 Wenkai Ren1,2,3†
Wenkai Ren1,2,3† Jie Yin1,2†
Jie Yin1,2† Hao Xiao1,2†Shuai Chen1,2Gang Liu1,2Bie Tan1,2*Nengzhang Li4Yuanyi Peng4Tiejun Li1,2*Benhua Zeng5Wenxia Li5Hong Wei5Zhinan Yin6Guoyao Wu7Philip R. Hardwidge8Yulong Yin1,9*
Hao Xiao1,2†Shuai Chen1,2Gang Liu1,2Bie Tan1,2*Nengzhang Li4Yuanyi Peng4Tiejun Li1,2*Benhua Zeng5Wenxia Li5Hong Wei5Zhinan Yin6Guoyao Wu7Philip R. Hardwidge8Yulong Yin1,9*
- 1National Engineering Laboratory for Pollution Control and Waste Utilization in Livestock and Poultry Production, Institute of Subtropical Agriculture, The Chinese Academy of Sciences, Changsha, China
- 2Key Laboratory of Agro-Ecology, Institute of Subtropical Agriculture, The Chinese Academy of Sciences, Changsha, China
- 3University of the Chinese Academy of Sciences, Beijing, China
- 4Chongqing Key Laboratory of Forage and Herbivorce, College of Animal Science and Technology, Southwest University, Chongqing, China
- 5Department of Laboratory Animal Science, College of Basic Medicine Science, Third Military Medical University, Chongqing, China
- 6Biomedical Translational Research Institute, Jinan University, Guangzhou, China
- 7Department of Animal Science, Texas A&M University, College Station, TX, USA
- 8Department of Diagnostic Medicine/Pathobiology, Kansas State University, Manhattan, KS, USA
- 9College of Animal Science, South China Agricultural University, Guangzhou, China
Intestinal microbiota has critical importance in pathogenesis of intestinal infection; however, the role of intestinal microbiota in intestinal immunity during enterotoxigenic Escherichia coli (ETEC) infection is poorly understood. The present study tested the hypothesis that the intestinal microbiota is associated with intestinal interleukin-17 (IL-17) expression in response to ETEC infection. Here, we found ETEC infection induced expression of intestinal IL-17 and dysbiosis of intestinal microbiota, increasing abundance of γ-aminobutyric acid (GABA)-producing Lactococcus lactis subsp. lactis. Antibiotics treatment in mice lowered the expression of intestinal IL-17 during ETEC infection, while GABA or L. lactis subsp. lactis administration restored the expression of intestinal IL-17. L. lactis subsp. lactis administration also promoted expression of intestinal IL-17 in germ-free mice during ETEC infection. GABA enhanced intestinal IL-17 expression in the context of ETEC infection through activating mechanistic target of rapamycin complex 1 (mTORC1)-ribosomal protein S6 kinase 1 (S6K1) signaling. GABA–mTORC1 signaling also affected intestinal IL-17 expression in response to Citrobacter rodentium infection and in drug-induced model of intestinal inflammation. These findings highlight the importance of intestinal GABA signaling in intestinal IL-17 expression during intestinal infection and indicate the potential of intestinal microbiota-GABA signaling in IL-17-associated intestinal diseases.
Introduction
The intestinal microbiota has critical importance in intestinal infections by increasing colonization resistance and promoting pathogen clearance after infection (1, 2). For example, Clostridium difficile infection, which is the leading health care-associated illness, usually follows the disruption of the indigenous gut microbiota after antibiotic treatment, leading to the loss of colonization resistance against C. difficile (2–4). The gut microbiota also affects intestinal infections by mediating the host innate and adaptive immune responses (5, 6). For example, germ-free mice are highly susceptible to Listeria monocytogenes infection because of impaired activation and accumulation of phagocytes to the site of infection (6).
Interleukin (IL)-17 promotes local chemokine production to recruit monocytes and neutrophils to sites of inflammation and is thus important in mediating protection against pathogens, especially against extracellular pathogens (7). IL-17 combats the microbes attacking epithelial layers and has critical functions in protecting against bacterial infection at mucosal sites (8). IL-17 is also thought to play major roles in the development and pathogenesis of various autoimmune diseases, including rheumatoid arthritis, psoriasis vulgaris, multiple sclerosis, and inflammatory bowel disease (9, 10).
Intestinal expression of IL-17 is induced after intestinal infection by most pathogens (11–13). For example, Citrobacter rodentium or Salmonella infection promotes intestinal IL-17 expression by enteric innate T helper type 17 (iTh17) cells (12). Enterotoxigenic Escherichia coli (ETEC) is a common cause of diarrhea in humans and livestock (14). Previous investigations have found that ETEC infection triggers intestinal IL-17 expression (15, 16). However, the underlying mechanisms are largely unknown. The present study tested the hypothesis that the intestinal microbiota is associated with intestinal IL-17 expression in response to ETEC infection.
We confirmed that ETEC promotes intestinal IL-17 expression in piglets and mice and showed that the activation of the mechanistic target of rapamycin complex 1 (mTORC1)-growth factor independence 1 (GFI-1) signaling mediates intestinal IL-17 expression in the context of ETEC infection. We clarified that γ-aminobutyric acid (GABA) signaling is critical to activating the mTORC1–GFI-1–IL-17 pathway during ETEC infection, and this signaling is largely dependent on the intestinal GABA-producing strain Lactococcus lactis subsp. lactis.
Materials and Methods
Bacterial Strains and Antibodies
This study involved the use of an E. coli F4-producing strain W25K (hereafter referred as ETEC; O149:K91, K88ac; LT, STb, EAST), which was isolated from a piglet with diarrhea (17). ETEC W470 (O4:F18; STa, STb, LT), W817 (O107:F18; STb), and W616 (F18; STa) were also isolated from piglets with diarrhea, while the Shiga-like toxin producing E. coli (W197, SLT-IIe) was isolated from a piglet with edema disease (18). These strains of bacteria were cultured in LB medium. L. lactis subsp. lactis (ATCC19435) was cultured in M17 medium. C. rodentium (DBS100) was cultured in LB medium. Antibodies against RAR-related orphan receptor gamma t (RORγt) (Sc-14196), forkhead box P3 (Foxp3) (Sc-28705), growth factor independent 1 (GFI-1) (Sc-8558), early growth response protein 2 (EGR-2) (Sc-20690), p85 (Sc-1637), phosphorylated protein kinase B (Akt) (Sc7985-R), GAT-2 (Sc-7668), actin (Sc-47778), and proliferating cell nuclear antigen (PCNA) (Sc-56) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Antibodies against mTOR (CST 2972), p-mTOR (CST 5536), p-p70 S6 Kinase (CST 9205), p-4E-BP1 (CST 9451), p-AMP-activated protein kinase (AMPK) (CST 2535), hypoxia-inducible factor 1 α (HIF-1α) (CST 14179), and p70 S6 Kinase 2 (CST 14130) were purchased from Cell Signaling Technology (Danvers, MA, USA).
ETEC Infection in Piglets
This study was approved and conducted according to the guidelines of the Institute of Subtropical Agriculture, Chinese Academy of Sciences and Southwest University. Piglets (Landrace Yorkshire; 18 days old) were purchased from ZhengDa Co., Chongqing, China. ETEC infection in piglets was established according to previous reports (19, 20). The jejunum samples were collected. Samples were stored at −80°C until processing.
Mice
TCR delta knockout mice were provided by Prof. Zhinan Yin, from Jinan University (Guangzhou, China). Rag 1 knockout mice were bought from Nanjing University (Nanjing, China). Germ-free mice were generated and provided by Prof. Hong Wei, from Third Military Medical University (Chongqing, China), and these mice were maintained in sterile Trexler-type isolators. ICR mice (6 weeks of age) were purchased from SLAC Laboratory Animal Central (Changsha, China). Experiments in mice were conducted according to the guidelines of the Laboratory Animal Ethical Commission of the Chinese Academy of Sciences.
ETEC Infection in Mice
Mice were orally gavaged with 108 CFUs of ETEC or other strains of E. coli. At 6-h postinfection, mice were sacrificed to collect the jejunum, ileum, and mesenteric lymph node. In some experiments, mice were treated with rapamycin (Fisher Scientific) at 2.5 mg/kg/d (i.p. injection) for six consecutive days prior to other manipulations. In experiments involving the inhibition of GABA signaling, mice were intraperitoneally injected with 20 mg/kg bicuculline (Dalian Meilun Bio. Tech. Co., Ltd., Dalian, China), 120 mg/kg CGP-35348 (Tocris Bioscience), 80 mg/kg l-allylglycine (Aladdin Industrial Corporation, China), or 100 mg/kg semicarbazide (Sangon Biotech Co., Ltd., Shanghai, China) at 30 min prior to other manipulations. For the inhibition of S6K1, mice were intraperitoneally injected with 75 mg/kg/d PF-4708671 (S2163, Selleck) for six consecutive days. In some experiments, mice were intraperitoneally injected with GABA (Aladdin Industrial Corporation, China) at dosages of 40 μg//kg to 40 mg/kg at 30 min prior to other manipulations [the physiologic level of GABA in mouse serum is about 2 mg/kg (21), while in human (adults), it is about 100–400 µg/kg]. For antibiotics treatment, mice received drinking water containing streptomycin (1.0 g/L, Sigma), ampicillin (1.0 g/L, Sigma), gentamicin (1.0 g/L, Sigma), and vancomycin (0.5 g/L, Sigma) for 1 week before ETEC infection. For the supplementation of GABA to antibiotics-treated mice, mice received drinking water containing antibiotics (1.0 g/L streptomycin, 1.0 g/L ampicillin, 1.0 g/L gentamicin, and 0.5 g/L vancomycin) and GABA (1.0 g/L or 5.0 g/L) for 1 week before ETEC infection. For L. lactis subsp. lactis inoculation into antibiotics-treated mice, mice received drinking water containing antibiotics (1.0 g/L streptomycin, 1.0 g/L ampicillin, 1.0 g/L gentamicin, and 0.5 g/L vancomycin) for 6 days, and then orally inoculated with L. lactis subsp. lactis at a dosage of 108 CFUs at 24 h before ETEC infection. For L. lactis subsp. lactis inoculation into germ-free mice, mice were orally inoculated with L. lactis subsp. lactis at a dosage of 108 CFUs at 7 days before ETEC infection.
L. lactis Subsp. lactis Inoculation in Mice
For L. lactis subsp. lactis inoculation into normal mice, mice were orally inoculated with L. lactis subsp. lactis at a dosage of 108 CFUs for five consecutive days. For GABA signaling inhibition, mice were intraperitoneally injected with 120 mg/kg CGP-35348 at 5 days postinoculation. The jejunum samples were collected for further analysis at 5 days postinoculation (6-h post-CGP-35348 treatment).
L. lactis Subsp. lactis Inoculation in Piglets
For L. lactis subsp. lactis inoculation in piglets, piglets were orally inoculated with L. lactis subsp. lactis at dose of 1010, while the control piglets received some volume of M17 medium. The jejunum samples were collected for analysis at 7 days postinoculation.
C. rodentium Infection in Mice
Mice (6 weeks of age) were orally gavaged with 108 CFUs of C. rodentium (DBS100). For rapamycin treatment, mice were treated with rapamycin at 2.5 mg/kg/d (i.p. injection) at 1–7 days postinfection. For semicarbazide treatment, mice were treated with semicarbazide at 100 mg/kg/d (i.p. injection) at 1, 3, 5, and 7 days postinfection. In some experiments, mice were intraperitoneally injected with 20 mg/kg bicuculline or 120 mg/kg CGP-35348 at 7 days postinfection. The colon samples from all groups were collected for further analysis at 7 days postinfection (6-h post-bicuculline or CGP-35348 treatment).
5-Fluorouracil Treatment in Mice
ICR mice (6 weeks of age) were intraperitoneally injected with 300 mg/kg 5-fluorouracil (Sigma-Aldrich). For the inhibition of GABA signaling, mice received intraperitoneal administration of 20 mg/kg bicuculline or 100 mg/kg semicarbazide at 30 min prior to 5-fluorouracil treatment. The jejunum was collected at 6-h posttreatment for further analysis.
Lymphocyte Isolation from Jejunum
Lamina propria lymphocytes from jejunum were obtained according to previous procedures (12, 22, 23). Briefly, jejunum tissue was extracted, opened longitudinally to wash luminal contents. Jejunum (cleared the visible PP) was cut into 0.5- to 1-cm segments that were incubated three times (37°C, 20 min, 250 rpm) in HBSS/HEPES medium (Life Technologies) containing 5 mM EDTA to remove the epithelial cells. Thereafter, the tissue segments were minced and digested in digestion buffer [HBSS + 5% FBS containing type VIII collagenase at 1.5 mg/mL (Sigma)] for 30-min incubations at 37°C. Digested material was passed through a 100-µm cell strainer, and the cells were collected by centrifugation, washed twice in DMEM, and then passed through a 40-µm cell strainer to obtain lamina propria lymphocytes from the jejunum.
Flow Cytometry Analysis
Lymphocytes isolated from mouse jejunal lamina propria were stained with cell surface markers of CD3 (FITC-CD3, 100203, Biolegend), CD4 (PE-CD4, 100407, Biolegend), or TCR γδ (PE-TCR γδ, 118107, Biolegend). For the analysis of intracellular cytokine, lymphocytes or CD4+ T cells were stimulated with PMA (50 ng/mL), ionomycin (1 µg/mL), and monensin (3 µg/mL) for 5 h. After staining with cell surface markers, intracellular cytokine staining was performed with a fixation and permeabilization kit (eBioscience) and IL-17A Ab (APC-IL-17A, 506195, Biolegend) in accordance with the manufacturer’s instructions. Flow cytometry was performed on a FACSCalibur (BD Biosciences) and data were analyzed using the FlowJo Software (Tree Star).
Metabolite Profiling Analysis
Metabolite concentrations in piglet jejunal samples were quantified using gas chromatography/mass spectrometry (GC–MS) according to the previous work (19).
RT-PCR
Real-time PCR was performed according to our previous study (20, 24). Primers (Table S1 in Supplementary Material) were selected according to previous references. β-Actin was used as an internal control to normalize target gene transcript levels.
Immunoblotting
Immunoblotting was performed according to our previous study (20, 24). Signal intensity was digitally quantified and normalized to actin or PCNA protein abundance.
Th17 Cell Differentiation
Naive CD4+ T cells were isolated from mouse splenocytes using a CD4+CD62L+ T cell isolation kit II (Miltenyi Biotec, purity > 95%). For Th17 differentiation, cells were stimulated with anti-CD3 (2 µg/mL, 100313, Biolegend) and anti-CD28 (2.0 µg/mL, 102111, Biolegend) supplemented with 5 ng/ml TGF-β1 (7666-MB-005, RD), 20 ng/mL IL-6 (575702, Biolegend), 10 µg/mL anti-IFN-γ (505812, Biolegend), and 10 µg/mL anti-IL-4 (504107, Biolegend) for 1–3 days. l-Allylglycine, semicarbazide, and rapamycin were used at 5 mM, 4 mM, and 3 µM, respectively.
Statistical Analyses
Data shown are the means ± SEM. Data between two groups were analyzed by unpaired t test (Prism 6.0) if the data were in Gaussian distribution and had equal variance, or by unpaired t test with Welch’s correction (Prism 6.0) if the data were in Gaussian distribution but with unequal variance, or by non-parametric test (Mann–Whitney U test, Prism 6.0) if the data were not normally distributed. Data among more than two groups were analyzed by the one-way ANOVA followed by Dunnett multiple comparisons (Prism 6.0) if the data were in Gaussian distribution and had equal variance, or analyzed by Kruskal–Wallis followed by Dunn’s multiple comparisons (Prism 6.0) if the data were not normally distributed. The Gaussian distribution of data was analyzed by D’Agostino–Pearson omnibus normality test (Prism 6.0) and Kolmogorov–Smirnov test (Prism 6.0). The variance of data was analyzed by homogeneity of variance test (SPSS 22.0) or Brown–Forsythe test (Prism 6.0). The Bonferroni correction was applied for multiple pairwise comparisons. Differences with p < 0.05 were considered significant.
Results
ETEC Increases IL-17 Expression in Piglets and Mice
Enterotoxigenic Escherichia coli infection is known to promote IL-17 expression in piglets (15, 16), a finding we also validated. We infected Landrace × Yorkshire piglets with ETEC W25K, a strain originally isolated from a piglet with diarrhea (17). IL-17 mRNA expression in the porcine jejunum was higher (about 17-fold) in piglets infected with ETEC, as compared with uninfected controls (Figure 1A). Increased mRNA expression of other proinflammatory cytokines, including IL-6, IL-8, and TNF-α, was also detected in the jejunum after ETEC infection (Figure 1A).
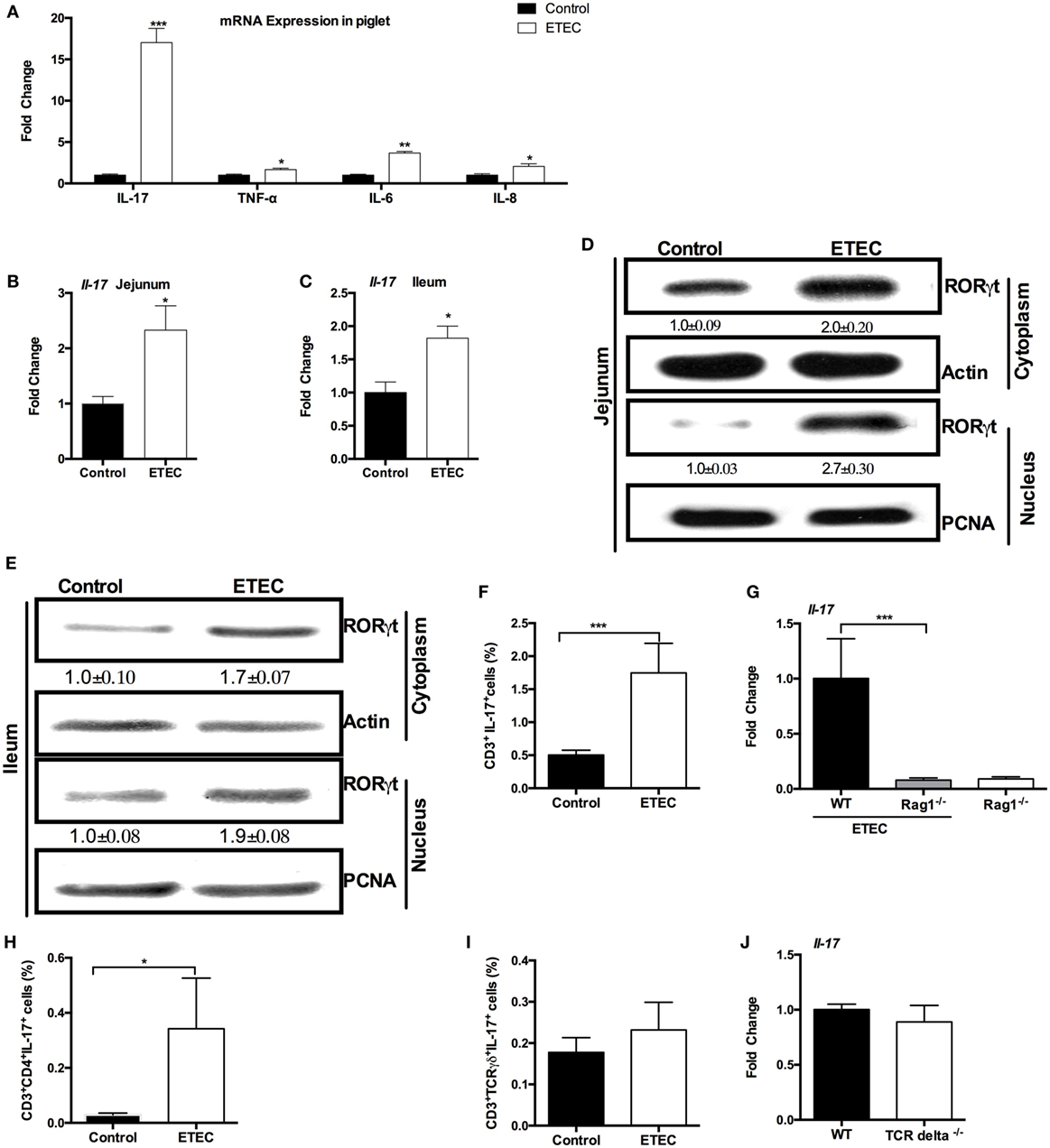
Figure 1. Enterotoxigenic Escherichia coli (ETEC) induces intestinal interleukin-17 (IL-17) expression. (A) Relative mRNA expression of indicated cytokines in piglet jejunum samples (n = 6). (B) Relative mRNA expression of IL-17 in mouse jejunum. (C) Relative mRNA expression of IL-17 in mouse ileum. (D) Nuclear and cytoplasmic abundance of RAR-related orphan receptor gamma t (RORγt) in mouse jejunum samples. In this, and in all subsequent figures, the statistical data (mean ± SEM; n = 5) below the image indicate the relative band amount of indicated protein obtained by dividing the actin or proliferating cell nuclear antigen (PCNA) band intensity by the indicated band intensity in each lane. (E) Nuclear and cytoplasmic abundance of RORγt in mouse ileum samples. (F,H,I) ICR mice were infected with ETEC for 6 h, and lymphocytes from the jejunal lamina propria were isolated and stimulated for 5 h with PMA, ionomycin, and monensin, followed by flow cytometry analysis of the frequency of CD3+IL-17+ cells (F), CD3+CD4+IL-17+ cells (H), and CD3+TCR γδ+IL-17+ cells (I). (G) Relative mRNA expression of IL-17 in Rag1−/− mouse jejunum at 6-h post-ETEC infection. (J) Relative mRNA expression of IL-17 in TCR delta−/− mouse jejunum at 6-h post-ETEC infection. *indicates a statistically significant difference between two groups (p < 0.05), while **indicates p < 0.01, and ***indicates p < 0.001 [unpaired t test (A,J); Mann–Whitney test (F,H,I); Kruskal–Wallis test (G); Bonferroni correction was also used for (A)]. Data shown are representative of three independent experiments with n = 10 (B,C) or 5 (D,E) in each experiment, or representatives of two independent experiments with n = 4–7 (F–J) in each experiment.
In mice, at 6-h postinfection, ETEC colonized in the jejunum with load of about 5.9 log10 CFU/g and promoted the expression of Il-17 in the jejunum (Figure 1B). ETEC also promoted the expression of Il-17 in the ileum (Figure 1C). ETEC infection enhanced the abundance of RORγt (a key transcriptional regulator of Th17 cells) in the cytoplasm and nuclei of mouse jejunum (Figure 1D) and ileum (Figure 1E).
Although the best-characterized cellular source of IL-17 is Th17 cells, a number of adaptive and innate immune cells, such as γδ T cells, natural killer T (NKT) cells, and even Paneth cells, are also the sources of IL-17 (11, 13). Similar to previous conclusion that the increased IL-17+ cells at 5-h post-E. coli infection are CD3+ cells (25), we found that most of IL-17+ cells from ETEC-infected mouse jejunum were CD3+ cells. Thus, the ETEC-infected mice had a higher percentage of CD3+IL-17+ cells in the jejunum compared with the controls (Figure 1F), suggesting that the increased expression of IL-17 at 6-h post-ETEC infection comes from T cells. Indeed, ETEC infection had little effect on the expression of Il-17 in the jejunum in Rag 1 knockout mice (mature T cell-depleted mice), while wild-type mice had higher expression of Il-17 in the jejunum compared to Rag 1 knockout mice during ETEC infection (Figure 1G). Unlike the previous study, which shows that the increased IL-17 is mainly produced by γδ T cells during early E. coli infection (25), we observed a higher percentage of CD3+CD4+IL-17+ cells in the jejunum from ETEC-infected mice as compared with uninfected mice, while the percentage of CD3+TCR γδ+IL-17+ cells in the jejunum did not differ between ETEC-infected mice and control mice (Figures 1H,I). Notably, Il-17 expression in the jejunum after 6 h of ETEC infection was similar between TCR delta−/− mice (γδ T cell-depleted mice) and wild-type mice (Figure 1J). In conclusion, ETEC infection promotes the intestinal mRNA expression of IL-17 in piglets and mice.
ETEC Promotes IL-17 Expression through mTORC1 Activation
A previous study has shown that ETEC infection inhibits the activation of NF-κB and mitogen-activated protein kinases pathways in the jejunum (20), indicating that new pathways are associated with expression of IL-17 during ETEC infection. Previous proteomic analysis indicated that ETEC infection upregulates ribosomal protein S6 kinase in the piglet jejunum (20), which suggests that ETEC infection may activate mTORC1 signaling. The mTORC1 has a critical role in Th17 responses and in IL-17 expression (26). Indeed, the mTORC1 pathway was activated in piglets infected with ETEC, based on the higher abundance of phosphorylated mTORC1 and its downstream target S6K (Figure 2A). Phosphorylated mTORC1 abundance was also higher in ETEC-infected mice than that in uninfected controls (Figure 2B). To validate the possible roles of mTORC1 signaling in the expression of IL-17 during ETEC infection, this study used rapamycin to inhibit the activation of mTORC1. Rapamycin treatment prior to ETEC infection prevented the increase in mTORC1 phosphorylation in the jejunum and ileum (Figure 2C) and reversed the increased Il-17 expression in mice infected with ETEC (Figure 2D). Similar to a previous report (27), rapamycin also inhibited Th17 cell differentiation from naïve T cells under Th17 polarization conditions in vitro (Figures 2E). In conclusion, ETEC infection promotes IL-17 expression through the activation of mTORC1 pathway.
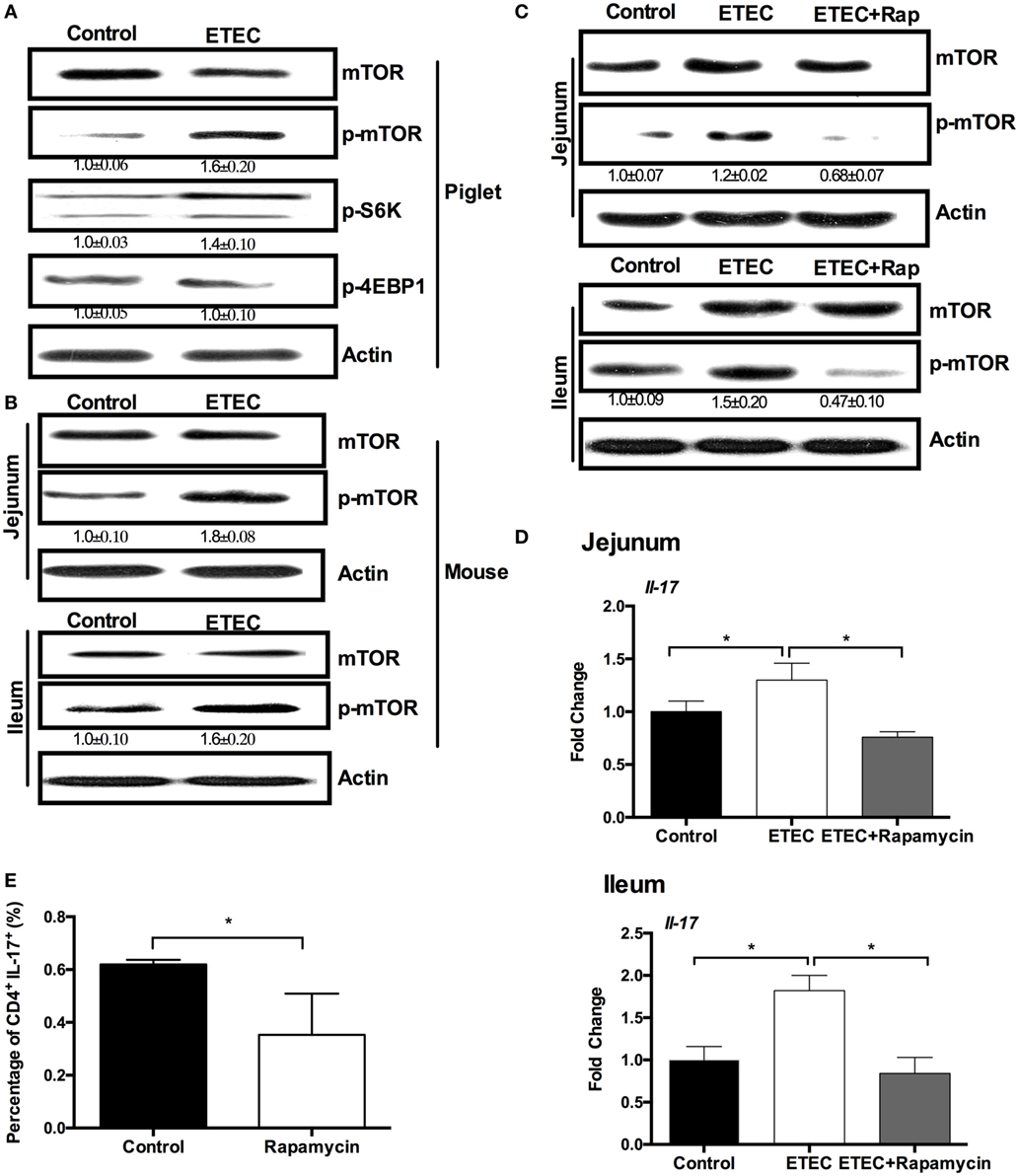
Figure 2. Enterotoxigenic Escherichia coli (ETEC) promotes intestinal interleukin-17 (IL-17) mRNA expression through mechanistic target of rapamycin complex 1 (mTORC1) activation. (A) The activation of mTOR pathway in piglet jejunum (n = 5). (B) The activation of mTOR pathways in mouse jejunum or ileum. (C) Rapamycin pretreatment inhibits mTOR activation in mouse jejunum and ileum. (D) mRNA expression of IL-17 in mouse jejunum and ileum after rapamycin treatment. (E) Intracellular staining of the expression of IL-17 by CD4+ T cells cultured under the Th17-inducing conditions with or without rapamycin for 24 h. *indicates a statistically significant difference between two groups (p < 0.05) [unpaired t test (A,B); one-way ANOVA (C,D); Mann–Whitney test (E)]. Data shown are representative of four independent experiments with n = 5 (B,C) or 10 (D) in each experiment, or representatives of three independent experiments with n = 3 (E) in each experiment.
ETEC Promotes IL-17 Expression through the mTORC1–EGR-2 Pathway
Among the downstream targets of mTORC1 signaling, HIF-1α and S6K (S6K1 and S6K2) can regulate IL-17 expression (26). mTORC1 signaling activates HIF-1α, which promotes IL-17 expression by activating RORγt and mediating Foxp3 degradation (26). ETEC infection decreased HIF-1α abundance and increased Foxp3 abundance in the cytoplasm of mouse jejunum compared to the controls (Figure 3A). Rapamycin treatment before ETEC infection increased HIF-1α abundance and lowered Foxp3 abundance (Figure 3A). S6K2 (the nuclear-localized counterpart of S6K1) binds to RORγt to promote the nuclear translocation of RORγt, which can complex with HIF-1α and p300 in the nucleus to promote IL-17 expression (26). Although ETEC infection increased RORγt abundance, rapamycin had no effect on RORγt abundance (Figure 3B, top). Similarly, rapamycin did not affect on the abundance of HIF-1α and S6K2 in the nucleus of the mouse jejunum (Figure 3B, bottom), although ETEC infection reduced the nuclear abundance of HIF-1α and S6K2 in mouse jejunum (Figure 3B, bottom).
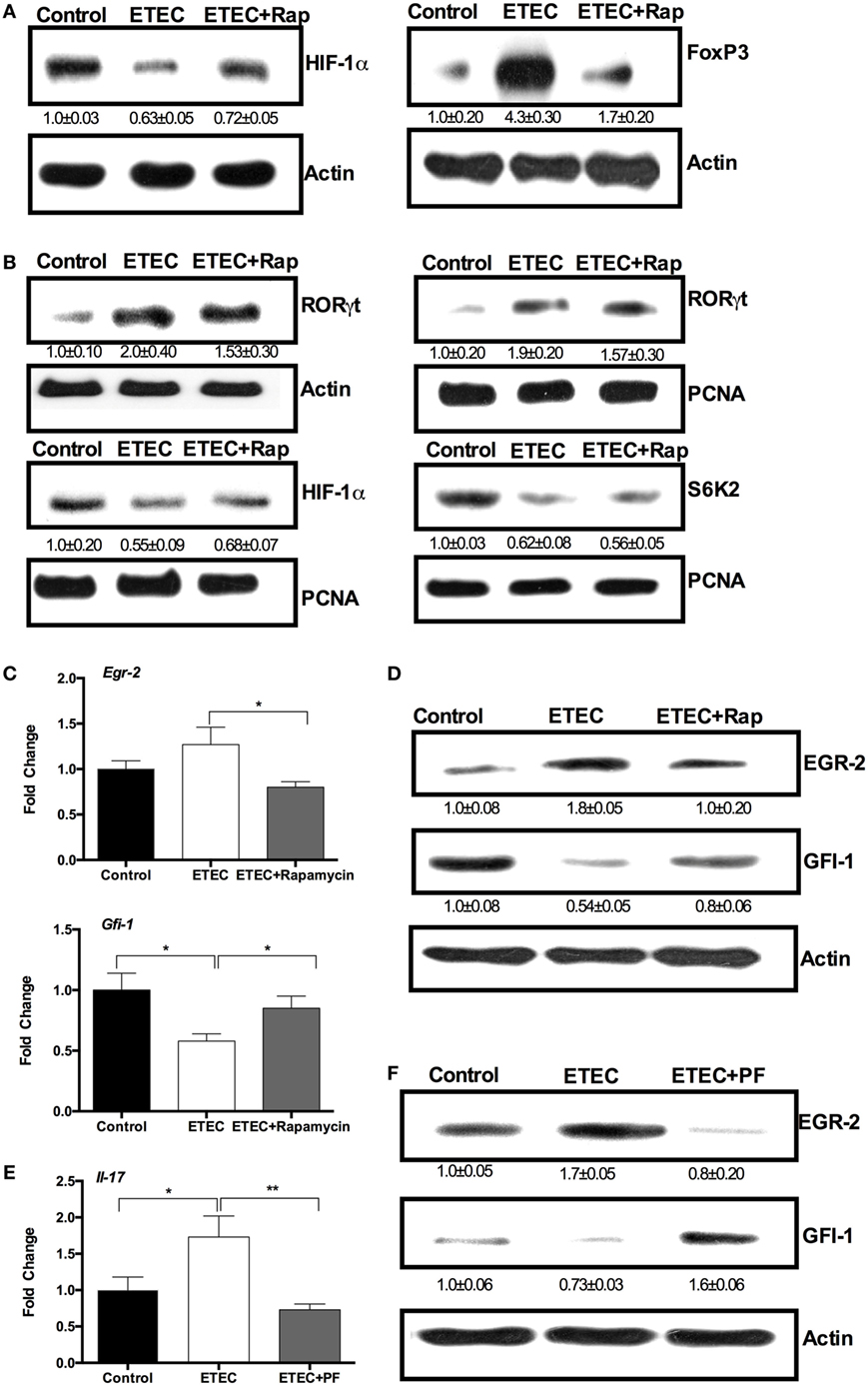
Figure 3. Enterotoxigenic Escherichia coli (ETEC) promotes interleukin-17 (IL-17) expression through the mechanistic target of rapamycin complex 1 (mTORC1)–early growth response protein 2 (EGR-2) pathway. (A) Hypoxia-inducible factor 1 α (HIF-1α) and Foxp3 protein abundance (n = 5). (B) RORγt, HIF-1α, and S6K2 abundance (n = 5). (C) EGR-2 and GFI-1 mRNA expression (n = 10). (D) EGR-2 and GFI-1 protein abundance (n = 5). (E) IL-17 mRNA expression in mouse jejunum (n = 10). (F) EGR-2 and GFI-1 abundance (n = 5). All these indicators were analyzed by RT-PCR (mRNA level) and immunblotting (protein level) in mouse jejunum at 6 h of post-ETEC infection. *indicates a statistically significant difference between two groups (p < 0.05), while **indicates p < 0.01 [one-way ANOVA (A–F)].
S6K1 promotes the expression of EGR-2, resulting in the inhibition of the GFI-1, which negatively regulates IL-17 expression without affecting Rorc expression (26). Although ETEC infection had little affect on Egr-2 expression in the mouse jejunum, rapamycin reduced Egr-2 expression in the mouse jejunum (Figure 3C). ETEC infection reduced Gfi-1 expression in the mouse jejunum, while rapamycin prevented this reduction (Figure 3C). At the protein level, ETEC infection increased the abundance of EGR-2 and lowered the abundance of GFI-1 in the mouse jejunum, while rapamycin abolished the effects of ETEC infection (Figure 3D). These results indicate that ETEC induces intestinal IL-17 expression may through the S6K1–EGR-2–GFI-1 axis. Intriguingly, ETEC infection increased Il-17 expression in the jejunum, while PF-4708671 (S6K1 specific inhibitor) reduced Il-17 expression in the jejunum in ETEC-infected mice (Figure 3E). ETEC infection increased the abundance of EGR-2 and decreased the abundance of GFI-1 in the mouse jejunum, while PF-4708671 treatment reversed this change during ETEC infection in mouse jejunum (Figure 3F). These data indicate ETEC promotes the IL-17 expression in the jejunum through the mTOR–S6K1–GFI-1 axis.
ETEC Promotes IL-17 Expression through GABA–mTORC1–EGR-2 Signaling
Mechanistic target of rapamycin complex 1 is activated by amino acids and by the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, while it is inactivated by the AMPK (28, 29). ETEC infection inhibited the PI3K/Akt pathway and activated AMPK in piglet jejunum (Figures 4A,B). Thus, the activation of mTORC1 in response to ETEC infection may result from changes in amino acid concentration in the jejunum. Using GC–MS analysis, we found that eight amino acids significantly changed in abundance after ETEC infection (Figure 4C). The relative abundance of spermidine, glutamine, asparagine, citrulline, and ornithine decreased, while the relative abundance of glycine, taurine, and GABA increased after ETEC infection (Figure 4C). However, there was little change in mRNA expression of the activating transcription factor 4 (Figure 4D), an indicator of amino acid starvation (30), suggesting there was no overall amino acid starvation after ETEC infection.
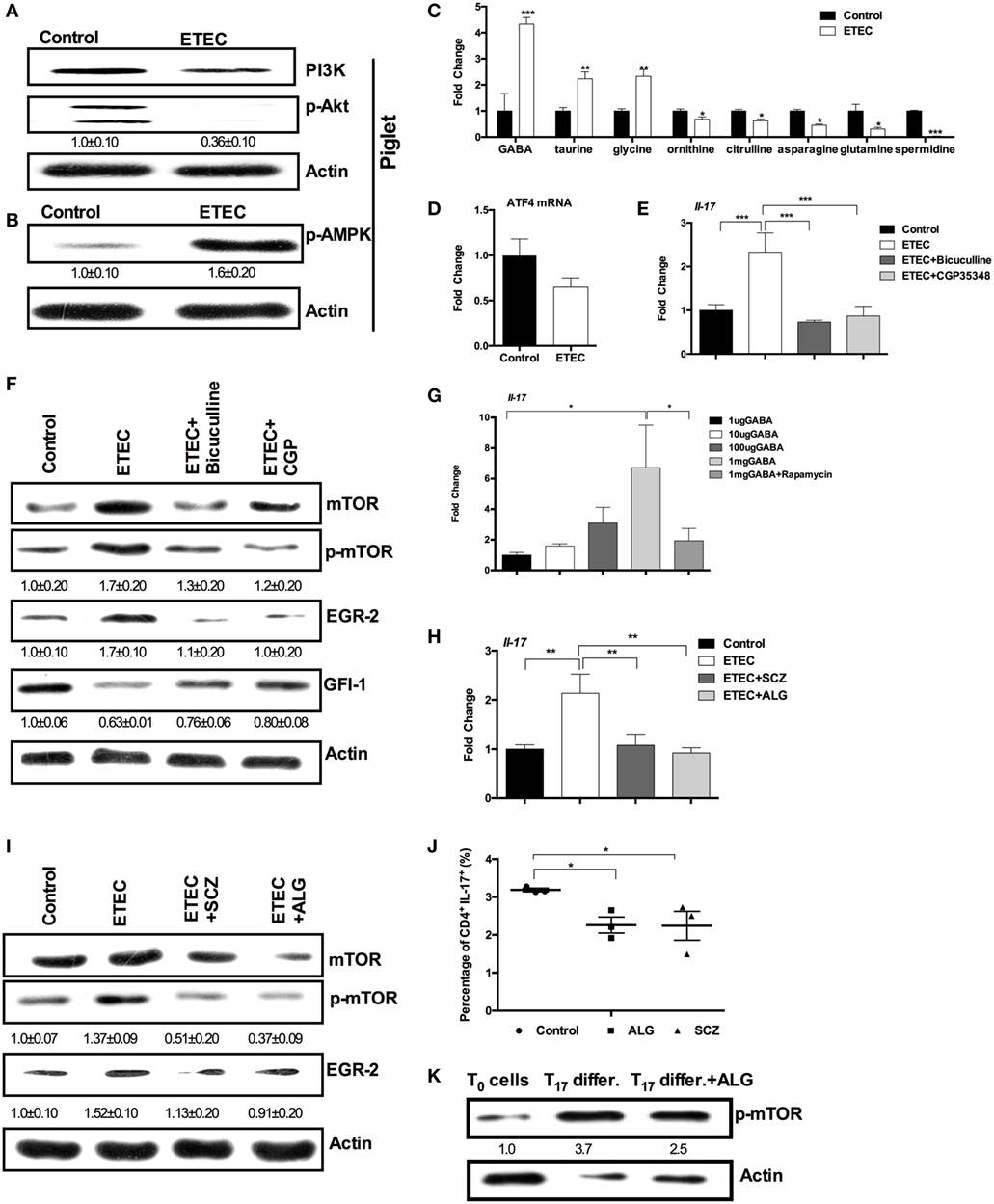
Figure 4. Enterotoxigenic Escherichia coli (ETEC) promotes interleukin-17 (IL-17) expression through γ-aminobutyric acid (GABA)-mechanistic target of rapamycin complex 1 (mTORC1) signaling. (A) The activation of PI3K-p-Akt pathway in piglet jejunum (n = 5). (B) The abundance of phosphorylated AMP-activated protein kinase (AMPK) in piglet jejunum (n = 5). (C) Relative amino acid concentrations in diarrheal piglets compared with uninfected piglets (n = 6). (D) Activating transcription factor 4 (ATF4) mRNA expression in piglet jejunum (n = 6). (E) mRNA expression of IL-17 after bicuculline or CGP-35348 treatment (n = 10). (F) mTORC1–GFI-1 activation after bicuculline or CGP-35348 treatment (n = 5). (G) mRNA expression of IL-17 after GABA injection (n = 10). (H) mRNA expression of IL-17 after l-allylglycine or semicarbazide treatment (n = 10). (I) mTORC1–early growth response protein 2 (EGR-2) activation after l-allylglycine or semicarbazide treatment (n = 5). (J) Intracellular staining of the expression of IL-17 by CD4+ T cells cultured under the Th17-inducing conditions with or without allyglycine or semicarbazide for 3 days. Results shown are representative of three independent experiments with n = 3 in each experiment. (K) The abundance of phosphorylated mTOR. Naïve CD4+ T cells were isolated (T0 cells) and cultured under the Th17-inducing conditions without (Th17 differ.) or with allyglycine (Th17 differ. + ALG) for 3 days. Results shown are representative of three independent experiments. The numbers indicate the relative band amount of p-mTOR protein in each sample obtained by dividing the actin band intensity by the p-mTOR band intensity in each lane. *indicates a statistically significant difference between two groups (p < 0.05), while **indicates p < 0.01, and ***indicates p < 0.001 [unpaired t test (A–D); one-way ANOVA (E–I); Kruskal–Wallis test (J); Bonferroni correction was also used for (C)].
As GABA was the most significantly increased amino acid (about fourfold), we hypothesized that GABA is responsible for increased IL-17 expression and mTORC1 activation during ETEC infection. Indeed, numerous investigations have shown that GABA signaling promotes mTORC1 activation (31–33). Treating mice with bicuculline (an antagonist of GABAA receptors) or with CGP-35348 (an antagonist of GABAB receptors) lowered Il-17 expression in the jejunum during ETEC infection (Figure 4E). Bicuculline and CGP-35348 treatment also reversed the activation of mTORC1–EGR-2 signaling in mouse jejunum induced by ETEC infection (Figure 4F). GABA injection promoted Il-17 expression in mouse jejunum during ETEC infection, while rapamycin prevented expression of Il-17 caused by GABA injection (Figure 4G). Furthermore, treatment with l-allylglycine or semicarbazide (blockers of synthetic enzyme for GABA) reduced Il-17 expression in mouse jejunum during ETEC infection (Figure 4H). l-Allylglycine or semicarbazide treatment also prevented the activation of mTORC1–EGR-2 signaling in mouse jejunum caused by ETEC infection (Figure 4I).
To investigate whether GABA signaling regulates Th17 cell differentiation, naïve CD4+ T cells were isolated and differentiated into Th17 cells under Th17 polarization conditions. In normal Th17 differentiation condition, about 3.0% of CD4+ T cells were differentiated into CD4+IL-17+ T cells after 3 days of differentiation; while l-allylglycine or semicarbazide decreased the percentage of CD4+IL-17+ T cells (Figure 4J). After 3 days of differentiation under normal Th17 polarization conditions, CD4+ T cells had higher abundance of phosphorylated mTORC1, compared to the naïve CD4+ T cells (Figure 4K). Also, l-allylglycine decreased the abundance of phosphorylated mTORC1 (Figure 4K). Collectively, GABA signaling promotes IL-17 expression during infection though mTORC1–EGR-2 signaling.
Microbiota-Derived GABA Mediates IL-17 Expression during ETEC Infection
Intestinal microbiota has critical roles in GABA production from glutamate (34, 35) and in regulation of intestinal IL-17 expression (36, 37). Our previous study has found that ETEC-infected piglets had a higher percentage of Lactococcus, compared to the controls (manuscript submitted). This result was supported by our current finding that ETEC-infected mice had higher numbers of L. lactis subsp. lactis, as compared with uninfected controls (Figure 5A). Within the intestinal microbiota, L. lactis subsp. lactis is one of the main GABA-producing strains (38, 39). Thus we hypothesized that the increased abundance of intestinal L. lactis subsp. lactis during ETEC infection is responsible for activated GABA–IL-17 signaling in the context of ETEC infection. Indeed, we found that piglets orally inoculated with L. lactis subsp. lactis (ATCC19435) has higher mRNA expression of IL-17 in the jejunum compared to the piglets without L. lactis subsp. lactis inoculation (Figure 5B). The GABA production activity of L. lactis subsp. lactis (ATCC19435) is about 210.4 µM (38, 39). Similar to the observation from piglets, L. lactis subsp. lactis inoculation in mice significantly promoted the expression of Il-17 in the jejunum compared to the mice without L. lactis subsp. lactis inoculation (Figure 5C). More importantly, inhibition of GABA signaling by CGP-35348 decreased the expression of Il-17 in the jejunum of mice with L. lactis subsp. lactis inoculation (Figure 5C). To further test this hypothesis, we treated mice with a mixture of antibiotics for 1 week to decrease the load of L. lactis subsp. lactis before ETEC infection. To increase the abundance of L. lactis subsp. lactis in antibiotics-treated mice, we orally inoculated the L. lactis subsp. lactis to antibiotics-treated mice. The antibiotics-treated mice had a lower numbers of L. lactis subsp. lactis than the mice without antibiotics treatment (Figure 5D). After ETEC infection, we found that antibiotics-treated mice had lower expression of Il-17 in the jejunum compared to the mice without antibiotics treatment (Figure 5E). Oral inoculation of L. lactis subsp. lactis before ETEC infection increased the numbers of L. lactis subsp. lactis in the content of jejunum and rescued the expression of Il-17 during ETEC infection (Figures 5D,E). Interestingly, in antibiotics-treated mice, higher dosage of GABA supplementation also rescued the expression of Il-17 in the jejunum during ETEC infection (Figure 5F). In germ-free mice, L. lactis subsp. lactis administration before ETEC infection also promoted expression of Il-17 (Figure 5G). Collectively, intestinal GABA-producing bacteria have vital importance in IL-17 expression during ETEC infection.
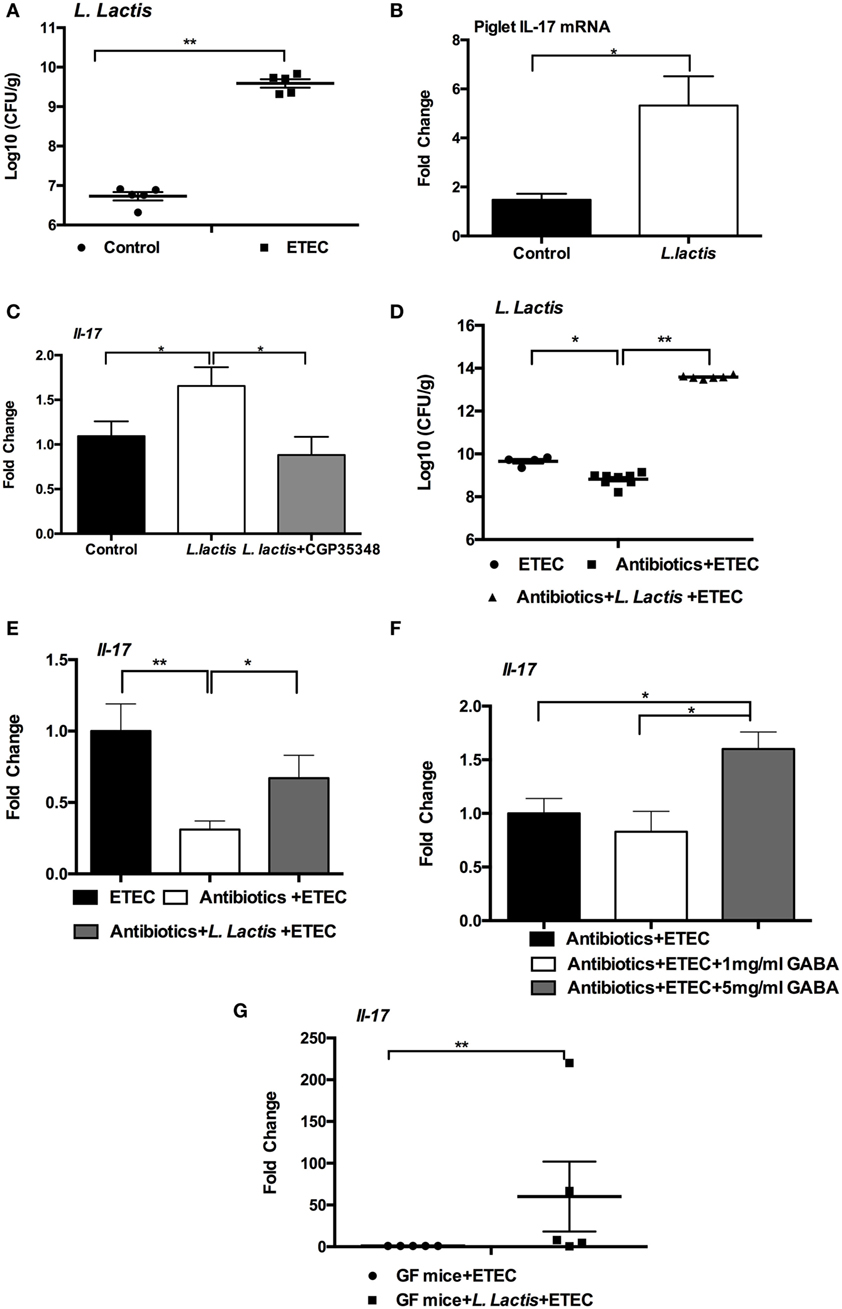
Figure 5. Intestinal microbiota affects interleukin-17 (IL-17) expression. (A) Bacterial counting of Lactococcus lactis subsp. lactis in enterotoxigenic Escherichia coli (ETEC)-infected mice and control mice. The jejunal contents were collect at 6-h post-ETEC infection for bacterial counting. (B) The mRNA expression of IL-17 in the jejunum was analyzed in piglets (n = 5). (C) The mRNA expression of IL-17 in the jejunum was analyzed in mice (n = 8). (D) Bacterial counting of L. lactis subsp. lactis in indicated models. (E) The mRNA expression of IL-17 in the jejunum was analyzed in indicated models. (F) The mRNA expression of IL-17 in the jejunum was analyzed in indicated models. (G) The mRNA expression of IL-17 in the mesenteric lymph node was analyzed in germ-free mice. Germ-free mice received L. lactis subsp. lactis or not at 1 week before ETEC infection. mRNA expression is relative to GF mice without L. lactis subsp. lactis administration (defined as 1; n = 5). Results shown are representative of two independent experiments with n = 5–8 (A,D,E) in each experiment, or of three independent experiments with n = 8 (F) in each experiment. *indicates a statistically significant difference between two groups (p < 0.05), while **indicates p < 0.01 [Mann–Whitney test (A,G); unpaired t test with Welch’s correction (B); one-way ANOVA (C–F)].
GABA–mTORC1 Signaling Mediates IL-17 Expression in Other Models
To validate the role of GABA–mTORC1 signaling in IL-17 expression in other intestinal infectious models, we infected mice with different ETEC strains and with Shiga-like toxin producing E. coli. In mouse model, E. coli W470, W197, W817, and W616 increased Il-17 expression in the jejunum at 6-h postinfection (Figure 6A). Pretreatment with rapamycin prevented expression of Il-17 in the jejunum caused by W470, W197, W817, and W616 at 6-h postinfection (Figure 6A). Similar to our previous W25K infection, bicuculline treatment reversed the increase in Il-17 expression caused by W470 in mouse jejunum (Figure 6B). In C. rodentium infected mice, the expression of Il-17 in the colon was higher than in control mice (Figure 6C). Interestingly, higher abundance of L. lactis subsp. lactis was also observed in the colon during C. rodentium infection (7.7 ± 0.03 log10 CFU/g in infected mice vs. 6.9 ± 0.08 log10 CFU/g in control mice). Rapamycin treatment lowered the colonic expression of Il-17 (Figure 6C), indicating that inhibition of mTORC1 pathway reduced the expression of Il-17 in the colon during C. rodentium infection. Furthermore, the inhibition of GABA signaling by semicarbazide, bicuculline, or CGP-35348 also lowered the expression of Il-17 in the colon in the context of C. rodentium infection (Figure 6C). 5-Fluorouracil (5-FU) is frequently used in cancer treatment and also induces intestinal inflammation (40, 41). 5-FU enhanced Il-17 expression in the jejunum at 6-h postinjection (Figure 6D). Semicarbazide or bicuculline treatment prevented the increase in Il-17 expression caused by 5-FU (Figure 6D). In summary, GABA–mTORC1 signaling also functions in intestinal IL-17 expression in other intestinal inflammatory models.
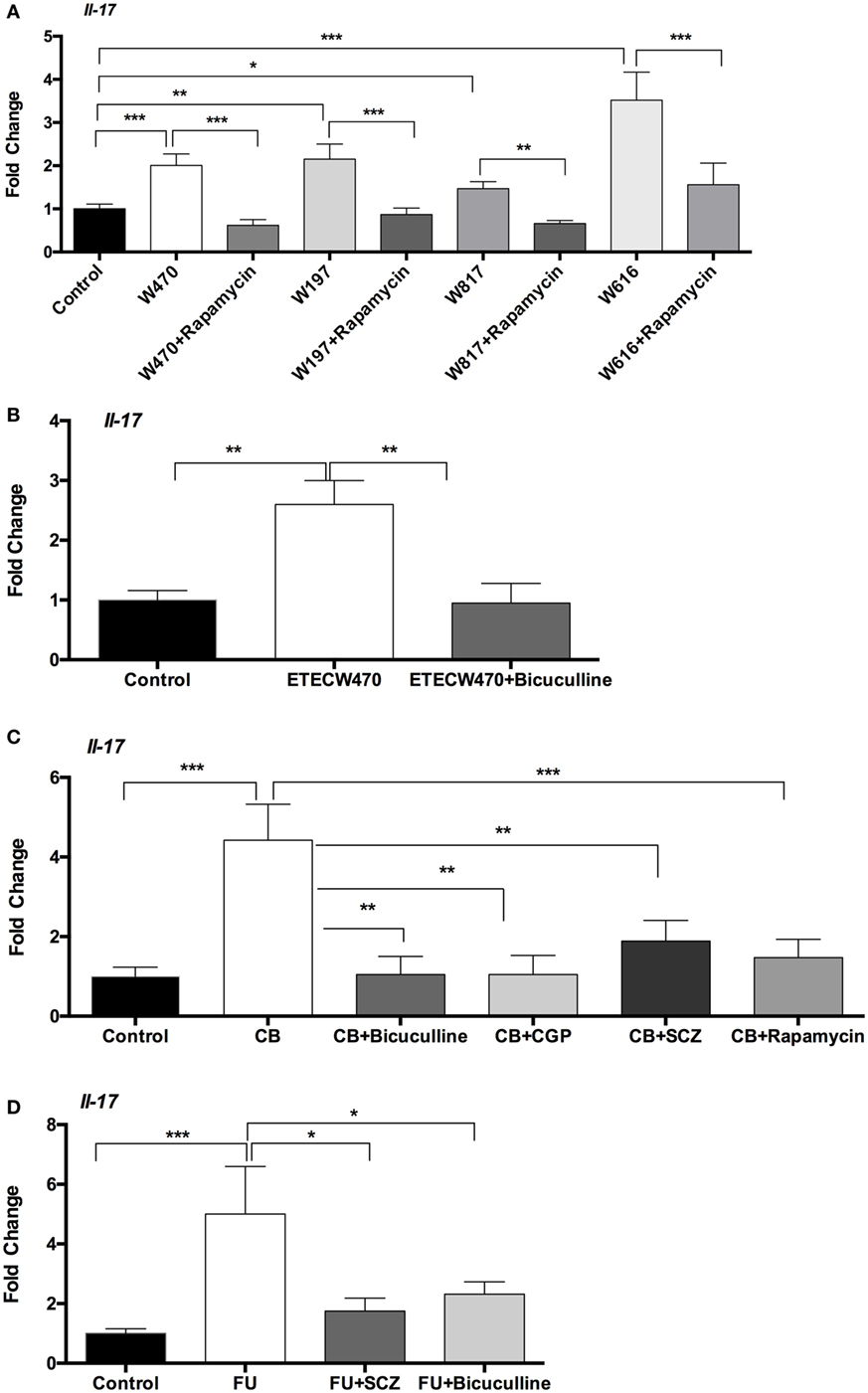
Figure 6. γ-Aminobutyric acid (GABA)-mechanistic target of rapamycin complex 1 (mTORC1) signaling mediates mRNA expression interleukin-17 (IL-17) in other models. (A) Rapamycin prevents mRNA expression of IL-17 induced by W470, W197, W817, or W616 (n = 10). (B) Bicuculline prevents mRNA expression of IL-17 induced by W470 (n = 10). (C) Inhibition of GABA–mTORC1 signaling inhibits mRNA expression of IL-17 in the colon during Citrobacter rodentium infection (n = 10). (D) Inhibition of GABA signaling inhibits the increase in mRNA expression of IL-17 induced by 5-fluorouracil (5-FU) (n = 10). *indicates a statistically significant difference between two groups (p < 0.05), while **indicates p < 0.01, and ***indicates p < 0.001 [one-way ANOVA (A–D)].
Discussion
Besides promoting local inflammation, emerging evidence shows that Th17 cells secrete IL-26, which kills extracellular bacteria through membrane-pore formation and triggers the production of interferon alpha through binding to bacterial DNA (42). Thus, Th17 response has a critical role in protection against extracellular bacterial infection. Similar to previous studies (15, 16), we observed that ETEC infection promotes intestinal IL-17 expression in piglet and mouse models. The increased expression of IL-17 during ETEC infection may come from adaptive cells (e.g., Th17 cells), or innate immune cells, such as innate lymphoid cells-3, γδ T cells, and NKT cells (11, 13). Unlike previous conclusion that increased IL-17 is mainly produced by γδ T cells at 5-h post-E. coli infection (25), this study indicates that a significant portion of the IL-17 comes from Th17 cells at 6-h post-ETEC infection. However, intestinal Th17 cell responses after bacterial infection (e.g., C. rodentium and E. coli O157) needs the epithelial adhesion of pathogens and is observed at 5–14 days postinfection (43, 44). Thus, the exact cellular source for increase IL-17 expression at 6-h post-ETEC infection needs further investigation. Indeed, enteric iTh17 has been observed at early time of Citrobacter and Salmonella infection (12).
Mechanistic target of rapamycin complex 1 is a conserved Ser/Thr protein kinase that functions as a sensor of growth factors, nutrients, and energy to promote anabolic cellular metabolism and functions as a master controller of cell growth (45). Recent studies have shown that mTORC1 signaling is crucially involved in Th17 cells differentiation and IL-17 expression (26). Inhibition of mTORC1 signaling with rapamycin reduces IL-17 expression in various inflammatory models (46–48). Similarly, we showed that mTORC1 signaling mediates the expression of IL-17 after intestinal ETEC infection, and this result is similar to the previous reports that mTORC1 signaling has a critical role in IL-17 expression (26). Likewise, another study has shown that rapamycin treatment inhibits the expression of IL-17 during Campylobacter jejuni infection (49). mTORC1–S6K1–EGR-2–GFI-1 axis is involved in IL-17 expression in ETEC infection. GFI-1, a downstream target of mTORC1–S6K1 axis (26), is a negative regulator of IL-17 expression and Th17 differentiation, and the downregulation of GFI-1 is important for IL-17 expression (26). However, the expression of IL-17 in the context of ETEC infection is independent of the S6K2 mediating nuclear translocation of RORγt, and HIF-1α pathway, which promotes IL-17 expression through: (a) transcriptional activation of RORγt in the cytoplasm, (b) collaboration with RORγt to promote IL-17 expression in the nucleus, through mechanisms involving p300 recruitment and histone acetylation, and (c) inhibition of Treg differentiation by targeting Foxp3 for ubiquitination and proteasomal degradation (26). Interestingly, ETEC infection reduces the abundance of HIF-1α. Although the underlying mechanisms are unknown, our results may indicate a complex regulatory role for mTORC1 in HIF-1α production, Th17 differentiation, and IL-17 expression.
We found that GABA signaling mediates intestinal IL-17 expression in ETEC infection through activation of mTORC1 signaling. In starvation conditions that normally inhibit mTORC1 signaling, the addition of GABA also increases S6 phosphorylation, suggesting that GABA signaling activates mTORC1 signaling even in starvation conditions (33). Recently, GABA has been suggested to exert a potential anti-inflammatory action by inhibiting major inflammatory events mediated by different immune cells. For example, increasing GABAergic activity ameliorates inflammation in experimental autoimmune encephalomyelitis (50). Oral GABA treatment ameliorates the inflammatory process both in non-obese diabetic mice (51) and in a mouse model of rheumatoid arthritis (52). Also, GABAA agonists are beneficial in experimental encephalomyelitis (50) and allergic asthma (53), and GABAB agonists exhibit a positive effect in dermatitis models (54). However, few investigations have addressed the possible action of GABA on intestinal inflammation during infection. Different from these previous reports (50–54), here we found that GABA promotes intestinal IL-17 expression. Similarly, dendritic cells increase GABA secretion in vitro after Toxoplasma gondii infection and exhibit a hyper-migratory phenotype because increased GABA activates GABAA receptor-mediated currents in T. gondii-infected dendritic cells (55). The inactivation of GABA signaling in T. gondii-infected dendritic cells by inhibition of GABA synthesis or transportation, or by blockade of GABAA receptor, results in impaired function of dendritic cells in vitro, including transmigration capacity, motility, and chemotactic response to CCL19 (55).
The gut microbiota affects numerous biological functions (56, 57) and is linked to the pathogenesis of various diseases, such as obesity (58), cancer (59), and liver cirrhosis (60). The influence of the gut microbiota on host physiological functions and the pathogenesis of disease may result from the activity of the microbiome and its metabolic products (56). Gut microbes produce various biologically active molecules, such as GABA, short-chain fatty acids, and biogenic amines (such as histamine), to actively regulate host health and disease states (56). For example, Lactobacillus rhamnosus JB-1 converts glutamate into GABA and alters GABA signaling in the brain, resulting in a reduction in stress-induced corticosterone, and in anxiety- and depression-related behavior (61). We found that ETEC infection induces a dysbiosis of the intestinal microbiota, especially promoting a higher percentage of GABA-producing L. lactis in the jejunum. Intestinal GABA-producing L. lactis has critical role in IL-17 expression during infection. However, the L. lactis used for GABA production in this study is not an inhabitant of the GI tract of either mice or piglets, thus it is interesting to test the function of GABA-producing L. lactis isolated from pigs. Also, it is interesting to explore the mechanism responsible for increased abundance of L. lactis during ETEC infection. Besides GABA, whether other products from L. lactis, or other members of intestinal microbiota are also involved in the pathogenesis of ETEC infection remain to know.
In conclusion, we propose the following scenario for how intestinal pathogens (i.e., ETEC) mediates intestinal IL-17 expression. During ETEC infection, ETEC induces the dysbiosis of gut microbiota, increasing GABA-producing L. lactis subsp. lactis. The increased content of GABA promotes IL-17 expression during infection through mTORC1–S6K1–EGR-2–GFI-1 pathway. This discovery has great potential to manipulate intestinal GABA–mTORC1 signaling and intestinal GABA-producing bacteria to treat intestinal chronic inflammatory diseases involving IL-17 expression. Our findings further support the notion that functional amino acids (e.g., glutamate and glutamine) play an important role in the immunity and health of animals (62–64).
Author Contributions
WR, PH, TL, BT, and YY designed the experiments; WR, JY, HX, and SC conducted the experiments; GL, NL, and YP helped with animal experiments; WR and JY analyzed the data; ZY provided the TCR delta knockout mice; BZ, WL, and HW provided the germ-free mice; WR wrote the manuscript; GW, PH, and YY revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by Key Programs of frontier scientific research of the Chinese Academy of Sciences (QYZDY-SSW-SMC008), the National Key Basic Research Program of China (2013CB127302, 2013CB127301, 2013CB532406), the National Natural Science Foundation of China (31330075, 31372326, 31110103909, 31272463, 81370906), the earmarked fund for China Agriculture Research System (Beef/Yak, CARS-38), Ministry of Science and Technology grants (2014AA022209) and Texas A&M AgriLife Research H-8200. The sponsor has no role in the study design, data collection, analysis, and interpretation of data.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00685/full#supplementary-material.
Abbreviations
EGR-2, early growth response protein 2; ETEC, enterotoxigenic Escherichia coli; Foxp3, forkhead box P3; GABA, γ-aminobutyric acid; GFI-1, growth factor independent 1; HIF-1α, hypoxia-inducible factor 1 α; IL-17, interleukin-17; mTORC1, mechanistic target of rapamycin complex 1; PCNA, proliferating cell nuclear antigen; RORγt, RAR-related orphan receptor gamma t; S6K, ribosomal protein S6 kinase.
References
1. Endt K, Stecher B, Chaffron S, Slack E, Tchitchek N, Benecke A, et al. The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog (2010) 6(9):e1001097. doi:10.1371/journal.ppat.1001097
2. Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest (2014) 124(10):4182–9. doi:10.1172/JCI72336
3. Britton RA, Young VB. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology (2014) 146(6):1547–53. doi:10.1053/j.gastro.2014.01.059
4. Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun (2014) 5:3114. doi:10.1038/ncomms4114
5. Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe (2014) 15(3):374–81. doi:10.1016/j.chom.2014.02.006
6. Mittrucker HW, Seidel D, Bland PW, Zarzycka A, Kaufmann SH, Visekruna A, et al. Lack of microbiota reduces innate responses and enhances adaptive immunity against Listeria monocytogenes infection. Eur J Immunol (2014) 44(6):1710–5. doi:10.1002/eji.201343927
7. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
8. Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol (2008) 38(10):2636–49. doi:10.1002/eji.200838535
9. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28(4):454–67. doi:10.1016/j.immuni.2008.03.004
10. Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest (2014) 124(5):2234–45. doi:10.1172/JCI73411
11. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10(7):479–89. doi:10.1038/nri2800
12. Geddes K, Rubino SJ, Magalhaes JG, Streutker C, Le Bourhis L, Cho JH, et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med (2011) 17(7):837–44. doi:10.1038/nm.2391
13. Kim JS, Jordan MS. Diversity of IL-17-producing T lymphocytes. Cell Mol Life Sci (2013) 70(13):2271–90. doi:10.1007/s00018-012-1163-6
14. Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect (2010) 12(2):89–98. doi:10.1016/j.micinf.2009.10.002
15. Loos M, Geens M, Schauvliege S, Gasthuys F, van der Meulen J, Dubreuil JD, et al. Role of heat-stable enterotoxins in the induction of early immune responses in piglets after infection with enterotoxigenic Escherichia coli. PLoS One (2012) 7(7):e41041. doi:10.1371/journal.pone.0041041
16. Zhu YH, Li XQ, Zhang W, Zhou D, Liu HY, Wang JF. Dose-dependent effects of Lactobacillus rhamnosus on serum interleukin-17 production and intestinal T-cell responses in pigs challenged with Escherichia coli. Appl Environ Microbiol (2014) 80(5):1787–98. doi:10.1128/AEM.03668-13
17. Ren W, Liu G, Yin J, Chen S, Li T, Kong X, et al. Draft genome sequence of enterotoxigenic Escherichia coli strain W25K. Genome Announc (2014) 2(3). doi:10.1128/genomeA.00593-14
18. Ren W, Yu R, Liu G, Li N, Peng Y, Wu M, et al. DNA vaccine encoding the major virulence factors of Shiga toxin type 2e (Stx2e)-expressing Escherichia coli induces protection in mice. Vaccine (2013) 31(2):367–72. doi:10.1016/j.vaccine.2012.10.107
19. Ren W, Yin J, Gao W, Chen S, Duan J, Liu G, et al. Metabolomics study of metabolic variations in enterotoxigenic Escherichia coli-infected piglets. RSC Adv (2015) 5(73):59550–5. doi:10.1039/C5RA09513A10.1039/c5ra09513a
20. Ren W, Yin J, Chen S, Duan J, Liu G, Li T, et al. Proteome analysis for the global proteins in the jejunum tissues of enterotoxigenic Escherichia coli-infected piglets. Sci Rep (2016) 6:25640. doi:10.1038/srep25640
21. Wang Y, Feng D, Liu G, Luo Q, Xu Y, Lin S, et al. Gamma-aminobutyric acid transporter 1 negatively regulates T cell-mediated immune responses and ameliorates autoimmune inflammation in the CNS. J Immunol (2008) 181(12):8226–36. doi:10.4049/jimmunol.181.12.8226
22. Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol (2011) 187(2):733–47. doi:10.4049/jimmunol.1002701
23. Agnello D, Denimal D, Lavaux A, Blondeau-Germe L, Lu B, Gerard NP, et al. Intrarectal immunization and IgA antibody-secreting cell homing to the small intestine. J Immunol (2013) 190(9):4836–47. doi:10.4049/jimmunol.1202979
24. Wu M, Xiao H, Liu G, Chen S, Tan B, Ren W, et al. Glutamine promotes intestinal SIgA secretion through intestinal microbiota and IL-13. Mol Nutr Food Res (2016) 60(7):1637–48. doi:10.1002/mnfr.201600026
25. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol (2007) 178(7):4466–72. doi:10.4049/jimmunol.178.7.4466
26. Ren W, Yin J, Duan J, Liu G, Tan B, Yang G, et al. mTORC1 signaling and IL-17 expression: defining pathways and possible therapeutic targets. Eur J Immunol (2015) 46(2):291–9. doi:10.1002/eji.201545886
27. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep (2012) 1(4):360–73. doi:10.1016/j.celrep.2012.02.007
28. Tsalikis J, Croitoru DO, Philpott DJ, Girardin SE. Nutrient sensing and metabolic stress pathways in innate immunity. Cell Microbiol (2013) 15(10):1632–41. doi:10.1111/cmi.12165
29. Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev (2014) 40(8):980–9. doi:10.1016/j.ctrv.2014.06.006
30. Bunpo P, Cundiff JK, Reinert RB, Wek RC, Aldrich CJ, Anthony TG. The eIF2 kinase GCN2 is essential for the murine immune system to adapt to amino acid deprivation by asparaginase. J Nutr (2010) 140(11):2020–7. doi:10.3945/jn.110.129197
31. Thanapreedawat P, Ohsumi M, Hayase K, Yoshizawa F, Yokogoshi H. Influence of GABA on brain protein synthesis mediated by the mammalian target on the rapamycin pathway. Biosci Biotechnol Biochem (2013) 77(3):660–2. doi:10.1271/bbb.120808
32. Workman ER, Niere F, Raab-Graham KF. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology (2013) 73:192–203. doi:10.1016/j.neuropharm.2013.05.037
33. Lakhani R, Vogel KR, Till A, Liu J, Burnett SF, Gibson KM, et al. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med (2014) 6(4):551–66. doi:10.1002/emmm.201303356
34. Matsumoto M, Kibe R, Ooga T, Aiba Y, Kurihara S, Sawaki E, et al. Impact of intestinal microbiota on intestinal luminal metabolome. Sci Rep (2012) 2:233. doi:10.1038/srep00233
35. Hemarajata P, Versalovic J. Effects of probiotics on gut microbiota: mechanisms of intestinal immunomodulation and neuromodulation. Therap Adv Gastroenterol (2013) 6(1):39–51. doi:10.1177/1756283X12459294
36. Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity (2009) 31(4):677–89. doi:10.1016/j.immuni.2009.08.020
37. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
38. Nomura M, Kimoto H, Someya Y, Suzuki I. Novel characteristic for distinguishing Lactococcus lactis subsp. lactis from subsp. cremoris. Int J Syst Bacteriol (1999) 49(Pt 1):163–6. doi:10.1099/00207713-49-1-163
39. Siragusa S, De Angelis M, Di Cagno R, Rizzello CG, Coda R, Gobbetti M. Synthesis of gamma-aminobutyric acid by lactic acid bacteria isolated from a variety of Italian cheeses. Appl Environ Microbiol (2007) 73(22):7283–90. doi:10.1128/AEM.01064-07
40. Liu Z, Xi J, Schroder S, Wang W, Xie T, Wang Z, et al. Chimonanthus nitens var. salicifolius aqueous extract protects against 5-fluorouracil induced gastrointestinal mucositis in a mouse model. Evid Based Complement Alternat Med (2013) 2013:789263. doi:10.1155/2013/789263
41. Justino PF, Melo LF, Nogueira AF, Costa JV, Silva LM, Santos CM, et al. Treatment with Saccharomyces boulardii reduces the inflammation and dysfunction of the gastrointestinal tract in 5-fluorouracil-induced intestinal mucositis in mice. Br J Nutr (2014) 111(9):1611–21. doi:10.1017/S0007114513004248
42. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol (2015) 16(9):970–9. doi:10.1038/ni.3211
43. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell (2015) 163(2):367–80. doi:10.1016/j.cell.2015.08.058
44. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell (2015) 163(2):381–93. doi:10.1016/j.cell.2015.08.061
45. Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol (2014) 36:79–90. doi:10.1016/j.semcdb.2014.09.011
46. Yoshizaki A, Yanaba K, Yoshizaki A, Iwata Y, Komura K, Ogawa F, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum (2010) 62(8):2476–87. doi:10.1002/art.27498
47. Yamaki K, Yoshino S. Preventive and therapeutic effects of rapamycin, a mammalian target of rapamycin inhibitor, on food allergy in mice. Allergy (2012) 67(10):1259–70. doi:10.1111/all.12000
48. Yurchenko E, Shio MT, Huang TC, Da Silva Martins M, Szyf M, Levings MK, et al. Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS One (2012) 7(4):e35572. doi:10.1371/journal.pone.0035572
49. Sun X, Threadgill D, Jobin C. Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology (2012) 142(1):86–95.e5. doi:10.1053/j.gastro.2011.09.042
50. Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, et al. Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci U S A (2010) 107(6):2580–5. doi:10.1073/pnas.0915139107
51. Tian J, Lu Y, Zhang H, Chau CH, Dang HN, Kaufman DL. Gamma-aminobutyric acid inhibits T cell autoimmunity and the development of inflammatory responses in a mouse type 1 diabetes model. J Immunol (2004) 173(8):5298–304. doi:10.4049/jimmunol.173.8.5298
52. Tian J, Yong J, Dang H, Kaufman DL. Oral GABA treatment downregulates inflammatory responses in a mouse model of rheumatoid arthritis. Autoimmunity (2011) 44(6):465–70. doi:10.3109/08916934.2011.571223
53. Munroe ME, Businga TR, Kline JN, Bishop GA. Anti-inflammatory effects of the neurotransmitter agonist Honokiol in a mouse model of allergic asthma. J Immunol (2010) 185(9):5586–97. doi:10.4049/jimmunol.1000630
54. Duthey B, Hubner A, Diehl S, Boehncke S, Pfeffer J, Boehncke WH. Anti-inflammatory effects of the GABA(B) receptor agonist baclofen in allergic contact dermatitis. Exp Dermatol (2010) 19(7):661–6. doi:10.1111/j.1600-0625.2010.01076.x
55. Fuks JM, Arrighi RB, Weidner JM, Kumar Mendu S, Jin Z, Wallin RP, et al. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog (2012) 8(12):e1003051. doi:10.1371/journal.ppat.1003051
56. Lee WJ, Hase K. Gut microbiota-generated metabolites in animal health and disease. Nat Chem Biol (2014) 10(6):416–24. doi:10.1038/nchembio.1535
57. Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature (2014) 510(7505):417–21. doi:10.1038/nature13421
58. Anhe FF, Roy D, Pilon G, Dudonne S, Matamoros S, Varin TV, et al. A polyphenol-rich cranberry extract protects from diet-induced obesity, insulin resistance and intestinal inflammation in association with increased Akkermansia spp. population in the gut microbiota of mice. Gut (2015) 64(6):872–83. doi:10.1136/gutjnl-2014-307142
59. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol (2014) 12(10):661–72. doi:10.1038/nrmicro3344
60. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature (2014) 513(7516):59–64. doi:10.1038/nature13568
61. Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A (2011) 108(38):16050–5. doi:10.1073/pnas.1102999108
62. Wu G. Functional amino acids in growth, reproduction and health. Adv Nutr (2010) 1:31–7. doi:10.3945/an.110.1008
63. Ren W, Wang K, Yin J, Chen S, Liu G, Tan B, et al. Glutamine induced secretion of intestinal secretory immunoglobulin a: a mechanistic perspective. Front. Immunol (2016) 7:503. doi:10.3389/fimmu.2016.00503
Keywords: intestinal microbiota, GABA, IL-17, mTORC1, ETEC, Lactococcus lactis
Citation: Ren W, Yin J, Xiao H, Chen S, Liu G, Tan B, Li N, Peng Y, Li T, Zeng B, Li W, Wei H, Yin Z, Wu G, Hardwidge PR and Yin Y (2017) Intestinal Microbiota-Derived GABA Mediates Interleukin-17 Expression during Enterotoxigenic Escherichia coli Infection. Front. Immunol. 7:685. doi: 10.3389/fimmu.2016.00685
Received: 16 August 2016; Accepted: 22 December 2016;
Published: 16 January 2017
Edited by:
Laurence Macia, University of Sydney, AustraliaReviewed by:
Douglas W. Bigwood, DBE Technologies, USAIan Antheni Myles, National Institutes of Health, USA
Copyright: © 2017 Ren, Yin, Xiao, Chen, Liu, Tan, Li, Peng, Li, Zeng, Li, Wei, Yin, Wu, Hardwidge and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bie Tan, YmlldGFuQGlzYS5hYy5jbg==;
Tiejun Li, dGpsaUBpc2EuYWMuY24=;
Yulong Yin, eWlueXVsb25nQGlzYS5hYy5jbg==
†These authors have contributed equally to this work.