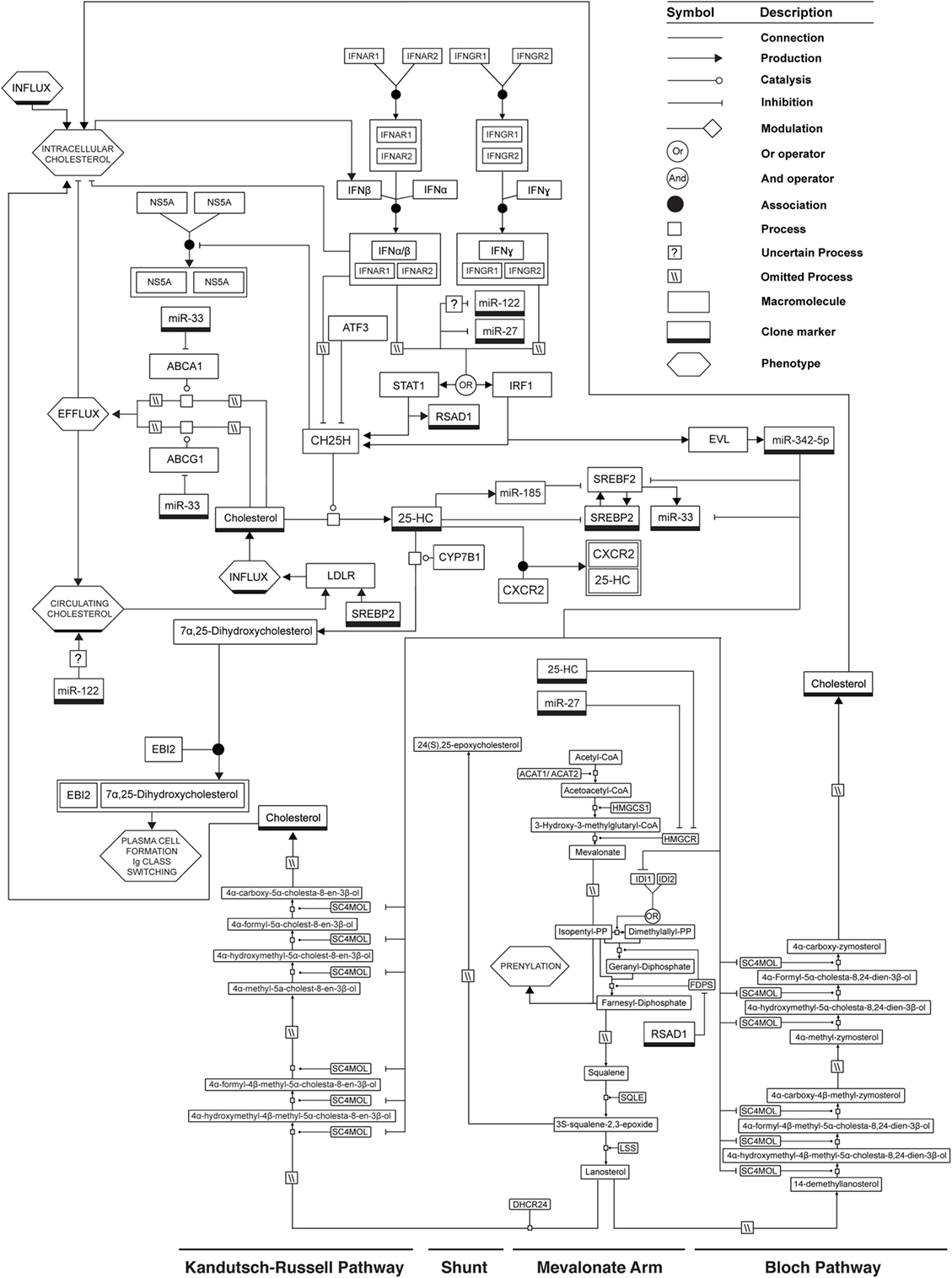
Figure 2. Mechanisms by which IFN can regulate the sterol metabolic network. See legend for glyph notation.
While in vitro data show that an inhibition of endogenous miR-342-5p can reduce the antiviral effects of exogenous IFN by 40–50%, the relative importance of this miRNA—and also 25-HC—in vivo are not known. A critical next step, therefore, will be the production of single miR-342-5p and combined miR-342-5p/CH25H KO murine models in which the individual and combined functions of the miRNA and oxysterol can be tested in the context of infection.
miR-122 Positively Regulates Cholesterol, Facilitates HCV Replication, and is Downregulated by IFN
Arguably, the first miRNA associated with IFN responses to infection and the regulation of cholesterol metabolism was miR-122. miR-122 is a tissue-specific miRNA highly expressed in hepatocytes in which it constitutes around 70% of all miRNA present in the cell. In agreement with this strict tissue-specific expression, we failed to detect miR-122 in resting or IFN-activated bone marrow-derived macrophages (7). In 2005, Jopling et al. (127) demonstrated a direct interaction between miR-122 and the 5′ region of the Hepatitis C genome and showed that this interaction results in the facilitation of viral replication (127). Subsequently, a role for miR-122 in the regulation of lipid metabolism was revealed when the administration of an antisense oligonucleotide (“antagomir”) to mice resulted in a significant reduction in circulating cholesterol levels (128, 129). Further analyses of miR-122 KO animals confirmed an absence of miR-122 results in reduced plasma cholesterol levels; however, it remains unclear what the specific sterol-related targets of miR-122 are and how this miRNA acts to regulate systemic cholesterol levels (130, 131). Notably, in 2007, miR-122 was identified as an IFN-regulated miRNA whose abundance decreased by around 80% in Huh cells treated with IFNβ (132). Pedersen et al. further showed that the transfection of a miR-122 inhibitor into cells could suppress HCV replication with a similar magnitude of regulation to that induced by IFNβ alone (132). This and other findings have led to the development and in vivo testing of therapeutic miR-122 inhibitors that show promise for the treatment of chronic HCV infection (133, 134).
In summary, while miR-122 couples IFN to the regulation of sterol metabolism and, by direct interaction with the viral genome, plays a fundamental role in the replication of HCV, it is currently unknown whether the IFN suppression of miR-122 directly influences circulating cholesterol levels and whether this plays a role in modulating host responses to infection.
miR-185 is Regulated by 25-HC and Inhibits Virus Replication by Targeting Lipid Metabolism
Recent evidence suggests that miR-185 functions to regulate sterol metabolism in the liver during an immune response to infection. In the absence of an infection or IFN treatment, data from hepatocytes show that miR-185 expression is downregulated when cholesterol is depleted in vitro and that expression of this miRNA is directly regulated by SREBP1c via a single sterol response element in its promoter (135). Multiple studies have further shown that this miRNA can regulate SR-BI, SREBP1c, SREBP2, HMGCR, and LDLR transcript abundance (126, 136, 137). Notably, in 2015, Li et al. showed that HCV can upregulate SREBP2 via a core protein-mediated suppression of miR-185 (138) while Singaravelu et al. demonstrated that miR-185 expression in hepatocytes is upregulated by 25-HC and restricts HCV replication via a repression of cellular lipid uptake and biosynthesis (111). Data suggest, therefore, that miR-185 is antiviral, indirectly upregulated by IFN through 25-HC and exerts its effects (at least in hepatocytes) via a suppression of the sterol metabolic network. This mode of regulation was not detected in activated or infected murine macrophages and the general antiviral significance of these observations in alternative cell types has yet to be tested (7). Importantly, however, these data strongly support previous findings demonstrating a membrane-independent antiviral mechanism for 25-HC.
miR-27 Integration of Immunity and Lipid Metabolism
Over the past decade, functional roles for miR-27 have been investigated in the context of several viral infections. In this regard, significant attention has focused on the ability of Herpesvirus saimiri and murine CMV to induce a reduction in miR-27 abundance and the virus-related mechanisms mediating this reduction are now relatively well characterized (139–142). Notably, the functional consequences and benefit to the Herpesviruses of this reduction are incompletely understood with studies focusing on a suppression of miR-27-inducing constitutive T cell activation (Herpesvirus saimiri) or the suggestion that this event enhances IL-10 production during MCMV infection (142). Since 2013, several studies have described a miR-27 regulation of lipid (including sterol) metabolism. In 2013, Vickers et al. described a miR-27-mediated reduction in HMGCR abundance and a sensitivity of miR-27 to lipid levels in vivo (143). Also at this time, Shirasaki et al. described a HCV (but not IFN) induction of miR-27a in hepatocytes and a repression of ABCA1, SREBP1, and SREBP2 by the miRNA. They further showed that an inhibition of miR-27 increased cellular lipids/viral replication and an over-expression of the miRNA resulted in a reduction in viral infectivity and enhanced IFN signaling (144). In 2014, Singaravelu et al. showed HCV induction of miR-27 is accompanied by the formation of large, abundant lipid droplets in hepatocytes. Zhang et al. further demonstrated this miRNA directly targets ABCA1, LPL, and ACAT1, and, in doing so, reduces cholesterol efflux/uptake and regulates the balance of free versus esterified cholesterol in THP1 cells (145). Notably, recent work from Zheng et al. (146) describes a type 1 IFN downregulation of miR-27 in macrophages resulting in enhanced SIGLEC1/TRIM27 expression. As a consequence, IFN signaling was suppressed and VSV replication enhanced (146). Taken together, the above studies suggest that miR-27 couples infection-induced IFN responses to the regulation of sterol metabolism. Importantly, however, the significance of miR-27 and the relative importance of its sterol-regulatory effects in the context of specific infections and cell types are, incompletely understood. In this regard, we and others have demonstrated that MCMV is dependent on the sterol metabolic network for its replication. It may be hypothesized, therefore, that a suppression of miR-27 functions to upregulate the sterol metabolic network and, in doing so, enhances viral replication.
Alongside the examples discussed above, several other miRNAs hold promise as IFN-regulated modulators of the sterol metabolic network. Others, and ourselves, have demonstrated an IFN-elicited downregulation of miR-33 (7, 147). Recent work from Lai and colleagues demonstrates that miR-33 promotes pro-inflammatory signaling via an ABCA1/ABCG1 augmentation of lipid raft microdomains (147). IFN-mediated downregulation of this miRNA, therefore, may serve to suppress the ongoing inflammatory response. Both viral infection and IFNγ can induce the expression of miR-19b—known to target ABCA1 (148, 149). Further, type 1 IFN (and hepatitis B virus) suppress the expression of miR-145—a miRNA known to target ABCA1 and HPV and play a role in the progression of atherosclerosis (150–154).
In summary, through their ability to simultaneously regulate multiple genes and propensity for fine-tuning rather than the induction of dramatic alterations in RNA expression, miRNA are ideally suited to the task of coordinating protective functions of the sterol metabolic network (on which the cell depends). Notably, work to-date supports cell- or tissue-specific expression of IFN-regulated miRNAs. In this regard, a great deal is still unknown about where and when IFN-regulated miRNA are expressed in vivo, how they are regulated by IFN and what their targets are in particular cell types in distinct species. In this regard, knockout miRNA models remain relatively scarce. There is a pressing need, therefore, for the development of new models to enhance our understanding of sterol regulatory miRNA and the roles they play in host protection against infection.
ISG Regulation of the Sterol Metabolic Network During Infection
While this review has focused on IFN-elicited oxysterol and miRNA-related mechanisms, several “conventional” ISG, integral to the cellular response to infection, also elicit their effects via the sterol metabolic network.
Viperin
Work characterizing the IFN-regulated gene Viperin (virus inhibitory protein, endoplasmic reticulum-associated, IFN-inducible, or RSAD2) has demonstrated that this protein can inhibit both RNA and DNA viruses (155–158). Viperin can decrease HCMV late gene expression, block the release of Influenza A and HIV-1 particles from the cell, and limit the replication of HCV, dengue virus, and WNV [reviewed in Ref. (159)]. Importantly, in vitro data show that Viperin inhibits Influenza A budding by disrupting cell membrane lipid raft integrity and increasing membrane fluidity. A key feature of this mechanism is the binding of Viperin to the sterol pathway enzyme farnesyl diphosphate synthase (FDPS)—an enzyme integral to sterol biosynthesis and the processes of farnesylation and geranylgeranylation (156, 158). Together, these observations suggest Viperin functions to inhibit Influenza A release via regulation of the sterol metabolic network; however, a precise mechanism has yet to be determined. Unlike Influenza A and other viruses such as HIV-1 and Ebola, HCV does not bud from lipid rafts. HCV does, however, have an intimate relationship with cellular lipid metabolism—in particular, a dependency on lipid droplets. It has been suggested, therefore, that Viperin may inhibit HCV replication by altering the lipid composition of these droplets via its interaction with FDPS. This has not, however, been confirmed (160). Very recently, a TLR4/IRF3-dependent Viperin-induced reduction in membrane cholesterol and sphingomyelin was found to be key to the inhibition of Rabies virus replication in RAW264.7 cells (161). Taken together, the above studies highlight the importance of Viperin as a very early IFN-induced antiviral protein. While our mechanistic understanding is incomplete, it is notable that Viperin exerts at least some of its effects via the specific targeting of a key enzyme in the sterol metabolic network and a profound alteration of cellular membrane composition. Further work is required to confirm a conclusive link between these two observations and investigate mechanisms by which pathogens can exploit this protein for their own benefit (162).
IFITM3
While the IFN-inducible transmembrane (IFITM) proteins were first described some 20 years ago, their antiviral properties remained unknown until 2009 when Brass et al. demonstrated a functional role in cellular resistance to Influenza A, WNV, and Dengue virus (163). Since 2009, a plethora of studies have demonstrated the importance of IFITM proteins in suppressing virus-related morbidity and mortality and have characterized roles for the IFITM proteins in responses to a range of enveloped and non-enveloped viruses [reviewed in Ref. (164)]. Much of this work has focused on the ability of IFITM proteins to inhibit viral entry and/or the very early stages of viral replication. In this regard, in 2013, Amini-Bavil-Olyaee et al. demonstrated that IFITM1, 2, and 3 interact with vesicle-associated membrane protein A (VAPA) (165). VAPA is a highly conserved protein, generally found in the ER and, importantly, known to play a role in cholesterol homeostasis via its interaction with OSBP. Under normal circumstances, OSBP is found in the cytoplasm where it serves as a cholesterol sensor and, together with VAPA, functions to redistribute cholesterol from the ER to other organelles in the cell. Amini-Bavil-Olyaee et al. (165) found that an IFITM-mediated disruption of the VAPA–OSBP interaction results in cholesterol levels increasing dramatically in late-endosomal compartments. They attributed a block in VSV release into the cytosol to this alteration in membrane composition (165). Notably, subsequent studies suggest an IFITM-mediated regulation of SNAREs and/or regulation of protein lateral mobility may explain the ability of these proteins to inhibit IAV entry to the cell (166). Interestingly, Munoz-Moreno et al., very recently, described a role for IFITM2 in protecting Vero cells against African Swine Fever Infection—a DNA virus (167). In agreement with previous work, they also described an IFITM-associated accumulation of cholesterol in cells, however, it remains unclear whether an IFN-induced IFITM-mediated regulation of the sterol metabolic network plays a direct role in the antiviral functions of this family of proteins.
Concluding Remarks and Future Perspectives
Three decades after Cantell and colleagues speculated that IFN regulates sterol metabolism, the first evidence for a molecular coupling of IFN to the sterol metabolic network was provided by a systems biology investigation of macrophage responses to infection (4, 13). Given the wealth of clinical and molecular data now available, it is clear that an IFN-mediated reshaping of the sterol metabolic network is an integral, core component of the immune response to infection. The functional outcomes of this event are, however, only beginning to emerge for the wide array of immune-related cells and tissues in the body and are likely to be complex and context dependent. For example, in secondary lymphoid organs, 25-HC is indispensible as an intermediate metabolite crucial to B cell, T cell, and DC migration and antibody class switching. At a cell-intrinsic level, however, this oxysterol can also inhibit viral entry and replication. The latter occurs via the regulation of sterol biosynthesis through SREBP2 and HMGCR in macrophages. A key question arising from work to-date is: how does the sterol metabolic network influence immunity? Studies now show that the molecular coupling between IFN and sterol metabolism is bidirectional. In this regard, the recent work of York et al. (33) is fascinating as it demonstrates the influence of sterol metabolic flux on antiviral IFN signaling (33). Similarly, Reboldi et al. recently showed that the transcription factor SREBP2, whose function is tightly coupled to cholesterol homeostasis, functions to regulate inflammatory responses to infection and it has been demonstrated that a cholesterol loading of macrophages leads to a reduction in miR-342-5p abundance (33, 100, 168). In this context, relatively little is known about how the microbiome and diet, in particular, cholesterol intake, affect IFN responses to infection and this is an important question for the future.
Work to-date emphasizes the complexity of the molecular circuitry governing the regulation of sterol metabolism by IFN and vice versa. Given IFN directly or indirectly regulates the expression of several hundred genes and the inherent complexity of the sterol metabolic network; it is likely that new bidirectional regulatory mechanisms will continue to appear. For example, we have shown that alongside the posttranscriptional and posttranslational effects of miR-342-5p and 25-HC, IFN can also repress SREBF2 transcription. The mechanism for this is unclear, however, epigenetic modifications may play a critical role and this will be important area to pursue in the future.
Given the many recent advances in our understanding of the role sterol metabolism plays in immunity to infection, how can we translate our new knowledge to clinical applications? While changes in systemic cholesterol levels may be of diagnostic value, the therapeutic targeting of host metabolic pathways for anti-infective treatment represents the most exciting application of our knowledge to-date. While statins are a widely utilized, clinically approved, therapy for regulating sterol metabolism and can inhibit a range of pathogens in vitro, in vivo utility in the context of infectious diseases remains inconclusive. The emergence of new pathogens and threat of antibiotic resistance means it is imperative that we develop new methods for treating infectious diseases. While studies have explored oxysterol and miRNA inhibitor regulation of sterol metabolism in a preclinical and clinical context, legitimate concerns have been raised about the pharmacokinetics and potential side-effects of both. For example, the miR-122 inhibitor Miraversin can be effectively delivered in vivo and substantially reduces HCV replication in a Chimpanzee model. Importantly, however, Miraversin administration is typically accompanied by an increase in circulating cholesterol leading to concerns that the cardiovascular health of recipients may be affected. Further, while miR-342-5p regulates sterol biosynthesis and, in doing so, can suppress viral replication it also targets AKT1 and, as a result, can promote inflammation (124). An important objective, therefore, will be to identify the specific mechanisms by which IFN-induced regulators of the sterol metabolic network function to suppress pathogen replication and specifically target these molecules. In doing so, undesired off-target effects will be reduced. In this context, several groups have already explored prenylation as a viable therapeutic target. Prenylation inhibitors are available as an oral medication and show promise in the treatment of, for example, HDV (169).
In conclusion, the sterol metabolic network has now moved center-stage in the context of IFN responses to infection and is increasingly recognized as a fundamentally important immune-metabolomic system holding great promise in the next decades as target for diagnostic and therapeutic intervention.
Author Contributions
KAR wrote first draft of manuscript. KAR and PG edited and revised final manuscript.
Conflict of Interest Statement
The authors declare that this document was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The Centre for Systems Biology at Edinburgh is a Centre for Integrative Systems Biology (CISB) supported by the BBSRC and EPSRC. This work was supported by the BBSRC (BB/K019112/1), the BBSRC/EPSRC (BB/D019621/1), and the Wellcome Trust (WT066784/Z/02/Z) to PG. The funders had no role in the preparation of the manuscript.
Abbreviations
7α,25-HC, 7α,25-dihydroxycholesterol; 25-HC, 25-hydroxycholesterol; CH25H, cholesterol 25-hydroxylase; EBI2, EBV-induced G-protein coupled receptor 2 (also known as GPR183); HCV, hepatitis C virus; HDL, high-density lipoprotein; HIV-1, human immunodeficiency virus 1; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HSV1, herpes simplex virus type 1; IAV, influenza A virus; IFN, interferon; IRF, interferon regulatory factor; LDL, low-density lipoprotein; LXR, liver X receptor; MCMV, murine cytomegalovirus; MHV-68, murine gammaherpesvirus 68; SREBP2, sterol regulatory-binding protein 2; SREBF2, sterol regulatory-binding transcription factor 2; TLR, toll-like receptor; VSV, vesicular stomatitis virus; VZV, varicella zoster virus; WNV, West Nile virus.
References
1. Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol (2008) 26(10):1179–86. doi:10.1038/nbt.1500
2. Spann N, Glass C. Sterols and oxysterols in immune cell function. Nat Immunol (2013) 14(9):893–900. doi:10.1038/ni.2681
3. Fessler MB. Regulation of adaptive immunity in health and disease by cholesterol metabolism. Curr Allergy Asthma Rep (2015) 15(8):48. doi:10.1007/s11882-015-0548-7
4. Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, et al. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol (2011) 9(3):e1000598. doi:10.1371/journal.pbio.1000598
5. Shibata N, Carlin A, Spann N, Saijo K, Morello C, McDonald J, et al. 25-Hydroxycholesterol activates the integrated stress response to reprogram transcription and translation in macrophages. J Biol Chem (2013) 288(50):35812–23. doi:10.1074/jbc.M113.519637
6. Lu H, Talbot S, Robertson K, Watterson S, Forster T, Roy D, et al. Rapid proteasomal elimination of 3-hydroxy-3-methylglutaryl-CoA reductase by interferon-gamma in primary macrophages requires endogenous 25-hydroxycholesterol synthesis. Steroids (2015) 99:219–29. doi:10.1016/j.steroids.2015.02.022
7. Robertson KA, Hsieh WY, Forster T, Blanc M, Lu H, Crick PJ, et al. An interferon regulated MicroRNA provides broad cell-intrinsic antiviral immunity through multihit host-directed targeting of the sterol pathway. PLoS Biol (2016) 14(3):e1002364. doi:10.1371/journal.pbio.1002364
8. Zilberstein A, Kimchi A, Schmidt A, Revel M. Isolation of two interferon-induced translational inhibitors: a protein kinase and an oligo-isoadenylate synthetase. Proc Natl Acad Sci U S A (1978) 75(10):4734–8. doi:10.1073/pnas.75.10.4734
9. Muller M, Winnacker EL, Brem G. Molecular cloning of porcine Mx cDNAs: new members of a family of interferon-inducible proteins with homology to GTP-binding proteins. J Interferon Res (1992) 12(2):119–29. doi:10.1089/jir.1992.12.119
10. Karupiah G, Xie QW, Buller RM, Nathan C, Duarte C, MacMicking JD. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science (1993) 261(5127):1445–8. doi:10.1126/science.7690156
11. Bailey CC, Zhong G, Huang IC, Farzan M. IFITM-family proteins: the cell’s first line of antiviral defense. Annu Rev Virol (2014) 1:261–83. doi:10.1146/annurev-virology-031413-085537
12. Baillie EE, Orr CW. Lowered high-density-lipoprotein cholesterol in viral illness. Clin Chem (1979) 25(5):817–8.
13. Cantell K, Ehnholm C, Mattila K, Kostiainen E. Interferon and high-density lipoproteins. N Engl J Med (1980) 302(18):1032–3. doi:10.1056/nejm198005013021817
14. Rosenzweig IB, Wiebe DA, Borden EC, Storer B, Shrago ES. Plasma lipoprotein changes in humans induced by beta-interferon. Atherosclerosis (1987) 67(2–3):261–7. doi:10.1016/0021-9150(87)90287-5
15. Borden EC, Rinehart JJ, Storer BE, Trump DL, Paulnock DM, Teitelbaum AP. Biological and clinical effects of interferon-beta ser at two doses. J Interferon Res (1990) 10(6):559–70. doi:10.1089/jir.1990.10.559
16. Schectman G, Kaul S, Mueller RA, Borden EC, Kissebah AH. The effect of interferon on the metabolism of LDLs. Arterioscler Thromb Vasc Biol (1992) 12(9):1053–62. doi:10.1161/01.ATV.12.9.1053
17. Ehnholm C, Aho K, Huttunen JK, Kostiainen E, Mattila K, Pakkarainen J, et al. Effect of interferon on plasma lipoproteins and on the activity of postheparin plasma lipases. Arteriosclerosis (1982) 2(1):68–73. doi:10.1161/01.ATV.2.1.68
18. Dixon RM, Borden EC, Keim NL, Anderson S, Spennetta TL, Tormey DC, et al. Decreases in serum high-density-lipoprotein cholesterol and total cholesterol resulting from naturally produced and recombinant DNA-derived leukocyte interferons. Metabolism (1984) 33(5):400–4. doi:10.1016/0026-0495(84)90136-7
19. Hawkins M, Horning S, Konrad M, Anderson S, Sielaff K, Rosno S, et al. Phase I evaluation of a synthetic mutant of beta-interferon. Cancer Res (1985) 45(11 Pt 2):5914–20.
20. Massaro ER, Borden EC, Hawkins MJ, Wiebe DA, Shrago E. Effects of recombinant interferon-alpha 2 treatment upon lipid concentrations and lipoprotein composition. J Interferon Res (1986) 6(6):655–62. doi:10.1089/jir.1986.6.655
21. Olsen EA, Lichtenstein GR, Wilkinson WE. Changes in serum lipids in patients with condylomata acuminata treated with interferon alfa-n1 (Wellferon). J Am Acad Dermatol (1988) 19(2 Pt 1):286–9. doi:10.1016/S0190-9622(88)70173-5
22. Boue F, Pastran Z, Spielmann M, Le Chevalier T, Subirana R, Sevin D, et al. A phase I trial with recombinant interferon gamma (Roussel UCLAF) in advanced cancer patients. Cancer Immunol Immunother (1990) 32(1):67–70. doi:10.1007/BF01741727
23. Soardo G, Pirisi M, Fonda M, Fabris C, Falleti E, Toniutto P, et al. Changes in blood lipid composition and response to interferon treatment in chronic hepatitis C. J Interferon Cytokine Res (1995) 15(8):705–12. doi:10.1089/jir.1995.15.705
24. Shinohara E, Yamashita S, Kihara S, Hirano K, Ishigami M, Arai T, et al. Interferon alpha induces disorder of lipid metabolism by lowering postheparin lipases and cholesteryl ester transfer protein activities in patients with chronic hepatitis C. Hepatology (1997) 25(6):1502–6. doi:10.1002/hep.510250632
25. Fernández-Miranda C, Castellano G, Guijarro C, Fernández I, Schöebel N, Larumbe S, et al. Lipoprotein changes in patients with chronic hepatitis C treated with interferon-alpha. Am J Gastroenterol (1998) 93(10):1901–4. doi:10.1111/j.1572-0241.1998.00546.x
26. Andrade RJ, García-Escaño MD, Valdivielso P, Alcántara R, Sánchez-Chaparro MA, González-Santos P. Effects of interferon-beta on plasma lipid and lipoprotein composition and post-heparin lipase activities in patients with chronic hepatitis C. Aliment Pharmacol Ther (2000) 14(7):929–35. doi:10.1046/j.1365-2036.2000.00792.x
27. Morra VB, Coppola G, Orefice G, De Michele G, Vacca G, Filla A, et al. Interferon-beta treatment decreases cholesterol plasma levels in multiple sclerosis patients. Neurology (2004) 62(5):829–30. doi:10.1212/01.WNL.0000113750.11090.67
28. Coppola G, Lanzillo R, Florio C, Orefice G, Vivo P, Ascione S, et al. Long-term clinical experience with weekly interferon beta-1a in relapsing multiple sclerosis. Eur J Neurol (2006) 13(9):1014–21. doi:10.1111/j.1468-1331.2006.01422.x
29. Mauss S, Berger F, Wehmeyer MH, Ingiliz P, Hueppe D, Lutz T, et al. Effect of antiviral therapy for HCV on lipid levels. Antivir Ther (2016). doi:10.3851/IMP3094
30. Kuo PT, Wilson AC, Goldstein RC, Schaub RG. Suppression of experimental atherosclerosis in rabbits by interferon-inducing agents. J Am Coll Cardiol (1984) 3(1):129–34. doi:10.1016/S0735-1097(84)80438-6
31. Pereira CA, Steffan AM, Koehren F, Douglas CR, Kirn A. Increased susceptibility of mice to MHV 3 infection induced by hypercholesterolemic diet: impairment of Kupffer cell function. Immunobiology (1987) 174(3):253–65. doi:10.1016/S0171-2985(87)80001-3
32. Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell (2003) 12(4):805–16. doi:10.1016/S1097-2765(03)00384-8
33. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell (2015) 163(7):1716–29. doi:10.1016/j.cell.2015.11.045
34. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell (2008) 134(1):97–111. doi:10.1016/j.cell.2008.04.052
35. Armstrong AJ, Gebre AK, Parks JS, Hedrick CC. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J Immunol (2010) 184(1):173–83. doi:10.4049/jimmunol.0902372
36. Maganto-García E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation (2011) 124(2):185–95. doi:10.1161/CIRCULATIONAHA.110.006411
37. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14(5):489–99. doi:10.1038/ni.2570
38. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499(7459):485–90. doi:10.1038/nature12297
39. Janes PW, Ley SC, Magee AI. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J Cell Biol (1999) 147(2):447–61. doi:10.1083/jcb.147.2.447
40. Swamy M, Beck-Garcia K, Beck-Garcia E, Hartl FA, Morath A, Yousefi OS, et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity (2016) 44(5):1091–101. doi:10.1016/j.immuni.2016.04.011
41. Rougerie P, Delon J. Rho GTPases: masters of T lymphocyte migration and activation. Immunol Lett (2012) 142(1–2):1–13. doi:10.1016/j.imlet.2011.12.003
42. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis (2004) 190(3):624–31. doi:10.1086/422329
43. Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med (2005) 202(6):761–9. doi:10.1084/jem.20050193
44. Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J Immunol (2006) 177(11):7980–9. doi:10.4049/jimmunol.177.11.7980
45. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14(3):275–81. doi:10.1038/nm1710
46. Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, et al. Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol (2015) 11(2):141–7. doi:10.1038/nchembio.1714
47. Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, et al. Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc Natl Acad Sci U S A (2014) 111(33):12163–8. doi:10.1073/pnas.1322807111
48. Pierce SK. Lipid rafts and B-cell activation. Nat Rev Immunol (2002) 2(2):96–105. doi:10.1038/nri726
49. Karnell FG, Brezski RJ, King LB, Silverman MA, Monroe JG. Membrane cholesterol content accounts for developmental differences in surface B cell receptor compartmentalization and signaling. J Biol Chem (2005) 280(27):25621–8. doi:10.1074/jbc.M503162200
50. Gupta N, DeFranco AL. Lipid rafts and B cell signaling. Semin Cell Dev Biol (2007) 18(5):616–26. doi:10.1016/j.semcdb.2007.07.009
51. Wilflingseder D, Stoiber H. Float on: lipid rafts in the lifecycle of HIV. Front Biosci (2007) 12:2124–35. doi:10.2741/2216
52. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity (2013) 38(1):106–18. doi:10.1016/j.immuni.2012.11.004
53. Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. Hepatitis C virus, cholesterol and lipoproteins – impact for the viral life cycle and pathogenesis of liver disease. Viruses (2013) 5(5):1292–324. doi:10.3390/v5051292
54. Seveau S. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Subcell Biochem (2014) 80:161–95. doi:10.1007/978-94-017-8881-6_9
55. Bah SY, Dickinson P, Forster T, Kampmann B, Ghazal P. Immune oxysterols: role in mycobacterial infection and inflammation. J Steroid Biochem Mol Biol (2016). doi:10.1016/j.jsbmb.2016.04.015
56. Gong X, Qian H, Zhou X, Wu J, Wan T, Cao P, et al. Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and Ebola infection. Cell (2016) 165(6):1467–78. doi:10.1016/j.cell.2016.05.022
57. Zawada KE, Wrona D, Rawle RJ, Kasson PM. Influenza viral membrane fusion is sensitive to sterol concentration but surprisingly robust to sterol chemical identity. Sci Rep (2016) 6:29842. doi:10.1038/srep29842
58. Manes S, del Real G, Martinez AC. Pathogens: raft hijackers. Nat Rev Immunol (2003) 3(7):557–68. doi:10.1038/nri1129
59. Gudleski-O’Regan N, Greco TM, Cristea IM, Shenk T. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe (2012) 12(1):86–96. doi:10.1016/j.chom.2012.05.012
60. Gower TL, Graham BS. Antiviral activity of lovastatin against respiratory syncytial virus in vivo and in vitro. Antimicrob Agents Chemother (2001) 45(4):1231–7. doi:10.1128/aac.45.4.1231-1237.2001
61. Bordier BB, Ohkanda J, Liu P, Lee SY, Salazar FH, Marion PL, et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J Clin Invest (2003) 112(3):407–14. doi:10.1172/jci17704
62. Wang C, Gale M Jr, Keller BC, Huang H, Brown MS, Goldstein JL, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol Cell (2005) 18(4):425–34. doi:10.1016/j.molcel.2005.04.004
63. Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol (2012) 19(2):218–27. doi:10.1016/j.chembiol.2011.12.016
64. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe (2007) 2(4):229–39. doi:10.1016/j.chom.2007.09.003
65. Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet (2007) 41:401–27. doi:10.1146/annurev.genet.41.110306.130315
66. Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem (2000) 275(36):28240–5. doi:10.1074/jbc.M003337200
67. Joseph SB, Tontonoz P. LXRs: new therapeutic targets in atherosclerosis? Curr Opin Pharmacol (2003) 3(2):192–7. doi:10.1016/S1471-4892(03)00009-2
68. Kandutsch AA, Chen HW. Regulation of sterol synthesis in cultured cells by oxygenated derivatives of cholesterol. J Cell Physiol (1975) 85(2 Pt 2 Suppl 1):415–24. doi:10.1002/jcp.1040850408
69. Miller SC, Melnykovych G. Regulation of cholesterol biosynthesis and esterification by 25-hydroxycholesterol in a macrophage-like cell line: uncoupling by progesterone. J Lipid Res (1984) 25(9):991–9.
70. Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J Lipid Res (2009) 50(Suppl):S15–27. doi:10.1194/jlr.R800054-JLR200
71. Diczfalusy U. On the formation and possible biological role of 25-hydroxycholesterol. Biochimie (2013) 95(3):455–60. doi:10.1016/j.biochi.2012.06.016
72. Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A (2009) 106(39):16764–9. doi:10.1073/pnas.0909142106
73. Diczfalusy U, Olofsson KE, Carlsson AM, Gong M, Golenbock DT, Rooyackers O, et al. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J Lipid Res (2009) 50(11):2258–64. doi:10.1194/jlr.M900107-JLR200
74. Park K, Scott AL. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol (2010) 88(6):1081–7. doi:10.1189/jlb.0610318
75. Kournikakis B, Murasko DM, Bozzola JJ. Ultrastructural study of biochemically modulated ADCC in HSV-1 infected and uninfected Chang liver cells. Microbios (1986) 48(195):81–91.
76. Moog C, Aubertin AM, Kirn A, Luu B. Oxysterols, but not cholesterol, inhibit human immunodeficiency virus replication in vitro. Antivir Chem Chemother (1998) 9(6):491–6. doi:10.1177/095632029800900605
77. Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A (2002) 99(24):15669–74. doi:10.1073/pnas.202608199
78. Ye J, Wang C, Sumpter R Jr, Brown MS, Goldstein JL, Gale M Jr. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci U S A (2003) 100(26):15865–70. doi:10.1073/pnas.2237238100
79. Molina S, Castet V, Fournier-Wirth C, Pichard-Garcia L, Avner R, Harats D, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol (2007) 46(3):411–9. doi:10.1016/j.jhep.2006.09.024
80. Pezacki JP, Sagan SM, Tonary AM, Rouleau Y, Belanger S, Supekova L, et al. Transcriptional profiling of the effects of 25-hydroxycholesterol on human hepatocyte metabolism and the antiviral state it conveys against the hepatitis C virus. BMC Chem Biol (2009) 9:2. doi:10.1186/1472-6769-9-2
81. Howe D, Heinzen RA. Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell Microbiol (2006) 8(3):496–507. doi:10.1111/j.1462-5822.2005.00641.x
82. Cyster JG, Dang EV, Reboldi A, Yi T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol (2014) 14(11):731–43. doi:10.1038/nri3755
83. Gold ES, Ramsey SA, Sartain MJ, Selinummi J, Podolsky I, Rodriguez DJ, et al. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med (2012) 209(4):807–17. doi:10.1084/jem.20111202
84. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity (2013) 38(1):92–105. doi:10.1016/j.immuni.2012.11.005
85. Roulin PS, Lotzerich M, Torta F, Tanner LB, van Kuppeveld FJ, Wenk MR, et al. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe (2014) 16(5):677–90. doi:10.1016/j.chom.2014.10.003
86. Faust JR, Luskey KL, Chin DJ, Goldstein JL, Brown MS. Regulation of synthesis and degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase by low density lipoprotein and 25-hydroxycholesterol in UT-1 cells. Proc Natl Acad Sci U S A (1982) 79(17):5205–9. doi:10.1073/pnas.79.17.5205
87. Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J Virol (2013) 87(8):4252–60. doi:10.1128/JVI.03546-12
88. Civra A, Cagno V, Donalisio M, Biasi F, Leonarduzzi G, Poli G, et al. Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol. Sci Rep (2014) 4:7487. doi:10.1038/srep07487
89. Iwamoto M, Watashi K, Tsukuda S, Aly HH, Fukasawa M, Fujimoto A, et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun (2014) 443(3):808–13. doi:10.1016/j.bbrc.2013.12.052
90. Anggakusuma, Romero-Brey I, Berger C, Colpitts CC, Boldanova T, Engelmann M, et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology (2015) 62(3):702–14. doi:10.1002/hep.27913
91. Dorobantu CM, Albulescu L, Harak C, Feng Q, van Kampen M, Strating JR, et al. Modulation of the host lipid landscape to promote RNA virus replication: the picornavirus encephalomyocarditis virus converges on the pathway used by hepatitis C virus. PLoS Pathog (2015) 11(9):e1005185. doi:10.1371/journal.ppat.1005185
92. Tani H, Shimojima M, Fukushi S, Yoshikawa T, Fukuma A, Taniguchi S, et al. Characterization of glycoprotein-mediated entry of severe fever with thrombocytopenia syndrome virus. J Virol (2016) 90(11):5292–301. doi:10.1128/JVI.00110-16
93. Mboko WP, Mounce BC, Emmer J, Darrah E, Patel SB, Tarakanova VL. Interferon regulatory factor 1 restricts gammaherpesvirus replication in primary immune cells. J Virol (2014) 88(12):6993–7004. doi:10.1128/JVI.00638-14
94. Xiang Y, Tang JJ, Tao W, Cao X, Song BL, Zhong J. Identification of cholesterol 25-hydroxylase as a novel host restriction factor and a part of the primary innate immune responses against hepatitis C virus infection. J Virol (2015) 89(13):6805–16. doi:10.1128/JVI.00587-15
95. Chen Y, Wang S, Yi Z, Tian H, Aliyari R, Li Y, et al. Interferon-inducible cholesterol-25-hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci Rep (2014) 4:7242. doi:10.1038/srep07242
96. Ren R, Zhou X, He Y, Ke M, Wu J, Liu X, et al. Protein structure. Crystal structure of a mycobacterial Insig homolog provides insight into how these sensors monitor sterol levels. Science (2015) 349(6244):187–91. doi:10.1126/science.aab1091
97. Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med (2010) 16(1):98–105. doi:10.1038/nm.2074
98. Wang F, Xia W, Liu F, Li J, Wang G, Gu J. Interferon regulator factor 1/retinoic inducible gene I (IRF1/RIG-I) axis mediates 25-hydroxycholesterol-induced interleukin-8 production in atherosclerosis. Cardiovasc Res (2012) 93(1):190–9. doi:10.1093/cvr/cvr260
99. Raccosta L, Fontana R, Maggioni D, Lanterna C, Villablanca EJ, Paniccia A, et al. The oxysterol-CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J Exp Med (2013) 210(9):1711–28. doi:10.1084/jem.20130440
100. Reboldi A, Dang E, McDonald J, Liang G, Russell D, Cyster J. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science (2014) 345(6197):679–84. doi:10.1126/science.1254790
101. Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, et al. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc Natl Acad Sci U S A (2014) 111(29):10666–71. doi:10.1073/pnas.1404271111
102. Jang J, Park S, Jin Hur H, Cho HJ, Hwang I, Pyo Kang Y, et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun (2016) 7:13129. doi:10.1038/ncomms13129
103. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature (2011) 475(7357):524–7. doi:10.1038/nature10280
104. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature (2011) 475(7357):519–23. doi:10.1038/nature10226
105. Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity (2012) 37(3):535–48. doi:10.1016/j.immuni.2012.06.015
106. Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and nonlymphoid tissues. J Immunol (2011) 187(6):3026–32. doi:10.4049/jimmunol.1101262
107. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity (2009) 31(2):259–69. doi:10.1016/j.immuni.2009.06.016
108. Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature (2009) 460(7259):1122–6. doi:10.1038/nature08226
109. Yi T, Cyster JG. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. Elife (2013) 2:e00757. doi:10.7554/eLife.00757
110. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature (2016) 533(7601):110–4. doi:10.1038/nature17947
111. Singaravelu R, O’Hara S, Jones DM, Chen R, Taylor NG, Srinivasan P, et al. MicroRNAs regulate the immunometabolic response to viral infection in the liver. Nat Chem Biol (2015) 11(12):988–93. doi:10.1038/nchembio.1940
112. Olsen BN, Schlesinger PH, Ory DS, Baker NA. 25-Hydroxycholesterol increases the availability of cholesterol in phospholipid membranes. Biophys J (2011) 100(4):948–56. doi:10.1016/j.bpj.2010.12.3728
113. Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, et al. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc Natl Acad Sci U S A (2008) 105(29):10209–14. doi:10.1073/pnas.0712144105
114. Bruce E, Digard P, Stuart A. The Rab11 pathway is required for influenza A virus budding and filament formation. J Virol (2010) 84(12):5848–59. doi:10.1128/JVI.00307-10
115. Bruce EA, Stuart A, McCaffrey MW, Digard P. Role of the Rab11 pathway in negative-strand virus assembly. Biochem Soc Trans (2012) 40(6):1409–15. doi:10.1042/BST20120166
116. Reinicke AT, Hutchinson JL, Magee AI, Mastroeni P, Trowsdale J, Kelly AP. A Salmonella typhimurium effector protein SifA is modified by host cell prenylation and S-acylation machinery. J Biol Chem (2005) 280(15):14620–7. doi:10.1074/jbc.M500076200
117. Price CT, Al-Quadan T, Santic M, Jones SC, Abu Kwaik Y. Exploitation of conserved eukaryotic host cell farnesylation machinery by an F-box effector of Legionella pneumophila. J Exp Med (2010) 207(8):1713–26. doi:10.1084/jem.20100771
118. Price CT, Jones SC, Amundson KE, Kwaik YA. Host-mediated post-translational prenylation of novel dot/icm-translocated effectors of Legionella pneumophila. Front Microbiol (2010) 1:131. doi:10.3389/fmicb.2010.00131
119. Ikegami T, Honda A, Miyazaki T, Kohjima M, Nakamuta M, Matsuzaki Y. Increased serum oxysterol concentrations in patients with chronic hepatitis C virus infection. Biochem Biophys Res Commun (2014) 446(3):736–40. doi:10.1016/j.bbrc.2014.01.176
120. Jeon TI, Osborne TF. miRNA and cholesterol homeostasis. Biochim Biophys Acta (2016) 1861(12 Pt B):2041–6. doi:10.1016/j.bbalip.2016.01.005
121. Grady W, Parkin R, Mitchell P, Lee J, Kim Y, Tsuchiya K, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene (2008) 27(27):3880–8. doi:10.1038/onc.2008.10
122. Careccia S, Mainardi S, Pelosi A, Gurtner A, Diverio D, Riccioni R, et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene (2009) 28(45):4034–40. doi:10.1038/onc.2009.255
123. De Marchis ML, Ballarino M, Salvatori B, Puzzolo MC, Bozzoni I, Fatica A. A new molecular network comprising PU.1, interferon regulatory factor proteins and miR-342 stimulates ATRA-mediated granulocytic differentiation of acute promyelocytic leukemia cells. Leukemia (2009) 23(5):856–62. doi:10.1038/leu.2008.372
124. Wei Y, Nazari-Jahantigh M, Chan L, Zhu M, Heyll K, Corbalan-Campos J, et al. The microRNA-342-5p fosters inflammatory macrophage activation through an Akt1- and microRNA-155-dependent pathway during atherosclerosis. Circulation (2013) 127(15):1609–19. doi:10.1161/circulationaha.112.000736
125. Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res (2013) 41(Database Issue):D561–5. doi:10.1093/nar/gks1114
126. Li X, Chen YT, Josson S, Mukhopadhyay NK, Kim J, Freeman MR, et al. MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS One (2013) 8(8):e70987. doi:10.1371/journal.pone.0070987
127. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science (2005) 309(5740):1577–81. doi:10.1126/science.1113329
128. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature (2005) 438(7068):685–9. doi:10.1038/nature04303
129. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab (2006) 3(2):87–98. doi:10.1016/j.cmet.2006.01.005
130. Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest (2012) 122(8):2871–83. doi:10.1172/JCI63539
131. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest (2012) 122(8):2884–97. doi:10.1172/JCI63455
132. Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature (2007) 449(7164):919–22. doi:10.1038/nature06205
133. Haussecker D, Kay MA. miR-122 continues to blaze the trail for microRNA therapeutics. Mol Ther (2010) 18(2):240–2. doi:10.1038/mt.2009.313
134. van der Ree MH, van der Meer AJ, van Nuenen AC, de Bruijne J, Ottosen S, Janssen HL, et al. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharmacol Ther (2016) 43(1):102–13. doi:10.1111/apt.13432
135. Yang M, Liu W, Pellicane C, Sahyoun C, Joseph B, Gallo-Ebert C, et al. Identification of miR-185 as a regulator of de novo cholesterol biosynthesis and low density lipoprotein uptake. J Lipid Res (2014) 55(2):226–38. doi:10.1194/jlr.M041335
136. Wang L, Jia XJ, Jiang HJ, Du Y, Yang F, Si SY, et al. MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol Cell Biol (2013) 33(10):1956–64. doi:10.1128/MCB.01580-12
137. Jiang H, Zhang J, Du Y, Jia X, Yang F, Si S, et al. microRNA-185 modulates low density lipoprotein receptor expression as a key posttranscriptional regulator. Atherosclerosis (2015) 243(2):523–32. doi:10.1016/j.atherosclerosis.2015.10.026
138. Li M, Wang Q, Liu SA, Zhang JQ, Ju W, Quan M, et al. MicroRNA-185-5p mediates regulation of SREBP2 expression by hepatitis C virus core protein. World J Gastroenterol (2015) 21(15):4517–25. doi:10.3748/wjg.v21.i15.4517
139. Buck AH, Perot J, Chisholm MA, Kumar DS, Tuddenham L, Cognat V, et al. Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA (2010) 16(2):307–15. doi:10.1261/rna.1819210
140. Cazalla D, Yario T, Steitz J. Down-regulation of a host MicroRNA by a Herpesvirus saimiri noncoding RNA. Science (2010) 328(5985):1563–6. doi:10.1126/science.1187197
141. Marcinowski L, Tanguy M, Krmpotic A, Radle B, Lisnic VJ, Tuddenham L, et al. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog (2012) 8(2):e1002510. doi:10.1371/journal.ppat.1002510
142. Guo YE, Riley KJ, Iwasaki A, Steitz JA. Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol Cell (2014) 54(1):67–79. doi:10.1016/j.molcel.2014.03.025
143. Vickers KC, Shoucri BM, Levin MG, Wu H, Pearson DS, Osei-Hwedieh D, et al. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology (2013) 57(2):533–42. doi:10.1002/hep.25846
144. Shirasaki T, Honda M, Shimakami T, Horii R, Yamashita T, Sakai Y, et al. MicroRNA-27a regulates lipid metabolism and inhibits hepatitis C virus replication in human hepatoma cells. J Virol (2013) 87(9):5270–86. doi:10.1128/JVI.03022-12
145. Singaravelu R, Chen R, Lyn RK, Jones DM, O’Hara S, Rouleau Y, et al. Hepatitis C virus induced up-regulation of microRNA-27: a novel mechanism for hepatic steatosis. Hepatology (2014) 59(1):98–108. doi:10.1002/hep.26634
146. Zheng Q, Hou J, Zhou Y, Yang Y, Cao X. Type I IFN-inducible downregulation of MicroRNA-27a feedback inhibits antiviral innate response by upregulating Siglec1/TRIM27. J Immunol (2016) 196(3):1317–26. doi:10.4049/jimmunol.1502134
147. Lai L, Azzam KM, Lin WC, Rai P, Lowe JM, Gabor KA, et al. MicroRNA-33 regulates the innate immune response via ATP binding cassette transporter-mediated remodeling of membrane microdomains. J Biol Chem (2016) 291(37):19651–60. doi:10.1074/jbc.M116.723056
148. Lv YC, Tang YY, Peng J, Zhao GJ, Yang J, Yao F, et al. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis (2014) 236(1):215–26. doi:10.1016/j.atherosclerosis.2014.07.005
149. Xue Y, Wei Z, Ding H, Wang Q, Zhou Z, Zheng S, et al. MicroRNA-19b/221/222 induces endothelial cell dysfunction via suppression of PGC-1alpha in the progression of atherosclerosis. Atherosclerosis (2015) 241(2):671–81. doi:10.1016/j.atherosclerosis.2015.06.031
150. Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol (2013) 87(10):6037–43. doi:10.1128/JVI.00153-13
151. Kang MH, Zhang LH, Wijesekara N, de Haan W, Butland S, Bhattacharjee A, et al. Regulation of ABCA1 protein expression and function in hepatic and pancreatic islet cells by miR-145. Arterioscler Thromb Vasc Biol (2013) 33(12):2724–32. doi:10.1161/atvbaha.113.302004
152. Lin L, Hou J, Ma F, Wang P, Liu X, Li N, et al. Type I IFN inhibits innate IL-10 production in macrophages through histone deacetylase 11 by downregulating microRNA-145. J Immunol (2013) 191(7):3896–904. doi:10.4049/jimmunol.1203450
153. Sala F, Aranda JF, Rotllan N, Ramirez CM, Aryal B, Elia L, et al. MiR-143/145 deficiency attenuates the progression of atherosclerosis in Ldlr-/-mice. Thromb Haemost (2014) 112(4):796–802. doi:10.1160/TH13-11-0905
154. Gao F, Sun X, Wang L, Tang S, Yan C. Downregulation of MicroRNA-145 caused by hepatitis B virus X protein promotes expression of CUL5 and contributes to pathogenesis of hepatitis B virus-associated hepatocellular carcinoma. Cell Physiol Biochem (2015) 37(4):1547–59. doi:10.1159/000438522
155. Chin KC, Cresswell P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc Natl Acad Sci U S A (2001) 98(26):15125–30. doi:10.1073/pnas.011593298
156. Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe (2007) 2(2):96–105. doi:10.1016/j.chom.2007.06.009
157. Nasr N, Maddocks S, Turville SG, Harman AN, Woolger N, Helbig KJ, et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood (2012) 120(4):778–88. doi:10.1182/blood-2012-01-407395
158. Tan KS, Olfat F, Phoon MC, Hsu JP, Howe JL, Seet JE, et al. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J Gen Virol (2012) 93(Pt 6):1269–77. doi:10.1099/vir.0.040824-0
159. Helbig KJ, Beard MR. The role of viperin in the innate antiviral response. J Mol Biol (2014) 426(6):1210–9. doi:10.1016/j.jmb.2013.10.019
160. Fitzgerald KA. The interferon inducible gene: viperin. J Interferon Cytokine Res (2011) 31(1):131–5. doi:10.1089/jir.2010.0127
161. Tang HB, Lu ZL, Wei XK, Zhong TZ, Zhong YZ, Ouyang LX, et al. Viperin inhibits rabies virus replication via reduced cholesterol and sphingomyelin and is regulated upstream by TLR4. Sci Rep (2016) 6:30529. doi:10.1038/srep30529
162. Seo JY, Yaneva R, Hinson ER, Cresswell P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science (2011) 332(6033):1093–7. doi:10.1126/science.1202007
163. Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell (2009) 139(7):1243–54. doi:10.1016/j.cell.2009.12.017
164. Smith S, Weston S, Kellam P, Marsh M. IFITM proteins-cellular inhibitors of viral entry. Curr Opin Virol (2014) 4:71–7. doi:10.1016/j.coviro.2013.11.004
165. Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, et al. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe (2013) 13(4):452–64. doi:10.1016/j.chom.2013.03.006
166. Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog (2014) 10(4):e1004048. doi:10.1371/journal.ppat.1004048
167. Munoz-Moreno R, Cuesta-Geijo MA, Martinez-Romero C, Barrado-Gil L, Galindo I, Garcia-Sastre A, et al. Antiviral role of IFITM proteins in African swine fever virus infection. PLoS One (2016) 11(4):e0154366. doi:10.1371/journal.pone.0154366
168. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science (2010) 328(5985):1570–3. doi:10.1126/science.1189862
Keywords: cholesterol, sterol, interferon, metabolism, miRNA, oxysterol, 25-hydroxycholesterol, miR-342-5p
Citation: Robertson KA and Ghazal P (2016) Interferon Control of the Sterol Metabolic Network: Bidirectional Molecular Circuitry-Mediating Host Protection. Front. Immunol. 7:634. doi: 10.3389/fimmu.2016.00634
Received: 31 October 2016; Accepted: 12 December 2016;
Published: 23 December 2016
Edited by:
Jorg Hermann Fritz, McGill University, CanadaReviewed by:
Michael B. Fessler, National Institute of Environmental Health Sciences, USAJason G. Cyster, University of California, San Francisco, USA
Copyright: © 2016 Robertson and Ghazal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin A. Robertson, a2V2aW4ucm9iZXJ0c29uQGVkLmFjLnVr;
Peter Ghazal, cC5naGF6YWxAZWQuYWMudWs=