 Pavel Kovarik
Pavel Kovarik Virginia Castiglia
Virginia Castiglia Masa Ivin
Masa Ivin Florian Ebner
Florian Ebner- Max F. Perutz Laboratories, University of Vienna, Vienna, Austria
Defense against bacterial infections requires activation of the immune response as well as timely reestablishment of tissue and immune homeostasis. Instauration of homeostasis is critical for tissue regeneration, wound healing, and host recovery. Recent studies revealed that severe infectious diseases frequently result from failures in homeostatic processes rather than from inefficient pathogen eradication. Type I interferons (IFN) appear to play a key role in such processes. Remarkably, the involvement of type I IFNs in the regulation of immune and tissue homeostasis upon bacterial insult may have beneficial or detrimental consequences for the host. The reasons for such ambivalent function of type I IFNs are not understood. The disparate effects of type I IFNs on bacterial infections are in marked contrast to their well-established protective roles in most viral infections. In this review, we will focus on type I IFN effector mechanisms which balance processes involved in immune and tissue homeostasis during specific bacterial infections and highlight the most important missing links in our understanding of type I IFN functions.
Introduction
Successful defense against pathogens requires both, the eradication of the infectious agent by the immune system as well as tissue protection against the damaging effects of the immune response. Increasing evidence indicates that many if not most infectious diseases result from insufficient resilience, i.e., from a failure of the infected host to repair and regenerate destroyed tissues, rather than from inefficient pathogen clearance (1–4). Mechanisms which preserve the integrity of host tissues during the intensive inflammatory response against the pathogen remain incompletely understood. Recent studies established that successful tissue protection during infection requires systems which balance the immune response as well as mechanisms which restore tissue homeostasis. These mechanisms are often interdependent and result from messengers like growth factors, cytokines, or lipids produced by immune cells (1–4). Examples include the anti-inflammatory cytokine IL-10, the tissue regeneration promoting IL-22 and amphiregulin, the tissue remodeler TGF-β, or the pro-resolving lipid lipoxin (5–8). Remarkably, several recent studies demonstrated that type I interferons (IFNs) can also act as critical resilience-promoting cytokines during infections with several streptococcal species (9–11). Such protective functions are in marked contrast to detrimental effects of type I IFN during infections with many other bacterial species (12, 13). The reasons for the ambivalent roles of type I IFNs in bacterial infections remain poorly understood. However, it appears that the ability of type I IFNs to both suppress and stimulate immune responses is of critical importance (Table 1; Figure 1). This review focuses on the role of type I IFNs in balancing pro- and anti-inflammatory processes as well as cell survival and cell death programs during antibacterial defense and discusses how these effects determine the outcome of an infection.
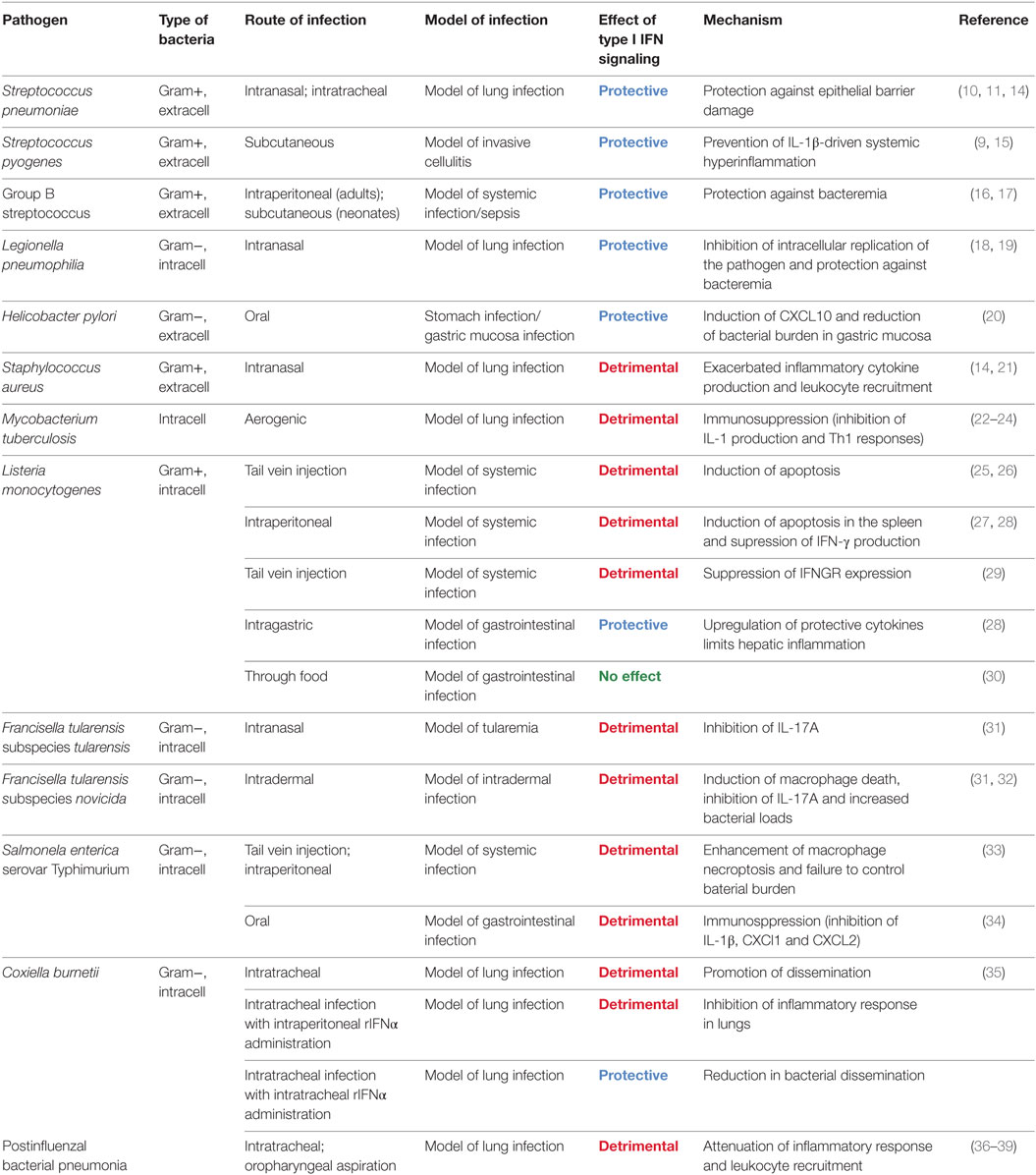
Table 1. Effects of type I interferons (IFN) signaling in bacterial infections.
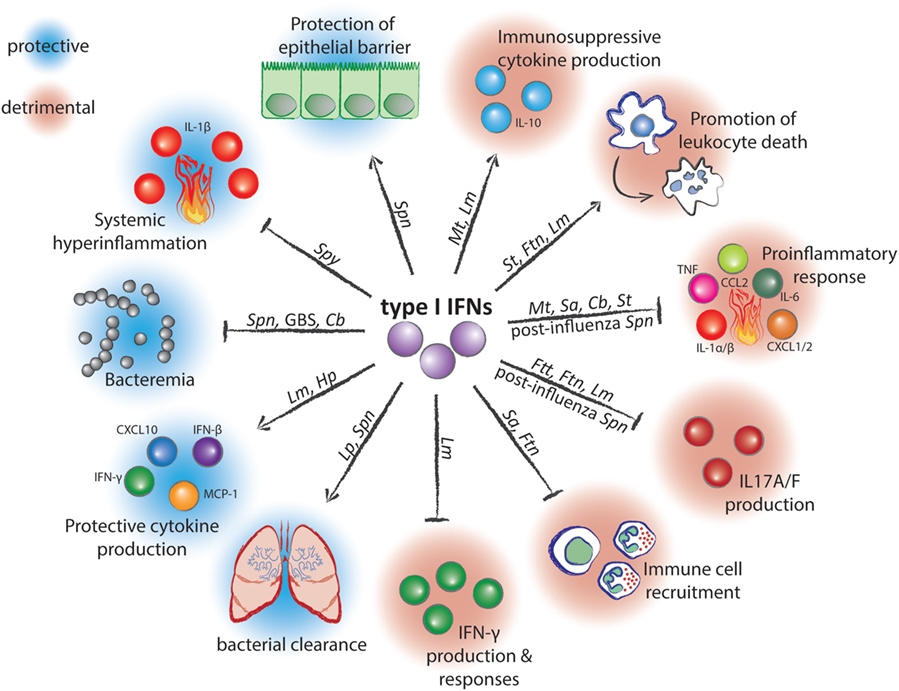
Figure 1. Mechanisms of action and effects of type I interferons (IFNs) during infection with bacterial pathogens. Arrow-headed lines represent stimulation and bar-headed lines represent inhibition by type I IFNs. Pathogen abbreviations: Spn, Streptococcus pneumoniae; Spy, Streptococcus pyogenes; GBS, Group B Streptococcus; Cb, Coxiella burnetii; Lm, Listeria monocytogenes; Hp, Helicobacter pylori; Lp, Legionella pneumophilia; Sa, Staphylococcus aureus; Ftn, Francisella tularensis subspecies novicida; Ftt, Francisella tularensis subspecies tularensis; Mt, Mycobacterium tuberculosis; St, Salmonella enterica serovar Typhimurium.
Type I IFN Induction by Bacteria
Type I IFNs were described more than a half century ago as products which are secreted by virus-infected cells and interfere with virus replication in autocrine and paracrine ways (40). It is now known that type I IFNs are cytokines produced in response to viral, bacterial, and fungal pathogens, as well as parasites. The effector mechanisms of type I IFNs mainly derive from products of genes which are transcriptionally regulated by type I IFN signaling. Type I IFNs induce hundreds of interferon-stimulated genes (ISGs) through activation of the homodimeric STAT1 and the heterotrimeric STAT1–STAT2–IRF9 (i.e., ISGF3) transcription factors (41). Bacteria trigger type I IFN production mostly following the recognition of bacterial nucleic acids or the Gram-negative cell wall component lipopolysaccharide (LPS) by innate immune receptors (12, 42). The induction mechanisms have been best studied for IFN-β which belongs together with IFNα4 to the first type I IFNs produced during infection and is the driver of other type I IFN genes (43, 44). The induction of IFN-β by bacterial DNA is complex and involves different pathways. The most common mode of IFN-β induction by bacterial DNA is through the cytosolic DNA sensor cyclic GMP-AMP synthase, as described for Francisella novicida, group B streptococcus (GBS) (Streptococcus agalactiae), Legionella pneumophila, Listeria monocytogenes, Mycobacterium tuberculosis, and Neisseria gonorrhoeae (45–51). The cytosolic DNA sensor Ifi204 (IFI16 in humans) contributes to IFN-β induction in the course of F. novicida and L. monocytogenes (48, 49). The signaling events downstream of these DNA sensors involve the STING–TBK1–IRF3 pathway driving the IFN-β gene transcription. L. monocytogenes can activate this pathway also independently of DNA sensing. This alternative mechanism is driven by binding of the bacterial product c-di-AMP to STING (51, 52). The recognition of bacterial DNA through unmethylated CpG motif-containing DNA by the endosomal Toll-like receptor 9 can also contribute to IFN-β induction, although the importance and relevance of this pathway in the context of the overall IFN-β production and host response have not been entirely clarified (42). Bacterial RNA has been recently established as another key IFN-β inducer. A short and highly conserved sequence found in the bacterial 23S rRNA is recognized by the mouse TLR13 leading to Myd88- and IRF5-dependent IFN-β induction (9, 53–56). Human cells employ TLR8 as IFN-β-inducing RNA sensor rather than TLR13 which is missing in humans (9, 53, 57–59). The precise nature of bacterial RNA triggering the human TLR8 remains to be identified.
IFN-β induction by LPS occurs after binding of this ligand to the TLR4 and the following internalization into the endosome (60). Subsequently, a TRIF-dependent activation of the kinase TBK1 causes phosphorylation of the transcription factor IRF3 to stimulate IFN-β gene transcription. Endosomal signaling has been also implicated in IFN-β induction by TLR2 in response to Gram-positive bacteria, although this mechanism appears to be restricted to specific immune cells and/or pathogens (61–63). The cytosolic receptors NOD1 and NOD2 were reported to trigger IFN-β production following infection with Helicobacter pylori and M. tuberculosis, respectively (20, 64). NOD2 engagement can induce IFN-β also in responses to Staphylococcus aureus (14).
While the pathways causing IFN-β induction by bacteria are relatively well understood, more studies are needed to assess the importance of individual pathways for the overall IFN-β production in whole organism rather than cells. One of the rare studies on this topic revealed that the dominant IFN-β-inducing pathway during infection with Streptococcus pyogenes (group A streptococcus) is the TLR13-mediated RNA recognition pathway (9).
Additional work is also needed to clarify the key IFN-β producing cells, as investigated during, e.g., L. monocytogenes infections (65–67) and the reported cell type-specific features of IFN-β induction (12, 15, 16).
Tipping the Balance I: Benefits of Immunomodulatory Effects of Type I IFN Signaling During Bacterial Infections
Type I IFNs’ ability to stimulate immune responses against viruses has been established very early after their discovery but it soon became clear that these cytokines exhibit also immunosuppressive activities. The first evidence for such immunosuppressive activities was provided in a study showing that type I IFNs were able to reduce carrageenin-induced footpad swelling (68). Thus, type I IFNs are now regarded as immunomodulatory cytokines capable of enhancing or dampening the immune response, depending on the context. This ambiguousness contributes to the disparate and still incompletely understood roles of type I IFNs during bacterial infections. Importantly, no unifying principles have been found to date: neither the beneficial nor detrimental effects of type I IFN signaling correlate with the broad pathogen classification into Gram-positive and -negative, extra- and intracellular pathogens, or the route of infection (Table 1; Figure 1).
Immunosuppressive effects of type I IFN signaling are beneficial during infection with the Gram-positive largely extracellular human pathogen S. pyogenes (9). S. pyogenes is the causative agent of mild (e.g., pharyngitis and scarlet fever) but also invasive and life-threatening infections (e.g., cellulitis, necrotizing fasciitis, and streptococcal toxic shock syndrome). Mice deficient in the type I IFN receptor IFNAR1 are more susceptible to subcutaneous S. pyogenes infection, which is a relevant model of severe invasive infection of the soft tissue (15). Type I IFN signaling promotes resistance against S. pyogenes by suppressing the transcription of the Il1b gene (9). The absence of type I IFN signaling results in an unrestricted production of IL-1β thereby causing a lethal hyperinflammation and organ damage. Importantly, type I IFN signaling balances rather than prevents Il1b transcription so that a controlled and life-saving IL-1β production is achieved (9). The key IFN-β producer and effector cells in this infection model are both LysM+ and CD11c+ myeloid cells (9).
Immunomodulation by type I IFN signaling is protective during infection with the human Gram-positive extracellular pathogen GBS (16, 17). GBS is regarded as commensal microbe asymptomatically colonizing the skin and mucosal tissues of 30% people, yet it is the leading cause of severe neonatal pneumonia and meningitis in developed countries. The absence of IFNAR1 results in increased bacterial loads during both subcutaneous GBS infection of neonate mice and intravenous infection of adult animals (16, 17). Similarly, type I IFN signaling is protective against uncontrolled bacteremia during infection with the Gram-negative intracellular bacterium L. pneumophila, which is a frequent cause of the severe pneumonia, Legionnaire’s disease (18). Type I IFN signaling inhibits in a cell-autonomous way replication of L. pneumophila inside the infected cell (18, 19). The organismal physiology of the protective effects of type I IFN signaling during infection with GBS and L. pneumophila remains to be elucidated so that it is presently unclear whether immunosuppressive or immunostimulatory effects of type I IFNs drive the resistance against these two pathogens.
Stimulation of the immune response by type I IFN signaling is advantageous in defense against the Gram-negative pathogen H. pylori (20). H. pylori is a frequent cause of chronic gastritis and is associated with increased risk of gastric ulcers and stomach cancer. Deficiency in type I IFN signaling causes increased H. pylori loads in the stomach of orally infected mice. The lack of type I IFN responses is associated with decreased levels of the chemokine CXCL10 suggesting that type I IFNs promote defense against H. pylori by stimulating CXCL10-driven inflammation (20). Immunostimulatory effects of type I IFNs are beneficial also during gastric infection with the food-borne Gram-positive intracellular pathogen L. monocytogenes (28). L. monocytogenes infects the gastrointestinal tract, where it traverses the epithelial barrier and spreads into distant organs. Deficiency in type I IFN signaling results in an increased bacterial dissemination and is accompanied by diminished production of several pro-inflammatory cytokines, including TNF and IL-6 upon gastric infection using oral gavage (28). Interestingly, type I IFN signaling plays no role in an infection model using food contaminated with L. monocytogenes (30).
Tipping the Balance II: Disadvantages of Immunomodulatory Effects of Type I IFN Signaling During Bacterial Infections
Immunosuppression by type I IFN signaling is detrimental during infection with the intracellular pathogen and causative agent of tuberculosis, M. tuberculosis (22–24). Type I IFN signaling-mediated inhibition of IL-1 cytokines during M. tuberculosis lung infection blunts the antimicrobial defense and results in increased local as well as systemic bacterial loads (24). The key type I IFN effector cells are transplantable inflammatory monocyte-macrophage cells and DCs (24). The precise mechanism of IL-1 inhibition by type I IFN signaling in this infection model is not resolved but includes both direct as well as indirect mechanisms (24). The indirect IL-1 inhibition appears to be mediated by the anti-inflammatory cytokine IL-10 which is known to be upregulated by type I IFNs (69). The importance of type I IFN signaling in M. tuberculosis infections is underlined by type I IFN signaling-associated gene expression pattern found in blood cells of patients with active tuberculosis (70). The detrimental function of type I IFN signaling during M. tuberculosis lung infection is converted into a protective one if IFN-γ signaling is missing (71). Under such conditions, type I IFNs inhibit the polarization of macrophages into infection-permissive alternatively activated macrophages.
Inhibition of immune response by type I IFNs is deleterious during infection with the facultative intracellular Gram-negative bacterium F. novicida (31). F. novicida is a subspecies of Francisella tularensis which infects humans through the skin or aerosol droplets and causes ulceroglandular or pneumonic tularemia, respectively. IFNAR1-deficient mice infected intradermally with F. novicida respond by an increased IL-17 production compared to WT animals and, correspondingly, are more resistant against infection (31). Similar increase in resistance is also observed during lung infection with F. tularensis (31). The key IL-17 producers during F. novicida infection are IL-17A+ γδ T cells which show enhanced expansion in the absence of type I IFN signaling.
Interferon-β exacerbates infection with S. typhimurium by reducing the ability of the host to launch a complete immune response (34). S. typhimurium is Gram-negative intracellular pathogen associated with gastroenteritis in humans and a severe disease resembling typhoid fever in mice. Mice deficient in IFN-β are more resistant against oral infection with S. typhimurium and display enhanced expression of IL-1β and the neutrophil chemoattractants, CXCL1 and CXCL2 (34). These changes are attributable to IFN-β-mediated inhibition of these genes in macrophages and are independent of S. typhimurium-induced macrophage death.
Listeria monocytogenes-induced type I IFN signaling downregulates the expression of both type II IFN receptor subunits, IFNGR1 and IFNGR2, thereby decreasing the responsiveness of macrophages and DCs to IFN-γ (29). The suppression of the IFN-γ signaling results in an increased susceptibility to L. monocytogenes infection. Increased susceptibility to L. monocytogenes infection is also caused by type I IFN-mediated induction of anti-inflammatory IL-10 (72).
Immunosuppressive effects of type I IFNs are harmful during postinfluenzal bacterial pneumonia (36–39). The immune response during a secondary postinfluenzal infection with the Gram-positive extracellular pathogen S. pneumoniae, a key causative agent of pneumonia, is impaired. This is caused by the ability of type I IFNs to suppress production of the neutrophil chemoattractants CXCL1 and CXCL2, the macrophage chemoattractant CCL2, and the inflammation-promoting cytokine IL-17 (36–38). The resulting reduction of leukocyte infiltration in the lung diminishes the capability of the host to control bacterial growth. Similar alterations in the immune response appear to be responsible for the increased susceptibility of mice to postinfluenzal infection with S. aureus and Pseudomonas aeruginosa (39). The mechanisms of the immunosuppressive effects of type I IFN signaling during postinfluenzal bacterial infection are not well understood but they might act downstream of the type I IFN-mediated inhibition of IL-1 cytokines.
Enhancement of the inflammatory response is associated with detrimental effects of type I IFN signaling during lung infection with the Gram-positive extracellular pathogen S. aureus (21). IFNAR1-deficient mice exhibit a lower TNF and IL-6 production and decreased leukocyte infiltration in the lung compared to WT animals suggesting that type I IFN signaling causes an exacerbated tissue damage (21). The pathogenicity of S. aureus strains correlates with the levels of type I IFNs induced during infection with differently virulent strains (14).
Tipping the Balance III: Regulation of Tissue and Cell Integrity by Type I IFNs During Bacterial Infections
Type I IFN signaling plays an indispensable role in the preservation of the epithelial barrier and epithelial integrity during lung infection with S. pneumoniae (10, 11). Type I IFN signaling promotes the maintenance of lung epithelial tight junctions during S. pneumoniae infection thereby reducing the passage of the pathogen from alveoli into the lung parenchyma (10). IFNAR1-deficient mice display increased permeability of the lung epithelium and enhanced invasiveness of S. pneumoniae infection associated with higher bacterial burden in distant organs. Type I IFN signaling protects the barrier function of the lung during S. pneumoniae infection also by promoting survival of the alveolar epithelial type II cells as revealed by IFNAR1 deletion specifically in this subtype of the barrier epithelium (11).
A common detrimental effect of type I IFN signaling during bacterial infections is the induction of various types of leukocyte cell death. Type I IFN-facilitated apoptosis of macrophages and lymphocytes appears to contribute to the increased susceptibility of WT mice compared to IFNAR1-deficient animals to intravenous and intraperitoneal infection with L. monocytogenes (25–27, 73). Type I IFN-facilitated death of macrophages is associated also with harmful effects of type I IFNs during infection with F. novicida and S. typhimurium (32, 33). F. novicida promotes macrophage death by type I IFN-mediated inflammasome-activation whereas S. typhimurium employs type I IFN induction to stimulate RIP-dependent macrophage necroptosis.
Conclusion and Future Directions
Ample evidence exists for the pivotal role of type I IFNs in regulation of defense against bacterial pathogens. The complex and often disparate effects of type I IFNs on the outcome of different bacterial infections provide chances to exploit type I IFNs and their inducers as well as effectors for adjuvant therapies tailored to specific infectious diseases. A prerequisite for the development of such therapies is a detailed understanding of the molecular, cellular, and organismal physiology of type IFNs in the course of bacterial infections. The following topics appear particularly important since they represent rather underexplored yet critically important areas.
Pathogen Species
The inconsistent roles of type I IFNs during infections with different bacteria remain a significant and challenging topic in the current research. Only few common principles of type I IFN action have been found to date. They include the cell death-promoting effects of type I IFNs which contribute mostly to detrimental functions of type I IFN signaling. Another frequent observation is the suppression of IL-1β and neutrophil chemoattractants—these effects are, however, associated with both beneficial and harmful consequences for the infection outcome. Future studies employing pathogens which have not yet been analyzed in detail, such as Klebsiella pneumoniae, uropathogenic E. coli, or Clostridium difficile, might reveal novel common principles.
Infection Route and Tissue-Specific Features of Type I IFN Signaling
The complexity of type I IFN function in bacterial infections is further increased by the distinct effects of type I IFNs in response to the same but differently administered pathogen. Type I IFNs are harmful followed intraperitoneal or intravenous infection with L. monocytogenes but protective in a physiologically more relevant intragastric infection (25–28). In contrast, type I IFN signaling has no impact on the overall outcome of L. monocytogenes infection after ingesting pathogen-contaminated food (30). These observations suggest that type I IFN signaling has, with regard to bacterial infections, distinct functions in different tissues/organs. This implication is supported by a recent study showing that exogenous type I IFN has, depending on the site of administration, disparate effects on the course of lung infection with Coxiella burnetii (35). Infections with C. burnetii, a Gram-negative intracellular bacterium, in humans occur after inhalation of bacteria and result in Q fever which can develop into an atypical pneumonia. Lung infection with C. burnetii in mice has a more severe course in WT compared to IFNAR1-deficient animals indicating that type I IFN signaling is disadvantageous in this infection model (35). Consistently, intraperitoneally administered type I IFN exacerbates C. burnetii infection. However, intratracheal delivery of type I IFN ameliorates the course of C. burnetii infection. The mechanisms of these distinct effects of type I IFN signaling in different tissues remain to be elucidated. Future studies should investigate other pathogens known of using various routes of infection and focus on physiologically most relevant routes.
Most Significant Type I IFN Inducers and Effectors
Modulation of type I IFN production during bacterial infection might represent a powerful approach in therapy of infectious diseases. Therapeutic targeting of type I IFN production requires the knowledge of the most important type I IFN-inducing pathway in a given infection. As most bacterial pathogens employ more than one pathway to stimulate type I IFN production, future efforts should focus on the identification of the most crucial bacterial and cellular components involved in type I IFN induction. These studies will need to use a combination of bacterial and animal host genetics for functional assessment and suitable reporter as well as imaging systems for type I IFN detection in vivo. On the effector side, recent studies provided a number of novel type I IFN-induced factors which interfere with pathogen replication and survival. Notably, various type I IFN-inducible small GTP-binding proteins have recently been showed to significantly contribute to the effects of type I IFNs [e.g., Ref. (74–77)]. These and yet to be discovered effectors represent potential targets for therapeutic intervention.
Human versus Mouse Systems
While many host defense mechanisms are well conserved among mice and men, important differences exist. For example, the mouse type I IFN inducer TLR13 is not expressed in humans (9, 53). Conversely, the human but not mouse TLR8 appears to be involved in type I IFN induction by bacterial RNA (59). Some antimicrobial functions of human neutrophils are enhanced by type I IFNs (78) whereas such stimulatory effects have so far not been described in mouse neutrophils. Future work should put more emphasis on studies of common and distinct features of type I IFN functions in bacterial infections.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Austrian Science Fund (FWF) grants P24540-B21 and I1621-B22 to PK, by funding from the European Union Seventh Framework Programme Marie Curie Initial Training Networks (FP7-PEOPLE-2012-ITN) for the project INBIONET under grant agreement PITN-GA-2012-316682, and by a joint research cluster initiative of the University of Vienna and the Medical University of Vienna.
References
1. Divangahi M, King IL, Pernet E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol (2015) 36:307–14. doi: 10.1016/j.it.2015.03.005
2. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell (2015) 26(160):816–27. doi:10.1016/j.cell.2015.02.010
3. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 24(335):936–41. doi:10.1126/science.1214935
4. Soares MP, Gozzelino R, Weis S. Tissue damage control in disease tolerance. Trends Immunol (2014) 35:483–94. doi:10.1016/j.it.2014.08.001
5. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 20(40):315–27. doi:10.1016/j.immuni.2014.02.009
6. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol (2015) 33:747–85. doi:10.1146/annurev-immunol-032414-112123
7. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 15(44):450–62. doi:10.1016/j.immuni.2016.02.015
8. Zaiss DM, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity (2015) 17(42):216–26. doi:10.1016/j.immuni.2015.01.020
9. Castiglia V, Piersigilli A, Ebner F, Janos M, Goldmann O, Dambock U, et al. Type I interferon signaling prevents IL-1beta-driven lethal systemic hyperinflammation during invasive bacterial infection of soft tissue. Cell Host Microbe (2016) 9(19):375–87. doi:10.1016/j.chom.2016.02.003
10. LeMessurier KS, Hacker H, Chi L, Tuomanen E, Redecke V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog (2013) 9:e1003727. doi:10.1371/journal.ppat.1003727
11. Maier BB, Hladik A, Lakovits K, Korosec A, Martins R, Kral JB, et al. Type I interferon promotes alveolar epithelial type II cell survival during pulmonary Streptococcus pneumoniae infection and sterile lung injury in mice. Eur J Immunol (2016) 46:2175–86. doi:10.1002/eji.201546201
12. Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe (2016) 8(19):760–9. doi:10.1016/j.chom.2016.05.016
13. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
14. Parker D, Planet PJ, Soong G, Narechania A, Prince A. Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog (2014) 10:e1003951. doi:10.1371/journal.ppat.1003951
15. Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S, et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog (2011) 7:e1001345. doi:10.1371/journal.ppat.1001345
16. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 1(178):3126–33. doi:10.4049/jimmunol.178.5.3126
17. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol (2009) 10:587–94. doi:10.1038/ni.1733
18. Lippmann J, Muller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M, et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol (2011) 13:1668–82. doi:10.1111/j.1462-5822.2011.01646.x
19. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog (2016) 12:e1005408. doi:10.1371/journal.ppat.1005408
20. Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest (2010) 120:1645–62. doi:10.1172/JCI39481
21. Martin FJ, Gomez MI, Wetzel DM, Memmi G, O’Seaghdha M, Soong G, et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest (2009) 119:1931–9. doi:10.1172/JCI35879
22. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25:694–701. doi:10.1089/jir.2005.25.694
23. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol (2007) 178:3143–52. doi:10.4049/jimmunol.178.5.3143
24. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 23(35):1023–34. doi:10.1016/j.immuni.2011.12.002
25. Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med (2004) 16(200):527–33. doi:10.1084/jem.20040976
26. O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med (2004) 16(200):437–45. doi:10.1084/jem.20040712
27. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med (2004) 16(200):535–40. doi:10.1084/jem.20040769
28. Kernbauer E, Maier V, Rauch I, Muller M, Decker T. Route of infection determines the impact of type I interferons on innate immunity to Listeria monocytogenes. PLoS One (2013) 8:e65007. doi:10.1371/journal.pone.0065007
29. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med (2010) 15(207):327–37. doi:10.1084/jem.20091746
30. Pitts MG, Myers-Morales T, D’Orazio SE. Type I IFN does not promote susceptibility to foodborne Listeria monocytogenes. J Immunol (2016) 1(196):3109–16. doi:10.4049/jimmunol.1502192
31. Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol (2010) 1(184):3755–67. doi:10.4049/jimmunol.0902065
32. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med (2007) 14(204):987–94. doi:10.1084/jem.20062665
33. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol (2012) 13(10):954–62. doi:10.1038/ni.2397
34. Perkins DJ, Rajaiah R, Tennant SM, Ramachandran G, Higginson EE, Dyson TN, et al. Salmonella Typhimurium co-opts the host type I IFN system to restrict macrophage innate immune transcriptional responses selectively. J Immunol (2015) 1(195):2461–71. doi:10.4049/jimmunol.1500105
35. Hedges JF, Robison A, Kimmel E, Christensen K, Lucas E, Ramstead A, et al. Type I interferon counters or promotes Coxiella burnetii replication dependent on tissue. Infect Immun (2016) 84:1815–25. doi:10.1128/IAI.01540-15
36. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest (2009) 119:1910–20. doi:10.1172/JCI35412
37. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest (2011) 121:3657–65. doi:10.1172/JCI57762
38. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol (2012) 86:12304–12. doi:10.1128/JVI.01269-12
39. Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol (2015) 15(309):L158–67. doi:10.1152/ajplung.00338.2014
40. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 12(147):258–67. doi:10.1098/rspb.1957.0048
41. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5(9):675–87. doi:10.1038/nri1684
42. Monroe KM, McWhirter SM, Vance RE. Induction of type I interferons by bacteria. Cell Microbiol (2010) 12:881–90. doi:10.1111/j.1462-5822.2010.01478.x
43. Juang YT, Lowther W, Kellum M, Au WC, Lin R, Hiscott J, et al. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc Natl Acad Sci U S A (1998) 18(95):9837–42. doi:10.1073/pnas.95.17.9837
44. Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J (1998) 17:6660–9. doi:10.1093/emboj/17.22.6660
45. Andrade WA, Agarwal S, Mo S, Shaffer SA, Dillard JP, Schmidt T, et al. Type I interferon induction by Neisseria gonorrhoeae: dual requirement of cyclic GMP-AMP synthase and Toll-like receptor 4. Cell Rep (2016) 14(15):2438–48. doi:10.1016/j.celrep.2016.05.030
46. Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, et al. Group B streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe (2016) 13(20):49–59. doi:10.1016/j.chom.2016.06.003
47. Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe (2015) 10(17):820–8. doi:10.1016/j.chom.2015.05.005
48. Hansen K, Prabakaran T, Laustsen A, Jorgensen SE, Rahbaek SH, Jensen SB, et al. Listeria monocytogenes induces IFNbeta expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J (2014) 1(33):1654–66. doi:10.15252/embj.201488029
49. Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J Immunol (2015) 1(194):3236–45. doi:10.4049/jimmunol.1402764
50. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe (2015) 10(17):799–810. doi:10.1016/j.chom.2015.05.003
51. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe (2015) 10(17):811–9. doi:10.1016/j.chom.2015.05.004
52. Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science (2010) 25(328):1703–5. doi:10.1126/science.1189801
53. Fieber C, Janos M, Koestler T, Gratz N, Li XD, Castiglia V, et al. Innate immune response to Streptococcus pyogenes depends on the combined activation of TLR13 and TLR2. PLoS One (2015) 10:e0119727. doi:10.1371/journal.pone.0119727
54. Hidmark A, von Saint Paul A, Dalpke AH. Cutting edge: TLR13 is a receptor for bacterial RNA. J Immunol (2012) 15(189):2717–21. doi:10.4049/jimmunol.1200898
55. Li XD, Chen ZJ. Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. Elife (2012) 1:e00102. doi:10.7554/eLife.00102
56. Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science (2012) 31(337):1111–5. doi:10.1126/science.1220363
57. Bergstrom B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, et al. TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-beta production via a TAK1-IKKbeta-IRF5 signaling pathway. J Immunol (2015) 1(195):1100–11. doi:10.4049/jimmunol.1403176
58. Eigenbrod T, Pelka K, Latz E, Kreikemeyer B, Dalpke AH. TLR8 senses bacterial RNA in human monocytes and plays a nonredundant role for recognition of Streptococcus pyogenes. J Immunol (2015) 1(195):1092–9. doi:10.4049/jimmunol.1403173
59. Kruger A, Oldenburg M, Chebrolu C, Beisser D, Kolter J, Sigmund AM, et al. Human TLR8 senses UR/URR motifs in bacterial and mitochondrial RNA. EMBO Rep (2015) 16(12):1656–63. doi:10.15252/embr.201540861
60. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol (2008) 9:361–8. doi:10.1038/ni1569
61. Aubry C, Corr SC, Wienerroither S, Goulard C, Jones R, Jamieson AM, et al. Both TLR2 and TRIF contribute to interferon-beta production during Listeria infection. PLoS One (2012) 7:e33299. doi:10.1371/journal.pone.0033299
62. Dietrich N, Lienenklaus S, Weiss S, Gekara NO. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One (2010) 20(5):e10250. doi:10.1371/journal.pone.0010250
63. Stack J, Doyle SL, Connolly DJ, Reinert LS, O’Keeffe KM, McLoughlin RM, et al. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J Immunol (2014) 15(193):6090–102. doi:10.4049/jimmunol.1401605
64. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog (2009) 5:e1000500. doi:10.1371/journal.ppat.1000500
65. Dresing P, Borkens S, Kocur M, Kropp S, Scheu S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon beta in murine listeriosis. PLoS One (2010) 5:e15567. doi:10.1371/journal.pone.0015567
66. Solodova E, Jablonska J, Weiss S, Lienenklaus S. Production of IFN-beta during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS One (2011) 6:e18543. doi:10.1371/journal.pone.0018543
67. Stockinger S, Kastner R, Kernbauer E, Pilz A, Westermayer S, Reutterer B, et al. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog (2009) 5:e1000355. doi:10.1371/journal.ppat.1000355
68. Koltai M, Meos E. Inhibition of the acute inflammatory response by interferon inducers. Nature (1973) 20(242):525–6. doi:10.1038/242525a0
69. Chang EY, Guo B, Doyle SE, Cheng G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol (2007) 1(178):6705–9. doi:10.4049/jimmunol.178.11.6705
70. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 19(466):973–7. doi:10.1038/nature09247
71. Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-gamma signaling. J Immunol (2016) 197(12):4714–26. doi:10.4049/jimmunol.1600584
72. Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med (2006) 17(203):933–40. doi:10.1084/jem.20060045
73. Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol (2002) 1(169):6522–9. doi:10.4049/jimmunol.169.11.6522
74. Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol (2016) 17:481–9. doi:10.1038/ni.3440
75. Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J Leukoc Biol (2016). doi:10.1189/jlb.4MR0516-223R
76. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 15(509):366–70. doi:10.1038/nature13157
77. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol (2015) 16:476–84. doi:10.1038/ni.3119
Keywords: type I interferon, bacterial infection, cytokines, chemokines, innate immunity, resilience to infections, immunomodulation, immunosuppression
Citation: Kovarik P, Castiglia V, Ivin M and Ebner F (2016) Type I Interferons in Bacterial Infections: A Balancing Act. Front. Immunol. 7:652. doi: 10.3389/fimmu.2016.00652
Received: 01 November 2016; Accepted: 14 December 2016;
Published: 26 December 2016
Edited by:
Claudia U. Duerr, McGill University, CanadaReviewed by:
Didier Soulat, Universitätsklinikum Erlangen, GermanyStefanie Scheu, University of Düsseldorf, Germany
Copyright: © 2016 Kovarik, Castiglia, Ivin and Ebner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pavel Kovarik, cGF2ZWwua292YXJpa0B1bml2aWUuYWMuYXQ=