 Qi Zhang
Qi Zhang Bo Zhu
Bo Zhu Yongsheng Li
Yongsheng Li- Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing, China
Inflammation is a protective response that eliminates harmful stimuli and restores tissue homeostasis, whereas the failure to resolve inflammation leads to the development of malignancies. Immune cells in the tumor inflammatory microenvironment endow cancer cells with their specific hallmarks, including mutations, metabolic reprograming, angiogenesis, invasion, and metastasis. Targeting the inflammatory microenvironment with anti-inflammatory drugs (e.g., aspirin) or by enhancing antitumor immunity (e.g., chimeric antigen receptor T cell therapy) has been extensively investigated and has achieved promising results in many cancers. Recently, a novel approach promoting antitumor immunity via a dual anti-inflammatory and pro-resolving strategy was proposed based on the discovery of potent, endogenous, specialized pro-resolving mediators, including lipoxins, resolvins, protectins, and maresins. In this review, we describe the updated principal cellular and molecular mechanisms of inflammation resolution and cancer immunity and discuss the pro-resolution strategy in cancer treatment and prevention.
Introduction
Inflammation is the protective immune response of a vascular organism that aids in the removal of internal and external harmful stimuli and the maintenance of tissue homeostasis (1). Acute inflammation seeks to repair injured tissues and eliminate unwanted elements. The ideal outcome of acute inflammation is complete and timely resolution with a return to homeostasis, which is actively programed by specialized pro-resolving mediators (SPM), including lipoxins (LXs), resolvins (Rvs), protectins, and maresins (MaRs) (1). SPM potently inhibit neutrophil infiltration and promote macrophage efferocytosis of apoptotic neutrophils in the inflammatory loci (2). However, persistent inflammation leads to chronic inflammation, which is categorized as either delayed-resolving or non-resolving (Figure 1). The symptoms and signs of chronic inflammation are not as serious as those of acute inflammation, but chronic inflammation is typically more risky since it can cause further damage (e.g., fibrosis, necrosis, organ dysfunction, and gene mutation) and an enormous proportion of refractory diseases [e.g., Alzheimer’s disease (AD) (3) and cancer (4)].
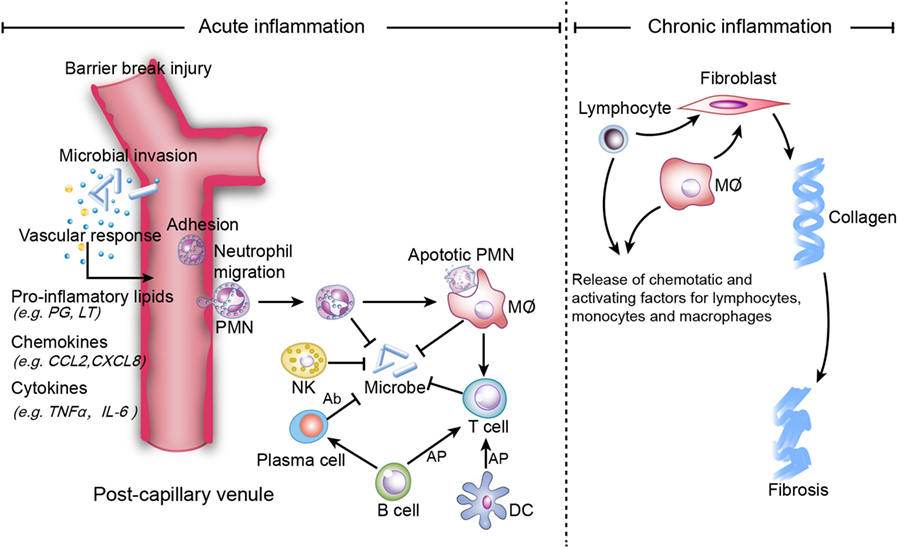
Figure 1. Mechanisms of acute and chronic inflammation. Within a few hours of stimulation (injury, trauma, stress, or infection), the release of pro-inflammatory lipids (e.g., prostaglandin (PG), leukotriene (LT), involved in vasodilation), chemokines (e.g., C-C motif chemokine ligand 2 (CCL2), C-X-C motif ligand 8 (CXCL8), involved in chemotaxis and adhesion), and cytokines [e.g., Tumor necrosis factor-α (TNF-α), interleukin (IL)-6] elicits the recruitment of neutrophils. Other immune cells [i.e., natural killer (NK) cells, macrophages, dendritic cells (DCs), B cells, and T cells] also participate in the process. NK cells kill microbes via complement-dependent cytotoxicity. Macrophages directly phagocytize organisms and apoptotic neutrophils, while B cells are converted to plasma cells to kill organisms via secreted antibodies, which are referred to as antibody-dependent cell-mediated cytotoxicity. Macrophages, B cells and DCs activate T cells via antigen cross presentation (AP). Homeostasis will be restored if inflammation is resolved completely, while non-resolution leads to chronic inflammation, which is characterized by persistent tissue infiltration by immune cells (e.g., macrophages, lymphocytes). In the extracellular zone, lymphocytes and macrophages release factors that result in the deposition of extracellular collagen and an excessive inflammatory response.
As early as the 1860s, Virchow indicated a link between cancer and inflammation by observing inflammatory cells in biopsied tumor tissues (4). Inflammatory stimuli, such as chronic infections, inhaled pollutants, smoking, and obesity (5), may result in DNA damage, somatic mutations, and tumorigenesis (6, 7). In the tumor microenvironment (TME), inflammatory cells are educated to accelerate cancer progression, metastasis, and immune responses against radiotherapy, chemotherapy, and immunotherapy (8). Therefore, targeting the inflammatory microenvironment is a reasonable direction for cancer treatment.
Indeed, anti-inflammatory drugs have exhibited efficacy by improving both prognosis and survival of patients (9, 10) and in cancer prevention (11). Enhancing tumor immunity by blocking inhibitory checkpoints or using chimeric antigen receptor T cell (CAR-T) immunotherapy has also shown promising efficacy in specific cancer types. However, the side effects of these therapies, such as coagulopathy and the “cytokine storm,” have hindered their full application to cancer therapy. Consequently, a better endogenous mechanism for improving the tumor inflammatory microenvironment is urgently needed.
Specialized pro-resolving mediator-driven inflammation resolution is an active process, which results in catabasis and homeostasis. To date, endogenous SPM have been applied in multiple models of cancer and achieved promising outcomes (12–15). In the present review, we highlight the role of inflammation in cancer development (e.g., tumor immunoediting) and suggest the immunomodulatory potential of SPM for cancer treatment in light of a brand-new strategy to remodel the TME by promoting inflammation resolution.
Cancer-Promoting Inflammation
It has been well established that pathogen-induced inflammation is a high-risk factor for cancer. For instance, persistent Helicobacter pylori infection is highly associated with gastric adenocarcinoma and lymphoma (16), human papilloma virus infection increases the risk of cervical cancer (17), hepatitis B and C virus infections increase the incidence of hepatocellular carcinoma (HCC) (18), and infection with Epstein–Barr virus is closely related to nasopharyngeal carcinoma (19). These causative agents lead to persistent infections associated with low levels of chronic inflammation. In addition, some autoimmune diseases also correlate with cancer development. Crohn’s disease and ulcerative colitis, also known as inflammatory bowel diseases, are highly associated with an increased risk of colorectal cancer (CRC) (20). Long-term exposure to irritants or obesity also induces tumor-promoting inflammation (21–23). Senescence-associated inflammation is postulated to be another promoter of most solid malignances (18). Moreover, cancer therapy (e.g., chemotherapy and radiotherapy)-induced inflammation can enhance antigen cross presentation and initiation of the antitumor immune response, whereas these therapies can also initiate inflammation by causing massive necrosis of malignant cells and pericarcinous tissue followed by tumor recurrence and resistance to therapy (18).
Tumor initiation and progression are finely immunoedited (24). Tumor immunoediting is divided into three phases (Figure 2): elimination, equilibrium, and escape (25). During the elimination phase, tumor cells with potent immunogenicity are removed by the immune system before they become clinically detectable. Activated NK cells and macrophages produce interferon (IFN)-γ and interleukin (IL)-12, which eliminate tumor cells by initiating cytotoxic responses, such as perforin, TNF-α and reactive oxygen species (ROS) (25). Antigen-presenting cells [such as DCs, macrophages, and B cells] take up and cross-present tumor antigens to T cells and activate T cells via co-stimulatory molecules (26). Therefore, antitumor inflammatory mediators (IM) predominantly participate in the elimination phase compared to pro-tumor IM. When a balance between pro-tumor and antitumor IM is established, tumors progress into the equilibrium stage. During this phase, variants that survived the elimination phase undergo various mutations but exhibit a weak-immunogenic phenotype (e.g., loss of antigenic tumor peptides and major histocompatibility complex components). Notably, some antitumor cytokines, such as TNF-α, become pro-tumorigenic. This phase may last for several years until new immune-resistant variants emerge, which are more likely to escape immunosurveillance (25). In this scenario, the IM balance is skewed toward pro-tumor IM since immunity fails to limit tumor outgrowth. The immune-resistant variants ultimately result in the formation of a clinically detectable solid tumor (25). In the tumor escape phase, pro-tumor immune cells, including myeloid-derived suppressor cells (MDSCs), tumor-associated dendritic cells (TADCs), tumor-associated macrophages (TAMs), Th17, and regulatory T cells (Tregs), along with cancer cells and cancer stem cells, induce immunosuppression via secretion of a variety of immunosuppressive cytokines and molecules. Furthermore, T cells express inhibitory checkpoint receptors, such as programed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which are activated by ligands expressed on pro-tumor immune cells (27). Altogether, these immunosuppressive mechanisms synergistically neutralize antitumor immunity and accelerate tumor progression.
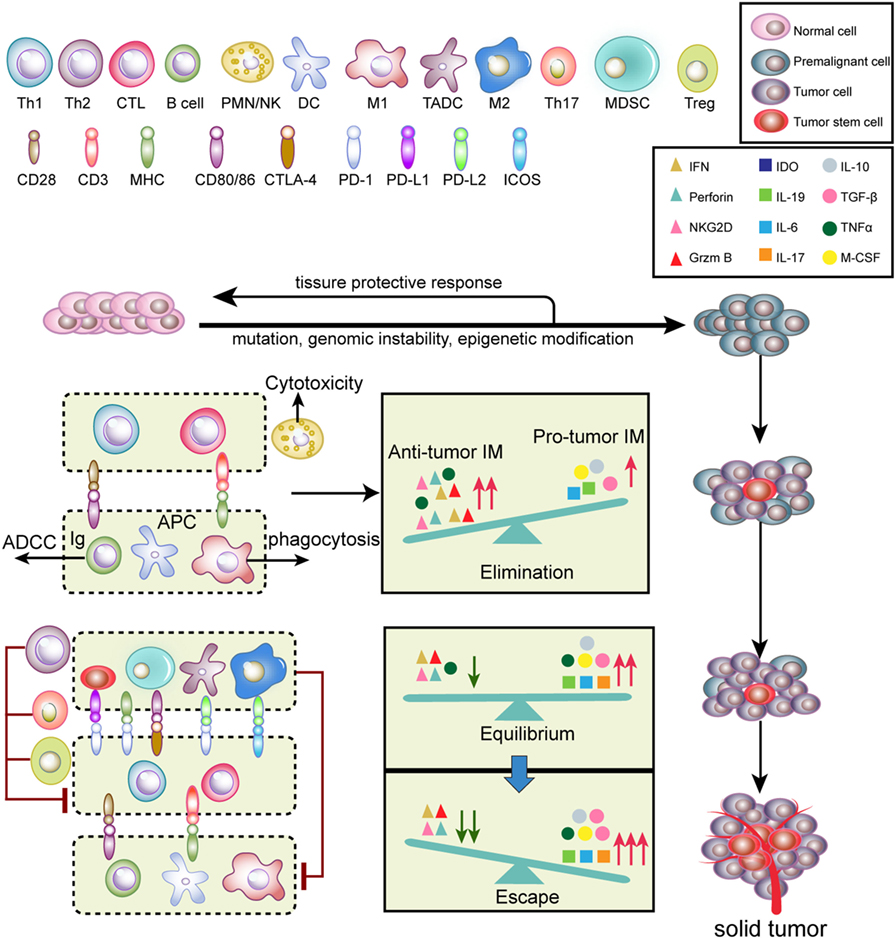
Figure 2. Tumor immunoediting. Normal cells are transformed into malignant cells by mutations, genomic instability, and epigenetic modification, during which innate and adaptive immunity regulate the tumor microenvironment. In the elimination phase, both innate and adaptive immunity synergistically detect and eliminate early tumor cells. Next, rare tumor cells that are not eliminated in the elimination phase can enter the equilibrium phase, where their outgrowth and elimination are controlled. Finally, the remaining tumor cell variants with weak immunogenicity escape from immune surveillance to form a clinically apparent neoplasm.
The typical underlying mechanisms through which inflammation promotes cancer include (1) mutations: DNA damage/mutation, genomic instability, epigenetic dysregulation, and DNA repair deficiency (28–30). DNA damage in turn promotes inflammation, generating a vicious cycle that synergistically initiates carcinogenesis (28); (2) angiogenesis: angiogenesis is crucial for solid tumor growth and invasion (6). Inflammatory cytokines, such as TNF-α and IL-1, activate chemokine receptor-4/chemokine (C-X-C motif) ligand 12 (CXCR4/CXCL12) signaling, which upregulates vascular endothelial growth factor (VEGF) expression via the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway. In addition to cytokines and chemokines, cyclooxygenases (COX)-2 and a portion of its metabolites are also engaged in vascular formation (31); (3) metastasis and invasion: inflammation also contributes to hypoxia, which further promotes angiogenesis, glycolysis, and invasion (31). Inflammatory cytokines secreted by immunosuppressive cells contribute to the progression of cancer. For example, MDSCs promote epithelial-to-mesenchymal transition (EMT) by secreting transforming growth factor β (TGF-β), epidermal growth factor (EGF), and hepatocyte growth factor (HGF) pathways (32) and shift M1 macrophages into TAMs (an M2 phenotype) (33). TAMs lose tumoricidal activity and contribute to immune suppression through the upregulation of inflammatory meditators [e.g., IL-10, TGF-β, and C–C motif chemokine ligand 22 (CCL22)], which promote T cell anergy and Treg recruitment (34). For further details, please refer to these current reviews that focus on inflammation in cancer development and progression (18, 29, 31).
Anticancer Strategies Targeting the Inflammatory Microenvironment
Antagonizing Inflammation
To date, several anti-inflammatory drugs have been used for prophylaxis and have shown efficacy in decreasing cancer morbidity (35), counteracting chemoresistance, suppressing tumor progression, and improving survival (10). Anti-inflammatory drugs are classified as non-steroidal anti-inflammatory drugs (NSAIDs) (e.g., aspirin), steroidal anti-inflammatory drugs (e.g., dexamethasone), or statins. In addition to its well-documented effects in CRC prevention (11), aspirin also reduces the incidence of several types of solid tumors, including melanoma (36), prostate cancer (37), and breast cancer (38). Mechanistically, aspirin inhibits the production of PGE2, a COX-metabolite derived from arachidonic acid (AA), which facilitates tumor growth through the enhancement of immune evasion (39). Aspirin was also adopted as a novel adjuvant to reverse chemoradiotherapy resistance (10, 40).
Steroids, such as dexamethasone and prednisolone, are widely used as monotherapies or combined with other therapeutic agents in various types of cancer. For instance, dexamethasone improves myeloma sensitivity to Venetoclax (a specific inhibitor of B-cell lymphoma-2) (41). In colon cancer, dexamethasone suppresses TGF-β1-induced migration via inhibition of AKT and extracellular signal-regulated kinase (ERK) phosphorylation (42). Dexamethasone is also used for the treatment of castration-refractory prostate cancer (43). The efficacy of statins has also been reported in a variety of cancers, such as HCC, CRC, and acute myelocytic leukemia (44).
Despite the multiple benefits of NSAIDs and steroids in cancer treatment, they have various adverse side effects, including gastrointestinal bleeding, liver and kidney dysfunction, Cushing’s syndrome, and osteoporosis (45–48). Some severe side effects of statins, such as necrotizing myopathy, increased risk of type 2 diabetes, and acute memory impairment, have also been reported (49–51). These negative side effects have restricted the full application of anti-inflammatory drugs to cancer therapy.
Enhancing Antitumor Immunity
The development of cancer immunotherapy was a major milestone in current cancer treatments and ranked first on the list of the top 10 breakthroughs of 2013 in the journal Science. Recent developments in cancer immunotherapy include vaccines, cytokines, checkpoint-blocking antibodies, and immune cell adoptive transfer therapies. Typical cancer vaccines include cancer antigen vaccines, DC vaccines, and nucleic acid vaccines, among others. The melanoma-associated antigen 3 vaccine Stimuvax (targeting Mucin 1) has entered phase III clinical trials (52). Viral vector-infected or peptide-based DCs have been widely used to treat prostate cancer, glioma, melanoma, and CRC (52). Other adoptive cell transfer therapies, including cytokine-induced killer cells (53), tumor-infiltrating lymphocytes (54), gamma delta T cells (γδ T cells) (55), and NKT cells (56), are also being used to enhance clinical antitumor immunity.
Cytokines, such as IL-2, IL-18, IL-21, and granulocyte colony-stimulating factor (G-CSF), are also common adjuvants for cancer therapy. However, combination therapies have shown better curative effects. A recent phase III clinical trial in advanced melanoma demonstrated a significantly improved prognosis by combining a high-dose of IL-2 with a peptide vaccine (gp100) (57). In patients with non-Hodgkin’s lymphoma, combined treatment with recombinant human (rh)IL-18 and rituximab appeared to increase the overall objective response rate by 26.3% (58). A multicenter phase II study of patients with metastatic melanoma showed that IL-21 has antitumor activity (59). The combination of G-CSF and paclitaxel/carboplatin was validated for the treatment of patients with recurrent platinum-resistant ovarian carcinoma or recurrent or advanced endometrial or cervical carcinoma (60).
TNFerade is a genetically engineered adenovector with a radiation-inducible promoter that specifically delivers the human TNF-α gene to cancer cells. Phase I trials in patients with various tumor types (e.g., liver, breast, CRC, melanoma, sarcomas) confirmed that the combination of TNFerade and radiation was more effective than TNFerade or radiation alone (61). The phase I trial of TNFerade plus chemoradiotherapy also improved survival in patients with advanced resectable esophageal cancer (62).
Immune-checkpoint blockade is a revolutionary approach to cancer immunotherapy. Overexpression of programed death-ligand 1 (PD-L1) in tumor cells is correlated with poor prognosis, and immunotherapies with anti-PD-1/PD-L1 and anti-CTLA-4 antibodies have shown promising results in a variety of cancers (63, 64). Thus far, four antibodies have been licensed: (1) ipilimumab, an antibody against CTLA-4, has been licensed for unresectable or metastatic melanoma; (2) two mAbs against PD-1, pembrolizumab and nivolumab, have been approved for unresectable metastatic melanoma and advanced metastatic non-small-cell lung carcinoma. Nivolumab has also been approved for advanced (metastatic) renal cell carcinoma; (3) atezolizumab, a PD-L1-blocking mAb, has been used in metastatic or advanced urothelial carcinoma with platinum chemotherapy resistance (63). However, only a few cancers, such as lymphoma and melanoma, are sensitive to these antibodies because of the heterogeneity of cancers.
Recently, CAR-T immunotherapy, an emerging immunotherapeutic strategy, has achieved unprecedented success in cancer treatment. In CAR-T immunotherapy, T cells are modified to express specific receptors for the various types of cancer. Therefore, these T cells gain the ability to recognize and eliminate cancer cells after reinfusion into patients. A recent study showed that CAR-T cell therapy could mediate valid anti-leukemic activity in patients with acute lymphoblastic leukemia with chemotherapy-resistant B precursors and also exhibited feasibility and invertible toxicity (65). CAR-T therapy is also designed to treat chronic lymphocytic leukemia or B cell lymphomas, breast carcinoma, and glioblastoma (66, 67). However, CAR-T therapy is not widely used because it can induce the life-threatening cytokine release syndrome (CRS) (68) and has low efficacy against solid tumors.
Owing to the limitations of the abovementioned anti-inflammatory drugs and antitumor immunotherapies, it is urgent and essential to develop a novel, safe potent approach to conquer inflammation, and synergize the effects of immunotherapy in the treatment of cancer.
The Potential Role of Inflammation Resolution in Remodeling the TME
Failure to Resolve Inflammation Can Result in Cancer
Whether inflammation is a friend or a foe of cancer has always been controversial (5). As mentioned earlier, a failure of resolution promotes tumorigenesis, progression, and metastasis. TME is a complex environment including tumor cells, immune cells, fibroblasts, blood vessels, and the extracellular matrix (69, 70). It is widely accepted that TME reprograms immune cells into pro-tumor phenotypes with distinct metabolic and biological functions, which are required for the establishment and maintenance of tumors (8).
For instance, MDSCs are immature and immunosuppressive cells that also promote EMT via the TGF-β, EGF, and HGF pathways (32). The immunosuppressive properties of MDSCs are mediated by the following mechanisms: (1) l-arginine deprivation via upregulation of arginases; (2) ROS and reactive nitrogen species (RNS) generation; (3) restricting lymphocyte trafficking and viability; (4) promoting activation and expansion of Tregs; (5) shifting M1 macrophages to TAMs by producing IL-10; (6) inhibiting DC maturation and antigen presentation (33); (7) secreting MMPs, which facilitate tumor cell invasion in vitro and in vivo (71); and (8) regulating miRNAs in cancer cells, leading to enhanced stemness and metastasis potential (72).
Macrophages in the TME lose tumoricidal activity and contribute to immunosuppression through the upregulation of IL-10, TGF-β, and CCL22, which promote T cell anergy and Treg recruitment (34). During tumor initiation, TAMs exhibit activated glycolysis and inhibited oxidative phosphorylation (OXPHOSP). RNS, ROS, IL-β, and TNF-α are generated to drive genetic instability and promote cancer-related inflammation. Intriguingly, at the later stages of tumor progression, the energy metabolism of TAMs is skewed toward OXPHOSP by adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) activation, lactate accumulation, IL-4 (from Th2 cells), and pyruvate kinase isozyme M2 activation (73). These metabolic changes drive the immunosuppressive phenotype in TAMs, which allows tumors to evade detection by the immune system.
In addition, nutrient exhaustion activates AMPK in TADCs, which promotes OXPHOSP, suppresses glycolysis and contributes to the immunosuppressive phenotype of DCs (74). TADCs elevate the numbers of Tregs and MDSCs in breast cancer, which in turn enhance bone metastasis by lowering the levels of CD8+ T cells (75). Recruitment of Tregs is also responsible for CD8+ T cell apoptosis and bone metastasis in breast cancer. Moreover, immune cell-derived TNF increases the infiltration of Tregs and MDSCs, which have been shown to enhance lung metastasis in a melanoma model (76). Recently, in experimental animal models of breast cancer, neutrophils have been identified as the main element and driver of metastatic formation within the premetastatic lung microenvironment, and neutrophil-derived LTs selectively expand the subpool of cancer cells with high tumorigenic potential in distant tissues (77). T cells carry out the bulk of immune surveillance; however, effector T cell activity is suppressed in the TME. The TME induces the loss of mitochondrial biogenesis in T cells, which drives metabolic insufficiency and dysfunction in tumor-infiltrating T cells (78). Moreover, the unresolved chronic inflammation aggregates low hydrogen ion concentration (pH) and hypoxia, accelerates extracellular acidosis, and further reprograms the metabolism of immune cells in the TME, which synergistically abrogates the efficacy of anticancer immunity (79). This evidence indicates a tight interaction between immune cells, and the TME that reprograms the plasticity of immune cells, suggesting that the failure of inflammation to resolve (chronic inflammation) can result in cancer.
Inflammation Resolution and SPM
Conventionally, inflammation is divided into two stages: initiation and resolution. The transition from resolution to homeostasis is an active process in acute inflammation orchestrated by SPM that possess versatile anti-inflammatory and pro-resolving properties (Figure 3) (1).
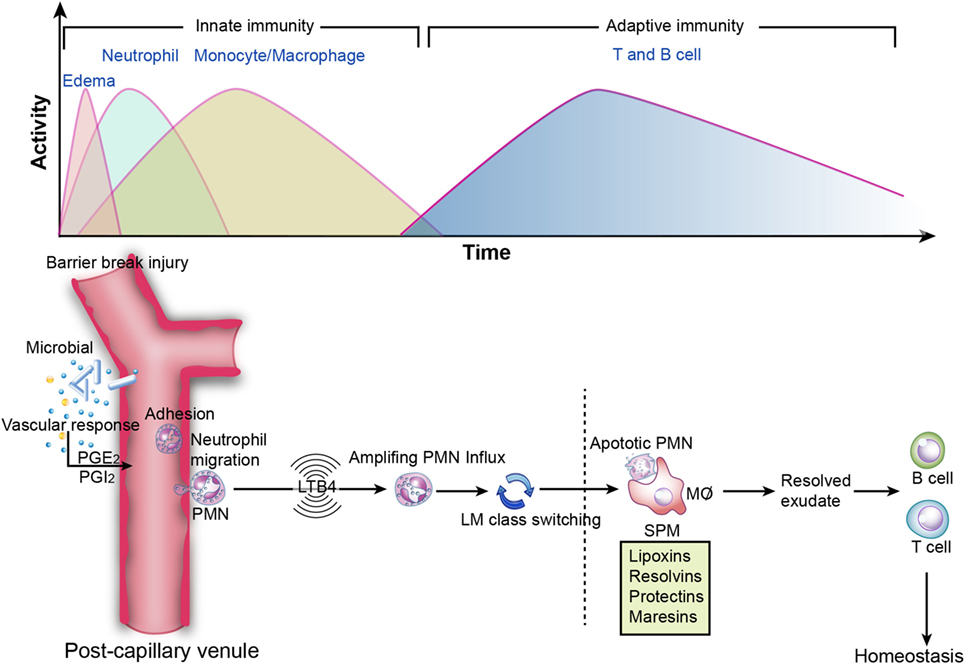
Figure 3. Inflammation resolution. Top image: innate and adaptive immunity in inflammation. During the initiation of inflammation, tissue edema is followed by polymorphonuclear neutrophil (PMN) influx and then a return to baseline, accompanied by the recruitment of monocytes and macrophages for resolution. Sequentially, effector T and B cells transform to memory T and B cells, which is essential for the secondary immune response. However, if resolution is not achieved, then the outcome is sustained inflammation (chronic inflammation). Bottom image: specialized pro-resolving mediators in the acute inflammatory response. PGE2 leads to vasodilation, and LTB4 stimulates PMN influx to the inflammatory loci. Subsequently, lipid mediator (LM) class switching converts pro-inflammatory signals to pro-resolving signals and triggers resolution. Lipoxins and resolvins restrict excessive PMN influx to the injury site, enhance efferocytosis, and stimulate pro-resolving signals and adaptive immunity.
SPM are biosynthesized temporally from ω-3 and ω-6 poly unsaturated fatty acids (PUFAs), such as AA, eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA), and docosahexaenoic acid (DHA), via the catabolism of lipoxygenases (LOX, e.g., 5-LOX and 12-LOX) and COX. LXs derived from AA were discovered by Serhan et al. (80). Later, LX epimers and aspirin-triggered LX (ATL) were identified (81). Omega-3 PUFAs, which are abundant in fish oils, can alter the expression of inflammatory genes and decrease the production of cytokines and the expression of adhesion molecules (82). The three major types of Rvs are series-D, Dp, and E-series. Specifically, RvD and RvDp are derived from DHA and DPA, respectively, RvE is generated from EPA and protectins and MaRs are derived from DHA. The protective actions of SPM have been demonstrated in acute inflammation [e.g., sepsis (83), lung injury (84), and ischemia reperfusion injury (85)] and chronic inflammation [e.g., asthma (86) and AD (87)]. The synthesis of SPM and their biofunctions in inflammation are summarized in Figure 4.
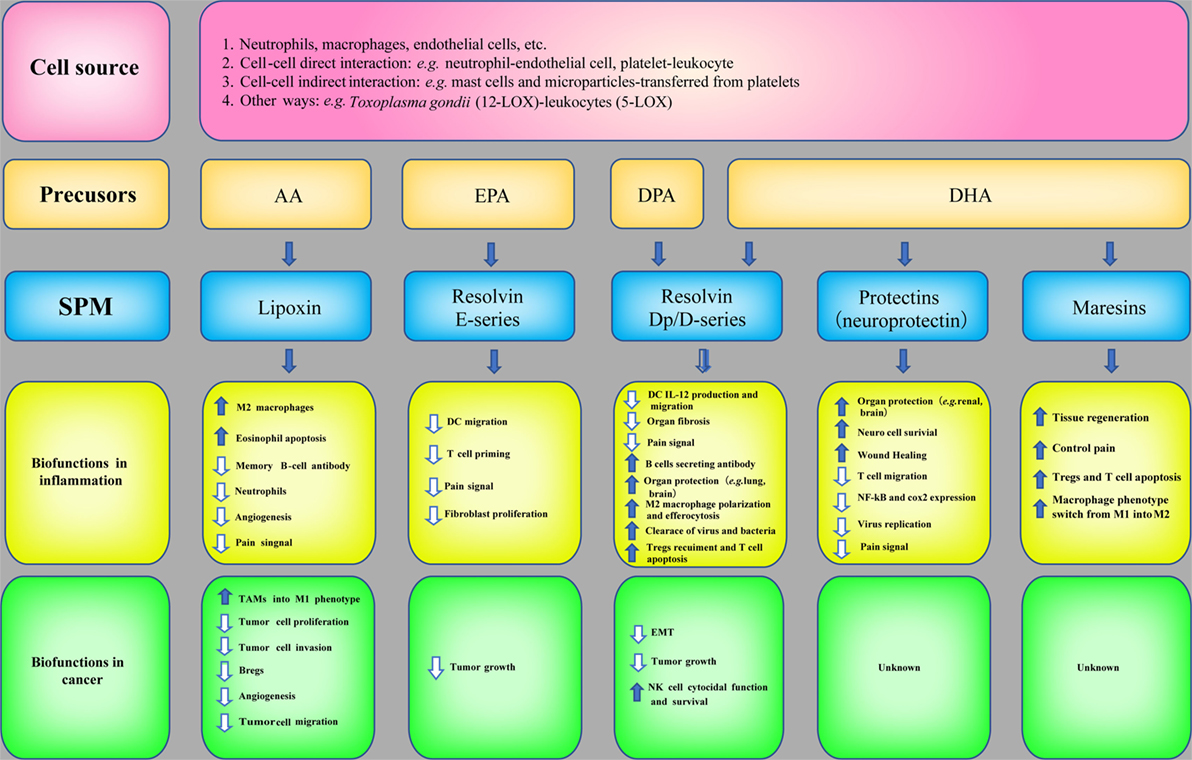
Figure 4. Specialized pro-resolving mediators (SPM) in inflammation and cancer. The biosynthetic pathways of SPM and their biofunctions in inflammation and anticancer immunity.
At the cellular level, the resolution of inflammation is characterized by the cessation of neutrophil infiltration and efferocytosis (macrophage clearance of apoptotic neutrophils). SPM restrict excessive PMN influx to the injury site and induce M1 macrophages to switch to the M2 phenotype, which confers improved phagocytosis abilities. Recent evidence has added a third phase of inflammation, termed post-resolution. During post-resolution, proliferation of memory T and B cells is increased. In this scenario, timely resolution of acute inflammation activates the priming and proliferation of T and B cells in the lymphatic tissues (88). Recent studies have reported that SPM regulate adaptive immunity in vitro. RvD1, RvD2, and MaR1 reduce the production of inflammatory cytokines (e.g., TNF-α and IFN-γ) in Th1 and Th17 cells while increasing the number of Tregs (89); LXA4 decreases memory but not naive B cell antibody production via an formyl peptide receptor 2 (FPR2/ALX)-dependent mechanism (90). These findings suggest that inflammation resolution links innate and adaptive immunity and that SPM play a role in both innate and acquired immunity.
Anticancer Actions of SPM
Owing to the potent bioactivities of SPM in inflammation resolution and the correlation between inflammation and cancer, the roles of SPM in cancer have also attracted attention and investigation (Figure 4). The mechanisms are as follows:
(1) Directly targeting tumor cells: LXA4 shares structural similarities with estrogen 17-estradiol (E2) and possesses antiestrogenic ability via regulating estrogen receptors, indicating the therapeutic potential of LX in estrogen-associated diseases, such as endometrial cancer (91). LXA4 can significantly inhibit the proliferation and migration of lipopolysaccharide-stimulated HeLa cells via the nuclear factor-κB pathway. These effects can be abrogated by inhibiting its receptor, FPR2/ALX (92). In lung cancer, both RvD1 and RvD2 suppress TGF-β1-induced EMT by reducing the expression of zinc finger E-box binding homeobox 1 to prevent tumor metastasis (93). RvD1 induces high caspase-3 activity in pancreatic ductal adenocarcinoma cells (PDAC) in vitro (94).
(2) Targeting the TME: LXA4 and its analog dramatically inhibit the proliferation, invasion, and angiogenesis of hepatocarcinoma via remodeling the TME (13, 95); LXA4 is decreased in papilloma, and administration of LXA4 accelerates papilloma regression in mice (14); ATL treatment reduces the proliferation of lymphangioleiomyoma cells by inhibiting COX-2 (96). In human Kaposi’s sarcoma cells, LXA4 and ATL decrease phosphorylation of the VEGF receptor, ephrin family receptor tyrosine kinases, and pro-inflammatory mediators, including PGE2, LT B4, IL-6, and IL-8, to exert dramatic antiangiogenic actions (97). In murine xenograft tumor models injected with hepatocarcinoma, melanoma or colorectal carcinoma cells, LXA4 is able to suppress tumor growth by targeting IL-10-producing regulatory B cells (Bregs) via dephosphorylation of signal transducer and activator of transcription 3 and ERK. Since Bregs can cause CD8+ T cell dysfunction in the TME (12), these results suggest that LXs may reverse the CD8+ T cell response and improve antitumor immunity. Moreover, LX analogs inhibit VEGF-induced endothelial permeability by stabilizing the VE-cadherin/β-catenin-dependent adherens junctions to protect patients from tumor extravasation across endothelial barriers (15). An interesting recent finding revealed that LXs selectively switch M2 TAMs to an M1 phenotype, which triggers tumor cell apoptosis and blunts tumor progression (98). RvD1 protects NK cells against deactivation and increases NK cell cytocidal function in PDAC (94).
(3) Targeting precancerous lesions: LXA4 analogs block intestinal pro-inflammatory gene expression and inhibit the severity of colitis in a mouse model (99). An LXA4 isomer (10S, 17S-DiHDoHE) exerts an inhibitory effect on neutrophil infiltration and reduces pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, thereby inhibiting the severity of colitis in mice (100). RvE1 increases survival and promotes resolution in a murine model of colitis (101). MaR1-induced attenuation of murine colitis was also recently observed in both dextran sulfate sodium and trinitro-benzene-sulfonic acid models (102). Furthermore, extensive clinical data have addressed the therapeutic role of omega-3 in various cancer types (e.g., breast cancer, CRC, leukemia, gastric cancer, pancreatic cancer, esophageal cancer, prostate cancer, lung cancer, head and neck cancer) and cancer cachexia (103). These trials suggest that SPM are the main mechanisms driving the antineoplastic effects of omega-3.
Together, the bioactions and mechanisms of SPM play important roles in attenuating tumor-promoting inflammation, which represents a synergistic principle that incorporates anti-inflammatory properties and enhances antitumor immunity. This new series of lipid mediators has created a potential new direction for cancer research.
Conclusion and Prospects
Tumor growth is closely connected to inflammation, and the crosstalk between the two processes is context dependent. Inflammatory cell plasticity in the TME can promote or inhibit cancer. Cancer immunology and immunotherapy targeting the inflammatory microenvironment is an exciting field because it is at the brink of mainstream clinical practice and shows promising benefits. However, there are several substantial issues that need to be resolved. Therapy-induced inflammation often endows residual cancer cells with resistance to subsequent courses of treatment (e.g., chemotherapy resistance and radiotherapy resistance) (18). Moreover, the efficacy of immunotherapy depends on cancer types or populations. For example, CAR-T therapy is more effective in hematological neoplasms (65) than in solid tumors and may even lead to the development of life-threatening CRS (104). Although blockade of PD-1 elicits significant clinical benefits in patients with melanoma, some patients are innately resistant to anti-PD-1 therapy because of individual genomic and transcriptomic features (105). Aspirin restores the susceptibility of pancreatic cancer to gemcitabine (10) and reduces the risk of mortality in patients treated with radical prostatectomy or radiation for prostate cancer (106). Aspirin also acts synergistically with anti-PD-1 in tumor models (39). This evidence indicates that anti-inflammatory drugs may serve as useful adjuvants to conventional and immune-based therapies.
Presently, both cancer researchers and doctors have an important social responsibility to combat the increased incidence of tumors. The rapid development of oncology basic research, clinical diagnosis, and treatment provides more opportunities to overcome cancer yet also supplies more rigorous challenges. Targeting the TME is the current research focus; however, our ancestors have already provided us with some philosophical hints as to where we should focus our efforts.
In the primitive society of China, the Great Flood occurred and led to great misery in the people for many times. A superman by the name of Gun, who commiserated with his suffering people, tried to control the flood by blocking and damming. However, he failed. After his death, his son, Yu, carried on his father’s unfulfilled task, fighting against the Great Flood. For thirteen arduous years, he devoted himself conscientiously to his work. Drawing a lesson from his father’s failure, he used the methods of channeling and dredging and finally succeeded in subduing the Great Flood. In honor of his work, Emperor Shun asked him to take over the throne. Yu the Great is the personification of wisdom, perseverance and selfless devotion and, as such, he is a popular figure in artistic creations.
Controlling the cancer inflammatory microenvironment may be similar to controlling the Great Flood: the best strategy is not blocking but dredging. Thus, promoting endogenous pro-resolution factors may be a more safe and potent method for controlling the TME. However, the current evidence for the use of endogenous SPM in animal models with cancer is sparse, and the use of SPM in patients with cancer has not yet been investigated. Based on the versatile pro-resolving properties of SPM and the key roles of inflammation resolution in innate and adaptive immunity, we speculate that SPM may pave the way for the development of novel monotherapies or combination therapies that may provide a breakthrough in anticancer interventions.
Author Contributions
YL conceived this topic and organized the manuscript. QZ and YL wrote the paper and drew the figures. BZ participated in the discussion and revision.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Dr. Lei Wu (Third Military Medical University) for his assistance in Figure 4 preparation. The work was supported by Youth 1000 Talents Project and the National Natural Science Foundation of China (81472435 and 81671573).
References
1. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi: 10.1038/nature13479
2. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40:315–27. doi:10.1016/j.immuni.2014.02.009
3. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci (2015) 16(6):358–72. doi:10.1038/nrn3880
4. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi:10.1038/nature07205
5. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res (2009) 15(2):425–30. doi:10.1158/1078-0432.CCR-08-0149
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
7. Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol (2012) 22(1):33–40. doi:10.1016/j.semcancer.2011.12.005
8. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi:10.1038/ni.2703
9. Solheim TS, Fearon KC, Blum D, Kaasa S. Non-steroidal anti-inflammatory treatment in cancer cachexia: a systematic literature review. Acta Oncol (2013) 52(1):6–17. doi:10.3109/0284186X.2012.724536
10. Zhang Y, Liu L, Fan P, Bauer N, Gladkich J, Ryschich E, et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget (2015) 6(12):9999–10015. doi:10.18632/oncotarget.3171
11. Chan AT, Detering E. An emerging role for anti-inflammatory agents for chemoprevention. Recent Results Cancer Res (2013) 191:1–5. doi:10.1007/978-3-642-30331-9_1
12. Wang Z, Cheng Q, Tang K, Sun Y, Zhang K, Zhang Y, et al. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett (2015) 364(2):118–24. doi:10.1016/j.canlet.2015.04.030
13. Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene (2011) 30(36):3887–99. doi:10.1038/onc.2011.112
14. Wang C, Xiao M, Liu X, Ni C, Liu J, Erben U, et al. IFN-gamma-mediated downregulation of LXA4 is necessary for the maintenance of nonresolving inflammation and papilloma persistence. Cancer Res (2013) 73(6):1742–51. doi:10.1158/0008-5472.CAN-12-2801
15. Vieira AM, Neto EH, Figueiredo CC, Barja Fidalgo C, Fierro IM, Morandi V. ATL-1, a synthetic analog of lipoxin, modulates endothelial permeability and interaction with tumor cells through a VEGF-dependent mechanism. Biochem Pharmacol (2014) 90(4):388–96. doi:10.1016/j.bcp.2014.05.019
16. Zeng M, Mao XH, Li JX, Tong WD, Wang B, Zhang YJ, et al. Efficacy, safety, and immunogenicity of an oral recombinant Helicobacter pylori vaccine in children in China: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2015) 386(10002):1457–64. doi:10.1016/S0140-6736(15)60310-5
17. Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet (2007) 370(9590):890–907. doi:10.1016/S0140-6736(07)61416-0
18. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi:10.1016/j.cell.2010.01.025
19. Chua ML, Wee JT, Hui EP, Chan AT. Nasopharyngeal carcinoma. Lancet (2016) 387(10022):1012–24. doi:10.1016/S0140-6736(15)00055-0
20. Jess T, Simonsen J, Jørgensen KT, Pedersen BV, Nielsen NM, Frisch M. Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology (2012) 143(2):e13–4. doi:10.1053/j.gastro.2012.04.016
21. Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell (2010) 17(1):89–97. doi:10.1016/j.ccr.2009.12.008
22. Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, et al. METABOLISM. S-nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science (2015) 349(6247):500–6. doi:10.1126/science.aaa0079
23. Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer – mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol (2014) 10(8):455–65. doi:10.1038/nrendo.2014.94
24. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482(7385):400–4. doi:10.1038/nature10755
25. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi:10.1111/j.1365-2567.2007.02587.x
26. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov (2015) 14(8):561–84. doi:10.1038/nrd4591
27. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi:10.1038/nri3862
28. Palmai-Pallag T, Bachrati CZ. Inflammation-induced DNA damage and damage-induced inflammation: a vicious cycle. Microbes Infect (2014) 16(10):822–32. doi:10.1016/j.micinf.2014.10.001
29. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30(7):1073–81. doi:10.1093/carcin/bgp127
30. Gasche JA, Hoffmann J, Boland CR, Goel A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int J Cancer (2011) 129(5):1053–63. doi:10.1002/ijc.25764
31. Vendramini-Costa DB, Carvalho JE. Molecular link mechanisms between inflammation and cancer. Curr Pharm Des (2012) 18(26):3831–52. doi:10.2174/138161212802083707
32. Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol (2011) 9(9):e1001162. doi:10.1371/journal.pbio.1001162
33. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
34. Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol (2013) 35(5):585–600. doi:10.1007/s00281-013-0367-7
35. Nelson N. On trial: evidence from using aspirin to prevent cancer. J Natl Cancer Inst (2015) 107(9):7–9. doi:10.1093/jnci/djv265
36. Gamba CA, Swetter SM, Stefanick ML, Kubo J, Desai M, Spaunhurst KM, et al. Aspirin is associated with lower melanoma risk among postmenopausal Caucasian women: the Women’s Health Initiative. Cancer (2013) 119(8):1562–9. doi:10.1002/cncr.27817
37. Cuzick J, Thorat MA, Andriole G, Brawley OW, Brown PH, Culig Z, et al. Prevention and early detection of prostate cancer. Lancet Oncol (2014) 15(11):e484–92. doi:10.1016/S1470-2045(14)70211-6
38. Bardia A, Keenan TE, Ebbert JO, Lazovich D, Wang AH, Vierkant RA, et al. Personalizing aspirin use for targeted breast cancer chemoprevention in postmenopausal women. Mayo Clin Proc (2015) 91(1):71–80. doi:10.1016/j.mayocp.2015.10.018
39. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162(6):1257–70. doi:10.1016/j.cell.2015.08.015
40. Restivo A, Cocco IM, Casula G, Scintu F, Cabras F, Scartozzi M, et al. Aspirin as a neoadjuvant agent during preoperative chemoradiation for rectal cancer. Br J Cancer (2015) 113(8):1133–9. doi:10.1038/bjc.2015.336
41. Matulis SM, Gupta VA, Nooka AK, Hollen HV, Kaufman JL, Lonial S, et al. Dexamethasone treatment promotes Bcl-2-dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia (2015) 30(5):1086–93. doi:10.1038/leu.2015.350
42. Han S, Bui NT, Ho MT, Kim YM, Cho M, Shin DB. Dexamethasone inhibits TGF-beta1-induced cell migration by regulating the ERK and AKT pathways in human colon cancer cells via CYR61. Cancer Res Treat (2015) 48(3):1141–53. doi:10.4143/crt.2015.209
43. Venkitaraman R, Lorente D, Murthy V, Thomas K, Parker L, Ahiabor R, et al. A randomised phase 2 trial of dexamethasone versus prednisolone in castration-resistant prostate cancer. Eur Urol (2015) 67(4):673–9. doi:10.1016/j.eururo.2014.10.004
44. Chae YK, Yousaf M, Malecek MK, Carneiro B, Chandra S, Kaplan J, et al. Statins as anti-cancer therapy; can we translate preclinical and epidemiologic data into clinical benefit? Discov Med (2015) 20(112):413–27.
45. McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med (2011) 8(9):e1001098. doi:10.1371/journal.pmed.1001098
46. Rafaniello C, Ferrajolo C, Sullo MG, Sessa M, Sportiello L, Balzano A, et al. Risk of gastrointestinal complications associated to NSAIDs, low-dose aspirin and their combinations: results of a regional spontaneous reporting system. Pharmacol Res (2015) 104:108–14. doi:10.1016/j.phrs.2015.12.026
47. Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab (2010) 11(6):517–31. doi:10.1016/j.cmet.2010.05.005
48. Bordag N, Klie S, Jürchott K, Vierheller J, Schiewe H, Albrecht V, et al. Glucocorticoid (dexamethasone)-induced metabolome changes in healthy males suggest prediction of response and side effects. Sci Rep (2015) 5:15954. doi:10.1038/srep15954
49. Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet (2015) 385(9965):351–61. doi:10.1016/S0140-6736(14)61183-1
50. Kipfer S, Frigerio S, Hench J, Aussy A, Boyer O. Immune-mediated necrotising myopathy linked to statin use. Lancet (2015) 386(10005):e26. doi:10.1016/S0140-6736(15)60068-X
51. Strom BL, Schinnar R, Karlawish J, Hennessy S, Teal V, Bilker WB. Statin therapy and risk of acute memory impairment. JAMA Intern Med (2015) 175(8):1399–405. doi:10.1001/jamainternmed.2015.2092
52. Schlom J. Therapeutic cancer vaccines: current status and moving forward. J Natl Cancer Inst (2012) 104(8):599–613. doi:10.1093/jnci/djs033
53. Pan K, Guan XX, Li YQ, Zhao JJ, Li JJ, Qiu HJ, et al. Clinical activity of adjuvant cytokine-induced killer cell immunotherapy in patients with post-mastectomy triple-negative breast cancer. Clin Cancer Res (2014) 20(11):3003–11. doi:10.1158/1078-0432.CCR-14-0082
54. Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother (2012) 35(8):615–20. doi:10.1097/CJI.0b013e31826e8f5f
55. Silva-Santos B, Serre K, Norell H. gammadelta T cells in cancer. Nat Rev Immunol (2015) 15(11):683–91. doi:10.1038/nri3904
56. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol (2016) 16(2):112–23. doi:10.1038/nri.2015.9
57. Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med (2011) 364(22):2119–27. doi:10.1056/NEJMoa1012863
58. Robertson MJ, Kline J, Struemper H, Koch KM, Bauman JW, Gardner OS, et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J Immunother (2013) 36(6):331–41. doi:10.1097/CJI.0b013e31829d7e2e
59. Petrella TM, Tozer R, Belanger K, Savage KJ, Wong R, Smylie M, et al. Interleukin-21 has activity in patients with metastatic melanoma: a phase II study. J Clin Oncol (2012) 30(27):3396–401. doi:10.1200/JCO.2011.40.0655
60. Vergote I, Debruyne P, Kridelka F, Berteloot P, Amant F, Honhon B, et al. Phase II study of weekly paclitaxel/carboplatin in combination with prophylactic G-CSF in the treatment of gynecologic cancers: a study in 108 patients by the Belgian Gynaecological Oncology Group. Gynecol Oncol (2015) 138(2):278–84. doi:10.1016/j.ygyno.2015.05.042
61. Liu L, Wang S, Shan B, Sang M, Liu S, Wang G. Advances in viral-vector systemic cytokine gene therapy against cancer. Vaccine (2010) 28(23):3883–7. doi:10.1016/j.vaccine.2010.03.041
62. Chang KJ, Reid T, Senzer N, Swisher S, Pinto H, Hanna N, et al. Phase I evaluation of TNFerade biologic plus chemoradiotherapy before esophagectomy for locally advanced resectable esophageal cancer. Gastrointest Endosc (2012) 75(6):1139–46. doi:10.1016/j.gie.2012.01.042
63. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity (2016) 44(6):1255–69. doi:10.1016/j.immuni.2016.06.001
64. Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest (2015) 125(9):3377–83. doi:10.1172/JCI80012
65. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3
66. Zhou F, Krishnamurthy J, Wei Y, Li M, Hunt K, Johanning GL, et al. Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology (2015) 4(11):e1047582. doi:10.1080/2162402X.2015.1047582
67. Maus MV. Designing CAR T cells for glioblastoma. Oncoimmunology (2015) 4(12):e1048956. doi:10.1080/2162402X.2015.1048956
68. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124(2):188–95. doi:10.1182/blood-2014-05-552729
69. Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol (2016) 40:41–8. doi:10.1016/j.copbio.2016.02.007
70. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi:10.1126/science.aaa6204
71. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13(1):23–35. doi:10.1016/j.ccr.2007.12.004
72. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity (2013) 39(3):611–21. doi:10.1016/j.immuni.2013.08.025
73. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43(3):435–49. doi:10.1016/j.immuni.2015.09.001
74. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol (2015) 194(11):5174–86. doi:10.4049/jimmunol.1303316
75. Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A, et al. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J Immunol (2012) 189(9):4258–65. doi:10.4049/jimmunol.1101855
76. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol (2015) 15(2):73–86. doi:10.1038/nri3789
77. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature (2015) 528(7582):413–7. doi:10.1038/nature16140
78. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45(2):374–88. doi:10.1016/j.immuni.2016.08.009
79. Faes S, Duval AP, Planche A, Uldry E, Santoro T, Pythoud C, et al. Acidic tumor microenvironment abrogates the efficacy of mTORC1 inhibitors. Mol Cancer (2016) 15(1):78. doi:10.1186/s12943-016-0562-y
80. Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A (1984) 81(17):5335–9. doi:10.1073/pnas.81.17.5335
81. Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A (1995) 92(21):9475–9. doi:10.1073/pnas.92.21.9475
82. Calder PC. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr (2006) 83(6 Suppl):1505S–19S.
83. Wu B, Capilato J, Pham MP, Walker J, Spur B, Rodriguez A, et al. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J (2016) 30(6):2400–10. doi:10.1096/fj.201500029R
84. Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol (2013) 6(2):256–66. doi:10.1038/mi.2012.66
85. Xian W, Wu Y, Xiong W, Li L, Li T, Pan S, et al. The pro-resolving lipid mediator maresin 1 protects against cerebral ischemia/reperfusion injury by attenuating the pro-inflammatory response. Biochem Biophys Res Commun (2016) 472(1):175–81. doi:10.1016/j.bbrc.2016.02.090
86. Flesher RP, Herbert C, Kumar RK. Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clin Sci (Lond) (2014) 126(11):805–14. doi:10.1042/CS20130623
87. Wang X, Zhu M, Hjorth E, Cortés-Toro V, Eyjolfsdottir H, Graff C, et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement (2015) 11(1):40–50. doi:10.1016/j.jalz.2013.12.024
88. Newson J, Stables M, Karra E, Arce-Vargas F, Quezada S, Motwani M, et al. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood (2014) 124(11):1748–64. doi:10.1182/blood-2014-03-562710
89. Chiurchiu V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med (2016) 8(353):353ra111. doi:10.1126/scitranslmed.aaf7483
90. Ramon S, Bancos S, Serhan CN, Phipps RP. Lipoxin A(4) modulates adaptive immunity by decreasing memory B-cell responses via an ALX/FPR2-dependent mechanism. Eur J Immunol (2014) 44(2):357–69. doi:10.1002/eji.201343316
91. Canny GO, Lessey BA. The role of lipoxin A4 in endometrial biology and endometriosis. Mucosal Immunol (2013) 6(3):439–50. doi:10.1038/mi.2013.9
92. Hao H, Xu F, Hao J, He YQ, Zhou XY, Dai H, et al. Lipoxin A4 suppresses lipopolysaccharide-induced HeLa cell proliferation and migration via NF-kappaB pathway. Inflammation (2015) 38(1):400–8. doi:10.1007/s10753-014-0044-6
93. Lee HJ, Park MK, Lee EJ, Lee CH. Resolvin D1 inhibits TGF-beta1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int J Biochem Cell Biol (2013) 45(12):2801–7. doi:10.1016/j.biocel.2013.09.018
94. Halder RC, Almasi A, Sagong B, Leung J, Jewett A, Fiala M. Curcuminoids and omega-3 fatty acids with anti-oxidants potentiate cytotoxicity of natural killer cells against pancreatic ductal adenocarcinoma cells and inhibit interferon gamma production. Front Physiol (2015) 6:129. doi:10.3389/fphys.2015.00129
95. Hao H, Liu M, Wu P, Cai L, Tang K, Yi P, et al. Lipoxin A4 and its analog suppress hepatocellular carcinoma via remodeling tumor microenvironment. Cancer Lett (2011) 309(1):85–94. doi:10.1016/j.canlet.2011.05.020
96. Li C, Lee PS, Sun Y, Gu X, Zhang E, Guo Y, et al. Estradiol and mTORC2 cooperate to enhance prostaglandin biosynthesis and tumorigenesis in TSC2-deficient LAM cells. J Exp Med (2014) 211(1):15–28. doi:10.1084/jem.20131080
97. Marginean A, Sharma-Walia N. Lipoxins exert antiangiogenic and anti-inflammatory effects on Kaposi’s sarcoma cells. Transl Res (2015) 166(2):111–33. doi:10.1016/j.trsl.2015.02.009
98. Simoes RL, Lee PS, Sun Y, Gu X, Zhang E, Guo Y, et al. Lipoxin A4 selectively programs the profile of M2 tumor-associated macrophages which favour control of tumor progression. Int J Cancer (2017) 140(2):346–57. doi:10.1002/ijc.30424
99. Gewirtz AT, Collier-Hyams LS, Young AN, Kucharzik T, Guilford WJ, Parkinson JF, et al. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J Immunol (2002) 168(10):5260–7. doi:10.4049/jimmunol.168.10.5260
100. Masterson JC, McNamee EN, Fillon SA, Hosford L, Harris R, Fernando SD, et al. Eosinophil-mediated signalling attenuates inflammatory responses in experimental colitis. Gut (2015) 64(8):1236–47. doi:10.1136/gutjnl-2014-306998
101. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med (2005) 201(5):713–22. doi:10.1084/jem.20042031
102. Marcon R, Bento AF, Dutra RC, Bicca MA, Leite DF, Calixto JB. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J Immunol (2013) 191(8):4288–98. doi:10.4049/jimmunol.1202743
103. Nabavi SF, Bilotto S, Russo GL, Orhan IE, Habtemariam S, Daglia M, et al. Omega-3 polyunsaturated fatty acids and cancer: lessons learned from clinical trials. Cancer Metastasis Rev (2015) 34(3):359–80. doi:10.1007/s10555-015-9572-2
104. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Cancer Discov (2016) 6(6):664–79. doi:10.1158/2159-8290.CD-16-0040
105. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165(1):35–44. doi:10.1016/j.cell.2016.02.065
Keywords: inflammation, cancer, lipoxins, resolvins, immunity
Citation: Zhang Q, Zhu B and Li Y (2017) Resolution of Cancer-Promoting Inflammation: A New Approach for Anticancer Therapy. Front. Immunol. 8:71. doi: 10.3389/fimmu.2017.00071
Received: 25 October 2016; Accepted: 17 January 2017;
Published: 02 February 2017
Edited by:
Fang-Ping Huang, University of Hong Kong, Hong KongReviewed by:
Lidija Klampfer, Albert Einstein College of Medicine, USAJoanne Lysaght, Trinity College Dublin, Ireland
Copyright: © 2017 Zhang, Zhu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongsheng Li, eWxpQHRtbXUuZWR1LmNu