 Mohamad El-Zaatari
Mohamad El-Zaatari John Y. Kao
John Y. Kao- Division of Gastroenterology, Department of Internal Medicine-Gastroenterology, University of Michigan, Ann Arbor, MI, USA
The host immune response to gastrointestinal (GI) infections, hypersensitivity reactions, or GI cancers comprises numerous pathways that elicit responses on different host cells. Some of these include (1) the stimulation of mast cells via their IgE receptor, (2) the production of antibodies leading to antibody-mediated cytotoxic T/natural killer cell killing, (3) the activation of the complement pathway, and (4) the activation of the adaptive immune response via antigen-presenting cell, T cell, and B cell interactions. Within the plethora of these different responses, several host immune cells represent major key players such as those of myeloid lineage (including neutrophils, macrophages, myeloid-derived suppressor cells) or lymphoid lineage (including T and B cells). In this review, we focus on newly identified metabolites and metabolite receptors that are expressed by either myeloid or lymphoid lineages. Irrespective of their source, these metabolites can in certain instances elicit responses on a wide range of cell types. The myeloid-expressed metabolic enzymes and receptors which we will discuss in this review include arginase 2 (Arg2), indoleamine-2,3-dioxygenase 1 (IDO1), hydroxycarboxylic acid receptor 2 (Hcar2; also called GPR109A), and immunoresponsive gene 1 (Irg1). We will also review the role of the lymphoid-expressed metabolite receptor that binds to the sphingosine-1-phosphate (S1P) sphingolipid. Moreover, we will describe the synthesis and metabolism of retinoic acid, and its effect on T cell activation. The review will then discuss the function of these metabolites in the context of GI disease. The review provides evidence that metabolic pathways operate in a disease- and context-dependent manner—either independently or concomitantly—in the GI tract. Therefore, an integrated approach and combinatorial analyses are necessary to devise new therapeutic strategies that can synergistically improve prognoses.
Introduction
The host immune response—during infections or carcinogenesis—comprises a plethora of stimulatory and inhibitory signals. These manifest in numerous cellular activities, which are underscored by extensively complicated, variegated, and overlapping molecular and cellular interactions. Metabolites—such as those generated by amino acid breakdown—comprise a subset of the above-mentioned signals, which can regulate the outcome of host immunopathology in a context-dependent (i.e., disease-specific) manner.
In this article, we will first briefly review several components of the host immune response in the gastrointestinal (GI) tract. Then, we will outline specific contexts in which metabolites and/or metabolite receptors influence these components of host immunity. We will describe the source of these metabolites, and the critical steps that can control the immunopathological outcome. By contrast, we will also discuss the manner in which failed approaches can arise in targeting these pathways. Due to our inability to review the entire metabolome, we will focus on eight specific metabolites and their receptors, which epitomize distinct and variegated responses. The review will focus on the roles of arginase 2 (Arg2), indoleamine-2,3-dioxygenase 1 (IDO1), hydroxycarboxylic acid receptor 2 (Hcar2; also called GPR109A), immunoresponsive gene 1 (Irg1), sphingosine-1-phosphate (S1P), and all-trans-retinoic acid (atRA).
Host Immunity in the GI Tract
The host immune response comprises the initiation of several reactions that are carried out by a subset of white blood cells (WBCs), or leukocytes. These WBCs are divided into two lineages, myeloid and lymphoid, which arise from a hematopoietic stem cell origin (1). The hematopoietic stem cell gives rise to common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs) (1). The lymphoid lineage arises from the CLP and comprises B cells (and mature plasma cells), T cells, and natural killer (NK) cells (2, 3). The myeloid lineage arises from the CMP and comprises monocytes, macrophages, dendritic cells, neutrophils, eosinophils, basophils, mast cells, megakaryocytes, and erythrocytes (2, 3).
After a foreign antigen is detected by a subset of myeloid cells (4) or B cells (5), these cells can either elicit an autonomous thymus-independent response (6), or otherwise interact with T cells to elicit an acquired T cell-dependent response (6). The cells that detect the antigen are known as antigen-presenting cells (APCs) because they are able to present antigen to T cells (7). As mentioned above, APCs are usually myeloid (mainly dendritic cells) (4), but can also be of the B cell lineage (5). These cells detect foreign antigen in the mucosa of the affected GI tissue (8). However, they can also detect antigen in tissue-draining lymph nodes if the foreign antigen was in circulation and reached the lymph nodes via afferent lymphatic vessels (9–11). Once APCs detect foreign antigen they can elicit an autonomous T cell-independent response; for example dendritic cells can produce cytokines (12) while B cells can produce antibody in a T cell-independent manner (13). Alternatively, APCs can travel to the lymph nodes via afferent lymphatic ducts (12, 14) and interact with T cell receptors on T cells via their major histocompatibility II (MHC-II) molecules, and as a result elicit a T cell-dependent response (15, 16). The APC:T cell interaction usually occurs in the paracortex of tissue-draining lymph nodes (16), although this interaction can also occur in tertiary lymphoid organs of the affected tissue in situ (17–19). Antigen presentation by dendritic cells to T cells sequentially leads to T cell–B cell interactions, thus triggering B cell maturation and antibody production (20, 21).
Numerous variegated—but coincidental—cellular activities regulate the outcome of host immunity in the GI tract, but some of the pathways are epitomized by the activities observed during hypersensitivity reactions (22–24). In type I hypersensitivity, a target antigen leads to the stimulation of mast cells via their IgE receptors leading to degranulation (22–24). In type II hypersensitivity, antibodies bind to target cells leading to direct cell to cell killing by NK or CD8+ T cells (22–24). Type III hypersensitivity comprises the binding of complement to the target antigen leading to chemotaxis and infiltration of neutrophils (22–24). Type IV hypersensitivity is mediated by cytokine release from helper T cells that stimulate macrophage or killer T cell activity against target cells (22–24). Overall, these pathways do not only operate in hypersensitivity or autoimmune reactions, but also in regular pathogenesis (25) in response to infection or carcinogenesis. For example, cytokines that exacerbate host immunopathology are induced in the cecum during Clostridium difficile infection (26) and in the stomach during chronic Helicobacter felis-induced gastritis (27). Both of these pathological phenomena are reminiscent of type IV autoimmunity (22–24). In addition, autoantibody production (28–38) and complement activation (39, 40) are detected during Helicobacter pylori gastritis, which are also reminiscent of type II and type III autoimmunity, respectively (22–24).
In summary, this paragraph outlines some of the complexity of host immunity and its coincidental activities of targeting pathogen versus host. The complexity of these pathways renders it unattainable to review all these processes. Hence, we will focus our review on two processes for which metabolite involvement has been characterized to a certain extent: (1) the role of metabolites in regulating myeloid cell function (namely the roles of Arg2, IDO1, Hcar2, and Irg1), and (2) the role of metabolites that exert a direct effect on CD8+ T cell migration and memory (namely S1P), or on CD8+ T cell activation via epithelial cell mediation (retinoic acid).
Arg2 in Suppressing Macrophage Cytotoxicity and Myeloid-Derived Suppressor Cell (MDSC) Function
Arginase 2 is an enzyme for which two previously defined mechanisms have been described: (1) Arg2 reduces macrophage cytotoxic activity (41) by depleting l-arginine availability for the production of nitric oxide (NO) by nitric oxide synthase (NOS) (42); and (2) Arg2 in MDSCs inhibits T cell immunity by depleting l-arginine availability for T cells in the inflamed microenvironment (43) (Figure 1A). l-arginine is a non-essential amino acid that is sufficiently produced by the human body, but becomes essential in disease contexts that upregulate Arg2 (44). Therefore, the use of l-arginine as a dietary supplement for treating disease is arguable since the pathway is regulated at the level of l-arginine breakdown. However, the metabolic enzyme Arg2 presents an attractive therapeutic target for modulating T cell immunity (45). In order to review the role of Arg2, it is important to outline the nature and function of MDSCs in tumor biology and autoimmune pathology.
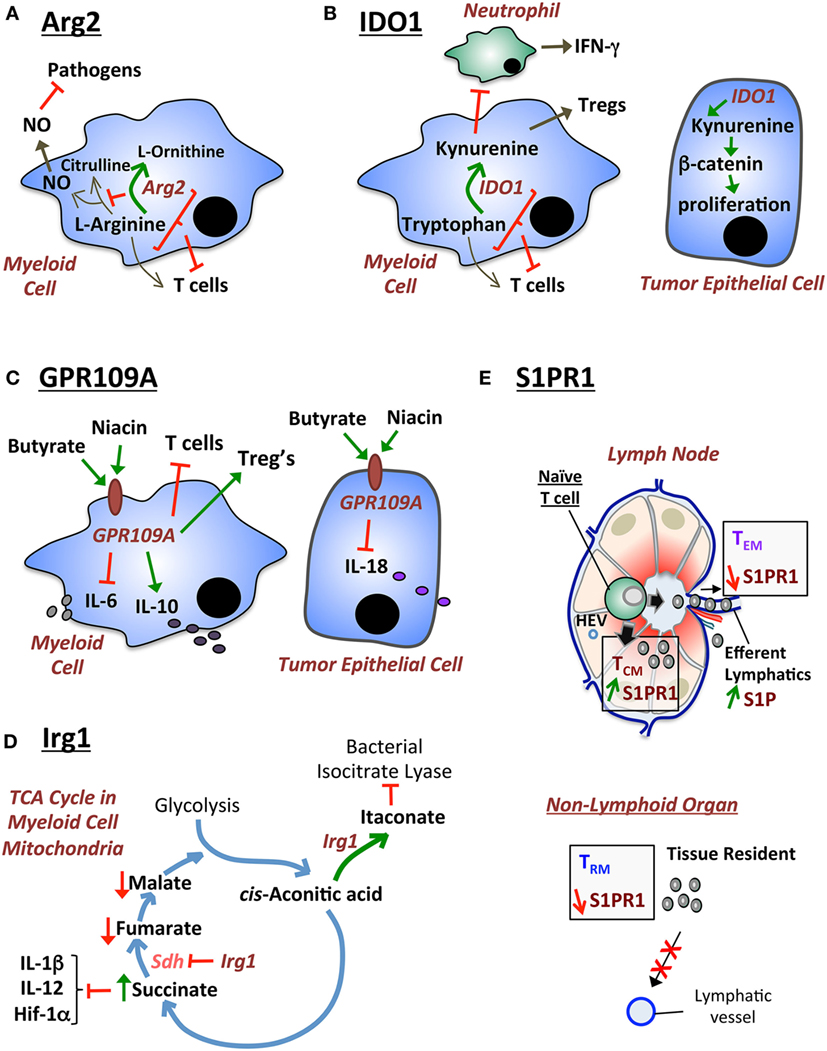
Figure 1. Diagrammatical modeling of exemplary metabolic pathways that regulate host immunity. (A) Arginase 2 (Arg2) functions by regulating two pathways: (i) depletion of l-arginine required for NO synthesis contributing to macrophage cytotoxic activity against pathogens; and (ii) depletion of l-arginine required for T cell immunity. (B) IDO1 has been traditionally described to suppress T cell immunity by depleting tryptophan. Moreover, IDO1 generates kynurenine that stimulates Tregs. However, new alternative mechanisms for IDO1 have recently been described: (i) IDO1 suppresses (potentially via kynurenine) IFN-γ-producing cecal neutrophils during Clostridium difficile colitis; and (ii) IDO1-produced metabolites (such as kynurenine) stimulate the β-catenin pathway and tumor epithelial cell proliferation in colorectal cancer. (C) GPR109A (also known as hydroxycarboxylic acid receptor 2) is the niacin/butyrate receptor which exhibits two previously described functions: (i) in myeloid cells GPR109A suppresses IL-6 and T cell immunity, while promoting IL-10 production and Treg differentiation; and (ii) in epithelial cells GPR109A suppresses IL-18 production. (D) Irg1 is an enzyme that regulates the tricarboxylic acid (TCA) (citric acid) cycle in the mitochondria of myeloid cells. Irg1 regulates two functions: (i) Irg1 generates itaconate, which exhibits antimicrobial activities by inhibiting the bacterial enzyme isocitrate lyase; and (ii) Irg1 inhibits succinate dehydrogenase (Sdh), which leads to an increase in succinate levels. Increased succinate suppresses pro-inflammatory cytokines (IL-1β, IL-12) and hypoxia-inducible factor 1 alpha (HIF-1α). (E) S1P provides a high gradient in efferent lymphatic vessels of the lymph node, which leads to the egress of memory T cells expressing high levels of the S1pr1 receptor. In non-lymphoid tissue, low levels of S1pr1 are necessary for sustaining tissue-resident memory T (TRM) cells by continuously preventing egress out of the tissue via lymphatic ducts.
Myeloid-derived suppressor cells are a myeloid population (that can be regulated by intrinsic Arg2 activity), which exerts immunosuppressive activities against T cells (46). The regulation of effector T cell function by MDSCs is critical in pathological situations (47, 48). The reason T cells are important is because, for example in cancer, CD8+ cytotoxic T cells exhibit antitumor activity by killing tumor cells (49). In viral infections, CD8+ T cells confer protection by eliminating virus-infected cells (49). However, the effect of MDSC on T cell function is also complicated by the fact that T cells exhibit variable phenotypes depending on two intertwined phenomena: (1) homing receptor expression, and (2) differentiation into specific T cell memory subtypes (50). The latter point will not be discussed here, but will be discussed in the last section of this article regarding S1P. Nevertheless, MDSCs are believed to play an unfavorable role in cancer and viral infections by suppressing T cell immunity (46). Hence, this suppressive activity of MDSCs against T cells is partly mediated by Arg2, which unravels an attractive therapeutic target against cancer (46). However, in vivo pathological situations warn of arguable outcomes for this strategy as will be described in the following paragraph.
The reason why the usefulness of Arg2 inhibitors against GI cancer is arguable is epitomized by a study in which Arg2 deficiency exacerbated gastric immunopathology during chronic gastritis (51, 52). The authors showed that this effect was not mediated by NOS (52). Even though the authors did not investigate the effect of reduced MDSC function on CD8+ T cell responses in this model, they did report a dramatic increase in tissue IFN-γ levels—a cytokine that is mainly produced by CD8+ T cells and NK cells (51, 52). Hence, if Arg2−/− leads to reduced MDSC function and increased CD8+ T cell response—as reported in the literature—the worsened immunopathology in Arg2−/− would suggest that CD8+ T cells are involved in an autoimmune etiology in the stomach. In this scenario, the use of Arg2 inhibitors to treat cancers becomes arguable since the inflamed normal tissue—adjacent to the tumor—might develop worsened immunopathology when Arg2 is inhibited. Therefore, one has to consider the unfavorable pathological outcome of Arg2 suppression, which might arise due to heightened inflammation in normal adjacent tissue to the tumor, which can exacerbate the development of preneoplastic metaplastic lesions.
An Unusual Role for IDO1 in Suppressing Cecal IFN-γ-Producing Neutrophils
IDO1 is a tryptophan-catabolizing enzyme that suppresses T cell immunity (53), but has recently been shown to play major alternative roles (other than T cell regulation) in the GI tract (26, 54) (Figure 1B). The role of IDO1 in regulating T cell immunity was first described in the prevention of allogeneic fetal rejection (53). In the latter study, IDO1 was proposed to deplete tryptophan pools for T cells by catabolizing tryptophan into kynurenine (53). In correlation with this finding, IDO1 inhibitor increased CD8+ T cells and reduced tumor growth in a transgenic mouse model of cecal gastrointestinal stromal tumors, which was reversed by CD8+ T cell depletion (55). Furthermore, IDO1 is expressed by MDSCs and regulates the immunosuppressive function of these cells against T cells (56). Alternatively, the mechanism of anti-T cell immunity of IDO1 can also be attributed to plasmocytoid dendritic cell expression of IDO1 in tumor-draining lymph nodes (57). In contrast to the latter observations, our recent finding demonstrates that IDO1 plays other distinct functions in non-tumor inflammatory environments that do not employ T cell immunity as a major component (26). Moreover, another recent study also showed that IDO1 plays a critical role in regulating epithelial cell function, which affects the outcome of colorectal cancer development in mouse models (54). The latter two observations indicate that the functions of IDO1 are not limited to regulating T cell immunity, but are subject to variability based on pathogenic context. These two studies will be reviewed in the following two paragraphs.
In our recent study, IDO1 suppressed IFN-γ-producing neutrophils in C. difficile colitis, but had no apparent effect on cecal CD4+ T cell number (26). It is important to note that C. difficile colitis is an acute neutrophilic disease (58), in which T cells do not affect immunopathology or survival in a significant manner (59). The latter observation was supported by the use of Rag1−/− mice, which succumbed to similar immunopathology relative to wild-type mice during C. difficile infection (59). Our observation that IDO1 deficiency led to increased cecal neutrophils (26) correlates with a recent finding that the IDO1 metabolite, kynurenine, suppresses neutrophil chemotaxis potentially via the aryl hydrocarbon receptor (60). The other surprising finding in our study was that cecal neutrophils—regulated by IDO1—were the major source (>90%) of IFN-γ (26). The latter observation also correlates with neutrophils being the major source of IFN-γ during Salmonella enterica Typhimurium-induced colitis (61). Therefore, the breakdown of the amino acid tryptophan into kynurenine in this context inhibits chemotaxis of IFN-γ-producing neutrophils during infectious colitis of the lower bowel. These findings unravel a novel role for IDO1 during acute infections of the GI tract, which warn of unfavorable side effects for IDO1 inhibitors. This is of special concern since cancer patients are more likely to succumb to C. difficile infections (62). Moreover, the findings also propose that IDO1 might potentially play alternative functions that regulate myeloid cells (e.g., neutrophils or MDSCs) in other contexts, such as tumor context. In support of the latter proposition, a recent paper showed that MDSCs were increased in IDO1-overexpressing B16 melanoma tumor models (63).
In addition to our observed effect of IDO1 on neutrophils, another study demonstrated a novel function of IDO1 in regulating epithelial cancer cell proliferation. The authors showed that IDO1 deficiency led to reduced tumor burden in the azoxymethane (AOM) and dextran sodium sulfate (DSS) model, which was mediated by a T cell-independent mechanism (54). The finding was similarly replicated in Rag1−/− mice that lack T and B lymphocytes, thus corroborating that T cells were not involved (54). The authors proposed that IDO1 influenced tumor development by regulating epithelial cell proliferation via β-catenin (54). Therefore, we conclude—based on the data from the last two paragraphs—that IDO1 plays additional T cell-independent roles, which are variable according to disease context. Hence, these pathways should be taken into consideration during the therapeutic design for IDO1 inhibitors.
The Niacin/Butyrate Receptor, GPR109A, Suppresses Inflammatory Responses in Myeloid Cells and the Colonic Epithelium
GPR109A (also known as the Hcar2) is a G-protein-coupled receptor (GPCR) that binds niacin (vitamin B3) and butyrate (64). Its first ligand, niacin, can be consumed from meats (e.g., fish, chicken, liver, turkey, and pork), vegetables (e.g., peas and mushrooms), cereal, and peanuts (65). Interestingly, some of the dietary requirement for niacin is provided by the production of nicotinic acid from the tryptophan/kynurenine pathway (66) mediated by IDO1. Its second ligand, butyrate, is produced by the gut microbiota via the fermentation of dietary fiber from plant products (67, 68).
Given the complexity of GPCR pathways (69), GPR109A plays variegated functions in different contexts. For example, GPR109A mediates the antidyslipidemic effect of nicotinic acid in decreasing low-density lipoprotein and triglycerides, and increasing high-density lipoprotein levels (70). This led to the use of nicotinic acid as an antihyperlipidemic agent (70), although its use is limited by the unfavorable side effect of cutaneous vasodilation, leading to skin flushing (70). This effect on the skin arises because, in addition to GPR109A expression by adipocytes (71), the receptor is also expressed by cutaneous immune cells (72) and epidermal Langerhan cells (73). Overall, GPR109A is generally highly expressed by myeloid cells such as neutrophils (74, 75), macrophages, monocytes, and dendritic cells (76, 77). It is also expressed by intestinal epithelial cells (78). Hence, it is not surprising for GPR109A to play an important role in regulating intestinal immunopathology and carcinogenesis.
The role of GPR109A has recently been described in suppressing colonic inflammation and cancer (77) (Figure 1C). In the latter study, the authors described GPR109A-deficient intestinal myeloid cells to express lower levels of IL-10 and exhibit a deficiency in their ability to stimulate Treg differentiation (77). The authors also showed that GPR109A−/− colonic epithelial cells were unable to produce IL-18 (77). Finally, the authors showed that GPR109A deficiency exacerbated tumor development in the AOM/DSS model (77). Bone marrow transplant experiments revealed that the antitumor GPR109A effect was mediated by both the epithelial and immune compartments (77). Interestingly, treatment with niacin suppressed colonic inflammation and carcinogenesis in this model (77). In addition, similar findings were observed in another study that utilized high- versus low-fiber diets and GPR109A-deficient mice (79). In conclusion, GPR109A is a metabolic receptor for niacin and butyrate for which the therapeutic value, in GI inflammation and carcinogenesis, should be evaluated. Since GPR109A is highly expressed in myeloid cells, it would be additionally interesting to evaluate the role of this receptor in MDSCs. This is especially important given the MDSC role in suppressing CD8+ T cell antitumor immunity.
The Itaconic Acid-Producing Enzyme, Irg1, in Regulating Succinate Levels and the Inflammatory Response in Macrophages
Immunoresponsive gene 1 (Irg1) is a mitochondria-associated metabolic enzyme that exhibits (1) an anti-inflammatory activity in host myeloid cells (80), and (2) an antimicrobial activity against pathogens (81, 82). Irg1 functions by decarboxylating cis-aconitic acid to produce itaconate, as part of the tricarboxylic acid (TCA) cycle (also known as the citric acid cycle) (Figure 1D) (81). The production of itaconate can generate an antimicrobial response by inhibiting the bacterial enzyme isocitrate lyase (81, 82). Moreover, in host myeloid cells, Irg1 functions by inhibiting succinate dehydrogenase (Sdh) (83–85), an enzyme that oxidizes succinic acid to fumaric acid within the TCA cycle. The inhibition of Sdh by Irg1-mediated itaconate sustains an accumulation of succinate during LPS stimulation and a decrease in the levels of fumarate and malate (80) (Figure 1D). Moreover, Irg1 suppresses inflammatory cytokine expression in myeloid cells such as IL-1β, IL-12, IL-6, IL-18, and the hypoxia-inducible factor 1 alpha (HIF-1α) (80). Hence, the mechanism of Irg1 and itaconate appears to be mediated by succinate inhibition, the latter of which can regulate major inflammatory pathways [such as HIF-1α (80)] in myeloid cells (86). In comparison to the previously described function of Irg1 against bacterial infections (81, 82), a recent study showed that the treatment with dimethyl itaconate did not affect the number of intracellular bacteria following Salmonella typhimurium infection of bone marrow-derived macrophages (80). Therefore, despite the established effect of Irg1 in myeloid cell responses (80), the antimicrobial effect of Irg1 remains debatable.
Given the above-described functions of Irg1, this molecule now represents an attractive target to investigate in GI diseases and cancer. To date, there have been no mechanistic reports on the function of Irg1 in the GI tract. However, two studies reported the induction of Irg1 in response to Salmonella infection in the chicken cecum. Hence, Irg1 represents a ripe topic for investigation in GI diseases and cancer (87–89).
The Sphingosine-1-Phosphate Sphingolipid in Regulating Egress of Memory T Cells
Sphingosine-1-phosphate (S1P) is a signaling sphingolipid that regulates lymphocyte egress from secondary lymphoid organs (such as lymph nodes) (90) or non-lymphoid tissue (91, 92) (Figure 1E). S1P levels are high in efferent lymphatic ducts (higher than interstitial fluid of secondary lymphoid organs) (93), which attract lymphocytes that express high levels of sphingosine-1-phosphate receptor 1 (S1pr1) to egress out of the organs into the lymphatic ducts (93). This leads to recirculation of these lymphocytes via the lymphatic ducts and/or blood vessels (94). Once these lymphocytes egress, they downregulate their S1pr1 expression (95). Moreover, in the case of lymphocytes that constantly reside in non-lymphoid organs, such as tissue-resident memory T (TRM) cells, the consistent low expression of S1pr1 is necessary to sustain these cells within the organ without recirculation (92). In discussing the role of S1P, it is important to consider its dietary sources and the effect of its intake on GI disease outcome.
S1P is derived from dietary sphingolipids (96) that can impact the outcome of GI disease and cancer (97). Sphingosine-1-phosphate (S1P) lyase is an enzyme that degrades S1P in enterocytes, and its intestinal deletion leads to S1P accumulation in the colon, and an increase in T cells and colon carcinogenesis (97). The dietary source of S1P is variable and high levels can be consumed from dairy products, meats, and eggs, whereas vegetables and fruits contain lower levels (98). However, it is unclear whether the amount of dietary intake of S1P is critical. This is because lymphocyte differentiation and homing can be regulated at the level of expression of the S1P receptor, S1pr1, in lymphocytes—as will be reviewed in the following paragraph.
S1pr1 regulates T cell homing, which directly impacts T cell memory differentiation (92, 99–101). There are three types of memory T cells, which include (i) central memory T cells (TCM), (ii) effector memory T cells (TEM), and (iii) TRM. TCM cells patrol lymph nodes and the white pulp of the spleen, and they express lymph node homing receptors (99–101). TEM circulate between the blood and non-lymphoid tissue, but do not express homing receptors (99–101). TRM are resident in the tissue and do not recirculate into the blood or secondary lymphoid organs (99–101). The homing of these cells is directly intertwined with their function and differentiation (99–101). As different types of memory T cells can exhibit different functions (99–101), the role of S1P and S1pr1 becomes critical in regulating disease outcome. This is especially important in regulating CD8+ T cell memory phenotypes to modulate their cytotoxic activities against tumors, or pro-inflammatory activities in autoimmune diseases. In support of the latter point, current drugs against S1pr1 in colon cancer have been considered (102) and are currently undergoing clinical trials for inflammatory bowel diseases and colorectal cancers.
The Vitamin A Metabolite, atRA, in Regulating Epithelial MHC-I Expression and CD8+ T Cell Activation
All-trans-retinoic acid is a vitamin A metabolite that induces epithelial MHC-I expression in mouse models of colon carcinogenesis, therefore, triggering CD8+ T cell antitumor immunity (103). Vitamin A can occur in two basic forms, which are, respectively, obtained from two different dietary sources: (i) retinoids are supplied by animal food, whereas (ii) carotenoids are obtained from plant products (104). Retinyl palmitate constitutes the major form of vitamin A from animal food, which is converted in the small intestine to retinol, then to retinaldehyde, and then to retinoic acid (104). The synthesis of retinoic acid in the small intestine is mediated by retinaldehyde dehydrogenase enzymes, such as Aldh1a1, Aldh1a2, and Aldh1a3 (105–107), which are expressed by both intestinal epithelial and immune cells (103). In contrast to animal food sources, plant products provide carotenoids (provitamin A), which can be broken down into beta-carotene and then retinol in the intestinal mucosa (106). In both scenarios, the metabolic enzymes that regulate the synthesis or breakdown of retinoic acid in the small intestine provide an important checkpoint that can determine the resulting abundance of intestinal retinoic acid. The recent study by Bhattacharya et al. (103) demonstrated that in the AOM/DSS model, colon carcinogenesis led to downregulation of the atRA-synthesizing enzyme Aldh1a1. The downregulation of this enzyme was observed in both the epithelial and immune cell compartments (103). This was accompanied by an upregulation of the atRA-catabolizing enzyme, Cyp26A1, in the epithelial compartment of the colon mucosa (103). The authors went on to show that atRA reduced tumor burden in the AOM/DSS model and that this was mediated by the stimulation of atRA for epithelial MHC-I expression. Overall, the authors showed that mucosal inflammation, triggered by the host microbiota during colitis-associated colorectal cancer, stimulated atRA metabolism to decrease its levels, leading to reduced MHC-I and CD8+ activation (103). It will additionally be insightful to determine the effect of retinoic acid on CD8+ T cell memory differentiation as will be discussed in the following paragraph.
Previous studies have shown that atRA inhibits TGF-β-mediated Th17 differentiation and stimulates Treg differentiation (108–110). However, those studies also showed that atRA stimulates gut homing receptor expression, of α4β7 integrin and CD103, on T cells (108, 109). Hence, given the role of TGF-β-mediated induction of α4β7 integrin and CD103 in TRM generation (111), it will be insightful to determine the mechanism in which atRA regulates TRM, TCM, and TEM differentiation. Such analyses will also unravel novel information about the distinct roles of these memory T cell subtypes in colon carcinogenesis.
Concluding Remarks
This article presents a number of scenarios in which metabolites and their receptors regulate the outcome of GI disease and cancer (Figure 1). In considering these scenarios, it is important to realize that several of these pathways can be triggered coincidentally within the same context of disease. Therefore, an integrated approach and combinatorial analyses are necessary to devise new therapeutic strategies that can synergistically improve prognoses. Theoretically, modulating these metabolic pathways to improve disease outcome should be feasible. However, what is not known are the “recipes” in which interrupting or altering these pathways would concomitantly lead to significantly improved outcomes.
Author Contributions
Both ME-Z and JK contributed equally to literature review and concept development in the preparation of this review article.
Conflict of Interest Statement
The authors declare that this review article was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the American Gastroenterological Association (AGA)/Gastric Cancer Foundation (GCF) Grant N017489 (ME-Z).
References
1. Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet (2000) 1(1):57–64. doi:10.1038/35049577
2. Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol (2004) 4(6):432–44. doi:10.1038/nri1375
3. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
4. Inaba K, Metlay JP, Crowley MT, Witmer-Pack M, Steinman RM. Dendritic cells as antigen presenting cells in vivo. Int Rev Immunol (1990) 6(2–3):197–206. doi:10.3109/08830189009056630
5. Chen X, Jensen PE. The role of B lymphocytes as antigen-presenting cells. Arch Immunol Ther Exp (2008) 56(2):77–83. doi:10.1007/s00005-008-0014-5
6. Stein KE. Thymus-independent and thymus-dependent responses to polysaccharide antigens. J Infect Dis (1992) 165(Suppl 1):S49–52. doi:10.1093/infdis/165-Supplement_1-S49
7. Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol (2008) 8(8):607–18. doi:10.1038/nri2368
8. Grainger JR, Askenase MH, Guimont-Desrochers F, da Fonseca DM, Belkaid Y. Contextual functions of antigen-presenting cells in the gastrointestinal tract. Immunol Rev (2014) 259(1):75–87. doi:10.1111/imr.12167
9. Gretz JE, Norbury CC, Anderson AO, Proudfoot AE, Shaw S. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med (2000) 192(10):1425–40. doi:10.1084/jem.192.10.1425
10. Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity (2005) 22(1):19–29. doi:10.1016/j.immuni.2004.11.013
11. Angel CE, Chen CJ, Horlacher OC, Winkler S, John T, Browning J, et al. Distinctive localization of antigen-presenting cells in human lymph nodes. Blood (2009) 113(6):1257–67. doi:10.1182/blood-2008-06-165266
12. Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol (2002) 2(3):151–61. doi:10.1038/nri746
13. Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev (2000) 176:154–70. doi:10.1034/j.1600-065X.2000.00607.x
14. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol (2005) 5(8):617–28. doi:10.1038/nri1670
15. Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol (2015) 15(3):185–9. doi:10.1038/nri3803
16. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427(6970):154–9. doi:10.1038/nature02238
17. Buckley CD, Barone F, Nayar S, Benezech C, Caamano J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol (2015) 33:715–45. doi:10.1146/annurev-immunol-032713-120252
18. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14(7):447–62. doi:10.1038/nri3700
19. Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol (2012) 33(6):297–305. doi:10.1016/j.it.2012.04.006
20. Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature (1985) 314(6011):537–9. doi:10.1038/314537a0
21. Mitchison NA. T-cell–B-cell cooperation. Nat Rev Immunol (2004) 4(4):308–12. doi:10.1038/nri1334
22. Uzzaman A, Cho SH. Chapter 28: classification of hypersensitivity reactions. Allergy Asthma Proc (2012) 33(Suppl 1):S96–9. doi:10.2500/aap.2012.33.3561
23. Rajan TV. The Gell–Coombs classification of hypersensitivity reactions: a re-interpretation. Trends Immunol (2003) 24(7):376–9. doi:10.1016/S1471-4906(03)00142-X
24. Gell PGH, Coombs RRA. The classification of allergic reactions underlying disease. In: Coombs RRA, Gells PGH, editors. Clinical Aspects of Immunology. Oxford: Blackwell (1963). p. 317–37.
26. El-Zaatari M, Chang YM, Zhang M, Franz M, Shreiner A, McDermott AJ, et al. Tryptophan catabolism restricts IFN-gamma-expressing neutrophils and Clostridium difficile immunopathology. J Immunol (2014) 193(2):807–16. doi:10.4049/jimmunol.1302913
27. El-Zaatari M, Kao JY, Tessier A, Bai L, Hayes MM, Fontaine C, et al. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS One (2013) 8(3):e58935. doi:10.1371/journal.pone.0058935
28. Ito M, Haruma K, Kaya S, Kamada T, Kim S, Sasaki A, et al. Serological comparison of serum pepsinogen and anti-parietal cell antibody levels between Japanese and German patients. Eur J Gastroenterol Hepatol (2002) 14(2):123–7. doi:10.1097/00042737-200202000-00005
29. Ito M, Haruma K, Kaya S, Kamada T, Kim S, Sasaki A, et al. Role of anti-parietal cell antibody in Helicobacter pylori-associated atrophic gastritis: evaluation in a country of high prevalence of atrophic gastritis. Scand J Gastroenterol (2002) 37(3):287–93. doi:10.1080/003655202317284183
30. Sugiu K, Kamada T, Ito M, Kaya S, Tanaka A, Kusunoki H, et al. Evaluation of an ELISA for detection of anti-parietal cell antibody. Hepatogastroenterology (2006) 53(67):11–4.
31. Sugiu K, Kamada T, Ito M, Kaya S, Tanaka A, Kusunoki H, et al. Anti-parietal cell antibody and serum pepsinogen assessment in screening for gastric carcinoma. Dig Liver Dis (2006) 38(5):303–7. doi:10.1016/j.dld.2005.10.021
32. Faller G, Steininger H, Eck M, Hensen J, Hann EG, Kirchner T. Antigastric autoantibodies in Helicobacter pylori gastritis: prevalence, in-situ binding sites and clues for clinical relevance. Virchows Arch (1996) 427(5):483–6. doi:10.1007/BF00199508
33. Appelmelk BJ, Simoons-Smit I, Negrini R, Moran AP, Aspinall GO, Forte JG, et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect Immun (1996) 64(6):2031–40.
34. Negrini R, Savio A, Poiesi C, Appelmelk BJ, Buffoli F, Paterlini A, et al. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterology (1996) 111(3):655–65. doi:10.1053/gast.1996.v111.pm8780570
35. Barrio R, Roldan MB, Alonso M, Canton R, Camarero C. Helicobacter pylori infection with parietal cell antibodies in children and adolescents with insulin dependent diabetes mellitus. J Pediatr Endocrinol Metab (1997) 10(5):511–6. doi:10.1515/JPEM.1997.10.5.511
36. Faller G, Steininger H, Kranzlein J, Maul H, Kerkau T, Hensen J, et al. Antigastric autoantibodies in Helicobacter pylori infection: implications of histological and clinical parameters of gastritis. Gut (1997) 41(5):619–23. doi:10.1136/gut.41.5.619
37. Faller G, Steininger H, Appelmelk B, Kirchner T. Evidence of novel pathogenic pathways for the formation of antigastric autoantibodies in Helicobacter pylori gastritis. J Clin Pathol (1998) 51(3):244–5. doi:10.1136/jcp.51.3.244
38. Claeys D, Faller G, Appelmelk BJ, Negrini R, Kirchner T. The gastric H+,K+-ATPase is a major autoantigen in chronic Helicobacter pylori gastritis with body mucosa atrophy. Gastroenterology (1998) 115(2):340–7. doi:10.1016/S0016-5085(98)70200-8
39. Berstad AE, Hogasen K, Bukholm G, Moran AP, Brandtzaeg P. Complement activation directly induced by Helicobacter pylori. Gastroenterology (2001) 120(5):1108–16. doi:10.1053/gast.2001.23248
40. Rautemaa R, Rautelin H, Puolakkainen P, Kokkola A, Karkkainen P, Meri S. Survival of Helicobacter pylori from complement lysis by binding of GPI-anchored protectin (CD59). Gastroenterology (2001) 120(2):470–9. doi:10.1053/gast.2001.21197
41. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50. doi:10.1146/annurev.immunol.15.1.323
42. Bronte V, Zanovello P. Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi:10.1038/nri1668
43. Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, et al. Suppression of T-cell functions by human granulocyte arginase. Blood (2006) 108(5):1627–34. doi:10.1182/blood-2006-11-010389
44. Patel JJ, Miller KR, Rosenthal C, Rosenthal MD. When is it appropriate to use arginine in critical illness? Nutr Clin Pract (2016) 31(4):438–44. doi:10.1177/0884533616652576
45. Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol (2009) 158(3):638–51. doi:10.1111/j.1476-5381.2009.00291.x
46. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
47. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
48. Gravano DM, Hoyer KK. Promotion and prevention of autoimmune disease by CD8+ T cells. J Autoimmun (2013) 45:68–79. doi:10.1016/j.jaut.2013.06.004
49. Tscharke DC, Croft NP, Doherty PC, La Gruta NL. Sizing up the key determinants of the CD8(+) T cell response. Nat Rev Immunol (2015) 15(11):705–16. doi:10.1038/nri3905
50. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol (2013) 13(5):309–20. doi:10.1038/nri3442
51. Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, et al. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol (2011) 186(6):3632–41. doi:10.4049/jimmunol.1003431
52. Hardbower DM, Asim M, Murray-Stewart T, Casero RA Jr, Verriere T, Lewis ND, et al. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to Helicobacter pylori infection. Amino Acids (2016) 48(10):2375–88. doi:10.1007/s00726-016-2231-2
53. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281(5380):1191–3. doi:10.1126/science.281.5380.1191
54. Thaker AI, Rao MS, Bishnupuri KS, Kerr TA, Foster L, Marinshaw JM, et al. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology (2013) 145(2):e1–4. doi:10.1053/j.gastro.2013.05.002
55. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med (2011) 17(9):1094–100. doi:10.1038/nm.2438
56. Yu J, Wang Y, Yan F, Li H, Ren X. Response to comment on “myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer”. J Immunol (2013) 190(11):5341–2. doi:10.4049/jimmunol.1390024
57. Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest (2004) 114(2):280–90. doi:10.1172/JCI21583
58. Jose S, Madan R. Neutrophil-mediated inflammation in the pathogenesis of Clostridium difficile infections. Anaerobe (2016) 41:85–90. doi:10.1016/j.anaerobe.2016.04.001
59. Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe (2015) 18(1):27–37. doi:10.1016/j.chom.2015.06.011
60. Loughman JA, Yarbrough ML, Tiemann KM, Hunstad DA. Local generation of kynurenines mediates inhibition of neutrophil chemotaxis by uropathogenic Escherichia coli. Infect Immun (2016) 84(4):1176–83. doi:10.1128/IAI.01202-15
61. Spees AM, Kingsbury DD, Wangdi T, Xavier MN, Tsolis RM, Baumler AJ. Neutrophils are a source of gamma interferon during acute Salmonella enterica serovar Typhimurium colitis. Infect Immun (2014) 82(4):1692–7. doi:10.1128/IAI.01508-13
62. Chopra T, Alangaden GJ, Chandrasekar P. Clostridium difficile infection in cancer patients and hematopoietic stem cell transplant recipients. Expert Rev Anti Infect Ther (2010) 8(10):1113–9. doi:10.1586/eri.10.95
63. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep (2015) 13(2):412–24. doi:10.1016/j.celrep.2015.08.077
64. Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov (2012) 11(8):603–19. doi:10.1038/nrd3777
65. Torres N, Guevara-Cruz M, Velazquez-Villegas LA, Tovar AR. Nutrition and atherosclerosis. Arch Med Res (2015) 46(5):408–26. doi:10.1016/j.arcmed.2015.05.010
66. Holman WI, De Lange DJ. Significance of tryptophane in human nicotinic acid metabolism. Nature (1950) 165(4186):112. doi:10.1038/165112a0
67. Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett (2002) 217(2):133–9. doi:10.1111/j.1574-6968.2002.tb11467.x
68. Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol (2000) 66(4):1654–61. doi:10.1128/AEM.66.4.1654-1661.2000
69. Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer (2007) 7(2):79–94. doi:10.1038/nrc2069
70. Offermanns S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharmacol Sci (2006) 27(7):384–90. doi:10.1016/j.tips.2006.05.008
71. Carlson LA. Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med Scand (1963) 173:719–22. doi:10.1111/j.0954-6820.1963.tb17457.x
72. Benyo Z, Gille A, Kero J, Csiky M, Suchankova MC, Nusing RM, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest (2005) 115(12):3634–40. doi:10.1172/JCI23626
73. Benyo Z, Gille A, Bennett CL, Clausen BE, Offermanns S. Nicotinic acid-induced flushing is mediated by activation of epidermal langerhans cells. Mol Pharmacol (2006) 70(6):1844–9. doi:10.1124/mol.106.030833
74. Kostylina G, Simon D, Fey MF, Yousefi S, Simon HU. Neutrophil apoptosis mediated by nicotinic acid receptors (GPR109A). Cell Death Differ (2008) 15(1):134–42. doi:10.1038/sj.cdd.4402238
75. Tang H, Lu JY, Zheng X, Yang Y, Reagan JD. The psoriasis drug monomethylfumarate is a potent nicotinic acid receptor agonist. Biochem Biophys Res Commun (2008) 375(4):562–5. doi:10.1016/j.bbrc.2008.08.041
76. Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism (2016) 65(2):102–13. doi:10.1016/j.metabol.2015.10.001
77. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40(1):128–39. doi:10.1016/j.immuni.2013.12.007
78. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res (2009) 69(7):2826–32. doi:10.1158/0008-5472.CAN-08-4466
79. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun (2015) 6:6734. doi:10.1038/ncomms7734
80. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab (2016) 24(1):158–66. doi:10.1016/j.cmet.2016.06.004
81. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A (2013) 110(19):7820–5. doi:10.1073/pnas.1218599110
82. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog (2016) 12(2):e1005408. doi:10.1371/journal.ppat.1005408
83. Ackermann WW, Potter VR. Enzyme inhibition in relation to chemotherapy. Proc Soc Exp Biol Med (1949) 72(1):1–9. doi:10.3181/00379727-72-17313
84. Booth AN, Taylor J, Wilson RH, Deeds F. The inhibitory effects of itaconic acid in vitro and in vivo. J Biol Chem (1952) 195(2):697–702.
85. Dervartanian DV, Veeger C. Studies on succinate dehydrogenase. I. Spectral properties of the purified enzyme and formation of enzyme-competitive inhibitor complexes. Biochim Biophys Acta (1964) 92:233–47.
86. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25(7):771–84. doi:10.1038/cr.2015.68
87. Matulova M, Rajova J, Vlasatikova L, Volf J, Stepanova H, Havlickova H, et al. Characterization of chicken spleen transcriptome after infection with Salmonella enterica serovar Enteritidis. PLoS One (2012) 7(10):e48101. doi:10.1371/journal.pone.0048101
88. Matulova M, Varmuzova K, Sisak F, Havlickova H, Babak V, Stejskal K, et al. Chicken innate immune response to oral infection with Salmonella enterica serovar Enteritidis. Vet Res (2013) 44:37. doi:10.1186/1297-9716-44-37
89. Rychlik I, Elsheimer-Matulova M, Kyrova K. Gene expression in the chicken caecum in response to infections with non-typhoid Salmonella. Vet Res (2014) 45:119. doi:10.1186/s13567-014-0119-2
90. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature (2004) 427(6972):355–60. doi:10.1038/nature02284
91. Zajac AJ, Harrington LE. Tissue-resident T cells lose their S1P1 exit visas. Cell Mol Immunol (2014) 11(3):221–3. doi:10.1038/cmi.2014.7
92. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14(12):1285–93. doi:10.1038/ni.2745
93. Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol (2012) 30:69–94. doi:10.1146/annurev-immunol-020711-075011
94. Garris CS, Blaho VA, Hla T, Han MH. Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology (2014) 142(3):347–53. doi:10.1111/imm.12272
95. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12(11):762–73. doi:10.1038/nri3298
96. Duan RD, Nilsson A. Sphingolipid hydrolyzing enzymes in the gastrointestinal tract. Methods Enzymol (2000) 311:276–86. doi:10.1016/S0076-6879(00)11089-4
97. Degagne E, Pandurangan A, Bandhuvula P, Kumar A, Eltanawy A, Zhang M, et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest (2014) 124(12):5368–84. doi:10.1172/JCI74188
98. Vesper H, Schmelz EM, Nikolova-Karakashian MN, Dillehay DL, Lynch DV, Merrill AH Jr. Sphingolipids in food and the emerging importance of sphingolipids to nutrition. J Nutr (1999) 129(7):1239–50.
99. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41(6):886–97. doi:10.1016/j.immuni.2014.12.007
100. Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med (2015) 21(7):688–97. doi:10.1038/nm.3883
101. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2016) 16(2):79–89. doi:10.1038/nri.2015.3
102. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell (2013) 23(1):107–20. doi:10.1016/j.ccr.2012.11.013
103. Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, et al. Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8(+) T cell-mediated immunity in colorectal cancer. Immunity (2016) 45(3):641–55. doi:10.1016/j.immuni.2016.08.008
104. Ruhl R. Effects of dietary retinoids and carotenoids on immune development. Proc Nutr Soc (2007) 66(3):458–69. doi:10.1017/S002966510600509X
105. Kumar S, Sandell LL, Trainor PA, Koentgen F, Duester G. Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim Biophys Acta (2012) 1821(1):198–205. doi:10.1016/j.bbalip.2011.04.004
106. Kedishvili NY. Enzymology of retinoic acid biosynthesis and degradation. J Lipid Res (2013) 54(7):1744–60. doi:10.1194/jlr.R037028
107. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity (2011) 35(1):13–22. doi:10.1016/j.immuni.2011.07.002
108. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317(5835):256–60. doi:10.1126/science.1145697
109. Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med (2007) 204(8):1765–74. doi:10.1084/jem.20070719
110. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204(8):1757–64. doi:10.1084/jem.20070590
Keywords: S1pr1, MDSC, CD8, cytotoxic, stomach, gastric, metaplasia, colorectal cancer
Citation: El-Zaatari M and Kao JY (2017) Role of Dietary Metabolites in Regulating the Host Immune Response in Gastrointestinal Disease. Front. Immunol. 8:51. doi: 10.3389/fimmu.2017.00051
Received: 28 October 2016; Accepted: 12 January 2017;
Published: 27 January 2017
Edited by:
Maritza Romero, Augusta University, USAReviewed by:
Christopher Alan Jolly, University of Texas at Austin, USAHelena Paidassi, CNRS, France
Copyright: © 2017 El-Zaatari and Kao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamad El-Zaatari, bW9oYW1hZGVAbWVkLnVtaWNoLmVkdQ==;
John Y. Kao, anlrYW9AbWVkLnVtaWNoLmVkdQ==