 Julius Brandenburg
Julius Brandenburg Norbert Reiling
Norbert Reiling- Microbial Interface Biology, Priority Research Area Infections, Research Center Borstel, Leibniz Center for Medicine and Biosciences, Borstel, Germany
In recent years, it has become apparent that the Wnt signaling pathway, known for its essential functions in embryonic development and tissue homeostasis, exerts immunomodulatory functions during inflammation and infection. Most functional studies indicate that Wnt5a exerts pro-inflammatory functions on its cellular targets, which include various types of immune and non-immune cells. Wnt5a expression has also been linked to the pathogenesis of chronic inflammatory diseases. Activation of beta-catenin-dependent Wnt signaling, e.g., by Wnt3a, has however been shown to limit inflammation by interfering with the nuclear factor kappa-light chain-enhancer of activated B-cells (NF-kappaB) pathway. This review focuses on the regulation of Wnt5a, Wnt3a, and the recently identified Wnt6 and their functional role in bacterial infections with a primary focus on pulmonary tuberculosis, a leading infectious cause of morbidity and mortality worldwide.
Tuberculosis (TB): Overview and Pathogenesis
With 1.5 million fatalities in 2014, TB ranks alongside with the human-immunodeficiency virus (HIV/AIDS) as the leading-cause of death from infectious disease worldwide (1). The causative agent of TB belongs to the genus Mycobacteria and to the Mycobacterium tuberculosis complex, which comprises of several closely related pathogenic sub-species (2). Mycobacterium tuberculosis (M. tuberculosis) is a non-motile, slow growing, gram-positive bacterium with a lipid-rich cell wall. The pathogen is adapted to an intracellular lifestyle, allowing it to survive and replicate within its main host cell, the macrophage (3, 4). TB primarily affects the lung (pulmonary TB) and is usually transmitted by inhalation of very small bacteria-containing droplets (aerosols).
Having entered the lung, M. tuberculosis is ingested by alveolar macrophages and elicits a localized inflammatory response, which, by production of a range of cytokines and chemokines, leads to early recruitment of mono- and polymorph nuclear cells from neighboring blood vessels to the infectious foci (5, 6). This leads to the formation of a cluster of immune cells termed granuloma, the hallmark of TB infection. Initial stages are mainly characterized by an amorphous mass of tissue resident- and monocyte-derived macrophages (7). Naïve T-cell differentiation is driven by antigen-presenting dendritic cells that have migrated to the local lung-draining lymph node (8). Subsequently, antigen-specific T-cells enter the site of infection and the granuloma lesion stratifies. During progression of disease, some cells differentiate into multinucleated giant cells and foamy macrophages. Infected macrophages in the core of the granuloma are enclosed by uninfected and foamy macrophages, which, in turn, are surrounded by varying fractions of T- and B-lymphocytes in the periphery (9–11). In “late” phase granulomas, a fibrous cuff may separate macrophages from lymphocytes, while the center of the granuloma necrotizes, generating the liquefied core termed “caseum.” A functional disintegration of the granuloma ultimately leads to the release of viable, extracellular mycobacteria into the airways. This facilitates transmission of disease by coughing and sneezing of the patient.
TB: Ligands, Receptors, and Signal Transduction Pathways
Cell wall components such as lipoproteins, lipoarabinomannans, and mycolic acids represent conserved mycobacterial structures, which are recognized by pattern recognition receptors (PRRs) of the immune system [recently extensively reviewed in Ref. (12)]. Among these, the family of Toll-like receptors (TLRs) significantly contributes to the initiation of the first line defense response against the invading pathogen. Engagement of TLRs initiates downstream signaling events, which are orchestrated by various adaptor proteins, including the myeloid differentiation primary response gene 88 (MyD88) (13, 14). TLR-MyD88-signaling leads to the translocation of nuclear factor kappa-light chain-enhancer of activated B-cells (NF-kappaB) into the nucleus, which induces the transcription of a variety of cytokines and chemokines, including interleukin (IL)-12 and tumor necrosis factor alpha (TNF-alpha). TNF-alpha is a pro-inflammatory cytokine mainly secreted by macrophages, which was shown to mediate early leukocyte recruitment and granuloma formation in mice (15–18). TNF-alpha expression is critically linked to host protection, since latently M. tuberculosis-infected individuals (19) that received anti-TNF reagents to treat inflammatory diseases (e.g., rheumatoid arthritis) showed an enhanced TB reactivation risk (20). TLR-induced macrophage activation and the release of cytokines and chemokines contribute to the initiation of adaptive immunity (21). Antigen-specific T-helper cells type 1 (Th1), migrating to the site of infection, secrete interferon-gamma (IFN-gamma), which is critical for the host defense against M. tuberculosis (22, 23). It induces a profound transcriptional remodeling of macrophages (24), which sensitizes cells for a more rapid and heightened response to TLR-ligands (25). Synergistic action of IFN-gamma and TLR-ligands “fully” activates macrophages (also referred to as classical activation of macrophages) (25) and enables cells to exert tuberculostatic functions (26, 27). Although it has been shown that TLR2 and Myd88 are not essentially required for the induction of adaptive T cell responses, MyD88 - but not TLR2, TLR4, and TLR9 - is critical for triggering macrophage effector mechanisms central to anti-mycobacterial defense (28, 29).
Wnt Signaling: Overview
The Wingless/Integrase 1 (Wnt) signaling pathway, which is evolutionary highly conserved in multicellular eukaryotic animals (metazoa), regulates basic cellular processes such as proliferation apoptosis, differentiation, polarization, and motility [reviewed in Ref. (30–32)]. Exerting fundamental functions on cells, Wnt signaling plays a crucial role during embryogenesis, but also throughout adult life by maintaining homeostasis in virtually every tissue and organ. Regarding the lung, as the organ primarily affected during TB, Wnt signaling regulates various processes during organogenesis including branching morphogenesis and regional specialization of the epithelium and mesenchyme thereby shaping the architecture of the airways not only in mice but also in other animal species [reviewed in Ref. (33, 34)]. Moreover, dysregulation of Wnt signaling has been linked to lung diseases such as lung cancer, pulmonary fibrosis, or pulmonary arterial hypertension (33, 34).
With regard to the immune system, there is evidence that Wnt signaling is necessary for normal hematopoiesis (35). Moreover, the work of the last years shows that Wnt signaling affects immune cell function during inflammation and infection, demonstrating that this ancient and conserved signaling pathway also regulates processes beyond development and homeostasis. Recent work indicates that Wnt ligands and downstream signaling molecules shape inflammation and the host response to pathogens by modulating various immune cell functions, including cytokine production, immune cell migration, or differentiation [reviewed in Ref. (36–38)]. The heterogeneity and complexity of Wnt regulated functions are reflected by the presence of numerous receptors, which integrate the signals of various ligands, altogether determining cellular functions of target cells.
Wnt Signaling: Ligands, Receptors, and Signal Transduction Pathways
The human and the mouse genome harbor 19 independent Wnt genes (39), which show distinct expression patterns depending on the cellular context. Wnt genes encode for Wnt ligands which are cysteine-rich, glycosylated, lipid-modified, and secreted proteins with a molecular weight of approximately 40 kDa (40). Wnts can act as short-range mediators as they stick to secreting cells or the extracellular matrix (41). To exert their action across a distance in tissues (e.g., as morphogen), it was proposed that Wnts are associated with lipoprotein particles (42). It has also been demonstrated that they are released in small extracellular micelles from endocytic origin, the exosomes (43). This extracellular vesicular transport enables Wnts to bypass long distances and, as they are exposed on the surface of the exosome, to efficiently bind to their receptors and induce signaling in their target cells. Wnt proteins mainly engage Frizzled (Fzd) family members to transduce signals into target cells. In addition, several coreceptors such as lipoprotein-receptor-related protein (LRP) participate in Wnt signaling (44). The 10 mammalian Fzd proteins are seven-transmembrane receptors, belonging to the family of G-protein-coupled receptors (45–47). Fzd receptors comprise of three different domains, the large N-terminal cysteine-rich extracellular region (CRD), the central core with seven hydrophobic membrane spanning alpha helices, and the cytoplasmatic region. Recently, the successful crystallization of Xenopus Wnt8 complexed to the CRD of Fzd8 gave first insights into the structural basis of ligand-receptor binding (48). For signal transduction inside the target cell, the cytoplasmatic region of Fzd receptors mediates binding of adaptor proteins such as dishevelled (Dvl) or heterotrimeric G-proteins (49, 50). To date, at least three distinct signaling cascades are known to be induced by ligand-receptor binding: the Wnt/beta-catenin pathway, the beta-catenin independent Wnt/Ca2+ pathway, and Wnt/Planar cell polarity pathway [reviewed in Ref. (51) and described below].
Wnt Signaling Promoting Inflammation - Wnt5a
Our group was the first who linked the activity of Wnt signaling to infectious disease mechanisms operative in pulmonary TB. We showed that Wnt5a and its putative receptor Frizzled5 (Fzd5) are present in lung biopsies of TB-patients, that mycobacteria induce Wnt5a in macrophages in a TLR-NF-kappaB dependent manner, and that Wnt5a exerts a distinct immunomodulatory function on immune cells (52). This observation was well in line with earlier findings by Sen et al., who had observed an enhanced presence of Wnt5a in inflamed synovial tissue of rheumatoid arthritis patients and had assigned a functional role for Wnt5a in the pathogenesis of this chronic inflammatory disease (53). During the last 10 years, an increased Wnt5a expression was observed in a variety of inflammatory disease settings such as skin lesions of patients with psoriasis vulgaris (54) or cutaneous lichen planus (55), obesity-induced adipose tissue inflammation (56, 57), atherosclerotic lesions (58), and in the sera of patients with septic shock (59). With regard to infections, Wnt5a induction has been described in response to a variety of pathogens (60, 61). While mostly human macrophages were studied, Bansal et al. could show that Wnt5 is expressed in murine macrophages after infection with Mycobacterium bovis BCG - but not with Mycobacterium smegmatis (62). Notably, the authors show that the induction of Wnt5a is also dependent on the presence of TLRs. Also engagement of the C-type lectin dectin-1 was shown to drive Wnt5a formation in macrophages (63). In addition to human and murine cells, an increased expression of Wnt5a was also observed in Mycobacterium marinum-infected adult zebrafish (64), demonstrating that induction of Wnt5a upon infection is a conserved mechanism.
Beside the abovementioned TLRs and dectin-1, a whole variety of PRRs contribute to host cell activation by pathogenic mycobacteria and were shown to be involved in mounting an anti-mycobacterial response. This includes the macrophage inducible C-type lectin (MINCLE), macrophage scavenger receptor A (SRA), macrophage receptor with collagenous structure (MARCO), and the intracellularly located nucleotide-binding oligomerization domain-containing protein 2 (NOD2), NLR family, pyrin domain-containing 3 (NLRP3), as well as the dsDNA sensor cyclic GMP-AMP synthase (12). It is currently not known whether ligand binding to these receptors and the resulting downstream signaling pathways contribute to the M. tuberculosis-induced expression of Wnt proteins in macrophages.
The work of many independent laboratories has shown that Wnt5a mainly exerts pro-inflammatory functions on its cellular targets, which include non-immune cells (65, 66) as well as various types of immune cells (52, 56, 59, 67–69). In macrophages, Wnt5a has been shown to activate the Wnt/Ca2+ pathway (59), which leads to an increase in cytoplasmic Ca2+ levels thereby activating Ca2+-dependent factors such as Proteinkinase C, Ca2+/calmodulin-dependent kinase II (CaMKII), and Calcineurin. Independent laboratories have shown that there is an intense crosstalk between the Wnt5a-induced signaling cascade and the NF-kappaB as well as certain mitogen-activated protein kinase pathways (56, 70, 71). Under homeostatic conditions, the Wnt5a–Fzd5–NF-kappaB (p65) signaling axis was shown to sustain macrophage immune functions (70). It was demonstrated that Wnt5a stimulates phagocytosis, but does not affect bacterial killing by macrophages (68). Wnt5a induces endothelial inflammation (65) and contributes to CXC chemokine 12-ligand mediated T-cell migration (67). In the context of mycobacterial infections, Wnt5a exerts important functions by bridging innate and adaptive immunity: the antigen response of peripheral blood mononuclear cells (PBMCs) of purified protein derivative of M. tuberculosis (PPD) reactive healthy donors is altered by blocking Wnt5a or its putative receptor Fzd5 (52). Both, the production of the cytokine IL-12, critical for dendritic cell migration and T cell priming during TB infection (72), and T cell derived IFN-gamma, critical for “full” activation macrophages, are significantly reduced in PBMCs incubated either with a neutralizing antibody or an antiserum against Wnt5a or Fzd5, respectively (52). These findings suggest that Wnt5a plays an important regulatory role during the host response to M. tuberculosis infection by activating innate immune cells, which in turn directly affects adaptive immune cell function. However, Wnt5a expression and its pro-inflammatory activity have also been linked to the pathogenesis of multiple chronic inflammatory diseases, including rheumatoid arthritis (66), psoriasis (73), colitis (69), atherosclerosis (74–76), and obesity (56, 57). With regard to TB, however, it remains elusive whether excessive Wnt5a formation may contribute to the M. tuberculosis-induced immunopathology.
Wnt Signaling Limiting Inflammation - Wnt/Beta-Catenin Signaling
Wnt/beta-catenin signaling is characterized by Wnt ligand binding to Fzd and LRP coreceptors, leading to the recruitment of heterotrimeric G-proteins and the adaptor protein Dvl to the membrane (77). Subsequently, the so-called “destruction complex,” which continuously tags beta-catenin for proteasomal degradation by the activity of kinases such as glycogen-synthase kinase-beta (GSK3beta), dissociates (78). The accumulation of beta-catenin in the cytoplasm and its translocation into the nucleus leads to activation of gene expression mediated by the transcription factors T-cell factor and lymphoid enhancer-binding factor (TCF/LEF).
In M. tuberculosis aerosol infection experiments, we systematically screened the mRNA expression of all 19 Wnt genes (79) and Fzd receptors (80) in lungs of infected mice. We were able to detect all 19 Wnt genes at all time points analyzed but found a very prominent expression pattern. The mRNA expression of Wnt2, 2a, 3a, 4, 5a, 7a, 8a, and 10b was significantly reduced during infection whereas Wnts 5b, 8b, 9a, 9b, 11, and Wnt16 were not regulated. These results complement data from an earlier study, which demonstrated that also the mRNA expression of the majority of Fzd receptors is downregulated after M. tuberculosis infection (80). The bacterial burden in the lung is characterized by a rapid increase of bacterial numbers between day 1 and day 21 postinfection (p.i.) followed by a plateau of M. tuberculosis replication until day 42. During the first 3 weeks, a massive inflammatory response in the lung in response to infection is established in order to limit the growth of the pathogen. This is characterized by the induction of a robust T cell response and the formation and release of pro- and anti-inflammatory mediators. Thus, when analyzing gene expression in the lung, we observe an inverse correlation of Wnt expression and the formation of pro-inflammatory factors. Although the individual signaling induced by a given Wnt protein needs to be studied on the cellular level, most of these downregulated Wnt factors were previously associated with Wnt/beta-catenin signaling. This is well reflected by a reduced expression of the known Wnt/beta-catenin target gene and feedback regulator Axin2 (81), which was reduced by approximately 50% at days 21 and 42 p.i. (80). In line with our data, in the lung of Streptococcus pneumoniae-infected mice, a reduced Wnt activity has been observed by kinase activity profiling (82).
Notably, M. tuberculosis-infected mouse lungs in vivo and -infected macrophages in vitro showed significantly enhanced expression levels of the receptor Frizzled1 (Fzd1) (80). Moreover, Fzd1 surface expression was increased by stimulation of macrophages with the key cytokine IFN-gamma. Initially, we were puzzled by this observation, because Fzd1 has been clearly linked to Wnt/beta-catenin signaling, while IFNgamma is known to boost pro-inflammatory responses of macrophages to TLR-ligands (25). In a variety of in vivo studies, it was shown that mimicking activation of Wnt/beta-catenin signaling by administration of a GSK3beta inhibitor potently suppresses pro-inflammatory responses and thereby protects animals from pathophysiological conditions (83–86). Due to the fact that epithelial cells in the lung did heavily stain for Wnt3a (80), we hypothesized that upregulation of Fzd1 renders the macrophages more sensitive to Wnt/beta-catenin signaling. And indeed we observed that macrophages reacted to Wnt3a stimulation or GSK3beta inhibition with beta-catenin stabilization and prominently enhanced Axin2 transcript levels, which was accompanied by a significant reduction of M. tuberculosis-induced TNF-alpha formation. This demonstrated that the Wnt3a-induced signal interferes with the M. tuberculosis-induced TLR2-dependent macrophage activation in vitro. To our knowledge, this was the first observation of a counter-regulation of TLR/NF-kappaB/IFNgamma and Wnt/beta-catenin signaling in infected macrophages. Interestingly, this phenotype of a Wnt/beta-catenin negative feedback loop, which represses TLR-triggered inflammatory responses, was also observed in alveolar epithelial cells (87). Along the same line, another beta-catenin-dependent Wnt, Wnt2 has been shown to inhibit enteric bacterial-induced inflammation also in intestinal epithelial cells (88).
Depending on the cellular context, other Wnt-independent pathways may also lead to inhibition of GSK3beta, as e.g., shown for the integrin-linked kinase, an enzyme regulated by integrin signaling, which was shown to stabilize beta-catenin and to activate its target genes (89). It also needs to be considered that infections with pathogenic microorganisms (including M. tuberculosis) are often associated with severe tissue damage. This leads to the release of various intracellular factors such as adenosin triphosphate (ATP), heat shock proteins, mitochondrial components and several alarmins, a group of proteins which include high-mobility group box 1 protein, IL-1α, IL-33, and the Ca2+-binding S100 proteins [reviewed in Ref. (90)]. Recent studies suggest that some of these factors indeed interfere with GSK3beta activity and affect beta-catenin stabilization and the expression of related target genes. Guo et al. observed that activation of P2X7R by BzATP caused the death of alveolar epithelial cells type I by suppressing Wnt/beta-catenin signaling through stimulating GSK3beta (91). In contrast, S100 A8/A9 proteins have been shown to induce beta-catenin signaling in macrophages (92).
Opposing Roles of Wnt5a and Wnt/Beta-Catenin Signaling in Inflammation
In a model of endotoxic shock, lung homogenates from rats that received lipopolysaccharide (LPS) exhibited a decrease in levels of Wnt3a, Fzd1, phosphorylated GSK3β at Ser9, total β-catenin, and nuclear beta-catenin, demonstrating that major constituents of the Wnt3a signaling machinery in the lung were downregulated. In contrast, Wnt5a, Fzd5, total CaMKII, and phosphorylated CaMKII were upregulated, indicating that Wnt5a dependent, pro-inflammatory Ca2+/calmodulin signaling is strongly activated in endotoxemic rats (93). These findings are corroborated by Villar et al. who observed that lungs of septic animals and humans are characterized by acute lung inflammation, collagen deposition, and marked increase of Wnt5a (94). These data show that in certain acute inflammatory situations pro-inflammatory Wnts, such as Wnt5a are induced, whereas Wnt/beta-catenin associated signals are downregulated. As mentioned above mimicking activation of Wnt/beta-catenin signaling by administration of a GSK3beta inhibitor protects animals from pathophysiological conditions. This was shown in mice receiving the TLR4 agonist LPS which were protected from endotoxin shock (84, 86), and in Francisella-infected mice, which were protected against tularemia (95). On a cellular level there is very likely a tightly controlled signal integration of both pathways. Not only in primary macrophages (37, 80), but also in dendritic cells beta-catenin has been shown to regulate the balance between inflammatory and regulatory responses. Beta-catenin in intestinal dendritic cells was required for the expression of anti-inflammatory mediators and the stimulation of regulatory T cell induction, while suppressing inflammatory effector T cells (96). Furthermore, ablation of beta-catenin expression in dendritic cells enhanced inflammatory responses and disease in a mouse model of inflammatory bowel disease. Thus, beta-catenin signaling programs dendritic cells to a tolerogenic state, regulating, and limiting the inflammatory response (96). During the process of infection, this balance is very likely to change. Once the acute phase of an infection has passed, it is likely that Wnt/beta-catenin signals regain more and more activity over time as it is known that its downstream effects are also associated with termination of infection or inflammation and the reestablishment of homeostasis. The latter has been shown in bacteria-induced injury of the urogenital tract (97) during which activation of Wnt/beta-catenin signaling promotes the regenerative response to bacterial injury. This is well in line with the observation that Wnt signaling induces epithelial differentiation during cutaneous wound healing (98).
Wnt6 and Its Role During Disease
In the murine lung, only 3 out of 19 Wnt ligands were upregulated after M. tuberculosis infection (79). Whereas Wnt1 and Wnt10a were expressed to a significantly higher extent only at day 42 p.i., mRNA expression of Wnt6 was increased significantly at days 21 and 42 p.i. (79). Performing immunohistochemical analyses of lung tissue sections, we found Wnt6 in the M. tuberculosis-infected lung at both time points analyzed, but not in the uninfected lung. To our knowledge, this was the first description of Wnt6 expression in the lung. Notably, Wnt6 expression was exclusively found in M. tuberculosis-infected cells within granulomatous lesions, which could be stained for CD68, a well known marker for myeloid cells such as monocytes and macrophages. Subsequent in vitro studies in primary cells showed that Wnt6 is macrophage-derived immunomodulatory factor, which impairs expression and release of the key cytokine TNF-alpha during mycobacterial infections (79). Remarkably, Wnt6-conditioned medium (Wnt6 CM) added to primary murine macrophages induced c-Myc expression, while c-Myc expression was significantly reduced in Wnt6-deficient cells when compared to respective control cells. The transcription factor c-Myc is a global regulator of chromatin which regulates diverse cellular processes including cell cycle (99). Indeed, Wnt6 CM also enhanced Ki-67 expression levels and 3[H]-Thymidin incorporation into cells, indicating that exogenous Wnt6 exerts proliferative effects on primary murine macrophages. However, exogenous Wnt6 did not enhance Axin2 mRNA levels or induce beta-catenin accumulation. These findings were surprising because (i) c-Myc is described as a well-known Wnt/beta-catenin-dependent target gene (100) and (ii) many reports describe that Wnt6-induced signaling is beta-catenin dependent (101, 102). Recent insights into the structural basis of ligand-receptor interaction (48), showing conservation of interacting binding structures, suggest that the induced signaling and function of a certain Wnt ligand might not be predicted by its structure per se. It is known that Wnt5a can initiate discrete signaling pathways through the activation of distinct receptors (103). Moreover, when studying binding affinities of purified Wnt ligands to different Fzd CRDs, Wnt3a, -4, and -5a are functional binding partners for Fzd2, 4, and 5 with appreciable affinity (104). Only Wnt3a has the same effect on downstream signaling with each of the three receptors (104, 105), while it is described that the Wnt ligands, Wnt4 and Wnt5a, are also able to induce beta-catenin-dependent signaling in fusion with specific Fzds and coreceptors (105). These findings indicate that, although Wnt ligands may have a putative bias for individual signaling pathways (104), downstream consequences depend on the presence of receptors, coreceptors, competing ligands, and signaling components in the target cell and a given tissue. The receptor and coreceptor context in which Wnt6 engages a particular signaling pathway in macrophages is, however, not known to date.
So far, Wnt6 was described in the context of early embryogenesis (106, 107), during mouse embryonic gut (108), stomach (109), kidney (110), and heart (111) organogenesis as well as in tooth morphogenesis (112). Dysregulation of Wnt6 has been shown to be related to carcinogenesis: single nucleotide polymorphisms (SNPs) in the Wnt6 gene could be associated with enhanced risk for developing colorectal adenomas (113). Wnt6 was also described to contribute to chemoresistance of gastric cancer cells (114). Both findings are in line with the observed proliferative effect of Wnt6 on macrophages.
The fact that Wnt6 is also induced by conserved microbial structures suggests that it may play a role not only during mycobacterial infections but also in the context of other infectious diseases. Indeed, Liu et al. reported that Salmonella infection induces Wnt6 expression in intestinal epithelial cells (115). In a non-infectious context, Choy et al. showed that Wnt6 expression positively correlates with the Th2 signature observed in mild-to-moderate asthmatics (116). Collectively, the currently available data demonstrate that Wnt6 is expressed during M. tuberculosis infection, but may also be expressed in the context of other inflammatory and infectious disease settings. With regard to function, our study identifies Wnt6 as a macrophage-derived anti-inflammatory and pro-proliferative factor, which is likely to influence immune cell function during TB infection (79).
How Much Wnt is in the M. Tuberculosis Granuloma?
The granuloma is the key pathologic feature of TB. Granulomas serve as an immunological microenvironment, which enable the host to contain microbes and control disease and, at the same time, act as a niche for the pathogen in which the bacteria can persist or even grow (117, 118). As a highly dynamic structure, the granuloma changes appearance during the course of disease. In addition, there is substantial granuloma heterogeneity, as granulomas with varying cellular composition exist within the same animal or patient (119, 120). It is widely accepted that the activation of robust Th1-dominated adaptive immune responses characterized by (i) efficient migration of antigen-specific T-cells into the lung, (ii) a profound IFNgamma release, and (iii) high numbers of classical activated macrophages at the site of infection are essential to efficiently restrict bacterial growth (22, 23, 121, 122). However, uncontrolled pro-inflammatory Th1 responses causing excessive tissue damage, which affects the functional integrity of the granuloma, may be detrimental for the host (123). Available data demonstrate that Wnt signaling is regulated during lung inflammation and suggest that the activity of beta-catenin-dependent Wnt signaling affects the inflammatory balance during inflammation and infection. Studies in non-human primates showing that even during active disease some lesions are sterile (119, 120), and recent results from patients showing that pro- and anti-inflammatory signals are physically segregated within each granuloma (124), suggest that local mechanisms rather than systemic inflammatory responses dictate the functional integrity of the granuloma. Moreover, it seems likely that other factors than cytokines present within the segregated microenvironments of the granuloma contribute to its “homeostasis.” Wnts act on immune cells in different ways as described above. This does not only include immunomodulatory functions as shown for Wnt5a and Wnt3a, but affects immune cell differentiation and proliferation as shown for Wnt6 (summarized in Figure 1). Although in vivo studies are missing, current data on Wnt6 imply that this factor, acting as an anti-inflammatory and pro-proliferative mediator in vitro, attenuates tissue inflammation and drives macrophage differentiation during TB infection. In addition Wnt6 is expressed a subset of lipid body-positive macrophages in M. tuberculosis-infected. (79). Foamy macrophages are considered as key cells, which constitute a nutrient-rich reservoir for M. tuberculosis persistence and contribute to tissue pathology (125, 126). There is increasing evidence that Wnt signaling is of critical importance for cellular lipid metabolism (127). Mani et al. identified Wnt signaling as a regulator of plasma lipid composition and as target for treatment of hyperlipidemia (128). Whether there is a link between Wnt6 and the formation of foamy macrophages in M. tuberculosis infection remains to be shown. The impact of Wnts may even target other important pathways in the host mycobacteria interaction. Activation by Wnt3a augments M. bovis BCG-induced apoptosis in RAW264.7 macrophages (129), indicating a modulation of cellular cell death pathways by Wnt/beta-catenin signaling in macrophages. In addition, the characterization of the microRNA profile circulating of patients with active pulmonary TB has led to the identification of increased levels of miR-29a (11.9- and 5.2-fold) in the TB serum and in the TB sputum, when compared to the expression of the controls (130). This is an important observation since MiR-29a directly targets negative regulators of Wnt signaling (131). Taken together, although Wnt expression is detected within tuberculous lesions, it is currently not clear how much of a given Wnt is “seen” by a macrophage and other immune cells in the granuloma and how this impacts the outcome of disease.
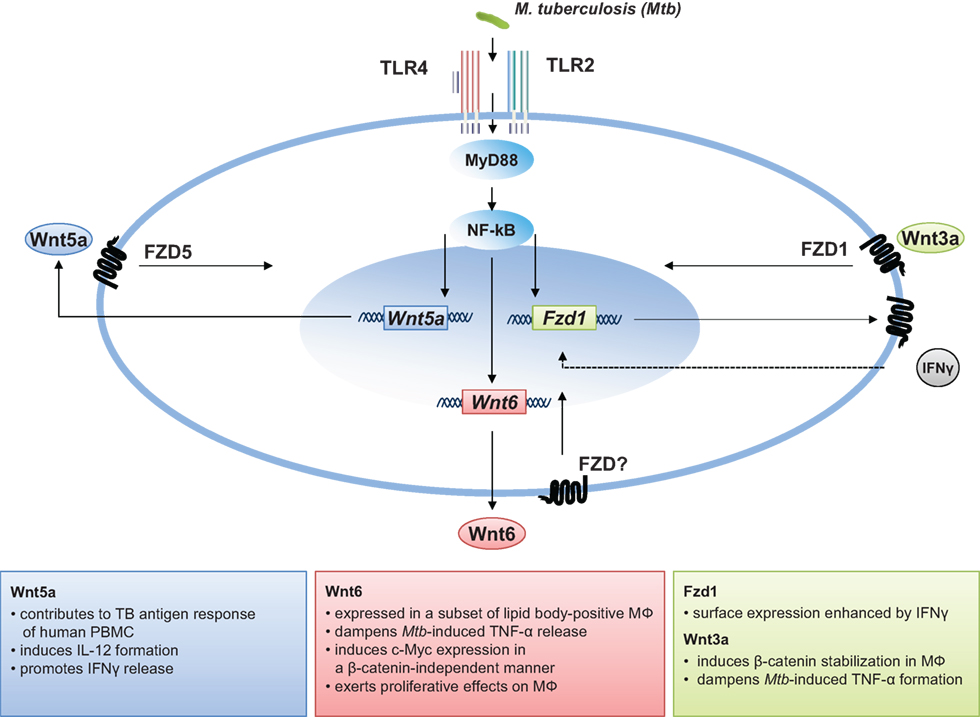
Figure 1. The role of Wnt signaling in M. tuberculosis infection.
Pathogens Exploiting Wnt Signaling
Whereas Wnt/beta-catenin signaling is host protective in many of the above mentioned infection models, it has also been shown that certain pathogens harness Wnt signaling components to promote infection. In a very recent study, Ehrlichia chaffeensis, an obligate intracellular bacterium responsible for the emerging life-threatening zoonosis human monocytotropic ehrlichiosis exploits beta-catenin-dependent and beta-catenin-independent host Wnt signaling pathways to stimulate phagocytosis and promote intracellular survival (132). Ehrlichial tandem repeat proteins were identified to be responsible for these effects. siRNA knockdown of several Wnt signaling pathway components or targets influences significantly reduced ehrlichial infection in human THP1 macrophages. A second example is Chlamydia trachomatis (Ctr) representing another gram-negative bacterium which exploits Wnt/beta-catenin signaling during infection. These bacteria disturb epithelial tissue homeostasis in fallopian tubes. The authors demonstrate that acute Ctr infection activates the paracrine Wnt signaling pathway, leading to profound disruption of epithelial cell structure and function, which facilitates the dissemination of damage beyond that of infected cells (133).
Also viruses have been shown to directly interfere with the Wnt signaling cascade. The activation of Wnt signaling has been demonstrated for the human polyoma virus JC (134), Epstein–Barr virus (135), and Kaposi’s sarcoma-associated herpesvirus (136), being just a few examples of viruses which are known to lead to tumor development, an issue very recently reviewed (137). With regard to viral infections of the airways, adenoviruses are known to strongly interfere with the Wnt signaling machinery as several factors are heavily regulated during infection (138). In a very recent study, the 1918 influenza virus PB2 protein was shown to be a virulence factor interfering with inflammatory and Wnt-mediated signaling in mice. Both, beta-catenin-dependent and -independent Wnt signaling pathways were repressed and associated with impaired lung regeneration and repair (139). Taken together, this illustrates that depending on the infection mechanism and the infected host cell, pathogens may exploit the Wnt signaling machinery in a very specific manner.
Wnts and the Susceptibility to Infectious Disease
With regard to infectious diseases, SNPs in the Wnt signaling cascade have been mainly associated with the risk of cancer development upon infection. It was shown that beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus (HCV) infection (140). In addition, WNT signaling pathway gene polymorphisms are related to the risk of hepatic fibrosis and inflammation in HCV-infected patients (141). In Helicobacter pylori infection, SNPs of MMP-9 -1562/TIMP-1 372 have been associated with an enhanced risk of gastric intestinal metaplasia (142). With regard to TB there are indications that certain SNPs are associated - not just with the consequences of the disease as described above but with disease susceptibility (143). Based on studies in two cohorts, the authors identified three SNPs in the CTNNB1 gene resulting in lower beta-catenin mRNA and protein levels, which were associated with a decreased risk of developing TB. By contrast, two SNPs associated with SFRP1, which were associated with higher C-reactive protein levels that were significantly associated with TB risk in a Chinese Han population, but not in a Tibetan group. These data are the first that suggest that Wnt pathway polymorphisms may influence TB susceptibility and host immune response. It will be interesting to see whether these observations will be seen in other, larger cohorts of different ethnicity, or whether this is a characteristic feature of these specific ethnic groups studied. To date, this recent study is to our knowledge the only one which directly links SNPs in Wnt signaling related genes to the susceptibility towards an infectious disease.
Concluding Remarks
Pulmonary TB is one of the most complex infectious diseases known, characterized by severe chronic inflammation being important for bacterial growth restriction but also being responsible for severe immunopathology. In the last 10 years, it has become clear that the immune function during M. tuberculosis infection is regulated by members of the Wnt signaling cascade. And still new factors and related functions are being identified. A simplified view on the systemic expression and functional role of each of the 19 individual Wnts in M. tuberculosis infection is currently not possible, since to our understanding local determinants define either the induction or repression of a given Wnt signaling factor and its related functions. The detailed characterization of the underlying signaling pathways and the identification of molecular interactions will lead to better understanding of the disease mechanisms and may even lead to the identification of new target structures in order to develop a shorter and more efficient therapy of pulmonary TB.
Author Contributions
All the authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
JB and NR are supported by grants of the Deutsche Forschungsgemeinschaft (Re1228/5-1 and Re1228/5-2 within the priority program SPP1580 and the Cluster of Excellence 306 “Inflammation at interfaces”) and a grant of the Deutsches Zentrum für Infektionsforschung (DZIF) within the “Thematic translational unit tuberculosis (TTU TB).” The authors thank Kolja Schaale for critical reading of the manuscript and helpful suggestions.
References
1. WHO. Global Tuberculosis Report 2015. WHO (2016). Available from: http://www.who.int/tb/publications/global_report/en/
2. Wirth T, Hildebrand F, Allix-Béguec C, Wölbeling F, Kubica T, Kremer K, et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog (2008) 4:e1000160. doi:10.1371/journal.ppat.1000160
3. Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science (1994) 263:678–81. doi:10.1126/science.8303277
4. Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med (1995) 181:257–70. doi:10.1084/jem.181.1.257
5. Scott HM, Flynn JL. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect Immun (2002) 70:5946–54. doi:10.1128/IAI.70.11.5946-5954.2002
6. Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2001) 98:7958–63. doi:10.1073/pnas.131207398
7. Russell DG. Who puts the tubercle in tuberculosis? Nat Rev Microbiol (2007) 5:39–47. doi:10.1038/nrmicro1538
8. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR. The immune response in tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi:10.1146/annurev-immunol-032712-095939
9. Orme IM, Roberts AD, Griffin JP, Abrams JS. Cytokine secretion by CD4 T lymphocytes acquired in response to Mycobacterium tuberculosis infection. J Immunol (1993) 151:518–25.
10. Ulrichs T, Kosmiadi GA, Trusov V, Jörg S, Pradl L, Titukhina M, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol (2004) 204:217–28. doi:10.1002/path.1628
11. Phuah JY, Mattila JT, Lin PL, Flynn JL. Activated B cells in the granulomas of nonhuman primates infected with Mycobacterium tuberculosis. Am J Pathol (2012) 181:508–14. doi:10.1016/j.ajpath.2012.05.009
12. Stamm CE, Collins AC, Shiloh MU. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunol Rev (2015) 264:204–19. doi:10.1111/imr.12263
13. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity (1998) 9:143–50. doi:10.1016/S1074-7613(00)80596-8
14. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity (1999) 11:115–22. doi:10.1016/S1074-7613(00)80086-2
15. Kaneko H, Yamada H, Mizuno S, Udagawa T, Kazumi Y, Sekikawa K, et al. Role of tumor necrosis factor-alpha in Mycobacterium-induced granuloma formation in tumor necrosis factor-alpha-deficient mice. Lab Invest (1999) 79:379–86.
16. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity (1995) 2:561–72. doi:10.1016/1074-7613(95)90001-2
17. Smith D, Hänsch H, Bancroft G, Ehlers S. T-cell-independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor-alpha and interferon-gamma. Immunology (1997) 92:413–21. doi:10.1046/j.1365-2567.1997.00384.x
18. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol (2002) 168:4620–7. doi:10.4049/jimmunol.168.9.4620
19. Raviglione M, Sulis G. Tuberculosis 2015: burden, challenges and strategy for control and elimination. Infect Dis Rep (2016) 8:6570. doi:10.4081/idr.2016.6570
20. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med (2001) 345:1098–104. doi:10.1056/NEJMoa011110
21. Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol (2010) 8:296–307. doi:10.1038/nrmicro2321
22. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med (1993) 178:2249–54. doi:10.1084/jem.178.6.2249
23. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med (1993) 178:2243–7. doi:10.1084/jem.178.6.2243
24. Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, et al. Reprogramming of the macrophage transcriptome in response to interferon-γ and Mycobacterium tuberculosis signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med (2001) 194:1123–40. doi:10.1084/jem.194.8.1123
25. Schroder K, Sweet MJ, Hume DA. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology (2006) 211:511–24. doi:10.1016/j.imbio.2006.05.007
26. Flesch IE, Kaufmann SH. Mechanisms involved in mycobacterial growth inhibition by gamma interferon-activated bone marrow macrophages: role of reactive nitrogen intermediates. Infect Immun (1991) 59:3213–8.
27. Flesch IE, Kaufmann SH. Activation of tuberculostatic macrophage functions by gamma interferon, interleukin-4, and tumor necrosis factor. Infect Immun (1990) 58:2675–7.
28. Reiling N, Hölscher C, Fehrenbach A, Kröger S, Kirschning CJ, Goyert S, et al. Cutting edge: toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol (2002) 169:3480–4. doi:10.4049/jimmunol.169.7.3480
29. Hölscher C, Reiling N, Schaible UE, Hölscher A, Bathmann C, Korbel D, et al. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol (2008) 38:680–94. doi:10.1002/eji.200736458
30. Holstein TW. The evolution of the Wnt pathway. Cold Spring Harb Perspect Biol (2012) 4:a007922–007922. doi:10.1101/cshperspect.a007922
31. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell (2012) 149:1192–205. doi:10.1016/j.cell.2012.05.012
32. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov (2014) 13:513–32. doi:10.1038/nrd4233
33. Königshoff M, Eickelberg O. WNT signaling in lung disease: a failure or a regeneration signal? Am J Respir Cell Mol Biol (2010) 42:21–31. doi:10.1165/rcmb.2008-0485TR
34. Grigoryan T, Wend P, Klaus A, Birchmeier W. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of -catenin in mice. Genes Dev (2008) 22:2308–41. doi:10.1101/gad.1686208
35. Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJT. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia (2012) 26:414–21. doi:10.1038/leu.2011.387
36. George SJ. Wnt pathway: a new role in regulation of inflammation. Arterioscler Thromb Vasc Biol (2008) 28:400–2. doi:10.1161/ATVBAHA.107.160952
37. Schaale K, Neumann J, Schneider D, Ehlers S, Reiling N. Wnt signaling in macrophages: augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur J Cell Biol (2011) 90:553–9. doi:10.1016/j.ejcb.2010.11.004
38. Ma B, Hottiger MO. Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front Immunol (2016) 7:378. doi:10.3389/fimmu.2016.00378
39. The Wnt Homepage. (2016). Available from: http://web.stanford.edu/group/nusselab/cgi-bin/wnt/
40. Willert K, Nusse R. Wnt proteins. Cold Spring Harb Perspect Biol (2012) 4:a007864. doi:10.1101/cshperspect.a007864
41. Port F, Basler K. Wnt trafficking: new insights into Wnt maturation, secretion and spreading. Traffic (2010) 11:1265–71. doi:10.1111/j.1600-0854.2010.01076.x
42. Panáková D, Sprong H, Marois E, Thiele C, Eaton S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature (2005) 435:58–65. doi:10.1038/nature03504
43. Gross JC, Chaudhary V, Bartscherer K, Boutros M. Active Wnt proteins are secreted on exosomes. Nat Cell Biol (2012) 14:1036–45. doi:10.1038/ncb2574
44. Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci (2006) 119:395–402. doi:10.1242/jcs.02826
45. Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin J-P, Davenport AP, et al. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev (2005) 57:279–88. doi:10.1124/pr.57.2.5
46. Katanaev VL, Ponzielli R, Sémériva M, Tomlinson A. Trimeric G protein-dependent frizzled signaling in Drosophila. Cell (2005) 120:111–22. doi:10.1016/j.cell.2004.11.014
47. Bikkavilli RK, Feigin ME, Malbon CC. G alpha o mediates WNT-JNK signaling through dishevelled 1 and 3, RhoA family members, and MEKK 1 and 4 in mammalian cells. J Cell Sci (2008) 121:234–45. doi:10.1242/jcs.021964
48. Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science (2012) 337:59–64. doi:10.1126/science.1222879
49. Schulte G, Bryja V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol Sci (2007) 28:518–25. doi:10.1016/j.tips.2007.09.001
50. Gao C, Chen Y-G. Dishevelled: the hub of Wnt signaling. Cell Signal (2010) 22:717–27. doi:10.1016/j.cellsig.2009.11.021
51. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol (2012) 13:767–79. doi:10.1038/nrm3470
52. Blumenthal A, Ehlers S, Lauber J, Buer J, Lange C, Goldmann T, et al. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood (2006) 108:965–73. doi:10.1182/blood-2005-12-5046
53. Sen M, Lauterbach K, El-Gabalawy H, Firestein GS, Corr M, Carson DA. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc Natl Acad Sci U S A (2000) 97:2791–6. doi:10.1073/pnas.050574297
54. Reischl J, Schwenke S, Beekman JM, Mrowietz U, Stürzebecher S, Heubach JF. Increased expression of Wnt5a in psoriatic plaques. J Invest Dermatol (2007) 127:163–9. doi:10.1038/sj.jid.5700488
55. Zhang Y, Zhang D, Tu C, Zhou P, Zheng Y, Peng Z, et al. Wnt5a is involved in the pathogenesis of cutaneous lichen planus. Clin Exp Dermatol (2015) 40:659–64. doi:10.1111/ced.12561
56. Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science (2010) 329:454–7. doi:10.1126/science.1188280
57. Fuster JJ, Zuriaga MA, Ngo DT-M, Farb MG, Aprahamian T, Yamaguchi TP, et al. Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes (2015) 64:1235–48. doi:10.2337/db14-1164
58. Christman MA, Goetz DJ, Dickerson E, McCall KD, Lewis CJ, Benencia F, et al. Wnt5a is expressed in murine and human atherosclerotic lesions. Am J Physiol Heart Circ Physiol (2008) 294:H2864–70. doi:10.1152/ajpheart.00982.2007
59. Pereira C, Schaer DJ, Bachli EB, Kurrer MO, Schoedon G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler Thromb Vasc Biol (2008) 28:504–10. doi:10.1161/ATVBAHA.107.157438
60. Chaussabel D, Semnani RT, McDowell MA, Sacks D, Sher A, Nutman TB. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood (2003) 102:672–81. doi:10.1182/blood-2002-10-3232
61. Nau GJ, Richmond JFL, Schlesinger A, Jennings EG, Lander ES, Young RA. Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci U S A (2002) 99:1503–8. doi:10.1073/pnas.022649799
62. Bansal K, Trinath J, Chakravortty D, Patil SA, Balaji KN. Pathogen-specific TLR2 protein activation programs macrophages to induce Wnt-β-catenin signaling. J Biol Chem (2011) 286:37032–44. doi:10.1074/jbc.M111.260414
63. Trinath J, Holla S, Mahadik K, Prakhar P, Singh V, Balaji KN. The WNT signaling pathway contributes to dectin-1-dependent inhibition of toll-like receptor-induced inflammatory signature. Mol Cell Biol (2014) 34:4301–14. doi:10.1128/MCB.00641-14
64. van der Sar AM, Spaink HP, Zakrzewska A, Bitter W, Meijer AH. Specificity of the zebrafish host transcriptome response to acute and chronic mycobacterial infection and the role of innate and adaptive immune components. Mol Immunol (2009) 46:2317–32. doi:10.1016/j.molimm.2009.03.024
65. Kim J, Kim J, Kim DW, Ha Y, Ihm MH, Kim H, et al. Wnt5a induces endothelial inflammation via β-catenin–independent signaling. J Immunol (2010) 185:1274–82. doi:10.4049/jimmunol.1000181
66. Sen M, Chamorro M, Reifert J, Corr M, Carson DA. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum (2001) 44:772–81. doi:10.1002/1529-0131(200104)44:4<772:AID-ANR133>3.0.CO;2-L
67. Ghosh MC, Collins GD, Vandanmagsar B, Patel K, Brill M, Carter A, et al. Activation of Wnt5A signaling is required for CXC chemokine ligand 12-mediated T-cell migration. Blood (2009) 114:1366–73. doi:10.1182/blood-2008-08-175869
68. Maiti G, Naskar D, Sen M. The Wingless homolog Wnt5a stimulates phagocytosis but not bacterial killing. Proc Natl Acad Sci U S A (2012) 109:16600–5. doi:10.1073/pnas.1207789109
69. Sato A, Kayama H, Shojima K, Matsumoto S, Koyama H, Minami Y, et al. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-γ in colitis. Sci Rep (2015) 5:10536. doi:10.1038/srep10536
70. Naskar D, Maiti G, Chakraborty A, Roy A, Chattopadhyay D, Sen M. Wnt5a–Rac1–NF-κB homeostatic circuitry sustains innate immune functions in macrophages. J Immunol (2014) 192:4386–97. doi:10.4049/jimmunol.1302817
71. Kim J, Chang W, Jung Y, Song K, Lee I. Wnt5a activates THP-1 monocytic cells via a β-catenin-independent pathway involving JNK and NF-κB activation. Cytokine (2012) 60:242–8. doi:10.1016/j.cyto.2012.06.013
72. Cooper AM, Roberts AD, Rhoades ER, Callahan JE, Getzy DM, Orme IM. The role of interleukin-12 in acquired immunity to Mycobacterium tuberculosis infection. Immunology (1995) 84:423–32.
73. Kim JE, Bang SH, Choi JH, Kim CD, Won CH, Lee MW, et al. Interaction of Wnt5a with notch1 is critical for the pathogenesis of psoriasis. Ann Dermatol (2016) 28:45–54. doi:10.5021/ad.2016.28.1.45
74. Woldt E, Terrand J, Mlih M, Matz RL, Bruban V, Coudane F, et al. The nuclear hormone receptor PPARγ counteracts vascular calcification by inhibiting Wnt5a signalling in vascular smooth muscle cells. Nat Commun (2012) 3:1077. doi:10.1038/ncomms2087
75. Kikuchi R, Nakamura K, MacLauchlan S, Ngo DT-M, Shimizu I, Fuster JJ, et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat Med (2014) 20:1464–71. doi:10.1038/nm.3703
76. Malgor R, Bhatt PM, Connolly BA, Jacoby DL, Feldmann KJ, Silver MJ, et al. Wnt5a, TLR2 and TLR4 are elevated in advanced human atherosclerotic lesions. Inflamm Res (2014) 63:277–85. doi:10.1007/s00011-013-0697-x
77. Liu X, Rubin JS, Kimmel AR. Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol (2005) 15:1989–97. doi:10.1016/j.cub.2005.10.050
78. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell (2002) 108:837–47. doi:10.1016/S0092-8674(02)00685-2
79. Schaale K, Brandenburg J, Kispert A, Leitges M, Ehlers S, Reiling N. Wnt6 is expressed in granulomatous lesions of Mycobacterium tuberculosis-infected mice and is involved in macrophage differentiation and proliferation. J Immunol (2013) 191:5182–95. doi:10.4049/jimmunol.1201819
80. Neumann J, Schaale K, Farhat K, Endermann T, Ulmer AJ, Ehlers S, et al. Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reprogramming Mycobacterium tuberculosis-infected macrophages. FASEB J (2010) 24:4599–612. doi:10.1096/fj.10-160994
81. Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, et al. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem (2002) 277:21657–65. doi:10.1074/jbc.M200139200
82. Hoogendijk AJ, Diks SH, van der Poll T, Peppelenbosch MP, Wieland CW. Kinase activity profiling of pneumococcal pneumonia. PLoS One (2011) 6:e18519. doi:10.1371/journal.pone.0018519
83. Stamos JL, Chu ML-H, Enos MD, Shah N, Weis WI. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. Elife (2014) 3:e01998. doi:10.7554/eLife.01998
84. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol (2005) 6:777–84. doi:10.1038/ni1221
85. Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol (2002) 283:G204–11. doi:10.1152/ajpgi.00016.2002
86. Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock (2007) 27:113–23. doi:10.1097/01.shk.0000238059.23837.68
87. Li Y, Shi J, Yang J, Ma Y, Cheng L, Zeng J, et al. A Wnt/β-catenin negative feedback loop represses TLR-triggered inflammatory responses in alveolar epithelial cells. Mol Immunol (2014) 59:128–35. doi:10.1016/j.molimm.2014.02.002
88. Liu X, Lu R, Wu S, Zhang Y, Xia Y, Sartor BR, et al. Wnt2 inhibits enteric bacterial-induced inflammation in intestinal epithelial cells. Inflamm Bowel Dis (2012) 18:418–29. doi:10.1002/ibd.21788
89. Tan C, Costello P, Sanghera J, Dominguez D, Baulida J, de Herreros AG, et al. Inhibition of integrin linked kinase (ILK) suppresses beta-catenin-Lef/Tcf-dependent transcription and expression of the E-cadherin repressor, snail, in APC-/- human colon carcinoma cells. Oncogene (2001) 20:133–40. doi:10.1038/sj.onc.1204052
90. Bertheloot D, Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol (2016). doi:10.1038/cmi.2016.34
91. Guo Y, Mishra A, Weng T, Chintagari NR, Wang Y, Zhao C, et al. Wnt3a mitigates acute lung injury by reducing P2X7 receptor-mediated alveolar epithelial type I cell death. Cell Death Dis (2014) 5:e1286. doi:10.1038/cddis.2014.254
92. van den Bosch MH, Blom AB, Schelbergen RFP, Vogl T, Roth JP, Slöetjes AW, et al. Induction of canonical Wnt signaling by the alarmins S100A8/A9 in murine knee joints: implications for osteoarthritis. Arthritis Rheumatol (2016) 68:152–63. doi:10.1002/art.39420
93. Hii H-P, Liao M-H, Chen S-J, Wu C-C, Shih C-C. Distinct patterns of Wnt3a and Wnt5a signaling pathway in the lung from rats with endotoxic shock. PLoS One (2015) 10:e0134492. doi:10.1371/journal.pone.0134492
94. Villar J, Cabrera-Benítez NE, Ramos-Nuez A, Flores C, García-Hernández S, Valladares F, et al. Early activation of pro-fibrotic WNT5A in sepsis-induced acute lung injury. Crit Care (2014) 18:1. doi:10.1186/s13054-014-0568-z
95. Zhang P, Katz J, Michalek SM. Glycogen synthase kinase-3beta (GSK3beta) inhibition suppresses the inflammatory response to Francisella infection and protects against tularemia in mice. Mol Immunol (2009) 46:677–87. doi:10.1016/j.molimm.2008.08.281
96. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science (2010) 329:849–53. doi:10.1126/science.1188510
97. Shin K, Lee J, Guo N, Kim J, Lim A, Qu L, et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature (2011) 472:110–4. doi:10.1038/nature09851
98. Houschyar KS, Momeni A, Pyles MN, Maan ZN, Whittam AJ, Siemers F. Wnt signaling induces epithelial differentiation during cutaneous wound healing. Organogenesis (2015) 11:95–104. doi:10.1080/15476278.2015.1086052
99. Varlakhanova NV, Knoepfler PS. Acting locally and globally: myc’s ever-expanding roles on chromatin. Cancer Res (2009) 69:7487–90. doi:10.1158/0008-5472.CAN-08-4832
100. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science (1998) 281:1509–12. doi:10.1126/science.281.5382.1509
101. Cawthorn WP, Bree AJ, Yao Y, Du B, Hemati N, Martinez-Santibañez G, et al. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone (2012) 50:477–89. doi:10.1016/j.bone.2011.08.010
102. Takase HM, Nusse R. Paracrine Wnt/β-catenin signaling mediates proliferation of undifferentiated spermatogonia in the adult mouse testis. Proc Natl Acad Sci U S A (2016) 113:E1489–97. doi:10.1073/pnas.1601461113
103. Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol (2006) 4:e115. doi:10.1371/journal.pbio.0040115
104. Dijksterhuis JP, Baljinnyam B, Stanger K, Sercan HO, Ji Y, Andres O, et al. Systematic mapping of WNT-FZD protein interactions reveals functional selectivity by distinct WNT-FZD pairs. J Biol Chem (2015) 290:6789–98. doi:10.1074/jbc.M114.612648
105. Ring L, Neth P, Weber C, Steffens S, Faussner A. β-Catenin-dependent pathway activation by both promiscuous “canonical” WNT3a-, and specific “noncanonical” WNT4- and WNT5a-FZD receptor combinations with strong differences in LRP5 and LRP6 dependency. Cell Signal (2014) 26:260–7. doi:10.1016/j.cellsig.2013.11.021
106. Geetha-Loganathan P, Nimmagadda S, Huang R, Christ B, Scaal M. Regulation of ectodermal Wnt6 expression by the neural tube is transduced by dermomyotomal Wnt11: a mechanism of dermomyotomal lip sustainment. Development (2006) 133:2897–904. doi:10.1242/dev.02464
107. Schmidt C, McGonnell IM, Allen S, Otto A, Patel K. Wnt6 controls amniote neural crest induction through the non-canonical signaling pathway. Dev Dyn (2007) 236:2502–11. doi:10.1002/dvdy.21260
108. Lickert H, Kispert A, Kutsch S, Kemler R. Expression patterns of Wnt genes in mouse gut development. Mech Dev (2001) 105:181–4. doi:10.1016/S0925-4773(01)00390-2
109. Nyeng P, Norgaard GA, Kobberup S, Jensen J. FGF10 signaling controls stomach morphogenesis. Dev Biol (2007) 303:295–310. doi:10.1016/j.ydbio.2006.11.017
110. Itäranta P, Lin Y, Peräsaari J, Roël G, Destrée O, Vainio S. Wnt-6 is expressed in the ureter bud and induces kidney tubule development in vitro. Genesis (2002) 32:259–68. doi:10.1002/gene.10079
111. Lavery DL, Martin J, Turnbull YD, Hoppler S. Wnt6 signaling regulates heart muscle development during organogenesis. Dev Biol (2008) 323:177–88. doi:10.1016/j.ydbio.2008.08.032
112. Laurikkala J, Mikkola M, Mustonen T, Åberg T, Koppinen P, Pispa J, et al. Signaling via the ligand–receptor pair ectodysplasin and edar controls the function of epithelial signaling centers and is regulated by Wnt and activin during tooth organogenesis. Dev Biol (2001) 229:443–55. doi:10.1006/dbio.2000.9955
113. Galbraith RL, Poole EM, Duggan D, Muehling J, Hsu L, Makar K, et al. Polymorphisms in WNT6 and WNT10A and colorectal adenoma risk. Nutr Cancer (2011) 63:558–64. doi:10.1080/01635581.2011.542539
114. Yuan G, Regel I, Lian F, Friedrich T, Hitkova I, Hofheinz RD, et al. WNT6 is a novel target gene of caveolin-1 promoting chemoresistance to epirubicin in human gastric cancer cells. Oncogene (2013) 32:375–87. doi:10.1038/onc.2012.40
115. Liu X, Lu R, Wu S, Sun J. Salmonella regulation of intestinal stem cells through the Wnt/beta-catenin pathway. FEBS Lett (2010) 584:911–6. doi:10.1016/j.febslet.2010.01.024
116. Choy DF, Modrek B, Abbas AR, Kummerfeld S, Clark HF, Wu LC, et al. Gene expression patterns of Th2 inflammation and intercellular communication in asthmatic airways. J Immunol (2011) 186:1861–9. doi:10.4049/jimmunol.1002568
117. Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol (2011) 4:271–8. doi:10.1038/mi.2011.14
118. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host–pathogen collusion. Front Immunol (2013) 3. doi:10.3389/fimmu.2012.00411
119. Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog (2015) 11:e1004603. doi:10.1371/journal.ppat.1004603
120. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med (2014) 20:75–9. doi:10.1038/nm.3412
121. Andersen P, Smedegaard B. CD4(+) T-cell subsets that mediate immunological memory to Mycobacterium tuberculosis infection in mice. Infect Immun (2000) 68:621–9. doi:10.1128/IAI.68.2.621-629.2000
122. Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol (2014) 192:2965–9. doi:10.4049/jimmunol.1400019
123. Hölscher C, Hölscher A, Rückerl D, Yoshimoto T, Yoshida H, Mak T, et al. The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J Immunol (2005) 174:3534–44. doi:10.4049/jimmunol.174.6.3534
124. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med (2016) 22:531–8. doi:10.1038/nm.4073
125. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog (2008) 4:e1000204. doi:10.1371/journal.ppat.1000204
126. Russell DG, Cardona P-J, Kim M-J, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol (2009) 10:943–8. doi:10.1038/ni.1781
127. Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J (2010) 427:1–17. doi:10.1042/BJ20091866
128. Mani A, Radhakrishnan J, Wang H, Mani A, Mani M-A, Nelson-Williams C, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science (2007) 315:1278–82. doi:10.1126/science.1136370
129. Wu X, Deng G, Hao X, Li Y, Zeng J, Ma C, et al. A caspase-dependent pathway is involved in Wnt/β-catenin signaling promoted apoptosis in Bacillus calmette-guerin infected RAW264.7 macrophages. Int J Mol Sci (2014) 15:5045–62. doi:10.3390/ijms15035045
130. Fu Y, Yi Z, Wu X, Li J, Xu F. Circulating microRNAs in patients with active pulmonary tuberculosis. J Clin Microbiol (2011) 49:4246–51. doi:10.1128/JCM.05459-11
131. Kapinas K, Kessler C, Ricks T, Gronowicz G, Delany AM. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J Biol Chem (2010) 285:25221–31. doi:10.1074/jbc.M110.116137
132. Luo T, Dunphy PS, Lina TT, McBride JW. Ehrlichia chaffeensis exploits canonical and noncanonical host Wnt signaling pathways to stimulate phagocytosis and promote intracellular survival. Infect Immun (2016) 84:686–700. doi:10.1128/IAI.01289-15
133. Kessler M, Zielecki J, Thieck O, Mollenkopf H-J, Fotopoulou C, Meyer TF. Chlamydia trachomatis disturbs epithelial tissue homeostasis in fallopian tubes via paracrine Wnt signaling. Am J Pathol (2012) 180:186–98. doi:10.1016/j.ajpath.2011.09.015
134. Enam S, Del Valle L, Lara C, Gan D-D, Ortiz-Hidalgo C, Palazzo JP, et al. Association of human polyomavirus JCV with colon cancer evidence for interaction of viral T-antigen and β-catenin. Cancer Res (2002) 62:7093–101.
135. Morrison JA, Klingelhutz AJ, Raab-Traub N. Epstein-Barr virus latent membrane protein 2A activates -catenin signaling in epithelial cells. J Virol (2003) 77:12276–84. doi:10.1128/JVI.77.22.12276-12284.2003
136. Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, et al. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat Med (2003) 9:300–6. doi:10.1038/nm829
137. van Zuylen WJ, Rawlinson WD, Ford CE. The Wnt pathway: a key network in cell signalling dysregulated by viruses. Rev Med Virol (2016) 26:340–55. doi:10.1002/rmv.1892
138. Zhao H, Granberg F, Pettersson U. How adenovirus strives to control cellular gene expression. Virology (2007) 363:357–75. doi:10.1016/j.virol.2007.02.013
139. Forero A, Tisoncik-Go J, Watanabe T, Zhong G, Hatta M, Tchitchek N, et al. The 1918 influenza virus PB2 protein enhances virulence through the disruption of inflammatory and Wnt-mediated signaling in mice. J Virol (2016) 90:2240–53. doi:10.1128/JVI.02974-15
140. Huang H, Sankiia HFA. B-Catenin Mutations Are Frequent in Human Hepatocellular Carcinomas Associated with Hepatitis. (2016). Available from: http://search.proquest.com/openview/f8cda34b83c9ef73d008c821c5efcf68/1.pdf?pq-origsite=gscholar&cbl=49196
141. Liu Y, El-Serag HB, Jiao L, Lee J, Moore D, Franco LM, et al. WNT signaling pathway gene polymorphisms and risk of hepatic fibrosis and inflammation in HCV-infected patients. PLoS One (2013) 8:e84407. doi:10.1371/journal.pone.0084407
142. Hung K-H, Hung H-W, Yang H-B, Lu C-C, Wu J-J, Sheu B-S. Host single nucleotide polymorphisms of MMP-9-1562/TIMP-1 372 have gender differences in the risk of gastric intestinal metaplasia after Helicobacter pylori infection. Helicobacter (2009) 14:580–7. doi:10.1111/j.1523-5378.2009.00717.x
Keywords: Mycobacterium tuberculosis, Wnt proteins, inflammation, macrophages, toll-like receptors, beta catenin, granuloma
Citation: Brandenburg J and Reiling N (2016) The Wnt Blows: On the Functional Role of Wnt Signaling in Mycobacterium tuberculosis Infection and Beyond. Front. Immunol. 7:635. doi: 10.3389/fimmu.2016.00635
Received: 15 October 2016; Accepted: 12 December 2016;
Published: 26 December 2016
Edited by:
Victor Manuel Baizabal-Aguirre, Universidad Michoacana de San Nicolás de Hidalgo, MexicoReviewed by:
Kushagra Bansal, Harvard Medical School, USATrinath Jamma, University of Massachusetts Boston, USA
Copyright: © 2016 Brandenburg and Reiling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norbert Reiling, bnJlaWxpbmdAZnotYm9yc3RlbC5kZQ==