 Dimitrios Balomenos
Dimitrios Balomenos Rahman Shokri
Rahman Shokri Lidia Daszkiewicz
Lidia Daszkiewicz Cristina Vázquez-Mateo
Cristina Vázquez-Mateo Carlos Martínez-A
Carlos Martínez-A- Department of Immunology and Oncology, Centro Nacional de Biotecnología (CNB-CSIC), UAM Campus de Cantoblanco, Madrid, Spain
Fas induces massive apoptosis in T cells after repeated in vitro T cell receptor (TCR) stimulation and is critical for lymphocyte homeostasis in Fas-deficient (lpr) mice. Although the in vitro Fas apoptotic mechanism has been defined, there is a large conceptual gap between this in vitro phenomenon and the pathway that leads to in vivo development of lymphadenopathy and autoimmunity. A striking abnormality in lpr mice is the excessive proliferation of CD4+ and CD8+ T cells, and more so of the double-negative TCR+CD4−CD8−B220+ T cells. The basis of lpr T cell hyperproliferation remains elusive, as it cannot be explained by Fas-deficient apoptosis. T cell-directed p21 overexpression reduces hyperactivation/hyperproliferation of all lpr T cell subtypes and lymphadenopathy in lpr mice. p21 controls expansion of repeatedly stimulated T cells without affecting apoptosis. These results confirm a direct link between hyperactivation/hyperproliferation, autoreactivity, and lymphadenopathy in lpr mice and, with earlier studies, suggest that Fas apoptosis-independent pathways control lpr T cell hyperproliferation. lpr T cell hyperproliferation could be an indirect result of the defective apoptosis of repeatedly stimulated lpr T cells. Nonetheless, in this perspective, we argue for an alternative setting, in which lack of Fas would directly cause lpr T cell hyperactivation/hyperproliferation in vivo. We propose that Fas/Fas ligand (FasL) acts as an activation inhibitor of recurrently stimulated T cells, and that its disruption causes overexpansion of T cells in lpr mice. Research to define the underlying mechanism of this Fas/FasL effect could resolve the phenotype of lpr mice and lead to therapeutics for related human syndromes.
Fas-Dependent Apoptosis of T Cells
Homeostasis regulates total lymphocyte number by balancing cell growth and death. Programmed cell death, referred to as apoptosis, eliminates activated or autoreactive lymphocytes and is thus important in immune system homeostasis. Interaction of Fas (CD95), a member of the tumor necrosis factor receptor family, with its ligand [(Fas ligand (FasL)] after in vitro recurrent T cell receptor (TCR) triggering, induces apoptosis via activation-induced cell death (AICD). Initiation of AICD requires IL-2 exposure prior to secondary TCR stimulation (1). Fas/FasL interaction recruits the adaptor protein Fas-associated death domain (FADD), which activates caspase-8 and initiates the apoptotic cascade. T cell apoptosis is considered central to lymphocyte homeostasis and tolerance induction (1).
Phenotype and Characteristics of Fas-Deficient Mice
lpr (lymphoproliferation spontaneous mutation) mice deficient in Fas show defective AICD of restimulated T cells in vitro. lpr mice present lymphadenopathy due to double-negative (DN) T cell (TCR+CD4−CD8−B220+) hyperproliferation and accumulation. They also develop lupus-like autoimmune disease, probably due to CD4+ T cell hyperactivation. The severity of these symptoms depends on genetic background. B6/lpr mice develop anti-DNA antibodies and mild, non-lethal glomerulonephritis, whereas lpr mice on the MRL background (MRL/lpr) show more pronounced autoimmune manifestations, thereby leading to kidney failure and death (1); this severe phenotype is a combined result of the autoimmune-prone background and the lpr mutation. lpr mice of both backgrounds develop severe lymphadenopathy and splenomegaly.
The hyperproliferative T cell phenotype of lpr mice is also observed in patients with autoimmune lymphoproliferative syndrome (ALPS) (2–4), an autoimmune disease also characterized by defective Fas/FasL signaling. ALPS patients are classified by distinct disease types, depending on the underlying genetic defect (5). The main characteristics of this syndrome are DN T cell accumulation and hyperproliferation, lymphadenopathy development, autoimmune manifestations, and increased risk of lymphomas.
The Controversy Over Fas-Dependent T Cell Apoptosis In Vivo
Although lack of Fas-triggered apoptosis in in vitro-activated T cells of lpr mice was initially suggested to be a direct cause of lymphadenopathy and lupus-like disease (6), the etiology of these symptoms remains enigmatic. While Fas-dependent apoptosis has been clearly established and extensively studied in IL-2-exposed and -restimulated T cells, in vivo Fas/FasL-induced apoptosis and T cell elimination have been questioned. All evidence for the Fas/FasL apoptosis pathway is based essentially on in vitro experiments of recurrent T cell activation, and the in vivo role of this system thus remains ill defined. For example, peptide-induced deletion of T cells in TCR transgenic mice was reported to be Fas dependent in one model, whereas it was Fas independent in other settings (7, 8). Superantigen-induced T cell deletion is dependent on Fas in some systems but not in others, and it was concluded that dissimilar experimental conditions might alter Fas effectiveness (9). Peripheral DN T cells appear to depend on Fas for superantigen-induced apoptosis (10). In addition, defective AICD was not identified following in vivo T cell activation in mice in which Fas was specifically deleted in T cells (11). In vivo Fas-dependent apoptosis is thus not clearly defined in in vivo systems (1), and the debate continues as to how Fas deficiency leads to DN T cell accumulation and lymphadenopathy development.
This debate was further fueled by research directed toward defining whether inactivation of components of the Fas/FasL apoptosis system could reproduce the disease-prone lpr phenotype. Transgenic mice were produced that overexpress the caspase-8 inhibitor CrmA in T cells, and AICD of these cells was efficiently inhibited in vitro (12, 13). The CrmA transgenic mice, nonetheless, showed no T cell abnormalities in vivo, no lymphadenopathy, and no predisposition to autoimmunity (12, 13); it was thus suggested that in addition to its role in apoptosis, Fas has other functions in the control of in vivo T cell homeostasis (12). In other studies, mice were rendered deficient in the major apoptotic regulators caspase-8 or FADD, but again, the lpr in vivo symptoms were not replicated (14, 15); instead, both caspase-8 and FADD were shown to be necessary for normal T cell proliferation. These studies predicted a possible Fas association not only to apoptosis but also to T cell proliferation, and it was proposed that the defective in vitro apoptosis of restimulated T cells might not be related to the lpr phenotype (16).
Hyperproliferation of all lpr Mouse T Cell Subsets
A critical characteristic of the lpr phenotype is extensive T cell hyperproliferation, which is not explained by the defective Fas/FasL apoptotic system. DN CD4+ and CD8+ T cells hyperproliferate in vivo (17–19) and memory T cells (CD44high/CD62Llow) expand massively in lymphoid organs. DN lpr T cells were initially considered inert or anergic cells that accumulate in lymph nodes (20, 21). This does not concur with their hyperproliferative state or with the fact that they secrete large amounts of inflammatory cytokines such as IFN-γ and IL-17 and can be pathogenic (22–24). Furthermore, IL-23 is essential for lymphadenopathy and DN T cell expansion, as disrupted signaling by this cytokine leads to lower numbers of DN T cells (22). The active state of DN cells is supported by recent findings that normal programmed cell death protein 1-expressing DN cells are pro-inflammatory and respond to self-antigens (25, 26) and by their association with human lupus (27, 28).
Lymph node lpr DN cells proliferate in vivo at much higher rates than CD8+ T cells (Figure 1A). lpr DN T cells might originate from activated single-positive T cells that have lost the CD8 receptor (29); however, they have a much higher proliferation rate than CD8+ cells. Alternatively, it was recently argued that lpr DN cells correspond to normal DN T cells with inherently increased proliferation than CD4+ or CD8+ T cells (10, 28). Regardless of their origin, DN cells proliferate at much higher rates and their homeostasis might be Fas/FasL pathway dependent (10, 28).
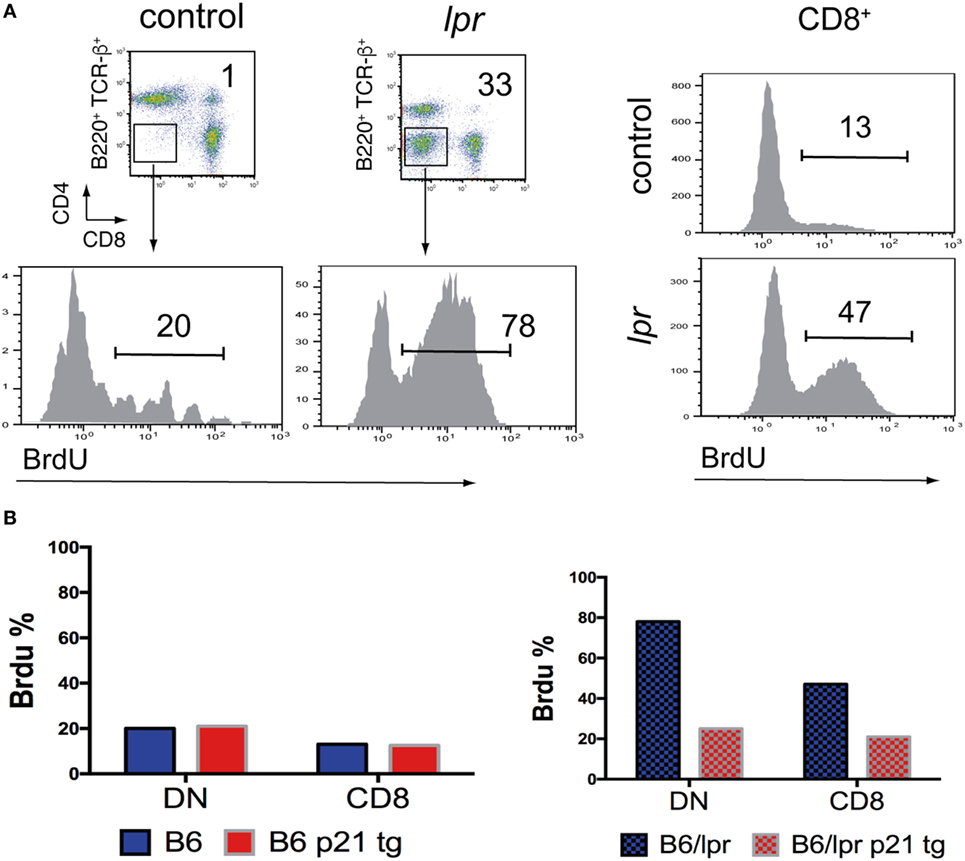
Figure 1. In vivo hyperproliferation of lpr T cells is moderated by p21. (A) Increased proliferation of B6/lpr double-negative (DN) and CD8+ T cells compared to control B6 cells. Lymph node cells were obtained from mice, which received BrdU in the drinking water for 8 days. FACS analysis was used to identify cell types and percentage of proliferating BrdUhi T cells. (B) Effect of a T cell-directed p21 transgene on the proliferation levels of DN and CD8+ T cells from lymph nodes of B6 and B6/lpr mice, detected by BrdU incorporation as in A. The p21 transgene did not affect the levels of low proliferating DN and CD8+ B6 T cells, while it reduced by more than 50% the BrdU uptake by the corresponding B6/lpr cells.
T cell hyperproliferation is also observed in patients with ALPS (2, 4), an autoimmune disease characterized by defective Fas/FasL signaling or by other genetic defects associated with the Fas/FasL pathway. The major features of this syndrome are DN T cell accumulation, lymphadenopathy development, autoimmunity, and increased lymphoma risk (5).
To explain the lymphoproliferative and autoimmune lpr phenotype, we suggested that Fas deficiency also leads to T cell proliferation abnormalities (30), a view previously proposed based on studies of ALPS patients (4). Indeed, treatment of ALPS patients includes drugs that target the activation/proliferation aspects of accumulating T cells, such as mycophenolate mofetil and sirolimus (rapamycin) (31).
While Fas functions other than apoptosis are well established (32), and lpr T cell hyperproliferation appears to be essential for development of the lpr phenotype, Fas-deficient apoptosis is still considered the prevailing cause of lymphadenopathy and autoimmunity development in lpr mice.
Reduction of T Cell Hyperproliferation Regulates Lymphadenopathy and Lupus Development in lpr Mice
To date, no direct evidence has correlated the defective in vitro apoptosis of lpr T cells with in vivo T cell hyperproliferation, lymphadenopathy, and autoimmune disease in lpr mice. The magnitude of hyperproliferation of all T cell subsets in lpr mice and ALPS patients suggests that this abnormality could be relevant in the development of lupus and lymphadenopathy.
Recent research from our laboratory addressed this point, demonstrating that reduction of lpr T cell hyperproliferation effectively suppressed lymphadenopathy and autoimmune disease development in these mice (30). In a previous study, T cell blasts from ALPS patients with higher responses than controls showed lower p21 expression (4). We therefore overexpressed the cell cycle inhibitor p21 in B6/lpr and MRL/lpr mice in a T cell-specific manner. p21 is an established negative regulator of repeatedly stimulated but not of naïve T cells and regulates CD4+ effector/memory T cell expansion (33, 34).
In general, overexpressed p21 diminished in vivo hyperproliferation of DN cells to almost normal levels and greatly reduced lymphadenopathy in lpr mice (Figure 1B). p21 also decreased CD4+ and CD8+ lpr T cell hyperproliferation and autoimmune disease development. Importantly, p21 reduced the activated phenotype of these cells as well as that of DN cells and downregulated their potential to produce IFN-γ and IL-17. p21 also detained lpr T cell hyperactivity, which suggests that p21 regulates activation pathways. In fact, p21 suppresses macrophage activation through the NF-κB activation pathway (35, 36) and inhibits inflammatory cytokine production (35–38) in a cell cycle-independent manner.
Overexpression of p21 had no effect on the in vivo proliferation/activation of normal background T cells (CD4+, CD8+, and DN) (Figure 1B), or their effector/memory T cell expansion. Therefore, p21 effects on lpr hyperproliferation and lymphadenopathy are not caused by p21 influence on essential normal T cell functions. It appears that lack of Fas interferes with the regulation of activation and proliferation, which is then adjusted by overexpressed p21. Recurrent in vitro activation of p21-overexpressing wt and lpr T cells showed no effect on apoptosis induction in wt T cells and did not restore the defective apoptosis of lpr T cells. This finding indicates that p21 reduced the lpr lymphoproliferation without interfering with apoptosis pathways.
p21 thus reduced lpr T cell hyperproliferation, lymphadenopathy, and autoimmune disease development in lpr mice, showing that this hyperproliferation is critical for lymphadenopathy and lupus-like disease development.
Etiology of In Vivo lpr T Cell Proliferation
Whereas T cell hyperproliferation is central to the development of lymphadenopathy and autoimmunity in lpr mice, the cause of the hyperproliferation/hyperactivation of all lpr T cell subsets remains unclear (17–19). Suppression of in vivo lpr T cell hyperproliferation by overexpressed p21 indicated that Fas deficiency alters regulation of the proliferation machinery in T cells, rendering lpr T cells prone to hyperactivation and hyperproliferation. This view is further supported by the finding that p21 overexpression has no effect on proliferation/activation of wt T cells with intact Fas/FasL signaling [Figure 1B; Ref. (30)].
The question that arises at this point is whether lpr T cell hyperproliferation is an indirect consequence of the Fas deficiency or whether Fas has a direct apoptosis-independent antiproliferative effect on T cells. Here, we will explore these two ideas.
The Possibility That Deficient Fas/FasL Apoptosis Drives lpr T Cell Hyperactivation and Hyperproliferation
To date, the in vivo proapoptotic role of Fas on T cells has been tested in various experimental models, but as mentioned above, there is no conclusive evidence to support an in vivo apoptotic effect for Fas. In the case that Fas would play such a role, however, it could be envisaged that activated apoptosis-deficient lpr T cells would accumulate (Figure 2, left). After reactivation, this population would secrete inflammatory cytokines such as IFN-γ, IL-17, and IL-23, and through a feedback mechanism could generate an environment that drives lpr T cell hyperactivation and hyperproliferation. Overexpressed p21 could reduce this lpr T cell hyperproliferation. Nonetheless, as the in vivo proapoptotic Fas/FasL effect remains unclear, the view that lpr T cell hyperproliferation is an indirect effect of deficient apoptosis remains hypothetical.
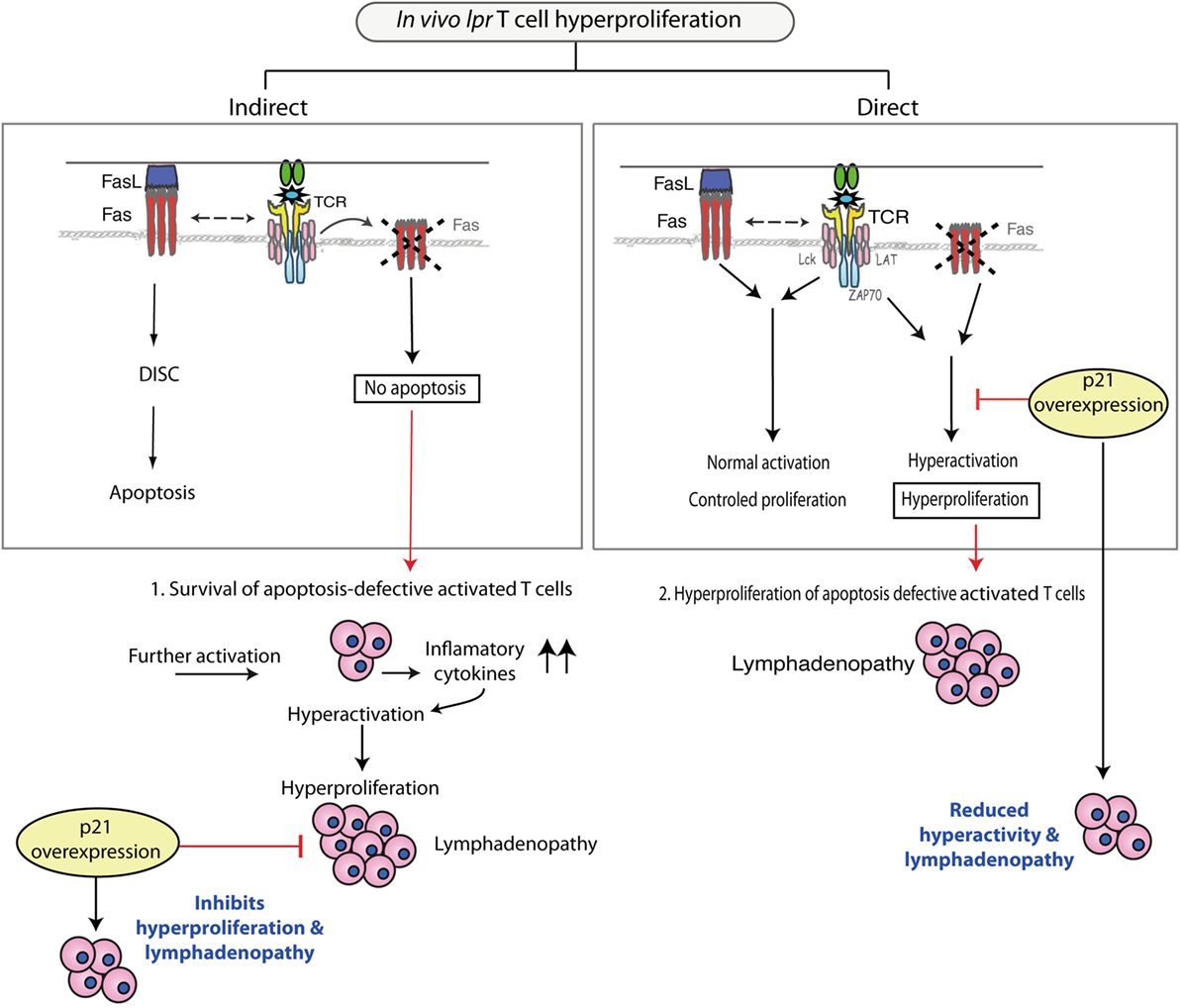
Figure 2. Pathways that could produce the in vivo hyperproliferation of lpr T cells. Left: Fas-deficient T cells that fail to undergo apoptosis might acquire an indirect increased capacity for proliferation, enhanced by inflammatory cytokines that create a feedback loop and result in hyperproliferating lpr T cells. p21 overexpression inhibits hyperproliferation of these cells, which reduces lymphadenopathy. Right: Fas-deficient T cells show a hyperproliferative phenotype due to lack of Fas, which after interacting with Fas ligand (FasL), has a direct regulatory effect on activation/proliferation. These hyperactivated lpr T cells accumulate in lymph nodes. p21 overexpression reduces hyperactivation/proliferation of lpr T cells, and lymphadenopathy development is minimal.
The Fas/FasL System As an Attenuator of T Cell Activation and Proliferation
A number of in vitro studies support a role for Fas/FasL in the regulation of T cell activation and proliferation. While in naïve T cell activation, Fas acts as a costimulatory molecule, in preactivated cells, the FasL/FasL interaction has the opposite effect and appears to inhibit T cell activation. Suggestive evidence for an in vivo Fas/FasL role as a negative regulator of T cell activation is derived from lpr mice that overexpress p21 in T cells, as high p21 levels controlled lpr T cell activation and proliferation as well as autoimmunity and lymphadenopathy. Here, we discuss and evaluate this facet of the Fas/FasL system.
The Dual Role of Fas in Regulating Naïve T Cell Activation
Experimental evidence from primary activation of Fas- and FasL-deficient T cells showed a role for Fas as a costimulatory molecule in naïve T cell activation (39), and other studies analyzed the mechanism of this costimulatory Fas effect (40).
In contrast to the role of Fas/FasL as enhancer of T cell activation, high-dose FasL inhibits TCR-dependent activation of human peripheral cells or naïve T cells (41, 42). It appears that FasL has an inhibitory effect on T cell activation only at high concentrations, while at low concentration, it acts as a costimulatory factor. The mechanisms of FasL interference with T cell activation and proliferation are reviewed by Paulsen and Jansen and Brint et al. (40, 43).
As large amounts of FasL were used in experiments that showed the inhibitory effect of Fas/FasL on T cell activation, it might be argued that the results imply a non-physiological effect of the Fas/FasL system. These findings nonetheless provide a basis for the hypothesis that Fas/FasL interactions inhibit in vivo activation and proliferation of T cells in certain conditions.
Fas Triggering Reduces the Response of Activated T Cells
Experiments using FasL treatment indicate a role for Fas/FasL signaling in naïve T cell activation and proliferation, as commented above. Previously activated T cells are also sensitive to Fas signaling, which reduces their activation and expansion potential. Signaling through Fas elicits antiproliferative effects, a finding first reported by the Tsokos laboratory in 1995 (44). In that study, Fas cross-linking inhibited subsequent anti-CD3 activation of human IL-2-dependent T cells. The same group confirmed this concept using Jurkat T cells (45). In a later study (4), Fas binding of human T cell blasts reduced their proliferation with a concomitant increase in p21, a finding that directly associates p21 with the antiproliferative mechanism of the Fas/FasL system. These studies reinforce the view that the Fas/FasL system attenuates proliferation, although Fas cross-linking in vitro could entail questions regarding the physiological relevance of these data.
Defective Fas/FasL Signaling Leads to lpr T Cell Hyperactivation and Is Counteracted by p21 Overexpression
One of the major autoimmune characteristics of lpr mice is hyperactivation of the immune response and specifically, of T cells. Use of lpr-cREL−/− mice (46) showed that lupus-like disease depends on immune response activation by c-REL, a component of the NF-κB system, whereas absence of NF-κB1 (p65) in lpr mice led to reduced lymphadenopathy. These data argue that hyperactivation is essential for lpr T cell hyperproliferation and corroborate previous results showing that reduction in DN cell activity leads to reduced lymphadenopathy (17).
Here, we propose a potential Fas/FasL role in negative regulation of T cells repeatedly stimulated by autoantigen that could control autoimmunity independently of the classical role of this system in in vitro T cell apoptosis. This possibility is reinforced by the abovementioned studies in which FasL treatment of T cells reduced their activation and proliferation capacity. This view would concur with the development of hyperactivated T cells, whose hyperproliferation leads to their accumulation in the lymphoid organs of lpr mice.
In the case of p21 overexpression in lpr mouse T cells, it can be considered that p21 directly suppresses the deregulated activation of lpr T cells. This would adjust the hyperproliferative state of lpr T cells and inhibit development of autoimmunity and lymphadenopathy (Figure 2, right). This view of a direct p21 effect on lpr T cell activity is supported by in vivo experiments in which p21 overexpression by lpr T cells reduced activation marker expression and IFN-γ production, which finally reduced proliferative capacity (30). In in vitro experiments, overexpressed p21 was detected shortly after secondary stimulation and coincided with reduction of T cell activation. These results link p21 to regulation of T cell activation, as p21 had no effect on Fas/FasL apoptosis. p21 overexpression leads to early reduction of extracellular signal-regulated kinase activity (30), a finding that further supports a role for p21 in controlling recurrent lpr T cell activation. This p21 function is further reinforced by studies showing that p21 attenuates NF-κB-dependent macrophage activation (35, 36). Unpublished data from our laboratory indicate a role for normally expressed p21 in T cell activation after secondary stimulation. Evidence that p21 regulates T cell activation after secondary apoptosis would further strengthen the view that lack of Fas/FasL signaling leads directly to T cell hyperactivation.
By introducing the idea that defective Fas/FasL signaling generates T cell hyperactivation/hyperproliferation in lpr mice independent of the apoptotic defect, we supply a model that explains autoimmunity and lymphadenopathy in these mice. It is our hope that this perspective will prompt research that clarifies the etiology of the lpr mouse phenotype and explains how Fas/FasL interaction modulates T cell activation and proliferation.
Emerging Views
Although Fas is considered a central molecule in T cell apoptosis, and the in vitro apoptosis effects of the Fas/FasL pathway have been studied extensively, the influence of this system on the lpr mouse phenotype remains unclear. With the exception of some recent studies, research in T cells has shifted mainly to other molecules that control homeostasis. Most of our understanding of Fas/FasL is based on analyses in other tissues and systems, and the role of Fas in lymphadenopathy and autoimmunity has remained undefined. We thus consider it imperative to determine how Fas contributes to T cell-driven autoimmunity, since MRL-lpr lupus-like disease is a prevalent model for autoimmunity studies. In this perspective, we explain how p21-based reduction in lpr T cell hyperactivation led us to link lpr T cell hyperactivation to lymphadenopathy and lupus development in lpr mice and ALPS patients. This hypothesis evokes the exciting possibility that impaired Fas/FasL signaling could control lpr T cell activation and proliferation directly. Further research is needed to address this prospect and draw to a close the debate as to how defective Fas apoptosis leads to the lpr phenotype. Perhaps, recent mechanistic evidence showing that Fas drives distinct activities (47, 48) will help to define the diverse Fas pathways involved in T cell function.
Ethics Statement
All animal experiments and protocols were designed in compliance with European Union directives and guidelines and were approved by the Centro Nacional de Biotecnología (CNB-CSIC) Ethics Committee.
Author Contributions
DB conceived and wrote the manuscript; RS composed the final versions of the figures and organized the argument and the manuscript; LD and CV-M contributed to the content and composition of the figures; CM-A contributed to the intellectual content of the manuscript. All the authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank C. Mark for editorial assistance. This work was supported by grants from the Ministry of Economy and Competitivity (MINECO/FEDER PI11/00950 and SAF2016-80803-R to DB, and SAF2013-42289-R and SAF2016-75456-R to CM-A), from the Fundación Alfonso Martin Escudero to CM-A, and from the Community of Madrid (MITIC S2011/BMD2502 to DB and CM-A).
References
1. Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol (2002) 2:11–9. doi: 10.1038/nri701
2. Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med (1999) 130:591–601. doi:10.7326/0003-4819-130-7-199904060-00020
3. Bidere N, Su HC, Lenardo MJ. Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol (2006) 24:321–52. doi:10.1146/annurev.immunol.24.021605.090513
4. Bosque A, Aguiló JI, Del Rey M, Paz-Artal E, Allende LM, Naval J, et al. Cell cycle regulation by FasL and Apo2L/TRAIL in human T-cell blasts. Implications for autoimmune lymphoproliferative syndromes. J Leukoc Biol (2008) 84:488–98. doi:10.1189/jlb.0108043
5. Madkaikar M, Mhatre S, Gupta M, Ghosh K. Advances in autoimmune lymphoproliferative syndromes. Eur J Haematol (2011) 87:1–9. doi:10.1111/j.1600-0609.2011.01617.x
6. Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature (1992) 356:314–7. doi:10.1038/356314a0
7. Singer GG, Abbas AK. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity (1994) 1:365–71. doi:10.1016/1074-7613(94)90067-1
8. Sytwu HK, Liblau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity (1994) 5:17–30. doi:10.1016/S1074-7613(00)80306-4
9. Hildernan DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity (2002) 16:759–67. doi:10.1016/S1074-7613(02)00322-9
10. Mohamood AS, Bargatze D, Xiao Z, Jie C, Yagita H, Ruben D, et al. Fas-mediated apoptosis regulates the composition of peripheral alphabeta T cell repertoire by constitutively purging out double negative T cells. PLoS One (2008) 3:e3465. doi:10.1371/journal.pone.0003465
11. Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity (2007) 26:629–41. doi:10.1016/j.immuni.2007.03.016
12. Smith KG, Strasser A, Vaux DL. CrmA expression in T lymphocytes of transgenic mice inhibits CD95 (Fas/APO-1)-transduced apoptosis, but does not cause lymphadenopathy or autoimmune disease. EMBO J (1996) 15:5167–76.
13. Newton K, Harris AW, Bath ML, Smith KG, Strasser A. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J (1998) 17:706–18. doi:10.1093/emboj/17.3.706
14. Zhang J, Cado D, Chen A, Kabra NH, Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature (1998) 392:296–300. doi:10.1038/32681
15. Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell (2002) 109:S97–107. doi:10.1016/S0092-8674(02)00704-3
16. Strasser A, Pellegrini M. T-lymphocyte death during shutdown of an immune response. Trends Immunol (2004) 25:610–5. doi:10.1016/j.it.2004.08.012
17. Balomenos D, Rumold R, Theofilopoulos AN. The proliferative in vivo activities of lpr double-negative T cells and the primary role of p59fyn in their activation and expansion. J Immunol (1997) 159:2265–73.
18. Fortner KA, Budd RC. The death receptor Fas (CD95/APO-1) mediates the deletion of T lymphocytes undergoing homeostatic proliferation. J Immunol (2005) 175:4374–82. doi:10.4049/jimmunol.175.7.4374
19. Gupta VA, Hermiston ML, Cassafer G, Daikh DI, Weiss AB. Cells drive lymphocyte activation and expansion in mice with the CD45 wedge mutation and Fas deficiency. J Exp Med (2008) 205:2755–61. doi:10.1084/jem.20081204
20. Giese T, Allison J, Davidson W. Functionally anergic lpr and gld B220+ T cell receptor (TCR)-ab double negative T cells express CD28 and respond to costimulation with phorbol myristate acetate and antibodies to CD28 and the TCR. J Immunol (1993) 151:597–609.
21. Ohteki T, Seki S, Abo T, Kumagai K. Liver is a possible site for the proliferation of abnormal CD3+4-8- double-negative lymphocytes in autoimmune MRL-lpr/lpr mice. J Exp Med (1990) 172:7–12. doi:10.1084/jem.172.1.7
22. Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol (2010) 184:4605–9. doi:10.4049/jimmunol.0903595
23. Crispin JC, Tsokos GC. Interleukin-17-producing T cells in lupus. Curr Opin Rheumatol (2010) 32:499–503. doi:10.1097/BOR.0b013e32833c62b0
24. Crispın JC, Tsokos GC. Human TCR-alpha beta CD4-CD8- T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J Immunol (2009) 183:4675–81. doi:10.4049/jimmunol.0901533
25. Rodríguez-Rodríguez N, Apostolidis SA, Penaloza-MacMaster P, Martín Villa JM, Barouch D, Tsokos GC, et al. Programmed cell death 1 and Helios distinguish TCR-αβ+ double-negative (CD4- CD8-) T cells that derive from self-reactive CD8 T cells. J Immunol (2015) 194:4207–14. doi:10.4049/jimmunol.1402775
26. Rodríguez-Rodríguez N, Apostolidis SA, Fitzgerald L, Meehan BS, Corbett AJ, Martín Villa JM, et al. Pro-inflammatory self-reactive T cells are found within murine TCR- αβ+ CD4- CD8- PD-1+ cells. J Immunol (2016) 46:1383–91. doi:10.1002/eji.201546056
27. Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol (2008) 181:8761–6. doi:10.4049/jimmunol.181.12.8761
28. Martina MN, Noel S, Saxena A, Rabb H, Hamed AR. Double negative (DN) αβ T cells: misperception and overdue recognition. Immunol Cell Biol (2015) 93:305–10. doi:10.1038/icb.2014.99
29. Landolfi M, van Houten N, Russell L, Scollay R, Parnes J, Budd R. CD2−CD4−CD8− lymph node T lymphocytes in MRL lpr mice are derived from a CD2+CD4+CD8+ thymic precursor. J Immunol (1993) 151:1086–96.
30. Daszkiewicz L, Vazquez-Mateo C, Rackov G, Ballesteros-Tato A, Weber K, Madrigal-Aviles A, et al. Distinct p21 requirements for regulating normal and self-reactive T cells through IFN-γ production. Sci Rep (2015) 5:7691. doi:10.1038/srep07691
31. Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr (2012) 24:1–8. doi:10.1097/MOP.0b013e32834ea739
32. Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, et al. The CD95 receptor: apoptosis revisited. Cell (2007) 129:447–50. doi:10.1016/j.cell.2007.04.031
33. Balomenos D, Martín-Caballero J, García MI, Prieto I, Flores JM, Serrano M, et al. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat Med (2000) 6:171–6. doi:10.1038/72272
34. Arias CF, Ballesteros-Tato A, Garcia MI, Martín-Caballero J, Flores JM, Martinez-A C, et al. p21CIP1/WAF1 controls proliferation of activated/memory T cells and affects homeostasis and memory T cell responses. J Immunol (2007) 4:2296–306. doi:10.4049/jimmunol.178.4.2296
35. Trakala M, Arias CF, Garcia MI, Moreno-Ortiz MC, Tsilingiri K, Fernandez PJ, et al. Regulation of macrophage activation and septic shock susceptibility via p21 (WAF1/CIP1). Eur J Immunol (2009) 39:810–9. doi:10.1002/eji.200838676
36. Rackov G, Hernández-Jiménez E, Shokri R, Carmona-Rodríguez L, Mañes S, Álvarez-Mon M, et al. p21 mediates macrophage reprogramming through regulation of p50-p50 NF-κB and IFN-β. J Clin Invest (2016) 126:3089–103. doi:10.1172/JCI83404
37. Scatizzi JC, Mavers M, Hutcheson J, Young B, Shi B, Pope RM, et al. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol (2009) 39:820–5. doi:10.1002/eji.200838683
38. Mavers M, Cuda CM, Misharin AV, Gierut AK, Agrawal H, Weber E, et al. Cyclin-dependent kinase inhibitor p21, via its C-terminal domain, is essential for resolution of murine inflammatory arthritis. Arthritis Rheum (2012) 64:141–52. doi:10.1002/art.33311
39. Alderson MR, Armitage RJ, Maraskovsky E, Tough TW, Roux E, Schooley K, et al. Fas transduces activation signals in normal human T lymphocytes. J Exp Med (1993) 178:2231–5. doi:10.1084/jem.178.6.2231
40. Paulsen M, Janssen O. Pro- and anti–appoptotic CD95 signaling in T cells. Cell Commun Signal (2011) 9:7. doi:10.1186/1478-811X-9-7
41. Paulsen M, Mathew B, Qian J, Lettau M, Kabelitz D, Janssen O. FasL cross-linking inhibits activation of human peripheral T cells. Int Immunol (2009) 21:587–98. doi:10.1093/intimm/dxp028
42. Strauss G, Lindquist JA, Arhel N, Felder E, Karl S, Haas TL, et al. CD95 co-stimulation blocks activation of naive T cells by inhibiting T cell receptor signaling. J Exp Med (2009) 206:1379–93. doi:10.1084/jem.20082363
43. Brint E, O’Callaghan G, Houston A. Life in the Fas lane: differential outcomes of Fas signaling. Cell Mol Life Sci (2013) 70:4085–99. doi:10.1007/s00018-013-1327-z
44. Kovacs B, Tsokos GC. Cross-linking of the Fas/APO-1 antigen suppresses the CD3-mediated signal transduction events in human T lymphocytes. J Immunol (1995) 155:5543–9.
45. Kovacs B, Liossis SN, Gist ID, Tsokos GC. Crosslinking of Fas/CD95 suppresses the CD3-mediated signaling events in Jurkat T cells by inhibiting the association of the T-cell receptor zeta chain with src-protein tyrosine kinases and ZAP70. Apoptosis (1999) 4:327–34. doi:10.1023/A:1009691120129
46. Low JT, Hughes P, Lin A, Siebenlist U, Jain R, Yaprianto K, et al. Impact of loss of NF-κB1, NF-κB2 or c-REL on SLE-like autoimmune disease and lymphadenopathy in Faslpr/lpr mutant mice. Immunol Cell Biol (2016) 94:66–78. doi:10.1038/icb.2015.66
47. Chakrabandhu K, Huault S, Durivault J, Lang K, Ngoc LT, Bole A, et al. An evolution-guided analysis reveals a multi-signaling regulation of Fas by tyrosine phosphorylation and its implication in human cancers. PLoS Biol (2016) 14(3):e1002401. doi:10.1371/journal.pbio.1002401
Keywords: Fas, p21, memory T cells, hyperproliferation, double-negative T cells, lupus autoimmunity, alternative functions, hyperactivation
Citation: Balomenos D, Shokri R, Daszkiewicz L, Vázquez-Mateo C and Martínez-A C (2017) On How Fas Apoptosis-Independent Pathways Drive T Cell Hyperproliferation and Lymphadenopathy in lpr Mice. Front. Immunol. 8:237. doi: 10.3389/fimmu.2017.00237
Received: 18 October 2016; Accepted: 20 February 2017;
Published: 10 March 2017
Edited by:
Abdel Rahim A. Hamad, Johns Hopkins University, USAReviewed by:
Alberto Anel, University of Zaragoza, SpainAmir Sharabi, Tel Aviv University, Israel; Harvard University, USA
Copyright: © 2017 Balomenos, Shokri, Daszkiewicz, Vázquez-Mateo and Martínez-A. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Balomenos, ZGJhbG9tZW5vc0BjbmIuY3NpYy5lcw==
†Present address: Lidia Daszkiewicz, OcellO B.V., Biopartner 2, Leiden, Netherlands;
Cristina Vázquez-Mateo, EMD Serono, Billerica, MA, USA