 Ping Ke
Ping Ke Bo-Zong Shao
Bo-Zong Shao Zhe-Qi Xu
Zhe-Qi Xu Xiong-Wen Chen*
Xiong-Wen Chen* Chong Liu
Chong Liu- Department of Pharmacology, Second Military Medical University, Shanghai, China
Intestinal mucosal barrier, mainly composed of the intestinal mucus layer and the epithelium, plays a critical role in nutrient absorption as well as protection from pathogenic microorganisms. It is widely acknowledged that the damage of intestinal mucosal barrier or the disturbance of microorganism balance in the intestinal tract contributes greatly to the pathogenesis and progression of inflammatory bowel disease (IBD), which mainly includes Crohn’s disease and ulcerative colitis. Autophagy is an evolutionarily conserved catabolic process that involves degradation of protein aggregates and damaged organelles for recycling. The roles of autophagy in the pathogenesis and progression of IBD have been increasingly studied. This present review mainly describes the roles of autophagy of Paneth cells, macrophages, and goblet cells in IBD, and finally, several potential therapeutic strategies for IBD taking advantage of autophagy.
Introduction
The intestinal tract mainly functions as the most important organ digesting and absorbing food and nutrients taken orally. Besides those obvious functions, the gut is also regarded as one of the largest immune organs in an organism since the gut lumen always harbors a great number of microorganisms. As a result, the host has to maintain the peaceful coexistence with this diverse microbial community and trigger the inflammatory and immune reaction for the detection and elimination of the pathogenic microorganisms (1, 2). During this process, an efficient intestinal mucosal barrier is critical for the maintenance of microbial homeostasis and fighting against pathogenic microorganisms, providing the first line of defense. In general, the intestinal mucosal barrier is composed of two layers, including the intestinal mucus layer and the epithelium, in combination with the equipment of diverse specific and unspecific protective mechanisms which collectively build up an effective intestinal mucosal barrier (3). However, the disturbance of microbial homeostasis and damage of intestinal mucosal barrier largely trigger the inflammatory responses, immune reaction, accumulation of reactive oxygen species (ROS), and mitochondrial dysfunction in the intestinal wall, finally leading to the pathogenesis and progression of inflammatory bowel disease (IBD). As a result, inhibiting the over-activation of those self-defensive processes may serve as a potential and effective strategy for the treatment of IBD. Autophagy is a popular self-protective mechanism, the function of which mainly relies on lysosome. There is much evidence to suggest that autophagy could effectively attenuate the over-triggering of several self-defensive pathways such as inflammatory reaction and immune responses (4–6). Based on the statement, autophagy has been increasingly studied by researchers for the development of novel and effective therapeutic strategies against inflammation- or immune-related disorders, including IBD. Here, in this review, we discuss the mechanisms underlying the pathogenesis of IBD, the signaling pathways of autophagy, as well as the roles of autophagy in three important cells, namely, Paneth cells, macrophages, and goblet cells, in IBD, and, last but not least, several pharmacological interventions of autophagy in the treatment of IBD.
Pathogenesis of IBD
We have mentioned above that the inflammatory and immune responses are triggered for the defense in the intestine. However, the over-triggered self-protective inflammatory and immune responses can lead to the damage of intestine itself as well as the intestinal mucosal barrier, contributing to the pathogenesis and progression of chronic inflammatory disorders, such as IBD.
Although the exact mechanisms underlying IBD have not yet been clarified, evidence has shown that the environment, genetics, and interactions between host defense and commensal microbiota are the main causes leading to the development of IBD (7). Smoking is an important environmental risk factor related to Crohn’s disease (CD) (8). Patients suffering from CD are responding badly to treatments and present more severe symptoms (9, 10). Another environmental factor attributing to IBD is air pollution (11). The particulate matter, ozone, or nitrous oxides in the air intrude into intestinal tract through food and water, which may cause an increased intestinal epithelial permeability, or induce pro-inflammatory response, thus leading to the occurrence of IBD (12, 13). In addition, it was noticed that antibiotics use was a potential factor of IBD onset as antibiotics could alter the amount and composition of the intestinal microflora, which ended up triggering abnormal inflammatory responses (14). The genetics of IBD were recognized by genome-wide association studies (GWAS) showing that people harboring variants in innate and adaptive immunity-related genes were more susceptible to both ulcerative colitis (UC) and CD, such as nucleotide-binding oligomerization domain-containing protein 2 (Nod2), immunity-related GTPase family protein M (IRGM), autophagy-related protein 16 like protein 1 (Atg16l1), interleukin 12B (IL-12B), Drosophila mothers against decapentaplegic protein 3 (SMAD3), and others (15–17). These susceptibility genes lead to dysregulation of the intestinal mucosal inflammatory and immune system that result in excessive immunologic responses to normal flora or impaired ability to clearance invasive bacteria (18, 19). Evidence has shown that virus-plus-susceptibility gene interactions induced colitis similar to CD (19). The interactions between the host and its diverse microbial community are complex and crucial in maintaining intestinal homeostasis. The intestinal defense system consists of the mucus layer, epithelium, and cells related to the innate immune system. In normal conditions, the mucus layer which contains large numbers of antimicrobial substance and mucins works together with the epithelium to separate microbes from the host, and if any pathogen intrudes through the epithelium, the dentritic cells and macrophages of the immune system in intestine will recognize it and generate an appropriate inflammatory or immune response by producing cytokines and antimicrobial agents (20). However, defects in mucin secretion (21) or epithelial integrity (22) and malfunction of the immune response to restrict pathogenic microbes alone or together contribute to the pathogenesis of IBD. The last factor for the pathogenesis and progression of IBD that deserves to be mentioned here is the adaptive immune response. It is generally acknowledged that increased pro-inflammatory cytokines produced by the T-helper cells or decreased anti-inflammatory cytokines induced by ineffective regulatory T-cells contribute to the pathogenesis of IBD (23, 24). Nevertheless, since the pathogenesis of IBD is complex and the exact underlying mechanisms still remain unclear, no effective therapeutic strategies have been developed for the treatment of IBD.
Autophagy and Its Functions
Autophagy is an evolutionarily conserved catabolic process that involves degradation of protein aggregates and damaged organelles for recycling (25). Its function of degrading and recycling mainly relies on lysosome. In general, autophagy is defined into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy (26, 27). Microautophagy belongs to a non-selective degradative mechanism through invagination of the lysosomal/vacuolar membranes for the engulfment of cytoplasmic components (28). Chaperone-mediated autophagy is the only autophagy style that allows the degradation of organelles and proteins relying on the presentation of chaperones. It has been reported previously that chaperone-mediated autophagy largely demands the presence of a targeting motifin which is the substrate protein, a set of cytosolic and lysosomal chaperones, and a receptor protein at the lysosomal membrane for the transportation into lysosome assisted by chaperone located in the lysosomal lumen (29). Macroautophagy is a catabolic process, during which intracellular components or invasive bacteria are surrounded by double-membrane-bound structures, widely acknowledged as autophagosome. After the confusion of autophagosome and lysosome, the degradative autolysosome with single-layer membrane is formed for the digestion of ingredients by lysozymes (30, 31). Since macroautophagy is, so far, the best-studied autophagy process, this review will mainly discuss the functions and roles of macroautophagy in IBD (hereafter referred to as “autophagy”).
In general, the induction of autophagy process is mainly involved in two steps. In the first step, the cup-shaped double-layer phagophores form in the cytoplasm, followed by the subsequent formation of spherical double-membraned autophagosomes that enclose misfolded proteins or damaged organelles for degradation. Autophagosomes are regarded to be shaped from the nucleation and membrane expansion of phagophores. In the second step, autophagosomes dispose of “coat proteins” named autophagy-related light chain 3 (LC3) on their surface followed by the integration with lysosomes for the formation of the single lipid layer membrane-surrounded autolysosomes, which are the functional units for degradation and digestion (32). The whole process is participated by more than 30 kinds of autophagy-related genes (Atgs) and autophagy-related proteins (32, 33).
So far, several signaling pathways have been uncovered for the involvement of autophagy process. As recently reviewed by us (32), there are two major signaling pathways participating in the induction and regulation of autophagy process, including the inhibitory Class I PI3K–mTOR signaling pathway and the inductive Class III PI3K–Beclin-1 signaling pathway. The inhibitory pathway is often activated by nutrient sufficiency or several growth factors stimulation. In this pathway, the Class I PI3K–mTOR signaling is largely activated by the phosphorylation of Akt pathway and formation of mTOR complex 1, a major anti-autophagy complex, which in turn induces the inhibition of Atg1 (ULK1) to prevent the initiation of autophagosome formation. The inductive signaling pathway often occurs in the opposite situations like nutrient insufficiency or stimulation of the inflammatory or ROS stress. In this process, the Class III PI3K–Beclin-1 complex is initially formed, which in turn promotes the assembly of the Atg12–Atg5–Atg16L complex and the Atg8/LC3. The triggering of this signaling pathway largely induces the formation of autophagosomes and autophagy process.
In the fundamental studies on autophagy, the application of autophagy inhibitors is commonly conducted as an effective strategy in the exploration of autophagy mechanisms and discussion on roles of autophagy in diseases. Traditionally, chemical inhibitors of autophagy were most widely used. According to the latest guidelines for the use and interpretation of assays for monitoring autophagy published in 2016, the chemical inhibitors of autophagy can be mainly divided into two categories based on the stages of autophagy blockade (34). The first one, including 3-methyladenine (3-MA), LY294002, and wortmannin, belongs to sequestration inhibitors, relying on the inhibition of PI3Ks as well as class III PtdIns3Ks, thus leading to the blocking of the formation of autophagosomes. The second one, including vinblastine, leupeptin, and bafilomycin A1, is post-sequestration inhibitors, which leads to the accumulation of sequestered materials in either autophagosomes or autolysosomes or both. However, researchers are increasingly aware that most chemical inhibitors are not entirely specific and show the dose- and time-dependent effects. For example, it was reported that in a certain condition for the application of 3-MA, autophagy might be promoted because of the inhibition of the class I enzyme (35). Recently, the specific loss-of-function Atg mutants are considered as the more effective approach for the blockade of autophagy process, but they may lead to the autophagy-independent effects and also be dispensable of autophagy (34). Consequently, more efforts must be made in order to seek the more effective and specific autophagy inhibitors.
So far, autophagy has been reported to be closely related with apoptosis, inflammatory response, immune reaction, ROS stress, and mitochondrial dysfunction, functioning as effective regulator of the occurrence and extent of those processes (31, 36–38). As a result, it is certain that autophagy is a vital factor in the pathogenesis and regulation of various kinds of inflammation- and immune-related diseases, serving as a potential and effective target for the intervention of those diseases.
The Roles of Autophagy in IBD
The roles of autophagy in the pathogenesis and progression of IBD has been increasingly studied by researchers recently. For example, GWAS have reported several Atgs like Atg16l1 and IRGM contributes to the susceptibility of IBD, suggesting that autophagy possibly mediated the pathophysiology of IBD (17, 39, 40). In fact, autophagy affects the pathogenesis of IBD in various pathways, one of which is to regulate the clearance of invading pathogens. When the host cells were infected by bacteria, cytoplasmic vesicles engulfed these pathogens to form autophagosome, thus confining them obtaining nutrients and promoting acidification of surroundings. The enhancement of autophagy promotes the integration of autophagosome and lysosome, leading to the degradation of pathogenic organisms (41–43). In addition, autophagy protects cells against the damage of bacterial toxins, thus promoting cell survival such as macrophages, neutrophils, and intestinal epithelial cells (44). It has also been reported that autophagy augments adaptive immune response. After degraded by autolysosome, antigens are presented to the major histocompatibility complex class II molecules (MHC II), and then recognized by T cells to prime the adaptive immune response. This process can be promoted by autophagy (45–47). Evidence has shown that impaired autophagy disturbs the function of intestinal epithelial cells and influences the innate and adaptive immune responses, ROS production, and endoplasmic reticulum (ER) stress, leading to abnormal inflammatory reaction, and ultimately promoting the occurrence and development of IBD (7, 48–50).
Moreover, it should be mentioned there exists the crosstalk between autophagy and the damage-associated molecular pattern molecules (DAMPs) release and degradation in IBD. DAMPs are defined as several kinds of endogenous molecules including the S100A calgranulins, chromatin-associated high-mobility group box 1 (HMGB1), heat shock proteins, interleukin (IL)-1 family members, histones, and adenosine triphosphate (ATP), DNA, RNA, uric acid, hyaluronan, and heparin sulfate which are released by dead, dying, injured, or stressed cells (51). Previous studies reported that high levels of DAMPs were detected in serum, fecal or mucosa of IBD patients as well as mice models (52–55). In addition, it was demonstrated that the regulation of IBD progression by DAMPs was majorly through several approaches: (1) affecting epithelial barrier function; (2) binding with pattern-recognition receptors (PRRs) to exert directly pro-inflammatory effect; and (3) assisting in antigen-presenting cells to regulate T cells function (56). Since DAMPs are highly relevant to IBD, it is extremely essential to monitor the release pattern of these molecules. It has been demonstrated that the levels of DAMPs can be decreased by autophagy through the promotion of DAMPs degradation (57–59). However, it was also noted that under certain conditions, such as starvation or the stimulation of cytotoxic drugs, the induced autophagy could substantially lead to the release of DAMPs, including HMGB1, ATP, and IL1B (60–62). Consequently, to ultimately take advantage of the inhibitory roles of autophagy on DAMPs, effective methods are demanded to get rid of the promoting effect of autophagy on the release of DAMPs.
The following parts of this paper will mainly focus on the influence of autophagy in three important kinds of cells, which are highly related to intestinal self-defense and inflammatory and immune reaction, namely Paneth cells, macrophages, and goblet cells, on IBD, and finally, several potential therapeutic strategies for the treatment of IBD taking advantage of autophagy.
Paneth Cell, IBD, and Autophagy
Paneth Cell and IBD
As mentioned above, the epithelium together with mucus layer forms a physical barrier against the invading pathogens, including bacteria, fungi, virus, and harmful antigens in food, while tolerating beneficial microbes, thus maintaining the intestinal homeostasis. Specifically speaking, intestinal mucus layer, a layer of sticky gel covering the surface of mucosa, is composed of proteins, lipids, and carbohydrates as well as the large amount of water (63, 64). In the colon or large intestine, the mucus layer is divided into two distinct sectors, including the inner and outer layers. The inner layer is densely and firmly attached to the epithelium, while the outer layer is relatively more removable (3, 65). Among all of the components of mucus layer, various kinds of antimicrobial peptides (AMPs) are the most important functional peptides, with the defensins and the cathelicidins as the two best featuring families of AMPs, protecting the intestinal against microorganisms (66). The epithelium is majorly composed of several kinds of intestinal epithelial cells, including absorptive enterocytes, goblet cells, enteroendocrine cells, and Paneth cells (67). The absorptive enterocytes are the most abundant cell type in the epithelium, since they are rich in intestinal villi (68). Goblet cells and enteroendocrine cells mainly functions in secretion and restoration of the intestinal tissue (2, 68). Paneth cells, named after the Austrian physiologist Joseph Paneth, are the target cells in this paper for discussion, since they contains various kinds of secretary granules with antimicrobial active substances like lysozyme and α-definsins, highly involved in the defensive inflammatory and immune response in the intestine as well as the pathogenesis and progression of chronic intestinal diseases (3, 69). A recent study demonstrated that Paneth cells were a site of origin for intestinal inflammation, which are situated in the base of crypts of Lieberkühn, harboring a large number of ER and Golgi apparatus. Studies conducted on them revealed their vital role in directing the balance between homeostasis and inflammation through releasing AMPs and peptides including lysozyme, lipopolysaccharide (LPS)-binding protein, matrix metalloproteinase-7, phospholipase A2, phospholipase B, and IgA, as well as inflammatory cytokines like transforming growth factor β1 (TGF-β1), tumor necrosis factor α (TNF-α), and prostaglandin E2 (70–72). These AMPs could regulate the composition and quantity of microbes that colonize in the intestinal lumen and clear the intracellular pathogens.
The Roles of Paneth Cell Autophagy in IBD
In recent years, researchers discovered that autophagy and the autophagy gene had a specific role in the biology and function of Paneth cells. Cadwell et al. revealed that Atg16l1-, Atg5-, and Atg7-deficient Paneth cells exhibited few granules and decreased amounts of antimicrobial proteins and peptides inside. These cells also defected in granule exocytosis pathway through which cytoplasmic granules containing AMPs and other proteins are secreted to the intestinal lumen. In addition, Atg16l1-deficient Paneth cells presented increased expression of lipoprotein lipase, apolipoprotein A-IV, adiponectin, leptin, adiponectin, resistin-like α, complement factor D (adipsin), and haptoglobin. Many of these genes were directly implicated in inflammation, and two of these transcripts, namely leptin and adiponectin, increased in CD patients. Likewise, CD patients who carried Atg16l1 risk allele showed morphology and granule abnormalities of Paneth cells similar to those observed in autophagy-gene-deficient mice and expressed increased levels of leptin protein (73–75). Different from decreased amounts of granules in Atg7-deficient Paneth cells reported by Cadwell et al., Wittkopf et al. found out that Atg7 deficiency induced increased number and smaller size of granules in Paneth cells. Despite the lack of Paneth cell biology, Atg7 deficiency did not lead to altered response toward dextran sodium sulfate (DSS)-induced colitis, suggesting that autophagy defects in intestinal epithelial cells alone was not potent enough to increase susceptibility to enteritis (76). This conclusion was further supported by Cadwell et al. who discovered that Atg16l1 mutation in Paneth cells alone does not induce more severe injury in colon of DSS-induced colitis but a specific virus-plus-Atg16l1 variant led to intestinal lesion in mice through secreting high levels of TNF-α and interferon-γ (IFN-γ) (19). Furthermore, global knockout of Atg4b caused alterations in Paneth cell, including less number and small size of granules and decreased lysozyme, and Atg4b-deficient mice were more susceptible to DSS-induced colitis (77). In addition to autophagy-related genes, some other molecules can regulate the level of autophagy in intestinal epithelial, like vitamin D receptor (VDR). The study showed that intestinal epithelial VDR deletion impaired antimicrobial function of Paneth cells by downregulation of Atg16l1 and lysozyme, thus causing increased susceptibility to DSS-induced colitis (78). Additional IBD risk gene, Nod2 or leucine-rich repeat kinase 2 (LRRK2) absence disturbs the lysozyme packaging and secretion of Paneth cells, thus leading to the failure in controlling pathogen invasion, which was shown by infection with Listeria monocytogenes (79).
It is well known that dysfunction of ER causes unfolded and misfolded proteins accumulating in the ER lumen, which is called ER stress (80). ER stress disturbs the normal development and functions of cells, especially Paneth cells, because they are the IEC subtypes with the strongest function of secretion, which synthesize and excrete mounts of AMPs (81). As shown in recent discussion showed (82), there was a crosstalk between ER stress and inflammatory reaction. They noted that ER stress-induced inflammation in Paneth cells, epithelial cells, and goblet cells could possibly contribute to the pathogenesis and progression of IBD. However, they also argued that ER stress-induced inflammation could impede tumorigenesis through the induction of immunogenic cellular death-based antitumor immune responses despite its already demonstrated contribution of tumorigenesis, thus indicating the complexity of relationship between ER stress-induced inflammation and cancer. It was recently demonstrated that autophagy could be induced by ER stress in Paneth cells. The augmentation of autophagy in Paneth cells attenuates the ER stress-induced intestinal inflammation and eases nuclear factor-kappaB (NF-κB)-induced inflammatory reaction (72). This study uncovers the connection between autophagy and ER stress in the occurrence of IBD. In addition, it was reported that the ROS-mediated antibacterial autophagy (well-known as “xenophagy”) as well as the mitochondrial autophagy (well-known as “mitophagy”) in Paneth cells contributed greatly to the attenuation of IBD, thus probably serving as potential strategies for the treatment of IBD (83, 84). Collectively, these findings indicate that autophagy in epithelial cells, especially in Paneth cells, contributes to the alleviation of IBD (illustrated in Figure 1A).
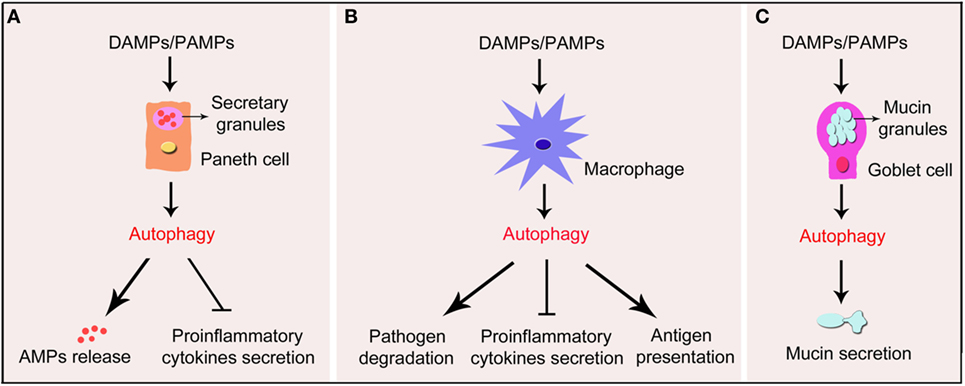
Figure 1. Schematic illustration of impact of autophagy in Paneth cells, macrophages, and goblet cells in IBD. Under the exposure of DAMPs or PAMPs, autophagy process is induced in Paneth cells, macrophages, and goblet cells in the gut wall. (A) Autophagy in Paneth cells triggers the formation of functional granules and substantial release of the AMPs. In addition, autophagy suppresses pro-inflammatory cytokines secreted by Paneth cells. (B) In macrophages, autophagy promotes the degradation of pathogens and the presentation of antigens. Autophagy also inhibits the secretion of pro-inflammatory cytokines by macrophages. (C) In goblet cells, autophagy promotes the formation of mucins granules and secretion of mucins. IBD, inflammatory bowel disease; DAMPs, damage-associated molecular pattern molecules; PAMPs, pathogen- associated molecular patterns; AMPs, antimicrobial peptides.
Macrophage, IBD, and Autophagy
Macrophage and IBD
The intestinal mucosa holds the largest population of macrophages in the body, which are vital and essential for the maintenance of mucosal homeostasis and epithelial renewal as well as protection against pathogenic microorganisms (85, 86). For the ontogeny of the gut-resident macrophages, it was traditionally believed that the majority of tissue macrophages were derived from the mononuclear phagocyte system in the bone marrow (87). So far, it is widely acknowledged that macrophages of the gut wall share the markers, including CD11c and MHC II, with other antigen-presenting cells like dendritic cells as well as “lineage-specifically” expressed CX3C chemokine receptor 1, F4/80, and CD64 (88, 89). Normally, macrophages are divided into M1 and M2 subtypes, the former displays pro-inflammatory characteristics and the later one has immunosuppressive and wound-healing properties (90). It was demonstrated that the gut-resident macrophages hold the features of both M1 and M2 subtypes. For instance, they have been reported to produce and secrete both the pro-inflammatory cytokines like TNF-α and the anti-inflammatory cytokines like IL-10, to adapt themselves to the environment of the gut wall (85, 91).
In the steady state, the gut-resident macrophages play an important role in the safeguarding of the intestine through the recognition and clearance of invading microorganisms and apoptotic cells as well as the regulation of tissue remodeling (92). The depletion of macrophages leads to the increasing susceptibility of DSS-induced colitis in mice (93). Intestinal macrophages can engulf and present the invading microorganisms to other immune cells like T cells, triggering the occurrence of immune reaction. Macrophages can also produce and secrete several pro-inflammatory cytokines and chemokines, including TNF-α, IL-6, IL-1β, and IL-12, leading to the further cascade amplification of inflammatory reaction (88, 94). It was demonstrated that during the occurrence of DSS-induced colitis in mice model, a large extent of infiltration of pro-inflammatory monocytes and macrophages was observed, leading to the further triggering of inflammatory and immune reaction, thus indicating that the regulation of macrophage-mediated inflammatory and immune responses might be vital in the treatment of IBD (94).
The Roles of Macrophage Autophagy in IBD
The roles of macrophage autophagy are generally illustrated in Figure 1B. It has been proven that pathogenesis and progression of IBD is highly related to the constant and overwhelming activation of innate immune reaction in gut wall, leading to the secretion of pro-inflammatory cytokines and chemokines, thus resulting in mucosal damage (95). Macrophages are regarded as important innate immune cells, since macrophages sense pathogen-associated molecular patterns of microorganisms and DAMPs through PRRs to initiate rapid and effective innate immune response to protect organism against pathogens along with DCs (96). What’s more, macrophages recognize antigens and present them to T cells to induce adaptive immune response (97). Given the key role of the interactions between host and microbe in the intestine, it is critical to regulate PRR signals and cytokine secretion properly. The NOD-like receptors (NLRs) in cytoplasm and Toll-like receptors (TLRs) on the surface are the two main types of PRRs in innate immune cells (95). It has been demonstrated that NLRs and TLRs in macrophages as well as other innate immune cells are closely associated with autophagy, and macrophage autophagy is highly related to the mediation of innate immune response in the gut wall (98, 99). For example, activating NOD2 using muramyldipeptide, a constituent of the bacterial peptidoglycan, recruited the autophagy-related protein ATG16L1 to the plasma membrane at the bacterial entry site to promote autophagy, but could not induce autophagy in cells with the Atg16l1 risk variant, thus indicating a functional connection between Nod2 and Atg16l1 (100–102). In addition, it was reported that macrophages with CD-associated Nod2 variants failed to induce autophagy, and the ability to kill pathogenic bacteria was impaired (103). Furthermore, there are much evidence of the connection between macrophage autophagy and the innate immune reaction in IBD. For instance, it was reported that impaired autophagy in macrophages transfected with siRNA targeted on Atg16l1 or IRGM and peritoneal macrophages from Nod2-deficient mice increased the amounts of intracellular adherent-invasive Escherichia coli (AIEC) and the levels of IL-6 and TNF-α stimulated by AIEC (104). Moreover, downregulation of autophagy caused by Atg4b deficiency increased the susceptibility to DSS-induced colitis, which is not only through affecting the function of Paneth cells but by promoting the production of pro-inflammatory cytokines in colon tissues from mice induced by DSS and in peritoneal macrophages stimulated with LPS (77). Those studies indicate that autophagy deficiency leads to the enhancement of the innate immune response and the production of pro-inflammatory cytokines in the gut wall with the inflammatory stress, thus demonstrating the close association between macrophage autophagy and the innate immune response in IBD.
Recently, a member of the NLR family, namely NLR family, pyrin domain-containing 3 (NLRP3) inflammasome, has been implicated in the pathogenesis and progression of IBD (105–108). Among all kinds of inflammasomes, the NLRP3 inflammasome is the best characterized one, containing NLRP3 protein, adapter protein apoptosis-associated speck-like protein, and pro-caspase-1 (109, 110). Interactions among these three proteins tightly regulate inflammasome functions through inducing the production and secretion of IL-1β and IL-18, whose maturity is triggered by caspase-1 from their “pro” forms (110). It has been reported that the polymorphism in the Nlrp3 gene is closely associated with the colitis severity and progression in patients with IBD (111, 112). In addition, studies in macrophages and mice models of IBD have uncovered the link between the abnormal activation of the NLRP3 inflammasome and colitis (113, 114). It was recently reported that oral administration of titanium dioxide (TiO2) nanoparticles aggravated the severity of IBD through a mechanism involving the activation of the NLRP3 inflammasome in DSS-induced colitis mice or UC patents (108). Studies have reported that the induction of macrophage autophagy produced an inhibitory effect on the activation of the NLRP3 inflammasome in different kinds of inflammation-related diseases, including multiple sclerosis and several kinds of cardiovascular diseases (115, 116). Impairing autophagy by Atg5 siRNA or 3-MA induced more robust initiation and activation of the NLRP3 inflammasome combined with increased caspase-1 activation and IL-1β production in peritoneal macrophages treated by LPS/DSS. Furthermore, using 3-MA to inhibit autophagy in vivo aggravated symptoms of DSS-induced colitis (117). Taken together, those results indicate that macrophage autophagy contributes to the alleviation of IBD through the inhibition of the NLRP3 inflammasome activation (illustrated in Figure 2). However, although the NLRP3 inflammasome has been shown to be highly related to the pathogenesis and progression of IBD and the crosstalk between autophagy and the NLRP3 inflammasome may probably provide a potential and promising therapeutic target for the treatment of IBD, yet the underlying mechanisms of IBD are not completely clear, and there is still a long way to go.
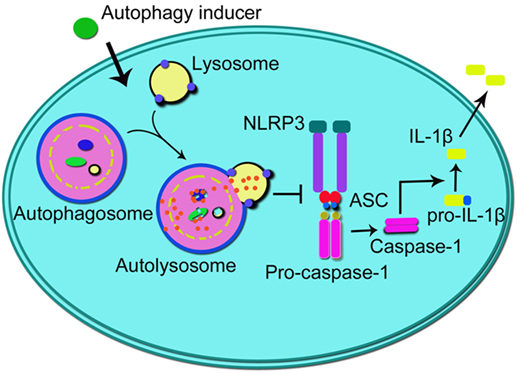
Figure 2. Schematic illustration of the impact of autophagy on NLRP3 inflammasome. Autophagy inducers promote autophagy process which inhibits the formation of NLRP3 inflammasome (integrated by NLRP3, ASC, and pro-caspase-1), thus suppressing the activation of caspase-1 and subsequent production of IL-1β. NLRP3, NLR family, pyrin domain-containing 3; ASC, adapter protein apoptosis-associated speck-like protein; IL-1β, interleukin-1β.
Goblet Cell, IBD, and Autophagy
Goblet Cell and IBD
Goblet cell is another critical kind of intestinal epithelial cell which is derived from the stem cells located in the crypt bottom. The main function of goblet cell is storage and secretion of large amount of mucin granules containing mucin glycoproteins like mucin 2, antimicrobial factors, and mucus cross-linking proteins like Fc-gamma binding protein, which combine with intestinal epithelium to form the first line of defense against gut flora from the host (118, 119). Since goblet cells are the most highly secretory cells among all kinds of intestinal epithelial cells, the increased mucins misfolding or decreased mucin glycoproteins secretion can induce ER stress and unfolded protein response. Both mucins depletion and ER stress give rise to the activation of inflammatory and immune responses and subsequent intestinal inflammation in mice (120–122). Consistently, dysfunction in mucins secretion and defective in mucus layer allow large quantities of bacteria to reach the epithelium and triggering excess host immune responses which has been demonstrated to be associated with IBD (123).
The Roles of Goblet Cell Autophagy in IBD
Recently, it has been revealed that autophagy could control the secretion function of goblet cells (illustrated in Figure 1C). Mice or cells deficiency in autophagy by depletion of autophagy-related proteins like ATG5, ATG7, or LC3 led to altered goblet cell morphology and significantly decreased mucins secretion (124, 125). In addition, mice expressing the ATG16l1 T300A variant exhibited the similar characters of goblet cells (126). Patel et al. found out that nicotinamide adenine dinucleotide phosphate oxidase-driven ROS production was required for autophagy-mediated secretion of goblet cells as Atg5-deficient spheroids produced significantly diminished ROS compared with those wild type, and these cells were overloaded with mucins. Furthermore, goblet cells from mice treated with ROS scavenger N-acetylcysteine were accumulated with mucins compared to controls (124). The accumulated mucins in goblet cells suggested dysfunction in granules exocytosis. The upstream pathway inducing autophagy in goblet cells is mainly through activation of the NLRP6 inflammasome, a member of the Nod-like receptor family of PRRs. The NLRP6 inflammasome promotes goblet cell mucin granules exocytosis by promoting autophagy and goblet cells from Nlrp6-deficient mice have impaired autophagy and are less effective in secreting mucins. This secretion alteration leads to significant impaired role of epithelial barrier in colonic host–microbial interactions (127, 128). These findings suggest that defected autophagy in goblet cells contribute to the pathogenesis of IBD. Regulating autophagy in goblet cells may provide new strategies for the treatment of IBD. However, since the precise mechanisms and molecules involving autophagy in regulating mucins secretion are not fully elucidated, further studies are needed for the final clarification.
The Roles of Autophagy in Colorectal Cancer (CRC)
Since IBD is likely to develop into CRC in the end and autophagy plays a pivotal role in the pathogenesis of IBD as discussed above, it is quite necessary to pay attention to the effects of autophagy on colorectal carcinogenesis. Autophagy is regarded as a double-edged sword during tumor progression, which suppresses tumor formation through inhibiting DNA injury, maintaining chromosomal stability, reducing local inflammation, and promoting immune system to eliminate potentially tumorigenic cells (129). However, on the other hand, autophagy can promote cancer cell survival and proliferation by escaping from intracellular and environmental stress as well as resisting to anticancer agents (129). Hence, the relationship between autophagy and CRC is complex. The first aspect lies in the expression profile of autophagy in CRC. Some evidence has shown that autophagy is highly induced in most CRC patients and CRC cells (130–135). However, there is also a small population of CRC patients with downregulated autophagy (136). Based on those reports, the expression profile of autophagy is inconsistent in CRC patients.
The second aspect is the complexity of the roles of autophagy in CRC. It was previously reported that overexpression of Beclin-1 in vitro inhibited CRC cells growth and enhanced the rapamycin-induced antitumor effects (136). Patients with high levels of Beclin-1 presented good prognosis and longer survival (131, 135), and ATG10 downregulation was associated with cell migration and invasion of CRC cells (137). In addition, several compounds were recently demonstrated to suppress colorectal carcinogenesis through the induction of autophagy, including Salvianolic acid B and IR-58 (138, 139). Those findings indicate that promoting autophagy may probably provide an effective strategy for the treatment of CRC. However, enhancing autophagy is not without its problems in the treatment of CRC. It was reported that promoting autophagy could enhance CRC cells tolerance to antitumor drugs (140, 141). For example, patients with high levels of Beclin-1 resist 5-FU-based adjuvant therapy and result in a poor prognosis and metastasis (142, 143). In addition, suppressing autophagy by deletion of Atg5 enhanced the sensitivity of CRC cells and CRC mice to oxaliplatin and INF-γ (144, 145). In conclusion, since the functions of autophagy in CRC are complicated, to ultimately develop autophagy as therapeutic strategy in the treatment of CRC, numerous studies are needed in seek of effective approaches to induce autophagy while getting rid of its side effects.
Pharmacological Intervention of Autophagy in the Treatment of IBD
In consideration of the importance of autophagy in the pathagenesis and progression of IBD, numerous studies have been conducted so far, focusing on the alleviation and treatment of IBD through the regulation of autophagy.
Herbal Extracts
Andrographolide belongs to diterpenoids extracted from a kind of herb called andrographolide. Andrographolide has anti-inflammatory and antitumor activity (146, 147), and it can effectively inhibit the growth of breast and colon cancer cells (148). The combination of andrographolide and radiation therapy can enhance the levels of apoptosis and autophagy in nude mice xenografted by human ovarian cancer skov3 (149). It was reported that andrographolide sulfonate could inhibit the p38 mitogen-activated protein kinase (p38 MAPK) and NF-κB signaling pathways, which contributed to the reduction of pro-inflammatory cytokines production in trinitrobenzene sulfonic acid (TNBS)-induced colitis mice model and further intervened the progression of colitis (150). In addition, Guo et al. (151) demonstrated that andrographolide could enhance mitophagy in macrophages, lead to the reverse of mitochondrial membrane potential, and thus alleviate the symptoms of DSS-induced colitis through the inhibition of NLRP3 inflammasome activity. However, several autophagy inhibitors, including 3-MA, chloroquine, bafilomycin A1, or Becline 1 siRNA abolished the inhibitory effects of andrographolide on the NLRP3 inflammasome and to a large extent reduced the therapeutic effects of andrographolide for colitis.
Another herbal extract worth mentioning is celastrol. It is a kind of triterpenoid extracted from the root of a traditional Chinese medicine called Tripterygium wilfordii. It was reported that celastrol could inhibit the development of IBD or colitis through several pathways, including: (1) regulating the oxidative stress and reducing lipid peroxide; (2) inhibiting the activation of the NLRP3 inflammasome; (3) increasing the levels of anti-inflammatory cytokines like IL-10; (4) enhancing the stability of intestinal epithelial barrier (152); (5) inhibiting the receptor interacting-protein 3/mixed lineage kinase domain-like protein-mediated programed necrosis pathway (153); and (6) inhibiting the production of NF-κB and its related inflammatory cytokines (154). Furthermore, it has been proven that celastrol can enhance the level of autophagy through the inhibition of the PI3K/Akt/mTOR signaling pathway in the colon tissue, and meanwhile reduce the production of several pro-inflammatory cytokines thus attenuating the inflammatory reaction in colon in IL-10-deficient mice (155). Based on those findings, it is reasonable to believe that those two herbal extracts may provide promising and effective therapies for IBD. However, since the clinical data for the treatment of IBD are poor, more detailed clinical studies are needed to explore their effectiveness and safety on IBD patients.
Receptor Regulators
As been discussed above, the activation of the NLRP3 inflammasome leads to the activation of caspase-1, which induces the transformation of proIL-1β and proIL-18 into the mature IL-1β and IL-18. The maturation of IL-1β and IL-18 contributes to the pathogenesis and progression of inflammatory diseases. Studies indicated that autophagy play an important role in the formation and activation of NLRP3 inflammasome, and autophagy deficiency led to the abnormal increase of IL-1β (156–158). Mice deficient in autophagy showed more severe symptoms of colitis (59). Recently, increasing studies are conducted to explore whether blocking IL-1β contributes to alleviating the progression of IBD. For example, De Luca et al. (159) found out that the blockade of IL-1β receptor using anakinra could effectively alleviate the progression of IBD through the reduction of neutrophils accumulation and Th17 cell response. Van De Veerdonk and Dinarello (49) demonstrated that anakinra could restore the autophagy level of patients with chronic granuloma and reduced the IL-1-mediated inflammatory reaction, thus playing a protective role in the TNBS-induced colitis and alleviating the severity of patients with chronic granuloma, promoting the recovery of rectal abscess.
Cannabinoid receptor 2 (CB2R) is a kind of seven transmembrane-spanning G protein coupled receptor, mainly distributing on immune cells (160). Activating CB2R has been reported to contribute to the alleviation of EAE through the increase of autophagy, which in turn led to the inhibition of the NLRP3 inflammasome (115). Nowadays, the role of CB2R in IBD or colitis has been widely recognized. For example, it has been reported that activating CB2R can attenuate the severity of colitis in wild-type mice (161). In addition, activating CB2R was also demonstrated to alleviate colitis through inhibiting T cell activation and promote apoptosis in IL-10-deficient mice (162). In the recent years, several kinds of CB2R activators have been successfully applied in the treatment of colitis animal model (117, 163, 164). For example, activating CB2R by HU-308, a selective CB2R agonist, contributed to the enhancement of macrophage autophagy through the AMPK-mTOR-P70S6K signaling pathway, and further inhibited the NLRP3 inflammasome activation, thus reducing the level of IL-1β and alleviating the progression of DSS-induced colitis in mice (117) (illustrated in Figure 2). So far, although studies uncovered the promising therapeutic value of CB2R activators in the treatment of IBD, there is still no related drug taking advantage of the beneficial effects of activating CB2R for IBD. Hence, further studies are demanded for the development of related drugs and in the other hand, more kinds of receptor regulators should be fully studied to serve for the treatment of IBD.
Nutrient Molecules
Vitamin D deficiency is regarded as an important pathological basis of IBD (165). Studies showed that the level of vitamin D is relatively low in patients with IBD (166, 167). Vitamin D leads to a series of physiological effects mainly through functioning on VDR, of which the gene polymorphism is closely connected with the susceptibility of IBD (168). It was reported that deficiency in vitamin D can lead to the reduction of ATG16L1 and the level of autophagy and further influenced the function of Paneth cells. After restoring the level of VDR by applying vitamin D3 in intestinal epithelial cells, the levels of autophagy is increased (78). Furthermore, a systematic review and meta-analysis conducted by Ma et al. (169) demonstrated that vitamin D intake as well as blood 25(OH)D levels were inversely associated with the risk of CRC. In consideration of the closely relationship between IBD and CRC, the antitumor feature of vitamin D provides further evidence to show the therapeutic value of vitamin D in the treatment of IBD.
Glutamine is a kind of free amino acid widely distributed in organisms. It is the respiratory material and metabolic precursors of many kinds of cells, including intestinal epithelial cells and immune cells (170–172). Under the physiological stress, glutamine reduces the permeability of intestinal epithelial cells, inhibits intestinal mucosa injury, and promotes the intestinal restoration (173–175). It was reported that in the intestinal epithelial cells glutamine contributed to the enhancement of autophagy level in the basal and stressing condition through the regulation of the mTOR and p38 MAPK signaling pathway, thus inhibiting the stress-induced cellular apoptosis (176).
Other Small-Molecular Compounds
Two kinds of rapamycin analogs, namely, sirolimus and everolimus, which have already been applied in clinic, contribute to the enhancement of autophagy level. It has been reported that continuous application of sirolimus for 6 months significantly led to the attenuation of symptoms in patients with severe recurrent CD (177). Sirolimus can not only benefit adult IBD patients, but also lead to the alleviation of IBD in the children patients. It was reported that after the application of sirolimus, 45% UC patients and 100% CD patients reached the clinical stable stage. Meanwhile, sirolimus contributes to the promotion of intestinal mucus restoration (178). In addition, after the occurrence of the spontaneous colitis in IL-10-deficient mice, researchers found out that treating with everolimus for 4 weeks significantly reduced the disease activity index as well as the infiltration of lymphocytes in spleen, mesenteric lymph nodes, and inherent layer (179). It was lately reported by Dumortier et al. (180) that after treated with everolimus in the original treatment therapy for 1½ years, the symptoms in a patient with refractory UC were effectively controlled. However, in a multicenter randomized double bind trial, everolimus was not beneficial for the maintenance of the stable stage treated with glucocorticoid in patients with activity stage in the present of placebo or azathioprine (181). This study indicates that the therapeutic strategy for IBD through enhancing autophagy process demands further assessment and the effectiveness and security needs further tested.
The drugs or molecules mentioned above that alleviate IBD through enhancing autophagy were listed in the Table 1, and some other drugs were also briefly mentioned.
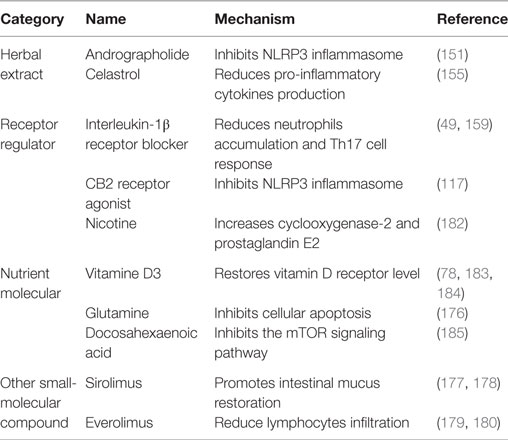
Table 1. The mechanisms of autophagy inducers in treatment of IBD.
Conclusion
Intestinal autophagy, especially autophagy in Paneth cells, macrophages, and goblet cells in the intestinal wall, produces an alleviative effect on the pathogenesis and progression of IBD. In the recent few years, we are pleased to have several kinds of drugs developed for the treatment of IBD taking advantage of the induction of autophagy, including herbal extracts, receptor regulators, nutrient molecules, and other small-molecular compounds, etc. However, despite the numerous therapeutic strategies available, there is still no effective way for the treatment of IBD. Furthermore, the specific mechanism of IBD still remains uncovered. Besides, there are several other hot-discussed issues worth mentioning in this field: first, since little literatures have reported the effects of autophagy inducers in the clinical treatment of IBD, proper doses demand further testing and side effects ought to be found out for the safe application. Second, based on the discussion above, although inducing autophagy contributes to the alleviation of IBD, yet it has already been reported that on certain conditions, the induction of autophagy may also lead to inverse effects on the attenuation of IBD (186–188). Third, as discussed above, although inducing autophagy contributes to the suppression of colorectal carcinogenesis, there are still side effects that autophagy inducers may lead to the enhancement of CRC cells tolerance to antitumor drugs. Therefore, more studies are needed to explore the mechanisms of IBD, and effective therapeutic strategies are demanded to be developed while getting rid of side effects of inducing autophagy.
Author Contributions
PK, B-ZS, and Z-QX retrieved and analyzed concerned literatures. PK and B-ZS wrote the manuscript. CL and X-WC revised the manuscript. All the authors agreed to be accountable for the content of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (81670260).
References
1. Wells JM, Rossi O, Meijerink M, Van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4607–14. doi:10.1073/pnas.1000092107
2. Goto Y, Kiyono H. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev (2012) 245:147–63. doi:10.1111/j.1600-065X.2011.01078.x
3. Antoni L, Nuding S, Wehkamp J, Stange EF. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol (2014) 20:1165–79. doi:10.3748/wjg.v20.i5.1165
4. Kabat AM, Pott J, Maloy KJ. The mucosal immune system and its regulation by autophagy. Front Immunol (2016) 7:240. doi:10.3389/fimmu.2016.00240
5. Lee HY, Kim J, Quan W, Lee JC, Kim MS, Kim SH, et al. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy (2016) 12:1390–403. doi:10.1080/15548627.2016.1184799
6. Negroni A, Colantoni E, Vitali R, Palone F, Pierdomenico M, Costanzo M, et al. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm Res (2016) 65:803–13. doi:10.1007/s00011-016-0964-8
7. Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol (2014) 20:6–21. doi:10.3748/wjg.v20.i1.6
8. Birrenbach T, Bocker U. Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm Bowel Dis (2004) 10:848–59. doi:10.1097/00054725-200411000-00019
9. Breuer-Katschinski BD, Hollander N, Goebell H. Effect of cigarette smoking on the course of Crohn’s disease. Eur J Gastroenterol Hepatol (1996) 8:225–8. doi:10.1097/00042737-199603000-00007
10. Cosnes J, Carbonnel F, Beaugerie L, Le Quintrec Y, Gendre JP. Effects of cigarette smoking on the long-term course of Crohn’s disease. Gastroenterology (1996) 110:424–31. doi:10.1053/gast.1996.v110.pm8566589
11. Ananthakrishnan AN, Mcginley EL, Binion DG, Saeian K. Ambient air pollution correlates with hospitalizations for inflammatory bowel disease: an ecologic analysis. Inflamm Bowel Dis (2011) 17:1138–45. doi:10.1002/ibd.21455
12. Kaplan GG, Hubbard J, Korzenik J, Sands BE, Panaccione R, Ghosh S, et al. The inflammatory bowel diseases and ambient air pollution: a novel association. Am J Gastroenterol (2010) 105:2412–9. doi:10.1038/ajg.2010.252
13. Salim SY, Kaplan GG, Madsen KL. Air pollution effects on the gut microbiota: a link between exposure and inflammatory disease. Gut Microbes (2014) 5:215–9. doi:10.4161/gmic.27251
14. Hviid A, Svanstrom H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut (2011) 60:49–54. doi:10.1136/gut.2010.219683
15. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314:1461–3. doi:10.1126/science.1135245
16. Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet (2007) 39:1329–37. doi:10.1038/ng.2007.17
17. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi:10.1038/ng1954
18. Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest (2007) 117:514–21. doi:10.1172/JCI30587
19. Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell (2010) 141:1135–45. doi:10.1016/j.cell.2010.05.009
20. Brandtzaeg P. Mucosal immunity: induction, dissemination, and effector functions. Scand J Immunol (2009) 70:505–15. doi:10.1111/j.1365-3083.2009.02319.x
21. Larsson JMH, Karlsson H, Crespo JG, Johansson MEV, Eklund L, Sjovall H, et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis (2011) 17:2299–307. doi:10.1002/ibd.21625
22. Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet (2004) 36:471–5. doi:10.1038/ng1339
23. Giles EM, Sanders TJ, Mccarthy NE, Lung J, Pathak M, Macdonald TT, et al. Regulation of human intestinal T-cell responses by type 1 interferon-STAT1 signaling is disrupted in inflammatory bowel disease. Mucosal Immunol (2016). doi:10.1038/mi.2016.44
24. Tosiek MJ, Fiette L, El Daker S, Eberl G, Freitas AA. IL-15-dependent balance between Foxp3 and ROR gamma t expression impacts inflammatory bowel disease. Nat Commun (2016) 7:10888. doi:10.1038/ncomms10888
25. Pavel M, Rubinsztein DC. Mammalian autophagy and the plasma membrane. FEBS J (2016). doi:10.1111/febs.13931
26. Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology (2011) 140:1738–47. doi:10.1053/j.gastro.2011.02.048
27. Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci (2011) 68:749–63. doi:10.1007/s00018-010-0565-6
28. Huber LA, Teis D. Lysosomal signaling in control of degradation pathways. Curr Opin Cell Biol (2016) 39:8–14. doi:10.1016/j.ceb.2016.01.006
29. Xilouri M, Stefanis L. Chaperone mediated autophagy in aging: starve to prosper. Ageing Res Rev (2016) 32:13–21. doi:10.1016/j.arr.2016.07.001
30. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet (2009) 43:67–93. doi:10.1146/annurev-genet-102808-114910
31. Deretic V. Autophagy in leukocytes and other cells: mechanisms, subsystem organization, selectivity, and links to innate immunity. J Leukoc Biol (2016) 100:969–78. doi:10.1189/jlb.4MR0216-079R
32. Shao BZ, Han BZ, Zeng YX, Su DF, Liu C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin (2016) 37:150–6. doi:10.1038/aps.2015.87
33. Liu N, Shi Y, Zhuang S. Autophagy in chronic kidney diseases. Kidney Dis (Basel) (2016) 2:37–45. doi:10.1159/000444841
34. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy (2016) 12:1–222. doi:10.1080/15548627.2015.1100356
35. Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem (2010) 285:10850–61. doi:10.1074/jbc.M109.080796
36. Canonico B, Cesarini E, Salucci S, Luchetti F, Falcieri E, Di Sario G, et al. Defective autophagy, mitochondrial clearance and lipophagy in Niemann-Pick type B lymphocytes. PLoS One (2016) 11:e0165780. doi:10.1371/journal.pone.0165780
37. Gao S, Sun D, Wang G, Zhang J, Jiang Y, Li G, et al. Growth inhibitory effect of paratocarpin E, a prenylated chalcone isolated from Euphorbia humifusa Wild., by induction of autophagy and apoptosis in human breast cancer cells. Bioorg Chem (2016) 69:121–8. doi:10.1016/j.bioorg.2016.10.005
38. Li Y, Yu G, Yuan S, Tan C, Xie J, Ding Y, et al. 14,15-Epoxyeicosatrienoic acid suppresses cigarette smoke condensate-induced inflammation in lung epithelial cells by inhibiting autophagy. Am J Physiol Lung Cell Mol Physiol (2016) 311:L970–80. doi:10.1152/ajplung.00161.2016
39. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet (2007) 39:830–2. doi:10.1038/ng2061
40. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39:596–604. doi:10.1038/ng2032
41. Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol (2003) 5:455–68. doi:10.1046/j.1462-5822.2003.00292.x
42. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119:753–66. doi:10.1016/j.cell.2004.11.038
43. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science (2004) 306:1037–40. doi:10.1126/science.1103966
44. Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, et al. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc Natl Acad Sci U S A (2007) 104:1829–34. doi:10.1073/pnas.0601437104
45. Crotzer VL, Blum JS. Autophagy and intracellular surveillance: modulating MHC class II antigen presentation with stress. Proc Natl Acad Sci U S A (2005) 102:7779–80. doi:10.1073/pnas.0503088102
46. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A (2005) 102:7922–7. doi:10.1073/pnas.0501190102
47. Dorfel D, Appel S, Grunebach F, Weck MM, Muller MR, Heine A, et al. Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood (2005) 105:3199–205. doi:10.1182/blood-2004-09-3556
48. Randall-Demllo S, Chieppa M, Eri R. Intestinal epithelium and autophagy: partners in gut homeostasis. Front Immunol (2013) 4:301. doi:10.3389/fimmu.2013.00301
49. Van De Veerdonk FL, Dinarello CA. Deficient autophagy unravels the ROS paradox in chronic granulomatous disease. Autophagy (2014) 10:1141–2. doi:10.4161/auto.28638
50. Hosomi S, Kaser A, Blumberg RS. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr Opin Gastroenterol (2015) 31:81–8. doi:10.1097/MOG.0000000000000144
51. Zhang Q, Kang R, Zeh HJ III, Lotze MT, Tang D. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy (2013) 9:451–8. doi:10.4161/auto.23691
52. Maeda S, Hikiba Y, Shibata W, Ohmae T, Yanai A, Ogura K, et al. Essential roles of high-mobility group box 1 in the development of murine colitis and colitis-associated cancer. Biochem Biophys Res Commun (2007) 360:394–400. doi:10.1016/j.bbrc.2007.06.065
53. Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis (2010) 16:1097–107. doi:10.1002/ibd.21175
54. Vitali R, Stronati L, Negroni A, Di Nardo G, Pierdomenico M, Del Giudice E, et al. Fecal HMGB1 is a novel marker of intestinal mucosal inflammation in pediatric inflammatory bowel disease. Am J Gastroenterol (2011) 106:2029–40. doi:10.1038/ajg.2011.231
55. Meuwis MA, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, Piver E, et al. Serum calprotectin as a biomarker for Crohn’s disease. J Crohns Colitis (2013) 7:e678–83. doi:10.1016/j.crohns.2013.06.008
56. Boyapati RK, Rossi AG, Satsangi J, Ho GT. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol (2016) 9:567–82. doi:10.1038/mi.2016.14
57. Li W, Zhu S, Li J, Assa A, Jundoria A, Xu J, et al. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem Pharmacol (2011) 81:1152–63. doi:10.1016/j.bcp.2011.02.015
58. Zhang Y, Li W, Zhu S, Jundoria A, Li J, Yang H, et al. Tanshinone IIA sodium sulfonate facilitates endocytic HMGB1 uptake. Biochem Pharmacol (2012) 84:1492–500. doi:10.1016/j.bcp.2012.09.015
59. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456:264–8. doi:10.1038/nature07383
60. Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ (2009) 16:175–83. doi:10.1038/cdd.2008.143
61. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J (2011) 30:4701–11. doi:10.1038/emboj.2011.398
62. Ayna G, Krysko DV, Kaczmarek A, Petrovski G, Vandenabeele P, Fesus L. ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One (2012) 7:e40069. doi:10.1371/journal.pone.0040069
63. Moran AP, Gupta A, Joshi L. Sweet-talk: role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut (2011) 60:1412–25. doi:10.1136/gut.2010.212704
64. Hansson GC. Role of mucus layers in gut infection and inflammation. Curr Opin Microbiol (2012) 15:57–62. doi:10.1016/j.mib.2011.11.002
65. Pearson JP, Brownlee IA. The interaction of large bowel microflora with the colonic mucus barrier. Int J Inflam (2010) 2010:321426. doi:10.4061/2010/321426
66. Cederlund A, Gudmundsson GH, Agerberth B. Antimicrobial peptides important in innate immunity. FEBS J (2011) 278:3942–51. doi:10.1111/j.1742-4658.2011.08302.x
67. Cheng H. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. IV. Paneth cells. Am J Anat (1974) 141:521–35. doi:10.1002/aja.1001410406
68. Roda G, Sartini A, Zambon E, Calafiore A, Marocchi M, Caponi A, et al. Intestinal epithelial cells in inflammatory bowel diseases. World J Gastroenterol (2010) 16:4264–71. doi:10.3748/wjg.v16.i34.4264
69. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol (2011) 9:356–68. doi:10.1038/nrmicro2546
70. Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted Paneth cell. Cell Mol Life Sci (2002) 59:156–70. doi:10.1007/s00018-002-8412-z
71. Ouellette AJ. Paneth cells and innate mucosal immunity. Curr Opin Gastroenterol (2010) 26:547–53. doi:10.1097/MOG.0b013e32833dccde
72. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
73. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
74. Cadwell K, Patel KK, Komatsu M, Virgin HWT, Stappenbeck TS. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy (2009) 5:250–2. doi:10.4161/auto.5.2.7560
75. Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, De Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut (2014) 63:1081–91. doi:10.1136/gutjnl-2012-303527
76. Wittkopf N, Gunther C, Martini E, Waldner M, Amann KU, Neurath MF, et al. Lack of intestinal epithelial atg7 affects Paneth cell granule formation but does not compromise immune homeostasis in the gut. Clin Dev Immunol (2012) 2012:278059. doi:10.1155/2012/278059
77. Cabrera S, Fernandez AF, Marino G, Aguirre A, Suarez MF, Espanol Y, et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy (2013) 9:1188–200. doi:10.4161/auto.24797
78. Wu S, Zhang YG, Lu R, Xia Y, Zhou D, Petrof EO, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut (2015) 64:1082–94. doi:10.1136/gutjnl-2014-307436
79. Rocha JD, Schlossmacher MG, Philpott DJ. LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat Immunol (2015) 16:898–900. doi:10.1038/ni.3255
80. Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol (2008) 8:663–74. doi:10.1038/nri2359
81. McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2010) 298:G820–32. doi:10.1152/ajpgi.00063.2010
82. Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med (2012) 18:589–98. doi:10.1016/j.molmed.2012.06.010
83. Billmann-Born S, Lipinski S, Bock J, Till A, Rosenstiel P, Schreiber S. The complex interplay of NOD-like receptors and the autophagy machinery in the pathophysiology of Crohn disease. Eur J Cell Biol (2011) 90:593–602. doi:10.1016/j.ejcb.2010.10.015
84. Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2013) 305:G573–84. doi:10.1152/ajpgi.00071.2013
85. Hume DA, Perry VH, Gordon S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localisation of antigen F4/80: macrophages associated with epithelia. Anat Rec (1984) 210:503–12. doi:10.1002/ar.1092100311
86. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev (2014) 260:102–17. doi:10.1111/imr.12192
87. Van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46:845–52.
88. Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol (2013) 6:498–510. doi:10.1038/mi.2012.89
89. Kc W, Satpathy AT, Rapaport AS, Briseno CG, Wu X, Albring JC, et al. L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature (2014) 507:243–7. doi:10.1038/nature12967
90. Zhuge F, Ni Y, Nagashimada M, Nagata N, Xu L, Mukaida N, et al. DPP-4 inhibition by linagliptin attenuates obesity-related inflammation and insulin resistance by regulating M1/M2 macrophage polarization. Diabetes (2016) 65:2966–79. doi:10.2337/db16-0317
91. Weber B, Saurer L, Schenk M, Dickgreber N, Mueller C. CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur J Immunol (2011) 41:773–9. doi:10.1002/eji.201040965
92. Muller AJ, Kaiser P, Dittmar KE, Weber TC, Haueter S, Endt K, et al. Salmonella gut invasion involves TTSS-2-dependent epithelial traversal, basolateral exit, and uptake by epithelium-sampling lamina propria phagocytes. Cell Host Microbe (2012) 11:19–32. doi:10.1016/j.chom.2011.11.013
93. Qualls JE, Kaplan AM, Van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol (2006) 80:802–15. doi:10.1189/jlb.1205734
94. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity (2012) 37:1076–90. doi:10.1016/j.immuni.2012.08.026
95. Talero E, Garcia-Maurino S, Motilva V. Melatonin, autophagy and intestinal bowel disease. Curr Pharm Des (2014) 20:4816–27. doi:10.2174/1381612819666131119110835
96. Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity (2014) 40:248–61. doi:10.1016/j.immuni.2013.12.012
97. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gamma delta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31:321–30. doi:10.1016/j.immuni.2009.06.020
98. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
99. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411:603–6. doi:10.1038/35079114
100. Homer CR, Richmond AL, Rebert NA, Achkar JP, Mcdonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology (2010) 139:1630–41. doi:10.1053/j.gastro.2010.07.006
101. Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol (2010) 12:99–113. doi:10.1111/j.1462-5822.2009.01381.x
102. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–U67. doi:10.1038/ni.1823
103. Strisciuglio C, Duijvestein M, Verhaar AP, Vos ACW, Van Den Brink GR, Hommes DW, et al. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. Journal Crohns Colitis (2013) 7:534–41. doi:10.1016/j.crohns.2012.08.009
104. Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol (2012) 14:791–807. doi:10.1111/j.1462-5822.2012.01768.x
105. Bisicchia E, Chiurchiu V, Viscomi MT, Latini L, Fezza F, Battistini L, et al. Activation of type-2 cannabinoid receptor inhibits neuroprotective and antiinflammatory actions of glucocorticoid receptor alpha: when one is better than two. Cell Mol Life Sci (2013) 70:2191–204. doi:10.1007/s00018-012-1253-5
106. He X, Wei Z, Wang J, Kou J, Liu W, Fu Y, et al. Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci Rep (2016) 6:28370. doi:10.1038/srep28370
107. Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L, et al. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem Pharmacol (2016) 112:37–49. doi:10.1016/j.bcp.2016.05.002
108. Ruiz PA, Moron B, Becker HM, Lang S, Atrott K, Spalinger MR, et al. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut (2016). doi:10.1136/gutjnl-2015-310297
109. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun (2015) 6. doi:10.1038/Ncomms8360
110. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi:10.3389/fphar.2015.00262
111. Lewis GJ, Massey DC, Zhang H, Bredin F, Tremelling M, Lee JC, et al. Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm Bowel Dis (2011) 17:1387–91. doi:10.1002/ibd.21499
112. Varghese GP, Uporova L, Halfvarson J, Sirsjo A, Fransen K. Polymorphism in the NLRP3 inflammasome-associated EIF2AK2 gene and inflammatory bowel disease. Mol Med Rep (2015) 11:4579–84. doi:10.3892/mmr.2015.3236
113. Cheng YL, Song LQ, Huang YM, Xiong YW, Zhang XA, Sun H, et al. Effect of enterohaemorrhagic Escherichia coli O157:H7-specific enterohaemolysin on interleukin-1 beta production differs between human and mouse macrophages due to the different sensitivity of NLRP3 activation. Immunology (2015) 145:258–67. doi:10.1111/imm.12442
114. Sun Y, Zhao Y, Yao J, Zhao L, Wu Z, Wang Y, et al. Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-kappaB and NLRP3 inflammasome activation. Biochem Pharmacol (2015) 94:142–54. doi:10.1016/j.bcp.2015.02.002
115. Shao BZ, Wei W, Ke P, Xu ZQ, Zhou JX, Liu C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther (2014) 20:1021–8. doi:10.1111/cns.12349
116. Abderrazak A, Couchie D, Mahmood DF, Elhage R, Vindis C, Laffargue M, et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation (2015) 131:1061–70. doi:10.1161/CIRCULATIONAHA.114.013730
117. Ke P, Shao BZ, Xu ZQ, Wei W, Han BZ, Chen XW, et al. Activation of cannabinoid receptor 2 ameliorates DSS-induced colitis through inhibiting NLRP3 inflammasome in macrophages. PLoS One (2016) 11:e0155076. doi:10.1371/journal.pone.0155076
118. Johansson ME. Fast renewal of the distal colonic mucus layers by the surface goblet cells as measured by in vivo labeling of mucin glycoproteins. PLoS One (2012) 7:e41009. doi:10.1371/journal.pone.0041009
119. El-Khider F, McDonald C. Links of autophagy dysfunction to inflammatory bowel disease onset. Dig Dis (2016) 34:27–34. doi:10.1159/000442921
120. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med (2008) 5:e54. doi:10.1371/journal.pmed.0050054
121. Kaser A, Blumberg RS. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin Immunol (2009) 21:156–63. doi:10.1016/j.smim.2009.01.001
122. Kaser A, Martinez-Naves E, Blumberg RS. Endoplasmic reticulum stress: implications for inflammatory bowel disease pathogenesis. Curr Opin Gastroenterol (2010) 26:318–26. doi:10.1097/MOG.0b013e32833a9ff1
123. Swidsinski A, Loening-Baucke V, Herber A. Mucosal flora in Crohn’s disease and ulcerative colitis – an overview. J Physiol Pharmacol (2009) 60(Suppl 6):61–71.
124. Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J (2013) 32:3130–44. doi:10.1038/emboj.2013.233
125. Chen GY, Stappenbeck TS. Mucus, it is not just a static barrier. Sci Signal (2014) 7:e11. doi:10.1126/scisignal.2005357
126. Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A (2014) 111:7741–6. doi:10.1073/pnas.1407001111
127. Johansson ME, Hansson GC. Is the intestinal goblet cell a major immune cell? Cell Host Microbe (2014) 15:251–2. doi:10.1016/j.chom.2014.02.014
128. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell (2014) 156:1045–59. doi:10.1016/j.cell.2014.01.026
129. Burada F, Nicoli ER, Ciurea ME, Uscatu DC, Ioana M, Gheonea DI. Autophagy in colorectal cancer: an important switch from physiology to pathology. World J Gastrointest Oncol (2015) 7:271–84. doi:10.4251/wjgo.v7.i11.271
130. Ahn CH, Jeong EG, Lee JW, Kim MS, Kim SH, Kim SS, et al. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS (2007) 115:1344–9. doi:10.1111/j.1600-0463.2007.00858.x
131. Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, Wan DS, et al. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy (2009) 5:303–6. doi:10.4161/auto.5.3.7491
132. Guo GF, Jiang WQ, Zhang B, Cai YC, Xu RH, Chen XX, et al. Autophagy-related proteins Beclin-1 and LC3 predict cetuximab efficacy in advanced colorectal cancer. World J Gastroenterol (2011) 17:4779–86. doi:10.3748/wjg.v17.i43.4779
133. Jo YK, Kim SC, Park IJ, Park SJ, Jin DH, Hong SW, et al. Increased expression of ATG10 in colorectal cancer is associated with lymphovascular invasion and lymph node metastasis. PLoS One (2012) 7:e52705. doi:10.1371/journal.pone.0052705
134. Zheng HY, Zhang XY, Wang XF, Sun BC. Autophagy enhances the aggressiveness of human colorectal cancer cells and their ability to adapt to apoptotic stimulus. Cancer Biol Med (2012) 9:105–10. doi:10.3969/j.issn.2095-3941.2012.02.004
135. Yang Z, Ghoorun RA, Fan X, Wu P, Bai Y, Li J, et al. High expression of Beclin-1 predicts favorable prognosis for patients with colorectal cancer. Clin Res Hepatol Gastroenterol (2015) 39:98–106. doi:10.1016/j.clinre.2014.06.014
136. Chen Z, Li Y, Zhang C, Yi H, Wu C, Wang J, et al. Downregulation of Beclin 1 and impairment of autophagy in a small population of colorectal cancer. Dig Dis Sci (2013) 58:2887–94. doi:10.1007/s10620-013-2732-8
137. Jo YK, Roh SA, Lee H, Park NY, Choi ES, Oh JH, et al. Polypyrimidine tract-binding protein 1-mediated downregulation of ATG10 facilitates metastasis of colorectal cancer cells. Cancer Lett (2016) 385:21–7. doi:10.1016/j.canlet.2016.11.002
138. Huang Y, Zhou J, Luo S, Wang Y, He J, Luo P, et al. Identification of a fluorescent small-molecule enhancer for therapeutic autophagy in colorectal cancer by targeting mitochondrial protein translocase TIM44. Gut (2016). doi:10.1136/gutjnl-2016-311909
139. Jing Z, Fei W, Zhou J, Zhang L, Chen L, Zhang X, et al. Salvianolic acid B, a novel autophagy inducer, exerts antitumor activity as a single agent in colorectal cancer cells. Oncotarget (2016). doi:10.18632/oncotarget.11385
140. Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer (2010) 46:1900–9. doi:10.1016/j.ejca.2010.02.021
141. Schonewolf CA, Mehta M, Schiff D, Wu H, Haffty BG, Karantza V, et al. Autophagy inhibition by chloroquine sensitizes HT-29 colorectal cancer cells to concurrent chemoradiation. World J Gastrointest Oncol (2014) 6:74–82. doi:10.4251/wjgo.v6.i3.74
142. Park JM, Huang S, Wu TT, Foster NR, Sinicrope FA. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther (2013) 14:100–7. doi:10.4161/cbt.22954
143. Han Y, Xue XF, Shen HG, Guo XB, Wang X, Yuan B, et al. Prognostic significance of Beclin-1 expression in colorectal cancer: a meta-analysis. Asian Pac J Cancer Prev (2014) 15:4583–7. doi:10.7314/APJCP.2014.15.11.4583
144. Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res (2013) 19:2995–3007. doi:10.1158/1078-0432.CCR-12-1542
145. Wang L, Wang Y, Lu Y, Zhang Q, Qu X. Heterozygous deletion of ATG5 in Apc(Min/+) mice promotes intestinal adenoma growth and enhances the antitumor efficacy of interferon-gamma. Cancer Biol Ther (2015) 16:383–91. doi:10.1080/15384047.2014.1002331
146. Xia YF, Ye BQ, Li YD, Wang JG, He XJ, Lin X, et al. Andrographolide attenuates inflammation by inhibition of NF-kappa B activation through covalent modification of reduced cysteine 62 of p50. J Immunol (2004) 173:4207–17. doi:10.4049/jimmunol.173.6.4207
147. Zhou J, Zhang S, Ong CN, Shen HM. Critical role of pro-apoptotic Bcl-2 family members in andrographolide-induced apoptosis in human cancer cells. Biochem Pharmacol (2006) 72:132–44. doi:10.1016/j.bcp.2006.04.019
148. Jada SR, Matthews C, Saad MS, Hamzah AS, Lajis NH, Stevens MF, et al. Benzylidene derivatives of andrographolide inhibit growth of breast and colon cancer cells in vitro by inducing G(1) arrest and apoptosis. Br J Pharmacol (2008) 155:641–54. doi:10.1038/bjp.2008.368
149. Zhang C, Qiu X. Andrographolide radiosensitizes human ovarian cancer SKOV3 xenografts due to an enhanced apoptosis and autophagy. Tumour Biol (2015) 36:8359–65. doi:10.1007/s13277-015-3578-9
150. Liu W, Guo W, Guo L, Gu Y, Cai P, Xie N, et al. Andrographolide sulfonate ameliorates experimental colitis in mice by inhibiting Th1/Th17 response. Int Immunopharmacol (2014) 20:337–45. doi:10.1016/j.intimp.2014.03.015
151. Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy (2014) 10:972–85. doi:10.4161/auto.28374
152. Shaker ME, Ashamallah SA, Houssen ME. Celastrol ameliorates murine colitis via modulating oxidative stress, inflammatory cytokines and intestinal homeostasis. Chem Biol Interact (2014) 210:26–33. doi:10.1016/j.cbi.2013.12.007
153. Jia Z, Xu C, Shen J, Xia T, Yang J, He Y. The natural compound celastrol inhibits necroptosis and alleviates ulcerative colitis in mice. Int Immunopharmacol (2015) 29:552–9. doi:10.1016/j.intimp.2015.09.029
154. Lin L, Sun Y, Wang D, Zheng S, Zhang J, Zheng C. Celastrol ameliorates ulcerative colitis-related colorectal cancer in mice via suppressing inflammatory responses and epithelial-mesenchymal transition. Front Pharmacol (2015) 6:320. doi:10.3389/fphar.2015.00320
155. Zhao J, Sun Y, Shi P, Dong JN, Zuo LG, Wang HG, et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int Immunopharmacol (2015) 26:221–8. doi:10.1016/j.intimp.2015.03.033
156. Crisan TO, Plantinga TS, Van De Veerdonk FL, Farcas MF, Stoffels M, Kullberg BJ, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One (2011) 6:e18666. doi:10.1371/journal.pone.0018666
157. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286:9587–97. doi:10.1074/jbc.M110.202911
158. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi:10.1038/ni.2215
159. De Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A (2014) 111:3526–31. doi:10.1073/pnas.1322831111
160. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature (1993) 365:61–5. doi:10.1038/365061a0
161. Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm Bowel Dis (2009) 15:1678–85. doi:10.1002/ibd.20960
162. Singh UP, Singh NP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Cannabinoid receptor-2 (CB2) agonist ameliorates colitis in IL-10(-/-) mice by attenuating the activation of T cells and promoting their apoptosis. Toxicol Appl Pharmacol (2012) 258:256–67. doi:10.1016/j.taap.2011.11.005
163. Tourteau A, Andrzejak V, Body-Malapel M, Lemaire L, Lemoine A, Mansouri R, et al. 3-Carboxamido-5-aryl-isoxazoles as new CB2 agonists for the treatment of colitis. Bioorg Med Chem (2013) 21:5383–94. doi:10.1016/j.bmc.2013.06.010
164. El Bakali J, Muccioli GG, Body-Malapel M, Djouina M, Klupsch F, Ghinet A, et al. Conformational restriction leading to a selective CB2 cannabinoid receptor agonist orally active against colitis. ACS Med Chem Lett (2015) 6:198–203. doi:10.1021/ml500439x
165. Blaney GP, Albert PJ, Proal AD. Vitamin D metabolites as clinical markers in autoimmune and chronic disease. Ann N Y Acad Sci (2009) 1173:384–90. doi:10.1111/j.1749-6632.2009.04875.x
166. Abreu MT, Kantorovich V, Vasiliauskas EA, Gruntmanis U, Matuk R, Daigle K, et al. Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn’s disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut (2004) 53:1129–36. doi:10.1136/gut.2003.036657
167. Lim WC, Hanauer SB, Li YC. Mechanisms of disease: vitamin D and inflammatory bowel disease. Nat Clin Pract Gastroenterol Hepatol (2005) 2:308–15. doi:10.1038/ncpgasthep0215
168. Simmons JD, Mullighan C, Welsh KI, Jewell DP. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut (2000) 47:211–4. doi:10.1136/gut.47.2.211
169. Ma Y, Zhang P, Wang F, Yang J, Liu Z, Qin H. Association between vitamin D and risk of colorectal cancer: a systematic review of prospective studies. J Clin Oncol (2011) 29:3775–82. doi:10.1200/JCO.2011.35.7566
171. Windmueller HG, Spaeth AE. Respiratory fuels and nitrogen metabolism in vivo in small intestine of fed rats. Quantitative importance of glutamine, glutamate, and aspartate. J Biol Chem (1980) 255:107–12.
172. Newsholme P. Why is l-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J Nutr (2001) 131:2515S–22S.
173. Kouznetsova L, Bijlsma PB, Van Leeuwen PA, Groot JA, Houdijk AP. Glutamine reduces phorbol-12,13-dibutyrate-induced macromolecular hyperpermeability in HT-29Cl.19A intestinal cells. JPEN J Parenter Enteral Nutr (1999) 23:136–9. doi:10.1177/0148607199023003136
174. Sukhotnik I, Khateeb K, Mogilner JG, Helou H, Lurie M, Coran AG, et al. Dietary glutamine supplementation prevents mucosal injury and modulates intestinal epithelial restitution following ischemia-reperfusion injury in the rat. Dig Dis Sci (2007) 52:1497–504. doi:10.1007/s10620-006-9629-8
175. Swaid F, Sukhotnik I, Matter I, Berkowitz D, Hadjittofi C, Pollak Y, et al. Dietary glutamine supplementation prevents mucosal injury and modulates intestinal epithelial restitution following acetic acid induced intestinal injury in rats. Nutr Metab (Lond) (2013) 10:53. doi:10.1186/1743-7075-10-53
176. Sakiyama T, Musch MW, Ropeleski MJ, Tsubouchi H, Chang EB. Glutamine increases autophagy under basal and stressed conditions in intestinal epithelial cells. Gastroenterology (2009) 136:924–32. doi:10.1053/j.gastro.2008.12.002
177. Massey DC, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut (2008) 57:1294–6. doi:10.1136/gut.2008.157297
178. Mutalib M, Borrelli O, Blackstock S, Kiparissi F, Elawad M, Shah N, et al. The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J Crohns Colitis (2014) 8:1730–4. doi:10.1016/j.crohns.2014.08.014
179. Matsuda C, Ito T, Song J, Mizushima T, Tamagawa H, Kai Y, et al. Therapeutic effect of a new immunosuppressive agent, everolimus, on interleukin-10 gene-deficient mice with colitis. Clin Exp Immunol (2007) 148:348–59. doi:10.1111/j.1365-2249.2007.03345.x
180. Dumortier J, Lapalus MG, Guillaud O, Poncet G, Gagnieu MC, Partensky C, et al. Everolimus for refractory Crohn’s disease: a case report. Inflamm Bowel Dis (2008) 14:874–7. doi:10.1002/ibd.20395
181. Reinisch W, Panes J, Lemann M, Schreiber S, Feagan B, Schmidt S, et al. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn’s disease. Am J Gastroenterol (2008) 103:2284–92. doi:10.1111/j.1572-0241.2008.02024.x
182. Pelissier-Rota MA, Pelosi L, Meresse P, Jacquier-Sarlin MR. Nicotine-induced cellular stresses and autophagy in human cancer colon cells: a supportive effect on cell homeostasis via up-regulation of Cox-2 and PGE(2) production. Int J Biochem Cell Biol (2015) 65:239–56. doi:10.1016/j.biocel.2015.06.013
183. Suh HW, Kim JK, Kim TS, Jo EK. New insights into vitamin D and autophagy in inflammatory bowel diseases. Curr Med Chem (2016). doi:10.2174/0929867323666161202151856
184. Sun J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy (2016) 12:1057–8. doi:10.1080/15548627.2015.1072670
185. Zhao J, Dong JN, Wang HG, Zhao M, Sun J, Zhu WM, et al. Docosahexaenoic acid attenuated experimental chronic colitis in interleukin 10-deficient mice by enhancing autophagy through inhibition of the mTOR pathway. JPEN J Parenter Enteral Nutr (2015). doi:10.1177/0148607115609308
186. Baxt LA, Xavier RJ. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology (2015) 149:553–62. doi:10.1053/j.gastro.2015.06.046
187. Lapaquette P, Guzzo J, Bretillon L, Bringer MA. Cellular and molecular connections between autophagy and inflammation. Mediators Inflamm (2015) 2015:398483. doi:10.1155/2015/398483
Keywords: autophagy, Paneth cell, macrophage, goblet cell, inflammatory bowel disease, immune reaction
Citation: Ke P, Shao B-Z, Xu Z-Q, Chen X-W and Liu C (2017) Intestinal Autophagy and Its Pharmacological Control in Inflammatory Bowel Disease. Front. Immunol. 7:695. doi: 10.3389/fimmu.2016.00695
Received: 19 November 2016; Accepted: 28 December 2016;
Published: 09 January 2017
Edited by:
Kai Fang, University of California Los Angeles, USACopyright: © 2017 Ke, Shao, Xu, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chong Liu, d2FubGMyMDA0QGFsaXl1bi5jb20=;
Xiong-Wen Chen, eGNoZW4wMDFAZ21haWwuY29t
†These authors have contributed equally to this work.