 Carlos de Torre-Minguela
Carlos de Torre-Minguela Pablo Mesa del Castillo1,2
Pablo Mesa del Castillo1,2 Pablo Pelegrín
Pablo Pelegrín- 1Unidad de Inflamación Molecular, Instituto Murciano de Investigación Biosanitaria-Virgen de la Arrixaca (IMIB-Arrixaca), CIBERehd, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain
- 2Unidad de Reumatología Pediátrica, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain
Inflammasomes are multiprotein complexes that critically control different aspects of innate and adaptive immunity. Among them we could highlight the release of pro-inflammatory cytokines that induce and maintain the inflammatory response. Usually, inflammasomes result from oligomerization of a nucleotide-binding domain-like receptor (NLR) after sensing different pathogenic or endogenous sterile dangerous signals; however, other proteins such as absent in melanoma 2, retinoic acid-inducible gene I, or pyrin could also form inflammasome platforms. Inflammasome oligomerization leads to caspase-1 activation and the processing and release of the pro-inflammatory cytokines, such as interleukin (IL)-1β and IL-18. Mutations in different inflammasomes are causative for multiple periodic hereditary syndromes or autoinflammatory diseases, characterized by acute systemic inflammatory flares not associated with infections, tumors, or autoimmunity. This review focuses on germline mutations that have been described in cryopyrin-associated periodic syndrome (CAPS) for NLRP3 or in familial Mediterranean fever (FMF) and pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND) for MEFV. Besides the implication of inflammasomes in autoinflammatory syndromes, these molecular platforms are involved in the pathophysiology of different illnesses, including chronic inflammatory diseases, degenerative processes, fibrosis, or metabolic diseases. Therefore, drug development targeting inflammasome activation is a promising field in expansion.
Danger Signals, Inflammasomes, and the Physiological Significance of the Inflammatory Response
Inflammation is the response of the innate immune system to a noxious stimulus, including infections or tissue damage (1, 2). Characterization of inflammasomes represents a considerable advance in the understanding of the inflammatory molecular events that occur in response to infections, and importantly, to tissue damage in the absence of pathogens. Furthermore, inflammasome activation has also been attributed to changes on physiological homeostatic parameters, such as changes in extracellular osmolarity (3, 4), and virtually, any perturbation in homeostasis could generate a local or systemic inflammatory response (1, 2). Tissue damage and alteration of the homeostatic parameters induce the release of danger signals from the cells that activate the inflammasome in innate immune cells (5). Danger signals are usually referred as danger or damage-associated molecular patterns (DAMPs). The dual use of the term “danger” or “damage” in the acronym DAMP denotes that danger signals are not only released after damaging conditions but also in response to dangerous situations, such as during cellular environment alterations. In homeostasis, cells in tissues are in a physiological “basal” state maintained by nutrients, oxygen, growth factors, and adherence to other cells and the extracellular matrix. Changes in environmental parameters (temperature, osmolarity, oxygen, or pH) induce a cellular stress response and the subsequent release of DAMPs. Stress is then recognized by tissue-resident macrophages, activating different signaling pathways, including inflammasomes, and inducing an inflammatory response aimed to restore tissue functionality during noxious conditions. This inflammatory response was termed para-inflammation by Medzhitov (1). Deregulation of para-inflammation is intimately related with immunity and involved in the pathogenesis of immune-mediated diseases, being the base for the chronic low-level inflammation associated, for example, to type 2 diabetes (6). If homeostasis imbalance continues or is complicated with infection, cells become necrotic inducing an acute inflammatory response that will damage the tissue (7).
Damage-associated molecular patterns are intracellular components released to the extracellular milieu in response to cell stress or necrosis that activates different inflammatory pathways, such as inflammasomes. Inflammasomes are multimeric complex of innate immune receptors, activating caspase-1 and proteolytic mechanisms involved in pro-inflammatory cytokines [interleukin (IL)-1β and IL-18] (8). During cell stress, plasma membrane becomes permeable to ions, such as K+, or to intracellular metabolites, such as the nucleotide adenosine triphosphate (ATP) or uric acid (1). One of the best characterized DAMP is ATP, since in physiological homeostatic conditions, ectonucleotidases maintain low extracellular ATP concentration, but during necrosis or inflammatory conditions, a high extracellular ATP concentration is reached, and the purinergic P2X7 receptor is activated in macrophages (9–12). P2X7 receptor is a potent activator of the inflammasome in macrophages and other innate immune cells (9). Leakage of cellular proteins with intracellular functions is another example of DAMPs; the release of these proteins usually follows secretory pathways independent of the endoplasmic reticulum (ER) and Golgi apparatus. Activation of caspase-1 by inflammasomes controls the release of these intracellular proteins by activating different unconventional release pathways, including a particular type of cell death called pyroptosis (1, 13, 14). Caspase-1 ultimately controls the release of inflammasome particles, a signal produced to amplify the release of DAMPs by activating caspase-1 in neighbor cells (11, 15). The high mobility group box 1 (HMGB1) nuclear protein is another example of DAMP released upon caspase-1 activation. HMGB1 presents histone-binding properties in the nucleus, and in the extracellular milieu, HMGB1 engages the advanced glycation end-product-specific receptor in conjunction with toll-like receptors (TLR) to induce an inflammatory response (16). In conclusion, innate immunity mechanisms converge in producing an inflammatory response as a consequence of infection, tissue damage, or loss of homeostasis.
Inflammasome Sensor Proteins
The nucleotide-binding domain-like receptor (NLR) family forms the main group of proteins considered as inflammasome sensors. These proteins contain a pyrin domain (PYD) or a caspase activation and recruitment domain (CARD). The presence of one of these domains in the sensor protein is required to assemble the inflammasome. Additionally, other proteins with some of these structural domains can also form functional inflammasomes, like absent in melanoma 2 (AIM2) protein, interferon-inducible protein 16 (IFI-16), retinoic acid-inducible gene I (RIG-I), and pyrin (17) (Figure 1).
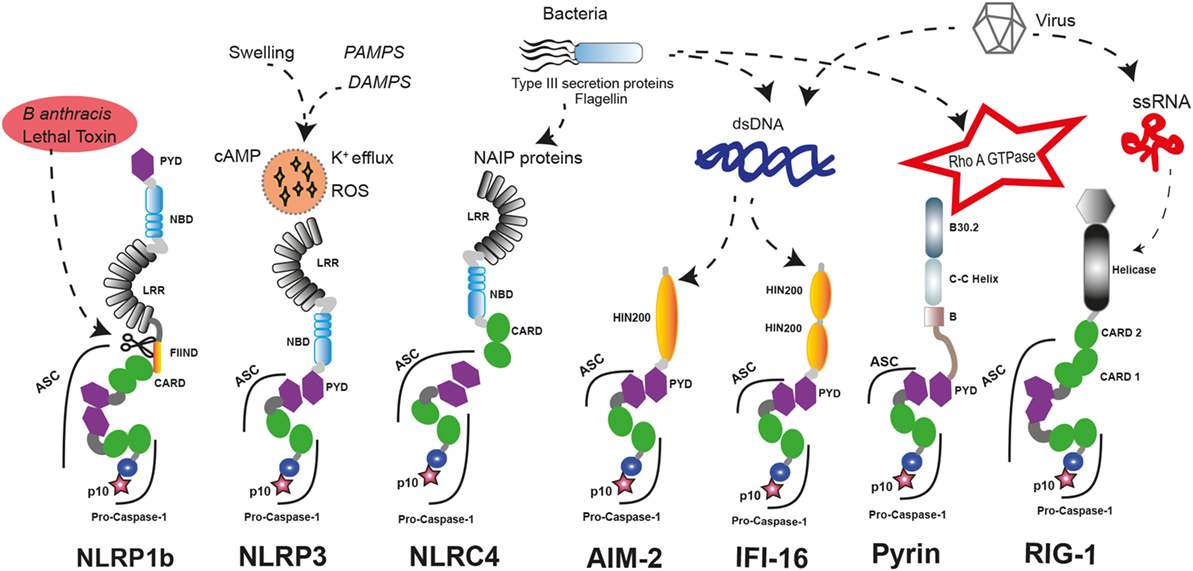
Figure 1. Inflammasome sensors and activators. A wide variety of pathogenic ligands and intracellular mediators are involved in inflammasome assembly. NLRP1b responds to proteolytic cleavage on their N-terminal induced by lethal toxin of Bacillus anthracis. NLRP3 is a general sensor of cellular damage that responds to intracellular harm induced by pathogenic or sterile insults. NLRC4 recognizes bacterial proteins via NLR family-apoptosis inhibitory proteins (NAIPs) and can assemble inflammasomes with or without recruiting ASC, similar to NLRP1b. Absent in melanoma 2 (AIM2) and interferon-inducible protein 16 (IFI-16) sense dsDNA through their HIN-200 domains; meanwhile, RIG-1 activates caspase-1 through an inflammasome assembly after it detects ssRNA. Pyrin inflammasome is induced by bacterial toxins that modify RhoA GTPase. DAMPs, danger-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; ssRNA, single strand RNA, dsDNA, double strand DNA.
There are different inflammasome sensors dedicated to recognize the presence of cytosolic nucleic acids. AIM2 presents an N-terminal PYD and a C-terminal hematopoietic interferon (IFN)-inducible nuclear protein with 200-amino acid repeat (HIN-200) domain. AIM2 is critical to respond against the infection of different pathogens by forming an inflammasome after recognition of double-stranded DNA (dsDNA) in the cytoplasm by the HIN-200 domain (18–20). Interestingly, other nucleic acid sensor protein called IFI-16 has two C-terminal HIN-200 domains and one N-terminal PYD. Upon detection of dsDNA, IFI-16 triggers the IFN response as a component of the signaling pathway (21) and can also induce the assembly of inflammasome with ulterior caspase-1 activation (22). RIG-I is also a sensor for viral RNA that contains two CARD domains and is able to assemble an inflammasome (23). However, it should be noted that additional studies are required to demonstrate that IFI-16 and RIG-I can form an inflammasome.
The structure of the sensor protein family NLR presents a central nucleotide-binding domain (NBD), and most of them have a C-terminal leucine-rich repeat (LRR) domain. The N-terminal protein domain is used to classify this group of proteins in NLRP if it contains a PYD domain or NLRC if it contains a CARD domain (24). Interestingly, the capacity for assembling inflammasome is a feature that has not been described for all members of the NLR family. These sensor proteins are also involved in other aspects of innate immune response by regulating diverse non-inflammasome pathways. Indeed, NLRP12 can play a role as a negative regulator of NF-κB signaling (25) or modulating IL-4 production in T cells (26), and NLRP6 is a negative regulator of mucosal immunity in the gut (27, 28).
The first sensor protein identified to form inflammasome was NLRP1 (29). Interestingly, human NLRP1 contains two additional protein domains compared to the canonical domains of the NLR family, such as a function-to-bind domain and a C-terminal CARD. These domains seem to play a critical role to assemble functional inflammasomes, as proteolytic cleavage of their N-terminal by pathogen components of Bacillus anthracis is required for their activation (30, 31). Furthermore, the presence of a CARD domain in the C-terminal allows the direct interaction and activation of caspase-1 without the presence of any other adaptor proteins like the apoptosis speck-like protein with a CARD domain (ASC), even though ASC incorporation to the platform enhances the processing of IL-1β (32), and in human THP-1 monocyte cell line, ASC is required for NLRP1 activation (33). In contrast, mouse NLRP1a could form an inflammasome independent of ASC (34).
A genetic study of families with vitiligo with or without other autoimmune diseases has revealed a link between these autoimmune disorders and the presence of polymorphisms in NLRP1 gene (35). Recently, a novel gain-of-function mutation in NLRP1 gene that predisposes to inflammasome activation has been associated with NLRP1-associated autoinflammation with arthritis and dyskeratosis autoinflammatory syndrome (36). This syndrome is characterized by diffuse skin dyskeratosis, autoinflammation, autoimmunity, arthritis, and elevated transitional B-cells (36) (Table 1). Furthermore, NLRP1 mutations have been implicated in non-fever inflammasome-related disorders, in particular with two overlapping skin disorders: multiple self-healing palmoplantar carcinoma and familial keratosis lichenoides chronica, demonstrating that NLRP1 has an important role controlling skin inflammation (33).
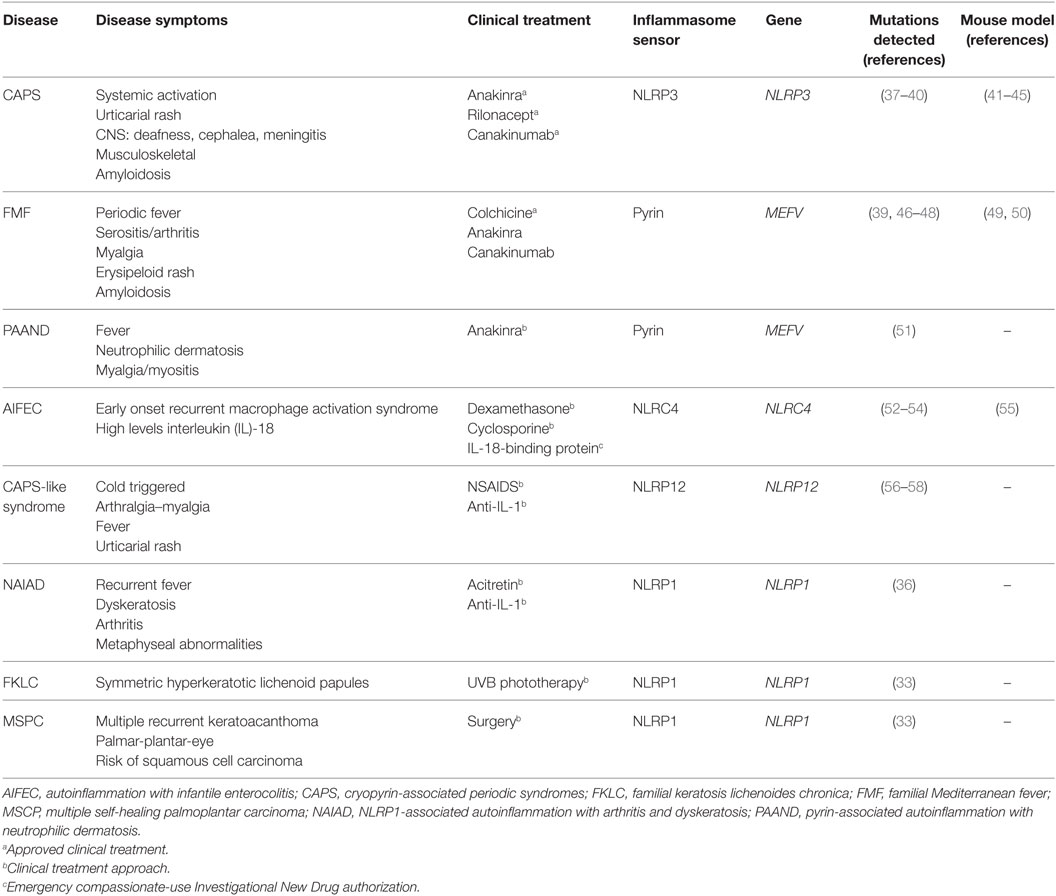
Table 1. Molecular and clinical features of autoinflammatory diseases associated with mutations in inflammasome sensor proteins.
The most prominent member of NLR family in the study of hereditary autoinflammatory syndromes is NLRP3. Indeed, gain-of-function mutations on NLRP3 gene have been identified in patients with cryopyrin-associated periodic syndromes (CAPS, see below) (59, 60) (Table 1). NLRP3 contains the three canonical domains described in the NLRP family: PYD, NBD, and LRR, and it is able to assemble a functional inflammasome in response to a wide variety of triggers, suggesting that it could be a global sensor of cellular damage and different pathogens (5).
Besides NLRP3, formation of active inflammasomes triggered by a bacterial infection has only been described in vitro for other two members of NLRP family: NLRP7 (61) and NLRP12 (62). Interestingly, NLRP12 displays a sequence similar to NLRP3, and it is predominantly expressed in myeloid-monocytic cells (63). In some cases, genetic studies of symptomatic patients with CAPS-like syndrome without mutations in NLRP3 revealed the presence of mutations in NLRP12 gene (56, 57). In vitro study of these NLRP12 variants has shown an increase in the activity of caspase-1 and the secretion of IL-1β, suggesting the potential role of NLRP12 mutations in CAPS-like syndrome-associated inflammation (Table 1) (58, 64).
NLRC4 is another well-known member of the NLR family assembling functional inflammasomes in response to pathogens. NLRC4 is a component of a detection system for bacterial proteins such as flagellin and several components of the type III secretion system (65, 66). As a member of the NLRC subgroup, NLRC4 contains a C-terminal CARD besides of NBD and LRR domains, but unlike other NLR sensor proteins, NLRC4 requires of sensors co-receptors, termed NLR family-apoptosis inhibitory proteins (NAIPs), that recognize the pathogen proteins in the cytoplasm and oligomerize NLRC4 (67, 68). Similar to NLRP1, NLRC4 could interact directly with pro-caspase-1 through their CARD domain generating an inflammasome with a less efficient state of activation, and the association with the adaptor protein ASC is important to amplify the activation of caspase-1 (69). Gain-of-function mutations in NLRC4 gene are associated with early onset autoinflammation with enterocolitis or recurrent macrophage activation syndrome depending on the mutation (Table 1) (52, 54). These patients are characterized by mutations in the NBD region of NLRC4 and benefits from recombinant human IL-18-binding protein therapy (53). The autoinflammatory-associated NLRC4 mutation H443P is able to constitutively activate caspase-8 and induce apoptosis via interaction with the component of the 26S proteasome Suppressor of Gal 1 and with ubiquitinated cellular proteins (70).
All inflammasome sensor proteins are activated in response to different pathogen and danger signals, suggesting that each activator triggers the formation of its own particular inflammasome complex. Interestingly, a recent work describes the recruitment of two sensor proteins (NLRC4 and NLRP3) to the same inflammasome complex as a result of the recognition of different danger signals from the same pathogenic infection (71).
Pyrin is another important inflammasome-forming protein (72). This protein contains an N-terminal PYD domain that is responsible for their interaction with ASC and later activation of caspase-1, a central coiled-coil domain and a C-terminal B30.2/SPRY domain that is not present in the mouse orthologous protein. The pyrin-inflammasome assembly could be triggered after sensing the activity of bacterial toxins from different species that covalently modify switch-I region of Rho family proteins (73). In addition, mutations in the gene that codify pyrin, MEFV gene, are found in symptomatic patients with hereditary autoinflammatory disorders (see below and Table 1) (46).
Inflammasome Adaptor and Effector Protein Assembly
Inflammasome sensor proteins are involved in the recognition of particular danger stimulus and then initiate the assembly of inflammasome multimeric complex; in most inflammasomes, the interaction with an adaptor protein is required to enhance the activation of caspase-1. The protein ASC (also known as Pycard) is the ubiquitous adaptor for inflammasomes, and its interaction with the active inflammasome sensor protein induces a prion-like oligomerization process essential for the final structural conformation of the inflammasome. ASC is composed by two death-fold domains, a N-terminus PYD and a C-terminus CARD (74, 75). For those inflammasome sensor proteins associated with autoinflammatory disorders, i.e., NLRP3 or pyrin, their PYD domain is responsible for ASC recruitment via PYD–PYD homotypic interactions inducing the formation of filamentous structures that assemble into a large protein aggregate (76). Caspase-1 activation occurs within this aggregate, and interestingly, the same process of polymerization for ASC and pro-caspase-1 has been shown independent of the inflammasome sensor protein activated (77).
Recent works have provided additional information about the interactions between the components of the inflammasome, suggesting an initial self-nucleation of the sensor protein (NLRP3 or AIM2) promoting the assembly of helical ASC filaments via PYD homotypic interaction (78, 79). These ASC filaments, generated after multiple PYD interactions, expose CARD domains in the outer part of the filament and consolidate the inflammasome aggregation with an appropriated cross-linking between filaments via CARD–CARD interactions (80). The multiple oligomerization of pro-caspase-1 with the ASC filaments also occurs via CARD–CARD interactions and amplifies the danger signal started by the sensor protein (81).
NLRP3 Inflammasome Activation Pathways
The activation of NLRP3 inflammasome appears in response to infection and is amplified by danger signals triggered during the infection, or by tissue injury or alterations in tissue homeostasis without infection. As it was described before, the majority of inflammasome sensor proteins are able to recognize different pathogen-associated molecules (bacterial proteins, toxins, and nucleic acids) and therefore activate inflammasome assembly in response to a microbial or viral infection. NLRP3 sensor is particularly able to oligomerize in response to a wide variety of stimuli that include pathogen molecules such as bacterial cell wall components or pore-forming toxins (nigericin and maitotoxin), endogenous danger signals like extracellular ATP, amyloid-β aggregates, uric acid crystals, or metabolic dysfunction, and pollutant particles as silica, asbestos, or alum (5, 82). The direct interaction between this broad range of activators and NLRP3 seems unlikely, and therefore it is suggested that NLRP3 is able to sense the cellular stress associated with the exposition to these agents. The precise molecular mechanism involved in the NLRP3 inflammasome activation remains elusive although recent studies begin to uncover the molecules and the cellular machinery responsible for this process (17, 83, 84).
Maintenance of ion gradients between different cellular compartments and between the cytosol and the extracellular environment is a feature of all living cells. Any alteration of this homeostasis will induce molecular mechanisms to respond and adapt to this aggression. Significant decrease of intracellular K+ is indeed detected during NLRP3 activation after the treatment with microbial pore-forming toxins or after P2X7 receptor engagement by extracellular ATP (85), where the hemichannel pannexin-1 plays a critical role (86). Interestingly, decrease of intracellular K+ is also detected during the NLRP3 inflammasome activation along with other sterile inductors as the decrease of osmolarity (3) or metabolic lipids (87), suggesting that intracellular K+ concentrations could be one of the common mechanisms involved in the activation of the NLRP3 inflammasome; however, its mechanism of function is not well understood (88–90).
In addition to the decrease of intracellular K+, a mobilization of Ca+2 in the cytosol is also detected in most of the stimulus that activates NLRP3. The ER is the main reservoir for intracellular Ca+2, and its mobilization as a consequence of the activation of inositol trisphosphate receptor has been observed during NLRP3 activation induced with different stimuli. The activation of P2X7 receptor also induces an influx of Ca+2 from the extracellular space; however, in this cellular context, the blockage of extracellular Ca+2 influx does not inhibit NLRP3 inflammasome, and artificial mobilization of Ca+2 is not sufficient to trigger the NLRP3 inflammasome activation in absence of K+ depletion (12, 14, 91). Cell swelling after hypotonic shock activates transient receptor potential cation channels (TRPM7 and TRPV2) involved in the modulation of intracellular Ca+2 that is crucial for the transforming growth factor beta-activated kinase 1 activation. These molecular events are required in combination with K+ efflux for NLRP3 inflammasome assembly (3). In addition, several works show evidences that extracellular Ca+2 can trigger mechanisms that activate inflammasome through G protein-coupled receptors (92, 93). The activation of these receptors leads to the mobilization of intracellular Ca+2 via phospholipase C activation with a concomitant reduction of cyclic AMP (cAMP) (92). The effect of this reduction in cAMP will be discussed later in the context of the negative regulation mechanisms of NLRP3. Interestingly, elevated concentrations of extracellular Ca+2 have been detected at infection sites or in ischemic injury, suggesting that extracellular Ca+2 would play a role as a DAMP (93).
Alteration of lysosomal function after phagocytosis of molecular crystals has been described as an additional activation process of NLRP3 inflammasome, possibly as a consequence of the activity of released lysosomal proteases altering the integrity of cellular organelles (94). Furthermore, other cellular stress associated with the intracellular ionic mobilization, as the induction of ER stress, is able to activate NLRP3 inflammasome in a K+ efflux-dependent manner. In this process, the endoribonuclease inositol-requiring enzyme 1α, an unfolded protein sensor expressed in ER, is required to activate the NLRP3 inflammasome (95, 96). Taken together, these data show that changes in intracellular ion concentration play a key role in the activation of NLRP3 inflammasome, although their precise molecular mechanism remains unclear.
Besides ion fluxes, changes in the cellular oxidative state is a common process detected during NLRP3 inflammasome activation, being mitochondrial damage one of the main source of reactive oxygen species (ROS) (97). Interestingly, several works link mitochondrial ROS production with changes in the intracellular concentration of K+ and Ca+2, which would induce depolarization of the mitochondrial membrane (91, 98). Mitochondrial ROS production has also been described as a novel NLRP3 activation mechanism involving a decrease of NADH levels after disruption of the glycolytic flux (82). Mitochondria have also been suggested as a cellular platform to assemble the NLRP3 inflammasome. The activation of NLRP3 induces its relocation from the ER to the proximity of the mitochondria in the perinuclear environment (97, 99). This recruitment requires the reorganization of the microtubule system (100). Moreover, the mitochondria may also release other molecules implicated in the activation of NLRP3 inflammasome as cardiolipin (101) or oxidized mitochondrial DNA (102, 103), and it has been shown that mitochondrial antiviral-signaling protein interacts with the PYD of NLRP3, being essential for their activation after the stimulation with ATP or nigericin but not with crystals (99). All these data point out the essential role of mitochondria in NLRP3 inflammasome activation.
Finally, caspase-4, and its mouse orthologous caspase-11, activates NLRP3 after recognition of cytosolic LPS (104, 105). This signaling is known as the non-canonical NLRP3 inflammasome activation pathway, and although the mechanism of caspase-4-inducing NLRP3 activation is not known, it is also dependent on the decrease of intracellular K+ (106–108).
Regulatory Mechanisms of NLRP3 Inflammasome
Several proteins have been described as positive or negative regulators of the NLRP3 inflammasome assembly (Figure 2). Guanylate-binding protein 5 binds via its GTPase domain to the PYD of NLRP3 during inflammasome activation by most of the stimuli except crystalline agents. This interaction promotes the oligomerization of NLRP3 with ASC (109). Furthermore, several works have described that, during ATP stimulation, NLRP3 deubiquitination mediated by the Lys63-specific deubiquitinase BRCC3 is an early process essential for inflammasome activation (110–112).
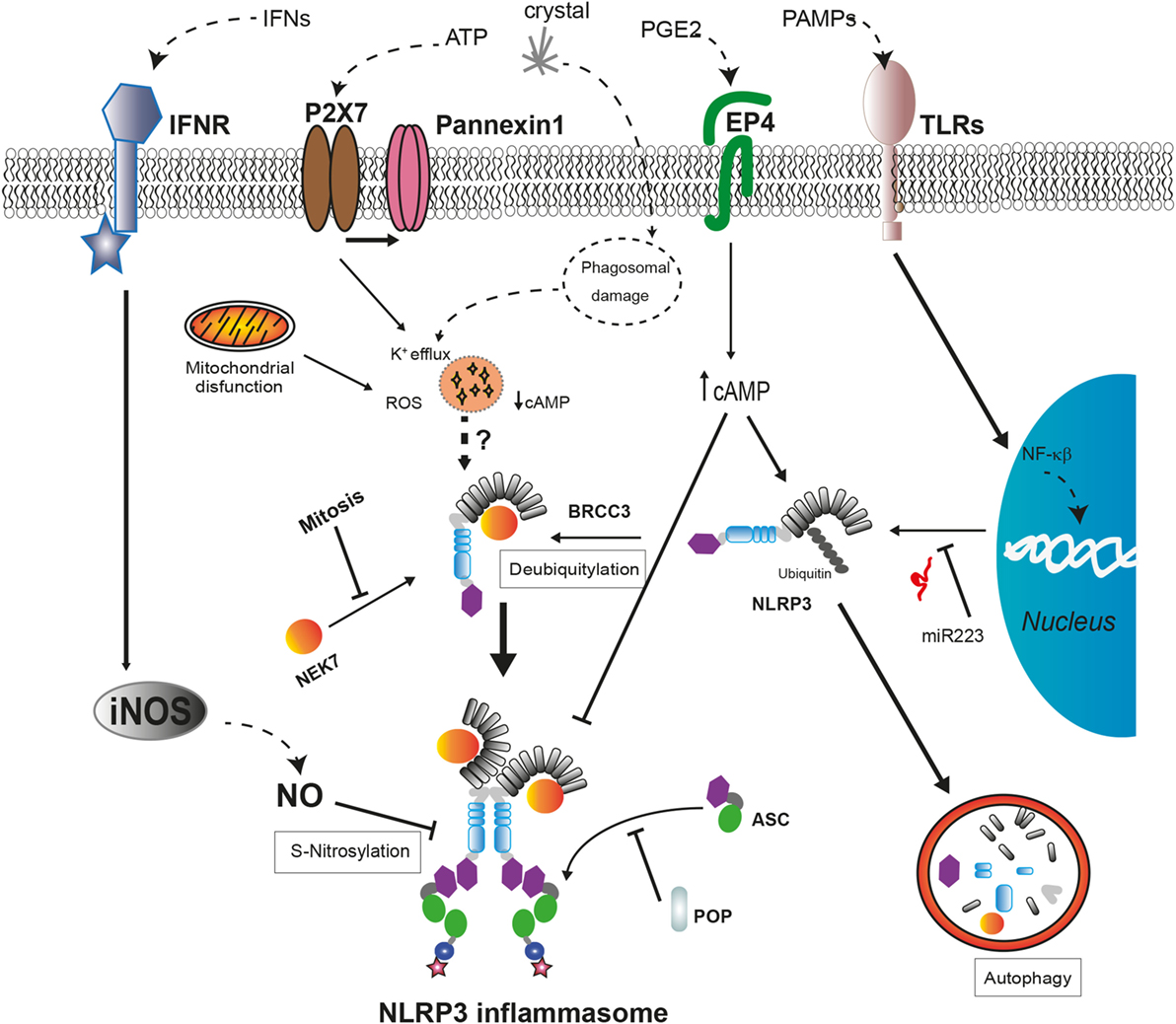
Figure 2. Regulatory mechanisms of NLRP3 inflammasome assembly. The expression levels of NLRP3 are regulated by miR-223 in basal conditions but can be upregulated after cell recognition of pathogen-associated molecular patterns that induce NF-κB signaling pathway. The activation of NLRP3 by cellular damage signals as intracellular K+ decrease or reactive oxygen species (ROS) production requires a deubiquitylation of NLRP3 by BRCC3 and also their interaction with NEK7, only available during interphase. External signals, such as interferons (IFNs) or prostaglandin E2 (PGE2), negatively regulate NLRP3 through different mechanisms. The increase in nitric oxide (NO) produced by inducible nitric oxide synthase (iNOS) leads to the S-nitrosylation of NLRP3 impairing the assembly of the NLRP3 inflammasome. The increase of cyclic AMP (cAMP) induced by PGE2 signaling via prostaglandin E2 receptor 4 (EP4) activates the phosphorylation of NLRP3 reducing its oligomerization and increasing its ubiquitination to be degraded in autophagosomes. The interaction with pyrin domain-only proteins (POPs) can regulate NLRP3 inflammasome assembly by sequestering ASC or NLRP3.
Recent works have revealed a new NLRP3 inflammasome regulatory molecule, the never-in-mitosis A-related kinase 7 (NEK7), a serine, and threonine kinase required for mitosis progression (113). This protein interacts with the LRR domain of NLRP3 upstream of NLRP3 inflammasome assembly independent of their kinase activity (114). This interaction is required for NLRP3 inflammasome oligomerization and introduces a new component of inflammasome regulation, the restriction of NLRP3 inflammasome formation to cells in interphase (115). Moreover, the absence of NEK7 in cellular models harboring frequent CAPS-associated mutations in NLRP3 reduces their ability to activate caspase-1, while the association between NEK7 and mutant NLRP3 is stronger (114, 115). Further investigation is required to elucidate the role of NEK7 in the auto-activation of NLRP3 inflammasome in autoinflammatory syndromes.
Maintenance of low NLRP3 protein levels avoids the auto-assembly of NLRP3 inflammasome in the absence of a danger stimulus; therefore, transcriptional regulation of NLRP3 is an additional control mechanism to avoid unexpected inflammasome activation. Transcriptional regulation of NLRP3 requires NF-κB activation by TLR or IL-1 receptor type I (IL-1RI) signaling to increase NLRP3 protein concentration to certain level that can be activated after sensing a triggering stimulus (116, 117). Furthermore, the amount of NLRP3 mRNA is tightly regulated in myeloid cells through the microRNA miR-223, although this miRNA is not regulated by pro-inflammatory signals (118). In addition, under unstimulated conditions, NLRP3 is inhibited by posttranslational modifications with ubiquitin chains that also target NLRP3 for its degradation through proteasome or autophagy as will be described later (112). Other mechanism involved in the inhibition of the NLRP3 activity is the posttranslational modification of NLRP3 generated by the activation of inducible nitric oxide synthase. The increase of nitric oxide leads to the S-nitrosylation of NLRP3 impairing the assembly of the inflammasome, and this mechanism is suggested as a protective mechanism (119, 120). Therefore, the control of functional NLRP3 concentration within the cell is crucial for the activation of the inflammasome.
In addition to these negative regulatory mechanisms, two families of proteins containing CARD (COPs) or PYD (POPs) that could sequester either sensor proteins or effector proteins through PYD–PYD and CARD–CARD interactions have been described (121). In the absence of mutations, pyrin is also suggested to be a key regulator for the degradation of several inflammasome components (caspase-1, NLRP1, and NLRP3), preventing an excessive release of pro-inflammatory cytokines (122, 123). However, a recent work shows that the absence of pyrin in a mouse model leads to an increase in the release of IL-1β without affecting different inflammasome assembly (124). Therefore, the role of pyrin as an inflammasome inhibitor remains to be determined, and different domains among human and mouse pyrin proteins should be taken into account.
Cellular damage implicated in the activation of the NLRP3 inflammasome also activates autophagy, a mechanism involved in the clearance of intracellular pathogens and damaged organelles (102, 125). Autophagy is a negative mechanism to control the induction of the inflammatory response given its involvement in the degradation of damaged mitochondria, including molecular NLRP3 inflammasome inductors as mitochondrial DNA or ROS (102, 126, 127), the clearance of ASC specks (125), and pro-IL-1β (128). Ubiquitinated NLRP3 could be directed to the autophagosome for degradation by a complex with cAMP that recruits the E3 ubiquitin ligase MARCH7 (129, 130). This molecular mechanism can be triggered by activators of the adenylate cyclase as the neurotransmitter dopamine (130). Furthermore, an alternative negative regulatory mechanism has been described for NLRP3 inflammasome involving cAMP. The increase of cAMP induced by prostaglandin E2 signaling via prostaglandin E2 receptor 4 activates protein kinase A that phosphorylates NLRP3 in their NBD domain reducing its ATPase activity and oligomerization (131). Interestingly, this negative regulation could be disrupted by certain CAPS-associated mutations in the NBD of NLRP3 (114).
Pyrin Inflammasome Activation Pathways and Regulatory Mechanisms
Recent data begin to unveil the mechanism involved in pyrin-inflammasome activation, as well as a protective mechanism concerned in blocking pyrin-inflammasome assembly. The inactivation of the RhoA GTPase by bacterial modification induces the activation of the pyrin inflammasome (73), suppressing a protective mechanism that avoids pyrin inflammasome activation through their downstream phosphorylation by serine/threonine-protein kinase N1 and N2 (132). This mechanism requires the phosphorylation of certain amino acids of pyrin (S208 and S242 in human) allowing their binding to regulatory protein 14-3-3 and blocking the formation of the pyrin inflammasome (51, 132). Pyrin inflammasome activation through bacterial toxins is detected in human and mice, indicating that the C-terminal B30.2/SPRY domain, present only in human, is not required for their activation (133). Interestingly, this domain harbors most of the mutations detected in familial Mediterranean fever (FMF) patients, although some mutations affect one of the serine, in other domain of the protein, described as a key amino acid in the protective mechanism against the uncontrolled activation of pyrin inflammasome (see below). The inhibition of microtubule polymerization by colchicine abolishes pyrin inflammasome assembly induced by bacterial toxins, without affecting pyrin dephosphorylation (133, 134). However, this control of pyrin inflammasome activation leaded by microtubules is not effective in FMF patients that harbor mutations in B30.2/SPRY domain (133), suggesting that these mutations may force a protein conformation that aids the assembly of pyrin inflammasome after their dephosphorylation.
Inflammasome-Associated Secretome
The formation of inflammasome leads to the activation of caspase-1, and this protease triggers a broad number of cellular events as a consequence of its catalytic activity, including the release of cytosolic proteins associated with a specific type of cell death termed pyroptosis. Specifically, the analysis of the secretome associated with caspase-1 has revealed the key role of this protease in the unconventional secretion of multiple essential molecules involved in the inflammatory process. From them, the cytokine IL-1β is one of the most prominent and critical products of inflammasome activation, since it is a key regulator of the inflammatory response and is the most important current target of therapeutic treatments for autoinflammatory syndromes. The synthesis of IL-1β mRNA and the production of the inactive precursor form of IL-1β are strongly induced by microbial products such as LPS signaling via TLR or by IL-1 itself signaling through the IL-1RI (135). Caspase-1 is able to process the inactive precursor form of IL-1β to its mature bioactive form and induce its release. Similarly, caspase-1 is also responsible to the maturation of the inactive IL-18 precursor, another IL-1 family cytokine, to its bioactive form (136).
Both IL-1β and IL-18 are the canonical cytokines signaling downstream inflammasome activation, but beyond these caspase-1 substrates, caspase-1 also controls the unconventional release of other cytosolic proteins (FGF-2, thioredoxin, and annexins), lysosomal proteins (cathepsins and cystatins), or nuclear proteins (HMBG1, IL-1α) that are not direct substrates of the protease (13, 14). In addition, caspase-1 also controls the release of large complex protein aggregates as ASC inflammasome oligomers that are now able to spread caspase-1 activation to adjacent cells and maintain inflammasome signaling (11, 15).
Unconventional protein release induced by caspase-1 has been widely studied for IL-1β, a cytokine that does not follow the conventional route of protein secretion through ER or Golgi. Different mechanisms of unconventional secretion have been explored to explain this process including the release through exocytosis of secretory lysosomes (137) or extracellular vesicles released after NLRP3 inflammasomes activation (138, 139). Caspase-1-induced pyroptosis is associated with an increase in plasma membrane permeation that may help a passive release of IL-1β (140). The destabilization of cell membrane integrity during pyroptosis is induced by the cleavage of the cytosolic substrate gasdermin D by caspase-1 or caspase-4; gasdermin D N-terminus integrates into the plasma membrane forming pores (141–143). The application of membrane stabilizing agents, as the complex polyphenol punicalagin, prevents the execution phase of pyroptosis and release of mature IL-1β from macrophages after NLRP3 inflammasome activation, suggesting that loss of plasma membrane is involved in this secretion in parallel with cell death (144). Interestingly, the stabilization of the plasma membrane by punicalagin inhibits the release of IL-1β in neutrophils in the absence of cell death (144). Therefore, the secretion of bioactive form of IL-1β requires membrane permeation and may occur in secreting cells as neutrophils where NLRP3 inflammasome does not induce pyroptosis (145, 146). Initially, these mechanisms are not mutually exclusive and may participate in the secretion of IL-1β depending on the intensity of the stimulus and cell type. The release of other caspase-1-dependent secretome proteins is less studied, and the involvement of pyroptotic cell death in this process is not known, neither its contribution to the pathophysiology of autoinflammatory syndromes.
Implications of NLRP3 Inflammasome in CAPS
Cryopyrin-associated periodic syndromes are rare hereditary autosomal-dominant autoinflammatory diseases with an estimated prevalence of 1–3 cases per million of inhabitants (147, 148). They include familial cold urticaria syndrome (FCAS) (59), Muckle–Wells syndrome (MWS) (149), and chronic infantile neurological cutaneous and articular (CINCA) syndrome also known as neonatal onset multisystemic inflammatory disease (NOMID) (150). All three syndromes were independently described and latterly found to be caused by gain-of-function mutations in the NLRP3 gene, located in the short arm of chromosome 1 (1q44) (37, 38). Mutant NLRP3 drives a constitutive hyperactivity of inflammasome, activation of caspase-1, and an excessive unregulated release of IL-1β, although systemic circulating levels of IL-1β during inflammatory flares are in most cases undetectable (11, 147).
Clinical features of CAPS are related to systemic effects of IL-1β-inducing fever, malaise, fatigue, and chronic pain along with a blood serum rise of acute-phase reactants, such as C-reactive protein and serum amyloid A. CINCA/NOMID is characterized for an almost continuous early onset inflammatory state with fever and non-pruritic migratory urticaria-like rash; central nervous system symptoms and arthropathy are common. MWS shows a moderate phenotype with latter onset of fever, rash, arthralgia, conjunctivitis, uveitis, sensorineural deafness, and potentially life-threatening amyloidosis. FCAS is a milder familial condition characterized by febrile urticarial rash with headache, arthralgia, and sometimes conjunctivitis but no central nervous system symptoms and is typically triggered by cold exposure. FCAS, MWS, and CINCA/NOMID are considered a clinical continuum than distinct diseases as intermediate phenotypes occur; being FCAS the mildest and CINCA/NOMID the most severe forms (151). Neurologic manifestations in CAPS are common including headache, sensorineural hearing loss, myalgia, chronic aseptic meningitis, increased intracranial pressure, cerebral atrophy, seizures, and mental retardation (152). Musculoskeletal symptoms in CAPS are also very frequent; up to 86% may have any musculoskeletal manifestation during follow up, 30% at onset. In a large cohort, these included arthralgia in 86% and arthritis in 58%; joint destruction and typical knee deformities appeared rarely (9% and 2%, respectively). Tendinopathies occurred in 21.5%, tender points in 16.5%, and myalgia in 33% of patients (153).
To date, a total of 177 variants of NLRP3 gene have been included in infevers database (39), most of them located in exons 3 or 4 and intron 4. Among them, the most frequent is R260W (40, 148) and are associated with a milder phenotype along with A439V. The variants, T348M and D303N, and low frequency mutations are associated with a severe phenotype; E311K accounts for a high rate of hearing loss. On the other hand, Q703K or V198M variants have little clinical significance and are considered a functional polymorphism and a low penetrance variant, respectively. Clinically affected patients with no germline mutations could have an NLRP3 somatic mosaicism (154–156). The highly heterogeneous phenotypes within identical genotypes show the need for advancing the underlying understanding of the pathophysiological mechanisms.
The relevance of NLRP3 mutations as key players in the induction of these autoinflammatory syndromes has been explored in animal models. Specifically, the development of knock-in mouse strains harboring some of the frequent mutations detected in CAPS syndrome has demonstrated the pivotal role of IL-1β and the innate immunity in the pathogenesis of this syndrome; meanwhile, the adaptive immune system seems not to be involved (41). These animal models exhibit an autoinflammatory disease similar to that in humans associated with an inflammasome hyperactivation and unregulated release of IL-1β (41–43). Humanized mice expressing CAPS-associated mutation D305N present an increased sensitivity to endotoxin and develop progressive and debilitating arthritis (44). Furthermore, the study of these knock-in animal models has revealed the critical role of microbiota as inducer of disease, and myeloid and mast cells as cellular sources of IL-1β in the development of the skin inflammation (42). In addition, the study of these CAPS-like animals has revealed the key role of IL-18 in the early tissue inflammation and suggests the presence of other players beyond IL-1β and IL-18 that are involved in inflammatory activities associated with the pyroptosis and possible by the caspase-1-associated secretome (45). A recent study with blood monocytes from patients affected by CAPS detected a high level of cellular stress including elevated levels of ROS compared with healthy subjects (157). Interestingly, associated with this oxidative stress, there is a reduction in the production of the anti-inflammatory cytokine, IL-1 receptor antagonist (IL-1Ra) (158), as well as a decrease in the threshold of inflammasome activation of CAPS monocytes (159). The exposure of this monocyte to inflammatory stimuli such as LPS induces an increase in the release of ATP that produces an increase in the secretion of IL-1β, IL-18, and IL-1α (159). These data collectively suggest the involvement of genetic and environmental factors beyond a single mutation that needs to be explored to obtain a more accurate clinical picture of this disease.
The Pyrin Inflammasome and Implications in FMF and Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis (PAAND)
The inflammasome sensor protein pyrin is primarily expressed in myeloid cells, and wild-type pyrin negatively modulates NLRP3 inflammasome-dependent IL-1β release (160). However, mutations in the MEFV gene (that codify for pyrin) are associated with two clinically different autoinflammatory syndromes: FMF and PAAND (51); in both diseases, mutated pyrin associates with high serum IL-1β levels during febrile episodes.
Familial Mediterranean fever is the most common inherited monogenic autoinflammatory disease worldwide and is caused by loss-of-function mutations in MEFV gene, mostly affecting eastern Mediterranean population (161). Classically considered an autosomal recessive condition, it is actually discussed if it should be considered an autosomal-dominant disease with variable penetrance, since heterozygosis mutations are associated with clinical autoinflammatory FMF manifestations (162). Nevertheless, homozygosis is associated with severe FMF phenotypes.
Familial Mediterranean fever patients typically show recurrent self-limited acute febrile attacks of 1–3 days of duration, accompanied by inflammation of serosa and/or synovial linings (90% abdominal pain, 40% arthritis, and 30% thoracic pain), myalgia (40%), and erysipeloid type rash (20%). Onset before the age of 18 is common and has been associated with higher rates of arthritis, arthralgia, myalgia, and erysipeloid-like rash (163). Pleuritis, pericarditis, scrotal pain, aseptic meningitis, thrombosis, and vasculitis may appear during flares, but FMF can also be associated with many other disorders in a non-canonical manner (164). The most severe complication of FMF is amyloidosis as a result of chronically uncontrolled inflammation that occurs in undiagnosed or untreated patients; it is more likely to occur in patients with recurrent arthritis (165). Renal amyloidosis seldom occurs as the first clinical manifestation of FMF, and these individuals are referred as phenotype II patients (166). Homozygosis in serum amyloid A gene 1 (alpha/alpha) and male sex have shown influence on the risk of developing amyloidosis in FMF patients (167, 168).
Over 300 MEFV gene variants have been described (39), but only 14 occur frequently in FMF (E148Q, E167D, T267I, P369S, F479L, I591T, M680I, I692del, M694I, M694V, K695R, V726A, A744S, and R761H). The majority of pathogenic mutations are located in exon 10, being M694V the most frequent MEFV mutation encountered in FMF patients; its presence in homozygosis or compound heterozygosis is related to severe phenotype. In exon 2, E148Q is the most frequent MEFV variant in asymptomatic carriers and in some population subsets, it may even be a benign polymorphism (47); it is present in FMF patients with a mild phenotype (48). Diagnosis of FMF is sometimes elusive and is made under clinical basis. Validated diagnostic criteria include typical clinical manifestations, family history, and response to colchicine therapy (169). Genetic testing leads to higher rates of diagnosis (170, 171), supporting but not excluding clinical diagnosis (172). Inconsistency among similar phenotypes may be explained by major histocompatibility class I chain-related gene A alleles, as shown in a study on homozygous M694V population (173).
Pyrin-associated autoinflammation with neutrophilic dermatosis is an inherited autosomal-dominant autoinflammatory disease characterized by childhood onset. This autoinflammatory syndrome is characterized by recurrent episodes of neutrophilic dermatosis, fever, elevated acute-phase reactants, arthralgia, and myalgia or myositis (51). PAAND is caused by a loss in guard mechanism of pyrin due to S242R mutation that leads to a non-phosphorylated pyrin in S242 (51). Dephosphorylated pyrin loses interaction with the protein 14-3-3 and thus forming a constitutive active inflammasome by recruiting ASC (51). Mutations and clinics of PAAND are distinct from FMF because of a clearly dominant inheritance pattern and for its longer fever episodes (lasting weeks), more prominent cutaneous features, and absence of serositis or amyloidosis (51). Currently, it is not fully understood how mutations in two regions of the same protein can induce different diseases. FMF-related mutations have recently been found to induce a pyrin-inflammasome that could be dephosphorylated by RhoA GTPase and not inhibited by colchicine, questioning the critical dependency on microtubules for ASC aggregation and inflammasome activation (133). PAAND-associated mutations in MEFV gene are associated with a reduction in the binding of pyrin to microtubules, decreasing the threshold to assemble pyrin inflammasome. It is not known if PAAND syndrome-associated pyrin inflammasome is dependent on microtubules, although the use of colchicine has shown partial clinical benefit in this patient (51). Cutaneous manifestations of PAAND resemble other autosomal-dominant monogenic autoinflammatory disease called pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome, in which arthritis is the distinct and prominent feature. PAPA is caused by mutations at proline–serine–threonine phosphatase-interacting protein 1 gene (174). This protein is a cytoskeleton-associated adaptor protein that interestingly binds pyrin and regulates IL-1β production (175). The generation of knock-in mice with FMF-associated pyrin mutation (harboring a human C-terminal B30.2/SPRY domain that is absent in mouse Mefv gene) has shown data supporting the activation of a pyrin-inflammasome and an increase of IL-1β in this animal model independent of NLRP3 (49). Furthermore, autoinflammation in this animal model is dependent on the ASC-caspase-1 axis and IL-1β, whereas IL-1α and caspase-8 are dispensable for the inflammation observed in this FMF model (50).
Current Therapeutics Targeting the Inflammasome Pathway
Inflammasomes are main drivers of autoinflammatory diseases as well as important regulators of innate immunity and inflammation. Although specific drugs that directly interfere with inflammasome activation are under development, current treatments used in clinic target upstream regulation process, in the case of colchicine, or downstream IL-1 signaling (176).
Colchicine is the classical mainstay treatment for FMF (177), decreasing attack frequency, improving quality of life, and preventing amyloidosis (178, 179). Clinical response to colchicine is considered a supportive diagnostic criterion for FMF, but it shows no benefit in CAPS patients. Colchicine is known to directly recover activity of the GTPase RhoA and therefore suppresses pyrin oligomerization but is also able to interfere with neutrophil migration and adhesion by downregulating the expression and distribution of selectins on neutrophils and endothelial cells (180). Interestingly, pyrin associates with microtubules and colocalizes with actin filaments (181). Thus, colchicine treatment may also prevent cytoskeletal changes that favor pyrin inflammasome assembly. However, recent data have shown that microtubule polymerization is not a requirement for pyrin inflammasome activation in FMF patients in contrast with wild-type pyrin carriers, providing a new concept for understanding the molecular mechanisms present in the activation of pyrin inflammasome (133). Nevertheless, some FMF patients are resistant to colchicine, and in this subset of patients, IL-1 blocking agents have shown efficacy (182–184). Anakinra therapy was also effective in a patient diagnosed with PAAND (51).
As exposed above, IL-1β is one of the main products of inflammasome and caspase-1 activation and exerts its inflammatory action by binding to the IL-1RI (185), this binding is antagonized by the IL-1Ra, a protein that binds IL-1RI without agonistic activity preventing IL-1β binding and signaling (185).
Therapies blocking IL-1 are available for the treatment of CAPS and other autoinflammatory syndromes (i.e., colchicine-unresponsive FMF patients). Anakinra is the recombinant form of IL-1Ra and was the first anti-IL1 agent clinically available. Due to its short half-life, it has to be administered by subcutaneous injection daily, and side effects are common at the site of injection; also liver enzymes need to be monitored regularly. There is a strong evidence of the effectiveness of anakinra for CAPS treatment (186, 187), with improvement of clinical features like hearing loss or amyloidosis with quick relapse of symptoms after withdrawal, demonstrating the requisite of daily injections in persistent and severe phenotypes. Despite its effectiveness, sore daily injections of anakinra are sometimes unpopular among patients, and in selected cases with mild phenotypes are possible to use it on demand basis during inflammatory attacks as in other autoinflammatory diseases (188, 189).
Other anti-IL-1 agents have been developed with a better pharmacokinetic profile and are actually approved for the treatment of CAPS. Canakinumab is a humanized monoclonal antibody against IL-1β administered intravenously or subcutaneously at a dose of 2–4 mg/kg every 8 weeks, and it is licensed for treatment of CAPS patients over 4 years of age. It has shown a very rapid and sustained response with little side effects, mainly infections, with stabilization of the majority of sequelae and potential improvement in clinical manifestations such as sensory-neural hearing loss (190). Abnormal bone formation in CAPS patients is unaffected by IL-1 blockage (191), revealing that other pathways downstream NLRP3 inflammasome play important roles in the clinical manifestations. Canakinumab up-titration may be needed and is actually encouraged in partial responders and severe phenotypes, rising the dose and shortening administration up to 8 mg/kg every 4 weeks (192).
Rilonacept is an engineered IL-1 trap that neutralizes circulating IL-1β and IL-1α, and it is administered subcutaneously with a load dose of 320 mg followed by 160 mg weekly (185). After initial pilot study and phase III studies, rilonacept was the first drug approved for treatment of CAPS, including FCAS and MWS in children of 12 years and older, due to its safety and effectiveness (193, 194). Benefits were obtained within hours of its administration with maximal effect within day 6 and 10, with mild or moderate adverse reactions.
Caspase-1 activation precedes IL-1β release after inflammasome activation; therefore, there have been advances in generating specific and clinical relevant caspase-1 inhibitors. The most developed caspase-1 inhibitor for therapeutic use is VX-765, an orally available pro-drug that is rapidly hydrolyzed by plasma and liver esterase into a potent and selective inhibitor of caspase-1 (195). In fact, VX-765 was able to reduce the release of IL-1β and IL-18 in monocytes of patients with FCAS treated with LPS (196). However, its clinical use is still under investigation.
The standard goal to treat autoinflammatory syndromes, specially CAPS patients, will be to directly target NLRP3 inflammasome using small compounds, in this respect a compound developed by Pfizer (CP-456773 or CRID3, recently renamed as MCC950) has been proved to block IL-1β release in CAPS monocytes after LPS treatment, being able to reduce clinical symptoms in an animal model of CAPS (197, 198). Furthermore, this compound has been recently found to reduce inflammation in animal models of renal, dermal, and pulmonary inflammation (199, 200). Therefore, CP-456773 represents a promising drug for the treatment of autoinflammatory syndromes.
Conclusion
Mutations in genes coding for inflammasome sensor proteins, such as NLRP3 or pyrin, accomplish for the development of different autoinflammatory diseases by uncontrolled activation of caspase-1 and the aberrant release of pro-inflammatory cytokines. In physiological conditions, the inflammasome pathway is activated in response to dangerous situations provoked by infections, tissue injury, or cellular stress, being the inflammasome formed by the sensor NLRP3 the most promiscuous inflammasome pathway activated in many different situations. Furthermore, non-mutated NLRP3 activation has been involved in different autoinflammatory syndromes, and, for example, patients with mutations in PLCG2 (autoinflammation and phospholipase Cγ2-associated antibody deficiency and immune dysregulation, APLAID syndrome) present an aberrant cytosolic Ca+2 signaling leading to NLRP3 activation, or patients with mutations in the deubiquitinase OTULIN (otulipenia) result in aberrant IL-1 production by NLRP3 activation (201, 202). NLRP3 has also been implicated as a key inflammasome sensor protein in different chronic diseases; in these circumstances, different endogenous danger signals activate NLRP3 and could contribute to the inflammatory response in metabolic and degenerative diseases, such as gout, type 2 diabetes, obesity atherosclerosis, or Alzheimer’s disease (6, 203, 204). Therefore, inflammasome is central in autoinflammatory diseases, and increasing our understanding on NLRP3 and pyrin activation may lead to development of more potent novel therapies for the treatment of not only autoinflammatory syndromes but also for chronic inflammatory, metabolic, and degenerative diseases.
Author Contributions
CT-M: figure preparation. CT-M, PC, and PP: literature search and manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MJ and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
This work was supported by the Spanish Ministry of Economy and Competitiveness ISCIII-FEDER “Una manera de hacer Europa” PI13/00174 (PP) and the European Research Council ERC-2013-CoG 614578 (PP).
References
1. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi:10.1038/nature07201
2. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140:771–6. doi:10.1016/j.cell.2010.03.006
3. Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity (2012) 37:487–500. doi:10.1016/j.immuni.2012.06.013
4. Ip WKE, Medzhitov R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun (2015) 6:6931. doi:10.1038/ncomms7931
5. Pelegrin P. Inflammasome activation by danger signals. In: Couillin I, Petrilli V, Martinon F, editors. The Inflammasomes. Basel: Springer (2011). p. 101–21.
6. Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol (2015) 10:395–424. doi:10.1146/annurev-pathol-012414-040431
7. Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev (2006) 20:1–15. doi:10.1101/gad.1376506
8. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi:10.1016/j.cell.2014.04.007
9. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509:310–7. doi:10.1038/nature13085
10. Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol (2009) 71:333–59. doi:10.1146/annurev.physiol.70.113006.100630
11. Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15:738–48. doi:10.1038/ni.2919
12. Barberà-Cremades M, Baroja-Mazo A, Gomez AI, Machado F, Di Virgilio F, Pelegrin P. P2X7 receptor-stimulation causes fever via PGE2 and IL-1β release. FASEB J (2012) 26:2951–62. doi:10.1096/fj.12-205765
13. Keller M, Rüegg A, Werner S, Beer H-D. Active caspase-1 is a regulator of unconventional protein secretion. Cell (2008) 132:818–31. doi:10.1016/j.cell.2007.12.040
14. de Torre-Minguela C, Barberà-Cremades M, Gomez AI, Martin-Sanchez F, Pelegrin P. Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep (2016) 6:22586. doi:10.1038/srep22586
15. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat Immunol (2014) 15:727–37. doi:10.1038/ni.2913
16. Yang D, Postnikov YV, Li Y, Tewary P, de la Rosa G, Wei F, et al. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J Exp Med (2012) 209:157–71. doi:10.1084/jem.20101354
17. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi:10.1038/nri3452
18. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi:10.1038/nature07710
19. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi:10.1038/nature07725
20. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323:1057–60. doi:10.1126/science.1169841
21. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi:10.1038/ni.1932
22. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe (2011) 9:363–75. doi:10.1016/j.chom.2011.04.008
23. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol (2010) 11:63–9. doi:10.1038/ni.1824
24. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity (2008) 28:285–7. doi:10.1016/j.immuni.2008.02.005
25. Zaki MH, Man SM, Vogel P, Lamkanfi M, Kanneganti TD. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci U S A (2014) 111:385–90. doi:10.1073/pnas.1317643111
26. Lukens JR, Lukens JR, Gurung P, Gurung P, Shaw PJ, Shaw PJ, et al. The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity (2015) 42:654–64. doi:10.1016/j.immuni.2015.03.006
27. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488:389–93. doi:10.1038/nature11250
28. Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163:1428–43. doi:10.1016/j.cell.2015.10.048
29. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
30. Frew BC, Joag VR, Mogridge J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog (2012) 8:e1002659. doi:10.1371/journal.ppat.1002659
31. Chavarria-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog (2013) 9:e1003452. doi:10.1371/journal.ppat.1003452
32. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25:713–24. doi:10.1016/j.molcel.2007.01.032
33. Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell (2016) 167:187–202.e17. doi:10.1016/j.cell.2016.09.001
34. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity (2012) 37:1009–23. doi:10.1016/j.immuni.2012.08.027
35. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med (2007) 356:1216–25. doi:10.1056/NEJMoa061592
36. Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis (2016). doi:10.1136/annrheumdis-2016-210021
37. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet (2001) 29:301–5. doi:10.1038/ng756
38. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum (2002) 46:3340–8. doi:10.1002/art.10688
39. de Menthière CS, Terrière S, Pugnère D, Ruiz M, Demaille J, Touitou I. INFEVERS: the registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res (2003) 31:282–5. doi:10.1093/nar/gkg031
40. Levy R, Gérard L, Kuemmerle-Deschner J, Lachmann HJ, Koné-Paut I, Cantarini L, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis (2015) 74:2043–9. doi:10.1136/annrheumdis-2013-204991
41. Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity (2009) 30:875–87. doi:10.1016/j.immuni.2009.05.005
42. Nakamura Y, Franchi L, Kambe N, Meng G, Strober W, Nunez G, et al. Critical role for mast cells in interleukin-1beta-driven skin inflammation associated with an activating mutation in the NLRP3 protein. Immunity (2012) 37:85–95. doi:10.1016/j.immuni.2012.04.013
43. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity (2009) 30:860–74. doi:10.1016/j.immuni.2009.04.012
44. Snouwaert JN, Nguyen M, Repenning PW, Dye R, Livingston EW, Kovarova M, et al. An NLRP3 mutation causes arthropathy and osteoporosis in humanized mice. Cell Rep (2016) 17:3077–88. doi:10.1016/j.celrep.2016.11.052
45. Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest (2013) 123:4695–705. doi:10.1172/JCI71543
46. Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat (2008) 29:803–8. doi:10.1002/humu.20720
47. Yilmaz E, Ozen S, Balci B, Duzova A, Topaloglu R, Besbas N, et al. Mutation frequency of familial Mediterranean fever and evidence for a high carrier rate in the Turkish population. Eur J Hum Genet (2001) 9:553–5. doi:10.1038/sj.ejhg.5200674
48. Topaloglu R, Batu ED, Yıldız Ç, Korkmaz E, Özen S, Beşbaş N, et al. Familial Mediterranean fever patients homozygous for E148Q variant. Int J Rheum Dis (2016). doi:10.1111/1756-185X.12929
49. Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity (2011) 34:755–68. doi:10.1016/j.immuni.2011.02.020
50. Sharma D, Sharma BR, Vogel P, Kanneganti T-D. IL-1β and caspase-1 drive autoinflammatory disease independently of IL-1α or caspase-8 in a mouse model of familial Mediterranean fever. Am J Pathol (2016). doi:10.1016/j.ajpath.2016.10.015
51. Masters SL, Lagou V, Jéru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med (2016) 8:332ra45. doi:10.1126/scitranslmed.aaf1471
52. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet (2014) 46:1135–9. doi:10.1038/ng.3066
53. Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with interleukin-18 inhibition. J Allergy Clin Immunol (2016). doi:10.1016/j.jaci.2016.10.022
54. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46:1140–6. doi:10.1038/ng.3089
55. Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med (2014) 211:2385–96. doi:10.1084/jem.20141091
56. Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A (2008) 105:1614–9. doi:10.1073/pnas.0708616105
57. Vitale A, Rigante D, Maggio MC, Emmi G, Romano M, Silvestri E, et al. Rare NLRP12 variants associated with the NLRP12-autoinflammatory disorder phenotype: an Italian case series. Clin Exp Rheumatol (2013) 31:155–6.
58. Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum (2011) 63:830–9. doi:10.1002/art.30170
59. Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol (2001) 108:615–20. doi:10.1067/mai.2001.118790
60. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol (2009) 27:621–68. doi:10.1146/annurev.immunol.25.022106.141627
61. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36:464–76. doi:10.1016/j.immuni.2012.02.001
62. Vladimer GI, Weng D, Paquette SWM, Vanaja SK, Rathinam VAK, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37:96–107. doi:10.1016/j.immuni.2012.07.006
63. Williams KL, Taxman DJ, Linhoff MW, Reed W, Ting JP. Cutting edge: monarch-1: a pyrin/nucleotide-binding domain/leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J Immunol (2003) 170:5354–8. doi:10.4049/jimmunol.170.11.5354
64. Jeru I, Le Borgne G, Cochet E, Hayrapetyan H, Duquesnoy P, Grateau G, et al. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum (2011) 63:1459–64. doi:10.1002/art.30241
65. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7:569–75. doi:10.1038/ni1344
66. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107:3076–80. doi:10.1073/pnas.0913087107
67. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. doi:10.1038/nature10394
68. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi:10.1038/nature10510
69. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
70. Raghawan AK, Sripada A, Gopinath G, Pushpanjali P, Kumar Y, Radha V, et al. A disease-associated mutant of NLRC4 shows enhanced interaction with SUG1 leading to constitutive FADD dependent caspase-8 activation and cell death. J Biol Chem (2016). doi:10.1074/jbc.M116.763979
71. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A (2014) 111:7403–8. doi:10.1073/pnas.1402911111
72. Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ (2006) 13:236–49. doi:10.1038/sj.cdd.4401734
73. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature (2014) 513:237–41. doi:10.1038/nature13449
74. Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem (1999) 274:33835–8. doi:10.1074/jbc.274.48.33835
75. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ (2007) 14:1590–604. doi:10.1038/sj.cdd.4402194
76. Vajjhala PR, Mirams RE, Hill JM. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J Biol Chem (2012) 287:41732–43. doi:10.1074/jbc.M112.381228
77. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156:1193–206. doi:10.1016/j.cell.2014.02.008
78. Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell (2014) 156:1207–22. doi:10.1016/j.cell.2014.01.063
79. Sborgi L, Ravotti F, Dandey VP, Dick MS, Mazur A, Reckel S, et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci U S A (2015) 112:13237–42. doi:10.1073/pnas.1507579112
80. Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med (2016) 213:771–90. doi:10.1084/jem.20151790
81. Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem (2002) 277:21119–22. doi:10.1074/jbc.C200179200
82. Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife (2016) 5:e13663. doi:10.7554/eLife.13663
83. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol (2016) 213:617–29. doi:10.1083/jcb.201602089
84. Próchnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research (2016) 5:1–15. doi:10.12688/f1000research.8614.1
85. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi:10.1038/nature04515
86. Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J (2006) 25:5071–82. doi:10.1038/sj.emboj.7601378
87. Luheshi NM, Giles JA, Lopez-Castejon G, Brough D. Sphingosine regulates the NLRP3-inflammasome and IL-1beta release from macrophages. Eur J Immunol (2012) 42:716–25. doi:10.1002/eji.201142079
88. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol (2015) 194:3937–52. doi:10.4049/jimmunol.1402658
89. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi:10.1016/j.immuni.2013.05.016
90. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi:10.1038/sj.cdd.4402195
91. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A (2012) 109:11282–7. doi:10.1073/pnas.1117765109
92. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492:123–7. doi:10.1038/nature11588
93. Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun (2012) 3:1329. doi:10.1038/ncomms2339
94. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi:10.1038/ni.1631
95. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis (2012) 3:e261. doi:10.1038/cddis.2011.132
96. Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity (2015) 43:451–62. doi:10.1016/j.immuni.2015.08.008
97. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
98. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis (2015) 6:e1954. doi:10.1038/cddis.2015.277
99. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi:10.1016/j.cell.2013.02.054
100. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol (2013) 14:454–60. doi:10.1038/ni.2550
101. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi:10.1016/j.immuni.2013.08.001
102. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
103. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi:10.1016/j.immuni.2012.01.009
104. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi:10.1038/nature10558
105. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi:10.1038/nature13683
106. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43:923–32. doi:10.1016/j.immuni.2015.10.009
107. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol (2015) 45:2911–7. doi:10.1002/eji.201545523
108. Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur J Immunol (2015) 45:2927–36. doi:10.1002/eji.201545772
109. Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science (2012) 336:481–5. doi:10.1126/science.1217141
110. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem (2012) 287:36617–22. doi:10.1074/jbc.M112.407130
111. Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, et al. Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J Biol Chem (2013) 288:2721–33. doi:10.1074/jbc.M112.422238
112. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49:331–8. doi:10.1016/j.molcel.2012.11.009
113. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem (2016) 291:103–9. doi:10.1074/jbc.C115.700492
114. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530:354–7. doi:10.1038/nature16959
115. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol (2016) 17:250–8. doi:10.1038/ni.3333
116. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi:10.4049/jimmunol.0901363
117. Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol (2009) 183:792–6. doi:10.4049/jimmunol.0900173
118. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol (2012) 189:4175–81. doi:10.4049/jimmunol.1201516
119. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol (2013) 14:52–60. doi:10.1038/ni.2474
120. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23:201–12. doi:10.1038/cr.2013.6
121. Le HT, Harton JA. Pyrin- and CARD-only proteins as regulators of NLR functions. Front Immunol (2013) 4:275. doi:10.3389/fimmu.2013.00275
122. Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A (2006) 103:9982–7. doi:10.1073/pnas.0602081103
123. Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer HD, et al. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ (2007) 14:1457–66. doi:10.1038/sj.cdd.4402142
124. Hesker PR, Nguyen M, Kovarova M, Ting JP, Koller BH. Genetic loss of murine pyrin, the familial Mediterranean fever protein, increases interleukin-1beta levels. PLoS One (2012) 7:e51105. doi:10.1371/journal.pone.0051105
125. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi:10.1038/ni.2215
126. Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy (2015) 11:166–82. doi:10.4161/15548627.2014.981915
127. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164:896–910. doi:10.1016/j.cell.2015.12.057
128. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286:9587–97. doi:10.1074/jbc.M110.202911
129. Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, et al. Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J Immunol (2015) 194:5472–87. doi:10.4049/jimmunol.1401343
130. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell (2015) 160:62–73. doi:10.1016/j.cell.2014.11.047
131. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol (2016) 17:1176–86. doi:10.1038/ni.3538
132. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol (2016) 17:914–21. doi:10.1038/ni.3457
133. Van Gorp H, Saavedra PHV, de Vasconcelos NM, Van Opdenbosch N, Vande Walle L, Matusiak M, et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci U S A (2016) 113:14384–9. doi:10.1073/pnas.1613156113
134. Gao W, Yang J, Liu W, Wang Y, Shao F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci U S A (2016) 113:E4857–66. doi:10.1073/pnas.1601700113
135. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity (2013) 39:1003–18. doi:10.1016/j.immuni.2013.11.010
136. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. doi:10.3389/fimmu.2013.00289
137. Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci U S A (2004) 101:9745–50. doi:10.1073/pnas.0308558101
138. Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol (2007) 179:1913–25. doi:10.4049/jimmunol.179.3.1913
139. MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1β by microvesicle shedding. Immunity (2001) 15:825–35. doi:10.1016/S1074-7613(01)00229-1
140. He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25:1285–98. doi:10.1038/cr.2015.139
141. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi:10.1038/nature18590
142. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi:10.1038/nature18629
143. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi:10.1038/nature15514
144. Martín-Sánchez F, Diamond C, Zeitler M, Gomez-Sánchez A, Peñalver M, Paszek P, et al. Inflammasome-dependent IL-1 β release depends upon membrane permeabilisation. Cell Death Differ (2016) 23:1219–31. doi:10.1038/cdd.2015.176
145. Chen KWW, Groß CJJ, Sotomayor FV, Stacey KJJ, Tschopp JJ, Sweet MJJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute salmonella challenge. Cell Rep (2014) 8:570–82. doi:10.1016/j.celrep.2014.06.028
146. Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat Commun (2016) 7:10555. doi:10.1038/ncomms10555
147. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol (2015) 33:823–74. doi:10.1146/annurev-immunol-032414-112227
148. Cuisset L, Jeru I, Dumont B, Fabre A, Cochet E, Le Bozec J, et al. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: epidemiological study and lessons from eight years of genetic analysis in France. Ann Rheum Dis (2011) 70:495–9. doi:10.1136/ard.2010.138420
149. Muckle TJ, Wells M. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q J Med (1962) 31:235–48.
150. Prieur AM, Griscelli C, Lampert F, Truckenbrodt H, Guggenheim MA, Lovell DJ, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl (1987) 66:57–68. doi:10.3109/03009748709102523
151. Aksentijevich I, D Putnam C, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum (2007) 56:1273–85. doi:10.1002/art.22491
152. Parker T, Keddie S, Kidd D, Lane T, Maviki M, Hawkins PN, et al. Neurology of the cryopyrin-associated periodic fever syndrome. Eur J Neurol (2016) 23:1145–51. doi:10.1111/ene.12965
153. Houx L, Hachulla E, Kone-Paut I, Quartier P, Touitou I, Guennoc X, et al. Musculoskeletal symptoms in patients with cryopyrin-associated periodic syndromes: a large database study. Arthritis Rheumatol (2015) 67:3027–36. doi:10.1002/art.39292
154. Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum (2011) 63:3625–32. doi:10.1002/art.30512
155. Nakagawa K, Gonzalez-Roca E, Souto A, Kawai T, Umebayashi H, Campistol JM, et al. Somatic NLRP3 mosaicism in Muckle-Wells syndrome. A genetic mechanism shared by different phenotypes of cryopyrin-associated periodic syndromes. Ann Rheum Dis (2015) 74:603–10. doi:10.1136/annrheumdis-2013-204361
156. Mensa-Vilaro A, Teresa Bosque M, Magri G, Honda Y, Martínez-Banaclocha H, Casorran-Berges M, et al. Late onset cryopyrin-associated periodic syndrome due to myeloid-restricted somatic NLRP3 mosaicism. Arthritis Rheumatol (2016) 68:3035–41. doi:10.1002/art.39770
157. Tassi S, Carta S, Delfino L, Caorsi R, Martini A, Gattorno M, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1 secretion. Proc Natl Acad Sci U S A (2010) 107:9789–94. doi:10.1073/pnas.1000779107
158. Carta S, Tassi S, Delfino L, Omenetti A, Raffa S, Torrisi MR, et al. Deficient production of IL-1 receptor antagonist and IL-6 coupled to oxidative stress in cryopyrin-associated periodic syndrome monocytes. Ann Rheum Dis (2012) 71:1577–81. doi:10.1136/annrheumdis-2012-201340
159. Carta S, Penco F, Lavieri R, Martini A, Dinarello CA, Gattorno M, et al. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci U S A (2015) 112:2835–40. doi:10.1073/pnas.1424741112
160. Omenetti A, Carta S, Delfino L, Martini A, Gattorno M, Rubartelli A. Increased NLRP3-dependent interleukin 1β secretion in patients with familial Mediterranean fever: correlation with MEFV genotype. Ann Rheum Dis (2014) 73:462–9. doi:10.1136/annrheumdis-2012-202774
161. Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet (1998) 351:659–64. doi:10.1016/S0140-6736(97)09408-7
162. Marek-Yagel D, Berkun Y, Padeh S, Abu A, Reznik-Wolf H, Livneh A, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum (2009) 60:1862–6. doi:10.1002/art.24570
163. Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, Yalcinkaya F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore) (2005) 84:1–11. doi:10.1097/01.md.0000152370.84628.0c
164. Rigante D, Lopalco G, Tarantino G, Compagnone A, Fastiggi M, Cantarini L. Non-canonical manifestations of familial Mediterranean fever: a changing paradigm. Clin Rheumatol (2015) 34:1503–11. doi:10.1007/s10067-015-2916-z
165. Mukhin NA, Kozlovskaya LV, Bogdanova MV, Rameev VV, Moiseev SV, Simonyan AK. Predictors of AA amyloidosis in familial Mediterranean fever. Rheumatol Int (2015) 35:1257–61. doi:10.1007/s00296-014-3205-x
166. Altunoğlu A, Erten Ş, Canoz MB, Yuksel A, Ceylan GG, Balci S, et al. Phenotype 2 familial Mediterranean fever: evaluation of 22 case series and review of the literature on phenotype 2 FMF. Ren Fail (2013) 35:226–30. doi:10.3109/0886022X.2012.745115
167. Bakkaloglu A, Duzova A, Ozen S, Balci B, Besbas N, Topaloglu R, et al. Influence of serum amyloid A (SAA1) and SAA2 gene polymorphisms on renal amyloidosis, and on SAA/C-reactive protein values in patients with familial Mediterranean fever in the Turkish population. J Rheumatol (2004) 31:1139–42.
168. Cazeneuve C, Ajrapetyan H, Papin S, Roudot-Thoraval F, Geneviève D, Mndjoyan E, et al. Identification of MEFV-independent modifying genetic factors for familial Mediterranean fever. Am J Hum Genet (2000) 67:1136–43. doi:10.1016/S0002-9297(07)62944-9
169. Demirkaya E, Saglam C, Turker T, Koné-Paut I, Woo P, Doglio M, et al. Performance of different diagnostic criteria for familial Mediterranean fever in children with periodic fevers: results from a Multicenter International Registry. J Rheumatol (2016) 43:154–60. doi:10.3899/jrheum.141249
170. Grateau G, Cuisset L, Dodé C, Delpech M. Breakthroughs in the genetics of hereditary fevers. Eur J Intern Med (2000) 11:242–4. doi:10.1016/S0953-6205(00)00104-7
171. Grateau G, Pêcheux C, Cazeneuve C, Cattan D, Dervichian M, Goossens M, et al. Clinical versus genetic diagnosis of familial Mediterranean fever. QJM (2000) 93:223–9. doi:10.1093/qjmed/93.4.223
172. Giancane G, Ter Haar NM, Wulffraat N, Vastert SJ, Barron K, Hentgen V, et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis (2015) 74:635–41. doi:10.1136/annrheumdis-2014-206844
173. Touitou I, Picot MC, Domingo C, Notarnicola C, Cattan D, Demaille J, et al. The MICA region determines the first modifier locus in familial Mediterranean fever. Arthritis Rheum (2001) 44:163–9. doi:10.1002/1529-0131(200101)44:1<163::AID-ANR20>3.0.CO;2-Z
174. Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet (2002) 11:961–9. doi:10.1093/hmg/11.8.961
175. Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA, et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci U S A (2003) 100:13501–6. doi:10.1073/pnas.2135380100
176. López-Castejón G, Pelegrin P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs (2012) 21:995–1007. doi:10.1517/13543784.2012.690032
177. Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med (1972) 287:1302. doi:10.1056/NEJM197212212872514
178. Hentgen V, Grateau G, Kone-Paut I, Livneh A, Padeh S, Rozenbaum M, et al. Evidence-based recommendations for the practical management of familial Mediterranean fever. Semin Arthritis Rheum (2013) 43:387–91. doi:10.1016/j.semarthrit.2013.04.011
179. Livneh A, Zemer D, Langevitz P, Shemer J, Sohar E, Pras M. Colchicine in the treatment of AA and AL amyloidosis. Semin Arthritis Rheum (1993) 23:206–14. doi:10.1016/S0049-0172(05)80042-3
180. Cronstein BN, Molad Y, Reibman J, Balakhane E, Levin RI, Weissmann G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest (1995) 96:994–1002. doi:10.1172/JCI118147
181. Mansfield E, Chae JJ, Komarow HD, Brotz TM, Frucht DM, Aksentijevich I, et al. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood (2001) 98:851–9. doi:10.1182/blood.V98.3.851
182. Hashkes PJ, Spalding SJ, Giannini EH, Huang B, Johnson A, Park G, et al. Rilonacept for colchicine-resistant or -intolerant familial Mediterranean fever: a randomized trial. Ann Intern Med (2012) 157:533–41. doi:10.7326/0003-4819-157-8-201210160-00003
183. Gül A, Ozdogan H, Erer B, Ugurlu S, Kasapcopur O, Davis N, et al. Efficacy and safety of canakinumab in adolescents and adults with colchicine-resistant familial Mediterranean fever. Arthritis Res Ther (2015) 17:243. doi:10.1186/s13075-015-0765-4
184. Alpay N, Sumnu A, Calışkan Y, Yazıcı H, Türkmen A, Gül A. Efficacy of anakinra treatment in a patient with colchicine-resistant familial Mediterranean fever. Rheumatol Int (2012) 32:3277–9. doi:10.1007/s00296-010-1474-6
185. Moltó A, Olivé A. Anti-IL-1 molecules: new comers and new indications. Joint Bone Spine (2010) 77:102–7. doi:10.1016/j.jbspin.2009.10.011
186. Leslie KS, Lachmann HJ, Bruning E, McGrath JA, Bybee A, Gallimore JR, et al. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Arch Dermatol (2006) 142:1591–7. doi:10.1001/archderm.142.12.1591
187. Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med (2006) 355:581–92. doi:10.1056/NEJMoa055137
188. Bodar EJ, Kuijk LM, Drenth JPH, van der Meer JWM, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis (2011) 70:2155–8. doi:10.1136/ard.2011.149922
189. Grimwood C, Despert V, Jeru I, Hentgen V. On-demand treatment with anakinra: a treatment option for selected TRAPS patients. Rheumatology (Oxford) (2015) 54:1749–51. doi:10.1093/rheumatology/kev111
190. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med (2009) 360:2416–25. doi:10.1056/NEJMoa0810787
191. Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep (2011) 13:123–31. doi:10.1007/s11926-011-0165-y
192. Caorsi R, Lepore L, Zulian F, Alessio M, Stabile A, Insalaco A, et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis Res Ther (2013) 15:R33. doi:10.1186/ar4184
193. Goldbach-Mansky R, Shroff SD, Wilson M, Snyder C, Plehn S, Barham B, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum (2008) 58:2432–42. doi:10.1002/art.23620
194. Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum (2008) 58:2443–52. doi:10.1002/art.23687
195. Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther (2007) 321:509–16. doi:10.1124/jpet.106.111344
196. Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JCR, et al. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol (2005) 175:2630–4. doi:10.4049/jimmunol.175.4.2630
197. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi:10.1038/nm.3806
198. Laliberte RE, Perregaux DG, Hoth LR, Rosner PJ, Jordan CK, Peese KM, et al. Glutathione S-transferase omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1beta posttranslational processing. J Biol Chem (2003) 278:16567–78. doi:10.1074/jbc.M211596200
199. Primiano MJ, Lefker BA, Bowman MR, Bree AG, Hubeau C, Bonin PD, et al. Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J Immunol (2016) 197:2421–33. doi:10.4049/jimmunol.1600035
200. Ludwig-Portugall I, Bartok E, Dhana E, Evers BDG, Primiano MJ, Hall JP, et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int (2016). doi:10.1016/j.kint.2016.03.035
201. Chae JJ, Park YH, Park C, Hwang I-Y, Hoffmann P, Kehrl JH, et al. Connecting two pathways through Ca2+ signaling: NLRP3 inflammasome activation induced by a hypermorphic PLCG2 mutation. Arthritis Rheumatol (2015) 67:563–7. doi:10.1002/art.38961
202. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci U S A (2016) 113:10127–32. doi:10.1073/pnas.1612594113
203. Masters SL, Latz E, O’Neill LAJ. The inflammasome in atherosclerosis and type 2 diabetes. Sci Transl Med (2011) 3:81s17. doi:10.1126/scitranslmed.3001902
Keywords: inflammation, NLRP3, pyrin, extracellular ATP, P2X7 receptor, cryopyrin-associated periodic syndrome, familial Mediterranean fever
Citation: de Torre-Minguela C, Mesa del Castillo P and Pelegrín P (2017) The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 8:43. doi: 10.3389/fimmu.2017.00043
Received: 01 December 2016; Accepted: 11 January 2017;
Published: 27 January 2017
Edited by:
José Hernández-Rodríguez, Hospital Clinic of Barcelona, SpainReviewed by:
Manel Juan, Hospital Clinic of Barcelona, SpainDominic De Nardo, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright: © 2017 de Torre-Minguela, Mesa del Castillo and Pelegrín. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pablo Pelegrín, cGFibG8ucGVsZWdyaW5AZmZpcy5lcw==