 Tomás Villaseñor
Tomás Villaseñor Edgardo Madrid-Paulino
Edgardo Madrid-Paulino Rafael Maldonado-Bravo
Rafael Maldonado-Bravo Antonio Urbán-Aragón
Antonio Urbán-Aragón Leonor Pérez-Martínez
Leonor Pérez-Martínez Gustavo Pedraza-Alva*
Gustavo Pedraza-Alva*
- Departamento de Medicina Molecular y Bioprocesos, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Morelos, Mexico
Mycobacterium tuberculosis (M. tuberculosis), an intracellular pathogenic Gram-positive bacterium, is the cause of tuberculosis (TB), a major worldwide human infectious disease. The innate immune system is the first host defense against M. tuberculosis. The recognition of this pathogen is mediated by several classes of pattern recognition receptors expressed on the host innate immune cells, including Toll-like receptors, Nod-like receptors, and C-type lectin receptors like Dectin-1, the Mannose receptor, and DC-SIGN. M. tuberculosis interaction with any of these receptors activates multiple signaling pathways among which the protein kinase C, the MAPK, and the NFκB pathways have been widely studied. These pathways have been implicated in macrophage invasion, M. tuberculosis survival, and impaired immune response, thus promoting a successful infection and disease. Interestingly, the Wnt signaling pathway, classically regarded as a pathway involved in the control of cell proliferation, migration, and differentiation in embryonic development, has recently been involved in immunoregulatory mechanisms in infectious and inflammatory diseases, such as TB, sepsis, psoriasis, rheumatoid arthritis, and atherosclerosis. In this review, we present the current knowledge supporting a role for the Wnt signaling pathway during macrophage infection by M. tuberculosis and the regulation of the immune response against M. tuberculosis. Understanding the cross talk between different signaling pathways activated by M. tuberculosis will impact on the search for new therapeutic targets to fuel the rational design of drugs aimed to restore the immunological response against M. tuberculosis.
Introduction
Mycobacterium tuberculosis (M. tuberculosis) is the cause of human tuberculosis (TB), which is regarded as one of the most harmful pathogen, just behind HIV, that is responsible for more deaths than any other microorganism. The WHO in 2015 estimated that 1/3 of the planet’s population harbor this bacterium, yet only 2–23% will develop disease during their lifespan. M. tuberculosis is a Gram-positive bacterium that spreads by aerosols. Upon entry into the lungs, M. tuberculosis infects alveolar macrophages, a process mediated by a variety of receptors expressed on the surface of these phagocytic cells (1). Receptors expressed in macrophages involved in M. tuberculosis internalization include complement receptors (2), C-type lectin receptors, dectin-1, mannose receptors, scavenger receptors (3–5), CD14 (6), CD43 (7), and lung surfactant protein A (8). However, results obtained from studies using null mice for the expression of any of these receptors indicate that none of them are essential for M. tuberculosis macrophage invasion (9). Likewise, none of these receptors confer a survival advantage for M. tuberculosis (10). The engagement of these receptors on the macrophage cell surface normally would result in the activation of different signaling pathways that lead to the destruction of the invading pathogen. However, the M. tuberculosis success as an intracellular pathogen resides in the manipulation of these signaling pathways by several mechanisms to avoid bactericidal activities of the host macrophages and survive inside the cell host (11–14). Accordingly with the fact that, most of the receptors engaged by M. tuberculosis upon the first contact with the macrophage activate proteins of the protein kinase C (PKC) family as well as calcium mobilization (Figure 1), among other known mechanisms involved in blocking phagosome-lysosome fusion, recently it was shown that Mycobacterium bovis (M. bovis) promotes the coating of the phagosome with coronin-1a, which leads to the induction of calcium fluxes activating the calcium-dependent phosphatase calcineurin. Upon M. bovis infection, coronin-1a-deficient macrophages show impaired calcium mobilization and calcineurin activation, resulting in mycobacterial destruction. Consistent with this, preventing calcium mobilization, with calcium chelators, or inhibiting calcineurin activation in wild-type macrophages reduces M. bovis proliferation and survival (1). Therefore, these data suggest that M. tuberculosis-dependent inhibition of phagosome–lysosome fusion involves the engagement of multiple receptors that lead to dephosphorylation of proteins regulating phagosome maturation (15). Nonetheless, the molecular mechanism by which coronin-1a leads to calcium mobilization or whether coronin-1a is subjected to regulation by any of the kinases activated by M. tuberculosis upon interaction with its plethora of receptors remain to be defined.
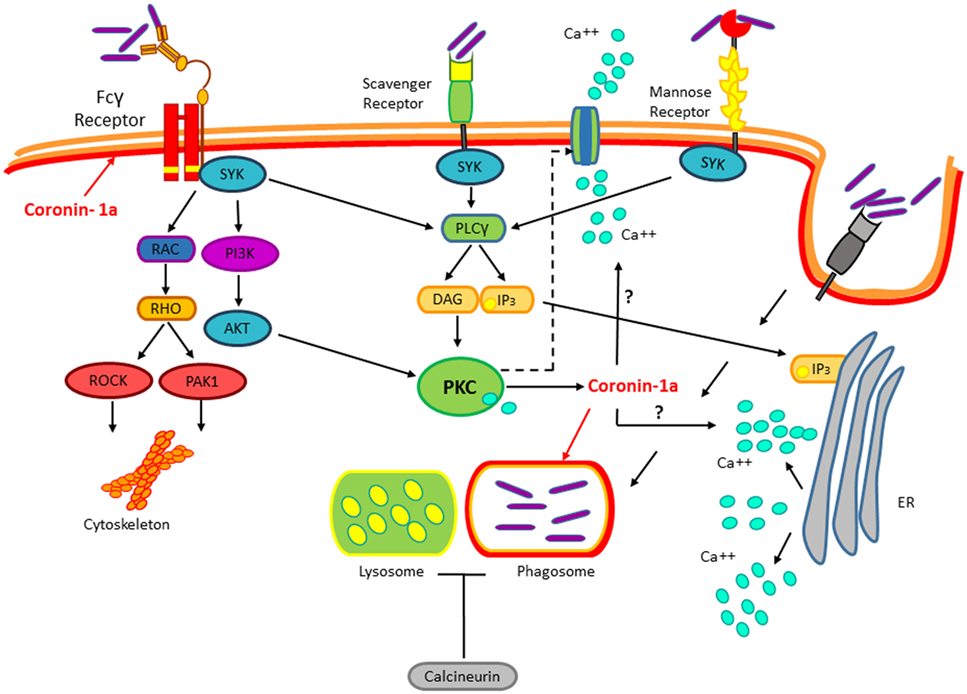
Figure 1. Mycobacterium tuberculosis (M. tuberculosis) recognition by immune receptors. During the infection, several classes of PPRs such as the Fcγ receptor, the Scavengers, and Mannose receptors recognize M. tuberculosis. Mycobacterium interaction with these receptors activates the protein kinase C pathway. This pathway is implicated in the cytoskeletal arrangements, macrophage invasion, and M. tuberculosis survival. Upon invasion of the macrophage by M. tuberculosis, coronin-1a is recruited to the mycobacterial phagosome leading to the induction of calcium fluxes, thus resulting in calcineurin activation, which blocks the phagosome–lysosome fusion by an unknown mechanism.
In addition to the signaling pathways activated by M. tuberculosis that allow macrophage invasion and survival, M. tuberculosis also activates numerous signaling pathways promoting cytokines and chemokines expression. Among those, pro-inflammatory cytokines like tumor necrosis factor (TNF), interleukin-1β (IL-1β), and interleukin-6 (IL-6) are secreted few hours after macrophage infection and precedes anti-inflammatory cytokine production, including transforming growth factor β (TGF-β) and interleukin-10 (IL-10) (Figure 2) (16). The production of the inflammatory cytokines and chemokines at early stages of the infectious process is crucial for the recruitment of neutrophils, macrophages to the lung, and later on for the recruitment of activated T cells. Depending on the final balance between pro- and anti-inflammatory cytokines, the host can either assemble a response that eliminates the infection or result in granuloma formation, thus controlling the infection without bacterial elimination (17). Different experimental evidence suggests that IL-10 expression is critical for mycobacterium survival, granuloma formation, and attenuation of the inflammatory response. Transgenic mice expressing human IL-10 specifically in the lungs showed increased bacterial burden in Mycobacterium avium (M. avium)-containing granulomas. This correlated with decreased pro-inflammatory cytokines’ levels, such as TNF and IL-12 [reviewed in Ref. (18)].
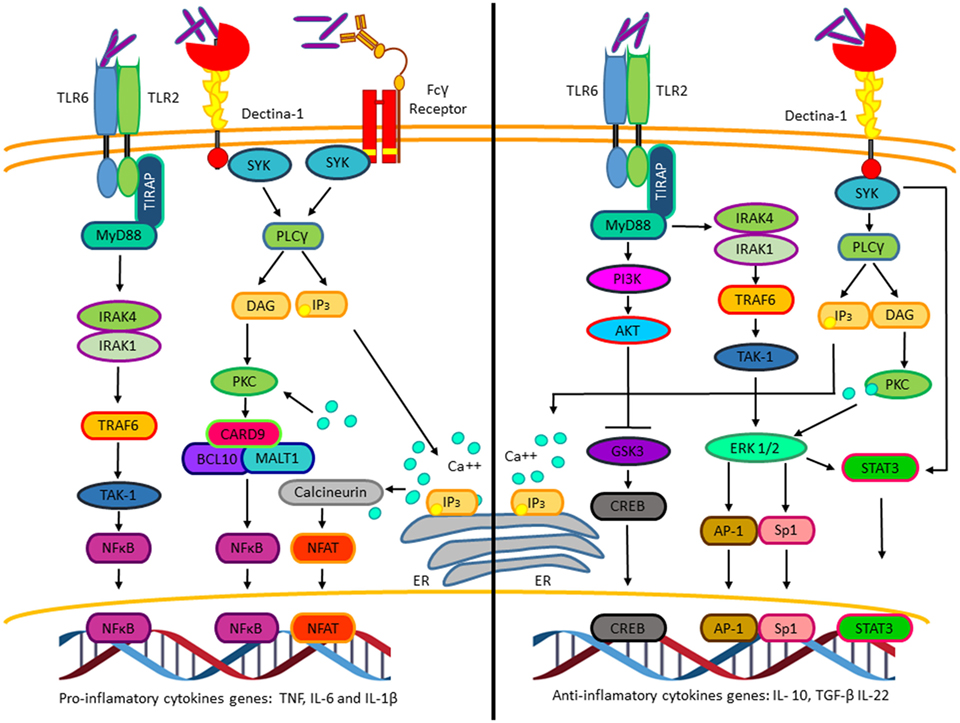
Figure 2. The pro-inflammatory and anti-inflammatory cytokines’ genes activated by Mycobacterium tuberculosis (M. tuberculosis). Pro-inflammatory (left). Interaction of M. tuberculosis with TLR2, Dectin-1, and Fcγ receptors activates NFκB and nuclear factor of activated T cells, which promotes transcription of genes such as tumor necrosis factor, IL-6, and IL-1β. Anti-inflammatory (right). Interaction of M. tuberculosis to TLR-2 and Dectin-1 through different mechanisms induces the activation of the transcription factors CREB, AP-1, Sp1, and STAT3, which initiates the transcription of genes such as IL-10, TGF-β, and IL-22.
Thus, M. tuberculosis infection depends on the mycobacterial ability to hijack different macrophage signaling pathways, at the early stages of infection to avoid its destruction, and at the later stages of infection to alter the cytokine profile to attenuate the adaptive immune response. In addition to the well-characterized pathways involved in these processes, like the PKC, MAPK, NFκB and JAK/STATs pathways (Figures 1 and 2), cumulative experimental evidence point out a role for the Wnt/β-catenin pathway during M. tuberculosis infection. Here, we will discus the molecular mechanism by which M. tuberculosis activates the Wnt/β-catenin pathway and its role during infection.
The Wnt Signaling Pathways
The Wnt signaling pathway has been extensively studied and reviewed (19–23). This signaling pathway is an ancient and highly conserved signaling mechanism and involves important molecular cascades that regulate cell fate throughout lifespan (19, 24). The Wnt signaling pathway has been generally associated with cellular proliferation, differentiation, apoptosis, motility, and polarization of cells, in invertebrates and mammals (25). Mutations in molecules controlling this pathway that result in constitutive activation of the pathway may lead to colon cancer, hair follicle tumors, and leukemia (26). However, the role of Wnt signaling in the immune response against pathological bacteria is poorly understood.
The Wnt/β-catenin network was first identified in 1982 with the discovery of the proto-oncogene Int1 in mice (27). Later in 1987, the segment polarity gene wingless, the Drosophila melanogaster homolog of Int1 was cloned and shown to be required for proper wing formation (28). Until now, there are three different characterized Wnt pathways: the canonical pathway or Wnt/β-catenin (cadherin-associated protein β), which involves β-catenin and members of T-cell factor (TCF)/lymphoid enhancer-binding factor (LEF) family of transcription factors (TCF/LEF); the planar cell polarity (PCP) pathway; and the Wnt/Ca++ pathway (29). The ligands involved in the Wnt pathway are soluble factors that can be divided into two subclasses: the Wnt1 and Wnt5a. The Wnt1 proteins (Wnt1, Wnt2, Wnt3, Wnt3a, Wnt7a, Wnt8a, and Wnt10b) (30) participate in the canonical Wnt pathway. On the other hand, the Wnt5a proteins (Wnt4, Wnt5a, and Wnt11) participate in non-canonical signaling pathway (31).
The Canonical Pathway
The canonical Wnt signaling or Wnt/β-catenin signaling is the most and best understood and characterized pathway (Figure 3) (29). In the absence of Wnt ligands, cytoplasmic β-catenin is phosphorylated on its N-terminal region, first on Ser 45 by casein kinase 1 (CK1), and then on Ser33, Ser37, and Thr41 by the glycogen synthase kinase 3 β (GSK3β) to create recognition sites for β-transducin-repeat-containing protein (βTRCP), leading to the β-catenin ubiquitylation and preteasomal breakdown, preventing in this way its nuclear translocation and Wnt-dependent gene expression (32). To activate the Wnt/β-catenin pathway, the Wnt ligands bind the receptor complex formed by the Frizzled (FZD) receptor and the low-density lipoprotein receptor-related protein (LRP), leading to the recruitment of the protein disheveled (DVL). This recruitment inactivates the β-catenin destruction complex, which is conformed by protein Axin, tumor-suppressor adenomatous polyposis coli gene product (APC), CK1, and GSK3β, thus preventing β-catenin phosphorylation by GSK3 and consequently, its proteasomal degradation. This, results in β-catenin accumulation and translocation to the nucleus where it drives the expression of TCF/LEF-dependent genes [c-Myc, CyclinD-1, Axin2, metalloproteinases, CD44, peroxisome proliferator-activated receptor gamma (PPARγ, etc.)], which are involved in cell proliferation, survival, and cell differentiation (31).
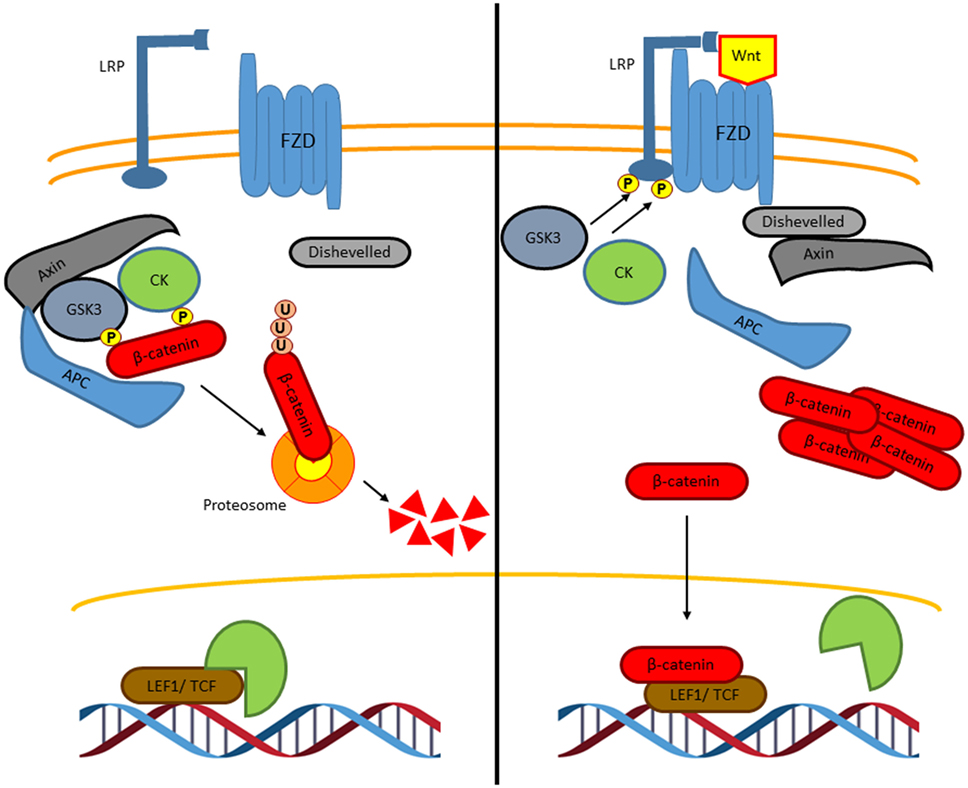
Figure 3. The canonical Wnt signaling pathway. In the absence of Wnt ligands, β-catenin interacts with a degradation complex, which is formed by Axin, APC, GSK3, and CK1. In this complex, β-catenin is phosphorylated by GSK3 and by CK1 leading to ubiquitylation and subsequently degradation of β-catenin in the proteasome. When β-catenin is degraded, Groucho, which is a co-repressor protein, interacts with LEF1 and TCF. The Groucho/LEF1/TCF complex represses the transcription of target genes (left). Wnt ligands interact with the Frizzled receptor, a G protein-coupled receptor, and with the co-receptor LRP. GSK3 and CK1 induce the phosphorylation of LRP leading to the recruitment of axin and disheveled to the LRP/G protein-coupled receptor complex releasing β-catenin. Then, β-catenin is accumulated in the nucleus where it binds to LEF1 and TCF to induce the transcription of specific genes (right).
Non-Canonical Pathways
The non-canonical Wnt pathway is defined as Wnt or FZD receptor-initiated signaling that is independent of β-catenin transcriptional function (33). So far, two different non-canonical Wnt pathways have been described, the Wnt–Ca++ pathway and the PCP pathway (Figure 4) (29). Such classifications are not strictly rigid and exclusive as these pathways overlap with or intersect one another (33). Wnt5 and Wnt11 participate in initializing PCP pathway through FZD and DVL (mammalian homolog of Drosophila disheveled) activating trimeric G proteins and DVL and DAAM (disheveled-associated activator of morphogenesis), which together activate the RHOA (RAS homolog gene-family member A)–ROCK (RHO-associated coiled-coil containing protein kinase) pathway, that mediates cytoskeletal re-organization (34). The DVL–DAAM–RHOA complex activates RHOA, resulting in ROCK kinase activation (33). In addition to RHOA activation, DVL also activates RAC1 (Ras-related C3 botulinum toxin substrate 1) leading to the activation of the JUN kinase stress response pathway, thus influencing cell shape (29). Hence, the PCP pathway, through regulating actin-dependent changes in cytoskeleton, controls cell adhesion and migration. Interestingly, in both T and B lymphocytes, it has been observed that this pathway can inhibit the canonical Wnt pathway by downregulating β-catenin levels (35, 36). Downstream pathways of DVL regulate the cytoskeleton and cell polarity; however, in immune cells, the molecular mechanism involved is not clear yet (29).
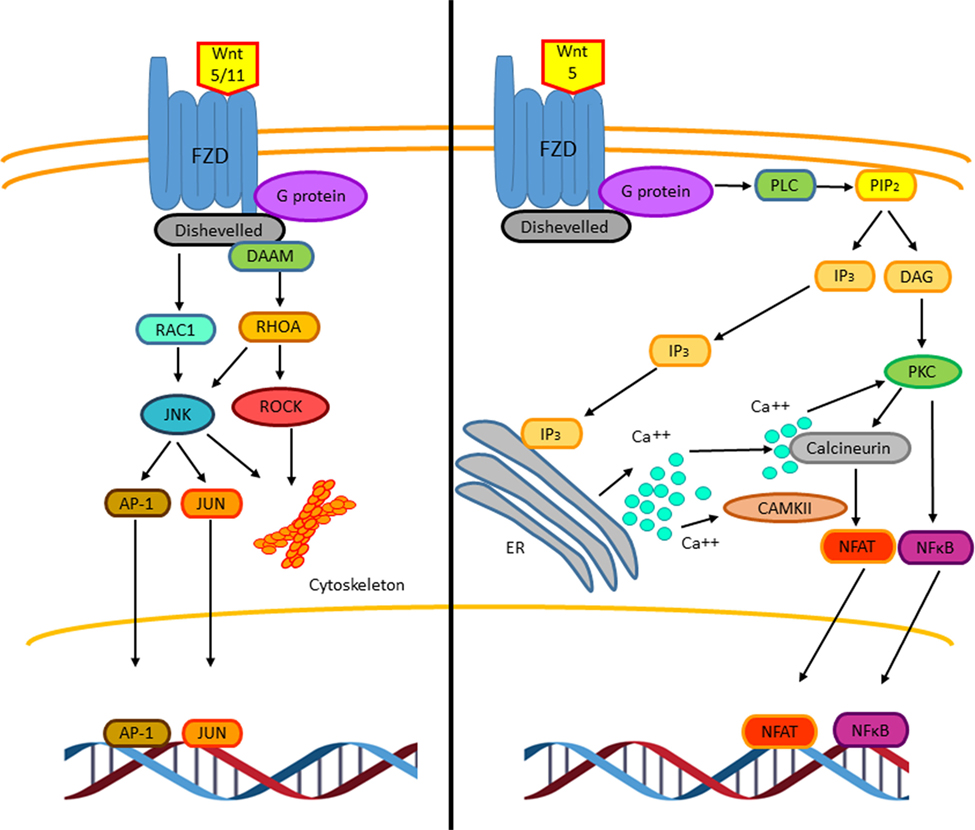
Figure 4. The non-canonical Wnt signaling pathway. Two β-catenin independent Wnt signaling pathways have been reported. The planar cell polarity (PCP) pathway is triggered by Wnt5 and Wnt11. Wnt5/11 binds Frizzled leading to the activation of a trimeric G protein that induces the activation of disheveled and DAAM. Together these proteins trigger the activation of the small GTPases RHOA and RAC-1 that leads to the activation of the kinases JUN kinase (JNK) and ROCK. These signaling pathways are involved in cell adhesion, migration, and cell cytoskeleton organization. The stress response pathway involves the activation of JNK, which phosphorylates the transcriptional factors AP-1 and JUN leading to their translocation to the nucleus to regulate gene expression (left). The Wnt/Ca2+ signaling pathway is triggered by Wnt5–FZD-2. The activation of this pathway is mediated through G proteins, which induces the phospholipase C activation, leading to the hydrolysis of PIP2 into DAG and IP3. The IP3 induces the release of intracellular Ca++, activation of calcineurin, and CAMKII. The active calcineurin induces the nuclear factor of activated T cells (NFAT) activation. Intracellular calcium as well as DAG activates protein kinase C (PKC) increasing the activity of calcineurin triggering the translocation of the transcriptional factor NFAT to the nucleus. Similarly, PKC induces the activation and translocation of NFκB to the nucleus (right).
The Wnt–Ca++ signaling pathway can influence the activation of both, canonical and non-canonical Wnt pathway. It has been proposed that Wnt5 and Frizzled-2 can initiate this pathway (37, 38). The Wnt–FZD complex activates phospholipase C through a small G protein. This activation leads to cleavage of phosphatidylinositol-4-5-biphosphate (PIP2) to diacylglycerol (DAG) and inositol triphosphate (IP3). IP3 binding to its receptor leads to an increase in intracellular calcium; DAG and calcium activates different members of the PKC family. Increased calcium levels also lead to activation of the phosphatase calcineurin, promoting the activation of the nuclear factor of activated T cells (NFAT) (39, 40), that is involved in T-cell receptor-mediated activation of interleukin-2 production in T-lymphocytes (29). Another connection between the non-canonical Wnt signaling pathway and NFAT function involves GSK3β, which through NFAT phosphorylation promotes its exit from the nucleus (41) and reduces NFAT-dependent gene expression. All together, these observations suggest that the non-canonical Wnt signaling pathways regulate NFAT-dependent gene expression by different mechanisms (29, 41).
Wnt Signaling and Inflammation
Recently, the role of Wnt signaling pathway in inflammatory processes started to be understood. Wnt signaling can prevent or promote inflammation. For example, exposing 3T3-L1 preadipocytes to TNF promotes Wnt10b expression and enhances activation of the canonical Wnt pathway, resulting in altered adipocyte differentiation toward an inflammatory phenotype (42). However, whether Wnt10b mediates TNF-induced preadipocyte differentiation toward the pro-inflammatory phenotype remains to be elucidated. Mouse microglial cells upregulate the expression and release of IL-6, IL-12, and IFNγ in response to Wnt3a (43). In contrast, Wnt3a has an anti-inflammatory effect on macrophages, while Wnt11 has the same effect on mouse colonic cells (44). These data indicate that the Wnt proteins promote pro- or anti-inflammatory responses and that this depends on the cellular context, the type of insult, and the cytokine environment (Figure 5). The fact that cytokines can upregulate Wnt expression raises the question of how many of the cellular responses triggered by cytokines are mediated by a paracrine effect mediated by Wnt factors and the activation of both the canonical and non-canonical Wnt pathways?
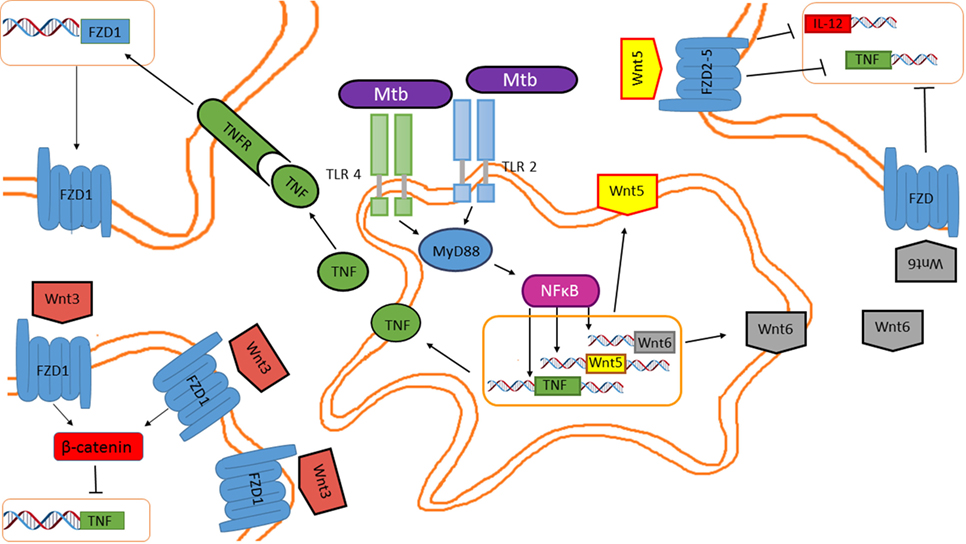
Figure 5. Mycobacterium tuberculosis (M. tuberculosis) inhibits tumor necrosis factor (TNF) through the Wnt pathway in a paracrine and autocrine manner. Wnt5, Wnt6, and pro-inflammatory cytokines are induced in macrophages in response to mycobacterial infection. Secreted Wnt5 and Wnt6 are recognized by FZD receptors expressed on the cell surface of neighboring macrophages, impairing TNF and IL-12 expression. Additionally, TNF released by the M. tuberculosis-infected macrophages promotes FZD1 expression in neighboring macrophages, rendering them susceptible to Wnt3 actions, which inhibits TNF expression.
There is very scant information regarding the role of different Wnt members in response to pathogenic bacterial infections and in particular on their role in M. tuberculosis infection. Table 1 summarizes our current knowledge on this matter.
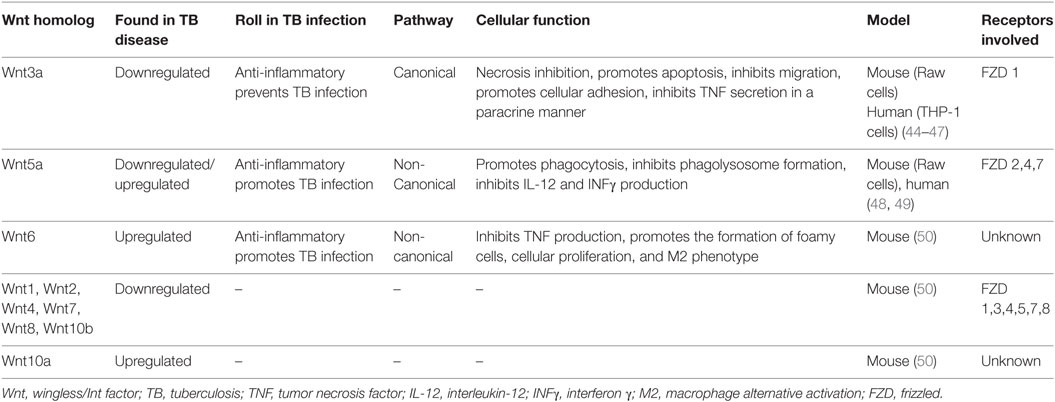
Table 1. The role of the Wnt family members in TB.
Additionally, the transcription factors that regulate the gene expression of the distinct Wnt and FZD family members in response to cytokines are largely unknown. Chromatin-immunoprecipitation-sequencing data from the Encode database revealed that transcription factors like FBJ murine osteosarcoma viral oncogene homolog (Fos), V-Jun avian sarcoma virus 17 oncogene homolog (Jun), signal transducer and activator of transcription 1 and 2 (STAT1, STAT2) interact with sequences located 1,000 base pairs upstream from the transcription start site for the genes encoding Wnt2, Fzd1, Fzd2, and Fzd7 (Table 2), and thus directly controlling the expression of these genes in response to cytokines. Currently, there is no information for the other Wnt or FZD family members.
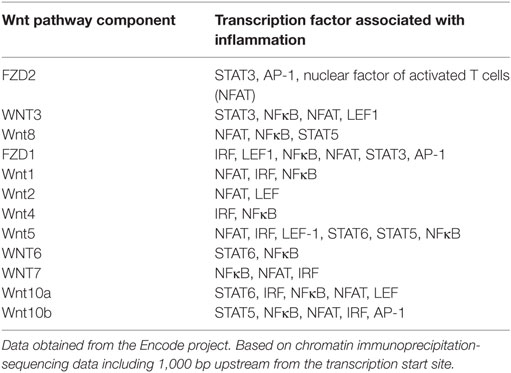
Table 2. Transcription factors activated by cytokine signaling that interact with 5′ regulatory sequences of genes encoding for Wnt or Fzd receptors.
Wnt3
Wnt3 is known as a canonical member of the Wnt family. Although monocytes and granulocytes express FZD receptors (FZD1, 5, and 7), when exposed to Wnt3 in vitro, only monocytes’ transendothelial migration capacity is reduced (51). This involves the activation of Wnt/β-catenin pathway and increased cell adhesion to endothelial cells (45), hence, suggesting an anti-inflammatory role for Wnt3. However, the Wnt/β-catenin regulated genes involved in enhanced endothelial cell adhesion remain to be identified. In the context of M. tuberculosis infection, the role of Wnt3 is controversial. In bone marrow-derived macrophages, M. tuberculosis H37rv promotes Fzd1 receptor gene expression through the TLR-2/MYD88/NFκB pathway; M. tuberculosis-induced Fzd1 expression also involves TNF (52). Interestingly, INFγ enhances Fzd1 levels on the cell surface of M. tuberculosis-infected macrophages. According to its proposed anti-inflammatory function, Wnt3 reduced TNF secretion but not Tnf gene expression in M. tuberculosis-infected macrophages. This effect is mediated through the canonical pathway, since Wnt3 induces β-catenin stabilization and Axin2 expression (52). Additionally, Wnt3 also promotes the expression of Arginase 1 in M. tuberculosis-infected macrophages, which has been associated with the anti-inflammatory M2 phenotype (44). In the lung epithelia, Wnt3 is expressed constitutively. Although M. tuberculosis infection does not alter Wnt3 expression in the lung epithelia, it promotes Fzd1 and Fzd5 gene expression while reducing the expression of Fzd 3,4, 6–10, this correlates with reduced β-catenin protein levels and thus β-catenin-dependent gene expression (52). However, whether in vivo M. tuberculosis-infected macrophages contain increased levels of the Fzd1 receptor on their cell surface and thus making them susceptible to Wnt3 activation remains to be determined, since the expression levels of the distinct Fzd receptors genes were determined using mRNA obtained from total lung homogenates. Nonetheless, the Wnt3 negative effect on TNF release by M. tuberculosis-infected macrophages suggests anti-inflammatory functions in murine macrophages (44). Moreover, it has been described that Wnt3 also inhibits IL-6 during mycobacteria infection (44). Interestingly, Wnt3 enhances apoptotic cell death in RAW 264.7 macrophage infected with M. bovis bacillus Calmette–Guerin (BCG), through a mechanism involving the regulation of pro-apoptotic and antiapoptotic protein levels. M. bovis BCG-infected macrophages showed increased levels of the pro-apoptotic protein Bax and reduced levels of the antiapoptotic protein Mcl-1 in response to Wnt3 exposure (46). Thus, by inducing apoptosis of infected macrophages, Wnt3 prevents bacterial dissemination. Consistent with this idea, another study showed that Wnt3 reduced the levels of reactive oxygen species (ROS) resulting from BCG infection, which correlates with the increase in glutathione levels (Figure 6) (46). However, whether the elevated glutathione levels induced by Wnt3 result from an increase in the enzymatic activity of the enzymes involved in glutathione synthesis (gamma-glutamylcysteine synthetase and glutathione synthetase) or from increased expression of the genes encoding these enzymes, remains to be determined. Together, these results suggest that at early stages of the infection, Wnt3 prevents the dissemination of infectious mycobacteria by promoting apoptosis and reducing necrosis of infected macrophages. However, Wnt3 promotes the differentiation toward an M2 anti-inflammatory phenotype of the infected macrophages that survive, thus attenuating the immune response against M. tuberculosis and therefore promoting TB.
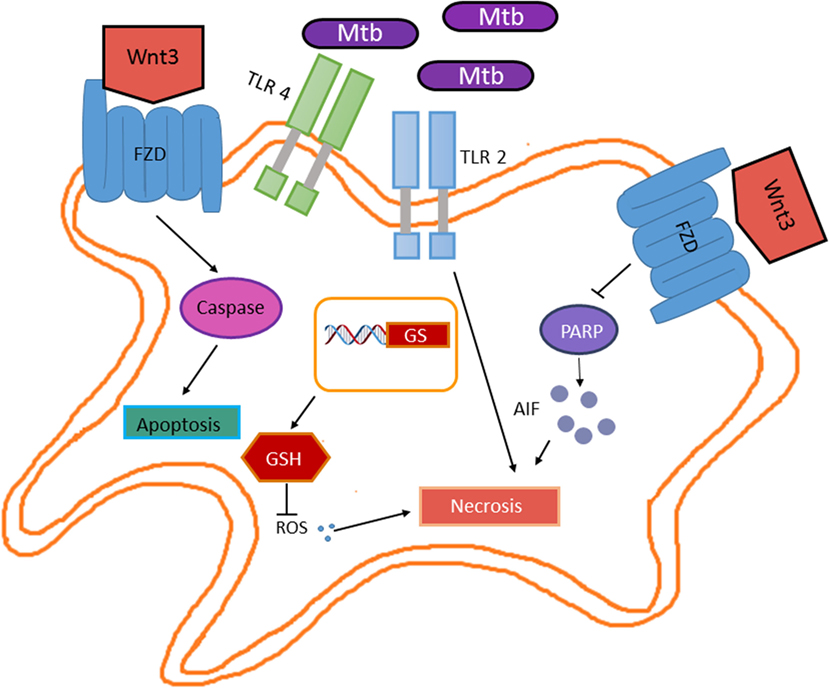
Figure 6. Wnt3 promotes apoptosis in response to Mycobacterium tuberculosis (M. tuberculosis) infection. Wnt3 plays a protective role in the M. tuberculosis infection by preventing bacterial dissemination. On one hand, Wnt3 promotes apoptosis in a caspase-dependent manner, while on the other hand inhibits necrosis by promoting glutathione synthase expression (GS) and thus increasing the levels of glutathione (GSH) that in turn results in reduced reactive oxygen species and by inhibiting the PARP/AIF pathway.
Wnt5
Genomic analyses have shown that macrophages and dendritic cells upregulate Wnt5a mRNA levels when exposed to pathogens (52, 53). Also, during the differentiation process of monocytes to macrophages or dendritic cells, granulocyte macrophage colony-stimulating factor and IL-4 promote Wnt5a gene expression (51). In Raw 264.7 macrophages, M. tuberculosis infection increases Wnt5 protein levels. Accordingly, WNT5A and WNT5A receptor FZD5 mRNA levels are induced in human macrophages in response to M. tuberculosis and M. avium infection (19). M. tuberculosis-dependent WNT5a expression requires TLR2 signaling and NFκB activation; however, whether M. tuberculosis uses the same signaling pathway to induce FZD5 gene expression remains to be confirmed. Interestingly, human peripheral blood mononuclear cells expressing WNT5A and FZD5 mRNA were detected in granulomatous lesions in the lungs of M. tuberculosis-infected patients (48). Given that the IL-12 production by mycobacterium-infected macrophages results from a Wnt5a autocrine loop and that activated T cell express FZD5 at the mRNA and protein levels (48), these experimental evidence indicates that INFγ production by mycobacterial antigen-stimulated T cells is induced not only by IL-12 but also by the interaction of WNT5A with FZD5 receptor expressed on the T cell surface. Whether the WNT5A-mediated INFγ production in T cells involves the non-canonical Wnt pathway remains to be determined. Nonetheless, the paramount role of INFγ in the adaptive immune response against M. tuberculosis suggest that Wnt5a produced by mycobacterium-infected macrophages also play a key role against M. tuberculosis control. However, recently, it was shown that M. bovis BCG prevents INFγ-induced macrophage autophagy through a mechanism that involves Wnt5a, which enhances the expression of ALOX5 and ALOX15 and thus the production of anti-inflammatory lipids (54). Together, these results place Wnt5a in the center of the fine balance that is required for maintaining the granuloma to avoid bacterial dissemination and also to prevent bacterial destruction. Whether Wnt5a is also required for granuloma formation remains to be determined.
Although the mechanism by which mycobacteria promotes Wnt5 expression remains largely unknown, recently, it has been demonstrated that dectin-1 activation through different dectin-1 ligands, including mycobacteria, results in Wnt5 expression (54). Upon dectin-1 engagement, activation of the tyrosine kinase SYK and ROS production lead to the inactivation of GSK3 resulting in β-catenin stabilization and thus Wnt5 expression (19). In addition to the functions described above, Wnt5 through an autocrine loop activates the non-canonical Wnt pathway, resulting in the activation of calcium-activated calmodulin kinase (Ca++/CAMKII) pathway and the expression of the protein inhibitors of activated STAT (PIAS1) and suppressor of cytokine signaling (SOCS1) expression (54). Overexpression of PIAS1 and SOCS1 in macrophages dampens TLR-induced pro-inflammatory response by reducing the protein levels of pivotal TLR adaptors such as IRAK1, IRAK4, and Myd88 (19). Accordingly, in vivo activation of dectin-1 receptor with pathogenic or fungi ligands resulted in an increased mycobacteria burden and a concomitant decrease in TLR-triggered pro-inflammatory cytokines (54). This might also involve impaired phagosome–lysosome fusion and bacterial destruction, since activation of the non-canonical Wnt pathway by Wnt5 involves Ca++ (37, 38), which is required for coronin-1 recruitment to the phagosome and calcineurin activation to prevent phagosome–lysosome fusion (55).
Wnt6
In a recent study, Schaale and coworkers showed that Wnt6 is expressed in granulomatous lesions in the lung of M. tuberculosis-infected mice (50). Wnt6 expression in response to mycobacteria is TLR-Myd88-NFκB-dependent. Interestingly, foamy macrophage-like cells are the principal source of Wnt6. Secreted Wnt6 acts as a paracrine signal on neighboring macrophages to promote proliferation and macrophages polarization toward an anti-inflammatory M2 like phenotype (50). The Wnt6-induced signal transduction driving macrophages proliferation and polarization does not involve β-catenin, but relay on G protein-dependent ERK activation and c-Myc expression, a pivotal regulator of cell proliferation. These observations are consistent with another study showing a key role for c-Myc in IL-4-induced alternative activated phenotype in human macrophages (56). In this scenario, c-Myc directly regulates the expression of genes associated with the M2 phenotype like scavenger receptor class B member 1, ALOX15, and mannose receptor, C-type 1 and indirectly regulates the expression of CD206 while strengthening IL-4 signaling by upregulating STAT6 and PPARγ expression (56). It remains to be determined whether these molecules are also involved in M2 polarization resulting from Wnt6 exposure. Nonetheless, these results point out an important role for Wnt6 in promoting macrophages alternative activation within the granulomatous lesions, thus providing M. tuberculosis a granuloma with an anti-inflammatory microenvironment that ensures its survival.
Another Wnt Homolog
Besides the roles of Wnt3, Wnt5, and Wnt6 in M. tuberculosis infection discussed above, the role of other Wnt homologs is unknown. In a recent study where Wnt6 was identified as a molecule expressed exclusively in granulomatous lesions, Schaale and coworkers monitored the expression of all Wnt homologs in mice infected with M. tuberculosis (50). They found that mRNA levels of most Wnt homologs were significantly reduced in the course of infection (Wnt2, Wnt2b, Wnt3a, Wnt4, Wnt5, Wnt7a, Wnt8a, and Wnt10b), while other homologs were upregulated (Wnt1, Wnt6, and Wnt10a) (50). The reduction in the expression levels of the majority of the Wnt homologs during mycobacterial infection is consistent with a study that indicates that FZD family of receptors and the Wnt/β-catenin target gene Axin2 are significantly downregulated in the lungs of M. tuberculosis-infected mice (44).
In a genetic study made in a Chinese population, 25 polymorphisms (SNPs) in the Wnt pathway related to TB risk were evaluated. Five polymorphisms were associated with TB susceptibility: secreted frizzled-related protein (SFRP1) rs4736958, CTNNB1 (catenin-β1), rs9859392, rs987055, and rs3864004 showed decreased TB risk, meanwhile, SFRP1 rs7832767 was associated with increased risk (57, 58). According to these results, the authors provide evidence that Wnt pathway polymorphisms may influence TB susceptibility and host immune response to M. tuberculosis, suggesting that these variations could serve as novel markers to identify the risk and susceptibility to develop TB (57, 58).
Nucleotide-Binding Oligomerization Domain (NOD2) Mediates the Activation of the Wnt Pathway in Response to M. tuberculosis Infection?
So far, we have discussed distinct experimental evidences that clearly indicate an important role of the Wnt factors on the biology of M. tuberculosis-infected macrophages and that M. tuberculosis modulates the expression of these factors and their receptors to ensure successful infection. However, whether M. tuberculosis triggers the Wnt pathway directly, is still an open question. The intracellular pathogen sensor NOD2, which is activated by bacterial cell wall components like peptidoglycan, specifically by muramyl dipeptide (MDP), has been shown to be activated during macrophage infection with M. tuberculosis and to synergize with the TLR signaling pathway to promote the production of pro-inflammatory cytokines (59–61). Interestingly, last year, it was suggested that the activation of NOD2 by MDP results in the activation of the Wnt pathway (62). In the presence of MDP, NOD2 is translocated to the membrane where it interacts with the Ly6/PLAUR domain-containing protein 6, which has been previously shown to be recruited to the Wnt receptor complex and promotes Wnt signaling (63) that results in β-catenin nuclear translocation and the expression of the X chromosome-linked inhibitor of apoptosis, that in turn activates the NLRP3 inflammasome leading to IL-1β maturation and secretion (62). Thus, it is possible that MDP, derived from the mycobacterial cell wall at early phases of the macrophages infection, triggers NOD2 that activates the Wnt pathway directly even in the absence of Wnt factors (Figure 7) to promote caspase-1 activation and IL-1β production, and probably some of the biological effects attributed to specific Wnt factors discussed above. Nonetheless, this hypothesis remains to be tested.
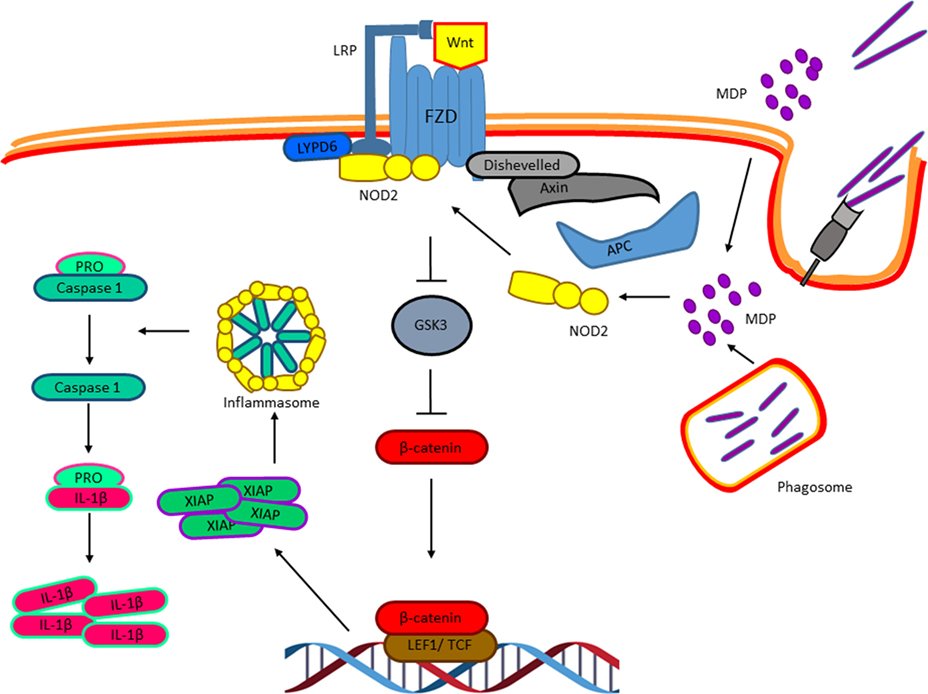
Figure 7. Nucleotide-binding oligomerization domain 2 (NOD2) and the activation of the Wnt pathway in response to Mycobacterium tuberculosis infection. Muramyl dipeptide, derived from the Mycobacterial cell wall at early phases of the macrophages’ infection, triggers NOD2 translocation to the membrane where it interacts with the Ly6/PLAUR domain-containing protein 6, which has been previously shown to be recruited to the Wnt receptor complex and promote Wnt signaling resulting in β-catenin nuclear translocation and the expression of the X chromosome-linked inhibitor of apoptosis, that in turn activates the NLRP3 inflammasome resulting in IL-1β maturation.
M. tuberculosis Fine Tuning of the Wnt Pathway through microRNAs (miRNAs) Regulation
A novel layer of complexity in M. tuberculosis regulatory networks controlling host infection, survival, and successful infection has recently emerged with the discovery that Mycobacterium regulates the expression of different miRNAs (64). miRNAs regulate the protein abundance in any given cell at any given time, thus influencing many cellular processes including differentiation, proliferation, apoptosis, DNA methylation, DNA repair, and provide anti-inflammatory or pro-inflammatory stimuli (65, 66). According to this, it is not surprising to find that M. tuberculosis positively modulates the expression of miRNAs that target key molecules involved in the destruction of the pathogen like apoptosis or autophagy and the setting of protective cytokine profiles while decreasing the expression of miRNAs whose targets are molecules involved in the negative control of these biological processes [reviewed in Ref. (67, 68)]. Although we know that the miRNA profile expressed by infected macrophages depends on the infecting Mycobacterium species (64, 69, 70), the virulence of the strain (71), or the stage of the disease (69), the role of those miRNAs and the signaling pathway that control their expression remains to be elucidated. Nonetheless, at early stage of infection, inhibition of autophagy and apoptosis and the control of the inflammatory response by M. tuberculosis involve different miRNAs, some of which could mediate their effect through the induction of the Wnt signaling pathway or be regulated directly by this pathway (Figure 8). At the onset of infection, miR155 expression increase in M. tuberculosis-infected macrophages (72). The induction of this miRNA may be mediated by the activation of NFκB (73); thus, miR155 could impair autophagy in M. tuberculosis-infected macrophages through its interaction with the 3′ UTRs of different mRNAs encoding for key proteins like ULK1, involved in the initiation phase; ATG14, part to the BECLIN1 complex that promotes phagophore nucleation; ATG5, ATG3, LC3, and GABARAPL1, key players in the elongation and maturation steps; and FOXO3, a transcription factor regulating the expression of autophagy-related genes, including MAP1LC3, ULK1, ATG14, and GABARAPL1 (74). Additionally, miR30a, another miRNA expressed in infected macrophages, through its interaction with BECLIN1 (75) could also impair autophagy. Interestingly miR155, through its negative effect on APC (76) and HMG-box transcription factor 1 (77), a strong Wnt pathway suppressor and miR30a by blocking PRDMI (78) could induce β-catenin/TCF-mediated gene expression, and thus regulate the expression of additional miRNAs, like miR21 and miR125b (79) and miR146b, which is positively regulated by Wnt5a (80) and miR29 (81, 82). miR21 by reducing BCL2 protein levels (83) and miR29 by targeting caspase-7 (84) might prevent apoptosis of infected macrophages. Additionally, miR29 could also further enhance the Wnt signaling pathway by targeting the negative regulators of Wnt signaling, Dikkopf-1, Kremen2, and secreted frizzled-related protein 2 (sFRP2) (81, 82). While miR146b, by targeting IL-1β, IL-6, IRAK1, TRAF6, TNF, TLR4, NFκB1 (85), and miR21 by negatively regulating IL-12 (86) damp the inflammatory response. Additionally, given that the lung epithelium constitutively produces Wnt3 and that the canonical Wnt pathway promotes miR125b expression (74), at early stages of M. tuberculosis infection, miR125b could also contribute to attenuate the inflammatory response by targeting the TNF 3′ UTR (87). Together, these results strongly indicate that, at early stages of infection, the Wnt pathway controls key macrophage functions by modulating miRNAs expression that allows M. tuberculosis survival.
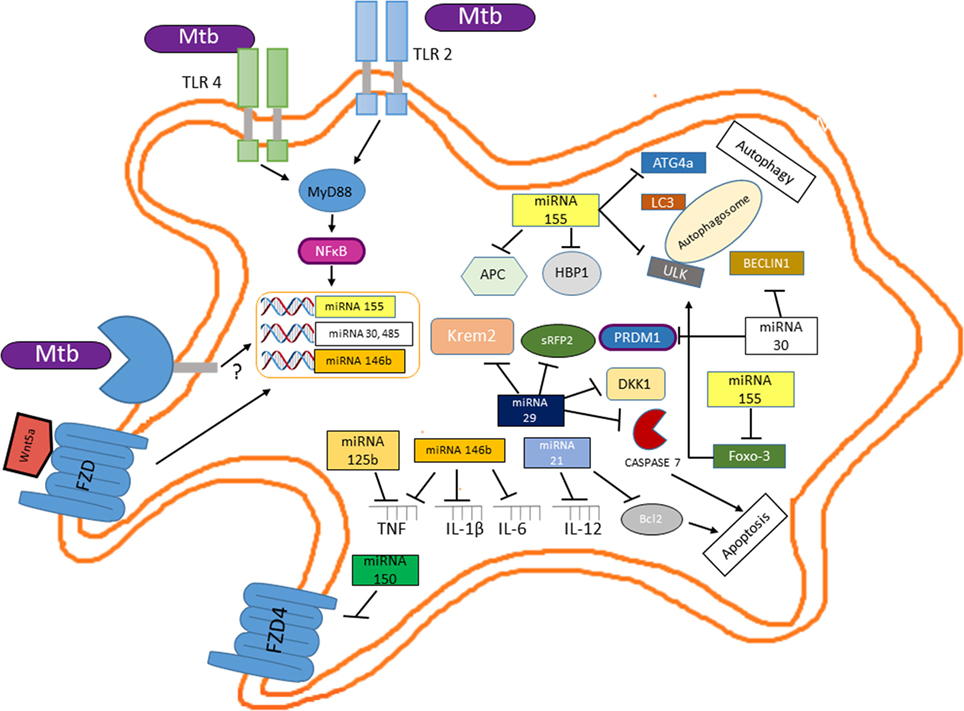
Figure 8. Mutual regulation between miRNAs and the Wnt pathway determine the survival of Mycobacterium tuberculosis (M. tuberculosis)-infected macrophage and the profile of the inflammatory response. Upon interaction of M. tuberculosis with TLR2 and TLR4, NFκB activation leads to increased miR155 levels which, besides its negative effect on autophagy and apoptosis, through the negative modulation of key inhibitors of the Wnt pathway (APC and HMG-box transcription factor 1) leads to β-catenin/TCF-mediated expression of miR2, miR125b, miR146b (a target of the Wnt5a signaling pathway), and miR29, thus wiping off the pro-inflammatory response. Additionally, miR29 further enhances Wnt signaling by targeting distinct set of Wnt negative regulators (Dikkopf-1, Kremen2, and sFRP2).
A recent study points out that different M. tuberculosis clinical strains induced specific macrophage miRNAs expression patterns (69). Macrophages infected with Beijing M. tuberculosis strains showed reduced expression of 13 miRNAs compared with macrophages infected with non-Beijing M. tuberculosis strains. Interestingly, only miR485 showed increased levels in the Beijing infected macrophage when compared with non-Beijing infected macrophages. Our bioinformatics analysis indicates that this miRNA has potential binding sites with thermodynamic values that allow specific miRNA/mRNA interactions within the 3′ UTR of mRNAs encoding for proteins involved in the Wnt pathway (Table 3). A gene ontology analysis revealed that through negatively modulating the levels of the SFRP1 and those of the Strabismus 1 (STB1), miR485 might positively regulate the canonical Wnt pathway. In contrast, miR485 might inhibit the non-canonical Wnt pathways by negatively modulating FZD4, RAC-1, and PKC (88) (Figure 9). The previous data indicate that the Beijing M. tuberculosis clinical strains upregulate the canonical Wnt pathway by inducing miR485 expression, to modulate the macrophage response in order to ensure a successful infection.

Table 3. Predicteda miRNA485 target genes.
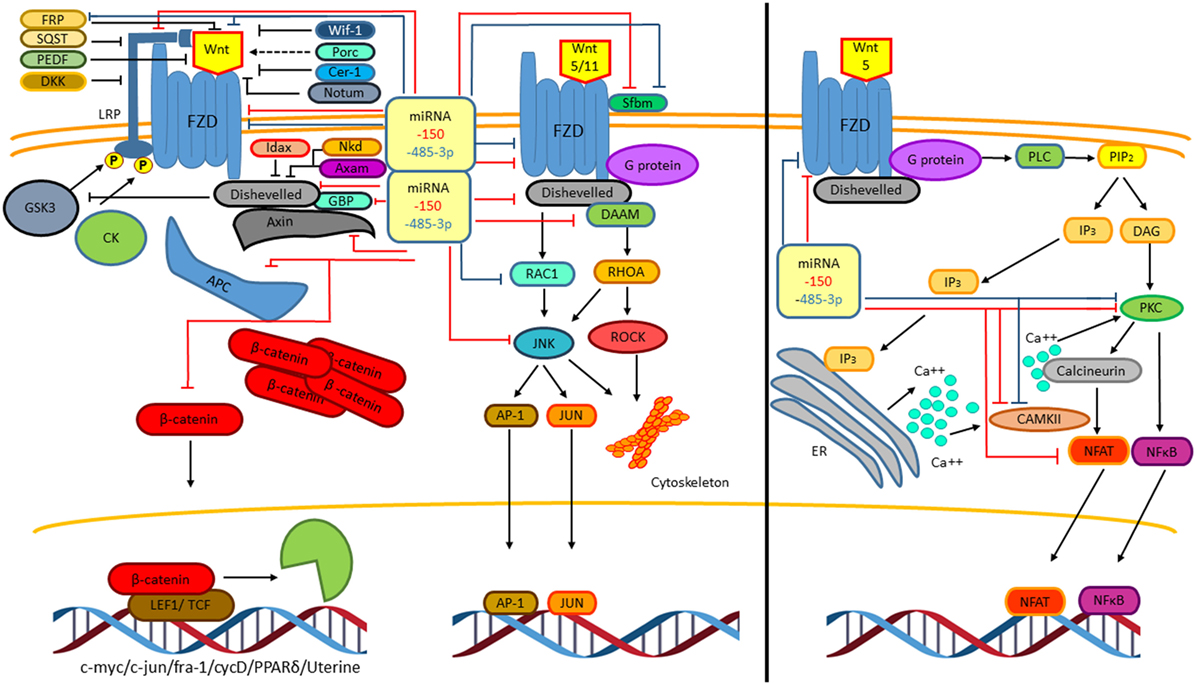
Figure 9. The Beijing Mycobacterium tuberculosis (M. tuberculosis) strains regulate the Wnt pathway during macrophage infection of healthy individuals or that of macrophages from patients with active tuberculosis by enhancing miR485 and reducing miR150 levels, respectively. An enrichment pathway analysis performed in the WebGestalt platform using the Kyoto Encyclopedia of Genes and Genomes indicates that the miRNA485 potentiates the canonical Wnt pathway through negatively regulating protein levels involved in the inhibition of these pathways, while by controlling the protein levels of key signaling molecules might inhibit the non-canonical Wnt pathway. In contrast, miR150 has clear inhibitory effect on both the canonical and non-canonical Wnt pathway. Thus, through differentially regulating the expression of these two miRNAs, the Beijing M. tuberculosis strains promote the Wnt canonical pathway.
On the other hand, from the miRNAs negatively regulated by the Beijing M. tuberculosis clinical strains in both THP-1 derived macrophages or in peripheral blood-derived macrophages obtained from TB patients with active disease, miR150 showed the most prominent reduction (69). Once again, our bioinformatic analysis indicated that, this miRNA has numerous potential targets in the Wnt pathway (Table 4). According to thermodynamic values indicating strong possibilities for specific miRNA/mRNAs interactions, from the predicted targets, FZD4 has been shown to be a bonafide miR150 target (89). The pathway analysis of the potential miR150 targets indicates that this mRNA might negatively regulate both the canonical and non-canonical Wnt pathways (Figure 9). Together, these results suggest that the upregulation of miR485 and the negative regulation of miR150 levels are key events triggered by Beijing M. tuberculosis clinical strains to sustain the activation of the canonical Wnt pathway during macrophage infection of healthy individual and that of individual with active disease. However, whether the opposite regulation of these two miRNAs by the Beijing M. tuberculosis is hallmark of the virulence phenotype remain to be determined. Nonetheless, it is clear that the regulation of miRNAs expression by M. tuberculosis constitutes a novel mechanism to modulate the Wnt pathway during infection and also that through modulating the levels of specific miRNAs, the Wnt pathway ensures the macrophage infection with M. tuberculosis and its survival by promoting a anti-inflammatory environment.

Table 4. Predicteda miRNA150 target genes.
Concluding Remarks
In the course of M. tuberculosis infection, several pathways are induced to promote inflammation to eliminate the pathogen and to attenuate the inflammatory response to avoid tissue damage. The data discussed above strongly suggest that the Wnt signaling pathway plays a key role at different stages of TB development, including M. tuberculosis survival in the macrophages, modulation of the inflammatory response, and later on granuloma formation and to control the adaptive immune response. However, further research is required to determine: (i) whether M. tuberculosis can directly activate the Wnt signaling pathway upon macrophage infection; (ii) which of the biological effects attributed to cytokines are mediated by Wnt factors; (iii) whether virulent strains are more dependent on the Wnt pathway to ensure infection; and (iv) define how the different miRNAs expressed in response to M. tuberculosis infection modulate the Wnt signaling pathway, among other important processes. Efforts to determine the mRNA and miRNAs profile of infected macrophages obtained from mouse or human granulomas using deep sequencing will provide a clear picture of which Wnt factors, FZD receptors, and Wnt-dependent genes are expressed among them miRNAs, the pathway targeted by these miRNAs and if all of this depend on the M. tuberculosis strain, the virulence or drugs resistance. This information may identify candidates for the development of new therapeutic strategies to promote mycobacterial destruction in the granuloma. Likewise, future clinical studies may provide new insights for therapeutic applications of Wnt factors.
Author Contributions
TV participated in the conception of the work, gathered information, and wrote the first draft of the paper. EM-P gathered information, performed bioinformatic analysis, and participated in the edition of the final draft. RM-B gathered information, performed artwork, and participated in the edition of the final draft. AU-A gathered information and performed the gene ontology and pathway enrichment analysis for the miRNA section. LP-M participated in the conception of the work, gathered information, and participated in the edition of the final draft. GP-A participated in the conception of the work, gathered information, and participated in the edition of the final draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partially supported by grants from ICGEB/Italy CRP/MEX11-01 to GP-A and grants from DGAPA/UNAM IN213316 to LP-M and IN227510 and IN212316 to GP-A. TV was postdoctoral fellow from DGAPA/UNAM. EM-P is a PhD student enrolled in the Biochemistry PhD program/UNAM and is supported by a scholarship from CONACYT/Mexico. RM-B is a master’s student enrolled in the Cells Biology Master program/UAEM and is supported by a scholarship from CONACYT/Mexico.
References
1. Jayachandran R, BoseDasgupta S, Pieters J. Surviving the macrophage: tools and tricks employed by Mycobacterium tuberculosis. Curr Top Microbiol Immunol (2013) 374:189–209. doi:10.1007/82_2012_273
2. Schlesinger LS, Horwitz MA. Phagocytosis of leprosy bacilli is mediated by complement receptors CR1 and CR3 on human monocytes and complement component C3 in serum. J Clin Invest (1990) 85(4):1304–14. doi:10.1172/JCI114568
3. Le Cabec V, Carreno S, Moisand A, Bordier C, Maridonneau-Parini I. Complement receptor 3 (CD11b/CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J Immunol (2002) 169(4):2003–9. doi:10.4049/jimmunol.169.4.2003
4. Schafer G, Jacobs M, Wilkinson RJ, Brown GD. Non-opsonic recognition of Mycobacterium tuberculosis by phagocytes. J Innate Immun (2009) 1(3):231–43. doi:10.1159/000173703
5. Ehlers S. DC-SIGN and mannosylated surface structures of Mycobacterium tuberculosis: a deceptive liaison. Eur J Cell Biol (2010) 89(1):95–101. doi:10.1016/j.ejcb.2009.10.004
6. Peterson PK, Gekker G, Hu S, Sheng WS, Anderson WR, Ulevitch RJ, et al. CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect Immun (1995) 63(4):1598–602.
7. Fratazzi C, Manjunath N, Arbeit RD, Carini C, Gerken TA, Ardman B, et al. A macrophage invasion mechanism for mycobacteria implicating the extracellular domain of CD43. J Exp Med (2000) 192(2):183–92. doi:10.1084/jem.192.2.183
8. Pasula R, Downing JF, Wright JR, Kachel DL, Davis TE Jr, Martin WJ II. Surfactant protein A (SP-A) mediates attachment of Mycobacterium tuberculosis to murine alveolar macrophages. Am J Respir Cell Mol Biol (1997) 17(2):209–17. doi:10.1165/ajrcmb.17.2.2469
9. Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J Clin Immunol (2007) 27(4):347–62. doi:10.1007/s10875-007-9084-0
10. Zimmerli S, Edwards S, Ernst JD. Selective receptor blockade during phagocytosis does not alter the survival and growth of Mycobacterium tuberculosis in human macrophages. Am J Respir Cell Mol Biol (1996) 15(6):760–70. doi:10.1165/ajrcmb.15.6.8969271
11. Pieters J. Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe (2008) 3(6):399–407. doi:10.1016/j.chom.2008.05.006
12. Gengenbacher M, Kaufmann SH. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev (2012) 36(3):514–32. doi:10.1111/j.1574-6976.2012.00331.x
13. Goldberg DE, Siliciano RF, Jacobs WR. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell (2012) 148(6):1271–83. doi:10.1016/j.cell.2012.02.021
14. Soldati T, Neyrolles O. Mycobacteria and the intraphagosomal environment: take it with a pinch of salt(s)! Traffic (2012) 13(8):1042–52. doi:10.1111/j.1600-0854.2012.01358.x
15. Cousin MA, Robinson PJ. The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci (2001) 24(11):659–65. doi:10.1016/S0166-2236(00)01930-5
16. Hossain MM, Norazmi MN. Pattern recognition receptors and cytokines in Mycobacterium tuberculosis infection – the double-edged sword? Biomed Res Int (2013) 2013:179174. doi:10.1155/2013/179174
17. Shaler CR, Horvath CN, Jeyanathan M, Xing Z. Within the Enemy’s Camp: contribution of the granuloma to the dissemination, persistence and transmission of Mycobacterium tuberculosis. Front Immunol (2013) 4:30. doi:10.3389/fimmu.2013.00030
18. Sasindran SJ, Torrelles JB. Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front Microbiol (2011) 2:2. doi:10.3389/fmicb.2011.00002
19. Schaale K, Neumann J, Schneider D, Ehlers S, Reiling N. Wnt signaling in macrophages: augmenting and inhibiting Mycobacteria-induced inflammatory responses. Eur J Cell Biol (2011) 90(6–7):553–9. doi:10.1016/j.ejcb.2010.11.004
20. Miki T, Yasuda SY, Kahn M. Wnt/beta-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev (2011) 7(4):836–46. doi:10.1007/s12015-011-9275-1
21. Voronkov A, Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des (2013) 19(4):634–64. doi:10.2174/1381612811306040634
22. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov (2014) 13(7):513–32. doi:10.1038/nrd4233
23. Coudreuse D, Korswagen HC. The making of Wnt: new insights into Wnt maturation, sorting and secretion. Development (2007) 134(1):3–12. doi:10.1242/dev.02699
24. Duan P, Bonewald LF. The role of the wnt/beta-catenin signaling pathway in formation and maintenance of bone and teeth. Int J Biochem Cell Biol (2016) 77(Pt A):23–9. doi:10.1016/j.biocel.2016.05.015
25. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev (1997) 11(24):3286–305. doi:10.1101/gad.11.24.3286
26. Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV, et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature (2016) 530(7590):340–3. doi:10.1038/nature16937
27. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell (1982) 31(1):99–109. doi:10.1016/0092-8674(82)90409-3
28. Baker NE. Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: the spatial distribution of a transcript in embryos. EMBO J (1987) 6(6):1765–73.
29. Staal FJT, Luis TC, Tiemessen MM. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol (2008) 8(8):581–93. doi:10.1038/nri2360
30. He S, Lu Y, Liu X, Huang X, Keller ET, Qian CN, et al. Wnt3a: functions and implications in cancer. Chin J Cancer (2015) 34(12):554–62. doi:10.1186/S40880-015-0052-4
31. Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes Dev (2006) 20(11):1394–404. doi:10.1101/Gad.1424006
32. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J (1997) 16(13):3797–804. doi:10.1093/Emboj/16.13.3797
33. Semenov MV, Habas R, MacDonald BT, He X. SnapShot: noncanonical Wnt signaling pathways. Cell (2007) 131(7):1378. doi:10.1016/j.cell.2007.12.011
34. Endo Y, Wolf V, Muraiso K, Kamijo K, Soon L, Uren A, et al. Wnt-3a-dependent cell motility involves RhoA activation and is specifically regulated by dishevelled-2. J Biol Chem (2005) 280(1):777–86. doi:10.1074/jbc.M406391200
35. Liang HL, Chen Q, Coles AH, Anderson SJ, Pihan G, Bradley A, et al. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell (2003) 4(5):349–60. doi:10.1016/S1535-6108(03)00268-X
36. Liang HL, Coles AH, Zhu ZQ, Zayas J, Jurecic R, Kang J, et al. Noncanonical Wnt signaling promotes apoptosis in thymocyte development. J Exp Med (2007) 204(13):3077–84. doi:10.1084/Jem.20062692
37. Kuhl M, Sheldahl LC, Park M, Miller JR, Moon RT. The Wnt/Ca2+ pathway – a new vertebrate Wnt signaling pathway takes shape. Trends Genet (2000) 16(7):279–83. doi:10.1016/S0168-9525(00)02028-X
38. Sheldahl LC, Slusarski DC, Pandur P, Miller JR, Kuhl M, Moon RT. Dishevelled activates Ca2+ flux, PKC, and CamKII in vertebrate embryos. J Cell Biol (2003) 161(4):769–77. doi:10.1083/jcb.200211094
39. Clipstone NA, Crabtree GR. Identification of calcineurin as a key signaling enzyme in lymphocyte-T activation. Nature (1992) 357(6380):695–7. doi:10.1038/357695a0
40. Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev (2001) 11(5):505–12. doi:10.1016/S0959-437x(00)00225-2
41. Murphy LLS, Hughes CCW. Endothelial cells stimulate T cell NFAT nuclear translocation in the presence of cyclosporin A: involvement of the wnt/glycogen synthase kinase-3 beta pathway. J Immunol (2002) 169(7):3717–25. doi:10.4049/jimmunol.169.7.3717
42. Gustafson B, Smith U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. J Biol Chem (2006) 281(14):9507–16. doi:10.1074/jbc.M512077200
43. Halleskog C, Mulder J, Dahlstrom J, Mackie K, Hortobagyi T, Tanila H, et al. WNT signaling in activated microglia is proinflammatory. Glia (2011) 59(1):119–31. doi:10.1002/glia.21081
44. Neumann J, Schaale K, Farhat K, Endermann T, Ulmer AJ, Ehlers S, et al. Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reprogramming Mycobacterium tuberculosis-infected macrophages. FASEB J (2010) 24(11):4599–612. doi:10.1096/fj.10-160994
45. Tickenbrock L, Schwable J, Strey A, Sargin B, Hehn S, Baas M, et al. Wnt signaling regulates transendothelial migration of monocytes. J Leukoc Biol (2006) 79(6):1306–13. doi:10.1189/jlb.0905539
46. Wu X, Deng G, Li M, Li Y, Ma C, Wang Y, et al. Wnt/beta-catenin signaling reduces bacillus Calmette-Guerin-induced macrophage necrosis through a ROS-mediated PARP/AIF-dependent pathway. BMC Immunol (2015) 16:16. doi:10.1186/s12865-015-0080-5
47. Wu X, Deng G, Hao X, Li Y, Zeng J, Ma C, et al. A caspase-dependent pathway is involved in Wnt/beta-catenin signaling promoted apoptosis in bacillus Calmette-Guerin infected RAW264.7 macrophages. Int J Mol Sci (2014) 15(3):5045–62. doi:10.3390/ijms15035045
48. Blumenthal A, Ehlers S, Lauber J, Buer J, Lange C, Goldmann T, et al. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood (2006) 108(3):965–73. doi:10.1182/blood-2005-12-5046
49. Maiti G, Naskar D, Sen M. The Wingless homolog Wnt5a stimulates phagocytosis but not bacterial killing. Proc Natl Acad Sci U S A (2012) 109(41):16600–5. doi:10.1073/pnas.1207789109
50. Schaale K, Brandenburg J, Kispert A, Leitges M, Ehlers S, Reiling N. Wnt6 is expressed in granulomatous lesions of Mycobacterium tuberculosis-infected mice and is involved in macrophage differentiation and proliferation. J Immunol (2013) 191(10):5182–95. doi:10.4049/jimmunol.1201819
51. Lehtonen A, Ahlfors H, Veckman V, Miettinen M, Lahesmaa R, Julkunen I. Gene expression profiling during differentiation of human monocytes to macrophages or dendritic cells. J Leukoc Biol (2007) 82(3):710–20. doi:10.1189/jlb.0307194
52. Chaussabel D, Semnani RT, McDowell MA, Sacks D, Sher A, Nutman TB. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood (2003) 102(2):672–81. doi:10.1182/blood-2002-10-3232
53. Nau GJ, Richmond JFL, Schlesinger A, Jennings EG, Lander ES, Young RA. Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci U S A (2002) 99(3):1503–8. doi:10.1073/Pnas.022649799
54. Trinath J, Holla S, Mahadik K, Prakhar P, Singh V, Balaji KN. The WNT signaling pathway contributes to dectin-1-dependent inhibition of Toll-like receptor-induced inflammatory signature. Mol Cell Biol (2014) 34(23):4301–14. doi:10.1128/MCB.00641-14
55. Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell (1999) 97(4):435–47. doi:10.1016/S0092-8674(00)80754-0
56. Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood (2012) 119(2):411–21. doi:10.1182/blood-2011-02-339911
57. Hu XJ, Zhou J, Chen XR, Zhou YH, Song XB, Cai B, et al. Pathway analyses identify novel variants in the WNT signaling pathway associated with tuberculosis in Chinese population. Sci Rep (2016) 6:28530. doi:10.1038/srep28530
58. Zhao ZZ, Peng W, Hu XJ, Zhang JY, Shang MQ, Zhou J, et al. SFRP1 variations influence susceptibility and immune response to Mycobacterium tuberculosis in a Chinese Han population. Infect Genet Evol (2016) 37:259–65. doi:10.1016/j.meegid.2015.11.031
59. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog (2005) 1(3):279–85. doi:10.1371/journal.ppat.0010034
60. Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol (2008) 181(10):7157–65. doi:10.4049/jimmunol.181.10.7157
61. Brooks MN, Rajaram MV, Azad AK, Amer AO, Valdivia-Arenas MA, Park JH, et al. NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell Microbiol (2011) 13(3):402–18. doi:10.1111/j.1462-5822.2010.01544.x
62. Singh V, Holla S, Ramachandra SG, Balaji KN. WNT-inflammasome signaling mediates NOD2-induced development of acute arthritis in mice. J Immunol (2015) 194(7):3351–60. doi:10.4049/jimmunol.1402498
63. Ozhan G, Sezgin E, Wehner D, Pfister AS, Kuhl SJ, Kagermeier-Schenk B, et al. Lypd6 enhances Wnt/beta-catenin signaling by promoting Lrp6 phosphorylation in raft plasma membrane domains. Dev Cell (2013) 26(4):331–45. doi:10.1016/j.devcel.2013.07.020
64. Sharbati J, Lewin A, Kutz-Lohroff B, Kamal E, Einspanier R, Sharbati S. Integrated MicroRNA-mRNA-analysis of human monocyte derived macrophages upon Mycobacterium avium subsp hominissuis infection. PLoS One (2011) 6(5):e20258. doi:10.1371/journal.pone.0020258
65. Belver L, de Yebenes VG, Ramiro AR. MicroRNAs prevent the generation of autoreactive antibodies. Immunity (2010) 33(5):713–22. doi:10.1016/j.immuni.2010.11.010
66. Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature (2011) 469(7330):336–42. doi:10.1038/nature09783
67. Abdalla AE, Duan X, Deng W, Zeng J, Xie J. MicroRNAs play big roles in modulating macrophages response toward mycobacteria infection. Infect Genet Evol (2016) 45:378–82. doi:10.1016/j.meegid.2016.09.023
68. Meza-Sosa KF, Pedraza-Alva G, Perez-Martinez L. microRNAs: key triggers of neuronal cell fate. Front Cell Neurosci (2014) 8:175. doi:10.3389/fncel.2014.00175
69. Zheng L, Leung E, Lee N, Lui G, To KF, Chan RCY, et al. Differential microRNA expression in human macrophages with Mycobacterium tuberculosis infection of Beijing/W and Non-Beijing/W strain types. PLoS One (2015) 10(6):e0126018. doi:10.1371/journal.pone.0126018
70. Ghorpade DS, Leyland R, Kurowska-Stolarska M, Patil SA, Balaji KN. MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Mol Cell Biol (2012) 32(12):2239–53. doi:10.1125/MCB.06597-11
71. Das K, Saikolappan S, Dhandayuthapani S. Differential expression of miRNAs by macrophages infected with virulent and avirulent Mycobacterium tuberculosis. Tuberculosis (2013) 93:S47–50. doi:10.1016/S1472-9792(13)70010-6
72. Rothchild AC, Sissons JR, Shafiani S, Plaisier C, Min D, Mai D, et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2016) 113(41):E6172–81. doi:10.1073/pnas.1608255113
73. Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol (2011) 3(3):159–66. doi:10.1093/jmcb/mjr007
74. D’Adamo S, Cetrullo S, Guidotti S, Borzi RM, Flamigni F. Hydroxytyrosol modulates the levels of microRNA-9 and its target sirtuin-1 thereby counteracting oxidative stress-induced chondrocyte death. Osteoarthritis Cartilage (2016) 32(12):2239–53. doi:10.1016/j.joca.2016.11.014
75. Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li A, et al. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One (2013) 8(1):e53950. doi:10.1371/journal.pone.0053950
76. Zhang X, Li M, Zuo K, Li D, Ye M, Ding L, et al. Upregulated miR-155 in papillary thyroid carcinoma promotes tumor growth by targeting APC and activating Wnt/beta-catenin signaling. J Clin Endocrinol Metab (2013) 98(8):E1305–13. doi:10.1210/jc.2012-3602
77. Sun X, Geng X, Zhang J, Zhao H, Liu Y. miR-155 promotes the growth of osteosarcoma in a HBP1-dependent mechanism. Mol Cell Biochem (2015) 403(1–2):139–47. doi:10.1007/s11010-015-2344-z
78. Wang X, Wang K, Han L, Zhang A, Shi Z, Zhang K, et al. PRDM1 is directly targeted by miR-30a-5p and modulates the Wnt/beta-catenin pathway in a Dkk1-dependent manner during glioma growth. Cancer Lett (2013) 331(2):211–9. doi:10.1016/j.canlet.2013.01.005
79. Peng Y, Zhang X, Feng X, Fan X, Jin Z. The crosstalk between microRNAs and the Wnt/beta-catenin signaling pathway in cancer. Oncotarget (2016) 8(1):e53950. doi:10.18632/oncotarget.12923
80. Codocedo JF, Inestrosa NC. Wnt-5a-regulated miR-101b controls COX2 expression in hippocampal neurons. Biol Res (2016) 49:9. doi:10.1186/s40659-016-0071-x
81. Kapinas K, Kessler C, Ricks T, Gronowicz G, Delany AM. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J Biol Chem (2010) 285(33):25221–31. doi:10.1074/jbc.M110.116137
82. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol (2011) 12(9):861–9. doi:10.1038/ni.2073
83. Xu LF, Wu ZP, Chen Y, Zhu QS, Hamidi S, Navab R. MicroRNA-21 (miR-21) regulates cellular proliferation, invasion, migration, and apoptosis by targeting PTEN, RECK and Bcl-2 in lung squamous carcinoma, Gejiu City, China. PLoS One (2014) 9(8):e103698. doi:10.1371/journal.pone.0103698
84. Fu Q, Shi HJ, Shi MT, Meng LP, Zhang H, Ren Y, et al. bta-miR-29b attenuates apoptosis by directly targeting caspase-7 and NAIF1 and suppresses bovine viral diarrhea virus replication in MDBK cells. Can J Microbiol (2014) 60(7):455–60. doi:10.1139/cjm-2014-0277
85. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103(33):12481–6. doi:10.1073/pnas.0605298103
86. Lu TX, Hartner J, Lim EJ, Fabry V, Mingler MK, Cole ET, et al. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol (2011) 187(6):3362–73. doi:10.4049/jimmunol.1101235
87. Rajaram MVS, Ni B, Morris JD, Brooks MN, Carlson TK, Bakthavachalu B, et al. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc Natl Acad Sci U S A (2011) 108(42):17408–13. doi:10.1073/pnas.1112660108
88. Heisenberg CP, Tada M. Wnt signalling: a moving picture emerges from van gogh. Curr Biol (2002) 12(4):R126–8. doi:10.1016/S0960-9822(02)00704-2
Keywords: tuberculosis, Wnt signaling, macrophage infection, inflammation, immune response, microRNAs
Citation: Villaseñor T, Madrid-Paulino E, Maldonado-Bravo R, Urbán-Aragón A, Pérez-Martínez L and Pedraza-Alva G (2017) Activation of the Wnt Pathway by Mycobacterium tuberculosis: A Wnt–Wnt Situation. Front. Immunol. 8:50. doi: 10.3389/fimmu.2017.00050
Received: 02 October 2016; Accepted: 12 January 2017;
Published: 01 February 2017
Edited by:
Victor Manuel Baizabal-Aguirre, Universidad Michoacana de San Nicolás de Hidalgo, MexicoReviewed by:
Jaya Talreja, Wayne State University School of Medicine, USASahana Holla, National Cancer Institute (NIH), USA
Copyright: © 2017 Villaseñor, Madrid-Paulino, Maldonado-Bravo, Urbán-Aragón, Pérez-Martínez and Pedraza-Alva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo Pedraza-Alva, Z3VzdGF2b0BpYnQudW5hbS5teA==