 Daniel A. Powell
Daniel A. Powell Jeffrey A. Frelinger
Jeffrey A. Frelinger- Department of Immunobiology, University of Arizona, Tucson, AZ, USA
Natural resistance-associated macrophage protein (NRAMP) encoded by the Slc11a1 gene is a membrane-associated transporter of divalent metal ions. Murine Slc11a1 has two known alleles, a functional Slc11a1Gly169, which is found in DBA2/J, NOD/LtJ, and 129p3/J and related mouse strains, and a non-functional Slc11a1Asp169, that is found in C56Bl/6J (B6) and BALB/cJ mice. B6 mice congenic for Slc11a1Gly169 (B6-Slc11a1G169) are markedly resistant to the intracellular pathogens Salmonella, Leishmania, and Mycobacterium tuberculosis. We examined the host cell response and replication of Francisella in B6-Slc11a1G169 mice. Bone marrow-derived macrophages from either B6-Slc11a1G169 or B6 mice were both effectively invaded by Francisella live vaccine strain (LVS). However, at 16 hours post-infection (hpi), the number of LVS bacteria recovered from B6 macrophages had increased roughly 100-fold, while in B6-Slc11a1G169 mice the number decreased 10-fold. When the mice were challenged intranasally (i.n.) B6 mice lost significant amounts (~15%) of weight, where as B6-Slc11a1G169 mice lost no weight. Three days after infection in B6-Slc11a1G169 mice, we failed to recover viable Francisella from the lungs, livers, or spleens. By contrast, B6 mice had bacterial burdens approaching 1 × 106 CFU/organ in all three organs. To further examine the degree of resistance imparted by Slc11a1Gly169 expression, we challenged mice deficient in TLR2, TLR4, and TLR9, but expressing the functional Slc11a1 (B6-Slc11a1G169Tlr2/4/9−/−). Surprisingly, B6-Slc11a1G169Tlr2/4/9−/− mice had no notable weight loss. Eighty percent of B6-Slc11a1G169Tlr2/4/9−/ − mice yielded no detectable Francisella in any organ tested. Additionally, Slc11a1G169 produced little detectable cytokine either in the lung or serum compared to B6 mice. Mice expressing Slc11a1Gly169 survived even high doses (~80 LD50) of LVS inoculation. These data taken together serve to highlight that functional Slc11a1Gly169 can compensate the lack of TLR2/4/9. Thus Slc11a1 is a critical player in murine resistance to pulmonary Francisella infection, but not footpad infection.
Introduction
Many host genes have been implicated in the control of intracellular bacterial infections. The Slc11a gene was originally described as responsible for immunity to Salmonella (Ity) and has been found to function in many other intracellular bacterial infections including Mycobacterium tuberculosis (1–3). Common inbred mice carry one of two different alleles for Sc11a1. C57BL/6 (B6) and BALB/c the most commonly used inbred mouse strains in immunology carry a non-functional variant (G169D) allele (Slc11a1−). C3H and 129 (and other strains) carry the functional allele 169G (Slc11a1+).
The innate immune response to Francisella tularensis (Ft) has been the subject of investigation for the past decade. Ft is a Gram-negative, facultative intracellular pathogen. In vivo and in vitro Ft largely infects myeloid cells. Based on its role in other intracellular bacterial infections, we hypothesized that Slc11a1 would impact Francisella infection. However, nearly all reports of Francisella immunity have used either B6 or BALB/c mice, eliminating the ability to detect the impact of Slca11a on the infection (4). There is one early report that investigated the role of Slc11a1, and compared B10 mice and a congenic line B10.A-slca11a1G169 following footpad challenge with Francisella strain live vaccine strain (LVS) (5). In that report they concluded that not only was a functional Slc11a not protective, but slightly increased susceptibility as measured by bacterial load in the spleen. However, no survival data were reported. One other report examined a sampling of inbred strains, including C3H and 129 that express functional Slca11a were not markedly more resistant to the most highly pathogenic Francisella strain, suggesting that Slc11a1 was not sufficient to mediate protection in that setting (6).
Francisella infection outcomes are markedly different depending on the route of infection. In our lab, we have demonstrated that the first cells infected, and the resulting immune response differs depending on the route of infection (7). Intranasal infection resulted in a high proportion of alveolar macrophages infected, whereas intradermal infection resulted in primarily neutrophils being infected. These changes resulted in a distinct adaptive immune response. Since Slc11a1 is preferentially expressed in macrophages and alveolar macrophages, we speculated that the impact of Slc11a1 might differ depending on the route of infection.
Pathogen-associated molecular patterns are stimulators of innate immune response and play a role in Francisella infection. Mice defective in TLR2, but not TLR4 are more susceptible to Francisella infection (8). It is striking that these experiments were all performed in the absence of a functional Slc11a1 gene. We speculated that Slc11a1 might compensate for a TLR2 defect. We tested TLR2-deficient mice with and without expression of Slc11a1G169 for their resistance to Ft LVS. Surprisingly, Slc11a1G169 expressing mice were nearly equivalent to TLR2+/Slc11a1G169 mice indicating that Slc11a1+ can compensate for the lack of TLR2 expression.
Materials and Methods
Bacteria
Francisella tularensis subsp. holarctica LVS was obtained from the Centers for Disease Control (Atlanta, GA, USA). All bacterial studies were carried out with approval from the University of Arizona Biosafety Committee. Bacteria were grown at 37°C on chocolate agar supplemented with 1% IsoVitalex (Becton Dickinson). Inocula were prepared from lawn grown Francisella. Bacteria were resuspended in sterile phosphate-buffered saline (PBS) at an optical density at 600 nm (OD600) of 1, equivalent of 1 × 1010 CFU/mL. Innocula were diluted in sterile PBS to obtain the desired bacterial dose. Challenge doses were quantified by serial dilution and plating on chocolate agar.
Bone Marrow-Derived Macrophage (BMDM) Generation
Primary BMDMs were cultured from femurs as previously described (9). Following differentiation, non-adherent cells were removed by multiple washes with PBS and BMDMs were removed from plates by scraping. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Atlas), l-glutamine (HyClone), sodium pyruvate (HyClone), and penicillin–streptomycin (Life Technologies). Medium was replaced with antibiotic-free medium 24 h prior to inoculation with Francisella.
Francisella Growth Assays
A total of 2 × 105 BMDM/well were seeded into 96-well flat bottom plates (~80% confluent) for Francisella intracellular growth assays and incubated 2 h to allow adherence to the plate. Cultures were inoculated with LVS at 1 or 100 bacteria per cell. Infection was facilitated by centrifugation at 300 × g for 5 min. Cells were incubated for 1 h with bacteria, and the medium was then removed. Fresh medium containing 50 μg/mL gentamicin (Sigma) was added to kill extracellular bacteria. One hour after gentamicin addition, medium was removed, and cells washed twice before the addition of fresh antibiotic-free medium. To determine intracellular growth, medium was removed at indicated time points post-infection, and 200 μL of PBS was added to the cultures. Cells were removed from the plate by vigorous pipetting. Cells were lysed by vortexing at maximal speed for 1 min. Serial 1:10 dilutions of the lysate were made and plated onto chocolate agar. The resulting colonies were counted 48 h later. For determination of reactive oxygen species (ROS), BMDM were harvested and processed with CellRox Green (ThermoFisher) per the manufacturers instructions.
Mice
C57Bl/6J (B6) and B6.129-Tlr2tm1Kir/J (TLR2−/−) mice were obtained from The Jackson Laboratories (Bar Harbor, ME) and bred in-house. B6 Slc11a1G169 (Slc11a1+) and B6 Slc11a1+ Tlr2/4/9−/ − (Slc11a1+ Tlr2/4/9−/ −) were obtained from Dr. Gregory M. Barton (University of California, Berkley) and bred in-house. The B6-Slc11a1+ mice are an incipient congenic, having been backcrossed to B6 five generations (10). This results in mice that are approximately 97% B6 background genes (excepting those linked to Slc11a1). The triple knockout has previously described and backcrossed to B6 (10). F1 mice (Slc11a1± Tlr2−/ −Tlr4± Tlr9±) were produced by crossing Slc11a1+ Tlr2/4/9−/ − with Tlr2−/ −. Loss of TLR2 expression was confirmed by flow cytometry. All animal protocols were approved by The University of Arizona IACUC.
Inoculation of Mice
For intranasal infection, mice were anesthetized with 575 mg/kg of body weight of tribromomethanol (Avertin; Sigma) administered intraperitoneally. Mice were then intranasally (i.n.) inoculated with various doses of LVS suspended in 25 μL PBS. For footpad infection, mice were restrained and inoculated with LVS suspended in 50 μL PBS in the right footpad. Mice were weighed daily following all inoculations. Mice were sacrificed if they lost more than 25% of their starting weight, as indicated in our University of Arizona IACUC protocol.
Determination of Bacterial Burdens
Spleens, livers, and lungs were homogenized in sterile PBS by using a Biojector (Bioject) as previously described (11). Tenfold serial dilutions were plated onto chocolate agar. The resulting colonies were counted 72 h later. The limit of detection (LOD) was 50 CFU per organ.
Preparation of Lung Cells
Lungs were perfused with PBS to remove blood and then finely minced. Minced lung was placed into 10 mL of digestion buffer containing 0.5 mg/mL collagenase I (Worthington Biochemical), 0.02 mg/mL DNase (Sigma), and 125 U/mL elastase (Worthington Biochemical) in RPMI 1640 (HyClone). Lungs were digested for 30 min at 37°C and then vigorously pipetted prior to filtering through a 100-μm filter. Red blood cells were lysed by using ammonium chloride–potassium carbonate lysis buffer. Viable cells were determined by trypan blue exclusion using a hemacytometer.
Determination of Serum and Lung Cytokine Production
Serum and lung cytokines (IL-6, IL-10, MCP-1, IFN-γ, TNF-α, and IL-12p70) were determined using a cytometric bead array (CBA) Mouse Inflammation Kit (Becton Dickinson) according to the manufacturers instructions. For lungs, single cell suspensions were incubated in DMEM with antibiotics for 24 h and the supernatant was harvested and used for the CBA. Serum was analyzed undiluted.
Antibodies and Flow Cytometry
The following directly conjugated antibodies were utilized for flow cytometry analysis: CD3 (clone 145-2C11; eBioscience), CD4 (clone GK1.5; Biolegend), CD8a (clone 53-6.7; BD Biosciences), CD11b (clone M1/70; Biolegend), CD11c (clone N418; Biolegend), CD19 (clone 6D5; Biolegend), F4/80 (clone BM8; Biolegend), and GR-1 (Ly-6G) (clone RB6-8C5; eBioscience). All antibodies were titrated on normal B6 splenocytes prior to use. Lung cells were prepared as above. After preparation of single cell suspensions, cells were blocked for 30 min at 4°C with 24G2 serum to block Fc receptors. Cells were then stained with indicated antibodies for 30 min at 4°C in the dark. Cells were then washed three times with PBS + 2%BSA and analyzed on a Becton Dickinson LSRII flow cytometer. FlowJo (Treestar) was used for all flow cytometry analysis.
Results
LVS Fails to Replicate in Slc11a1+ Macrophages
Since Slc11a1 has been previously implicated in control of intracellular bacterial infections and has only been superficially tested in Francisella infection, we tested the ability of LVS to replicate in BMDMs. We cultured BMDM from both B6 and Slc11a1+ mice and confirmed their macrophage phenotype (CD11b+ F4/80+) by flow cytometry. The BMDMs were then infected with LVS at either a multiplicity of infection (MOI) of 1 (Figure 1A) or 100 (Figure 1B). Two MOIs were used to ensure adequate infection. At 1, 4, and 16 hours post-infection (hpi), macrophages were lysed and viable bacterial enumerated. One hour after infection there were slightly fewer LVS in Slc11a1+ than B6 BMDM at both 100 and 1 MOI. By 4 h, there was a decrease in the number of LVS recovered from the Slc11a1+ BMDM at an MOI of 100, at the MOI of 1 there was no detectable LVS at 4 h in Slc11a1+ BMDM. The B6 BMDM had little change in burden at MOI of 100 and limited growth at the MOI of 1 at 4 hpi. By 16 h, LVS was still undetectable in Slc11a1+ BMDM at an MOI of 1, at the MOI of 100 there was a 100-fold decrease in bacteria recovered compared to the B6 BMDM and 10-fold compared to the Slc11a1+ 1 h after infection (Figure 1). Taken together these data indicate that LVS is slightly impaired in its ability to invade Slc11a1+ BMDM, but more importantly is unable to proliferate after invasion.
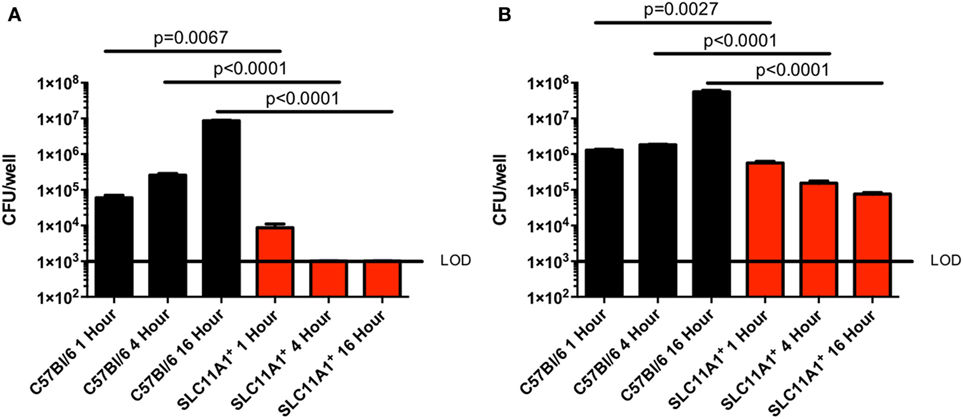
Figure 1. Live vaccine strain (LVS) fails to replicate in Slc11a1+ bone marrow-derived macrophage (BMDM). BMDMs from either B6 (black bars) mice or Slc11a1+ (red bars) mice were infected with LVS at a multiplicity of infection of 1 (A) or 100 (B). At the indicated times, macrophages were lysed and bacteria were enumerated by plating serial dilutions on chocolate agar. The solid line represents the limit of detection (LOD). A Student’s t-test on log-transformed data was used to calculate significance. N = 3 wells per time point, data are representative of three experiments of similar design. Error bars indicate SEM.
Francisella LVS Replicates to Similar Levels in following Foot Pad Inoculation in Slc11a1+ and Slc11a1− Mice
Our in vitro experiments seemed to conflict with the earlier report that Slc11a1 increased susceptibility to LVS infection in vivo (5). Those experiments used sublethal footpad infection. The report of Kovarova used different mouse strains and we needed to test their route of infection with our strains. We therefore examined the effect of Slc11a1 expression on footpad infection. Slc11a1+ and B6 mice were inoculated with a sublethal dose of 1,100 CFU of LVS. On days 3 and 7 post-infection, these mice were sacrificed and total bacterial burden was determined for the lung, liver, and spleen. In contrast to the previous report (5) where there was increased bacterial load in Slc11a1+ mice, we observed no significant difference in the burdens between Slc11a1+ and B6 mice in any organ tested at either time point (Figure 2).
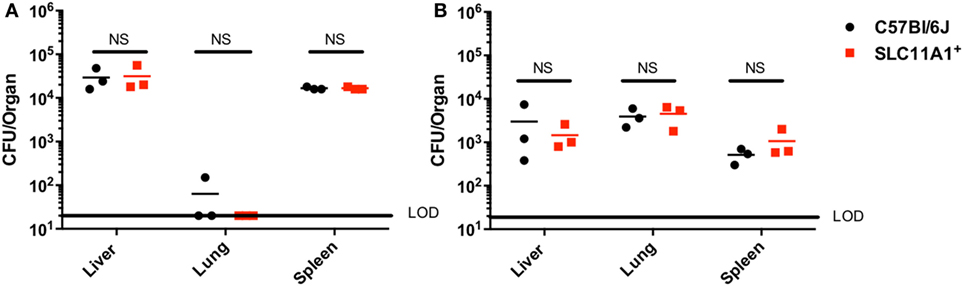
Figure 2. Slc11a1+ mice show no difference compared to B6 in footpad infections with live vaccine strain (LVS). B6 (black circles) or Slc11a1+ (red squares) mice were infected with 1,100 CFU of LVS via the footpad. At day 3 (A) or 5 (B) post-infection organs were harvested and bacterial burdens were enumerated by plating serial dilutions on chocolate agar. The solid line represents the limit of detection (LOD). A Student’s t-test on log-transformed data was used to calculate significance. No significant differences were found. N = 3 mice per time point, data are representative of two independent experiments of similar design.
Slc11a1+ Mice Are Resistant to Intranasal LVS
Since respiratory infection with Francisella alters the immune response compared to cutaneous infection, we sought to determine the role of Slc11a1 in intranasal Francisella infection (7). Slc11a1+ and B6 mice were infected with ~1,300 CFU LVS by the intranasal route. Mice were monitored for weight loss and survival and a subset were sacrificed at day 3 post-infection to determine organ burdens. Surprisingly, Slc11a1+ mice did not lose weight compared to their starting weight (Figure 3A). By contrast, B6 mice lost weight starting at day 3. At day 7, 2/5 surviving B6 mice had lost greater than 25% of their starting weight and were euthanized as required by our IACUC protocol. The mice who survived started regaining weight around day 10 (Figure 3A). In addition to maintaining weight, we were not able to recover any viable LVS from Slc11a1+ mice (LOD, 50 CFU) in any of the organs tested at day 3 post-infection. We recovered high levels of Francisella (105–106 CFU) from every organ tested in B6 mice (Figure 3B). These data demonstrate that Slc11a1 plays an important role in the resistance of mice to intranasal LVS infection. This is relevant to our previous results that demonstrated important differences in immune responses between intradermal and intranasal infection (7).
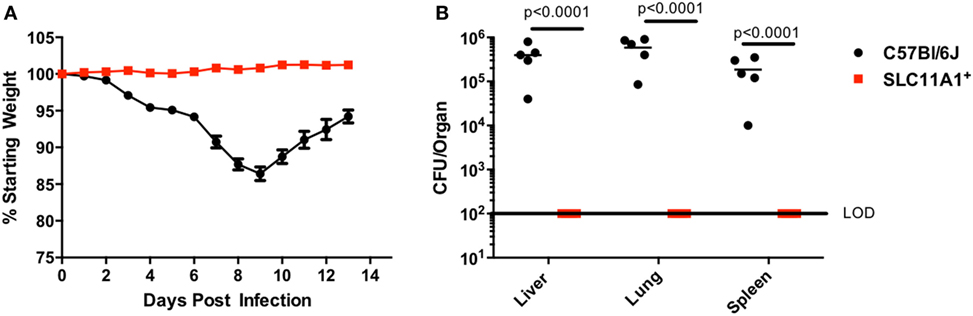
Figure 3. Slc11a1+mice show no weight loss or detectable bacteria after intranasal live vaccine strain (LVS) inoculation. B6 (black circles) or Slc11a1+ (red squares) mice were infected with 1,300 CFU of LVS via the intranasal route. (A) Mice were weighed daily as an indication of disease progression. (B) At 3 days post-infection organs were harvested from five mice and bacterial burdens were enumerated by plating serial dilutions on chocolate agar. The solid line represents the limit of detection (LOD). A Student’s t-test on log-transformed data was used to calculate significance. N = 5 mice per time point, data are representative of three experiments of similar design.
Slc11a1 Expression Can Compensate for Lack of TLR Expression
TLR2 has been reported to play a critical role in Ft resistance. TLR2-deficient mice are more sensitive to Ft infection that B6 mice (8, 12). Since SLC11A1+ B6 mice were resistant to intranasal LVS infection, we wanted to determine if Slc11a1 expression could compensate for the loss of TLR2. We reasoned that Slc11a1+ Tlr2/4/9−/ − mice would be at least as sensitive as TLR2 KO mice, as TLR2 has been strongly linked to resistance to Ft infection. We challenged Slc11a1+ Tlr2/4/9−/ − mice along with Slc11a1+ and B6 mice with ~1,300 CFU of LVS i.n. as above. Similar to earlier experiments (Figure 3), most Slc11a1+ mice yielded no detectable LVS in the organs (LOD, 20 CFU) while we recovered mice high levels of bacteria on day 3 that increased into day 5 from B6 mice (Figures 4A,B). Surprisingly, Slc11a1+ Tlr2/4/9−/ −, like Slc11a1+ mice, yielded 1,000-fold lower organ burdens than B6 in all organs tested, in spite of lacking TLR2/4/9 (Figures 4A,B). TLR4 has little role in Francisella pathogenesis as its unique LPS structure fails to bind TLR4. While EF-Tu and DNAK have been identified as ligands for TLR4, TLR4-deficient mice show no increase in sensitivity Francisella infection compared to B6 mice (13–16). TLR2 has been previously demonstrated to be important in survival of LVS infection in B6 genetic background mice. These experiments demonstrate that the presence of functional Slc11a1 can compensate for the TLR2 defect in resistance to Ft infection, although they are higher than Tlr2+ Slc11a1+ mice.
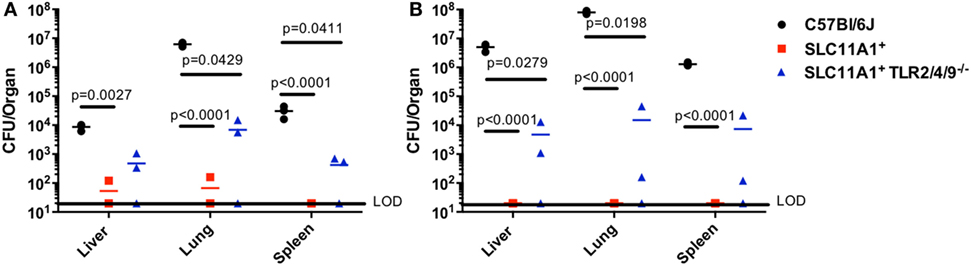
Figure 4. Fewer bacteria are recovered from Slc11a1+ Tlr2/4/9− /− mice than B6 mice following intranasal live vaccine strain (LVS) inoculation. C57Bl/6 (black circles), Slc11a1+ (red squares), or Slc11a1+ Tlr2/4/9−/− (blue triangles) mice were intranasally infected with 1,300 CFU of LVS. At 3 (A) and 5 (B) days post-infection organs were harvested and bacterial burdens were enumerated by plating serial dilutions on chocolate agar. The solid line represents the limit of detection (LOD). A Student’s t-test on log-transformed data corrected for multiple comparisons was used to calculate significance. Unless indicated, differences are not significant. N = 3 mice per time point, data are representative of two experiments of similar design.
Slc11a1+ Mice Produce Little Cytokine Compared to B6 Mice following Intranasal LVS Inoculation
To examine the impact of functional Slc11a1 expression on the innate immune response to LVS infection, we measured cytokine production in both the serum and lung homogenates of Ft infected mice. B6, Slc11a1+, and Slc11a1+ Tlr2/4/9−/ − were i.n. infected with 1,300 CFU of LVS. Additional B6 and Slc11a1+ mice were infected with 1,000 CFU of LVS by the footpad route and serum cytokines measured. As a negative control, naïve mice received PBS by either the intranasal or footpad route. On day 3 and 5 post-infection, mice were sacrificed and cytokine production determined. Following the footpad infection, there were no significant differences between the B6 and Slc11a1+ mice (Figure 5). Both mouse strains produced high levels of IFN-γ, MCP-1, and IL-6. This is not surprising since they had similar LVS organ burdens in previous experiments (Figure 2). By contrast, following intranasal infection, the Slc11a1+ and Slc11a1+ Tlr2/4/9−/ − mice accumulate very little cytokine in either in serum (Figure 6) or lung (Figure 7) after intranasal infection. B6 mice had significantly increased levels of IFN-γ and elevated levels of MCP-1, and IL-6 in the serum compared to Slc11a1+ and Slc11a1+ Tlr2/4/9−/ − mice (Figure 6). Lung cultures from B6 mice produced significantly more IFN-γ, MCP-1, and IL-6 compared to Slc11a1+ and Slc11a1+ Tlr2/4/9−/ − cultures (Figure 7). The lack of cytokine response seen in Slc11a1+ mice demonstrates it is not an increased innate cytokine response that protects Slc11a1+ mice and mediates bacterial clearance.
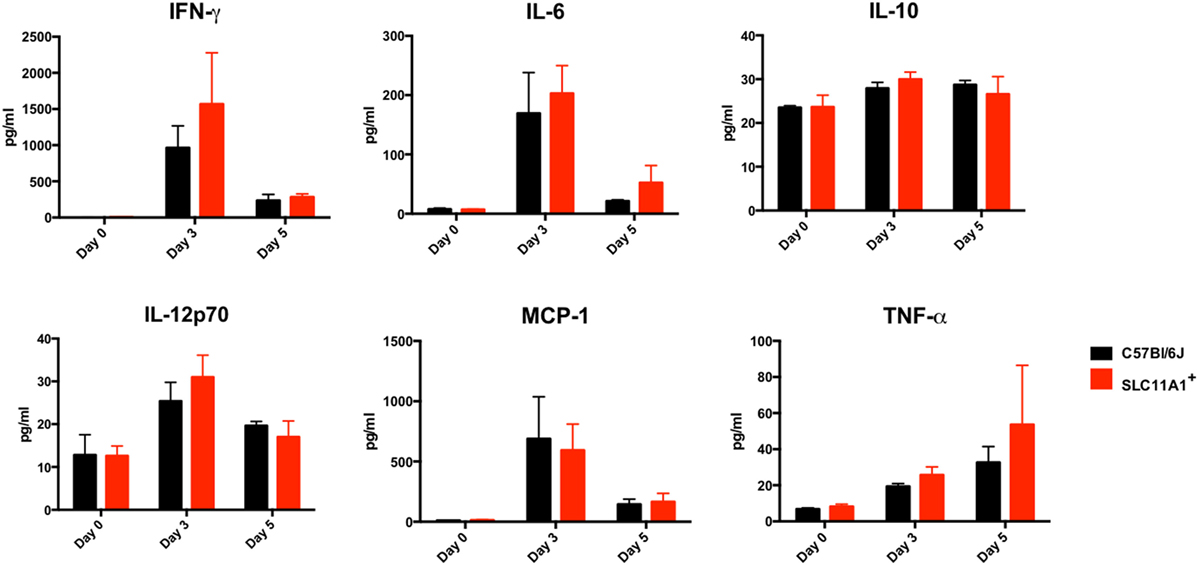
Figure 5. Slc11a1+ and B6 mice produce similar cytokine following live vaccine strain (LVS) inoculation in the footpad. B6 (black bars) or Slc11a1+ (red bars) mice were inoculated with 1,300 or 1,100 CFU of LVS in the footpad. Serum was harvested after 3 and 5 days, and cytokine was quantified by cytometric bead array. A Student’s t-test was used to calculate significance. Unless indicated, differences are not significant. N = 3 mice per time point, data are representative of two independent experiments of similar design.
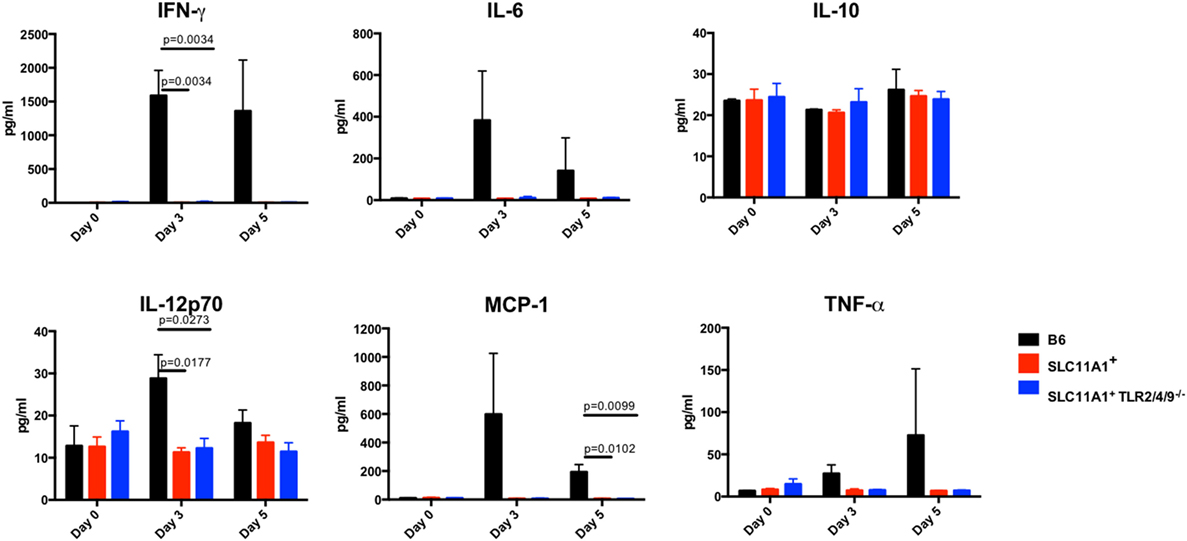
Figure 6. Slc11a1+ mice show little detectable changes in serum cytokines after live vaccine strain (LVS) intranasal inoculation. C57Bl/6 (black bars), Slc11a1+ (red bars), or Slc11a1+ Tlr2/4/9−/− (blue bars) mice were infected with 1,300 CFU of LVS intranasally. Serum was harvested and cytokines were quantified by cytometric bead array. A Student’s t-test corrected for multiple comparisons was used to calculate significance. Unless indicated, differences are not significant. N = 3 mice per time point, data are representative of two independent experiments of similar design.
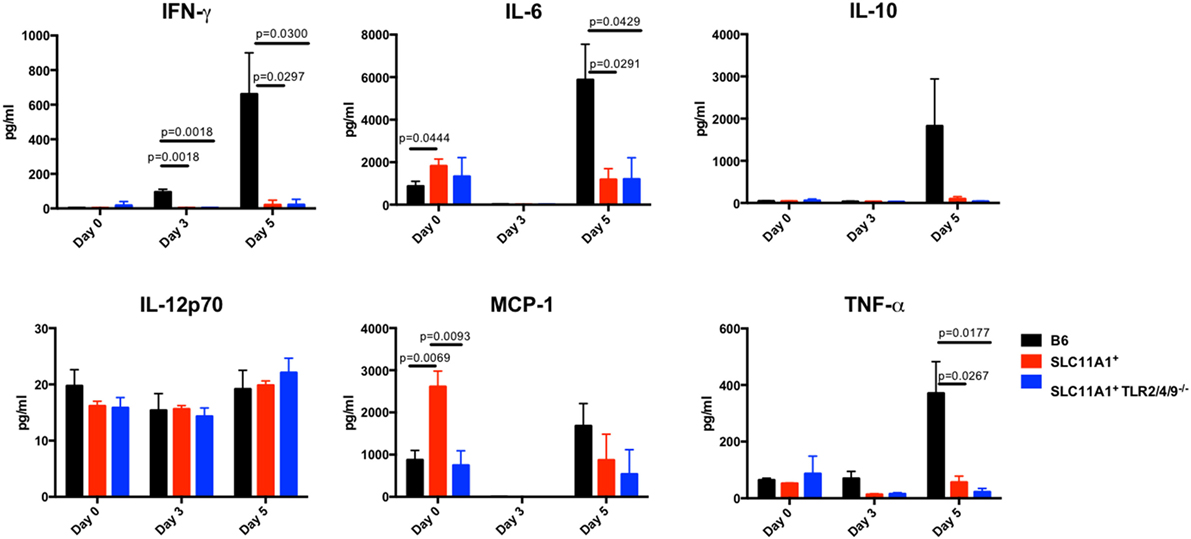
Figure 7. Slc11a1+ mice show little detectable changes in lung cytokine after live vaccine strain (LVS) intranasal infection. B6 (black bars), Slc11a1+ (red bars), or Slc11a1+ Tlr2/4/9−/− (blue bars) mice were inoculated with 1,300 CFU of LVS via the intranasal route. At the indicated time point, lungs were harvested and processed into a single cellular suspension. After 24 h incubation, supernatants were harvested and cytokines were quantified by cytometric bead array. A Student’s t-test corrected for multiple comparisons was used to calculate significance. Unless indicated, differences are not significant. N = 3 mice per time point, data are representative of two independent experiments of similar design.
B6 Mice Have Increased Lung Cellularity after Intranasal Infection Driven by Neutrophil Recruitment
Because we found so little cytokine in Slc11a1+ mice we hypothesized the cellular milieu in the lung would differ after infection. To determine the total cellularity of lungs B6, Slc11a1+, and Slc11a1+ Tlr2/4/9−/ − were i.n. infected with 1,300 CFU of LVS, naïve mice received PBS as a control. At day 3 and 5 post-infection, mice were sacrificed and lungs were harvested, the right lobe was used for CFU determination (Figure 4) and the left lobe was used for lung cellularity counts and flow cytometry (Table 1). The naïve mice had no significant changes in total lung cellularity among strains. Consistent with previously published data, B6 mice demonstrated a significant increase in total lung cellularity at both days 3 and 5 (17). Strikingly in Slc11a1+ mice, we detected no increased cellularity compared to naïve mice at either 3 or 5 days post-infection. The lack of cellular infiltrate in the Slc11a1+ is striking, but consistent with the low levels of bacteria present. We detected only a moderate increase of cellularity in the Slc11a1+ Tlr2/4/9−/ − mice at day 3 although still significantly lower than the B6 mice. By day 5, the cellularity of all Slc11a1+ Tlr2/4/9−/ − mice had decreased close to naïve levels although B6 levels remained high (Table 1).
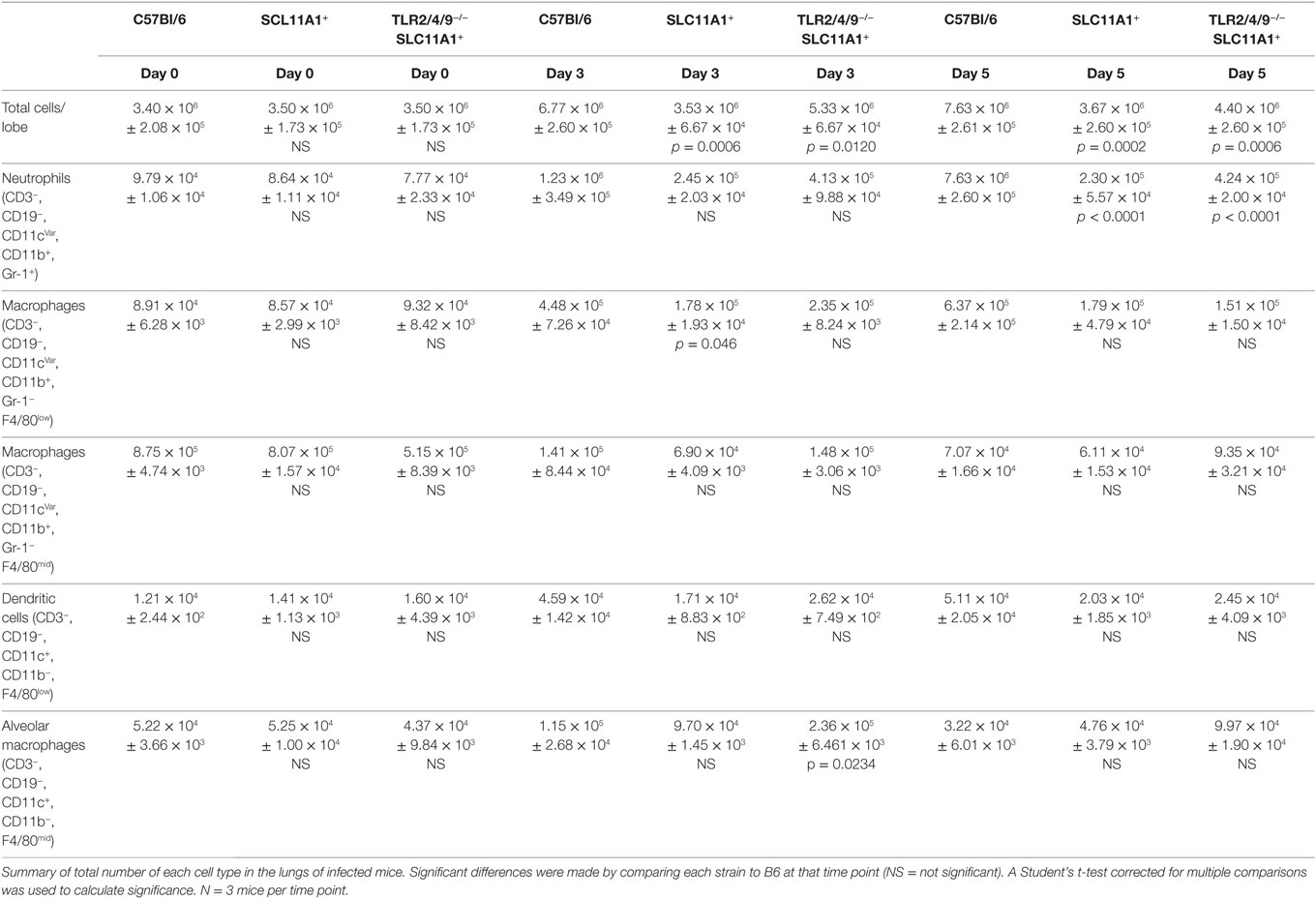
Table 1. Cell subtypes in lung after live vaccine strain infection.
To determine which cells are responsible for this increased cellularity, samples were stained for flow cytometric analysis. The gating strategy for this analysis is in Figure S1 in Supplementary Material. Both the total number and the percentage of macrophages, dendritic cells, neutrophils, T, and B lymphocytes were determined. In B6 mice, two cell types were significantly increased, in both number and percentage. Neutrophils (CD3−, CD19−, CD11b+, Gr-1+) and monocyte/macrophages (CD3−, CD19−, CD11b+, Gr-1−, F4/80−) (Table 1) were increased. There was no significant difference in any other population in either Slc11a1+ or Slc11a1+ Tlr2/4/9−/ − mice. This is consistent with Figure 1 showing the inability of Francisella to replicate in the myeloid cells of Slc11a1+ mice. Thus, the resistance to infection is not due to increased recruitment of innate cells, but rather a cell intrinsic property of the resident infected cells of Slc11a1+ mice.
Following LVS Infection Slc11a1+ Mice Produce More ROS than B6 Mice
One mechanism used by Francisella for controlling the innate immune response is the reduction of host produced ROS (18, 19). To examine the ability of Slc11a1+ cells to produce ROS in response to LVS infection, BMDMs were cultured from B6 and Slc11a1+ mice and infected. We measured ROS production every 10 min. Control cells were treated with 70% tert-butyl hydroperoxide as a positive control or left untreated. Control cultures were harvested at 50 min after the infection. Both untreated B6 and Slc11a1+ BMDM produced similar low levels of ROS. In response to the positive control, tert-butyl hydroperoxide, the Slc11a1+ BMDM made significantly more ROS than the B6 cells. As seen previously, the B6 cells made little ROS in response to LVS infection. Strikingly, the Slc11a1+ BMDM made significantly more ROS in response to LVS starting at 20 min after infection and increasing throughout the assay (Figure 8). Thus the expression of functional Slc11a1 overrides the inhibition of ROS production by Francisella presumably mediating better early pathogen control both in vitro and in vivo.
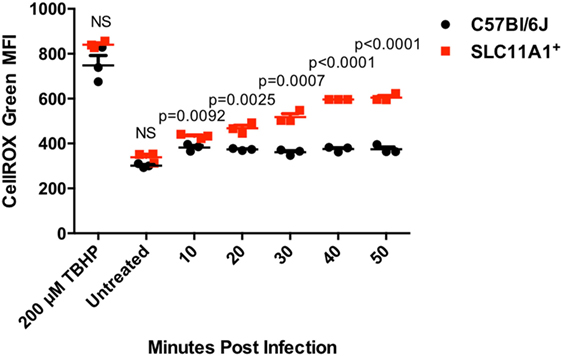
Figure 8. Slc11a1+ bone marrow-derived macrophage (BMDM) show increased reactive oxygen species (ROS) compared to B6 in response to live vaccine strain (LVS) infection. BMDMs were generated from either B6 (black circles) mice or Slc11a1+ (red circles) mice and infected with LVS at a multiplicity of infection 10. Control cells were treated for 50 min with 200μM tert-butyl hyperoxide (TBHP). At the indicated time point, macrophages were harvested and processed for determination of ROS by CellROX green on the flow cytometer. A Student’s t-test corrected for multiple comparisons was used to calculate significance. N = 3 wells per time point. Data are representative of two independent experiments of similar design.
Slc11a1+ Mice Are Resistant to High-Dose Intranasal LVS Challenge
Because of the low bacterial proliferation in Slc11a1+ mice, we hypothesized that Slc11a1+ mice would be resistant to high-dose LVS challenge. In order to determine the extent of protection conferred by a functional Slc11a1—B6, Slc11a1+, Slc11a1+ Tlr2/4/9−/ −, and Slc11a1± Tlr2−/ −Tlr4± Tlr9± mice were challenged i.n. with increasing doses of LVS and monitored for survival. B6 mice only received 8,700 CFU of LVS (~8 LD50). All B6 mice succumbed to infection by day 8 as expected. Slc11a1+ mice received either 8,700 CFU (~8 LD50) or 87,000 (~80 LD50) of LVS. All mice (5/5) survived the 8,700 CFU challenge and 80% (4/5) survived the 87,000 CFU challenge. Slc11a1+ Tlr2/4/9−/ −, and Slc11a1± Tlr2−/ −Tlr4± Tlr9± had similar survival with all mice surviving the 8,700 CFU challenge and 60% of Slc11a1+ Tlr2/4/9−/ −, and 100% of Slc11a1± Tlr2−/ −Tlr4± Tlr9± survived the 87,000 CFU challenge (Figure 9). This resistance to even very high-dose challenges of LVS, even in the absence of TLR2, highlights the strong effect functional Slc11a1 has the outcome of LVS infection.
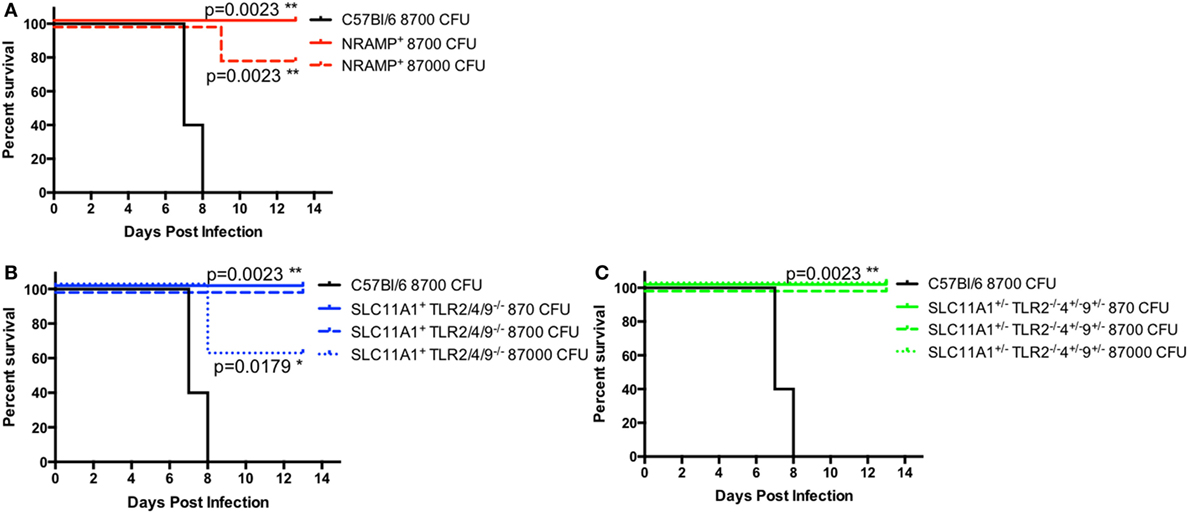
Figure 9. Slc11a1+ mice are resistant to high-dose challenges of live vaccine strain (LVS) by the intranasal route. B6 [black lines (A–C)], Slc11a1+ [red lines (A)], Slc11a1+ Tlr2/4/9−/− [blue lines (B)], or Slc11a1± Tlr2−/−/4±/9± [green lines (C)] mice were infected with the indicated doses of LVS via the intranasal route. Mice were monitored for survival and signs of disease. Significance was determined using a Mantel–Cox log rank test. N = 5 mice per group.
Discussion
Slc11a1 has been implicated in a variety of intracellular infections including Salmonella and Mycobacterium (20). The exact mechanism of Slc11a1 restriction of pathogen replication is unclear, but some work seems to indicate that it is iron restriction outside of the vesicle. Slc11a1 is a transporter of both ferrous iron (Fe2+) and manganese (Mn2+) (21), both of which are important in ROS production and suggest a mechanism for the effect of Slc11a1 expression. In Mycobacterium infections excess iron can overwhelm the Slc11a1 restriction (22–24). It is clear that Slc11a1 restriction is dependent on pathogens passing through a LAMP-1+ vesicle, intracellular pathogens that avoid LAMP-1+ vesicles such as Listeria are not restricted by Slc11a1 (20). Francisella is sensitive to iron starvation when cells are treated with bafilomycin A to limit Francisella escape into the cytosol, sequestration of iron by Slc11a1 may play a role in the resistance to LVS infection (25, 26). Initial studies in BMDM indicated that LVS was only slightly deficient in invasion of Slc11a1+ macrophages and supported no discernable growth. By contrast, LVS replicated robustly in B6 BMDM, indicating an early event after entry that restricts growth in Slc11a1+ BMDM. There is evidence that Francisella is able to spread cell to cell through trogocytosis (27), while not specifically examined in our experiments, the fact that LVS is unable to replicate in Slc11a1+ cells, either in vivo or in vitro, indicates cell to cell spread does not allow LVS to escape Slc11a1-mediated resistance.
Previous work had indicated that expression of Slc11a1 enhanced LVS proliferation following footpad inoculation (5). In our hands, we saw no significant difference between B6 and Slc11a1+ mice following footpad infection (Figure 2). One difference in our study design was using a higher dose of LVS in the footpad (103 vs. 102). This increased dose may account for why we were unable to see the small difference that was seen in the study by Kovarova et al. Additionally, the background of the mouse strains is different (B6 vs. B10), which may also contribute to the fact we saw no difference in the footpad infection. Other genes on chromosome 1 that differ between the Slc11a1 donor in the A/J and the 129 and could be passengers in the congenics. In our experiments they could have epistatic effects that could also contribute to these differences. By contrast, when we challenged mice i.n., Slc11a1+ mice were significantly more resistant to LVS than B6 mice. Functional Slc11a1 expression in C3H/HeN and 129/J mice is not sufficient to survive a highly virulent type A (Strain 33) challenge (28, 29). This might be due to differences in either the genetic background of the mice or differences between Francisella Strain 33 and LVS. Other host genetic differences among mouse strains remain to be explored. A recent study by Fink et al. examined quantitate trait loci (QTL) following LVS infection (6). They reported that A/J mice, which are Slc11a1+, are more sensitive to LVS infection as compared to B6 mice. Recombinant inbred (RI) mice between these two strains were tested and QTL analysis performed. Three QTL were found to be significant, one region on chromosome 1 that includes the Slc11a1 gene, one on chromosome 2, and a large region of chromosome 19. Further analysis demonstrated the QTL on chromosomes 1 and 2 was epistatic. This indicates that the genetic variation on chromosome 2 of A/J mice acts in concert with the Slc11a1 region on chromosome 1. Our Slc11a1+ mice, being incipient congenics, are highly likely to be same as B6 mice on chromosome 2, lacking whatever element from A/J is interacting with the Slc11a1 region on chromosome 1. Combined with our data it seems that functional Slc11a1 expression in a B6 background in beneficial for LVS infection, whereas with the interactions from chromosome 2 Slc11a1 expression can be detrimental. Further RI strains, with more founder strains such as the Collaborative Cross, will be needed to narrow the region on chromosome 2 that interacts with Slc11a1 (30). While the B6 Slc11a1+ strain does still contain 129 DNA surrounding the Slc11a1 allele (10), the F1 cross (B6-Tlr2−/ −Slc11a1D/D X B6-Tlr2,4,9−/ −Slc11a1G/G) we have made should be heterozygous in that region of chromosome 1. Since this F1 strain behaves similar to the parental strain B6-Tlr2,4,9−/ −Slc11a1G/G we feel confident that the remaining 129 DNA is not confounding our results (Figure 9A). In our hands, all Slc11a1+ mice showed marked resistance to intranasal LVS infections. Slc11a1+ mice had little to no detectable bacteria by day 3 post-infection. Additionally, when Slc11a1+ were challenged i.n. with escalating doses of LVS they were able to survive challenges of 87,000 CFU (80 LD50). B6-Slc11a1+ were also resistant to a fully virulent murine Francisella novicida challenge by the intranasal route (data not shown). The Slc11a1+ mice also had no change in total lung cellularity. B6 mice show a large influx of macrophages and neutrophils as well as increased production of IFN-γ, MCP-1, and IL-6. This lack of change in cellularity may be linked with the lack of cytokine production in Slc11a1+ mice. Taken together it would seem that resident lung cells are carrying out the rapid clearance of Francisella. Previous work by our laboratory and others has demonstrated that Francisella can infect a variety of cells and these infected cells differ by route of infection (17, 31–33). Particularly of interest is the fact that alveolar macrophages are the primary infected cell in the lung after intranasal infection whereas after intradermal infection the primary infected lung cells are interstitial macrophages and neutrophils (7). Experiments on PMNs, which do not express Slc11a1, indicate that LVS infection of PMNs prolongs their life span, whereas LVS has been shown to induce cell death in macrophages (34–37). These differential effects depending on cell type may help explain the differences seen in Slc11a1+ mice depending on the route of infection.
The rescue of the TLR2 defect was surprising. TLR2 has been reported by a variety of groups to be critical in survival of Francisella infection (8, 11, 12, 15, 38). This work has been carried out in the Slc11a1− B6 background. Functional Slc11a1 can not only overcome a lack of TLR2, but Slc11a1+ Tlr2−/ − mice are more resistant to intranasal LVS infection than B6 mice. B6-Tlr2/4/9−/ − (Slc11a1−) mice are not available to used as a control, though we expect they would behave like B6-Tlr2−/ − mice as TLR4 and TLR9 have little role in Francisella infection (39, 40). Further work is needed to determine if Slc11a1+ mice are resistant to fully virulent human Francisella strains such as SchuS4. A variety of live attenuated mutants have been generated in both LVS and the type A strain SchuS4. While attenuation has been relatively easily achieved, full protection against a type A challenge has been rare and even successful vaccines only have protected against limited doses of highly virulent bacteria [reviewed in Ref. (41)]. Since most humans express a functional Slc11a1, retesting of these vaccine candidates in a Slc11a1+ background may yield more applicable results.
Author Contributions
DP designed and carried out the experiments. JF and DP analyzed the data and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by NIAID Southeast Regional Center of Excellence for Emerging Infections and Biodefense grant U54 AI057157 and University of Arizona Flow Cytometry Shared Resource funded by the National Cancer Institute of the National Institutes of Health under award number P30 CA023074.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00206/full#supplementary-material.
References
1. Plant J, Glynn AA. Natural resistance to Salmonella infection, delayed hypersensitivity and Ir genes in different strains of mice. Nature (1974) 248:345–7. doi:10.1038/248345a0
2. Skamene E, Gros P, Forget A, Kongshavn PA, St Charles C, Taylor BA. Genetic regulation of resistance to intracellular pathogens. Nature (1982) 297:506–9. doi:10.1038/297506a0
3. Blackwell JM. The macrophage resistance gene Lsh/Ity/Bcg. Res Immunol (1989) 140:767–9. doi:10.1016/0923-2494(89)90029-1
4. Mclendon MK, Apicella MA, Allen LA. Francisella tularensis: taxonomy, genetics, and immunopathogenesis of a potential agent of biowarfare. Annu Rev Microbiol (2006) 60:167–85. doi:10.1146/annurev.micro.60.080805.142126
5. Kovarova H, Hernychova L, Hajduch M, Sirova M, Macela A. Influence of the bcg locus on natural resistance to primary infection with the facultative intracellular bacterium Francisella tularensis in mice. Infect Immun (2000) 68:1480–4. doi:10.1128/IAI.68.3.1480-1484.2000
6. Fink A, Hassan MA, Okan NA, Sheffer M, Camejo A, Saeij JP, et al. Early interactions of murine macrophages with Francisella tularensis map to mouse chromosome 19. MBio (2016) 7:e02243. doi:10.1128/mBio.02243-15
7. Roberts LM, Tuladhar S, Steele SP, Riebe KJ, Chen CJ, Cumming RI, et al. Identification of early interactions between Francisella and the host. Infect Immun (2014) 82:2504–10. doi:10.1128/IAI.01654-13
8. Cole LE, Shirey KA, Barry E, Santiago A, Rallabhandi P, Elkins KL, et al. Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect Immun (2007) 75:4127–37. doi:10.1128/IAI.01868-06
9. Powell DA, Roberts LM, Ledvina HE, Sempowski GD, Curtiss R III, Frelinger JA. Distinct innate responses are induced by attenuated Salmonella enterica serovar Typhimurium mutants. Cell Immunol (2016) 299:42–9. doi:10.1016/j.cellimm.2015.10.002
10. Arpaia N, Godec J, Lau L, Sivick KE, Mclaughlin LM, Jones MB, et al. TLR signaling is required for Salmonella typhimurium virulence. Cell (2011) 144:675–88. doi:10.1016/j.cell.2011.01.031
11. Roberts LM, Ledvina HE, Sempowski GD, Frelinger JA. TLR2 signaling is required for the innate, but not adaptive response to LVS clpB. Front Immunol (2014) 5:426. doi:10.3389/fimmu.2014.00426
12. Katz J, Zhang P, Martin M, Vogel SN, Michalek SM. Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect Immun (2006) 74:2809–16. doi:10.1128/IAI.74.5.2809-2816.2006
13. Chen W, Kuolee R, Shen H, Busa M, Conlan JW. Toll-like receptor 4 (TLR4) does not confer a resistance advantage on mice against low-dose aerosol infection with virulent type A Francisella tularensis. Microb Pathog (2004) 37:185–91. doi:10.1016/j.micpath.2004.06.010
14. Ashtekar AR, Zhang P, Katz J, Deivanayagam CC, Rallabhandi P, Vogel SN, et al. TLR4-mediated activation of dendritic cells by the heat shock protein DnaK from Francisella tularensis. J Leukoc Biol (2008) 84:1434–46. doi:10.1189/jlb.0308215
15. Abplanalp AL, Morris IR, Parida BK, Teale JM, Berton MT. TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS One (2009) 4:e7920. doi:10.1371/journal.pone.0007920
16. Sharma J, Mishra BB, Li Q, Teale JM. TLR4-dependent activation of inflammatory cytokine response in macrophages by Francisella elongation factor Tu. Cell Immunol (2011) 269:69–73. doi:10.1016/j.cellimm.2011.03.023
17. Barrigan LM, Tuladhar S, Brunton JC, Woolard MD, Chen CJ, Saini D, et al. Infection with Francisella tularensis LVS clpB leads to an altered yet protective immune response. Infect Immun (2013) 81:2028–42. doi:10.1128/IAI.00207-13
18. Mccaffrey RL, Allen LA. Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol (2006) 80:1224–30. doi:10.1189/jlb.0406287
19. Mohapatra NP, Soni S, Rajaram MV, Strandberg KL, Gunn JS. Type A Francisella tularensis acid phosphatases contribute to pathogenesis. PLoS One (2013) 8:e56834. doi:10.1371/journal.pone.0056834
20. Govoni G, Gros P. Macrophage NRAMP1 and its role in resistance to microbial infections. Inflamm Res (1998) 47:277–84. doi:10.1007/s000110050330
21. Schlessinger A, Matsson P, Shima JE, Pieper U, Yee SW, Kelly L, et al. Comparison of human solute carriers. Protein Sci (2010) 19:412–28. doi:10.1002/pro.320
22. Kuhn DE, Baker BD, Lafuse WP, Zwilling BS. Differential iron transport into phagosomes isolated from the RAW264.7 macrophage cell lines transfected with Nramp1Gly169 or Nramp1Asp169. J Leukoc Biol (1999) 66:113–9.
23. Zwilling BS, Kuhn DE, Wikoff L, Brown D, Lafuse W. Role of iron in Nramp1-mediated inhibition of mycobacterial growth. Infect Immun (1999) 67:1386–92.
24. Kuhn DE, Lafuse WP, Zwilling BS. Iron transport into mycobacterium avium-containing phagosomes from an Nramp1(Gly169)-transfected RAW264.7 macrophage cell line. J Leukoc Biol (2001) 69:43–9.
25. Byrd TF, Horwitz MA. Chloroquine inhibits the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. A potential new mechanism for the therapeutic effect of chloroquine against intracellular pathogens. J Clin Invest (1991) 88:351–7. doi:10.1172/JCI115301
26. Clemens DL, Lee BY, Horwitz MA. Francisella tularensis phagosomal escape does not require acidification of the phagosome. Infect Immun (2009) 77:1757–73. doi:10.1128/IAI.01485-08
27. Steele S, Radlinski L, Taft-Benz S, Brunton J, Kawula TH. Trogocytosis-associated cell to cell spread of intracellular bacterial pathogens. Elife (2016) 5:e10625. doi:10.7554/eLife.10625
28. Chen W, Kuolee R, Shen H, Conlan JW. Susceptibility of immunodeficient mice to aerosol and systemic infection with virulent strains of Francisella tularensis. Microb Pathog (2004) 36:311–8. doi:10.1016/j.micpath.2004.02.003
29. Shen H, Chen W, Conlan JW. Susceptibility of various mouse strains to systemically- or aerosol-initiated tularemia by virulent type A Francisella tularensis before and after immunization with the attenuated live vaccine strain of the pathogen. Vaccine (2004) 22:2116–21. doi:10.1016/j.vaccine.2003.12.003
30. Aylor DL, Valdar W, Foulds-Mathes W, Buus RJ, Verdugo RA, Baric RS, et al. Genetic analysis of complex traits in the emerging collaborative cross. Genome Res (2011) 21:1213–22. doi:10.1101/gr.111310.110
31. Bosio CM, Dow SW. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J Immunol (2005) 175:6792–801. doi:10.4049/jimmunol.175.10.6792
32. Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol (2007) 178:4538–47. doi:10.4049/jimmunol.178.7.4538
33. Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, et al. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun (2008) 76:5843–52. doi:10.1128/IAI.01176-08
34. Lai XH, Golovliov I, Sjostedt A. Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect Immun (2001) 69:4691–4. doi:10.1128/IAI.69.7.4691-4694.2001
35. Lai XH, Sjostedt A. Delineation of the molecular mechanisms of Francisella tularensis-induced apoptosis in murine macrophages. Infect Immun (2003) 71:4642–6. doi:10.1128/IAI.71.8.4642-4646.2003
36. Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, Monack DM. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A (2007) 104:6037–42. doi:10.1073/pnas.0609675104
37. Rajaram MV, Butchar JP, Parsa KV, Cremer TJ, Amer A, Schlesinger LS, et al. Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS One (2009) 4:e7919. doi:10.1371/journal.pone.0007919
38. Malik M, Bakshi CS, Sahay B, Shah A, Lotz SA, Sellati TJ. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect Immun (2006) 74:3657–62. doi:10.1128/IAI.02030-05
39. Collazo CM, Sher A, Meierovics AI, Elkins KL. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect (2006) 8:779–90. doi:10.1016/j.micinf.2005.09.014
40. Jones CL, Napier BA, Sampson TR, Llewellyn AC, Schroeder MR, Weiss DS. Subversion of host recognition and defense systems by Francisella spp. Microbiol Mol Biol Rev (2012) 76:383–404. doi:10.1128/MMBR.05027-11
Keywords: SLC11A1, natural resistance-associated macrophage protein, host response, Francisella tularensis, mouse genetics
Citation: Powell DA and Frelinger JA (2017) Efficacy of Resistance to Francisella Imparted by ITY/NRAMP/SLC11A1 Depends on Route of Infection. Front. Immunol. 8:206. doi: 10.3389/fimmu.2017.00206
Received: 14 November 2016; Accepted: 15 February 2017;
Published: 15 March 2017
Edited by:
Heinrich Korner, University of Tasmania, AustraliaReviewed by:
Manuel Vilanova, University of Porto, PortugalChristian Engwerda, QIMR Berghofer Medical Research Institute, Australia
Copyright: © 2017 Powell and Frelinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel A. Powell, ZGFuaWVscG93ZWxsQGVtYWlsLmFyaXpvbmEuZWR1