 Sophie Laffont
Sophie Laffont Cyril Seillet
Cyril Seillet Jean-Charles Guéry
Jean-Charles Guéry- 1Centre de Physiopathologie de Toulouse Purpan (CPTP), Université de Toulouse, INSERM, CNRS, UPS, Toulouse, France
- 2Division of Molecular Immunology, The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia
- 3Department of Medical Biology, University of Melbourne, Melbourne, VIC, Australia
Autoimmunity, infectious diseases and cancer affect women and men differently. Because they tend to develop more vigorous adaptive immune responses than men, women are less susceptible to some infectious diseases but also at higher risk of autoimmunity. The regulation of immune responses by sex-dependent factors probably involves several non-redundant mechanisms. A privileged area of study, however, concerns the role of sex steroid hormones in the biology of innate immune cells, especially dendritic cells (DCs). In recent years, our understanding of the lineage origin of DC populations has expanded, and the lineage-committing transcription factors shaping peripheral DC subsets have been identified. Both progenitor cells and mature DC subsets express estrogen receptors (ERs), which are ligand-dependent transcription factors. This suggests that estrogens may contribute to the reported sex differences in immunity by regulating DC biology. Here, we review the recent literature and highlight evidence that estrogen-dependent activation of ERα regulates the development or the functional responses of particular DC subsets. The in vitro model of GM-CSF-induced DC differentiation shows that CD11c+ CD11bint Ly6cneg cells depend on ERα activation by estrogen for their development, and for the acquisition of competence to activate naive CD4+ T lymphocytes and mount a robust pro-inflammatory cytokine response to CD40 stimulation. In this model, estrogen signaling in conjunction with GM-CSF is necessary to promote early interferon regulatory factor (Irf)-4 expression in macrophage-DC progenitors and their subsequent differentiation into IRF-4hi CD11c+ CD11bint Ly6cneg cells, closely related to the cDC2 subset. The Flt3L-induced model of DC differentiation in turn shows that ERα signaling promotes the development of conventional DC (cDC) and plasmacytoid DC (pDC) with higher capability of pro-inflammatory cytokine production in response to TLR stimulation. Likewise, cell-intrinsic ER signaling positively regulates the TLR-driven production of type I interferons (IFNs) in mouse pDCs in vivo. This effect of estrogens likely contributes to the greater proficiency of women’s pDCs than men’s as regards the production of type I IFNs elicited by TLR7 ligands. In summary, evidence is emerging in support of the notion that estrogen signaling regulates important aspects of cDC and pDC development and/or effector functions, in both mice and humans.
Introduction
The pro-inflammatory role of estrogens, particularly through their actions on the innate immune system, has been proposed to contribute to the higher immune responses that develop in women (1, 2). Estrogen mediates their effects through two receptors, estrogen receptors (ERs) ERα and ERβ, which are members of the nuclear receptor super family (3). ERα and ERβ function in the nucleus as transcription factors in a ligand-dependent manner. Many studies have shown that mature cells both from the innate and the adaptive immune system express ERs, particularly ERα in mouse (4–10) suggesting that estrogens could regulate their effector functions. Estrogens could also directly act, through their receptors, on progenitor cells to influence the development of myeloid and lymphoid cells (11–17).
Dendritic cells (DCs) are professional antigen-presenting cells that bridge innate and adaptive immunity. They are essential for the activation of naive T cells specific to self or non-self protein antigens and for their subsequent differentiation into effector T (Teff) cells through the secretion of specific cytokines (18). Ontogeny and global gene expression studies indicate that DCs form a separate lineage of mononuclear phagocytes or mononuclear-derived cells, notably on the basis of their continual replenishment from common DC precursors that are distinct from the precursors of monocytes and macrophages (19). DCs are subdivided into three main subtypes: conventional DCs (cDCs), monocyte-derived DCs and plasmacytoid DCs (pDCs) (19). While cDCs are specialized in antigen processing and presentation, thereby converting naive T cells into effector CD4+ and CD8+ Teff cells, blood monocytes can be rapidly mobilized from the circulation and differentiate into monocyte-derived cells (MCs) with distinct functions such as local reactivation of Teff cells, and superior inflammatory and chemokine production relative to cDCs (18). By contrast, pDCs produce massive amounts of type I interferons (IFNs) in response to viral infections (20).
This review surveys the state of the knowledge regarding estrogen-dependent regulation of DC biology and revisits previously published experiments in light of recent advances in the DC field (18, 19, 21). We will focus on key experimental studies supporting a direct regulatory role of sex hormone estrogens vis-à-vis cDCs and pDCs, which hinges on the ligand-dependent activation of their nuclear receptor, ERα (summarized in Table 1).
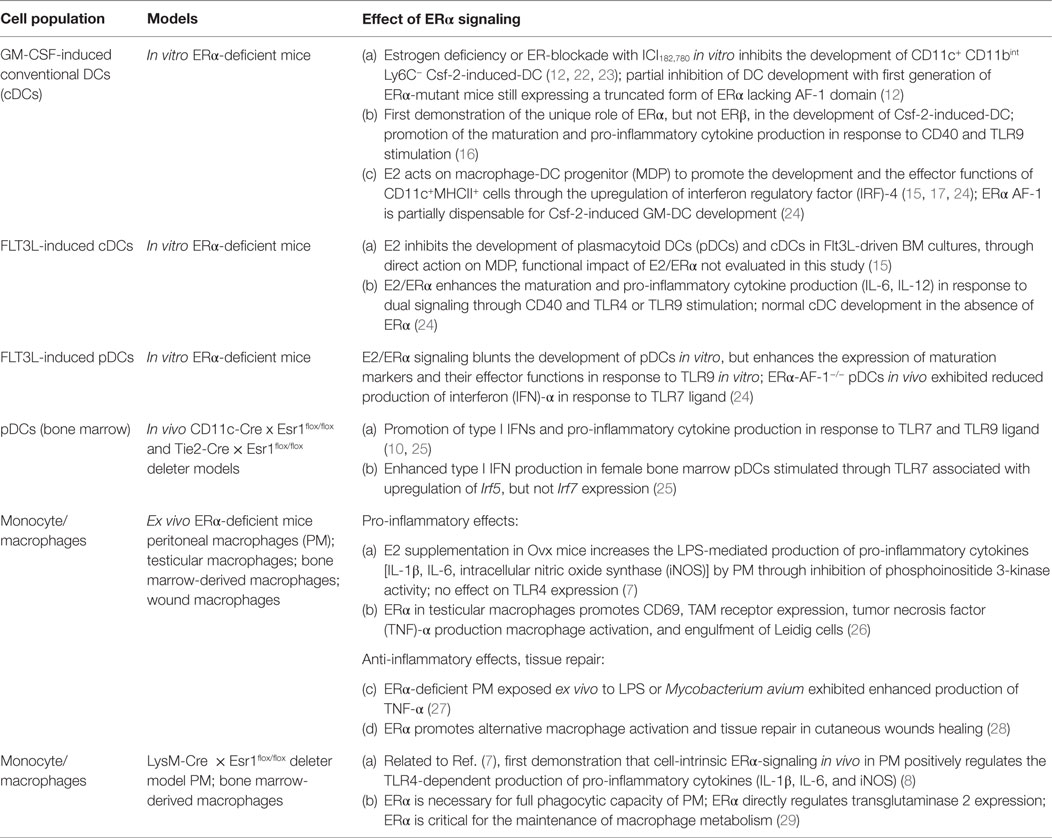
Table 1. Effect of estrogen receptor (ER)α signaling on dendritic cells (DCs) and monocyte-derived cells in mouse models.
ER Expression in Hematopoietic Cells
Estrogen receptors are ligand-inducible transcription factors whose activity is regulated by endogenous estrogens naturally produced over the estrus cycle or during pregnancy. The main endogenous estrogens are estrone (E1), 17β-estradiol (E2), estriol (E3) and estetrol (E4), with E2 as the predominant and most biologically active of the four. In humans, E3 and E4 are selectively produced during pregnancy, by the placenta or by the fetal liver, respectively. ER activities can be modulated also by exogenous ligands, such as synthetic, non-steroidal, non-hormonal agonist, and antagonist ligands known as selective ER modulators (SERMs) (30). SERMs such as tamoxifen and raloxifen are in current clinical use for the treatment of breast cancer and osteoporosis, respectively. It is, therefore, important to understand how such molecules, through ER signaling, may interfere with the development and/or function of immune cells. Despite a growing body of evidence that ER-dependent mechanisms influence both the development and the function of innate immune cells, few studies have sought to determine the relative expression of ER isotypes in different steady-state populations of immune cells. Estrogens exert their effects through two known nuclear receptor family members: ERα, which is encoded by ESR1 on human chromosome 6, and ERβ, which is encoded by the ESR2 locus on chromosome 14 (30).
Estrogen receptors are widely expressed in most tissues of both women and men (31). They have also been identified in many different immune cell types including hematopoietic precursors (32). Both ER are expressed in CD34+ human hematopoietic progenitor cells (HPCs) found in the bone marrow, but not in HPCs from cord blood (33–36). These findings indicate that hormone-receptor expression is highly regulated in hematopoietic precursors during development. The absence of ER expression in HPCs from cord-blood may protect the fetus from high hormone levels that suppress maternal lymphopoiesis (33) while promoting hematopoietic stem cell (HSC) self-renewal and myelopoiesis (35). Indeed, E2 signaling through ERα was recently reported to promote the cycling of HSCs and multipotent progenitors, thereby supporting the increased demand for erythropoiesis during pregnancy (35).
In mature human cells, monocyte-derived DCs express ESR1 transcripts and low levels of ESR2 mRNA (37, 38). ESR1 expression was comparable between CD4+, CD8+ T cells, and natural killer cells, which also expressed ESR2 although at lower levels. B lymphocytes expressed the highest levels of ESR1 mRNA relative to other leukocytes. Of note, B cells and pDCs displayed the highest levels of ESR2 mRNA in comparison with any other cell type examined (38). As previously reported (39), expression levels of both ER isotypes were low in blood monocytes, despite evidence in mouse that E2 promoted the TLR4-mediated production of pro-inflammatory cytokines by peritoneal monocyte and macrophage cells, through cell-intrinsic ERα-signaling in vivo (7, 8) (Table 1).
In mice, Esr1, but not Esr2, is expressed in bone marrow progenitor cells, including the common lymphoid progenitor (CLP) and myeloid progenitor (MP) compartments (15). Most of the mature immune cells likewise express Esr1, with so far little evidence for Esr2-dependent regulation. A notable exception is microglia, the brain-resident macrophages, where TLR-mediated response can be differentially regulated through ERα- or ERβ-dependent mechanisms (40, 41). Murine splenic cDCs and pDCs, as well as bone marrow-derived DCs (BMDCs), also express Esr1, but only negligible levels of Esr2 (32). However, ER expression can be modulated by the tissue microenvironment or by the activation status of the cells, as DCs infiltrating the brain during experimental autoimmune encephalomyelitis (EAE) express ERβ, unlike splenic DCs (42). Moreover, evidence exists that regulatory cytokines, such as IL-27, can induce a dramatic increase in Esr1 gene expression in cDCs, although the functional consequences of this upregulation are presently unknown (43). ERα appears thus to be the most prominent ER in the human and murine dendritic cell lineages, even though human pDCs express both ESR1 and ESR2 transcripts (38).
Mechanisms of ER Signaling
The ERα and ERβ paralogs are conserved across vertebrate lineages (44). The proteins share five functional and structural domains: two transcriptional activation function domains, AF-1 (A/B domain) and AF-2 (F domain); a DNA-binding domain (DBD; C domain); a hinge domain (D); and a ligand-binding domain (LBD; E/F domain) (45–47).
Following interaction with its estrogen ligand, an ER can regulate cellular function through either of two different mechanisms: the nuclear genomic mechanism, implying direct or indirect binding of the receptor to transcription control regions of targeted genes, and the non-genomic mechanism, initiated by receptors at the cell membrane, which signal through kinase pathways (47).
Occupation of the ligand cavity of the LBD results in conformational changes in the receptor allowing interaction with coactivators, if the ligand is an agonist, or preventing such interactions, if the ligand is an antagonist (48). Transcriptional responses to estrogens were initially recognized to depend on specific interaction of homo/heterodimeric ERs with estrogen-response elements (EREs, GGTCANNNTGACC) found in the promoters of target genes. In addition to this “classical” pathway, a “tethered” signaling pathway has been described in which ligand-activated ERs can interact with other transcription factor complexes and bind to non-ERE sequences. ERs can thus regulate a number of key transcriptional pathways including those of NF-κB, AP-1, Sp-1, and C/EBPβ (30). Of note, the AP-1 and Sp-1 transcription factor family members are ubiquitously expressed and are known to regulate the expression of several genes important for immune cell development and function (49, 50).
In cancer cells, it has been shown that estrogen-bound ERα can regulate gene expression through its direct or indirect recruitment to target gene promoters, or through its binding to distant regulatory elements or enhancers. ERα molecules can associate with other cofactors such as GATA-3, P2A-g, and FOXA1 to the establishment of chromatin loops, bringing distal ERα-bound elements to the vicinity of the transcription start site in target genes (51, 52). Once bound, ERα associates with coactivator proteins displaying chromatin-modification properties potentiating the recruitment of RNA polymerase II and chromatin remodeling enzymes, such as histone modifying enzymes (HATs, HDACs, and HMTs) (53, 54). It is unknown, however, whether such mechanisms occur in non-reproductive tissues, such as immune cells, in which ER expression is 100-fold lower than in breast cancer cells. Moreover, limited information exists regarding the expression of ER at the protein level in immune cell subsets, as control experiments using appropriate knockout cells are often missing in many experimental settings. However, ER protein levels do not necessarily correlate with mRNA expression, as shown for ER- cancer cells, owing to the coupling of ER cytoplasmic signaling cascades and target gene transcription with rapid ER proteolysis effected by the ubiquitin-proteasome system (55).
Full ligand-dependent ERα activation entails interactions between the AF domains, but the AF-1 and AF-2 transactivation domains can independently activate transcription in a promoter- and cell type-specific manner (46, 56, 57). The relative contribution of AF-1 and AF-2 in ERα activity may depend upon the differentiation stage of the cells (58) or the tissue examined (59). Mice lacking either AF-1 (ERαAF-10) or AF-2 (ERαAF-20) have been recently generated (60–62). Using these animal models, it has been shown that ERαAF-1 was required neither for the E2-dependent vasculoprotective effects (60) nor for osteoporosis prevention (62). AF1 was, however, necessary for breast cancer cell proliferation (63) and uterine growth (60). Unlike AF-1, AF-2 was required for all E2-mediated actions with the exception of the acceleration of endothelial healing (61, 62).
Apart from the “genomic/nuclear” actions that classically rely on AF-1 and/or AF-2 activation, E2 modulates also the activation of several kinases [MAPK, phosphoinositide 3-kinase (PI3K) or PKC], phosphatases, and adenylyl cyclase as well as changes in intracellular calcium (30). These membrane-initiated steroid signaling (MISS) actions are mediated by a pool of intracellular receptors localized to caveolae or rafts at the plasma membrane (64). The palmitoylation of human ERα cysteine 447 appears to be crucial for the targeting of ERα to the plasma membrane through physical interaction with caveolin-1 (65, 66). Recently, a mouse model where ERα Cys451, the putative palmitoylation site, was mutated into alanine has provided the first evidence for a tissue-specific physiological role of MISS effects of ERα in vivo (67).
These new tools should boost our understanding of the ER-dependent signaling mechanisms by which estrogens regulate the development and function of DC subpopulations in vivo. In vitro studies using mice mutant for AF-1 and AF-2 domains have already led to advances in this field (24), but we still lack a detailed understanding of the respective contributions of the genomic and MISS effects of ERα signaling to DC biology in physiological settings in vivo.
DC Development and Lineages
Murine DCs are generally categorized into three main groups: cDCs, pDCs and monocyte-derived inflammatory DCs (moDCs). These different populations arise from the common MPs (CMPs) found in the bone marrow (21), which become progressively restricted into macrophage-DC progenitors (MDPs) (68). Although the MDP has been identified as the first progenitor committed to the DC and monocyte lineage, the clonal potential of MDPs to give rise to both monocytes and DCs is still debated (69). Subsequently, a common DC progenitor (CDP), expressing high levels of Flt3 has been identified, which gives rise to both the cDC and pDC lineages (70).
Conventional DCs
Conventional DCs are subdivided into two main lineages, cDC1 and cDC2 (19). Conventional cDC1s express the chemokine receptor XCR1 (71, 72) and express either CD8α or CD103 (also known as αE integrin), but do not express CD11b. The cDC1 subset is highly efficient at cross-presenting exogenous antigens on MHC class I molecules (73–75) and cDC1s further depend on a common set of transcription factors (Irf-8, Id2, Nfil3, and Batf3) for their development. DCs from the cDC2 subset found in the spleen express CD4 and CD172α and cross-present antigens poorly. They are, however, highly competent at presenting MHC class II-restricted antigens to CD4+ T cells. In peripheral tissues, the homologous subset, CD11b+ DCs (either CD103+ or CD103−), do not express CD4 but functionally resemble their splenic counterparts (76, 77). CDC2s mainly depend on Irf-4 (78) and Relb (79) for their development.
Plasmacytoid DCs
Plasmacytoid DCs are a distinct lineage separated from the cDCs by their transcriptional dependency (particularly on E2-2), their capacity to secrete abundant type 1 IFN, their morphology, and their poor antigen presentation capacity (80–82). They are characterized by their expression of marker molecules B220, Ly49Q, Siglec-H, and Bst2 (CD317, bone marrow stromal cell antigen 2, also known as PDCA-1) (83) in mouse, and BDCA-2 (CD303) and BDCA-4 (CD304) in humans (84).
Monocyte-derived DCs
Monocyte-derived cells were initially considered an important reservoir for DC development based on the observation that murine and human monocytes can differentiate in vitro into “DC-like” cells in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF, Csf-2) (85, 86). The in vivo equivalent of GM-CSF-derived cells has been difficult to identify, and it is now clear that they represent a distinct type of highly plastic cells able to acquire multiple functions in response to environmental cues (87). MCs were initially described as a subset of DCs emerging during inflammation induced by Listeria monocytogenes infection and characterized as cells producing large amounts of tumor necrosis factor α (TNF-α) and intracellular nitric oxide synthase (iNOS); as such, they were called Tip-DC (TNF-α and iNOS-producing DCs) (88). MCs were subsequently identified in the response to infection by other pathogens (87, 89) and were shown to differentiate from CCR2+ Ly6Chi monocytes recruited to the site of inflammation where they can present antigens to both CD4+ and CD8+ T cells (89). MCs were shown to be crucial for the induction of Th1 and Th17 immunity in response to pathogens and following immunization in Freund’s complete adjuvant (90, 91). In addition, human MCs have been identified in the joints of rheumatoid arthritis patients and in tumor ascites and preferentially primed Th17 responses (92). Another subset of MCs has been reported to accumulate in inflammatory lymph nodes in response to LPS and to express CD14 and the c-type lectin DC-SIGN (CD209a) (93). However, these cells were subsequently found to express the DC-specific transcription factor Zbtb46 (94). Unlike Tip-DCs, LPS-induced MCs were lost in Zbtb46-DTR deleter mice treated with diphtheria toxin and failed to accumulate in Flt3L−/− mice, suggesting that these inflammatory DCs were more closely related to the cDC lineage (94).
Critical Role of E2/ERα-Signaling in the Generation of CSF-2-Induced CDCs
Most of the evidence regarding the capacity of estrogens to regulate DC development and function has been obtained using in vitro models of DC development, particularly the culture of bone marrow cells in the presence of GM-CSF (Csf-2), a cytokine involved in the development and homeostasis of the myeloid lineage (95). It was initially suggested that the CD11c+ MHCII+ cells that develop under these conditions were equivalent to the inflammatory DCs arising from monocytes during inflammation in vivo (96), as mice lacking Csf-2 or its receptor do not carry any major defects in the development of lymph node and spleen cDCs (21). Csf-2 deficiency has, however, been associated with a reduction in the numbers of CD103+ cDCs and CD11b+ cDCs in the intestine, dermis, and lung (97). Thus, in vivo Csf-2 appears dispensable for lymphoid DC homeostasis, but necessary for the development and maintenance of some tissue-resident DCs, suggesting that Csf-2 is a critical regulator of cDC survival in non-lymphoid tissues (97). Although some studies have suggested that inflammatory or mo-DCs can in vivo develop from progenitor cells constitutively lacking Csf-2 receptor (91, 97), evidence also exists for a Csf-2-dependent regulation of mo-DC accumulation in tissues during injury and response to pathogens (98). For instance, Csf-2 neutralization has been recently reported to inhibit the development of a cycling mo-DC population (99). On the other hand, CD4+ T cell-derived Csf-2 production can drive the generation of mo-DC during inflammatory diseases (100, 101) and is essential for the pathogenicity of Th17 cells in CNS autoimmunity (102, 103). After subcutaneous immunization with antigen in complete Freund’s adjuvant, Csf-2 has been reported to promote the differentiation of Th1 or Th17 effector cells, by acting on CD103+ dermal DCs (104) or mo-DCs (91), respectively. Indeed, Csf-2 is critical for IL-6 and IL-23 production by DCs and for Th17 induction in autoimmune myocarditis (105) and EAE (91). Thus, besides its potential role in promoting the expansion of mo-DCs during inflammation (99–101), Csf-2 appears also critical in assigning the Th17-inducing potential of mo-DCs in vivo (91, 105).
Application of this in vitro model of Csf-2-induced differentiation to either bulk bone marrow cells or highly purified progenitors, such as MDPs, has shown the presence of E2 in the culture medium to be critical in promoting the development of CD11c+ MHCII+ cells (106) (Table 1). This was initially demonstrated by using either steroid-free culture medium supplemented with estrogens, pure ER antagonists, or SERMs shown to block ER signaling by estrogens originating from fetal calf serum (FCS) supplementation (12, 15, 16, 22, 107). These studies concluded that estrogens in standard FCS-supplemented medium, probably in the 10−10–10−9 M range, are sufficient to promote the development of CD11c+ DC-like cells. Such estrogen doses are compatible with physiological concentrations in female mice during diestrus and estrus (108). It was then formally established that estrogen-mediated activation of ERα, but not ERβ, was necessary to promote BMDC development in the presence of Csf-2 (16). This was not related to a progenitor deficiency in ERα-deficient mice, as the frequency of MDPs and CDPs in the bone marrow was not affected (24).
Time-course experiments using the pure ER-antagonist ICI182,780 showed that the essential effect of E2 in promoting Csf-2-mediated DC development occurred within the first 48 h of culture (17). This suggested that E2-dependent activation of ERα could regulate the activation state or expression level of transcription factors implicated in DC lineage commitment in the early stages of the differentiation of bone marrow precursors. Indeed, it was subsequently reported that E2 acts directly on highly purified MPs, including MDPs, to regulate GM-CSF-induced differentiation of CD11c+ DC-like cells through the upregulation of the transcription factor Irf-4 (15, 17, 24) (Figure 1).
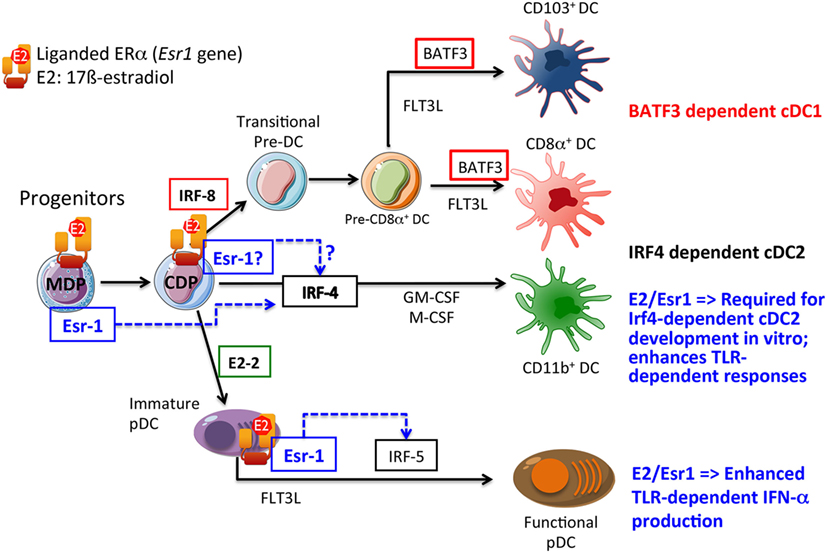
Figure 1. Growth factors and transcription factors regulating conventional dendritic cell (cDC) and plasmacytoid DC (pDC) development. A simplified view of the current transcriptional network in DC development is depicted. The plausible developmental stages where E2/ERα-signaling is likely to regulate DC development and/or functions are shown. The common DC progenitors (CDPs) that originate from the common progenitor for monocytes and DCs [macrophage-DC progenitor (MDP)] produce exclusively the cDC and pDC subsets, but have lost the capacity to generate monocytes. Previous works demonstrated that E2 may act directly in MDP to promote interferon regulatory factor (Irf)-4 expression and GM-DC development. Whether E2 may also signal through ERα at later stage in CDP to promote cDC2 development still remains to be established. In the pDC lineage, E2 is likely to signal through ERα to promote optimal TLR-dependent production of type I interferons (IFNs) through the upregulation of signaling intermediate such as Irf-5, and also possibly through additional unknown mechanisms. Solid arrows indicate connections that have been proven, while dotted arrows indicate direct or indirect relationships.
Even though Csf-2 has been widely used to promote the differentiation of mouse and human hematopoietic progenitors and monocytes into cells resembling mouse splenic DCs (85, 109), it was appreciated from the beginning that this culture system harbors heterogeneous cell populations. These include monocyte/macrophage-like cells (85, 109) and CD11chi CD86hi MHCIIhi DCs, which arise spontaneously in the absence of exogenous danger signals (110). A clarification has been provided recently regarding the origin and phenotypic characterization of the cells generated in such in vitro model system (111).
Based on their hematopoietic origin and the expression of phenotypic markers, it was reported that the CD11c+ MHCII+ cells arising in these cultures were comprised of: (i) cMoP-derived macrophages (GM-Macs) defined as CD11bhi MHCIIint CD115+, and (ii) CDP-derived classical DCs (GM-DCs) expressing high levels of MHCII, PDL-2, and Irf-4 and defined as CD115− CD135+ cells. The latter subset was enriched in BMDC cultures generated with GM-CSF and IL-4, and its development was dependent on Irf-4 expression in CD11c+ cells (112). Thus, GM-DCs may be more closely related to the CD8α− cDC2 subset, consistent with their expression of CD11b and SIRP-α (111).
These results have important implications for the interpretation of earlier studies on the origin (GM-Macs or GM-DCs) of the estrogen-dependent CD11c+ MHCII+ cells that preferentially develop in Csf-2-stimulated bone marrow cultures. In previous investigations by different laboratories, the relative expression of CD11b and Ly6C among CD11c+ cells was used to better define the CD11c+ cell subsets in this model (16, 22), as Ly6C expression appears to represent a reliable marker of inflammatory macrophages, both in vitro and in vivo (21). From initial studies, it was clear that the CD11bhi Ly6C+ population was mostly unchanged by the absence of E2/ERα-signaling. E2, however, preferentially promoted the differentiation of CD11c+ CD11bint cells lacking Ly6C expression, likely including the GM-DC population (16, 22). We corroborated this effect in competition experiments using wild-type and ERα-deficient bone marrow cells, which provided direct evidence for a cell-autonomous function of ERα-signaling in promoting the development of CD11c+ CD11bint Ly6C− cells. By contrast, the CD11bhi Ly6C+ population was reciprocally enriched in estrogen-deprived cultures or in the absence of ERα function in the bone marrow precursors. This cell population may correspond to the “GM-Macs-like” monocyte/macrophage population described by Helft et al. (111). Indeed, we observed a twofold to threefold increase in the occurrence of macrophage-like cells in ERα−/− BMDC cultures (16). This observation correlated with an increased frequency of cells expressing high levels of TLR4-MD2 active complexes and CD11b among ERα−/− CD11c+ cells and estrogen-deprived wild-type BMDCs (16, 22). Accordingly, these cells produced massive amounts of IL-6 and IL-12 upon stimulation with LPS, but not in response to TLR9 or CD40 ligands (16, 24). In striking contrast, ERα-signaling promoted the development of CD11bint Ly6C− CD11c+ cells capable of producing higher levels of pro-inflammatory cytokines (IL-16 and IL-12) upon TLR9 or CD40 activation than ERα−/− CD11c+ cells (16). Moreover, ERα−/− DCs, enriched in GM-Macs, were markedly inferior at stimulating naive CD4+ T cells, when compared to BMDCs from E2-stimulated cultures, when given preprocessed antigenic peptides or unprocessed protein antigen (16). Further studies are warranted to explain how E2 promotes the development of GM-DCs in preference to GM-Macs in this culture model. As suggested in Figure 1, E2 could promote GM-DC generation by selectively acting in CDPs and/or by regulating the earlier transition from MDPs to CDPs.
While canceling estrogen signaling in the GM-CSF culture model had no apparent effect on the development of monocyte/macrophage-like cells, this does not necessarily indicate that monocytes or monocyte-derived cells are insensitive to estrogen in vivo. In fact, by using the LysM-cre deleter model, we provided compelling evidence that ERα signaling in murine monocytes/macrophages was associated in vivo with an enhanced production of pro-inflammatory cytokines by peritoneal macrophages (8). This ERα-dependent effect of E2 on the LPS-driven inflammatory response of macrophages was mediated by inhibition of PI3K activity acting as a negative feedback mechanism of TLR4-induced activation (7, 8). This observation is at odds with other studies indicating that in vivo, estrogens exert pro-inflammatory actions on murine macrophages and microglia (40, 41), whereas ERβ ligands are anti-inflammatory (41). This conclusion, however, does not seem to hold for human monocytes/macrophages, in light of reports describing in vitro E2-mediated inhibition of LPS-driven production of pro-inflammatory cytokines such as IL-6 (113) through the blockade of NF-κB signaling (114). Whether this effect involves classical ERα is unclear. A recent study showed that the 36-kDa isoform of ERα, which excludes the AF-1 and AF-2 domains owing to alternative mRNA splicing, was strongly expressed in human monocytes and could contribute to the inhibition of NF-κB transcriptional activity through direct interaction in the nucleus (115). Besides macrophages, the effects of estrogen signaling on MCs such as monocyte-derived DCs or TipDCs deserve further investigation. Table 1 summarizes known ERα-mediated modulatory effects of estrogen on the biological responses and functions of DC subsets and monocytes-macrophages in different contexts.
IRF-4 is a Key Target of ERα Signaling in the Regulation of DC Biology
Interferon regulatory factor-4 is a key transcription factor that plays an essential role in the regulation of Th2 responses through its function in Th2 cells themselves (116, 117) but also in Treg cells (118), M2 macrophages (119) and, as shown more recently, the cDC2 subset (110, 120). Irf-4 was initially shown to regulate the development of CD11bhi CD8α− DCs in the spleen (121), though it was unclear whether Irf-4 was required early during development, or for continued survival or functional competence of Irf-4+ DCs in tissues. Recently, DC-specific deletion of Irf-4 has been associated with the absence of CD11b+ CD8α− DCs from barrier tissues such as lamina propria, the lung, and the respective draining lymph nodes (122, 123). Mice carrying a DC-specific deficiency for Irf-4 correctly develop dermal CD11b+ CD301b+ PDL2+ DCs in vivo, but these cells fail to localize to the draining lymph nodes (110). This is consistent with the known dependency on Irf-4 for DC migration from the skin to the draining lymph nodes (124).
Kovats and colleagues identified Irf-4 as a key transcription factor whose expression was strongly up-regulated by E2 in Csf-2/Stat-5-stimulated MDPs (17). Retrovirally delivered Irf-4 expression in ERα-/- MDPs restored their potential to develop into CD11b+ Ly6C- GM-DCs, indicating that high levels of Irf-4 can substitute for the requirement for E2/ERα-signaling during Csf-2-mediated DC differentiation (17). Data from our laboratory, however, point to further ERα-dependent pathways contributing to Csf-2-driven DC development (24). Using mice expressing AF-1- or AF-2 domain-defective ERα, we showed that both AF domains were required for early Irf-4 expression in Csf-2-stimulated MDPs (24). Intriguingly, in spite of the absence of Irf-4 upregulation in ERα-AF-10 MDPs, E2 was able to sustain GM-DC development and functional responses to near-equivalent levels relative to wild-type cells at later time points. This observation may explain previous results by others showing substantial E2 effects in GM-DC cultures derived from the first-generation strain of ERα-mutant mice (12), which expressed a truncated form of ERα lacking the AF-1 domain (125), similar to the ERαAF-10 mice used in our study (24). That AF-1 synergizes with AF-2 to enhance Csf-2-dependent expression of Irf-4 in early stage MDPs, but is dispensable at later stages of Irf-4-dependent DC development, suggests indirect mechanisms of Irf-4 regulation by E2-ERα signaling. AF-2 critically controls ERα interactions with most transcription cofactors, but AF-1-specific coactivators have also been described (126). ERα signaling may regulate Irf-4 through the rapid activation of coactivators or corepressors (127), and the induction of other transcription factors whose early expression may require synergistic interactions between both ERα AFs. In the absence of ERα AF1, a longer signaling period could be required for alternative pathways of activation or for the accumulation of rate-limiting factors above the threshold level required to transactivate Irf-4 expression. This would fit in with the delayed upregulation of Irf-4 observed in ERαAF-10 MP cells as compared to ERα-deficient ones, although many other mechanisms could conceivably be at play (24).
Relevance of E2/ERα Signaling in CDCs in vivo
Little is known regarding the role of ERα-signaling in the homeostasis of cDC subsets in lymphoid and non-lymphoid tissues in vivo. Competition experiments in irradiated bone marrow chimeras suggested that newly differentiated splenic CD11c CD11b+ cDCs arose from ERα-proficient bone marrow cells in a ~2:1 ratio to cells derived from the ERα−/− donor (17). This model likely represents a pro-inflammatory environment where both GM-CSF and Flt3L may be elevated, suggesting that ERα-signaling in vivo may promote the development of CD11b+ cDC2 subsets. This conclusion, however, needs further confirmation as few phenotypic and functional markers were used in this study (17).
Experiments by Kovats and colleagues have shown unexpectedly that the skin-derived CD103+ cDCs were enriched in the cutaneous lymph nodes of female mice as compared to males (124). Whether this sex bias was due to estrogens was not addressed specifically, but it was independent from Irf-4 expression, in agreement with the currently accepted notion that CD103+ cDCs in the skin belong to the Batf-3-dependent cDC1 subset. By contrast, the Irf-4-dependent CD11b+ cDC subsets showed no such sex bias (124). Thus, despite strong evidence that E2-ERα-signaling strongly regulates Irf-4-dependent cDC subsets in the in vitro Csf-2 differentiation model, it is still unclear how this relates to particular cDC subsets found in lymphoid or non-lymphoid tissues in vivo, at the steady state, or during inflammation.
Our search of the ImmGen database at http://www.immgen.org (128) showed that lung CD11b+ CD103neg DCs display the highest levels of Esr1 expression among DC subsets. As mentioned above, several studies have shown that a subset of CD11b+ DCs-expressing Irf-4, present in the skin and at the mucosal surfaces in the lung and gut, is crucial in driving CD4+ T cell responses and effector T cell development (129). As ERα-signaling controls the level of Irf-4 in Csf-2-stimulated MDPs, and thereby promotes efficient development of the Irf4-dependent CD11b+ cDC subset in vitro, further investigations are warranted on the impact of cell-intrinsic ERα signaling in cDCs in vivo in the context of immune responses against allergens or viruses. A sex bias has in fact been reported for respiratory pathologies such as allergic asthma and influenza virus infection, which are more severe in females than in males (130, 131). It was also reported recently that E2 treatment increased the frequency of Irf-4+ DCs in the murine vaginal mucosa, associated with enhanced competence of vaginal DCs to prime Th17 cells in vitro. It is unresolved whether this effect of E2 hinged on ERα signaling in DCs (132).
E2-ERα-Signaling Regulates the Flt3L-Dependent DC Subsets, CDCs and PDCs: In vitro Evidence
As mentioned earlier, bone marrow progenitors cultured in the presence of cytokine Flt3L generate the two main conventional DC subsets (cDC1 and cDC2) as well as pDCs. E2 was initially reported to downregulate the development of these Flt3L-driven CD11c+ DCs (FL-DCs) through ERα-signaling in MPs (15). Later studies used new strains of ERα mutant mice that allowed the relative contribution of each AF domain of ERα to be examined (24). It was thus shown that this effect of E2 depended on the AF-1 domain, but not on AF-2. Precise analysis of the DC subsets differentiated in the presence of Flt3L confirmed the previous observation that ERα signaling negatively regulates the absolute numbers of FL-DCs in vitro. This concerned mainly the development of pDCs, whereas cDC numbers were largely unchanged (24). Importantly, cDCs differentiated in the presence of E2 exhibited an enhanced expression of MHC class II and costimulatory molecules, and an increased capacity to produce IL-12 and IL-6 upon combined stimulation through TLR-4 and CD40. Although E2 likewise decreased the absolute numbers of pDCs, these cells exhibited a more mature phenotype and an enhanced capacity to produce the pro-inflammatory cytokine IL-12 in response to TLR9 stimulation. Similar to cDCs, these effects of E2 on pDC biology were missing in ERα-deficient mice as well as in mice defective for the AF-1 function of ERα (24). This requirement for AF-1 may point to a dominant role for the genomic actions of ERα signaling on target genes, but non-genomic mechanisms could also be at play. Studies in mice lacking the MISS function of ERα will help to resolve this issue.
Thus, in contrast to the Csf2-model of cDC2 development, which is strictly dependent of E2-ERα signaling, the development of Flt3L-driven cDCs and pDCs is not abolished in the absence of estrogens or Esr1 expression. Instead, our data are consistent with a key role of estrogens in promoting the development of cDCs and pDCs endowed with a more mature functional phenotype, notably characterized by enhanced TLR-mediated responses.
Functional Impact of E2-ERα Signaling on TLR-Dependent Response of PDCs in Humans
An important issue regards the in vivo relevance of the effects of E2 observed in vitro on the development and effector functions of cDCs and pDCs, and its translation to human DCs. Addressing the hormonal regulation of immune cells in humans is challenging, and very few studies have as yet been reported. The most remarquable observation probably concerns the human pDC subsets, whose capacity to produce IFN-α in response to TLR ligands has been independently reported to be highly regulated by sex-dependent factors (10, 25, 38, 133, 134). Studying large cohorts of healthy donors, Berghöfer and colleagues demonstrated that pDCs in the peripheral blood of women produced more type I IFNs in response to TLR7 ligands than pDCs from men (133). Subsequent studies showed that this sex bias was in fact due to an increased frequency of pDCs capable to secreting IFN-α among PBMCs from women, rather than a sex bias in IFN-α production on a single cell-basis (10, 134). Support for a key role of estrogens in regulating the TLR-mediated response of female pDCs came from observations that pDCs from postmenopausal women exhibited a reduced TLR7-mediated response by comparison with premenopausal women, which was partially reversed by hormone replacement therapy with E2 (10). Indeed, E2-supplementation of postmenopausal women was found to substantially enhance IFN-α production by blood pDCs not only in response to TLR7 stimulation but also to TLR9 ligands. Importantly, enhanced cytokine production by pDCs was elicited not only by synthetic ligands of TLR7 or TLR9 but also by natural ligands such as self nucleic acid-containing immune complexes present in the sera of systemic lupus erythematosus (SLE) patients (10).
Using an in vitro model of Flt3L/IL-7-driven human pDC differentiation from CD34+ HPCs, we showed that blockade of ER signaling by the pure ER antagonist ICI182,780 in developing human pDCs blunted the IFN-α response to TLR7 ligands (38). Unlike their mouse counterparts, however, human pDCs expressed both ER genes, ESR1 and ESR2 (38). It will, therefore, be of importance to investigate the respective contributions of ERα and ERβ to the TLR7-dependent response of human pDCs. The role of either receptor is complex and often mutually antagonistic when expressed in the same cells (135).
Mechanisms of ERα-Mediated Regulation of Type I IFNs in PDCs
Because they target virtually any cell type in the organism, understanding the mechanisms of immune cell regulation by estrogens is often not straightforward. New mouse models have been generated recently to induce conditional deletions of ER in order to discriminate between direct and indirect in vivo effects of estrogens on DC functions (8–10). Using such models, the impact of ER deficiency in specific immune cell types in the context of physiological hormone levels has been assessed (8, 10, 25). These studies pinpointing the cellular targets of estrogens are critical to understanding the molecular mechanisms of ER-regulated immune responses in vivo.
Using mice lacking ERα in CD11c-expressing cells specifically, it was established that ERα expression in pDCs was required for the estrogen-mediated increase of IFN-α (10, 25). This in vivo cell-intrinsic modulation by estrogen of the TLR7- and TLR9-dependent responses in mouse pDCs (10, 25) was corroborated by evidence from in vitro models of murine and human pDC development (24, 38). Altogether, these studies provided new insights into the mechanism of sex bias in the TLR-driven IFN-α production of female pDCs and identified ERα as an attractive target for specific modulation of this pathway.
Interferon-α induction is regulated primarily at the transcriptional level by members of the interferon regulatory factor (Irf) family (136). Human pDCs constitutively express high levels of Irf-5 and Irf-7, and TLR9 engagement in pDCs leads to the activation and phosphorylation of both Irf-5 and Irf-7 (20). Irf-7 is widely recognized as the “master regulator” of type I IFN production (137), while Irf-5 has been shown to be a central mediator of TLR7 signaling in mice (138, 139). In human pDCs, the production of IFN-α through TLR7 or TLR9 stimulation relies on PI3K activities and the phosphorylation of Akt, leading to nuclear translocation of Irf-7 (140). However, a recent study in human pDC leukemic cells challenged the notion of a key regulatory role of Irf-7 in type I IFN production by human pDCs. It was reported that Irf-5, rather than Irf-7, played a non-redundant role in IFN-β and IL-6 production following cell stimulation with TLR9 ligands (36). Thus, while Irf-7 is the master regulator of type I IFN production upon TLR7 or TLR9 stimulation in mouse pDCs, the respective contribution of Irf-7 and Irf-5 to IFN-α induction in human cells is yet to be clarified.
The role of Irf-5 in the differential IFN-α production between females and males was investigated recently (25). Basal levels of Irf-5 were significantly higher in the pDCs from women and positively correlated with the frequency of IFN-α-secreting pDCs upon TLR7 stimulation. Genetic ablation of the Esr1 gene in the hematopoietic compartment or the DC lineage of female mice reduced Irf-5 mRNA expression in pDCs and IFN-α production upon stimulation with TLR7 ligand-containing vesicles. Of note, Irf-7 expression in bone marrow pDCs was not affected by ERα deficiency (25). These results suggest that estrogens regulate pDC type I IFN production through Irf-5, which may act by enhancing IFN-α production in synergy with Irf-7 (141). This would be particularly relevant to human pDCs, whose capacity to produce IFN-β upon TLR9 engagement has been shown to depend strongly on Irf-5, rather than on Irf-7 (36, 141). However, a direct relationship between E2-ERα signaling and Irf-5 expression has yet to be established. In parallel, it has been reported that estrogen signaling in immune cells upregulated the expression of the endoplasmic reticulum transmembrane protein Unc93b1 (142). This chaperone molecule regulates the trafficking of TLR3, TLR7, and TLR9 from the endoplasmic reticulum to the endosomal compartments, and Unc93b1 knockdown abrogates cytokine production (143). However, estrogen-mediated regulation of Unc93b1 was not assessed in pDCs, and it was unclear whether this regulation involved cell-intrinsic ERα signaling (142). The contribution of Unc93b1 to the sex-bias in the TLR-mediated response of pDCs remains an unsettled question.
While a study has suggested a direct regulation of TLR expression through ERα signaling in human monocyte-derived macrophages (144), we and others have failed to detect sex-dependent modulation of TLR7 or TLR9 gene expression in pDCs, at least at the mRNA level in bulk cell populations (38, 133). Confirmation of these results in various TLR7-expressing cells both in mice and human will be necessary, along with analyses of protein expression levels using highly specific antibodies.
Other than TLRs and proteins implicated in their intracellular trafficking, a recent study pointed to ERα-dependent regulation of the cell surface molecule PDC-triggering receptor expressed on myeloid cells (TREM) in pDCs from lupus-prone mice. PDC-TREM is a pDC-specific receptor, a member of the TREM family, which is induced by TLR signaling and mediates IFN-α production (145). ERα deficiency reduced the TLR9-dependent expression of PDC-TREM in pDCs from mice of NZM2410 (lupus-prone) or C57BL/6 genetic background (146). Although PDC-TREM expression is reportedly required for optimal production of type I IFNs by TLR9-stimulated pDCs (145), further studies are needed for any functional links to be established between ERα-dependent regulation of PDC-TREM and changes in the TLR-responsiveness of pDCs.
Despite recent inroads, the mechanistic details of the functional impact of E2-ERα signaling on the TLR-dependent responses of pDCs remain largely unexplored. Apart from the direct regulation of genes implicated in the TLR signaling pathway, ERα could control the expression of key pDC transcription factors, such as E2-2, Spi-B, Irf-8, Irf-7, and Ikaros. One would also expect major effects on the development of pDCs in the bone marrow. This raises exciting questions for investigators, because understanding the mechanisms that promote pDC development and regulate their peripheral function will be key to manipulating this cellular compartment through ER signaling to enforce homeostasis and fight chronic inflammation. The sexual dimorphism in type I IFN production by pDCs could indeed be implicated in the increased susceptibility of women to autoimmunity, and in the sex differences vis-à-vis the acquisition and clinical course of certain viral diseases (1).
Concluding Remarks
Evidence is emerging that estrogens, through their nuclear receptor ERα, regulate various facets of cDC and pDC develoment and TLR-dependent responses in humans and mice. However, the full demonstration of a direct involvement of this pathway in the sex-biased susceptibility to autoimmunity or infection remains a work in progress. Genetic and cell type-specific Esr1 deletion strategies have come to the fore as critical tools, and further investigations regarding genes regulated by estrogens in DCs are eagerly awaited. Such unbiased approaches should shed light on the molecular mechanisms set in motion by cell-intrinsic E2-ERα signaling in DCs in the physiological context of the female environment. Another important issue concerns the optimal experimental models to study the role of sex hormones. Although standard estrogen supplementation is useful in addressing certain questions, it does not mirror the estrus cycle and, because of the pleiotropic actions of sexual hormones, may introduce confounding factors, especially in integrated models of autoimmune or infectious diseases. The physiological context of natural estrogen production during the estrus cycle should, therefore, be preferred over experimental models of E2-supplementation, as the only natural instance of sustained steroid hormone production is pregnancy.
The pDC subset appears to be subjected to estrogen-dependent regulation, notably in regard of its functional capacity to produce type I IFNs in response to TLR7 ligands (133, 134). Evidence has arisen that E2-ERα signaling and X-linked genetic factors independently contribute to the strong sex bias in the TLR7-driven response of pDCs (10, 38). Cell type-specific strategies of genetic ablation have recently demonstrated the pivotal role of pDCs in the development of autoantibodies and in the progression of lupus-like syndrome (147, 148). Targeting the ER signaling pathway in pDCs could represent a valuable therapeutic approach to limit the pathogenic production of type I IFN, particularly during the initial stages of SLE. Indeed, early studies suggested beneficial effects of RE antagonists or SERMs in mouse models of lupus (149, 150). Encouraging results were reported from a clinical double-blind trial of fulvestrant (ICI182,780) in women with SLE (151). In patients treated for 12 months, an improvement in clinical signs was observed, associated with a reduction of medication dosage (151). Together with our in vitro results showing ICI182,780 to downregulate the TLR7-type I IFN pathway in developing human pDCs, these clinical outcomes strongly suggest that ER signaling could be amenable to pharmacological manipulation in pDCs. Along this line, we reported earlier that E2 supplementation in postmenopausal women enhances the TLR7- and TLR9-dependent production of type I IFNs (10).
In addition to the well-established sex bias in the incidence and severity of autoimmune diseases, sex differences in the response to viral vaccines have been also clearly documented in humans and in mice (130, 152). Given the central role of DCs in the initiation of adaptive immunity, studying how sex-related factors could regulate the homeostasis and responses of tissue-resident DC subsets at steady state and in the context of vaccination using attenuated viruses or vaccine sub-units is of critical importance. For instance, prophylactic vaccination with HSV-2 glycoprotein D in adjuvant exhibited efficacy in women, but not in men (153). More recently, systems biology approaches have shown that expression of genes associated with TLR and IFN pathways immediately after vaccination with yellow fever virus 17D predicted subsequent adaptive immune responses (154, 155). Interestingly, re-analysis of these data according to the sex of the subjects demonstrated that most of the TLR-associated genes that activate the IFN pathway were upregulated to a greater extent in women as compared to men during the first 10 days after vaccination (130). Although this may suggest that disease protection might be greater in women than in men, this also indicates that the enhanced innate immune response to YF17FD vaccine might also underlie the increased incidence of side-effects in women (130). These examples illustrate the importance of understanding how sex-linked factors could contribute to regulate innate immunity, and particularly DC biology. This may help to explain and predict the heterogeneous responses to vaccines in humans according to sex, not only in the context of infectious diseases but also in cancer immunotherapy.
Author Contributions
SL, CS, and J-CG participated in the conception and the writing of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Arthritis Fondation Courtin, Fondation ARC pour la Recherche sur le Cancer, Fondation pour la Recherche Médicale (DEQ2000329169), Conseil Régional Midi-Pyrénées, and the French National Agency for Research on AIDS and Viral Hepatitis (ANRS). SL has been supported by fellowships from Association pour la Recherche sur la Sclérose en Plaques (ARSEP). CS is supported by fellowship and grant from the National Health and Medical Research Council (NHMCR, APP1123000 and APP1098832). We wish to thank Dr. JE Méjia for critical readings of the manuscript.
References
1. Markle JG, Fish EN. Sex matters in immunity. Trends Immunol (2014) 35:97–104. doi: 10.1016/j.it.2013.10.006
2. Whitacre CC. Sex differences in autoimmune disease. Nat Immunol (2001) 2:777–80. doi:10.1038/ni0901-777
3. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev (2007) 87:905–31. doi:10.1152/physrev.00026.2006
4. Maret A, Coudert JD, Garidou L, Foucras G, Gourdy P, Krust A, et al. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor a expression in hematopoietic cells. Eur J Immunol (2003) 33:512–21. doi:10.1002/immu.200310027
5. Gourdy P, Araujo LM, Zhu R, Garmy-Susini B, Diem S, Laurell H, et al. Relevance of sexual dimorphism to regulatory T cells: estradiol promotes IFN-{gamma} production by invariant natural killer T cells. Blood (2005) 105:2415–20. doi:10.1182/blood-2004-07-2819
6. Delpy L, Douin-Echinard V, Garidou L, Bruand C, Saoudi A, Guery JC. Estrogen enhances susceptibility to experimental autoimmune myasthenia gravis by promoting type 1-polarized immune responses. J Immunol (2005) 175:5050–7. doi:10.4049/jimmunol.175.8.5050
7. Calippe B, Douin-Echinard V, Laffargue M, Laurell H, Rana-Poussine V, Pipy B, et al. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol 3-kinase pathway. J Immunol (2008) 180:7980–8. doi:10.4049/jimmunol.180.12.7980
8. Calippe B, Douin-Echinard V, Delpy L, Laffargue M, Lelu K, Krust A, et al. 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. J Immunol (2010) 185:1169–76. doi:10.4049/jimmunol.0902383
9. Lelu K, Laffont S, Delpy L, Paulet PE, Perinat T, Tschanz SA, et al. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol (2011) 187:2386–93. doi:10.4049/jimmunol.1101578
10. Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood (2012) 119:454–64. doi:10.1182/blood-2011-08-371831
11. Medina KL, Garrett KP, Thompson LF, Rossi MI, Payne KJ, Kincade PW. Identification of very early lymphoid precursors in bone marrow and their regulation by estrogen. Nat Immunol (2001) 2:718–24. doi:10.1038/90659
12. Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c(+) CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol (2004) 172:1426–36. doi:10.4049/jimmunol.172.3.1426
13. Harman BC, Miller JP, Nikbakht N, Gerstein R, Allman D. Mouse plasmacytoid dendritic cells derive exclusively from estrogen-resistant myeloid progenitors. Blood (2006) 108:878–85. doi:10.1182/blood-2005-11-4545
14. Welner RS, Pelayo R, Garrett KP, Chen X, Perry SS, Sun XH, et al. Interferon-producing killer dendritic cells (IKDCs) arise via a unique differentiation pathway from primitive c-kitHiCD62L+ lymphoid progenitors. Blood (2007) 109:4825–931. doi:10.1182/blood-2006-08-043810
15. Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligand-mediated dendritic cell differentiation. J Immunol (2008) 180:727–38. doi:10.4049/jimmunol.180.2.727
16. Douin-Echinard V, Laffont S, Seillet C, Delpy L, Krust A, Chambon P, et al. Estrogen receptor {alpha}, but not {beta}, is required for optimal dendritic cell differentiation and CD40-induced cytokine production. J Immunol (2008) 180:3661–9. doi:10.4049/jimmunol.180.10.7047-b
17. Carreras E, Turner S, Frank MB, Knowlton N, Osban J, Centola M, et al. Estrogen receptor signaling promotes dendritic cell differentiation by increasing expression of the transcription factor IRF4. Blood (2010) 115:238–46. doi:10.1182/blood-2009-08-236935
18. Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nat Immunol (2012) 13:1145–54. doi:10.1038/ni.2467
19. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14:571–8. doi:10.1038/nri3712
20. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol (2008) 8:594–606. doi:10.1038/nri2358
21. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
22. Mao A, Paharkova-Vatchkova V, Hardy J, Miller MM, Kovats S. Estrogen selectively promotes the differentiation of dendritic cells with characteristics of Langerhans cells. J Immunol (2005) 175:5146–51. doi:10.4049/jimmunol.175.8.5146
23. Nalbandian G, Paharkova-Vatchkova V, Mao A, Nale S, Kovats S. The selective estrogen receptor modulators, tamoxifen and raloxifene, impair dendritic cell differentiation and activation. J Immunol (2005) 175:2666–75. doi:10.4049/jimmunol.175.4.2666
24. Seillet C, Rouquie N, Foulon E, Douin-Echinard V, Krust A, Chambon P, et al. Estradiol promotes functional responses in inflammatory and steady-state dendritic cells through differential requirement for activation function-1 of estrogen receptor alpha. J Immunol (2013) 190:5459–70. doi:10.4049/jimmunol.1203312
25. Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, et al. Sex differences in plasmacytoid dendritic cell levels of IRF5 drive higher IFN-alpha production in women. J Immunol (2015) 195:5327–36. doi:10.4049/jimmunol.1501684
26. Yu W, Zheng H, Lin W, Tajima A, Zhang Y, Zhang X, et al. Estrogen promotes leydig cell engulfment by macrophages in male infertility. J Clin Invest (2014) 124:2709–21. doi:10.1172/JCI59901
27. Lambert KC, Curran EM, Judy BM, Lubahn DB, Estes DM. Estrogen receptor-alpha deficiency promotes increased TNF-alpha secretion and bacterial killing by murine macrophages in response to microbial stimuli in vitro. J Leukoc Biol (2004) 75:1166–72. doi:10.1189/jlb.1103589
28. Campbell L, Emmerson E, Williams H, Saville CR, Krust A, Chambon P, et al. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J Invest Dermatol (2014) 134:2447–57. doi:10.1038/jid.2014.175
29. Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, et al. Myeloid-specific estrogen receptor alpha deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci U S A (2011) 108:16457–62. doi:10.1073/pnas.1104533108
30. Nilsson S, Koehler KF, Gustafsson JA. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov (2011) 10:778–92. doi:10.1038/nrd3551
31. Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem (2001) 276:36869–72. doi:10.1074/jbc.R100029200
32. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol (2015) 294:63–9. doi:10.1016/j.cellimm.2015.01.018
33. Igarashi H, Kouro T, Yokota T, Comp PC, Kincade PW. Age and stage dependency of estrogen receptor expression by lymphocyte precursors. Proc Natl Acad Sci U S A (2001) 98:15131–6. doi:10.1073/pnas.011513098
34. Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood (2009) 114:2236–43. doi:10.1182/blood-2008-09-178871
35. Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, et al. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature (2014) 505:555–8. doi:10.1038/nature12932
36. Steinhagen F, McFarland AP, Rodriguez LG, Tewary P, Jarret A, Savan R, et al. IRF-5 and NF-kappaB p50 co-regulate IFN-beta and IL-6 expression in TLR9-stimulated human plasmacytoid dendritic cells. Eur J Immunol (2013) 43:1896–906. doi:10.1002/eji.201242792
37. Komi J, Lassila O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood (2000) 95:2875–82.
38. Laffont S, Rouquie N, Azar P, Seillet C, Plumas J, Aspord C, et al. X-chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-alpha production of plasmacytoid dendritic cells from women. J Immunol (2014) 193:5444–52. doi:10.4049/jimmunol.1303400
39. Phiel KL, Henderson RA, Adelman SJ, Elloso MM. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol Lett (2005) 97:107–13. doi:10.1016/j.imlet.2004.10.007
40. Soucy G, Boivin G, Labrie F, Rivest S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J Immunol (2005) 174:6391–8. doi:10.4049/jimmunol.174.10.6391
41. Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell (2011) 145:584–95. doi:10.1016/j.cell.2011.03.050
42. Du S, Sandoval F, Trinh P, Umeda E, Voskuhl R. Estrogen receptor-beta ligand treatment modulates dendritic cells in the target organ during autoimmune demyelinating disease. Eur J Immunol (2011) 41:140–50. doi:10.1002/eji.201040796
43. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol (2013) 14:1054–63. doi:10.1038/ni.2695
44. Kelley ST, Thackray VG. Phylogenetic analyses reveal ancient duplication of estrogen receptor isoforms. J Mol Evol (1999) 49:609–14. doi:10.1007/PL00006582
45. Krust A, Green S, Argos P, Kumar V, Walter P, Bornert JM, et al. The chicken oestrogen receptor sequence: homology with v-erbA and the human oestrogen and glucocorticoid receptors. EMBO J (1986) 5:891–7.
46. Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell (1989) 59:477–87. doi:10.1016/0092-8674(89)90031-7
47. Hewitt SC, Winuthayanon W, Korach KS. What’s new in estrogen receptor action in the female reproductive tract. J Mol Endocrinol (2016) 56:R55–71. doi:10.1530/JME-15-0254
48. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest (2006) 116:561–70. doi:10.1172/JCI27987
49. Chen HM, Pahl HL, Scheibe RJ, Zhang DE, Tenen DG. The Sp1 transcription factor binds the CD11b promoter specifically in myeloid cells in vivo and is essential for myeloid-specific promoter activity. J Biol Chem (1993) 268:8230–9.
50. Friedman AD. Transcriptional regulation of granulocyte and monocyte development. Oncogene (2002) 21:3377–90. doi:10.1038/sj.onc.1205324
51. Liu MH, Cheung E. Estrogen receptor-mediated long-range chromatin interactions and transcription in breast cancer. Mol Cell Endocrinol (2014) 382:624–32. doi:10.1016/j.mce.2013.09.019
52. Hah N, Kraus WL. Hormone-regulated transcriptomes: lessons learned from estrogen signaling pathways in breast cancer cells. Mol Cell Endocrinol (2014) 382:652–64. doi:10.1016/j.mce.2013.06.021
53. Mann M, Cortez V, Vadlamudi RK. Epigenetics of estrogen receptor signaling: role in hormonal cancer progression and therapy. Cancers (Basel) (2011) 3:1691–707. doi:10.3390/cancers3021691
54. Magnani L, Lupien M. Chromatin and epigenetic determinants of estrogen receptor alpha (ESR1) signaling. Mol Cell Endocrinol (2014) 382:633–41. doi:10.1016/j.mce.2013.04.026
55. Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Canc (2013) 14:26–38. doi:10.1038/nrc3671
56. Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J (1990) 9:2811–8.
57. Metzger D, Losson R, Bornert JM, Lemoine Y, Chambon P. Promoter specificity of the two transcriptional activation functions of the human oestrogen receptor in yeast. Nucleic Acids Res (1992) 20:2813–7. doi:10.1093/nar/20.11.2813
58. Merot Y, Metivier R, Penot G, Manu D, Saligaut C, Gannon F, et al. The relative contribution exerted by AF-1 and AF-2 transactivation functions in estrogen receptor alpha transcriptional activity depends upon the differentiation stage of the cell. J Biol Chem (2004) 279:26184–91. doi:10.1074/jbc.M402148200
59. Arnal JF, Lenfant F, Flouriot G, Tremollieres F, Laurell H, Fontaine C, et al. From in vivo gene targeting of oestrogen receptors to optimization of their modulation in menopause. Br J Pharmacol (2012) 165:57–66. doi:10.1111/j.1476-5381.2011.01538.x
60. Billon-Gales A, Fontaine C, Filipe C, Douin-Echinard V, Fouque MJ, Flouriot G, et al. The transactivating function 1 of estrogen receptor alpha is dispensable for the vasculoprotective actions of 17beta-estradiol. Proc Natl Acad Sci U S A (2009) 106:2053–8. doi:10.1073/pnas.0808742106
61. Billon-Gales A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, et al. Activation function 2 (AF2) of estrogen receptor-{alpha} is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci U S A (2011) 108:13311–6. doi:10.1073/pnas.1105632108
62. Borjesson AE, Windahl SH, Lagerquist MK, Engdahl C, Frenkel B, Moverare-Skrtic S, et al. Roles of transactivating functions 1 and 2 of estrogen receptor-alpha in bone. Proc Natl Acad Sci U S A (2011) 108:6288–93. doi:10.1073/pnas.1100454108
63. Fujita T, Kobayashi Y, Wada O, Tateishi Y, Kitada L, Yamamoto Y, et al. Full activation of estrogen receptor alpha activation function-1 induces proliferation of breast cancer cells. J Biol Chem (2003) 278:26704–14. doi:10.1074/jbc.M301031200
64. Kim KH, Bender JR. Membrane-initiated actions of estrogen on the endothelium. Mol Cell Endocrinol (2009) 308:3–8. doi:10.1016/j.mce.2009.03.025
65. Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER. Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol (2003) 23:1633–46. doi:10.1128/MCB.23.5.1633-1646.2003
66. Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, et al. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell (2005) 16:231–7. doi:10.1091/mbc.E04-07-0547
67. Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A (2014) 111:E283–90. doi:10.1073/pnas.1322057111
68. Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science (2006) 311:83–7. doi:10.1126/science.1117729
69. Sathe P, Metcalf D, Vremec D, Naik SH, Langdon WY, Huntington ND, et al. Lymphoid tissue and plasmacytoid dendritic cells and macrophages do not share a common macrophage-dendritic cell-restricted progenitor. Immunity (2014) 41:104–15. doi:10.1016/j.immuni.2014.05.020
70. Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol (2007) 8:1217–26. doi:10.1038/ni1522
71. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity (2009) 31:823–33. doi:10.1016/j.immuni.2009.08.027
72. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1283–92. doi:10.1084/jem.20100223
73. den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(-) dendritic cells in vivo. J Exp Med (2002) 196:817–27. doi:10.1084/jem.20020295
74. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi:10.1126/science.1164206
75. Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med (2010) 207:823–36. doi:10.1084/jem.20091627
76. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206:3115–30. doi:10.1084/jem.20091756
77. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity (2009) 31:513–25. doi:10.1016/j.immuni.2009.08.010
78. Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol (2005) 174:2573–81. doi:10.4049/jimmunol.174.5.2573
79. Wu L, D’Amico A, Winkel KD, Suter M, Lo D, Shortman K. RelB is essential for the development of myeloid-related CD8alpha- dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity (1998) 9:839–47. doi:10.1016/S1074-7613(00)80649-4
80. Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med (1997) 185:1101–11. doi:10.1084/jem.185.6.1101
81. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284:1835–7. doi:10.1126/science.284.5421.1835
82. Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol (2001) 2:1144–50. doi:10.1038/ni736
83. Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol (2003) 171:6466–77. doi:10.4049/jimmunol.171.12.6466
84. Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol (2000) 165:6037–46. doi:10.4049/jimmunol.165.11.6037
85. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med (1992) 176:1693–702. doi:10.1084/jem.176.6.1693
86. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med (1994) 179:1109–18. doi:10.1084/jem.179.4.1109
87. Schlitzer A, McGovern N, Ginhoux F. Dendritic cells and monocyte-derived cells: two complementary and integrated functional systems. Semin Cell Dev Biol (2015) 41:9–22. doi:10.1016/j.semcdb.2015.03.011
88. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity (2003) 19:59–70. doi:10.1016/S1074-7613(03)00171-7
89. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against leishmania. Immunity (2007) 26:519–31. doi:10.1016/j.immuni.2007.01.017
90. Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol (2009) 10:394–402. doi:10.1038/ni.1707
91. Ko HJ, Brady JL, Ryg-Cornejo V, Hansen DS, Vremec D, Shortman K, et al. GM-CSF-responsive monocyte-derived dendritic cells are pivotal in Th17 pathogenesis. J Immunol (2014) 192:2202–9. doi:10.4049/jimmunol.1302040
92. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38:336–48. doi:10.1016/j.immuni.2012.10.018
93. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143:416–29. doi:10.1016/j.cell.2010.09.039
94. Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med (2012) 209:1153–65. doi:10.1084/jem.20112675
96. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol (2007) 7:19–30. doi:10.1038/nri1996
97. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity (2012) 36:1031–46. doi:10.1016/j.immuni.2012.03.027
98. Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol (2013) 34:81–9. doi:10.1016/j.it.2012.08.006
99. Louis C, Cook AD, Lacey D, Fleetwood AJ, Vlahos R, Anderson GP, et al. Specific contributions of CSF-1 and GM-CSF to the dynamics of the mononuclear phagocyte system. J Immunol (2015) 195:134–44. doi:10.4049/jimmunol.1500369
100. Campbell IK, van Nieuwenhuijze A, Segura E, O’Donnell K, Coghill E, Hommel M, et al. Differentiation of inflammatory dendritic cells is mediated by NF-kappaB1-dependent GM-CSF production in CD4 T cells. J Immunol (2011) 186:5468–77. doi:10.4049/jimmunol.1002923
101. van Nieuwenhuijze AE, Coghill E, Gray D, Prato S, Metcalf D, Alexander WS, et al. Transgenic expression of GM-CSF in T cells causes disseminated histiocytosis. Am J Pathol (2014) 184:184–99. doi:10.1016/j.ajpath.2013.09.014
102. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12:560–7. doi:10.1038/ni.2027
103. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12:568–75. doi:10.1038/ni.2031
104. King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med (2010) 207:953–61. doi:10.1084/jem.20091844
105. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med (2008) 205:2281–94. doi:10.1084/jem.20071119
106. Kovats S, Carreras E. Regulation of dendritic cell differentiation and function by estrogen receptor ligands. Cell Immunol (2008) 252:81–90. doi:10.1016/j.cellimm.2007.10.008
107. Nalbandian G, Kovats S. Understanding sex biases in immunity: effects of estrogen on the differentiation and function of antigen-presenting cells. Immunol Res (2005) 31:91–106. doi:10.1385/IR:31:2:091
108. Bebo BF Jr, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol (2001) 166:2080–9. doi:10.4049/jimmunol.166.3.2080
109. Lutz MB, Kukutsch N, Ogilvie AL, Rößner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Meth (1999) 223:77–92. doi:10.1016/S0022-1759(98)00204-X
110. Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity (2013) 39:722–32. doi:10.1016/j.immuni.2013.08.028
111. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity (2015) 42:1197–211. doi:10.1016/j.immuni.2015.05.018
112. Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol (2014) 15:161–7. doi:10.1038/ni.2795
113. Vegeto E, Ghisletti S, Meda C, Etteri S, Belcredito S, Maggi A. Regulation of the lipopolysaccharide signal transduction pathway by 17beta-estradiol in macrophage cells. J Steroid Biochem Mol Biol (2004) 91:59–66. doi:10.1016/j.jsbmb.2004.02.004
114. Ghisletti S, Meda C, Maggi A, Vegeto E. 17{beta}-estradiol inhibits inflammatory gene expression by controlling NF-{kappa}B intracellular localization. Mol Cell Biol (2005) 25:2957–68. doi:10.1128/MCB.25.8.2957-2968.2005
115. Pelekanou V, Kampa M, Kiagiadaki F, Deli A, Theodoropoulos P, Agrogiannis G, et al. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERalpha36 and GPR30/GPER1. J Leukoc Biol (2016) 99:333–47. doi:10.1189/jlb.3A0914-430RR
116. Lohoff M, Mittrucker HW, Prechtl S, Bischof S, Sommer F, Kock S, et al. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc Natl Acad Sci U S A (2002) 99:11808–12. doi:10.1073/pnas.182425099
117. Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med (2002) 195:1003–12. doi:10.1084/jem.20011128
118. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458:351–6. doi:10.1038/nature07674
119. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol (2010) 11:936–44. doi:10.1038/ni.1920
120. Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity (2013) 39:733–43. doi:10.1016/j.immuni.2013.08.029
121. Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha-dendritic cell development. Proc Natl Acad Sci U S A (2004) 101:8981–6. doi:10.1073/pnas.0402139101
122. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38:970–83. doi:10.1016/j.immuni.2013.04.011
123. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hagerbrand K, Marsal J, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity (2013) 38:958–69. doi:10.1016/j.immuni.2013.03.009
124. Bajana S, Roach K, Turner S, Paul J, Kovats S. IRF4 promotes cutaneous dendritic cell migration to lymph nodes during homeostasis and inflammation. J Immunol (2012) 189:3368–77. doi:10.4049/jimmunol.1102613
125. Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, et al. The AF-1 activation-function of ERalpha may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci U S A (2002) 99:2205–10. doi:10.1073/pnas.042688499
126. Arnal JF, Valera MC, Payrastre B, Lenfant F, Gourdy P. Structure-function relationship of estrogen receptors in cardiovascular pathophysiological models. Thromb Res (2012) 130(Suppl 1):S7–11. doi:10.1016/j.thromres.2012.08.261
127. Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell (2007) 27:691–700. doi:10.1016/j.molcel.2007.08.012
128. Heng TS, Painter MW; Immunological Genome Project Consortium. The immunological genome project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
129. Flutter B, Nestle FO. What on “irf” is this gene 4? Irf4 transcription-factor-dependent dendritic cells are required for T helper 2 cell responses in murine skin. Immunity (2013) 39:625–7. doi:10.1016/j.immuni.2013.09.008
130. Klein SL, Jedlicka A, Pekosz A. The Xs and Y of immune responses to viral vaccines. Lancet Infect Dis (2010) 10:338–49. doi:10.1016/S1473-3099(10)70049-9
131. Keselman A, Heller N. Estrogen signaling modulates allergic inflammation and contributes to sex differences in asthma. Front Immunol (2015) 6:568. doi:10.3389/fimmu.2015.00568
132. Anipindi VC, Bagri P, Roth K, Dizzell SE, Nguyen PV, Shaler CR, et al. Estradiol enhances CD4+ T-cell anti-viral immunity by priming vaginal DCs to induce Th17 responses via an IL-1-dependent pathway. PLoS Pathog (2016) 12:e1005589. doi:10.1371/journal.ppat.1005589
133. Berghöfer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN-alpha production in females. J Immunol (2006) 177:2088–96. doi:10.4049/jimmunol.177.4.2088
134. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med (2009) 15:955–9. doi:10.1038/nm.2004
135. Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer (2011) 11:597–608. doi:10.1038/nrc3093
136. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25:349–60. doi:10.1016/j.immuni.2006.08.009
137. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7. doi:10.1038/nature03464
138. Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem (2005) 280:17005–12. doi:10.1074/jbc.M412584200
139. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by toll-like receptors. Nature (2005) 434:243–9. doi:10.1038/nature03308
140. Guiducci C, Ghirelli C, Marloie-Provost MA, Matray T, Coffman RL, Liu YJ, et al. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J Exp Med (2008) 205:315–22. doi:10.1084/jem.20070763
141. Pelka K, Latz E. IRF5, IRF8, and IRF7 in human pDCs – the good, the bad, and the insignificant? Eur J Immunol (2013) 43:1693–7. doi:10.1002/eji.201343739
142. Panchanathan R, Liu H, Choubey D. Expression of murine Unc93b1 is up-regulated by interferon and estrogen signaling: implications for sex bias in the development of autoimmunity. Int Immunol (2013) 25:521–9. doi:10.1093/intimm/dxt015
143. Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via toll-like receptors 3, 7 and 9. Nat Immunol (2006) 7:156–64. doi:10.1038/ni1297
144. Young NA, Wu LC, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MV, et al. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNalpha-independent mechanism of sex-bias in systemic lupus erythematosus. Clin Immunol (2014) 151:66–77. doi:10.1016/j.clim.2014.01.006
145. Watarai H, Sekine E, Inoue S, Nakagawa R, Kaisho T, Taniguchi M. PDC-TREM, a plasmacytoid dendritic cell-specific receptor, is responsible for augmented production of type I interferon. Proc Natl Acad Sci U S A (2008) 105:2993–8. doi:10.1073/pnas.0710351105
146. Scott JL, Cunningham MA, Naga OS, Wirth JR, Eudaly JG, Gilkeson GS. Estrogen receptor alpha deficiency modulates TLR ligand-mediated PDC-TREM expression in plasmacytoid dendritic cells in lupus-prone mice. J Immunol (2015) 195(12):5561–71. doi:10.4049/jimmunol.1500315
147. Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med (2014) 211:1969–76. doi:10.1084/jem.20132522
148. Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med (2014) 211:1977–91. doi:10.1084/jem.20132620
149. Sthoeger ZM, Zinger H, Mozes E. Beneficial effects of the anti-oestrogen tamoxifen on systemic lupus erythematosus of (NZBxNZW)F1 female mice are associated with specific reduction of IgG3 autoantibodies. Ann Rheum Dis (2003) 62:341–6. doi:10.1136/ard.62.4.341
150. Wu WM, Lin BF, Su YC, Suen JL, Chiang BL. Tamoxifen decreases renal inflammation and alleviates disease severity in autoimmune NZB/W F1 mice. Scand J Immunol (2000) 52:393–400. doi:10.1046/j.1365-3083.2000.00789.x
151. Abdou NI, Rider V, Greenwell C, Li X, Kimler BF. Fulvestrant (Faslodex), an estrogen selective receptor downregulator, in therapy of women with systemic lupus erythematosus. clinical, serologic, bone density, and T cell activation marker studies: a double-blind placebo-controlled trial. J Rheumatol (2008) 35:797.
152. Klein SL. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays (2012) 34:1050–9. doi:10.1002/bies.201200099
153. Stanberry LR, Spruance SL, Cunningham AL, Bernstein DI, Mindel A, Sacks S, et al. Glycoprotein-d-adjuvant vaccine to prevent genital herpes. N Engl J Med (2002) 347:1652–61. doi:10.1056/NEJMoa011915
154. Gaucher D, Therrien R, Kettaf N, Angermann BR, Boucher G, Filali-Mouhim A, et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med (2008) 205:3119–31. doi:10.1084/jem.20082292
Keywords: sex differences, dendritic cells, estrogens, estrogen receptors, toll-like receptors
Citation: Laffont S, Seillet C and Guéry J-C (2017) Estrogen Receptor-Dependent Regulation of Dendritic Cell Development and Function. Front. Immunol. 8:108. doi: 10.3389/fimmu.2017.00108
Received: 14 September 2016; Accepted: 23 January 2017;
Published: 10 February 2017
Edited by:
Manfred B. Lutz, University of Würzburg, GermanyReviewed by:
Meredith O’Keeffe, Monash University, AustraliaPieter J. M. Leenen, Erasmus University Rotterdam, Netherlands
Copyright: © 2017 Laffont, Seillet and Guéry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Laffont, c29waGllLmxhZmZvbnQtcHJhZGluZXNAaW5zZXJtLmZy;
Jean-Charles Guéry, amVhbi1jaGFybGVzLmd1ZXJ5QGluc2VybS5mcg==