 Jenny A. Hyde
Jenny A. Hyde- Department of Microbial Pathogenesis and Immunology, College of Medicine, Texas A&M Health Science Center, Bryan, TX, USA
Borrelia burgdorferi is the etiological agent of Lyme disease, a multisystemic, multistage, inflammatory infection resulting in patients experiencing cardiac, neurological, and arthritic complications when not treated with antibiotics shortly after exposure. The spirochetal bacterium transmits through the Ixodes vector colonizing the dermis of a mammalian host prior to hematogenous dissemination and invasion of distal tissues all the while combating the immune response as it traverses through its pathogenic lifecycle. The innate immune response controls the borrelial burden in the dermis, but is unable to clear the infection and thereby prevent progression of disease. Dissemination in the mammalian host requires temporal regulation of virulence determinants to allow for vascular interactions, invasion, and colonization of distal tissues. Virulence determinants and/or adhesins are highly heterogenetic among environmental B. burgdorferi strains with particular genotypes being associated with the ability to disseminate to specific tissues and the severity of disease, but fail to generate cross-protective immunity between borrelial strains. The unique motility of B. burgdorferi rendered by the endoflagella serves a vital function for dissemination and protection from immune recognition. Progress has been made toward understanding the chemotactic regulation coordinating the activity of the two polar localized flagellar motors and their role in borrelial virulence, but this regulation is not yet fully understood. Distinct states of motility allow for dynamic interactions between several B. burgdorferi adhesins and host targets that play roles in transendothelial migration. Transmigration across endothelial and blood–brain barriers allows for the invasion of tissues and elicits localized immune responses. The invasive nature of B. burgdorferi is lacking in proactive mechanisms to modulate disease, such as secretion systems and toxins, but recent work has shown degradation of host extracellular matrices by B. burgdorferi contributes to the invasive capabilities of the pathogen. Additionally, B. burgdorferi may use invasion of eukaryotic cells for immune evasion and protection against environmental stresses. This review provides an overview of B. burgdorferi mechanisms for dissemination and invasion in the mammalian host, which are essential for pathogenesis and the development of persistent infection.
Introduction
Lyme disease, the leading tick-borne infection in the United States, occurs in multiple stages and is a multisystemic disease due to the etiologic agent Borrelia burgdorferi (1–4). This disease was first recognized during the 1970s when a cluster of rheumatoid-like arthritis cases occurred in patients who were predominantly children in Lyme, Connecticut (5). Lyme disease has become a significant emerging infectious disease with the Centers for Disease Control (CDC) estimating approximately 300,000 new cases in 2013 in the United States (6). The spirochetal bacterium is transmitted when an Ixodes tick vector colonized with B. burgdorferi takes a blood meal on reservoir mammals, such as small rodents and birds, or accidental human hosts, resulting in the colonization of dermal tissue and develops into a localized infection (7). This earliest stage of Lyme disease is characterized by a painless bulls-eye rash, called an erythema migrans, experienced by approximately 70–80% of patients at the site of the tick bite (1–3). In the absence of the distinctive erythema migrans, Lyme disease can be difficult to diagnose due to non-specific flu-like symptoms including headache, neck stiffness, malaise, fatigue, myalgia, and fever. During localized infection, the number of B. burgdorferi cells increases in the dermal tissue in preparation for dissemination to sites of secondary colonization. Days to weeks after infection, B. burgdorferi progresses onto a disseminated stage when the pathogen travels away from the site of the tick bite through the bloodstream and/or lymphatic system to invade and colonize various tissues, such as the heart, synovial fluid of joints, and the nervous system. Patients can develop secondary erythema migrans at distal locations on the skin from the original site of infection. Late infection, the third stage of disease, develops months to years after exposure to B. burgdorferi and patients can experience different manifestations including neuroborreliosis, Lyme carditis, and/or arthritis. Following antibiotic treatment, a subset of patients continues to present with arthritic symptoms that has been designated postinfectious, antibiotic-refractory Lyme arthritis (8).
Borrelia burgdorferi is a gram-negative “like” bacterium that is a member of the Spirochaetaceae family known by its distinctive spiral morphology and motility propelled by the endoflagellum (9, 10). Motility of B. burgdorferi is essential for pathogenesis throughout the enzootic cycle, though swimming traits differ greatly in the Ixodes tick than that observed in the mammalian host (11, 12). The segmented genome of B. burgdorferi is unlike most bacterial pathogens consisting of a 910,725 bp linear chromosome along with up to 23 circular and linear plasmids ranging in size from 5 to 56 kb (13, 14). The borrelial genome is minimal in size relative to other bacterial pathogens but is also limited in genes annotated to encode metabolic capabilities, virulence, and defense mechanisms. The glycolytic pathway is the only complete metabolic pathway identified in the annotated genome while pathways for generating energy, amino acids, and lipid synthesis, to name a few, are noticeably incomplete or absent (15, 16). B. burgdorferi scavenges from its environment to fulfill basic needs for survival and the pathogen goes as far as incorporating host lipids into the outer membrane (OM) that could also serve as an evasion mechanism (17). The simple bilayer membrane structure and limited number of detoxification genes provides little protection to B. burgdorferi from the onslaught of the host immune response, specifically oxidative and nitrosative stress (17–22). Borrelial oxidative stress regulator (BosR) has been associated with regulating genes involved in combating oxidative stress and is important for establishing mammalian infection (23–31). A single superoxide dismutase (sodA) aids in the resolution of reactive oxygen species (ROS), but to date a functional catalase has not been identified (32, 33). The reduced form of coenzyme A (CoASH) has been shown to serve in the place of catalase in B. burgdorferi by reducing hydrogen peroxide (22). Dynamic temporal and spatial regulation of borrelial genes is crucial for successful colonization, dissemination, and invasion of B. burgdorferi in the tick vector and mammalian host (7, 34, 35). Borrelial two-component pathways HK1/Rrp1 and Rrp2-RpoN-RpoS in the tick vector and mammalian host, respectively, regulate genes with a variety of functions including metabolism, chemotaxis, antigenic variation, and adhesion (7, 34). B. burgdorferi gene regulation also contributes to immune evasion as it progresses through the different stages of murine infection by inducing the recombination events of an antigenic variation gene (vlsE) and the activation or repression of lipoprotein genes in a tissue specific and temporal manner (36). The lack of secretion systems or toxins limits the ability of B. burgdorferi to modulate the resulting inflammatory disease.
Escape of B. burgdorferi from the tick midgut to the hemolymph during a blood meal is an important step for transmission through the salivary glands to a mammalian host providing the necessary environmental cues, such as temperature and pH, for adaptation (7, 34). B. burgdorferi gene regulation in response to environmental cues is the best characterized for the early localized stage of infection, but much remains to be understood about the mechanisms for dissemination and invasion necessary for the later stages of Lyme disease (7, 35, 37). Dissemination is a necessary step in pathogenesis, and this process inhibits clearance by the immune system, but the specific mechanisms and host–pathogen events that initiate and complete this stage of infection are not well understood. Invasion of B. burgdorferi allows the pathogen to reach immunoprotected niches in the mammalian host where the pathogen is not cleared, but induces inflammation. The intimate interaction between B. burgdorferi lipoproteins and vascular tissue, in particular with extracellular matrices (ECM), has been shown to be integral to borrelial pathogenesis (38–41). B. burgdorferi adhesin interactions with the ECM and/or requirements for mammalian infection have been thoroughly reviewed by several contributors in the field (38–40), but the specific roles for many of these adhesins in dissemination and/or invasion has not been evaluated, therefore, were not addressed in this review. The purpose of this review is to highlight the mechanisms utilized by B. burgdorferi for dissemination and invasion of tissues of human and reservoir host.
Morphology, Motility, and Chemotaxis Regulation
A discussion of dissemination and invasion would be incomplete without briefly describing the unique morphology, motility, and chemotaxis of B. burgdorferi that has been previously thoroughly reviewed (10, 12, 42). B. burgdorferi is often referred to as a gram-negative “like” bacterium because it is void of lipopolysaccharide (LPS) in its OM, but has a similar membrane organization. The unique helical shape of spirochetes is due to its composition of the lipid bilayer OM, a periplasmic space containing peptidoglycan and endoflagella, and a second lipid bilayer inner membrane (IM) forming the inner most compartment of the cell structure (also called the protoplasmic cell cylinder). B. burgdorferi cells are long in length and thin in diameter with dimensions of 10–20 µm and approximately 0.3 µm, respectively. We have limited knowledge about the role of peptidoglycan in the morphology and motility of B. burgdorferi, but recent work has demonstrated that the temporal and spatial regulation of peptidoglycan synthesis occurs separately from cell elongation and septum formation take place separately, thus potentially contributes to the unique cell shape (43). In the periplasmic space, a flagellum is attached to each pole then travels toward the midline of the cell and potentially overlaps with the flagellum anchored at the opposite end of the cell (10, 12). Multiple flagellum, ranging from 7 to 11 individual flagellum, cluster at the poles of the cell and arrange themselves into a ribbon to form the borrelial endoflagella that wraps around the periplasmic cylinder generating the characteristic helical or planar flat-wave morphology. The concealment of the flagella within the periplasmic space preventing recognition by the host innate immune response is a passive means of immune evasion by B. burgdorferi.
The intricacy of the borrelial cell structure and complexity of its motility program makes the accompanying regulation astoundingly complex. Motility of each flagella in the cluster requires the coordinated activities of the basal body, hook, and filament (10, 12) (Figure 1). The basal body includes the export apparatus (FliH and FliI), C-ring switch complex (FliG and FliM), MS ring (FliF), collar structure, FliL, rod (FliE), P-ring, and the motor stator (MotA and MotB). The export apparatus and C-ring are located on the cytoplasmic face of the IM and associates with the MS ring that is flanked by FliL and stator embedded in the IM. FliL is important for the orientation of the motor and localizes between the stator and rotor. MotB, a motor protein, is required for in vivo helical morphology and motility (44). The rod extends from the MS ring into the P ring prior to transitioning into the flagellar hook. The rod and P-ring flank the collar and genes involved in collar formation have not been identified in B. burgdorferi. Recently identified, FlbB is a novel flagellar motor protein that associates with the basal body to achieve the proper orientation of the flagella and collar assembly (45). The primary contributor to the flagellar hook is FlgE that connects to the filament composed of FlaA and FlaB that are exported by a SecA-mediated pathway and Type III flagellin-specific pathway, respectively (10, 12). Coordinated motility of polarized endoflagella is a complex process for directional swimming or non-directional flex modality of borrelial cells. Endoflagella anchored on opposite ends rotation is coordinated to drive swimming in a single direction by one flagella moving clockwise and the second counterclockwise. Reversal of swimming occurs when both endoflagella change direction of rotation. The non-directional mode of motility occurs when the both endoflagella move in a clockwise or counterclockwise rotation resulting in a flex state that is similar to the tumble pattern of movement observed for other bacteria (46). B. burgdorferi motility in conventional liquid cultivation is widely homogeneous with the cells in a constant swim state. However, the in vivo environments in which the pathogen traverses throughout the enzootic cycle are rarely consistent. In vitro cultivated B. burgdorferi in a media complex with 3% gelatin displayed heterogeneous motility patterns not unlike that observed in mammalian dermis (47). Another morphology taken on by B. burgdorferi is a round body, also referred to as a cystic formation, which is a proposed reversible survival mode when under conditions of stress (48–53). The mechanism for round body formation and the role of borrelial endoflagella in this process is unknown.
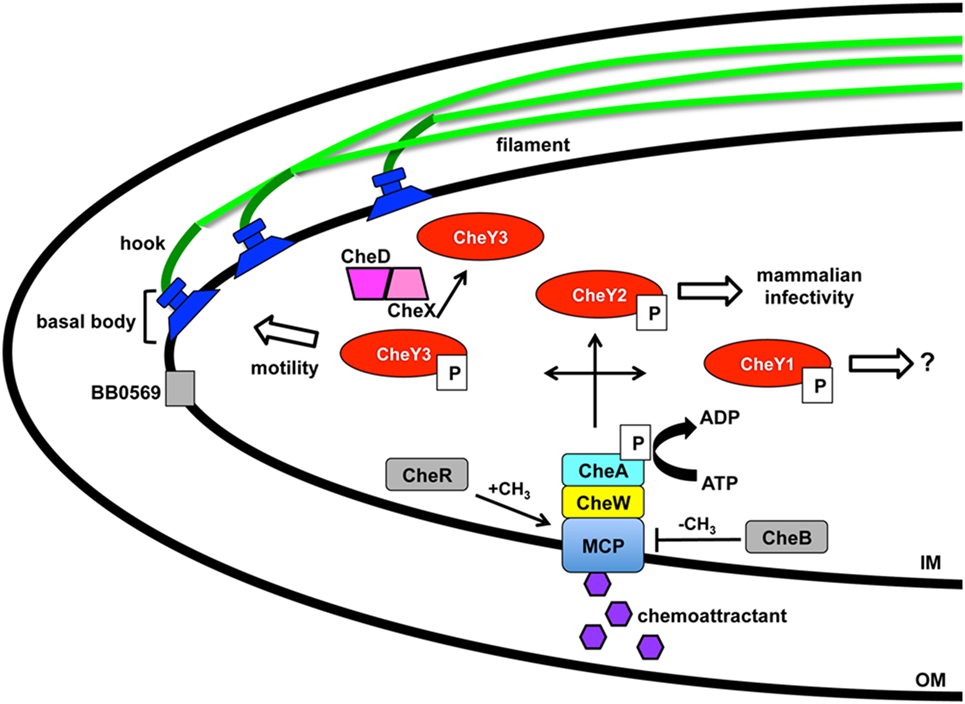
Figure 1. Borrelia burgdorferi motility and associated regulation. Motility of B. burgdorferi is driven by multiple flagellum in the periplasmic space that are comprised of an inner membrane anchored basal body, hook, and filament. Coordinated rotation of the flagellar motors drive the direction of swimming or causes flexing of the spirochete. A two component regulatory system controls the motility in response to chemoattractant signals through the membrane-bound chemoreceptor and methyl-accepting chemotaxis protein (MCP). A complex is formed between MCP, linker protein CheW, and histidine kinase CheA with the latter able to autophosphorylate and transfer the phosphate to response regulators CheY1, CheY2, and CheY3 resulting in distinct outcomes. Levels of CheY-P can be reduced by the phosphatase CheX that forms a complex with CheD and increases its activity. The borrelial proteins designated in gray are chemotaxis homologs that have not been characterized to date. BB0569 localizes to the poles of the cell near flagellar motors and influences motility, but its specific function is unknown. CheR and CheB are potential methyltransferase and methylesterase proteins, respectively, which may modify MCP. Modification of Xu et al. (54).
Borrelial motility is required for virulence, but more specifically is essential for the invasion of tissues in response to environmental signals (10, 12). A two-component signal transduction pathway regulates B. burgdorferi chemotaxis and utilizes a membrane-bound chemoreceptor and methyl-accepting chemotaxis protein (MCP) that forms a complex with linker protein CheW and histidine kinase CheA to sense environmental cues (Figure 1). CheA autophosphorylates prior to transferring the phosphate to three response regulators, CheY1, CheY2, and CheY3, encoded by B. burgdorferi. A low concentration of CheY-P initiates a run motility and high CheY-P concentrations causes B. burgdorferi to flex. CheX phosphorylase rapidly reduces the amount of CheY-P and is enhanced by forming a complex with CheD (55, 56). CheY3 is the only CheY response regulator that has a role in motility and CheY1 or CheY2 are not able to complement the function of CheY3 (57). Phosphorylated CheY3 interacts with the flagellar switch proteins to regulate the rotation of the motors. Xu et al. recently demonstrated a requirement of CheY2 for infectivity. However, this protein does not play a role in chemotaxis or motility (54). B. burgdorferi encodes homologs for chemotaxis adaptation proteins CheB and CheR, but to date, these proteins have not been characterized. In E. coli, CheR and CheB alter the methylation of MCP by methyltransferase and methylesterase activity, respectively, in response to positive and negative stimuli (46). The number of chemotaxis homologs encoded in the B. burgdorferi genome is greater and far more complex than other motile pathogens, such as E. coli or S. enterica (13, 14). Additional genes, csrA and bb0569, influence the motility regulation of B. burgdorferi (58, 59). Carbon storage regulator, CsrA, is involved in posttranscriptional regulation of flagellar genes and contributes to cell morphology (59, 60). BB0569, annotated as a hypothetical protein, has sequence similarity to MCPs and was found to localize to the poles of the borrelial cell and important for motility and chemotaxis (58). B. burgdorferi motility is regulated throughout the enzootic cycle with the distinct behavior of intermittent swimming in the tick vector that includes periods of a non-motile state relative to the persistent swimming observed in the murine host (44, 61).
Dissemination
Borrelia burgdorferi disseminates from the site of the tick bite within the mammalian host to secondary colonization sites requiring dramatic adaptation of the pathogen in response to environmental changes and obstacles presented by the host innate immune response (7, 62–65). A bottleneck occurs during the B. burgdorferi initial colonization of dermal tissue when establishing localized infection. This process is in part mediated through the MyD88 pathway, which potentially alters the spirochetal population progressing into the dissemination stage when it infects distal tissues (66–70). B. burgdorferi dissemination in mammals occurs by hematogenous and non-hematogenous routes, such as through the lymphatic system or direct spread through the tissues, with the former being the most well understood (71, 72). B. burgdorferi hematogenous dissemination has been the focus of much research identifying distinct infectivity classifications of environmental or human isolates by the ribosomal spacer type (RST) or ospC sequence heterogeneity (73). Three RST genotypes have been defined for B. burgdorferi as RST1, RST2, and RST3 in descending order of dissemination capabilities (71, 74). At least 16 different ospC genotypes that vary in the ability to disseminate and cause disease have been recognized (73, 75–80). More human infections have been associated with ospC types A, B, I, H, and K, but there is no correlation with the frequency distribution of ospC genotypes in infected Ixodes scapularis (73, 81). Genotypes A and B are part of RST1, type I is in RST2 and H and K in RST3 (73). Genotypic differences can predict the ability of a B. burgdorferi strain to disseminate and cause infection. The effectiveness or lack of physical barriers, host factors, and immune response can also influence the outcome of borrelial infection.
B. burgdorferi Hematogenous Vascular Interactions
After the establishment of a localized dermal infection, B. burgdorferi begins to disseminate throughout the mammalian host via the hematogenous route (3, 71, 82). Borrelia hermsii, etiologic agent of relapsing fever, reaches high bacterial loads in the blood. However, B. burgdorferi, which also disseminates by hematogenous route, is difficult to isolate from blood due to the low number of bacterial cells present. The monitoring of bioluminescent B. burgdorferi in real time during murine experimental infection with an in vivo imaging system (IVIS) demonstrated the pathogen established a strong localized infection signified by elevated emission of light centered around the site of inoculation followed by dissemination throughout the skin (83). The inoculum dose altered the spatial and temporal dissemination of B. burgdorferi constitutively expressing codon-optimized firefly luciferase, but the general pattern of dissemination was the same independent of dose (83, 84). Upon reaching specific tissues or target sites, B. burgdorferi traverses dense extracellular matrix (ECM) and crosses tissue barriers through yet to be elucidated mechanisms.
Hematogenous dissemination of pathogens within mammals is poorly understood due to the difficulty of evaluating bacteria under the shear stress conditions in the blood stream. Shear stress is a type of tangential stress that acts along a parallel surface due to friction caused by fluid viscosity that is overcome by bacteria for dissemination and invasion (85). The utilization of intravital microscopy (IVM) and spinning disk confocal intravital IVM allowed the evaluation of biofluorescent B. burgdorferi in real time under in vivo shear stress conditions that characterized the multiple stages of vascular interaction prior to transmigration (86–88). GFP-expressing B. burgdorferi visualized by IVM in murine ears approximately 3–4 weeks after infection demonstrated swimming and flexing motility commonly observed under in vitro cultivation (86). Microvascular interactions visualized in the murine skin flank by epifluorescence and spinning disk confocal IVM following intravenous injection of biofluorescent B. burgdorferi found that bacterial cells did not localize to arterioles, but localized to capillaries, postcapillary venules, and large veins. The study defined B. burgdorferi dissemination interactions as transient tethering-type associations, dragging interactions, and stationary adhesion (Figure 2). It is important to note that not all of the B. burgdorferi cells associated with the vasculature and thus were not directly assessed (86). Rapid extravasation of B. burgdorferi that did not form prior interactions was observed from postcapillary venules. Transient tethering-type interactions occurred briefly when B. burgdorferi slowed down relative to the velocity of the blood stream to associate with the endothelium at one region or tip of the cell in a manner that appeared to tether the cell to the surface followed by a detachment. Tethering stabilizes interactions by reducing the dissociation rates of B. burgdorferi from the endothelium (89). Dragging, another type of short-term interaction, was distinguished from tethering by B. burgdorferi coming into contact with the endothelium over the length of the cell and being dragged along the vascular surface in the direction of blood flow (86). The final stage of vascular interaction prior to transmigration is stationary adhesion where the B. burgdorferi cell is at a complete stop and no longer progressing in the direction of the blood stream. Borrelial cells maintained stationary adhesion for an average of 10 min. 3D visualization was performed to determine the localization of interactions in relation to endothelial junctions labeled with PECAM-1 that found stationary adhesion primarily occurred at the junctions while tethering and dragging took place along the endothelial cell surface (86). Non-infectious borrelial cells lacking genes for OM lipoproteins did not display the same interactions with the endothelium as infectious B. burgdorferi indicating pathogen ligands that can bind to specific host receptors (87). An in vitro flow chamber model was developed to evaluate B. burgdorferi interactions with human endothelia under a controlled environment for shear force (89). B. burgdorferi endothelial interactions in postcapillary venules by IVM and those in the flow chamber were consistent, thus providing an opportunity to understand the specific mechanics behind tethering, dragging, and stationary adhesion in controlled conditions.
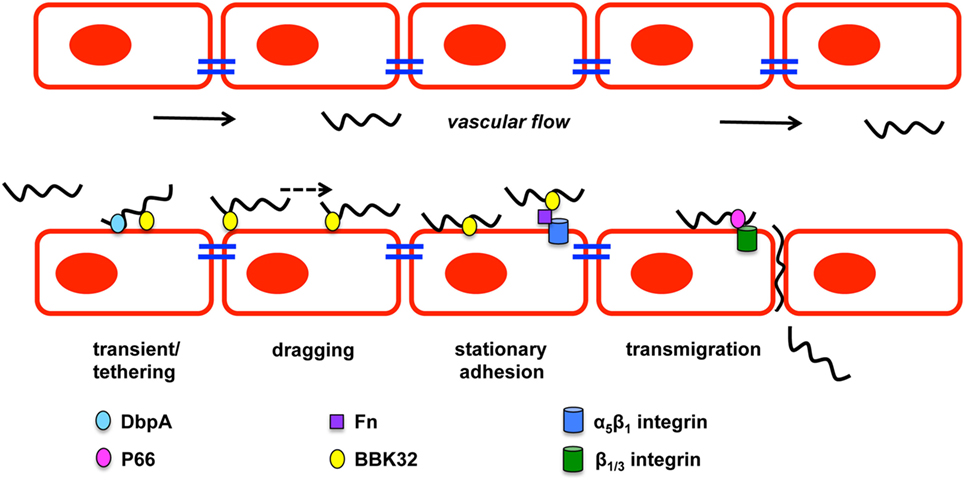
Figure 2. Borrelia burgdorferi vascular interactions and transmigration. B. burgdorferi forms three types of vascular interactions: transient/tethering, dragging, and stationary adhesion. Decorin-binding protein A (DbpA) promotes transient/tethering with endothelial cells. Fibronectin-binding protein (BBK32) is involved in all three types of interactions. Stationary interaction through BBK32 may be through direct interaction with the endothelial surface or through a fibronectin bridge (Fn) to a host integrin. P66 also interacts with integrins and is important for transmigration of B. burgdorferi. Figure is a modification of Moriarty et al. (86).
Numerous borrelial lipoproteins have previously been shown to bind components of the ECM (39, 41, 90–94). Norman et al. examined potential mechanisms mediating the interactions with the vasculature through fibronectin (Fn) and glycosaminoglycans (GAGs) (87). Biofluorescent B. burgdorferi was incubated with dextran sulfate to block potential GAG interactions and evaluated in vivo by epifluorescence IVM resulting in a 30 and 80% reduction in transient and dragging interactions, respectively. Stationary adhesion was reduced to a similar level as dragging when cells were treated with dextran sulfate (87). This indicates that GAGs are involved in mediating the host–pathogen interaction of B. burgdorferi in the vasculature. The role of Fn in B. burgdorferi binding to the vasculature was assessed by treating B. burgdorferi that expresses fibronectin-binding protein, BBK32, with anti-Fn IgG prior to IV injection. Short-lived reduction in transient and dragging interactions were observed, suggesting that Fn plays an important role in the ability of B. burgdorferi to interact with the host vascular endothelium (87). Fibronectin C-terminal heparin-binding domain and integrin-interacting cell-binding domain that were responsible for B. burgdorferi vascular interaction was evaluated by competition with peptides representing the binding domains under in vivo conditions. B. burgdorferi was able to readily interact with the endothelium at all stages when the RGD sequence of the integrin-interacting cell-binding domain was in circulation to compete with plasma Fn for binding. Heparin-binding domain peptide decreased B. burgdorferi transient interactions by 52% and further reduced dragging and stationary adhesion by 84%, suggesting that host GAGs are responsible the observed vascular interactions. The heparin-binding domain of Fn and GAGs are directly or indirectly important for B. burgdorferi interactions with the host endothelial surface.
BBK32 is one of several B. burgdorferi proteins shown to bind Fn and its role in mammalian infectivity has been assessed as a ligand under in vivo conditions (83, 87, 88, 95–97). Fibronectin binding by B. burgdorferi may build a bridge for binding integrins allowing further host–pathogen interactions to occur (92, 98, 99). B. burgdorferi lacking bbk32 attenuates the infectivity phenotype and increases the ID50 about 15-fold as determined by cultivation of murine tissues 3 weeks after inoculation (95). High dose infection, specifically 105, with the bbk32 mutant strain results in colonization of disseminated tissues at a slightly lower level than wild-type when bacterial loads are quantitatively determined (97). In vivo imaging of bioluminescent bbk32 deletion strain demonstrated a similar dose dependent phenotype, but also that the role for bbk32 may be more important for earlier stages of infection (83). Light emission from mice infected with this strain was significantly less 1 week after inoculation when compared to wild-type infected controls. Gain-of-function experiments utilized non-infectious B. burgdorferi lacking several plasmids and an adhesin protein necessary for dissemination or invasion (86–88). Non-infectious B. burgdorferi that was complemented with bbk32 driven by the ospC promoter caused increased vascular interactions relative to the non-infectious parent strain lacking the transgene (87). Mutagenesis of bbk32 in infectious B. burgdorferi allowed the specific role of this fibronectin-binding protein to be characterized for Fn binding and vascular interactions (88) (Figure 2). Biofluorescent bbk32 deletion strain displayed a 20 and 50% reduction in the murine skin flank and joint, respectively, in tethering and dragging. Epifluorescence IVM requires a high dose inoculation by IV injection and may contribute to the different outcomes of bbk32 infectivity studies using lower dose intradermal inoculum that resulted in more pronounced virulence attenuation. The disruption of two regions of bbk32 (Fn-binding, Δ158–182, or GAG-binding sequences, Δ45–68) caused a reduction of tethering and dragging, respectively, indicating these binding domains serve distinct mechanistic functions for B. burgdorferi endothelial interactions. BBK32 has a fast binding association and disassociation with Fn, but GAG binding is longer lasting and more stable (88). These differences in binding kinetics may explain the distinct binding interactions observed by in vivo IVM. Flow chamber studies evaluated the specific mechanism by which BBK32 associates with the endothelium and determined it contributes to stabilization of interactions and increases the range of force and shear stress through catch bond mechanism (89). A catch bond occurs when the higher the tensile force the longer the life of a non-covalent bond between a receptor and ligand (100). It is not clear if BBK32 is directly binding the endothelium utilizing lectin-binding sites or if the interactions are occurring through host ligands. Other possible fibronectin-binding proteins, RevA, RevB, and BB0347, were not able to restore vascular interactions in non-infectious B. burgdorferi (88). It is likely that a yet to be identified borrelial proteins that are able to bind fibronectin contribute to vascular interactions in addition to BBK32.
It is likely that many more players are involved in vascular interactions and possibly do so in a tissue specific or spatial manner. A prime candidate is borrelial lipoprotein decorin-binding protein A (DbpA) that is able to bind decorin, heparin, and dermatan sulfate GAG (101–103). dbpA is co-transcribed with highly conserved dbpB that has a decreased decorin-binding ability and does not impact infection to the degree observed for dbpA (13, 14, 101, 104–106). Environmental signals, such as temperature, pH, and CO2, regulate dbpBA expression in a RpoS-dependent manner (107–116). Incubation of B. burgdorferi with neuroglial or human umbilical vein endothelial cell (HUVEC) cells induced the expression of dbpBA among other virulence associated lipoproteins (117). B. burgdorferi lacking dbpBA has a severely attenuated infectivity phenotype with reduced colonization of the skin, heart, and joint when needle inoculated, but tick transmission of the dbpBA mutant resulted in an infectivity phenotype similar to wild-type (83, 118–122). Over time high dose inoculum of the dbpBA mutant can overcome the limitations to dissemination and reach wild-type bacterial burden in tissues despite reduced pathology in the heart and joint (118). Expression of dbpA is detectable up to 8 weeks after inoculation in several tissues, which is dissimilar to another RpoS-regulated gene ospC that is downregulated about a week after infection (123, 124). The importance of decorin binding by B. burgdorferi is further demonstrated in the resistance of decorin-deficient mice to infection (125). Transcripts of dbpA are readily detectable from blood during infection, but there is also some indication that DbpA contributes to lymphatic dissemination of B. burgdorferi (118). The dbpBA mutant has reduced colonization of lymph nodes of immunocompetent mice relative to wild-type B. burgdorferi, but there was no difference observed in immunodeficient mice (118). The ability of DbpBA to aid in vascular interactions was assessed using an in vitro flow chamber seeded with HUVECs to mimic shear stress of the blood stream (126). Non-infectious B. burgdorferi B313 missing numerous plasmids was complemented with dbpBA on a shuttle vector and formed the short-lived tethering interaction (126) (Figure 2). DbpA has not been evaluated under in vivo conditions with IVM. In addition, its specific contribution to invasion as assessed by its ability to cross cell culture monolayers or to move in in vivo transmigration assays has not been determined. Vascular interaction is merely one step in the borrelial pathogenic process with stage each building upon the other for the end result of persistent infection in several mammalian tissues.
Invasion
Borrelial dissemination and subsequent invasion of mammalian tissues in the latter stages of disease is an essential step for pathogenesis and immune evasion. Invasion of B. burgdorferi by entering into a tissue through transmigration or extravasation results in the colonization of secondary infection sites that become the foci of disease. B. burgdorferi has the ability to preferentially escape into heart or joint tissue and cross the blood–brain barrier (BBB) stimulating an inflammatory immune response to cause carditis, arthritis, and neuroborreliosis, respectively (1). The endothelial surface of the BBB has an unique organization and increased level of protection in comparison to other endothelial tissues targeted for invasion by B. burgdorferi. Invasion of the BBB by B. burgdorferi results in activation of the host plasminogen activation system (PAS), matrix metalloproteases (MMP), and host calcium signaling (127–129). It is likely that B. burgdorferi utilizes unique invasion strategies in a tissue-specific manner in addition to mechanisms common to other pathogens for entry into immunoprotected niches.
Early host–pathogen studies evaluated the ability of B. burgdorferi to cross cell culture monolayers of various types (130–135). Infectious B. burgdorferi is able to adhere and penetrate through tight junctions from the apical surface to traverse the monolayer. This interaction with the host and subsequent transmigration is not without consequence as it can result in the enhanced response of neutrophils and T lymphocytes in HUVEC culture (136, 137). Evidence limited to in vitro studies indicate that B. burgdorferi has the ability to undergo intracellular invasion of human fibroblast, umbilical vein endothelial, synovial, neuronal, and glial cells without the loss of viability (49, 133, 138, 139). These findings lead to the speculation that borrelial cellular invasion is a mechanism for immune evasion and disease modulation, but the significance is unknown, as this has not been observed using an in vivo model system. Furthermore, an important effect of B. burgdorferi interacting with the host is the alteration of virulence determinants, including Dbp, OspA, and BBA64, which are important for mammalian infection (117, 140). The host–pathogen interactions and the resulting responses of B. burgdorferi and host tissue-specific pathways are important for the advancement of the spirochete into secondary colonization sites. The targeting of immunoprotective niches requires non-specific and specific vascular interactions, as described above, followed by transmigration that is likely mediated by several distinct borrelial adhesins.
B. burgdorferi-Elicited Transmigration in the Mammalian Host: The Role of P66
P66 (bb0603) is encoded on the borrelial chromosome and forms an OM β-barrel porin with adhesive capabilities to host integrins that contribute to mammalian infection (141–148). B. burgdorferi lacking P66 is able to persist in ticks through molting, but is unable to establish infection in the mammalian dermis and is cleared at the site of inoculation within 48 h following inoculation (149, 150). Expression of p66 is not observed in the tick vector until exposure to a blood meal and continues well into mammalian infection providing further support for the importance of P66 during mammalian infection. P66 engages the innate and adaptive immune response in the murine host as it is required for infection of mice deficient in the innate immune signaling molecules MyD88 and TLR-4 and elicits a specific antibody response against p66 expressing B. burgdorferi. A short-term infection model with intravenously infected Balb/c mice was used to evaluate the tissue localization of wild-type or p66 mutant B. burgdorferi 1 h after inoculation and resulted in a P66-dependent tissue tropism for the ear and heart (94). Disseminated B. burgdorferi requires specific adhesins dependent upon the tissue for colonization of distal sites.
The requirement for infectivity could be due to one or both of P66 identified functions: porin and/or integrin-binding activity. P66 was initially identified as an integral membrane porin in B. burgdorferi and was further shown to possess channel conductance (141, 142). Recent work by Kenedy et al. characterized the β-barrel structure of P66 and found that it associates with lipoproteins OspA and OspB (143). The second identified function of P66 is its interaction with β1 chain and β3 chain integrins that are homodimeric proteins with the function of host cellular signaling and interactions with host cell matrices (147, 149, 151, 152). Traditional mutational analysis and a phage-display screen verified β3 intergrin binding by P66 (147, 153). The specific binding region for integrin interaction was localized to amino acids 203–209 of P66 (151). Transcriptional analysis of human embryonic kidney cells and endothelial cells exposed to B. burgdorferi with or without p66 indicated significant changes among host genes involved in several pathways including cellular interactions and actin arrangement indicating a potential role in pathogen invasion (152). Interaction of P66 with the host causing potential cellular arrangements may support the transmigration of the pathogen through tight junctions or possibly the intracellular invasion of B. burgdorferi. An in vitro gentamicin protection study demonstrated B. burgdorferi utilizes β1 integrin to invade endothelial or fibroblast cells requiring the rearrangement of actin filaments and Src kinase activity (49). Internalized B. burgdorferi displayed the round body morphology previously observed when the pathogen is under stress conditions, but the change in shape could also be due to spatial limitations within the host cell. At this time, the physiologic relevance of intracellular B. burgdorferi is unclear because evidence that this event also occurs during a natural infection has not been reported.
The requirement for borrelial P66 binding of host β3 integrin for infection in the murine experimental model was assessed by Ristow et al. (154). Mice expressing or devoid of β3 integrin had the same ID50 as wild-type B. burgdorferi and were also not infected by the p66 mutant strain, indicating that β3 integrin is not essential for infection and redundant integrin binding by borrelial cells may also support tissue invasion. Two p66 integrin-binding targeted mutant strains were generated consisting of a regional deletion of amino acids 202 to 208 (BDel202-208) and double point mutation (BD205A,D207A). Phenotypes of the p66 targeted mutant strains were characterized for infectivity, in vitro integrin binding, porin activity, and penetration of endothelial monolayers. BDel202-208 had an attenuated infectivity phenotype with significant reduced bacterial loads in the heart and tibiotarsal joint, while BD205A,D207A decreased in the heart relative to wild-type B. burgdorferi when mice were subcutaneously infected. Intravenous infection slightly alters the kinetics of borrelial infection and only BDel202-208 had a reduced bacterial burden in the heart. BDel202-208 and BD205A,D207A significantly reduced αVβ3 integrin binding relative to wild-type, but did not disrupt channel conductance and maintained porin activity (147, 154). In correspondence with integrin binding, BDel202-208 and BD205A-D207A had an impaired ability to cross endothelial monolayer when compared to the wild-type strain. It is well established that P66 and, more specifically, the region encompassing the integrin-binding region is important for B. burgdorferi infection and possibly invasion. The route of host interaction is possibly through β1 integrin in addition to β3 integrin. β1 integrin could not be directly evaluated in vivo due to lethality associated with β1 deficiency.
The potential that P66 was involved in the dissemination or invasion of B. burgdorferi was evaluated by in vivo IVM for vascular interactions and in a newly developed high resolution intravital transmigration assay (155). Previous studies utilizing in vivo IVM focused on vascular interactions in the skin (86–88). Cd1d−/− mice are a preferred strain to examine B. burgdorferi infection in the joint because the lack of invariant natural killer T cells (iNKT) increases the bacterial burden and the likelihood of visualizing spirochetal vascular interactions or transmigration (156, 157). Kumar et al. focused on the vascular interactions in joint proximal tissue of Cd1d−/− mice that were capable of recapitulating tethering, dragging, and stationary adhesion interactions of wild-type B. burgdorferi observed in murine skin (155). Spinning disk laser confocal microscopy captured a back and forth motility of B. burgdorferi as it transmigrates in the joint one day after high dose intravenous inoculation of Cd1d−/− mice (155). B. burgdorferi lacking p66 showed no difference in tethering, dragging, or stationary adhesion relative to wild-type. Therefore, this adhesive porin is not involved in vasculature interactions. Transmigration and clearance from the bloodstream of p66 mutants was dramatically reduced when compared to wild-type or complemented p66 mutant strains raising the question whether the inability of B. burgdorferi to invade was due to clearance by the host immune response rather than the ability to cross the epithelial layer. Targeted p66 mutant strains were also not able to transmigrate, but interestingly, bloodstream clearance is at a similar rate to wild-type indicating that P66 serves an active role in a borrelial transmigration mechanism. In vivo localization of β3 integrin in the vasculature in relation to B. burgdorferi was determined that P66-β3 integrin interaction associates with transmigration events at cellular junctions where stationary adhesion most often occurs. B. burgdorferi localized to areas in the vasculature with higher concentrations of β3 integrin indicating this colocalization is taking place and contributing to transmigration. Studies with β3 integrin-deficient mice showed B. burgdorferi was able to disseminate to various tissues, suggesting that β3 integrin binding is not the sole mechanism for borrelial invasion. It is likely that the pathogen utilizes redundant invasion mechanisms through the binding of other integrins, such as β1, or host receptors. Active transmigration in the joint occurs above background levels 24 h after inoculation, which may allow the time for activation of cell signaling pathways in endothelial cells and borrelial adaptation to support transmigration for successful invasion (158). The contribution of BBK32 to endothelial invasion, in addition to the previously noted vascular interactions, is determined not to be involved as the bbk32 mutant and wild-type B. burgdorferi displayed similar rates of transmigration. The role of P66 porin activity in vascular interactions, transmigration, and survival following epithelial invasion is unknown at this time. Taken together, P66 is involved in a potential invade-to-evade mechanism in B. burgdorferi that may have additional pathogenic functions (154, 155). Further studies are needed to elucidate possible P66 initiated actin and ECM rearrangements and the impact on the inflammatory response characteristic of Lyme disease.
B. burgdorferi Protease HtrA Role in Invasion
The high-temperature requirement (HtrA) family of ATP-independent serine proteases serve as chaperones, support membrane integrity by degrading damaged or improperly folded proteins, and pathogenesis by processing virulence determinants or degrading components of the ECM in other bacterial pathogens (159, 160). B. burgdorferi chromosomally encodes a single HtrA protease (BbHtrA) at bb0104 (13, 14). BbHtrA exhibits proteolytic activity and the potential of this protein to support borrelial pathogenesis and physiology was the subject of a MicroCommentary in Molecular Microbiology and a perspectives article in Frontiers in Cellular and Infection Microbiology (161–165). BbHtrA is a surface exposed protein that localizes to both the soluble and membrane fractions and can also form oligomeric structures (163, 164, 166). Production levels of BbHtrA are higher at 37°C relative to 34°C correlating with higher activity levels and induction during the stationary phase of in vitro cultivation conditions (167). It was the first HtrA in which proteolytic activity inhibited by zinc was described (166). Cell fission protein BB0323 is required for mammalian infection and proteolytically cleaved by BbHtrA demonstrating that B. burgdorferi is able to modify its own proteins (168). The B. burgdorferi htrA mutant strain does not contain processed BB0323 at elevated temperatures validating the proteolytic function of BbHtrA in vivo (167). BbHtrA targets proteins for proteolysis that vary in function including cell fission (BB0323), chemotaxis (CheX), laminin binding (BmpD), and integrin binding (P66) (163, 168, 169). Additional borrelial proteins are bound by BbHtrA without undergoing degradation, indicating that BbHtrA may also function as a chaperone (163). Transcripts of p66 in a borrelial strain overexpressing BbhtrA were reduced demonstrating another layer of regulation employed by this borrelial protease (169). The impact of BbHtrA on pathogenesis is widespread in regards its diverse mechanisms of regulation.
Another host target of borrelial proteolysis is aggrecan, which is a large aggregating proteoglycan found in cartilage with multiple functional domains consisting of two GAG-attachment domains and three globular domains (170). Behera et al. found that B. burgdorferi-induced aggrecanase 1 and 2 (ADAMITS 4 and 5) in human chrondrocyte cells causes the cleavage of aggrecan, thus possibly explaining cartilage damage associated with Lyme disease (171). The binding ability of B. burgdorferi aggrecan was attributed to borrelial GAG-binding protein (Bgp) (encoded by bb0588) and BbHtrA (164). The aggrecan interglobular domain (IGD), which resides between globular domains 1 and 2 (G1 and G2), is specifically cleaved by BbHtrA, thereby disrupting its ability to associate with GAG (164, 170). Proteolytic assays were performed to identify other host ECM proteins degraded by BbHtrA (165). The hyalectin family of chondroitin sulfate proteoglycans, including aggrecan, brevican, neurocan, and versican, were also degraded by BbHtrA (Figure 3). Additional BbHtrA degraded ECM proteins were identified as fibronectin, biglycan, and decorin. B. burgdorferi is known to encode specific adhesins with the ability to bind these proteoglycans. Cell junction protein E-cadherin was only moderately degraded while collagen II and tenascin C were unaffected by incubation with recombinant BbHtrA. Together, these findings indicate that BbHtrA also has target specificity for degradation of host ECM. A point mutation was generated in the active site of BbHtrA, designated BbHtrAS226A, eliminating the protease activity and resulting in the loss of degradation of host ECM proteins (164, 165). The potential physiologic significance of BbHtrA proteolytic and chaperone activity was confirmed when Ye et al. who generated an htrA deletion mutant strain that was not recoverable by cultivation from murine tissues two weeks following inoculation (167). It cannot be determined if the loss of infectivity was due to lack of proper borrelial protein processing through degradation and chaperone activity, a reduction in host ECM degradation that may inhibit invasion, or a compounding effect of both functions. The absence of htrA results in B. burgdorferi cells with in vitro growth deficiencies, cell membrane blebbing, and clustering of cells at elevated temperature when BbHtrA has the highest level of activity (167). These described in vitro phenotypes of B. burgdorferi would significantly impair infection if also observed in vivo. BbHtrA is highly immunogenic causing the stimulation of inflammatory signaling and possibly causes further damage of the host ECM (164, 165). The immunogenicity of BbHtrA did not translate into protection against infection in the murine model (172). B. burgdorferi may utilize the multifunctional BbHtrA to promote invasion through its ability to regulate and/or process virulence determinants along with the degradation of the host ECM.
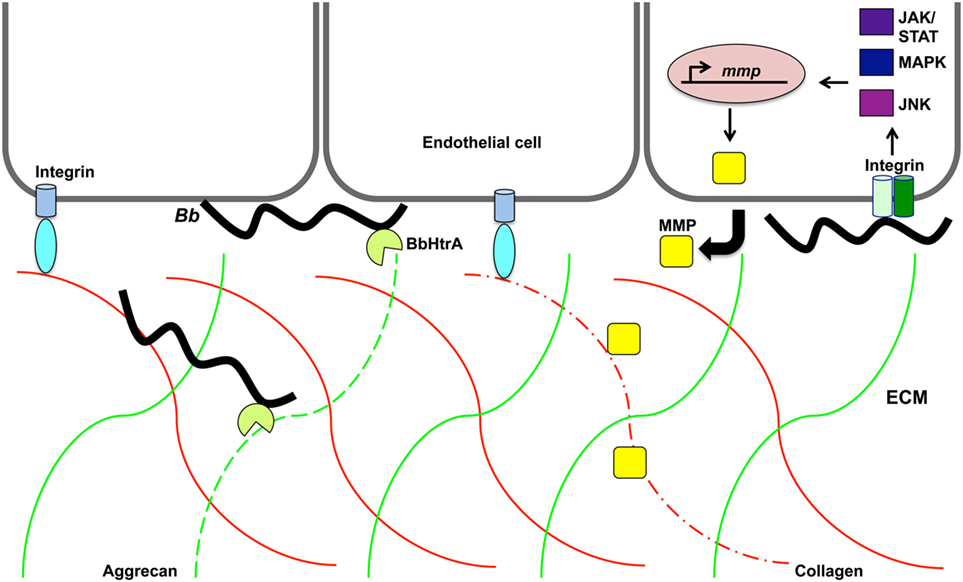
Figure 3. Proteolytic activity may promote Borrelia burgdorferi invasion of the extracellular matrix. B. burgdorferi (Bb) protease BbHtrA cleaves the family of hyalectin proteoglycans that includes hyalectin family of chondroitin sulfate proteoglycans, including aggrecan, brevican, neurocan, and versican. Specifically, BbHtrA protease activity degrades aggrecan (green lines) as indicated by dotted green line. This activity may contribute to ECM degradation and allow invasion of the pathogen. The presence of B. burgdorferi induces host matrix metalloproteases (MMP) from endothelial cells, chondrocytes, neutrophils, lymphoblast, and keratinocytes. Collagen (red lines) is degraded (dotted red line) by MMP-1 that is transcriptionally regulated along with MMP-3 through the JNK, mitogen-activated protein kinase, and JAK/STAT pathways.
Induction of Host Proteases in Response to B. burgdorferi
Borrelia burgdorferi induction of aggrecanase activity was first observed in the synovial fluid of patients with Lyme arthritis and was thought to possibly be caused by host MMPs (173). MMPs are secreted zinc and calcium-dependent host proteases that act to remodel the ECM by specific degradation of aggrecan, collagen, elastin, fibronectin, and gelatin (174). MMPs aid in tissue growth, repair, and remodeling of healthy tissues. These host proteases also cleave chemokines, cytokines, and the accompanying receptors causing an inactivation or activation of these immunological molecules. The degraded ECM fragments released stimulate a localized inflammatory response similar to that observed in Lyme disease manifestations. B. burgdorferi induces a range of MMPs that are dependent upon the types of cell stimulated and infecting borrelial strain (173, 175–177). Human neutrophils, lymphoblast, and keratinocytes, secrete pro-MMP-9 when exposed to B. burgdorferi (175). HUVECs were able to release pro-MMP-9 and pro-MMP-2 under the same conditions. Borrelial infection induces the production of collagenase-1 (MMP-1), gelatinase-2 (MMP-2), stromelysin-1 (MMP-3), and gelatinase B (MMP-9) in synovial fluid from patients with erythema migrans in addition to aggrecanase activation (173, 178) (Figure 3). The aggrecanase activity observed in synovial fluid of Lyme disease patients was found not to be associated with the stimulated MMP-2, MMP-3, or MMP-9, but may have been due to the more recently characterized BbHtrA activity described above (173). Cartilage explants from rhesus monkey and bovine were used as an in vitro model of Lyme arthritis and incubation with B. burgdorferi resulted in cartilage degradation that was prevented in the presence of MMP inhibitors (173). This could provide an explanation for the damage observed with Lyme arthritis and shed light on mechanisms utilized by B. burgdorferi for invasion and modulation of disease. The B. burgdorferi-induced profile of MMPs is specific to the tissue type and host species (173, 175). It was shown that both similarities and differences exist in the types of MMPs induced by B. burgdorferi in mice and humans and could be the reason for differences in susceptibility to borrelial infection and the development of arthritis. Different MMPs activated in response to B. burgdorferi are regulated through distinct host pathways (179–181). Specifically, MMP-1 and MMP-3 are regulated through c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), and Janus kinase/signal transducer and activation of transcription (JAK/STAT) pathway (179) (Figure 3). MMP-1, MMP-3, and MMP-9 regulation occurs through toll-like receptor 2 (TLR-2) (180, 181). MMP-1 and MMP-3 are also induced through the interaction of borrelial cells with α3 integrins (181). B. burgdorferi may utilize MMPs to manipulate ECM remodeling to promote invasion of the pathogen for secondary colonization of tissues with the diversity of induced MMPs and regulatory pathways ensuring progression of disease.
The idea that MMPs could promote invasion was supported by the increased transmigration of B. burgdorferi through ECM components under in vitro conditions when incubated with MMP-9 (175). MMP-9 is able to cleave collagen type I and the resulting fragments chemoattract peripheral blood mononuclear cells; thus, this activity also elicits an immune response that could limit borrelial pathogenesis (182). The specific role of MMP-9 in B. burgdorferi invasion and inflammatory response under in vivo conditions is assessed by infecting MMP-9−/− mice and comparing to wild-type mice in regards to bacterial burden, arthritis, and carditis. Murine hearts and joints were colonized at similar levels of bacterial burden indicating that MMP-9 alone is not responsible for invasion. This observation does not completely rule out the involvement of MMP-9 in aiding B. burgdorferi invasion of tissues through ECM remodeling. It is likely redundant mechanisms that are utilized to guarantee successful pathogenesis, which may occur through other MMPs or BbHtrA. Infected MMP-9−/− mice develop less arthritis relative to wild-type mice with an unexpected similarity in cytokine and chemokine expression, indicating that the ability of MMP-9 to modulate an inflammatory response is independent of B. burgdorferi infection. Further investigation of other MMPs individually or in combination is needed to fully understand the contribution of MMPs to B. burgdorferi invasion.
Plasmin has been observed in multiple studies to be activated by B. burgdorferi and is able to degrade ECM components and activate MMPs (173, 175, 183). The PAS, also referred to as the fibrinolytic system, involves the induction of a series of proteases and accompanying regulatory inhibitors for the purpose of fibrin cleavage that support tissue remodeling, healing, and cell migration (129, 184). It was suspected that the activation of plasmin by the presence of B. burgdorferi could serve as a potential mechanism for spirochetal invasion. B. burgdorferi readily binds plasminogen and is converted to plasmin by host-derived urokinase plasminogen activator (uPA) (38, 185, 186). This tightly regulated process is kept in check by plasminogen inhibitors type 1 and 2 (PAI-1 and PAI-2). Plasminogen binds B. burgdorferi through numerous identified receptors and promotes the transmigration of the pathogen across HUVEC monolayers (187). MMP-9 induction was observed in the presence of plasminogen when human PMBC cells were co-incubated with B. burgdorferi (175). During a tick blood meal, plasminogen is activated, but is not required for borrelial transmission to a murine host (188). uPA is induced in monocytes of mice and humans in the presence of B. burgdorferi (189, 190). Hovius et al. demonstrated that borrelia induces the PAS and, more specifically, showed that the receptor of uPA controls bacterial burden and promotes the phagocytosis of B. burgdorferi by leukocytes (191). uPAR-deficient mice had increased B. burgdorferi colonization relative to the C56BL/6 wild-type mice. However, this increase was not observed in uPA, tPA, or PAI-1 knockout mice. In vitro phagocytosis assays display limited leukocyte phagocytosis in the absence of uPAR, but not the other members of the PAS system. Therefore, the host PAS is not hijacked by B. burgdorferi for ECM remodeling leading to the conclusion that this system does not contribute to invasion. However, this conclusion does not rule out the contributions of tick salivary glands or other supporting pathogenic mechanisms in mediating transmigration.
Summary
The success of B. burgdorferi as a pathogen involves dynamic adaptation and interactions of specific tissues of the mammalian host with the pathogen during each stage of disease. B. burgdorferi engages numerous adhesins that are important for mammalian infection, but the specific contributions of these adhesins for dissemination or invasion are not fully elucidated. The specific mechanisms known to contribute to borrelial dissemination in the form of vascular interaction and invasion by transmigration under in vitro conditions or during mammalian infection has been the focus of this review. It is clear that B. burgdorferi is a resilient bacterium as it relies on redundant functions for both dissemination and invasion to ensure progression of pathogenesis. This presents a challenge to clearly understand these mechanisms. Significant advances in our understanding of B. burgdorferi vascular interactions and invasion have been made through the development of in vivo imaging technologies allowing the identification of stages of interactions. Borrelial adhesins are responsible for interactions with the host that promote dissemination or invasion in a tissue specific manner. It is unclear the degree to which B. burgdorferi interaction with the endothelium can directly induce cellular rearrangement allowing for the invasion of host tissues. Furthermore, pathogen and host proteases combat a barrier to B. burgdorferi invasion by proteolytically cleaving the ECM.
Author Contributions
The only contributor to this manuscript is JH.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Appreciation is extended to Dr. Paul deFigueiredo, Dr. Robert Gilmore, and Dr. Tara Moriarty for their time and helpful comments in the preparation of this manuscript.
Funding
This work was supported by National Institute of Health Grants AI101740A. The funders had no role preparation of the manuscript or decision to publish.
References
1. Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest (2004) 113:1093–101. doi:10.1172/JCI21681
2. Stanek G, Wormser GP, Gray J, Strle F. Lyme borreliosis. Lancet (2012) 379:461–73. doi:10.1016/S0140-6736(11)60103-7
3. Shapiro ED. Clinical practice. Lyme disease. N Engl J Med (2014) 370:1724–31. doi:10.1056/NEJMcp1314325
4. Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JWR, et al. Lyme borreliosis. Nat Rev Dis Primers (2016) 2:16090. doi:10.1038/nrdp.2016.90
5. Steere AC, Malawista SE, Snydman DR, Shope RE, Andiman WA, Ross MR, et al. Lyme arthritis: an epidemic of oligoarticular arthritis in children and adults in three Connecticut communities. Arthritis Rheum (1977) 20:7–17. doi:10.1002/art.1780200102
6. Mead PS. Epidemiology of Lyme disease. Infect Dis Clin North Am (2015) 29:187–210. doi:10.1016/j.idc.2015.02.010
7. Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol (2012) 10:87–99. doi:10.1038/nrmicro2714
8. Steere AC, Angelis SM. Therapy for Lyme arthritis: strategies for the treatment of antibiotic-refractory arthritis. Arthritis Rheum (2006) 54:3079–86. doi:10.1002/art.22131
9. Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease-a tick-borne spirochetosis? Science (1982) 216:1317–9. doi:10.1126/science.7043737
10. Charon NW, Cockburn A, Li C, Liu J, Miller KA, Miller MR, et al. The unique paradigm of spirochete motility and chemotaxis. Annu Rev Microbiol (2012) 66:349–70. doi:10.1146/annurev-micro-092611-150145
11. Goldstein S, Li C, Liu J, Miller M, Motaleb MA, Norris S, et al. The chic motility and chemotaxis of Borrelia burgdorferi. In: Samuels D, Radolf J, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk: Caister Academic Press (2010). p. 167–88.
12. Motaleb MA, Liu J, Wooten RM. Spirochetal motility and chemotaxis in the natural enzootic cycle and development of Lyme disease. Curr Opin Microbiol (2015) 28:106–13. doi:10.1016/j.mib.2015.09.006
13. Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature (1997) 390:580–6. doi:10.1038/37551
14. Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, et al. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol (2000) 35:490–516. doi:10.1046/j.1365-2958.2000.01698.x
15. Gherardini F, Boylan J, Lawrence K, Skare J. Metabolism and physiology of Borrelia. In: Samuels D, Radolf J, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk: Caister Academic Press (2010). p. 103–38.
16. Zhang K, Bian J, Deng Y, Smith A, Nunez RE, Li MB, et al. Lyme disease spirochaete Borrelia burgdorferi does not require thiamin. Nat Microbiol (2016) 2:16213. doi:10.1038/nmicrobiol.2016.213
17. Boylan JA, Lawrence KA, Downey JS, Gherardini FC. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol Microbiol (2008) 68:786–99. doi:10.1111/j.1365-2958.2008.06204.x
18. Bergström S, Zuckert WR. Structure, function and biogenesis of the Borrelia cell envelope. Borrelia: Molecular Biology, Host Interaction, and Pathogenesis. Norfolk, UK: Caister Academic Press (2000). p. 139–66.
19. Bourret TJ, Boylan JA, Lawrence KA, Gherardini FC. Nitrosative damage to free and zinc-bound cysteine thiols underlies nitric oxide toxicity in wild-type Borrelia burgdorferi. Mol Microbiol (2011) 81:259–73. doi:10.1111/j.1365-2958.2011.07691.x
20. Bourret TJ, Lawrence KA, Shaw JA, Lin T, Norris SJ, Gherardini FC. The nucleotide excision repair pathway protects Borrelia burgdorferi from nitrosative stress in Ixodes scapularis ticks. Front Microbiol (2016) 7:1397. doi:10.3389/fmicb.2016.01397
21. Seshu J, Boylan JA, Gherardini FC, Skare JT. Dissolved oxygen levels alter gene expression and antigen profiles in Borrelia burgdorferi. Infect Immun (2004) 72:1580–6. doi:10.1128/IAI.72.4.2456.2004
22. Boylan JA, Hummel CS, Benoit S, Garcia-Lara J, Treglown-Downey J, Crane EJ, et al. Borrelia burgdorferi bb0728 encodes a coenzyme A disulphide reductase whose function suggests a role in intracellular redox and the oxidative stress response. Mol Microbiol (2006) 59:475–86. doi:10.1111/j.1365-2958.2005.04963.x
23. Boylan JA, Posey JE, Gherardini FC. Borrelia oxidative stress response regulator, BosR: a distinctive Zn-dependent transcriptional activator. Proc Natl Acad Sci U S A (2003) 100:11684–9. doi:10.1073/pnas.2032956100
24. Hyde JA, Seshu J, Skare JT. Transcriptional profiling of Borrelia burgdorferi containing a unique bosR allele identifies a putative oxidative stress regulon. Microbiology (2006) 152:2599–609. doi:10.1099/mic.0.28996-0
25. Hyde JA, Shaw DK, Smith R, Trzeciakowski JP, Skare JT. Characterization of a conditional bosR mutant in Borrelia burgdorferi. Infect Immun (2010) 78:265–74. doi:10.1128/IAI.01018-09
26. Hyde JA, Shaw DK, Smith R III, Trzeciakowski JP, Skare JT. The BosR regulatory protein of Borrelia burgdorferi interfaces with the RpoS regulatory pathway and modulates both the oxidative stress response and pathogenic properties of the Lyme disease spirochete. Mol Microbiol (2009) 74:1344–55. doi:10.1111/j.1365-2958.2009.06951.x
27. Seshu J, Boylan JA, Hyde JA, Swingle KL, Gherardini FC, Skare JT. A conservative amino acid change alters the function of BosR, the redox regulator of Borrelia burgdorferi. Mol Microbiol (2004) 54:1352–63. doi:10.1111/j.1365-2958.2004.04352.x
28. Ouyang Z, Kumar M, Kariu T, Haq S, Goldberg M, Pal U, et al. BosR (BB0647) governs virulence expression in Borrelia burgdorferi. Mol Microbiol (2009) 74:1331–43. doi:10.1111/j.1365-2958.2009.06945.x
29. Ouyang Z, Deka RK, Norgard MV. BosR (BB0647) controls the RpoN-RpoS regulatory pathway and virulence expression in Borrelia burgdorferi by a novel DNA-binding mechanism. PLoS Pathog (2011) 7:e1001272. doi:10.1371/journal.ppat.1001272
30. Shi Y, Dadhwal P, Li X, Liang FT. BosR functions as a repressor of the ospAB operon in Borrelia burgdorferi. PLoS One (2014) 9:e109307. doi:10.1371/journal.pone.0109307
31. Wang P, Dadhwal P, Cheng Z, Zianni MR, Rikihisa Y, Liang FT, et al. Borrelia burgdorferi oxidative stress regulator BosR directly represses lipoproteins primarily expressed in the tick during mammalian infection. Mol Microbiol (2013) 89:1140–53. doi:10.1111/mmi.12337
32. Esteve-Gassent MD, Elliott NL, Seshu J. sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Mol Microbiol (2009) 71:594–612. doi:10.1111/j.1365-2958.2008.06549.x
33. Whitehouse CA, Williams LR, Austin FE. Identification of superoxide dismutase activity in Borrelia burgdorferi. Infect Immun (1997) 65:4865–8.
34. Caimano MJ, Drecktrah D, Kung F, Samuels DS. Interaction of the Lyme disease spirochete with its tick vector. Cell Microbiol (2016) 18:919–27. doi:10.1111/cmi.12609
35. Groshong AM, Blevins JS. Insights into the biology of Borrelia burgdorferi gained through the application of molecular genetics. Adv Appl Microbiol (2014) 86:41–143. doi:10.1016/B978-0-12-800262-9.00002-0
36. Norris SJ. vls antigenic variation systems of Lyme disease Borrelia: eluding host immunity through both random, segmental gene conversion and framework heterogeneity. Microbiol Spectr (2014) 2:1–18. doi:10.1128/microbiolspec.MDNA3-0038-2014
37. Brisson D, Drecktrah D, Eggers CH, Samuels DS. Genetics of Borrelia burgdorferi. Annu Rev Genet (2012) 46:515–36. doi:10.1146/annurev-genet-011112-112140
38. Caine JA, Coburn J. Multifunctional and redundant roles of Borrelia burgdorferi outer surface proteins in tissue adhesion, colonization, and complement evasion. Front Immunol (2016) 7:442. doi:10.3389/fimmu.2016.00442
39. Kenedy MR, Lenhart TR, Akins DR. The role of Borrelia burgdorferi outer surface proteins. FEMS Immunol Med Microbiol (2012) 66:1–19. doi:10.1111/j.1574-695X.2012.00980.x
40. Brissette CA, Gaultney RA. That’s my story, and I’m sticking to it – an update on B. burgdorferi adhesins. Front Cell Infect Microbiol (2014) 4:41. doi:10.3389/fcimb.2014.00041
41. Antonara S, Ristow L, Coburn J. Adhesion mechanisms of Borrelia burgdorferi. Adv Exp Med Biol (2011) 715:35–49. doi:10.1007/978-94-007-0940-9_3
42. Zhao X, Norris SJ, Liu J. Molecular architecture of the bacterial flagellar motor in cells. Biochemistry (Mosc) (2014) 53:4323–33. doi:10.1021/bi500059y
43. Jutras BL, Scott M, Parry B, Biboy J, Gray J, Vollmer W, et al. Lyme disease and relapsing fever Borrelia elongate through zones of peptidoglycan synthesis that mark division sites of daughter cells. Proc Natl Acad Sci U S A (2016) 113:9162–70. doi:10.1073/pnas.1610805113
44. Sultan SZ, Sekar P, Zhao X, Manne A, Liu J, Wooten RM, et al. Motor rotation is essential for the formation of the periplasmic flagellar ribbon, cellular morphology, and Borrelia burgdorferi persistence within Ixodes scapularis tick and murine hosts. Infect Immun (2015) 83:1765–77. doi:10.1128/IAI.03097-14
45. Moon KH, Zhao X, Manne A, Wang J, Yu Z, Liu J, et al. Spirochetes flagellar collar protein FlbB has astounding effects in orientation of periplasmic flagella, bacterial shape, motility, and assembly of motors in Borrelia burgdorferi. Mol Microbiol (2016) 102:336–48. doi:10.1111/mmi.13463
46. Sourjik V, Wingreen NS. Responding to chemical gradients: bacterial chemotaxis. Curr Opin Cell Biol (2012) 24:262–8. doi:10.1016/j.ceb.2011.11.008
47. Harman MW, Dunham-Ems SM, Caimano MJ, Belperron AA, Bockenstedt LK, Fu HC, et al. The heterogeneous motility of the Lyme disease spirochete in gelatin mimics dissemination through tissue. Proc Natl Acad Sci U S A (2012) 109:3059–64. doi:10.1073/pnas.1114362109
48. Dunham-Ems SM, Caimano MJ, Eggers CH, Radolf JD. Borrelia burgdorferi requires the alternative sigma factor RpoS for dissemination within the vector during tick-to-mammal transmission. PLoS Pathog (2012) 8:e1002532. doi:10.1371/journal.ppat.1002532
49. Wu J, Weening EH, Faske JB, Höök M, Skare JT. Invasion of eukaryotic cells by Borrelia burgdorferi requires β(1) integrins and Src kinase activity. Infect Immun (2011) 79:1338–48. doi:10.1128/IAI.01188-10
50. Alban PS, Johnson PW, Nelson DR. Serum-starvation-induced changes in protein synthesis and morphology of Borrelia burgdorferi. Microbiology (2000) 146(Pt 1):119–27. doi:10.1099/00221287-146-1-119
51. Brorson O, Brorson SH. Transformation of cystic forms of Borrelia burgdorferi to normal, mobile spirochetes. Infection (1997) 25:240–6. doi:10.1007/BF01713153
52. Brorson O, Brorson SH. In vitro conversion of Borrelia burgdorferi to cystic forms in spinal fluid, and transformation to mobile spirochetes by incubation in BSK-H medium. Infection (1998) 26:144–50. doi:10.1007/BF02771839
53. Miklossy J, Kasas S, Zurn AD, McCall S, Yu S, McGeer PL. Persisting atypical and cystic forms of Borrelia burgdorferi and local inflammation in Lyme neuroborreliosis. J Neuroinflammation (2008) 5:40. doi:10.1186/1742-2094-5-40
54. Xu H, Sultan S, Yerke A, Moon KH, Wooten RM, Motaleb MA. Borrelia burgdorferi CheY2 is dispensable for chemotaxis or motility but crucial for the infectious life cycle of the spirochete. Infect Immun (2016) 85(1):e264–216. doi:10.1128/IAI.00264-16
55. Moon KH, Hobbs G, Motaleb MA. Borrelia burgdorferi CheD promotes various functions in chemotaxis and the pathogenic life cycle of the spirochete. Infect Immun (2016) 84:1743–52. doi:10.1128/IAI.01347-15
56. Motaleb MA, Miller MR, Li C, Bakker RG, Goldstein SF, Silversmith RE, et al. CheX is a phosphorylated CheY phosphatase essential for Borrelia burgdorferi chemotaxis. J Bacteriol (2005) 187:7963–9. doi:10.1128/JB.187.23.7963-7969.2005
57. Novak EA, Sekar P, Xu H, Moon KH, Manne A, Wooten RM, et al. The Borrelia burgdorferi CheY3 response regulator is essential for chemotaxis and completion of its natural infection cycle. Cell Microbiol (2016) 18(12):1782–99. doi:10.1111/cmi.12617
58. Zhang K, Liu J, Charon NW, Li C. Hypothetical protein BB0569 is essential for chemotaxis of the Lyme disease spirochete Borrelia burgdorferi. J Bacteriol (2016) 198:664–72. doi:10.1128/JB.00877-15
59. Sze CW, Morado DR, Liu J, Charon NW, Xu H, Li C. Carbon storage regulator A (CsrA(Bb)) is a repressor of Borrelia burgdorferi flagellin protein FlaB. Mol Microbiol (2011) 82:851–64. doi:10.1111/j.1365-2958.2011.07853.x
60. Sanjuan E, Esteve-Gassent MD, Maruskova M, Seshu J. Overexpression of CsrA (BB0184) alters the morphology and antigen profiles of Borrelia burgdorferi. Infect Immun (2009) 77:5149–62. doi:10.1128/IAI.00673-09
61. Sultan SZ, Manne A, Stewart PE, Bestor A, Rosa PA, Charon NW, et al. Motility is crucial for the infectious life cycle of Borrelia burgdorferi. Infect Immun (2013) 81:2012–21. doi:10.1128/IAI.01228-12
62. Revel AT, Talaat AM, Norgard MV. DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci U S A (2002) 99:1562–7. doi:10.1073/pnas.032667699
63. Brooks CS, Hefty PS, Jolliff SE, Akins DR. Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect Immun (2003) 71:3371–83. doi:10.1128/IAI.71.6.3371-3383.2003
64. Iyer R, Caimano MJ, Luthra A, Axline D, Corona A, Iacobas DA, et al. Stage-specific global alterations in the transcriptomes of Lyme disease spirochetes during tick feeding and following mammalian host adaptation. Mol Microbiol (2015) 95:509–38. doi:10.1111/mmi.12882
65. Coutte L, Botkin DJ, Gao L, Norris SJ. Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Pathog (2009) 5:e1000293. doi:10.1371/journal.ppat.1000293
66. Troy EB, Lin T, Gao L, Lazinski DW, Camilli A, Norris SJ, et al. Understanding barriers to Borrelia burgdorferi dissemination during infection using massively parallel sequencing. Infect Immun (2013) 81:2347–57. doi:10.1128/IAI.00266-13
67. Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, et al. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol (2004) 173:2003–10. doi:10.4049/jimmunol.173.3.2003
68. Bockenstedt LK, Liu N, Schwartz I, Fish D. MyD88 deficiency enhances acquisition and transmission of Borrelia burgdorferi by Ixodes scapularis ticks. Infect Immun (2006) 74:2154–60. doi:10.1128/IAI.74.4.2154-2160.2006
69. Liveris D, Varde S, Iyer R, Koenig S, Bittker S, Cooper D, et al. Genetic diversity of Borrelia burgdorferi in Lyme disease patients as determined by culture versus direct PCR with clinical specimens. J Clin Microbiol (1999) 37:565–9.
70. Ruzic-Sabljic E, Arnez M, Logar M, Maraspin V, Lotric-Furlan S, Cimperman J, et al. Comparison of Borrelia burgdorferi sensu lato strains isolated from specimens obtained simultaneously from two different sites of infection in individual patients. J Clin Microbiol (2005) 43:2194–200. doi:10.1128/JCM.43.5.2194-2200.2005
71. Wormser GP. Hematogenous dissemination in early Lyme disease. Wien Klin Wochenschr (2006) 118:634–7. doi:10.1007/s00508-006-0688-9
73. Wormser GP, Brisson D, Liveris D, Hanincová K, Sandigursky S, Nowakowski J, et al. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J Infect Dis (2008) 198:1358–64. doi:10.1086/592279
74. Wormser GP, Liveris D, Nowakowski J, Nadelman RB, Cavaliere LF, McKenna D, et al. Association of specific subtypes of Borrelia burgdorferi with hematogenous dissemination in early Lyme disease. J Infect Dis (1999) 180:720–5. doi:10.1086/314922
75. Brisson D, Dykhuizen DE. ospC diversity in Borrelia burgdorferi: different hosts are different niches. Genetics (2004) 168:713–22. doi:10.1534/genetics.104.028738
76. Brisson D, Baxamusa N, Schwartz I, Wormser GP. Biodiversity of Borrelia burgdorferi strains in tissues of Lyme disease patients. PLoS One (2011) 6:e22926. doi:10.1371/journal.pone.0022926
77. Wang IN, Dykhuizen DE, Qiu W, Dunn JJ, Bosler EM, Luft BJ. Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics (1999) 151:15–30.
78. Rhodes DVL, Earnhart CG, Mather TN, Meeus PFM, Marconi RT. Identification of Borrelia burgdorferi ospC genotypes in canine tissue following tick infestation: implications for Lyme disease vaccine and diagnostic assay design. Vet J (2013) 198:412–8. doi:10.1016/j.tvjl.2013.07.019
79. Earnhart CG, Buckles EL, Dumler JS, Marconi RT. Demonstration of OspC type diversity in invasive human Lyme disease isolates and identification of previously uncharacterized epitopes that define the specificity of the OspC murine antibody response. Infect Immun (2005) 73:7869–77. doi:10.1128/IAI.73.12.7869-7877.2005
80. Dykhuizen DE, Brisson D, Sandigursky S, Wormser GP, Nowakowski J, Nadelman RB, et al. The propensity of different Borrelia burgdorferi sensu stricto genotypes to cause disseminated infections in humans. Am J Trop Med Hyg (2008) 78:806–10.
81. Seinost G, Dykhuizen DE, Dattwyler RJ, Golde WT, Dunn JJ, Wang IN, et al. Four clones of Borrelia burgdorferi sensu stricto cause invasive infection in humans. Infect Immun (1999) 67:3518–24.
83. Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Höök M, Cirillo JD, et al. Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that the fibronectin-binding protein BBK32 is required for optimal infectivity. Mol Microbiol (2011) 82:99–113. doi:10.1111/j.1365-2958.2011.07801.x
84. Blevins JS, Revel AT, Smith AH, Bachlani GN, Norgard MV. Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi. Appl Environ Microbiol (2007) 73:1501–13. doi:10.1128/AEM.02454-06
85. Persat A, Nadell CD, Kim MK, Ingremeau F, Siryaporn A, Drescher K, et al. The mechanical world of bacteria. Cell (2015) 161:988–97. doi:10.1016/j.cell.2015.05.005
86. Moriarty TJ, Norman MU, Colarusso P, Bankhead T, Kubes P, Chaconas G. Real-time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog (2008) 4:e1000090. doi:10.1371/journal.ppat.1000090
87. Norman MU, Moriarty TJ, Dresser AR, Millen B, Kubes P, Chaconas G. Molecular mechanisms involved in vascular interactions of the Lyme disease pathogen in a living host. PLoS Pathog (2008) 4:e1000169. doi:10.1371/journal.ppat.1000169
88. Moriarty TJ, Shi M, Lin Y-P, Ebady R, Zhou H, Odisho T, et al. Vascular binding of a pathogen under shear force through mechanistically distinct sequential interactions with host macromolecules. Mol Microbiol (2012) 86:1116–31. doi:10.1111/mmi.12045
89. Ebady R, Niddam AF, Boczula AE, Kim YR, Gupta N, Tang TT, et al. Biomechanics of Borrelia burgdorferi vascular interactions. Cell Rep (2016) 16:2593–604. doi:10.1016/j.celrep.2016.08.013
90. Kim JH, Singvall J, Schwarz-Linek U, Johnson BJ, Potts JR, Hook M. BBK32, a fibronectin binding MSCRAMM from Borrelia burgdorferi, contains a disordered region that undergoes a conformational change on ligand binding. J Biol Chem (2004) 279:41706–14. doi:10.1074/jbc.M401691200
91. Fischer JR, LeBlanc KT, Leong JM. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect Immun (2006) 74:435–41. doi:10.1128/IAI.74.1.435-441.2006
92. Probert WS, Kim JH, Hook M, Johnson BJ. Mapping the ligand-binding region of Borrelia burgdorferi fibronectin-binding protein BBK32. Infect Immun (2001) 69:4129–33. doi:10.1128/IAI.69.6.4129-4133.2001
93. Benoit VM, Fischer JR, Lin YP, Parveen N, Leong JM. Allelic variation of the Lyme disease spirochete adhesin DbpA influences spirochetal binding to decorin, dermatan sulfate, and mammalian cells. Infect Immun (2011) 79:3501–9. doi:10.1128/IAI.00163-11
94. Caine JA, Coburn J. A short-term Borrelia burgdorferi infection model identifies tissue tropisms and bloodstream survival conferred by adhesion proteins. Infect Immun (2015) 83:3184–94. doi:10.1128/IAI.00349-15
95. Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Höök M, et al. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol Microbiol (2006) 59:1591–601. doi:10.1111/j.1365-2958.2005.05042.x
96. Fikrig E, Feng W, Barthold SW, Telford SR, Flavell RA. Arthropod- and host-specific Borrelia burgdorferi bbk32 expression and the inhibition of spirochete transmission. J Immunol (2000) 164:5344–51. doi:10.4049/jimmunol.164.10.5344
97. Li X, Liu X, Beck DS, Kantor FS, Fikrig E. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect Immun (2006) 74:3305–13. doi:10.1128/IAI.02035-05
98. Raibaud S, Schwarz-Linek U, Kim JH, Jenkins HT, Baines ER, Gurusiddappa S, et al. Borrelia burgdorferi binds fibronectin through a tandem beta-zipper, a common mechanism of fibronectin binding in staphylococci, streptococci, and spirochetes. J Biol Chem (2005) 280:18803–9. doi:10.1074/jbc.M501731200
99. Liang X, Garcia BL, Visai L, Prabhakaran S, Meenan NA, Potts JR, et al. Allosteric regulation of fibronectin/α5β1 interaction by fibronectin-binding MSCRAMMs. PLoS One (2016) 11:e0159118. doi:10.1371/journal.pone.0159118
100. Thomas W. Catch bonds in adhesion. Annu Rev Biomed Eng (2008) 10:39–57. doi:10.1146/annurev.bioeng.10.061807.160427
101. Guo BP, Brown EL, Dorward DW, Rosenberg LC, Hook M. Decorin-binding adhesins from Borrelia burgdorferi. Mol Microbiol (1998) 30:711–23. doi:10.1046/j.1365-2958.1998.01103.x
102. Guo BP, Norris SJ, Rosenberg LC, Hook M. Adherence of Borrelia burgdorferi to the proteoglycan decorin. Infect Immun (1995) 63:3467–72.
103. Fortune DE, Lin Y-P, Deka RK, Groshong AM, Moore BP, Hagman KE, et al. Identification of lysine residues in the Borrelia burgdorferi DbpA adhesin required for murine infection. Infect Immun (2014) 82:3186–98. doi:10.1128/IAI.02036-14
104. Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, et al. Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PLoS One (2012) 7:e47532. doi:10.1371/journal.pone.0047532
105. Fischer JR, Parveen N, Magoun L, Leong JM. Decorin-binding proteins A and B confer distinct mammalian cell type-specific attachment by Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci U S A (2003) 100:7307–12. doi:10.1073/pnas.1231043100
106. Roberts WC, Mullikin BA, Lathigra R, Hanson MS. Molecular analysis of sequence heterogeneity among genes encoding decorin binding proteins A and B of Borrelia burgdorferi sensu lato. Infect Immun (1998) 66:5275–85.
107. Hyde JA, Trzeciakowski JP, Skare JT. Borrelia burgdorferi alters its gene expression and antigenic profile in response to CO2 levels. J Bacteriol (2007) 189:437–45. doi:10.1128/JB.01109-06
108. Carroll JA, Garon CF, Schwan TG. Effects of environmental pH on membrane proteins in Borrelia burgdorferi. Infect Immun (1999) 67:3181–7.
109. Carroll JA, Cordova RM, Garon CF. Identification of 11 pH-regulated genes in Borrelia burgdorferi localizing to linear plasmids. Infect Immun (2000) 68:6677–84. doi:10.1128/IAI.68.12.6677-6684.2000
110. Ouyang Z, Haq S, Norgard MV. Analysis of the dbpBA upstream regulatory region controlled by RpoS in Borrelia burgdorferi. J Bacteriol (2010) 192:1965–74. doi:10.1128/JB.01616-09
111. Hubner A, Yang X, Nolen DM, Popova TG, Cabello FC, Norgard MV. Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN-RpoS regulatory pathway. Proc Natl Acad Sci U S A (2001) 98:12724–9. doi:10.1073/pnas.231442498
112. Ojaimi C, Brooks C, Casjens S, Rosa P, Elias A, Barbour A, et al. Profiling of temperature-induced changes in Borrelia burgdorferi gene expression by using whole genome arrays. Infect Immun (2003) 71:1689–705. doi:10.1128/IAI.71.4.1689-1705.2003
113. Yang X, Goldberg MS, Popova TG, Schoeler GB, Wikel SK, Hagman KE, et al. Interdependence of environmental factors influencing reciprocal patterns of gene expression in virulent Borrelia burgdorferi. Mol Microbiol (2000) 37:1470–9. doi:10.1046/j.1365-2958.2000.02104.x
114. Caimano MJ, Iyer R, Eggers CH, Gonzalez C, Morton EA, Gilbert MA, et al. Analysis of the RpoS regulon in Borrelia burgdorferi in response to mammalian host signals provides insight into RpoS function during the enzootic cycle. Mol Microbiol (2007) 65:1193–217. doi:10.1111/j.1365-2958.2007.05860.x
115. Fisher MA, Grimm D, Henion AK, Elias AF, Stewart PE, Rosa PA, et al. Borrelia burgdorferi sigma54 is required for mammalian infection and vector transmission but not for tick colonization. Proc Natl Acad Sci U S A (2005) 102:5162–7. doi:10.1073/pnas.0408536102
116. Ouyang Z, Blevins JS, Norgard MV. Transcriptional interplay among the regulators Rrp2, RpoN and RpoS in Borrelia burgdorferi. Microbiology (2008) 154:2641–8. doi:10.1099/mic.0.2008/019992-0
117. Schmit VL, Patton TG, Gilmore RD. Analysis of Borrelia burgdorferi surface proteins as determinants in establishing host cell interactions. Front Microbiol (2011) 2:141. doi:10.3389/fmicb.2011.00141
118. Imai DM, Samuels DS, Feng S, Hodzic E, Olsen K, Barthold SW. The early dissemination defect attributed to disruption of decorin-binding proteins is abolished in chronic murine Lyme borreliosis. Infect Immun (2013) 81:1663–73. doi:10.1128/IAI.01359-12
119. Weening EH, Parveen N, Trzeciakowski JP, Leong JM, Hook M, Skare JT. Borrelia burgdorferi lacking DbpBA exhibits an early survival defect during experimental infection. Infect Immun (2008) 76:5694–705. doi:10.1128/IAI.00690-08
120. Blevins JS, Hagman KE, Norgard MV. Assessment of decorin-binding protein A to the infectivity of Borrelia burgdorferi in the murine models of needle and tick infection. BMC Microbiol (2008) 8:82. doi:10.1186/1471-2180-8-82
121. Shi Y, Xu Q, Seemanapalli SV, McShan K, Liang FT. The dbpBA locus of Borrelia burgdorferi is not essential for infection of mice. Infect Immun (2006) 74:6509–12. doi:10.1128/IAI.00740-06
122. Shi Y, Xu Q, McShan K, Liang FT. Both decorin-binding proteins A and B are critical for the overall virulence of Borrelia burgdorferi. Infect Immun (2008) 76:1239–46. doi:10.1128/IAI.00897-07
123. Hodzic E, Feng S, Freet KJ, Borjesson DL, Barthold SW. Borrelia burgdorferi population kinetics and selected gene expression at the host-vector interface. Infect Immun (2002) 70:3382–8. doi:10.1128/IAI.70.7.3382-3388.2002
124. Hodzic E, Feng S, Freet KJ, Barthold SW. Borrelia burgdorferi population dynamics and prototype gene expression during infection of immunocompetent and immunodeficient mice. Infect Immun (2003) 71:5042–55. doi:10.1128/IAI.71.9.5042-5055.2003
125. Brown EL, Wooten RM, Johnson BJ, Iozzo RV, Smith A, Dolan MC, et al. Resistance to Lyme disease in decorin-deficient mice. J Clin Invest (2001) 107:845–52. doi:10.1172/JCI11692
126. Salo J, Pietikäinen A, Söderström M, Auvinen K, Salmi M, Ebady R, et al. Flow-tolerant adhesion of a bacterial pathogen to human endothelial cells through interaction with biglycan. J Infect Dis (2016) 213:1623–31. doi:10.1093/infdis/jiw003
127. Grab DJ, Perides G, Dumler JS, Kim KJ, Park J, Kim YV, et al. Borrelia burgdorferi, host-derived proteases, and the blood-brain barrier. Infect Immun (2005) 73:1014–22. doi:10.1128/IAI.73.2.1014-1022.2005
128. Grab DJ, Nyarko E, Nikolskaia OV, Kim YV, Dumler JS. Human brain microvascular endothelial cell traversal by Borrelia burgdorferi requires calcium signaling. Clin Microbiol Infect (2009) 15:422–6. doi:10.1111/j.1469-0691.2009.02869.x
129. Vieira ML, Nascimento AL. Interaction of spirochetes with the host fibrinolytic system and potential roles in pathogenesis. Crit Rev Microbiol (2016) 42:573–87. doi:10.3109/1040841X.2014.972336
130. Comstock LE, Thomas DD. Penetration of endothelial cell monolayers by Borrelia burgdorferi. Infect Immun (1989) 57:1626–8.
131. Comstock LE, Thomas DD. Characterization of Borrelia burgdorferi invasion of cultured endothelial cells. Microb Pathog (1991) 10:137–48. doi:10.1016/0882-4010(91)90074-K
132. Thomas DD, Comstock LE. Interaction of Lyme disease spirochetes with cultured eucaryotic cells. Infect Immun (1989) 57:1324–6.
133. Ma Y, Sturrock A, Weis JJ. Intracellular localization of Borrelia burgdorferi within human endothelial cells. Infect Immun (1991) 59:671–8.
134. Szczepanski A, Furie MB, Benach JL, Lane BP, Fleit HB. Interaction between Borrelia burgdorferi and endothelium in vitro. J Clin Invest (1990) 85:1637–47. doi:10.1172/JCI114615
135. Klempner MS, Noring R, Rogers RA. Invasion of human skin fibroblasts by the Lyme disease spirochete, Borrelia burgdorferi. J Infect Dis (1993) 167:1074–81. doi:10.1093/infdis/167.5.1074
136. Sellati TJ, Burns MJ, Ficazzola MA, Furie MB. Borrelia burgdorferi upregulates expression of adhesion molecules on endothelial cells and promotes transendothelial migration of neutrophils in vitro. Infect Immun (1995) 63:4439–47.
137. Gergel EI, Furie MB. Activation of endothelium by Borrelia burgdorferi in vitro enhances transmigration of specific subsets of T lymphocytes. Infect Immun (2001) 69:2190–7. doi:10.1128/IAI.69.4.2190-2197.2001
138. Livengood JA, Gilmore J. Invasion of human neuronal and glial cells by an infectious strain of Borrelia burgdorferi. Microbes Infect (2006) 8:2832–40. doi:10.1016/j.micinf.2006.08.014
139. Girschick HJ, Huppertz HI, Rüssmann H, Krenn V, Karch H. Intracellular persistence of Borrelia burgdorferi in human synovial cells. Rheumatol Int (1996) 16:125–32. doi:10.1007/BF01409985
140. Livengood JA, Schmit VL, Gilmore RD. Global transcriptome analysis of Borrelia burgdorferi during association with human neuroglial cells. Infect Immun (2008) 76:298–307. doi:10.1128/IAI.00866-07
141. Skare JT, Mirzabekov TA, Shang ES, Blanco DR, Erdjument-Bromage H, Bunikis J, et al. The Oms66 (p66) protein is a Borrelia burgdorferi porin. Infect Immun (1997) 65:3654–61.
142. Pinne M, Thein M, Denker K, Benz R, Coburn J, Bergstrom S. Elimination of channel-forming activity by insertional inactivation of the p66 gene in Borrelia burgdorferi. FEMS Microbiol Lett (2007) 266:241–9. doi:10.1111/j.1574-6968.2006.00529.x
143. Kenedy MR, Luthra A, Anand A, Dunn JP, Radolf JD, Akins DR. Structural modeling and physicochemical characterization provide evidence that P66 forms a β-barrel in the Borrelia burgdorferi outer membrane. J Bacteriol (2014) 196:859–72. doi:10.1128/JB.01236-13
144. Bárcena-Uribarri I, Thein M, Sacher A, Bunikis I, Bonde M, Bergström S, et al. P66 porins are present in both Lyme disease and relapsing fever spirochetes: a comparison of the biophysical properties of P66 porins from six Borrelia species. Biochim Biophys Acta (2010) 1798:1197–203. doi:10.1016/j.bbamem.2010.02.011
145. Bárcena-Uribarri I, Thein M, Maier E, Bonde M, Bergström S, Benz R. Use of nonelectrolytes reveals the channel size and oligomeric constitution of the Borrelia burgdorferi P66 porin. PLoS One (2013) 8:e78272. doi:10.1371/journal.pone.0078272
146. Coburn J, Leong JM, Erban JK. Integrin alpha IIb beta 3 mediates binding of the Lyme disease agent Borrelia burgdorferi to human platelets. Proc Natl Acad Sci U S A (1993) 90:7059–63. doi:10.1073/pnas.90.15.7059
147. Coburn J, Cugini C. Targeted mutation of the outer membrane protein P66 disrupts attachment of the Lyme disease agent, Borrelia burgdorferi, to integrin alpha(v)beta3. Proc Natl Acad Sci U S A (2003) 100:7301–6. doi:10.1073/pnas.1131117100
148. Coburn J, Magoun L, Bodary SC, Leong JM. Integrins alpha(v)beta3 and alpha5beta1 mediate attachment of Lyme disease spirochetes to human cells. Infect Immun (1998) 66:1946–52.
149. Ristow LC, Miller HE, Padmore LJ, Chettri R, Salzman N, Caimano MJ, et al. The β3-integrin ligand of Borrelia burgdorferi is critical for infection of mice but not ticks. Mol Microbiol (2012) 85:1105–18. doi:10.1111/j.1365-2958.2012.08160.x
150. Cugini C, Medrano M, Schwan TG, Coburn J. Regulation of expression of the Borrelia burgdorferi beta(3)-chain integrin ligand, P66, in ticks and in culture. Infect Immun (2003) 71:1001–7. doi:10.1128/IAI.71.2.1001-1007.2003
151. Defoe G, Coburn J. Delineation of Borrelia burgdorferi p66 sequences required for integrin alpha(IIb)beta(3) recognition. Infect Immun (2001) 69:3455–9. doi:10.1128/IAI.69.5.3455-3459.2001
152. LaFrance ME, Pierce JV, Antonara S, Coburn J. The Borrelia burgdorferi integrin ligand P66 affects gene expression by human cells in culture. Infect Immun (2011) 79:3249–61. doi:10.1128/IAI.05122-11
153. Antonara S, Chafel RM, LaFrance M, Coburn J. Borrelia burgdorferi adhesins identified using in vivo phage display. Mol Microbiol (2007) 66:262–76. doi:10.1111/j.1365-2958.2007.05924.x
154. Ristow LC, Bonde M, Lin Y-P, Sato H, Curtis M, Wesley E, et al. Integrin binding by Borrelia burgdorferi P66 facilitates dissemination but is not required for infectivity. Cell Microbiol (2015) 17:1021–36. doi:10.1111/cmi.12418
155. Kumar D, Ristow LC, Shi M, Mukherjee P, Caine JA, Lee W-Y, et al. Intravital imaging of vascular transmigration by the Lyme spirochete: requirement for the integrin binding residues of the B. burgdorferi P66 protein. PLoS Pathog (2015) 11:e1005333. doi:10.1371/journal.ppat.1005333
156. Lee W-Y, Moriarty TJ, Wong CHY, Zhou H, Strieter RM, van Rooijen N, et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol (2010) 11:295–302. doi:10.1038/ni.1855
157. Lee WY, Sanz MJ, Wong CH, Hardy PO, Salman-Dilgimen A, Moriarty TJ, et al. Invariant natural killer T cells act as an extravascular cytotoxic barrier for joint-invading Lyme Borrelia. Proc Natl Acad Sci U S A (2014) 111:13936–41. doi:10.1073/pnas.1404769111
158. Sellati TJ, Abrescia LD, Radolf JD, Furie MB. Outer surface lipoproteins of Borrelia burgdorferi activate vascular endothelium in vitro. Infect Immun (1996) 64:3180–7.
159. Singh N, Kuppili RR, Bose K. The structural basis of mode of activation and functional diversity: a case study with HtrA family of serine proteases. Arch Biochem Biophys (2011) 516:85–96. doi:10.1016/j.abb.2011.10.007
160. Hansen G, Hilgenfeld R. Architecture and regulation of HtrA-family proteins involved in protein quality control and stress response. Cell Mol Life Sci (2013) 70:761–75. doi:10.1007/s00018-012-1076-4
161. Gherardini FC. Borrelia burgdorferi HtrA may promote dissemination and irritation. Mol Microbiol (2013) 90:209–13. doi:10.1111/mmi.12390
162. Stricker RB, Johnson L. Borrelia burgdorferi aggrecanase activity: more evidence for persistent infection in Lyme disease. Front Cell Infect Microbiol (2013) 3:40. doi:10.3389/fcimb.2013.00040
163. Coleman JL, Crowley JT, Toledo AM, Benach JL. The HtrA protease of Borrelia burgdorferi degrades outer membrane protein BmpD and chemotaxis phosphatase CheX. Mol Microbiol (2013) 88:619–33. doi:10.1111/mmi.12213
164. Russell TM, Johnson BJB. Lyme disease spirochaetes possess an aggrecan-binding protease with aggrecanase activity. Mol Microbiol (2013) 90:228–40. doi:10.1111/mmi.12276
165. Russell TM, Delorey MJ, Johnson BJB. Borrelia burgdorferi BbHtrA degrades host ECM proteins and stimulates release of inflammatory cytokines in vitro. Mol Microbiol (2013) 90:241–51. doi:10.1111/mmi.12377
166. Russell TM, Tang X, Goldstein JM, Bagarozzi D, Johnson BJB. The salt-sensitive structure and zinc inhibition of Borrelia burgdorferi protease BbHtrA. Mol Microbiol (2016) 99:586–96. doi:10.1111/mmi.13251
167. Ye M, Sharma K, Thakur M, Smith AA, Buyuktanir O, Xiang X, et al. HtrA, a temperature- and stationary phase-activated protease involved in maturation of a key microbial virulence determinant, facilitates Borrelia burgdorferi infection in mammalian hosts. Infect Immun (2016) 84:2372–81. doi:10.1128/IAI.00360-16
168. Kariu T, Sharma K, Singh P, Smith AA, Backstedt B, Buyuktanir O, et al. BB0323 and novel virulence determinant BB0238: Borrelia burgdorferi proteins that interact with and stabilize each other and are critical for infectivity. J Infect Dis (2015) 211:462–71. doi:10.1093/infdis/jiu460
169. Coleman JL, Toledo A, Benach JL. Borrelia burgdorferi HtrA: evidence for twofold proteolysis of outer membrane protein p66. Mol Microbiol (2016) 99:135–50. doi:10.1111/mmi.13221
170. Watanabe H, Yamada Y, Kimata K. Roles of aggrecan, a large chondroitin sulfate proteoglycan, in cartilage structure and function. J Biochem (1998) 124:687–93. doi:10.1093/oxfordjournals.jbchem.a022166
171. Behera AK, Hildebrand E, Szafranski J, Hung H-H, Grodzinsky AJ, Lafyatis R, et al. Role of aggrecanase 1 in Lyme arthritis. Arthritis Rheum (2006) 54:3319–29. doi:10.1002/art.22128
172. Ullmann AJ, Russell TM, Dolan MC, Williams M, Hojgaard A, Weiner ZP, et al. Evaluation of Borrelia burgdorferi BbHtrA protease as a vaccine candidate for Lyme borreliosis in mice. PLoS One (2015) 10:e0128868. doi:10.1371/journal.pone.0128868
173. Hu LT, Eskildsen MA, Masgala C, Steere AC, Arner EC, Pratta MA, et al. Host metalloproteinases in Lyme arthritis. Arthritis Rheum (2001) 44:1401–10. doi:10.1002/1529-0131(200106)44:6<1401:AID-ART234>3.0.CO;2-S
174. Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem (1999) 274:21491–4. doi:10.1074/jbc.274.31.21491
175. Gebbia JA, Coleman JL, Benach JL. Borrelia spirochetes upregulate release and activation of matrix metalloproteinase gelatinase B (MMP-9) and collagenase 1 (MMP-1) in human cells. Infect Immun (2001) 69:456–62. doi:10.1128/IAI.69.1.456-462.2001
176. Singh SK, Morbach H, Nanki T, Faber C, Baar V, Girschick HJ. Differential expression of matrix metalloproteinases and cyclooxygenases in synovial cells exposed to Borrelia burgdorferi. Inflamm Res (2004) 53:689–96. doi:10.1007/s00011-004-1313-x
177. Bramwell KKC, Mock K, Ma Y, Weis JH, Teuscher C, Weis JJ. β-Glucuronidase, a regulator of Lyme arthritis severity, modulates lysosomal trafficking and MMP-9 secretion in response to inflammatory stimuli. J Immunol (2015) 195:1647–56. doi:10.4049/jimmunol.1500212
178. Zhao Z, Chang H, Trevino RP, Whren K, Bhawan J, Klempner MS. Selective up-regulation of matrix metalloproteinase-9 expression in human erythema migrans skin lesions of acute Lyme disease. J Infect Dis (2003) 188:1098–104. doi:10.1086/379039
179. Behera AK, Thorpe CM, Kidder JM, Smith W, Hildebrand E, Hu LT. Borrelia burgdorferi-induced expression of matrix metalloproteinases from human chondrocytes requires mitogen-activated protein kinase and Janus kinase/signal transducer and activator of transcription signaling pathways. Infect Immun (2004) 72:2864–71. doi:10.1128/IAI.72.5.2864-2871.2004
180. Gebbia JA, Coleman JL, Benach JL. Selective induction of matrix metalloproteinases by Borrelia burgdorferi via toll-like receptor 2 in monocytes. J Infect Dis (2004) 189:113–9. doi:10.1086/380414
181. Behera AK, Hildebrand E, Uematsu S, Akira S, Coburn J, Hu LT. Identification of a TLR-independent pathway for Borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin alpha 3 beta 1. J Immunol (2006) 177:657–64. doi:10.4049/jimmunol.177.1.657
182. Heilpern AJ, Wertheim W, He J, Perides G, Bronson RT, Hu LT. Matrix metalloproteinase 9 plays a key role in Lyme arthritis but not in dissemination of Borrelia burgdorferi. Infect Immun (2009) 77:2643–9. doi:10.1128/IAI.00214-09
183. Coleman JL, Roemer EJ, Benach JL. Plasmin-coated Borrelia burgdorferi degrades soluble and insoluble components of the mammalian extracellular matrix. Infect Immun (1999) 67:3929–36.
184. Coleman JL, Benach JL. Use of the plasminogen activation system by microorganisms. J Lab Clin Med (1999) 134:567–76. doi:10.1016/S0022-2143(99)90095-1
185. Klempner MS, Noring R, Epstein MP, McCloud B, Hu R, Limentani SA, et al. Binding of human plasminogen and urokinase-type plasminogen activator to the Lyme disease spirochete, Borrelia burgdorferi. J Infect Dis (1995) 171:1258–65. doi:10.1093/infdis/171.5.1258
186. Klempner MS, Noring R, Epstein MP, McCloud B, Rogers RA. Binding of human urokinase type plasminogen activator and plasminogen to Borrelia species. J Infect Dis (1996) 174:97–104. doi:10.1093/infdis/174.1.97
187. Coleman JL, Sellati TJ, Testa JE, Kew RR, Furie MB, Benach JL. Borrelia burgdorferi binds plasminogen, resulting in enhanced penetration of endothelial monolayers. Infect Immun (1995) 63:2478–84.
188. Coleman JL, Gebbia JA, Piesman J, Degen JL, Bugge TH, Benach JL. Plasminogen is required for efficient dissemination of B. burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell (1997) 89:1111–9. doi:10.1016/S0092-8674(00)80298-6
189. Coleman JL, Gebbia JA, Benach JL. Borrelia burgdorferi and other bacterial products induce expression and release of the urokinase receptor (CD87). J Immunol (2001) 166:473–80. doi:10.4049/jimmunol.166.1.473
190. Fuchs H, Simon MM, Wallich R, Bechtel M, Kramer MD. Borrelia burgdorferi induces secretion of pro-urokinase-type plasminogen activator by human monocytes. Infect Immun (1996) 64:4307–12.
Keywords: Borrelia burgdorferi, Lyme disease, dissemination, invasion, vascular interaction, protease, motility, chemotaxis
Citation: Hyde JA (2017) Borrelia burgdorferi Keeps Moving and Carries on: A Review of Borrelial Dissemination and Invasion. Front. Immunol. 8:114. doi: 10.3389/fimmu.2017.00114
Received: 30 November 2016; Accepted: 25 January 2017;
Published: 21 February 2017
Edited by:
Monica E. Embers, Tulane University, USAReviewed by:
Sukanya Narasimhan, Yale School of Medicine, USAJanis J. Weis, University of Utah, USA
Copyright: © 2017 Hyde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny A. Hyde, anNoeWRlQG1lZGljaW5lLnRhbWhzYy5lZHU=