 Jelena Milovanovic1,2
Jelena Milovanovic1,2 Branka Popovic3
Branka Popovic3 Marija Milovanovic1
Marija Milovanovic1 Daria Kvestak3Aleksandar Arsenijevic1Bojana Stojanovic1,4Irena Tanaskovic2
Daria Kvestak3Aleksandar Arsenijevic1Bojana Stojanovic1,4Irena Tanaskovic2 Astrid Krmpotic3Nebojsa Arsenijevic1Stipan Jonjic3
Astrid Krmpotic3Nebojsa Arsenijevic1Stipan Jonjic3 Miodrag L. Lukic1*
Miodrag L. Lukic1*
- 1Center for Molecular Medicine and Stem Cell Research, Faculty of Medical Sciences, University of Kragujevac, Kragujevac, Serbia
- 2Faculty of Medical Sciences, Institute of Histology, University of Kragujevac, Kragujevac, Serbia
- 3Center for Proteomics, Faculty of Medicine, Department for Histology and Embryology, University of Rijeka, Rijeka, Croatia
- 4Faculty of Medical Sciences, Institute of Pathophysiology, University of Kragujevac, Kragujevac, Serbia
In contrast to C57BL/6 mice, BALB/c mice are relatively resistant to the induction of experimental autoimmune encephalomyelitis (EAE) after challenge with MOG35–55 peptide. Here, we provide the first evidence that infection with murine cytomegalovirus (MCMV) in adulthood abrogates this resistance. Infected BALB/c mice developed clinical and histological signs similar to those seen in susceptible C57BL/6 mice. In addition to CD4+ cells, large proportion of cells in the infiltrate of diseased BALB/c mice was CD8+, similar with findings in multiple sclerosis. CD8+ cells that responded to ex vivo restimulation with MOG35–55 were not specific for viral epitopes pp89 and m164. MCMV infection favors proinflammatory type of dendritic cells (CD86+CD40+CD11c+) in the peripheral lymph organs, M1 type of microglia in central nervous system, and increases development of Th1/Th17 encephalitogenic cells. This study indicates that MCMV may enhance autoimmune neuropathology and abrogate inherent resistance to EAE in mouse strain by enhancing proinflammatory phenotype of antigen-presenting cells, Th1/Th17, and CD8 response to MOG35–55.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating disease of the central nervous system (CNS) with axonal injury, characterized by varying clinical course, pathology, and inflammatory patterns (1). It develops in susceptible hosts after interaction with environmental factors which trigger the disease by promoting the activation of myelin-specific T cells that normally circulate in the peripheral lymph organs of all individuals (2). It has been suggested that some infectious agents, in particular viruses, may be potential triggers of MS (2–4). Among different infective agents, Epstein–Barr virus (EBV) has been mostly associated with increased MS risk (5). Recently, it has been shown increased CD8+ T cell response to EBV lytic antigens in active MS and also in relapses (6). Infection with murine gamma herpesvirus 68 (γHV-68), the murine homolog to EBV, polarizes the adaptive immune response and heightens CNS pathology following experimental autoimmune encephalomyelitis (EAE) induction and likely, influences MS pathogenesis (7).
Experimental autoimmune encephalomyelitis is the experimental model of MS, induced in susceptible animals by active immunization with myelin antigens mixed with adjuvant (8). Immunized mice develop ascending paralysis with CD4+ T cells and macrophages in infiltrations in the white matter of the spinal cord, and with minimal brain inflammation in the majority of experimental models. However, in MS, the vast majority of myelin lesions are found within the brain parenchyma with infiltrations that contain equivalent numbers of CD8+ T and CD4+ T cells (9, 10). Despite these differences, EAE is considered as a valuable tool for research of MS pathogenesis. Moreover, several therapeutics that are now being used to treat MS were developed in EAE (11). BALB/c mice are found partially or completely resistant to the induction of EAE with encephalitogenic peptide, myelin oligodendrocyte glycoprotein (MOG35–55).
Cytomegalovirus (CMV) classified within the Betaherpesvirinae subfamily establishes life-long latent infections in 70–100% of the human population (12). After a primary infection of fibroblasts, epithelial, endothelial, and smooth muscle cells (6), mostly asymptomatic in the immunocompetent host, CMV persists in myeloid precursor cells (7). During latency periodic asymptomatic reactivations occur (13). CMV contains a large number of latent and lytic genes, many of which code proteins that have the role in immunoregulation (5). When monocytes that carry CMV enter visceral parenchyma and differentiate into macrophages and myeloid dendritic cells, virus reactivates and through expression of different genes can modulate the immune response of the host (14).
Data on the role of CMV infection in etiopathogenesis of MS are controversial. CMV has been found in demyelinating plaques and the cerebrospinal fluid of MS patients (15) and causes demyelination mainly in the CNS of immunocompromised hosts (16). Further, enhancement of numbers of EBV and CMV-specific CD8+ T cells among T cells in chronic inflammatory lesions of brain of MS patients was reported (17). Several studies involving human subjects indicate correlation between CMV infection and MS development, greater rate of relapses and greater brain atrophy (18–20). Other studies indicate that CMV seropositivity is associated with a decreased MS risk and predicts a better clinical and radiological outcome in MS patient (21), suggesting a protective effect of CMV on autoimmune neuropathology (22). Furthermore, CMV encodes multiple factors that trigger immunomodulatory or evasion mechanisms, which can decrease the immune response in MS patients (23, 24).
We have recently shown that deletion of an immunoregulatory pathway, IL-33/ST2 axis, may enhance susceptibility to EAE in resistant BALB/c strain (25, 26). The present study was done with the aim to explore whether infection with murine CMV (MCMV) create “fertile field” (27, 28) that facilitates the expansion and activation of encephalitogenic cells leading to autoimmune disease of CNS.
Here, we show that MCMV infected and MOG35–55 immunized BALB/c mice develop very pronounced neuroinflammation with extensive infiltrations in brain and spinal cord parenchyma containing large proportion of CD8+ cells in infiltrates in addition to accentuation of Th1 and Th17 immune response and skewing microglia to M1 phenotype. Our results are compatible with the notion that MCMV abrogates inherent resistance of BALB/c mice to EAE induction with MOG35–55 peptide through enhancement of inflammatory dendritic cells in the periphery, M1 type of microglia and recruitment of MOG35–55 responsive CD8+ T cells in the CNS. Thus, CMV-induced inflammatory environment may enhance autoimmunity in CNS.
Materials and Methods
Infection, Induction, and Scoring of EAE
Female 8-week-old BALB/c mice were used throughout this study. Mice were infected subcutaneously (footped) with 105 plaque-forming units of tissue culture MCMV, strain MW97.01 (29). EAE was induced 10 days after infection by subcutaneous administration of 200 µL suspension at two sites over the hind flanks. Depletion of CD4+ lymphocytes, where indicated, was performed with intraperitoneal injection of 100 µg of anti-CD4 mAb, 1 day prior to and 5 days after MOG35–55 immunization. The suspension consisted of 300 µg MOG35–55 peptide (Sigma-Aldrich, Germany) in 100 µL of PBS, emulsified with 100 µL complete Freund’s adjuvant (Sigma-Aldrich, Germany) with 0.7 mg heat-inactivated Mycobacterium tuberculosis (strain H37 RA; Difco Laboratories, Detroit, MI, USA). Each mouse was immediately thereafter, injected intraperitoneally and 48 h later with 300 ng pertussis toxin (List Biological Laboratories, Campbell, CA, USA) in 100 µL 0.9% NaCl. Clinical signs of EAE were assessed daily by the following scoring system: grade 0, no signs; grade 1, paralyzed tail; grade 2, ataxic; grade 2.5, one hind leg paralyzed; grade 3, both hind legs paralyzed; grade 3.5, three legs paralyzed; grade 4, both hind legs and front limbs completely paralyzed; grade 5, moribund as previously described (30, 31). Mice were monitored daily with fluid administration and mashed chow on the base of cages for all mice displaying a clinical score of 3. Mice were maintained in our animal facilities in a temperature-controlled environment with a 12-h light/12-h dark cycle and were administered standard laboratory food and water ad libitum. All experiments were approved by and conducted in accordance with the Guidelines of the Animal Ethics Committee of Faculty of Medical Sciences, University of Kragujevac, Serbia. Endangered animal species were not used in this study.
Isolation of Mononuclear Cells from CNS and Lymph Nodes
At day 15 post-EAE induction (mean clinical score of 3 for MCMV EAE mice), mice were perfused with PBS, and brain and spinal cord were carefully removed. The mononuclear cells from CNS were isolated as described previously (25). Briefly, the brains and spinal cords were homogenized in RPMI 1640 (Sigma-Aldrich) with 10% FBS and 1 mg/mL collagenase type I (Sigma-Aldrich) and incubated at 37°C for 60 min. After digestion, the tissue was passed through a 40-µm mesh, pelleted, resuspended in 10 mL 30% Percoll (Sigma-Aldrich), overlaid onto 5 mL 70% Percoll, and centrifuged at 390 g for 20 min. The myelin layer was removed, and the mononuclear cells accumulated in the intermediate phase were collected, washed twice in PBS, and resuspended in RPMI 1640 containing 10% FBS. Total cell numbers were determined by counting on a hemocytometer, and viability was assessed by trypan blue exclusion. Lymph nodes were minced in RPMI 1640 (Sigma-Aldrich) and forced gently through 40-µm cell-strainer nylon mesh (Falcon) using a sterile syringe plunger and centrifuged at 400 g for 5 min. Pellet from lymph nodes was resuspended in RPMI 1640 containing 10% FBS.
Flow Cytometry
Single-cell suspensions of brain and spinal cord tissue were prepared according to standard protocols. For cytofluorometry, following antibodies were used: CD4, CD8, CD45, CCR6, CXCR3, TCRβ, CD11c, CD11b, CD49b, CCR2, CD86, T-bet, RORγt, IL-17, IFN-γ, TNF-α, and IL-12 with conjugated fluorochromes (BD Biosciences). Antibodies were incubated with cells in PBS with 2% FBS for 30 min at 4°C, and then cells were analyzed. For intracellular staining of cytokines, cells were stimulated for 4 h in RPMI 1640 containing 10% FBS (Gibco), 10 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich), and 500 ng/mL ionomycin (Sigma-Aldrich) with addition of Brefeldin A (BD Biosciences). Antibodies for the cell surface markers were added to the cells in PBS with 2% FBS for 30 min on ice. After wash, cells were resuspended in Cytofix/Cytoperm buffer (BD Biosciences) for 20 min on ice, washed twice, and incubated with Abs for intracellular antigens (cytokines) in Perm buffer (30 min, on ice). For staining of transcriptional factors, unstimulated cells were used. Data were acquired using FACSCalibur (BD Biosciences) and analyzed with FlowJo software (Tree Star).
Tetramer Staining
Immune cells were isolated as described above. Cells isolated from CNS were incubated with H-2L(d)/IE-1/pp89 (168–176 YPHFMPTNL) and H-2D(d)/m164 (257–265 AGPPRYSRI) tetramers provided by NIH tetramer core facility. Cells stained with tetramer and anti-CD8 and anti-CD3 antibodies were incubated for 30 min at room temperature and then washed. Data were acquired using a FACSCalibur (BD Biosciences) and analyzed with FlowJo software (Tree Star).
Immunohistochemistry and Evaluation of Brain and Spinal Cord Pathology
Brains and spinal cords were fixed in 4% buffered formalin fixative overnight. Paraffin wax embedded sections (5 µm) were stained with hematoxylin and eosin and CD3 (ab699; Abcam) immunohistochemical staining. The slides were analyzed on light microscope (BX51; Olympus), and digital images were acquired by digital camera. The level of infiltration was graded using the following score: 0, no inflammatory cells; 1, a few scattered inflammatory cells; 2, organization of inflammatory infiltrates into perivascular cuffs; 3, extensive perivascular cuffing with extension into adjacent subarachnoid space and CNS parenchyma, and 4, extensive perivascular cuffing with increasing subarachnoid and parenchymal inflammation (32). Slides were analyzed on Olympus BX51 microscope, and digital images were acquired by Olympus digital camera (DP71).
Interferon-γ Assay
Mononuclear cells isolated from CNS, 105 in 100 µL complete media were put on 96-well plate in duplicates, and 100 µL of media, or MOG35–55 (1 μg/well) were added. After incubation for 1 h on 37°C, 0.2 µL of Brefeldin A in 10 µL of medium was added to each well and incubated for 4 h on 37°C. Cells were washed and then incubated with anti-CD8 and anti-CD4 antibodies on +4°C for 15 min. After washing, cells were resuspended in Cytofix/Cytoperm buffer for 30 min on ice, washed twice, and incubated with anti-IFNγ antibodies diluted in Perm wash buffer (30 min, on ice) and resuspended in FACS media.
Statistical Analysis
All statistics were carried out using SPSS 18.0 for Windows software. Results were analyzed using the Student’s t-test or Mann–Whitney test and ANOVA or Kruskal–Wallis. Data in this study were expressed as the mean + SEM. Values of P < 0.05 were considered significant.
Results
MCMV Infection in Adult Life Abrogates Resistance to EAE in BALB/c Mice
BALB/c mice immunized with MOG35–55 did not develop clinical signs of EAE, while BALB/c mice infected with MCMV 8 weeks after birth and 10 days later challenged with MOG35–55 in CFA and pertussis toxin (MCMV + MOG35–55) developed clinical signs that correspond to EAE manifestations seen in C57BL/6 mice (Figure 1A). Based on evaluation of clinical course (Figure 1A) and mean maximal clinical score (Figure 1B), MCMV-infected BALB/c mice developed disease indistinguishable from disease in susceptible C57BL/6 mice. Infiltration in CNS of MCMV + MOG35–55, expressed by mean histological score (Figure 1D) and total cell number (Figure 1C), was significantly higher compared with BALB/c mice immunized with MOG35–55 only mice. BALB/c mice infected with MCMV and 10 days later immunized with MOG35–55 developed subarachnoid and perivascular infiltrations in the brain cortex, perivascular infiltrations in brainstem and cerebellum with spreading to parenchyma (Figure 1E), and white matter spinal cord infiltrations (Figure 1F). Single-cell infiltrates were detected in brains and spinal cords of MOG35–55-immunized mice only, and mild perivascular infiltrations were detected in brainstem of MCMV-infected mice (Figures 1E,F). Immunostaining of spinal cord sections showed presence of CD3+ cells in the infiltrates (Figure 1G).
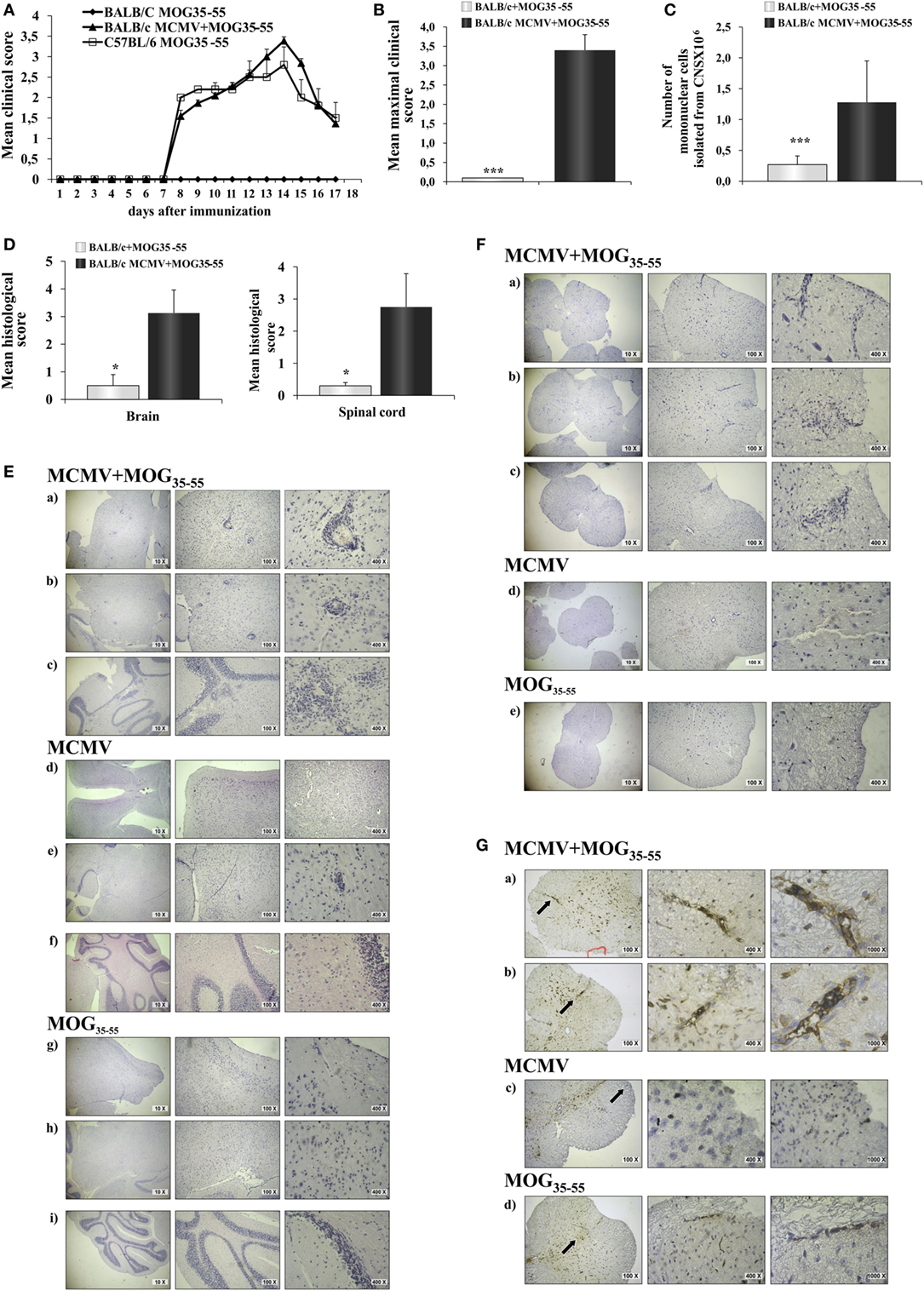
Figure 1. BALB/c mice infected with murine cytomegalovirus (MCMV) and immunized with MOG35–55 develop experimental autoimmune encephalomyelitis (EAE). Eight-week-old BALB/c mice were infected (foot-pad injection) with MCMV and 10 days after were immunized with MOG35–55 peptide in CFA and pertussis toxin (BALB/c MCMV + MOG35–55). Control mice C57BL/6 and BALB/c were immunized with MOG35–55 peptide in CFA and pertussis toxin without previous infection (C57BL/6 MOG35–55 and BALB/c MOG35–55). (A) EAE scores up to day 15 post-EAE induction (four separate experiments, n = 29/group). (B) Mean maximal clinical score up to day 15 post-EAE induction (four separate experiments, n = 29/group). (C) The mean value of the mononuclear cells isolated from central nervous system (CNS) of BALB/c MCMV + MOG35–55 and BALB/c MOG35–55 mice (three independent experiments, n = 24/group). (D) Mean histological scores were calculated from a total of five sections per group (two separate experiments, n = 8/group). (E) The representative images of brain cortex (a), brain stem (b), cerebellum (c) of BALB/c MCMV + MOG35–55; brain cortex (d), brain stem (e), cerebellum (f) of BALB/c MCMV-infected mice (BALB/c MCMV); and brain cortex (g), brain stem (h), cerebellum (i) of BALB/c MOG35–55 mice. (F) The representative images of spinal cords of BALB/c MCMV + MOG35–55 (a–c); BALB/c MCMV (d); and BALB/c MOG35–55 mice (e). (G) Representative sections of CD3 spinal cord immunohistochemistry of BALB/c MCMV + MOG35–55 (a,b); BALB/c MCMV (c); and BALB/c MOG35–55 mice (d), arrows in left panels indicate the area presented in magnified sections in right panels. All pictures are representative of two separate experiments (n = 16/group). Data were analyzed by Student’s t-test and presented as mean + SE: *P < 0.05 and ***P < 0.001.
CNS Infiltrates of MCMV + MOG35–55 Mice Contain Higher Amounts of T1/T17 CD4+ and CD8+ T Cells
Further analysis showed significantly higher number of CD4+ and CD8+ T cells in the infiltrates of MCMV + MOG35–55 mice compared with MOG35–55 mice (Figure 2A). In the CNS of MCMV + MOG35–55 mice, there was higher number of CD4+ T cells then CD8+ T cells, similar to typical EAE in C57BL/6 mice. MCMV + MOG35–55 mice had increased percentage (Figure 2B) and number (Figure 2C) of both Th1 and Th17 cells, as well as Tc1 and Tc17 cells compared to MOG35–55 mice. Similarly, significantly higher number of CD4+ cell- and CD8+ cell-expressing transcriptional factors, T-bet and RORγt (Figure 2D), and chemokine receptors CCR6 and CXCR3 (Figure 2E) was noticed in CNS of MCMV + MOG35–55 mice compared with MOG35–55 mice.
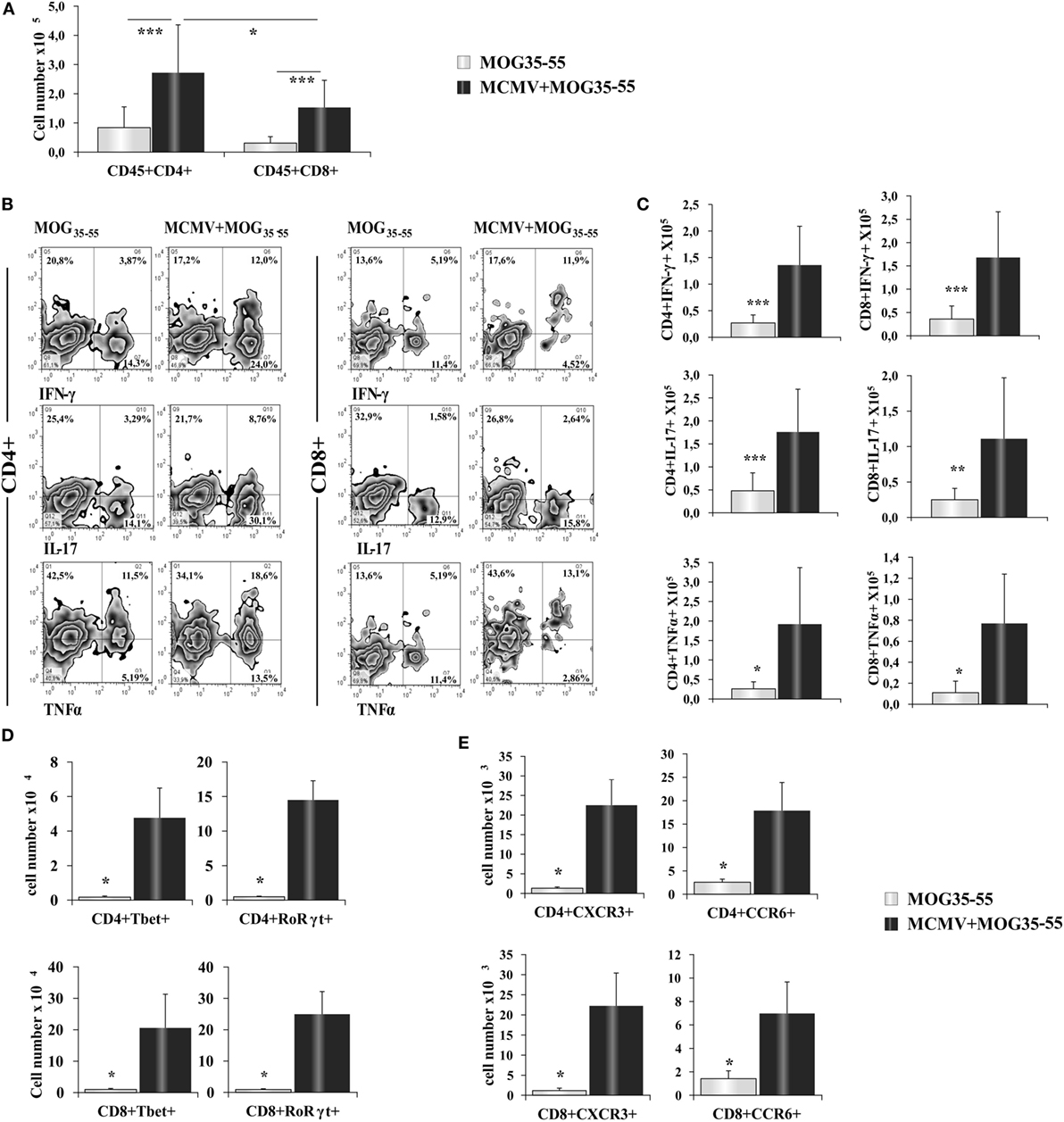
Figure 2. BALB/c murine cytomegalovirus (MCMV) + MOG35–55 mice have increased number of inflammatory CD4+ and CD8+ cells. Eight-week-old BALB/c mice were infected with MCMV and 2 weeks after were immunized with MOG35–55 peptide. After 15 days, mice were perfused, central nervous system was harvested, and mononuclear cells were isolated and restimulated ex vivo with PMA and ionomycin before performing intra cellular staining. (A) Total cell numbers of CD45+CD4+, CD45+CD8+ cells. (B) Representative FACS images of percentages and (C) total cell numbers of CD4+IFN-γ+, CD4+TNF-α+, CD4+IL-17+, CD8+IFN-γ+, CD8+TNF-α+ and CD8+IL-17+ cells. Total cell numbers of CD8+ cell- and CD4+ cell-expressing transcriptional factors T-bet and RORγt (D) and chemokine receptors CXCR3 and CCR6 (E). Data from three separate experiments with 22 mice/group are presented as mean + SE. Data were analyzed with Student’s t-test: *P < 0.05, **P < 0.005, and ***P < 0.001.
CD8+ Cells Take a Role in Autoimmune Neuropathology in BALB/c Mice with MCMV Infection
Depletion of CD4+ cells in MCMV-infected mice abrogated susceptibility to MOG35–55-induced disease (Figure 3A) indicating autoimmune disease. Given the significant number of CD8+ cells in the infiltrates that were not seen in classical EAE, we explored in more details these cells found in CNS. There was significantly higher percentage and number of CD8+ cell-expressing markers of cytolytic activity in the CNS of MCMV + MOG35–55 BALB/c mice compared to MOG35–55 mice (Figure 3B). To indirectly determine the percentage of MOG35–55-specific CD4+ and CD8+ cells, mononuclear cells were isolated from CNS of MCMV + MOG35–55 BALB/c, MOG35–55 BALB/c, and MOG35–55 C57BL/6 mice and ex vivo restimulated with MOG35–55 peptide, and IFN-γ+ cells were enumerated. Significantly higher percentage of CD4+ and CD8+ cells from CNS of MCMV + MOG35–55 mice responded to ex vivo restimulation compared with MOG35–55 only BALB/c mice (Figure 3C). Further, significantly higher percentage of CD8+ cells isolated from CNS of MCMV + MOG35–55 BALB/c mice contained IFN-γ after ex vivo restimulation with MOG35–55 compared to C57BL/6 mice immunized with MOG35–55. Even more importantly, IFN-γ-containing CD8+ cells in MCMV + MOG35–55 BALB/c mice after restimulation with MOG35–55 were not specific for viral epitopes pp89 and m164, suggesting that inflammatory CD8+ cells in the CNS are autoimmune (Figure 3D).
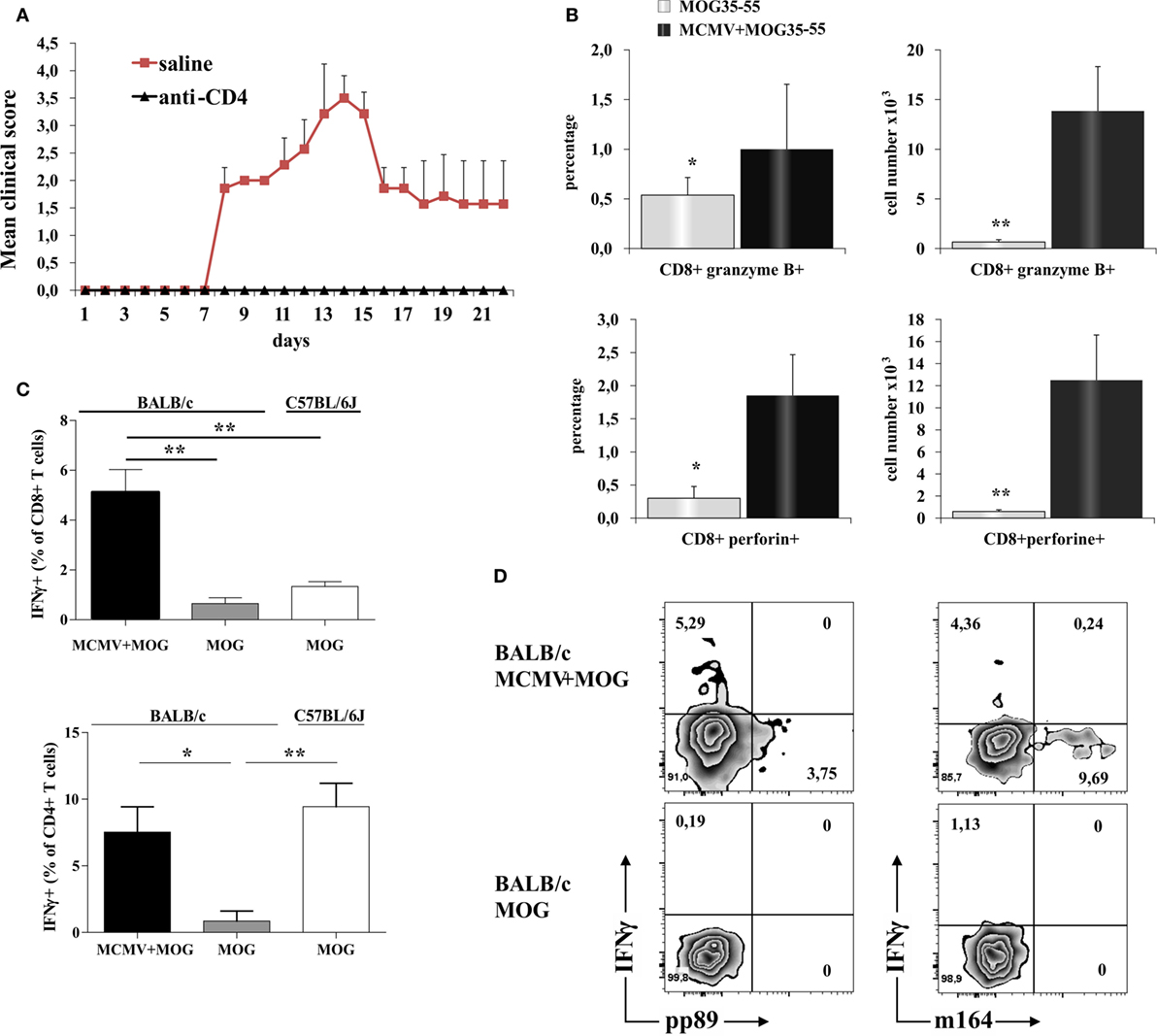
Figure 3. CD8 T cells in the central nervous system (CNS) of BALB/c murine cytomegalovirus (MCMV) + MOG35–55 mice express markers of cytolytic activity and contribute to autoimmune reactions in CNS. CD4+ cells were depleted 10 days after MCMV infection and 5 days before immunization with MOG35–55 (anti-CD4), control mice were infected and immunized but received saline instead of depleting antibody (saline). (A) Experimental autoimmune encephalomyelitis (EAE) scores up to day 22 post-EAE induction in anti-CD4 and saline mice, data are presented as mean + SE from one experiment with five mice per group. (B) Percentages and absolute numbers of CD8+granzyme B+ and CD8+perforin+ cells among mononuclear cells isolated from CNS 15 days post-immunization with MOG35–55 peptide of BALB/c mice infected with MCMV 10 days earlier (MCMV + MOG35–55) and previously untreated BALB/c mice (MOG35–55). Data are presented as mean + SE, from two separate experiments with 14 mice/group. (C) Percentages of CD4+IFN-γ+ and CD8+IFN-γ+ cells and (D) representative flow images of IFN-γ, pp89, and m164 expression in CD8+ population among mononuclear cells isolated from CNS of MCMV-infected and MOG35–55-immunized BALB/c mice and MOG35–55-immunized BALB/c and C57BL/6 mice in vitro restimulated with MOG35–55 peptide. Percentages are presented as mean + SE (representative experiment with six mice per group). Data were analyzed with Student’s t-test and Kruskal–Wallis: *P < 0.05, **P < 0.005.
Chronic Non-Productive MCMV Infection Also Facilitates EAE Development in BALB/c Mice
In order to test whether the chronic non-productive MCMV infection could facilitate EAE development, we immunized BALB/c mice with MOG35–55 peptide 3 months after MCMV infection in adult life. As shown in Figure 4, infected mice developed clinical signs of EAE while age-matched mice immunized with encephalitogen only, did not. Mice with chronic non-productive MCMV infection started to manifest signs of EAE 6 days after immunization with MOG35–55 peptide; maximal clinical score reached 15 days after immunization and had very mild signs of the disease 60 days after immunization (Figure 4A). Chronic disease was confirmed with histological analysis. Perivascular infiltrates were detected in the spinal cords of mice with latent MCMV infection 2 months after challenge with MOG35–55 peptide (Figure 4B). Among cells isolated from brains of MCMV-infected BALB/c mice 2 months after MOG35–55 immunization, higher percentage of CD4+ and CD8+ cells contained inflammatory cytokines IL-17 and IFN-γ, after in vitro restimulation with MOG35–55 peptide compared to stimulated cells isolated from MOG35–55-immunized mice (Figure 4C). Our findings indicate that BALB/c mice with latent MCMV infection develop disease with long-lasting infiltrates in the CNS that contains lymphocytes specific for neuroantigens.
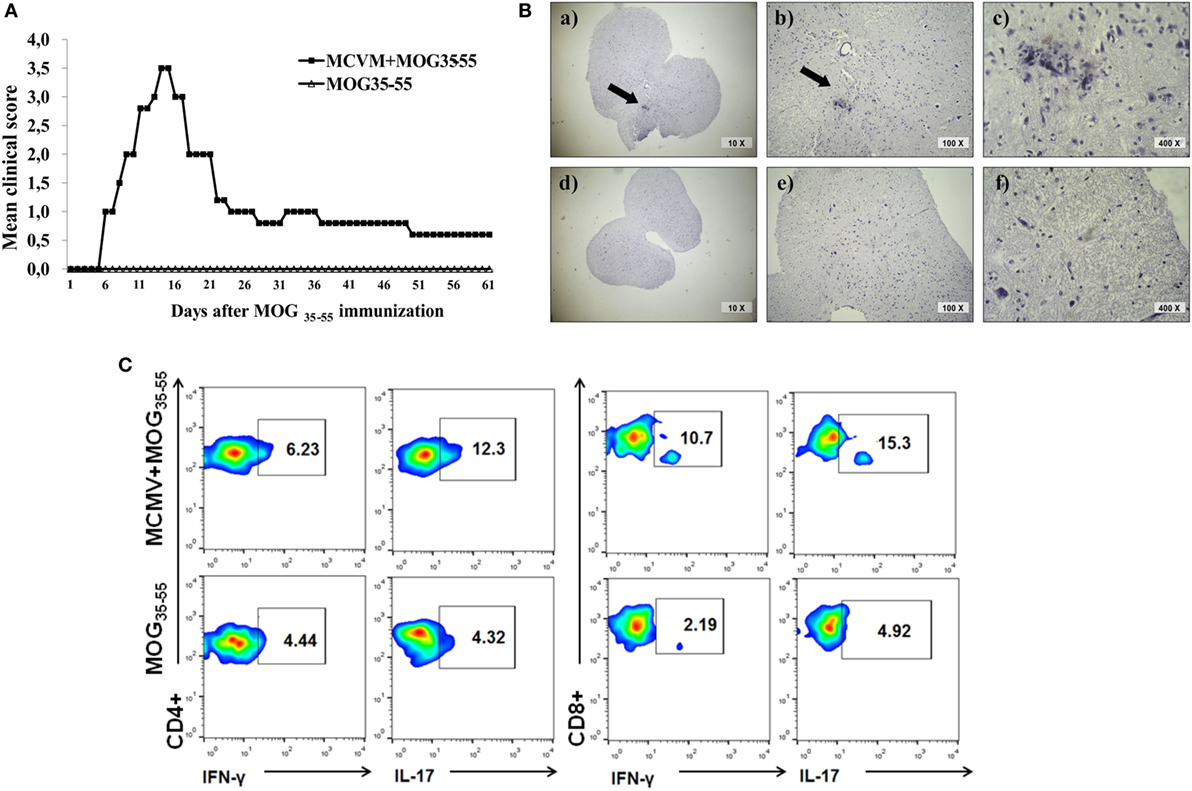
Figure 4. BALB/c mice with latent murine cytomegalovirus (MCMV) infection develop experimental autoimmune encephalomyelitis and longlasting infiltrates in central nervous system. Mice were infected with MCMV, and 3 months later they were immunized with MOG35–55 peptide, and disease was evaluated for 60 days. (A) Mean clinical score and (B) representative images of spinal cord sections 60 days after immunization with MOG35–55 peptide in MCMV + MOG35–55 group (a–c) and MOG35–55 group (d–f). (C) Representative flow cytometric images presenting percentages of IL-17- and IFN-γ-expressing CD4+ and CD8+ cells among mononuclear cells isolated from brains 60 days after immunization with MOG35–55 peptide. Presented data are from representative experiment with seven mice per group.
MCMV Infection Induces Inflammatory Phenotype of Antigen-Presenting Cells in Periphery and in CNS
It is known that viral infection induces antiviral immune response mediated by NK cells, CD8+ and CD4+ lymphocytes (33). Such inflammatory microenvironment in peripheral lymph organs can affect activation of antigen-presenting cells and thus indirectly contribute to development of inflammatory lymphocytes. Therefore, we explored possible influence of viral infection on changes of phenotype of antigen-presenting cells. To this end, mononuclear cells were isolated from inguinal lymph node 12 days after MCMV was administered in foot pad and compared with cells isolated from mice immunized with MOG35–55 only. Lymph nodes of MCMV-infected and MCMV + MOG35–55 mice had significantly higher percentage of CD11c+ dendritic cells and CD11c+PDCA1+plasmocitoid dendritic cells in inguinal lymph nodes compared to MOG35–55-immunized mice (Figure 5A). Also higher percentage of dendritic cell-expressing CCR2 chemokine receptor was found in both groups of MCMV-infected mice (Figure 5B). More importantly, lymph nodes of MCMV-infected and MCMV + MOG35–55 mice contained higher percentage of activated CD86+ and CD40+ (Figure 5C) and inflammatory IL-12+ dendritic cells (Figure 5E). Higher expression of markers of activation, CD86+ and CD40+ was noticed in MCMV-infected mice compared with MOG35–55-immunized mice (Figure 5D). These data suggest that inflammatory phenotype of dendritic cells is achieved in BALB/c mice with viral infection but not with encephalitogenic challenge only, as it was seen in C57BL/6 mice.
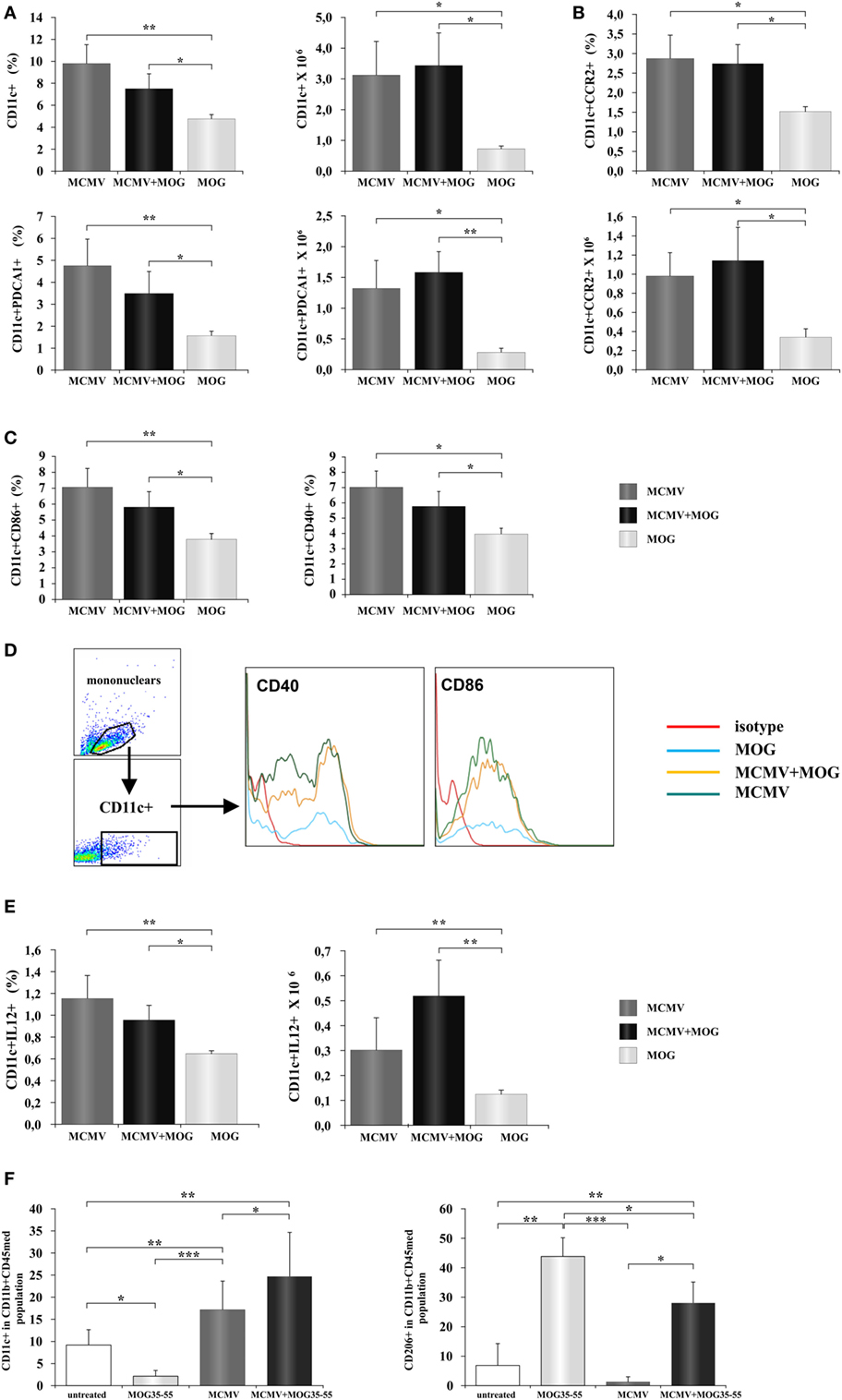
Figure 5. Murine cytomegalovirus (MCMV) infection favors inflammatory phenotype of antigen-presenting cells. Mononuclear cells were isolated from inguinal lymph nodes 2 days after immunization with MOG35–55 peptide (MOG35–55) and from lymph nodes of mice 10 days after their MCMV infection (MCMV). Flow cytometric analysis of dendritic cells phenotype was done. (A) Percentages and absolute numbers of CD11c+ dendritic cells and CD11c+CD11b−PDCA1+ dendritic cells, (B) CCR2-expressing CD11c+ cells, and (C) CD86+ and CD40+ dendritic cells are presented as mean + SE (10 mice per group). (D) Representative histograms of CD40 and CD86 expression in CD11c+ population. (E) Percentages and absolute numbers of IL-12+ dendritic cells presented as mean + SE (10 mice per group). (F) Mononuclear cells were isolated from central nervous system of saline-treated, MOG35–55-immunized, MCMV-infected, and MCMV-infected and MOG35–55-immunized mice, 15 days after MOG35–55 immunization percentages of classically (CD11c+) and alternatively (CD206+) microglia are presented as mean + SE (8–10 mice per group). Data were analyzed with Student’s t-test and ANOVA: *P < 0.05, **P < 0.005, and ***P < 0.001.
It is known that systemic inflammation in mice causes activation of microglia that persists for months (34, 35). Since MCMV infection in BALB/c mice causes systemic inflammation, we wanted to see effect of MCMV infection on phenotype of antigen-presenting cells in CNS. We analyzed expression of markers of classical (CD11c) and alternative activation (CD206) in the population of microglia (CD45intCD11b+) in healthy mice, MOG35–55-immunized mice, MCMV-infected, and MCMV + MOG35–55 mice. As shown in Figure 5F, microglia of mice with viral infection in adult life, with and without EAE, had proinflammatory, M1 phenotype. Significantly higher percentage of CD11c-expressing cells in microglia population was found in both groups of infected mice compared to healthy and MOG35–55-immunized mice. On the other hand, the highest percentage of (type 2) CD206-expressing microglia was found in MOG35–55 BALB/c mice. Higher percentage of M2 microglia was also found in MCMV + MOG35–55-immunized mice compared to healthy and MCMV-infected mice but lower compared to MOG35–55-immunized mice. Thus, high percentage of M2 microglia in mice at the peak of EAE could be also the compensatory mechanism that precedes disease attenuation.
Discussion
Here, we provide the first evidence that MCMV infection results in breaking resistance of BALB/c mice to EAE induction with MOG35–55 peptide, as indicated by typical clinical manifestations and massive inflammatory infiltration in the CNS (Figure 1).
The role of MCMV infection in EAE is not studied, while its significance in MS is controversial. There are several prospective clinical studies that indicate protective effect of CMV infection on MS risk (21, 22, 36), while recent well-powered meta-analysis found no significant difference in the rate of CMV seropositivity between MS patients and healthy controls based on pooled samples from all studies to date (37). On the other hand, CMV has been found in demyelinating plaques and the liquor of MS patients (15), and several clinical studies support the role of CMV in MS pathogenesis (18–20). Additionally, CMV infection induces expansion of inflammation-seeking/proinflammatory effector-memory CD4+CD28null T cells that are attracted to MS lesions via a CX3CL1 gradient (38, 39) and are mostly found in MS patients (40). Stimulation of these cells with myelin autoantigens results in their proliferation and release of cytotoxic granules, and thus may contribute to MS pathology (41). Some experimental animal studies support the role of CMV in EAE development. Cross-reactivity between CMV981–1,003 and MOG35–55 peptides was found in Lewis rats immunized with MOG35–55 (42), while cross-reactivity between CMV981–1,003 and MOG34–56 was found in rhesus monkeys (43). Immunization of rhesus monkeys with human CMV981–1,003 peptide induced expansion of MOG34–56-specific T cells (43). Female SJL/J mice primed with vaccinia virus that contain PLP gene and later challenged with MCMV-developed lesions in white matter regions in the brains (28). Other studies on primates also support the role of CMV in MS pathogenesis (44, 45). Recently, Juranic Lisnic et al. demonstrated that MCMV infection of murine fibroblasts induced highest expression of interferon β, transcriptional factor T-bet, chemokine CXCL10 (46), and the role of these markers of Th1 cells, in EAE pathogenesis is well known (47–49).
However, there was no evidence that MCMV infection may directly facilitate EAE. Here, we show that adult MCMV infection overcomes resistance of BALB/c mice to induction of EAE with MOG35–55 peptide. The disease is characterized by typical clinical signs (Figure 1A) seen in susceptible C57BL/6 mice and massive brain and spinal cord infiltrations (Figures 1E,F). It should be noted that brain infiltrates are more significant in BALB/c mice treated with encephalitogen then in “classical” EAE in C57BL/6 mice. It appears that the disease after infection + MOG35–55 challenge in otherwise resistant mice is more similar to MS than classical EAE (50). While in C57BL/6 mice, CD4+ cells dominate in CNS infiltrates in the experiments presented here, there was similar number of CD4+ and CD8+ cells in the infiltrates (Figure 1).
Encephalitogenity of CD4+ T cells in the infiltrates in BALB/c mice is further documented by their expression of chemokine receptors CXCR3 (Figure 2E), whose blockade during EAE induction attenuated the disease (51), and CCR6 receptor known to have the key role in development of initial autoimmune infiltration in the CNS (52, 53). While γHV-68 infection in C57BL/6 mice with EAE leads to almost exclusive infiltration with Th1 cells (7), in the CNS of MCMV-infected BALB/c mouse with EAE there is almost equal participation of Th1 and Th17 cell-expressing IFN-γ and T-bet (Th1) and IL-17 and RORγt (Th17) (Figures 2C,D). Moreover, inflammatory infiltrates in MCMV-pretreated BALB/c mice immunized with MOG35–55 contained CD8+ cell-expressing T1 and T17 transcriptional factors and corresponding cytokines TNF-α and IFN-γ (Tc1) and IL-17 (Tc17 cells) (Figures 2C,D). Interestingly, it was suggested that Tc17 cells are required for Th17 accumulation and development of MS (54). Patients with early-stage MS harbor a greater number of Tc17 cells in the cerebrospinal fluid than in peripheral blood that contribute to the initiation of CNS autoimmunity (54). Since in the CNS of BALB/c mice with neonatal MCMV infection but without immunization with MOG35–55 dominates Tc1 cells (IFN-γ and T-bet+) (55), it appears that the newly developing autoimmune process attracts a different population of CD8+ cells (IL-17 producing) that contribute to autoimmune process (54).
Significant accumulation of CD8+ cells in the infiltrates and their role in autoimmune pathogenesis is documented by clinical signs and typical pathology complemented by specificity of infiltrating cells. Ex vivo restimulation with MOG35–55 leads to significant increase of CD8+ cell-producing IFN-γ in MCMV + MOG35–55 BALB/c mice (Figure 3C). This finding is at variance with our finding in C57BL/6 mice. CD8+ cells from EAE C57BL/6 mice did not responded to restimulation with MOG35–55 (Figure 3C). Further, MOG35–55 responsive CD8+ cells in MCMV + MOG35–55 BALB/c mice were not specific for viral epitopes pp89 and m164 (Figure 3D) implicating that CD8+ cell contributes to autoimmune process in this model of EAE. This finding is in line with previous report that initiation of autoimmune process in CNS with CD4+ T cells is followed with spreading to myelin-specific CD8+ T cells that are capable of direct recognition of oligodendrocytes and contribute to tissue damage (56). Similarly, heighten EAE in C57BL/6 mice infected with γHV-68 is accompanied with infiltration of brain parenchyma with CD8+IFN-γ+granzyme+ cells (7). However, specificity for autoantigen of these inflammatory and cytolytic CD8+ cells in γHV-68-infected C57BL/6 mice was not studied (7). Finding that mice with depletion of CD4+ cells after MCMV infection and before MOG35–55 immunization did not develop the disease (Figure 3A) contributes to the conclusion that MCMV-infected BALB/c mice developed autoimmune neuropathology. Persistence of CNS infiltrations and MOG35–55-specific CD4+ and CD8+ cells in CNS of BALB/c mice 2 months after MOG35–55 immunization that were infected 3 months previously (Figure 4) also proving autoimmune nature of the disease. Thus although CD4+ cells are required, it appears that CD8+ cells are the main effector cells.
MCMV infection significantly increases proportion of dendritic cells (CD11c+), plasmocitoid dendritic cells (CD11c+PDCA1+) in peripheral lymph nodes (Figure 5A) compared to immunization with MOG35–55. Further higher percentage of dendritic cells in lymph nodes in MCMV-infected mice is accompanied with higher percentage of CCR2+ dendritic cells (Figure 5B). It is known that MCMV encodes proinflammatory factor (MCK-2), analog of chemokine CCL2 (57) that enhances monocyte recruitment and viral dissemination (58). Then, higher percentage of dendritic cells in inguinal lymph nodes of MCMV-infected mice could be the consequence of CCL2 analog production, since CCL2 goes to lymph nodes where is presented on the surface of high endothelial venules for recruitment of monocytes (59). Although it was previously shown that MCMV attracts monocytes that have the immunosuppressive role (60), here, we found higher percentage of dendritic cell-expressing markers of activation CD86 and CD40 (Figure 5C) and containing Th1 promoting cytokine, IL-12 (Figures 5D,E). Our results indicate that MCMV infection of BALB/c mice induces increase of inflammatory dendritic cells in peripheral lymph nodes and thus enables development of encephalitogenic T cells. This finding is in correlation with previous report that MCMV-infected mice are resistant to bacterial infection due to prolonged production of the antiviral cytokine IFN-γ and systemic activation of macrophages (61). Significantly, there was an increase of classically activated microglia (CD45medCD11b+CD11c+) in the CNS of BALB/c mice 25 days after MCMV infection compared to MOG35–55-immunized mice without previous infection that had mostly alternatively activated microglia (CD45medCD11b+CD206+) (Figure 5F). Lower percentage of alternatively activated microglia in MCMV + MOG35–55 mice observed on day 15 after immunization and day 25 after infection in compared to MOG35–55 treated mice, and prevalence of M1 microglia in virus-infected mice may contribute to chronic disease in MCMV-infected MOG35–55-immunized BALB/c mice. Previously, it was shown that systemic MCMV infection elicited a significant increase in the number of microglia with morphological signs of activation and M1 phenotype (62). Thus, it appears that at least one level of resistance of BALB/c mice to EAE is the inability to convert microglia into M1 phenotype.
In summing up, we report here that MCMV infection may promote autoimmune neuropathology and convert resistant mice into susceptible to EAE induction. This was achieved by activation of antigen-presenting cells and promoting M1 phenotype of microglia as well as participation of CD8+ encephalitogen-specific T cells in the autoimmune pathogenesis.
Author Contributions
Conceived and designed the experiments: JM, MM, NA, SJ, and ML. Performed the experiments: JM, BP, MM, AA, BS, and DK. Analyzed the data: JM, BP, MM, DK, IT, and AK. Wrote the paper: JM and ML.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by grants from the Serbian Ministry of Science and Technological Development (Grants No. ON175071, ON175069, and ON175103), Serbia, and the Faculty of Medical Sciences, University of Kragujevac (MP 01/14 and MP 02/14). The authors thank Milan Milojevic for excellent technical assistance.
References
1. Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung HP. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol (2006) 2(4):201–11. doi:10.1038/ncpneuro0154
2. Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet (1985) 2(8441):1313–5.
4. Soldan SS, Jacobson S. Role of viruses in etiology and pathogenesis of multiple sclerosis. Adv Virus Res (2001) 56:517–55. doi:10.1016/S0065-3527(01)56037-6
5. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol (2007) 61(4):288–99. doi:10.1002/ana.21117
6. Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, et al. Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog (2013) 9(4):e1003220. doi:10.1371/journal.ppat.1003220
7. Casiraghi C, Shanina I, Cho S, Freeman ML, Blackman MA, Horwitz MS. Gammaherpesvirus latency accentuates EAE pathogenesis: relevance to Epstein–Barr virus and multiple sclerosis. PLoS Pathog (2012) 8(5):e1002715. doi:10.1371/journal.ppat.1002715
8. Krishnamoorthy G, Wekerle H. EAE: an immunologist’s magic eye. Eur J Immunol (2009) 39(8):2031–5. doi:10.1002/eji.200939568
9. Johnson TA, Jirik FR, Fournier S. Exploring the roles of CD8(+) T lymphocytes in the pathogenesis of autoimmune demyelination. Semin Immunopathol (2010) 32:197–209. doi:10.1007/s00281-010-0199-7
10. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol (2015) 15(9):545–58. doi:10.1038/nri3871
11. Mix E, Meyer-Rienecker H, Zettl UK. Animal models of multiple sclerosis for the development and validation of novel therapies – potential and limitations. J Neurol (2008) 255(Suppl 6):7–14. doi:10.1007/s00415-008-6003-0
12. Hanley PJ, Bollard CM. Controlling cytomegalovirus: helping the immune system take the lead. Viruses (2014) 6(6):2242–58. doi:10.3390/v6062242
13. Koch S, Larbi A, Ozcelik D, Solana R, Gouttefangeas C, Attig S, et al. Cytomegalovirus infection: a driving force in human T cell immunosenescence. Ann N Y Acad Sci (2007) 1114:23–35. doi:10.1196/annals.1396.043
14. Sinclair J. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J Clin Virol (2008) 41(3):180–5. doi:10.1016/j.jcv.2007.11.014
15. Smyk DS, Alexander AK, Walker M, Walker M. Acute disseminated encephalomyelitis progressing to multiple sclerosis: are infectious triggers involved? Immunol Res (2014) 60(1):16–22. doi:10.1007/s12026-014-8499-y
16. ’t Hart BA, Hintzen RQ, Laman JD. Multiple sclerosis – a response-to-damage model. Trends Mol Med (2009) 15(6):235–44. doi:10.1016/j.molmed.2009.04.001
17. Scotet E, Peyrat MA, Saulquin X, Retiere C, Couedel C, Davodeau F, et al. Frequent enrichment for CD8 T cells reactive against common herpes viruses in chronic inflammatory lesions: towards a reassessment of the physiopathological significance of T cell clonal expansions found in autoimmune inflammatory processes. Eur J Immunol (1999) 29(3):973–85.
18. Sanadgol N, Ramroodi N, Ahmadi GA, Komijani M, Moghtaderi A, Bouzari M, et al. Prevalence of cytomegalovirus infection and its role in total immunoglobulin pattern in Iranian patients with different subtypes of multiple sclerosis. New Microbiol (2011) 34(3):263–74.
19. Horakova D, Zivadinov R, Weinstock-Guttman B, Havrdova E, Qu J, Tamaño-Blanco M, et al. Environmental factors associated with disease progression after the first demyelinating event: results from the multi-center SET study. PLoS One (2013) 8(1):e53996. doi:10.1371/journal.pone.0053996
20. Weinstock-Guttman B, Horakova D, Zivadinov R, Tamaño-Blanco M, Badgett D, Tyblova M, et al. Interactions of serum cholesterol with anti-herpesvirus responses affect disease progression in clinically isolated syndromes. J Neuroimmunol (2013) 263(1–2):121–7. doi:10.1016/j.jneuroim.2013.07.010
21. Sundqvist E, Bergström T, Daialhosein H, Nyström M, Sundström P, Hillert J, et al. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult Scler (2014) 20(2):165–73. doi:10.1177/1352458513494489
22. Zivadinov R, Nasuelli D, Tommasi MA, Serafin M, Bratina A, Ukmar M, et al. Positivity of cytomegalovirus antibodies predicts a better clinical and radiological outcome in multiple sclerosis patients. Neurol Res (2006) 28(3):262–9. doi:10.1179/016164106X98134
23. Jackson SE, Mason GM, Wills MR. Human cytomegalovirus immunity and immune evasion. Virus Res (2011) 157(2):151–60. doi:10.1016/j.virusres.2010.10.031
24. Noriega V, Redmann V, Gardner T, Tortorella D. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res (2012) 54(1–3):140–51. doi:10.1007/s12026-012-8304-8
25. Milovanovic M, Volarevic V, Ljujic B, Radosavljevic G, Jovanovic I, Arsenijevic N, et al. Deletion of IL-33R (ST2) abrogates resistance to EAE in BALB/C mice by enhancing polarization of APC to inflammatory phenotype. PLoS One (2012) 7(9):e45225. doi:10.1371/journal.pone.0045225
26. Jiang HR, Milovanović M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur J Immunol (2012) 42(7):1804–14. doi:10.1002/eji.201141947
27. Vanheusden M, Stinissen P, ’t Hart BA, Hellings N. Cytomegalovirus: a culprit or protector in multiple sclerosis? Trends Mol Med (2015) 21(1):16–23. doi:10.1016/j.molmed.2014.11.002
28. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev (2006) 19(1):80–94. doi:10.1128/CMR.19.1.80-94.2006
29. Wagner M, Jonjic S, Koszinowski UH, Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol (1999) 73(8):7056–60.
30. Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc (2006) 1(4):1810–9. doi:10.1038/nprot.2006.285
31. Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc (2006) 1(4):1952–60. doi:10.1038/nprot.2006.284
32. Wraith DC, Pope R, Butzkueven H, Holder H, Vanderplank P, Lowrey P, et al. A role for galanin in human and experimental inflammatory demyelination. Proc Natl Acad Sci U S A (2009) 106(36):15466–71. doi:10.1073/pnas.0903360106
33. Halenius A, Hengel H. Human cytomegalovirus and autoimmune disease. Biomed Res Int (2014) 2014:472978. doi:10.1155/2014/472978
34. Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci (2004) 20(2):467–73. doi:10.1111/j.1460-9568.2004.03514.x
35. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia (2007) 55(5):453–62. doi:10.1002/glia.20467
36. Waubant E, Mowry EM, Krupp L, Chitnis T, Yeh EA, Kuntz N, et al. Common viruses associated with lower pediatric multiple sclerosis risk. Neurology (2011) 76(23):1989–95. doi:10.1212/WNL.0b013e31821e552a
37. Pakpoor J, Pakpoor J, Disanto G, Giovannoni G, Ramagopalan SV. Cytomegalovirus and multiple sclerosis risk. J Neurol (2013) 260(6):1658–60. doi:10.1007/s00415-013-6912-4
38. Broux B, Pannemans K, Zhang X, Markovic-Plese S, Broekmans T, Eijnde BO, et al. CX(3)CR1 drives cytotoxic CD4(+)CD28(-) T cells into the brain of multiple sclerosis patients. J Autoimmun (2012) 38(1):10–9. doi:10.1016/j.jaut.2011.11.006
39. Pinto-Medel MJ, García-León JA, Oliver-Martos B, López-Gómez C, Luque G, Arnáiz-Urrutia C, et al. The CD4+ T-cell subset lacking expression of the CD28 costimulatory molecule is expanded and shows a higher activation state in multiple sclerosis. J Neuroimmunol (2012) 243(1–2):1–11. doi:10.1016/j.jneuroim.2011.11.008
40. Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P. CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol (2007) 179(10):6514–23. doi:10.4049/jimmunol.179.10.6514
41. Broux B, Markovic-Plese S, Stinissen P, Hellings N. Pathogenic features of CD4+CD28- T cells in immune disorders. Trends Mol Med (2012) 18(8):446–53. doi:10.1016/j.molmed.2012.06.003
42. Zheng MM, Zhang XH. Cross-reactivity between human cytomegalovirus peptide 981-1003 and myelin oligodendroglia glycoprotein peptide 35-55 in experimental autoimmune encephalomyelitis in Lewis rats. Biochem Biophys Res Commun (2014) 443(3):1118–23. doi:10.1016/j.bbrc.2013.12.122
43. Brok HP, Boven L, van Meurs M, Kerlero de Rosbo N, Celebi-Paul L, Kap YS, et al. The human CMV-UL86 peptide 981-1003 shares a crossreactive T-cell epitope with the encephalitogenic MOG peptide 34-56, but lacks the capacity to induce EAE in rhesus monkeys. J Neuroimmunol (2007) 182(1–2):135–52. doi:10.1016/j.jneuroim.2006.10.010
44. Wroblewska Z, Gilden D, Devlin M, Huang ES, Rorke LB, Hamada T, et al. Cytomegalovirus isolation from a chimpanzee with acute demyelinating disease after inoculation of multiple sclerosis brain cells. Infect Immun (1979) 25(3):1008–15.
45. Jagessar SA, Kap YS, Heijmans N, van Driel N, van Straalen L, Bajramovic JJ, et al. Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete Freund adjuvant. J Neuropathol Exp Neurol (2010) 69(4):372–85. doi:10.1097/NEN.0b013e3181d5d053
46. Juranic Lisnic V, Babic Cac M, Lisnic B, Trsan T, Mefferd A, Das Mukhopadhyay C, et al. Dual analysis of the murine cytomegalovirus and host cell transcriptomes reveal new aspects of the virus-host cell interface. PLoS Pathog (2013) 9(9):e1003611. doi:10.1371/journal.ppat.1003611
47. Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med (2009) 206(7):1549–64. doi:10.1084/jem.20082584
48. Lalor SJ, Segal BM. Th1-mediated experimental autoimmune encephalomyelitis is CXCR3 independent. Eur J Immunol (2013) 43(11):2866–74. doi:10.1002/eji.201343499
49. Carter SL, Müller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-gamma but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia (2007) 55(16):1728–39. doi:10.1002/glia.20587
50. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med (2000) 192(3):393–404. doi:10.1084/jem.192.3.393
51. Sporici R, Issekutz TB. CXCR3 blockade inhibits T-cell migration into the CNS during EAE and prevents development of adoptively transferred, but not actively induced, disease. Eur J Immunol (2010) 40(10):2751–61. doi:10.1002/eji.200939975
52. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol (2009) 10(5):514–23. doi:10.1038/ni.1716
53. Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, et al. Inhibition of CCR6 function reduces the severity of experimental autoimmune encephalomyelitis via effects on the priming phase of the immune response. J Immunol (2009) 182(5):3121–30. doi:10.4049/jimmunol.0713169
54. Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest (2013) 123(1):247–60. doi:10.1172/JCI63681
55. Bantug GR, Cekinovic D, Bradford R, Koontz T, Jonjic S, Britt WJ. CD8+ T lymphocytes control murine cytomegalovirus replication in the central nervous system of newborn animals. J Immunol (2008) 181(3):2111–23. doi:10.4049/jimmunol.181.3.2111
56. Ji Q, Castelli L, Goverman JM. MHC class I-restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8? T cells. Nat Immunol (2013) 14(3):254–61. doi:10.1038/ni.2513
57. Saederup N, Aguirre SA, Sparer TE, Bouley DM, Mocarski ES. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol (2001) 75:9966–76. doi:10.1128/JVI.75.20.9966-9976.2001
58. Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, et al. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood (2006) 107:30–8. doi:10.1182/blood-2005-05-1833
59. Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, et al. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med (2001) 194:1361–73. doi:10.1084/jem.194.9.1361
60. Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity (2012) 37(1):122–33. doi:10.1016/j.immuni.2012.04.014
61. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature (2007) 447(7142):326–9. doi:10.1038/nature05762
Keywords: experimental autoimmune encephalomyelitis, BALB/c mice, murine cytomegalovirus infection, antigen-presenting cells, T cells
Citation: Milovanovic J, Popovic B, Milovanovic M, Kvestak D, Arsenijevic A, Stojanovic B, Tanaskovic I, Krmpotic A, Arsenijevic N, Jonjic S and Lukic ML (2017) Murine Cytomegalovirus Infection Induces Susceptibility to EAE in Resistant BALB/c Mice. Front. Immunol. 8:192. doi: 10.3389/fimmu.2017.00192
Received: 30 July 2016; Accepted: 09 February 2017;
Published: 27 February 2017
Edited by:
Luigi Daniele Notarangelo, Harvard Medical School, USAReviewed by:
Silva Markovic-Plese, University of North Carolina at Chapel Hill, USAAnne Kathrin Mausberg, Essen University Hospital, Germany
Copyright: © 2017 Milovanovic, Popovic, Milovanovic, Kvestak, Arsenijevic, Stojanovic, Tanaskovic, Krmpotic, Arsenijevic, Jonjic and Lukic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miodrag L. Lukic, bWlvZHJhZy5sdWtpY0BtZWRmLmtnLmFjLnJz