 Soumaya Kouidhi
Soumaya Kouidhi Amel Benammar Elgaaied2
Amel Benammar Elgaaied2 Salem Chouaib
Salem Chouaib- 1ISBST, Laboratory BVBGR, LR11ES31, Higher Institute of Biotechnology of Sidi Thabet, University of Manouba, Sidi Thabet, Tunisia
- 2Laboratory of Genetics, Immunology and Human Pathology, Faculty of Sciences of Tunis, University Tunis El Manar, Tunis, Tunisia
- 3Institut National de la Santé et de la Recherche Médicale (INSERM) UMR1186, Laboratory «Integrative Tumor Immunology and Genetic Oncology», Equipe Labellisée LIGUE 2015, Villejuif, France
- 4Institut National de la Santé et de la Recherche Médicale (INSERM), Gustave Roussy, University of Paris-Sud, Villejuif, France
- 5Institut National de la Santé et de la Recherche Médicale (INSERM), Gustave Roussy, Université Paris-Saclay, Villejuif, France
The immune system and metabolism are highly integrated and multilevel interactions between metabolic system and T lymphocyte signaling and fate exist. Accumulating evidence indicates that the regulation of nutrient uptake and utilization in T cells is critically important for the control of their differentiation and manipulating metabolic pathways in these cells can shape their function and survival. This review will discuss some potential cell metabolism pathways involved in shaping T lymphocyte function and differentiation. It will also describe show subsets of T cells have specific metabolic requirements and signaling pathways that contribute to their respective function. Examples showing the apparent similarity between cancer cell metabolism and T cells during activation are illustrated and finally some mechanisms being used by tumor microenvironment to orchestrate T-cell metabolic dysregulation and the subsequent emergence of immune suppression are discussed. We believe that targeting T-cell metabolism may provide an additional opportunity to manipulate T-cell function in the development of novel therapeutics.
Introduction
It is well admitted that one of the mechanisms by which immune cells integrate the signals required for their proliferation, migration, differentiation, and effector functions is through the modulation of their metabolic activity (1). In this regard, T cells metabolically reprogram and upregulate glucose and amino acid, to allow the synthesis of the new macromolecules required for their proliferation and effector function (2, 3). Furthermore, beyond these key nutrients, iron uptake is also critical for T-cell function (4). Indeed, development and differentiation of antigen-specific T cells depend on iron uptake and internalization via type I transferrin receptor (5). Several previous studies suggested that iron deficiency impaired T-cell proliferation and cytokine production in activated T cells. Conversely, less is known about the effect of iron overload on T-cell function (6).
Nevertheless, how metabolism regulates immune T-cell differentiation, function, and plasticity remains very challenging and how immune cells function in terms of their intracellular metabolism and how these metabolic pathways affect the phenotype and activation of immune cells is attracting a lot of attention at present. Tumor progression is characterized by a tangled network of relationships among different cell types that collectively exploit a metabolic reprogramming and mutually influence their functionality and, in particular, T-cell functions. Our recent knowledge of T-cell molecules involved in the regulation of antitumor T-cell responses has led to the development of several monoclonal antibody-based therapies, against molecules like cytotoxic T-lymphocyte antigen (CTLA-4) or programmed death-1 (PD-1) (7). Although these treatments have shown unprecedented responses in some patients suffering from several cancers (8–10), the response rates are usually low and transient. This is likely due to multiple mechanisms suppressing antitumor immune functions within an unfavorable tumor milieu and metabolism. The metabolic activity of T cells in the context of tumor microenvironment could be one of the key mechanisms.
It should be noted that the dynamic and reciprocal interactions between tumor cells, metabolites, and a variety of cells including immune cells from the tumor microenvironment orchestrate several events, which are critical for tumor evolution toward metastasis. In this context, many cellular and molecular elements of the tumor ecosystem are emerging as attractive targets for therapeutic approaches. Among these targets, hypoxia, which is a hallmark of solid tumors, is strongly associated with advanced disease stage and poor clinical outcome. This is, in part, due to inappropriate local immune reaction and resistance of hypoxic tumor cells to cytotoxic treatments. In fact, most human tumors develop a pathophysiological microenvironment during growth, characterized by an irregular microvascular network and regions of chronically and transiently hypoxic cells. We and others provided evidence that hypoxia plays a crucial role in tumor promotion and immune escape by conferring tumor resistance (11) immunosuppression (12) and tumor heterogeneity (13), which contributes to the generation of diverse cancer invasion programs and enhanced stroma plasticity (11, 14). Therefore, it is of major interest to understand how immune cell intracellular metabolism and some metabolic pathways influence the acquisition of their phenotype, the regulation of their activation and effector function. The metabolic activity of T cells in the context of tumor microenvironment, its heterogeneity, and complexity is therefore an important consideration in immunotherapy. Clearly, if T cells play the music during an adaptive immune response, the metabolic tumor microenvironment calls the tune. Indeed, a better understanding of these metabolic related issues in relationship with T-cell activity may offer new therapeutic strategies in future to better control their plasticity and effector function and boost their efficacy and potential use in cancer immunotherapy approaches.
Basic Overview of Metabolism in T Cells
Metabolism is the process whereby cells can either break down molecules to generate energy in the form of adenosine triphosphate (ATP) or synthesize several macromolecules. Metabolism could be divided into two complex pathways: the catabolic processes, critical for cellular proliferation and functions and the anabolic process, important for cellular growth.
Consistent studies focused on the molecular mechanisms that dictate metabolic reprogramming in the immune cells (15). It is now widely appreciated that T-cell metabolic remodeling plays a key role to shape immune response, in particular, antitumor immunity. Profound metabolic changes occur under tight regulation allowing T cells to maintain energy balance between anabolic and catabolic metabolism, which support adequate immune responses (16, 17).
During quiescence, T cells require energy-oriented oxidative metabolism and relatively small amounts of glucose, amino acids and fatty acids to maintain basic energetic, primarily anabolic and minimal replacement biosynthesis demands. Encounter with cognate antigen activation, T-cell stimulation by T-cell receptor (TCR) ligation and binding with costimulatory molecules induce metabolic remodeling (18, 19). In fact, metabolism shifts to glycolysis to support rapid growth and to biosynthesis for differentiation into effector T cells (Teff) (1, 20, 21) (Figure 1A). Albeit, aerobic glycolysis is less efficient than oxidative phosphorylation (OXPHOS) at yielding ATP, it generates metabolic intermediates which are important for cell growth and proliferation as well as for cytotoxicity and cytokine production. Nevertheless, glycolytic pathway generates macromolecule precursors required in the pentose phosphate pathway (PPP) for cell growth and NAD phosphate (NADPH) production important for anabolic pathways and maintaining redox balance (22).
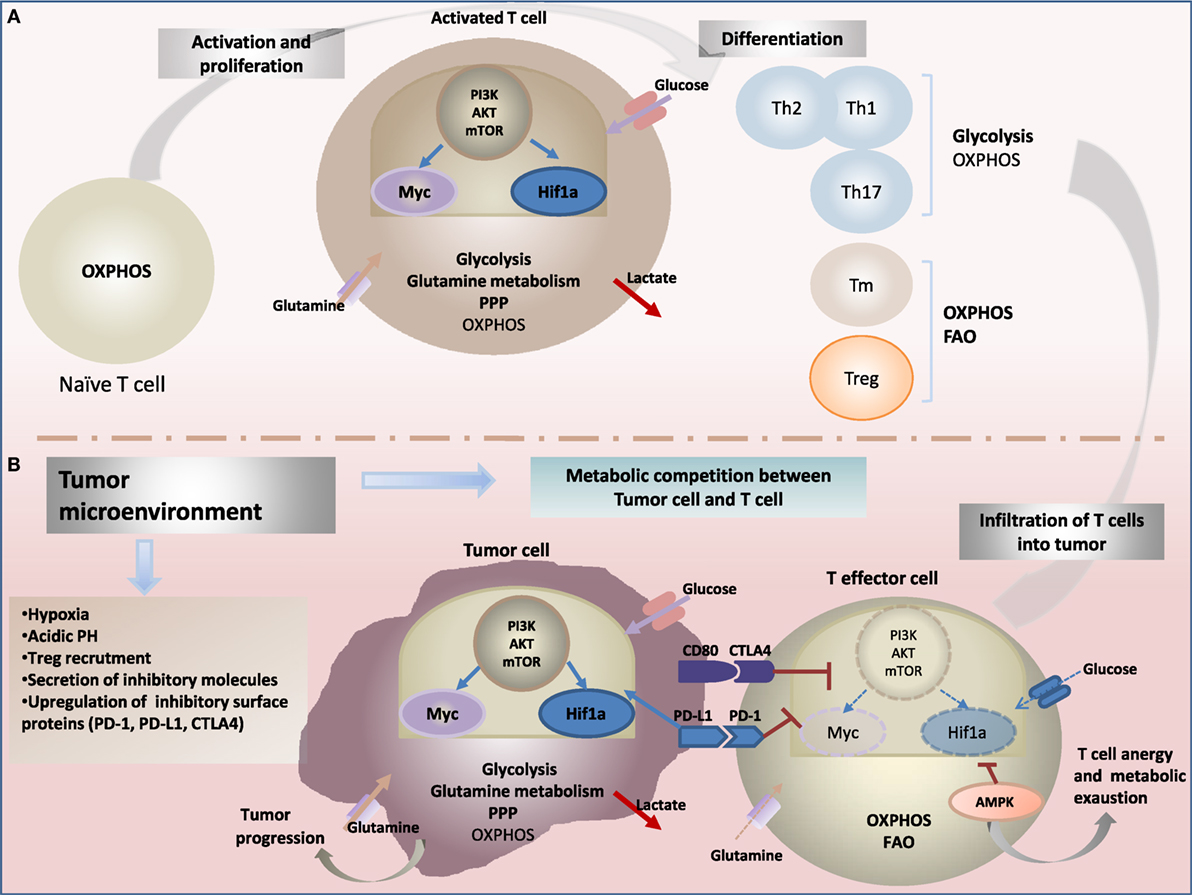
Figure 1. Metabolic reprogramming drives: (A) T-cell fate and function and (B) antitumor-immune response. (A) Upon activation through T-cell receptor (TCR) and costimulatory signals, T cells engage in growth, and differentiation into different cytotoxic, regulatory T cells (Treg), helper T (Th), and memory T (Tm) subsets cells. Metabolic reprogramming has been shown to intimately support T-cell activation and differentiation. While naïve T cells rely on oxidative phosphorylation (OXPHOS) to maintain energy demand; activated T cells engage increased aerobic glycolysis and glutaminolysis consuming massive amount of glucose and glutamine, enabling to generate effector cytokines, including interferon-γ (IFN-γ) and IL-2. In contrast to cytotoxic and effector Th cells, the metabolic profile of Treg and Tm cells rely on OXPHOS and fatty acid oxidation (FAO) to support their survival and differentiation. The central energy-monitoring system underlying this metabolic remodeling is the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway that boosts glycolytic activity in T cells via activation of transcription factors hypoxia-inducible factor-1α (HIF-1α) and Myc pathways. (B) Tumor-specific T cells are often rendered dysfunctional due to an immunosuppressive microenvironment. Infiltrating T cells are reprogrammed by the tumor favoring its survival and immune escape. Cancer cells induce several metabolic changes in the microenvironment. Tumor-mediated decreased extracellular nutrients levels cause impaired glycolysis capacity and IFN-γ production in T cells. Cancer cells also generate a hypoxic microenvironment. Hypoxia stabilizes HIF-1α and enhance glycolysis in tumor cells, a phenomenon recognized as “the Warburg effect.” HIF-1α also enhances constitutive expression of programmed death ligand-1 (PD-L1) leading to activation of Akt/mTOR pathway. Activating immune checkpoints and PD-L1 signaling through binding to its receptor programmed death-1 (PD-1) leads to dampening of the Akt-mTOR pathway and reduced T-cell glycolysis. Collectively, tumor environment affects metabolic fitness of infiltrating immune cells and drives impairment of antitumor effector function and increased tumor progression.
After pathogen clearance, most T cells go through apoptosis while few of them remain as long-lived memory cells responsible for enhanced immunity against upcoming pathogens or tumors re-exposure (23).
Distinct Metabolic Programs for T Cells Differentiation and Function
T lymphocytes (T cells) that undergo an immune response constitute an ideal system to study the rapid shift from quiescent to active state that belongs to growth, proliferation, and differentiation into largely heterogeneous T-cell subsets. Emerging concepts in immunology suggest that lymphocyte activation is intricately linked to metabolic reprogramming (24–26). In fact, metabolism fundamentally underpins T-cell function and lymphocytes metabolism is dynamically regulated depending on their separate phases of development: (1) naïve or resting, (2) effector or activated, and (3) memory T (Tm) cells (27).
Naïve T cells are activated to rapidly respond to foreign pathogens or inflammation through a tight interaction with the TCR and major-histocompatibility complex. Further, T cells enter the effector phase of rapid growth, proliferation, and differentiation. Teff could be divided into cytolytic T cells, secreting granzyme B, perforin, interferon-γ (IFN-γ), into helper T cells (Th) including the type-1 (Th1), type-2 (Th2), and type-17 (Th17) producing characteristic cytokines or into regulatory T cells (Treg) (28, 29). Teff play a pivotal role to mediate antitumor immunity. Hence, Treg obstruct Teff activity and suppress immunity, showing a poor prognosis in many cancers (30). After expansion phase and antigen clearance, most clonally differentiated T cells undergo apoptosis, while a small fraction become quiescent Tm cells, responsible for enhanced immunity after re-exposure to the pathogen (31, 32). The differences in functional and phenotypic characteristics of quiescent T cells and activated T cells are supported by differential metabolic requirements (17). Each subset of T-cell demonstrates unique metabolic demands and signaling pathways that contribute to its fate and function (25).
Quiescent T cells and activated T-cell fate are characterized by different metabolic pathways (33–35). Globally, activated T cells adopt an anabolic metabolism supporting rapid proliferation whereas quiescent T cells engage catabolic metabolism (36). Teff subtypes switch their metabolic program to robust aerobic glycolysis, but increased glycolytic rates occurred much higher in Th1, Th2, and Th17 cells than in Treg cells (24). Treg cells sustain enhanced fatty acid oxidation (FAO) metabolism as a major source of energy to maintain their survival (37–39). Upon antigen encounter, upregulation of aerobic glycolysis in extensive proliferating T cells is accompanied with glutaminolysis, PPP, not only to support ATP generation, but also to enhance biosynthesis of crucial intermediates and precursors necessary for subsequent macromolecules that are incorporated into cellular biomass (40, 41) (Figure 1A). Th17 cells rely, in particular, on increased glycolysis. Hence, inhibiting glycolysis during Th17 cell differentiation re-enforce Treg generation (42). Nevertheless, consistent data suggest that mitochondrial reactive oxygen species (ROS) produced during OXPHOS is also crucial to activate T-cell and to enhance antigen-specific proliferation. However, excessive ROS levels are toxic for T cells and leads to apoptosis (43). CD4 + regulatory T lineage cells exhibit a mixed metabolic program involving mainly FAO and OXPHOS and low level of glycolysis (44). Treg favor FA catabolism via b-oxidation and prioritize oxidative ATP to meet their energetic demands, an important metabolic phenotype for the differentiation of Treg (38).
After the clearance of pathogens, the remaining antigen-specific T cells (Tm cells) as a quiescent T-cell population share common metabolic requirements with other nonproliferating cells. Tm cells maintain catabolic profile with lower nutrient uptake and biomass synthesis and predominantly engage mitochondrial OXPHOS and FAO metabolism for long-term persistence, ATP production and the capacity to vigorously respond to antigen stimulation (45). Several studies revealed that maintaining mitochondrial mass is critical for Tm cells development since it offers the opportunity to use a wide range of substrates responsible for energy generation, like fatty acids (46, 47). FAO constitute a preferred fuel source for Tm cells as this lipid oxidation generates intermediate of tricarboxylic acid (TCA) cycle related to OXPHOS metabolism. However, their detailed metabolic profiles remain to be explored (25).
Metabolic regulation of T-cell fate and function involves a network of molecular regulators. The main induced signaling pathways underlying the activation through the TCR with CD28 costimulation, is the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) (48–50). Increasing evidences suggest that the mammalian target of rapamycin (mTOR) is a central regulator of cell metabolism. Interestingly, T-cell differentiation to effectors or Tm cells is governed in part by asymmetric partitioning of fate determining proteins (51, 52). Recent study demonstrates that asymmetric division of T cells generates two sets of daughter cells with differential mTORC1 activity (53). The first set exhibits increased activity mTORC1, as well as high levels of glycolysis and effector molecules expression. Besides, the second T-cell set shows decrease in mTORC1 activity associated with enhanced rates of lipid metabolism and antiapoptotic molecules. Behind, the latter daughter cells display enhanced long-term survival and differentiate to Tm cells (53). This pathway plays key transcriptional and post-transcriptional roles to promote anabolic gene expression and intracellular trafficking of nutrient transporters (54). mTOR is the downstream target of the PI3K–AKT signaling and a central player governing metabolic reprogramming and fate of T-cell (55–57). Two major transcription factors are upregulated by mTOR: avian myelocytomatosis virus oncogene cellular homolog (c-Myc) and hypoxia-inducible factor-1α (HIF-1α) (58, 59) (Figure 1A). It has been shown that c-Myc is crucial to activate glucose transporters (GLUT) and key enzymes for enhancing glucose influx and glycolysis that accompany early stage of T-cell growth, proliferation, and the transition from a naïve T-cell to a Teff cell (60). Furthermore, c-Myc is responsible for enhanced glutaminolysis by inducing glutamine transporters and glutaminase1 expression to sustain cell growth and proliferation (60, 61).
Hypoxia-inducible factor-1α is another master transcription factor monitoring glycolytic enzymes expression (62, 63). HIF-1α acts also to downregulate mitochondrial oxygen consumption and inhibit TCA cycle. At later times of differentiation, the role of HIF-1α appears more complex to mediate T-cell fate and function (64). HIF-1α is reported to play a more selective role in inflammatory Th17 CD4 T-cell subsets (42) and cytolytic CD8 T (58). In addition, HIF-1α appears to influence the balance of Th17:Treg cells (65). Indeed, it directly promotes glycolysis in differentiating Th17 cells and reciprocally increases Th17 differentiation and decreases Treg differentiation (66). However, in vitro and in vivo studies demonstrate that a lack of HIF-1α strongly impair Th17 cell development and drives Treg cell differentiation and FAO. Treg cells unlike other Teff cells mainly display increased FAO metabolism and enhance AMP-activated protein kinase (AMPK) activation (67). The utilization of lipid oxidation by Treg cells might play a central role in their survival advantage over Teff cells and in the maintenance of a stable pool of pro-tumor (68, 69).
Finally, the mechanisms regulating the transition of T cells from effector to memory states remain to be elucidated. Recent studies demonstrated that mitochondrial FAO in Tm cells require stimulation of tumor necrosis factor receptor-associated factor 6 pathway (70). Further, memory CD8+ T-cell development is also supported by activating the energy sensor AMPK pathway (71, 72). FAO has clinical implications for memory CD8+ T as well as for Treg cells (73). In fact, administration of metformin or the mTOR inhibitor rapamycin, reduce mTOR activity and induce AMPK phophorylation that in turn perform lipid oxidation and enhance the formation of Tm cells after infection and increase Treg responses in asthma model (74, 75).
Fueling T-Cell Proliferation
Increasing data suggest that regulation of metabolic fuels uptake is a critical component of T-cell activation to accomplish their functional requirements. Yet, limiting conditions could suppress the suitable access to nutrients, causing a barrier to T-cell function. To maintain a proper response, T-cell activation requires the upregulation of both glucose and amino acid transporters (1, 76). Several metabolic pathways that are imminent for lymphocyte proliferation are supported by the availability of these fuels (24).
Glucose
Glucose is the most used nutrient predominantly existing in the surrounding environment, and glucose metabolism, in particular, is essential for T cells for normal survival and function. Glucose is a critical substrate for energy production, and its deprivation prevents T-cell function despite the presence of other alternative carbon source (77, 78). When Teff are activated, glucose uptake raises to maintain aerobic glycolysis and subsequently to support growth and proliferation, whereas glucose use via OXPHOS is decreased (79). Further, the expression and trafficking patterns of GLUT are upregulated allowing T cells to enrich their intracellular glucose. The GLUT consists of 14 different members (GLUT1–14) relying on diverse substrate specificities (80). GLUT2 and GLUT3 are expressed in resting human peripheral blood T cells, while GLUT1 is expressed at a low level in naïve T cells, but rapidly induced upon T-cell activation. Consequently, overexpression of GLUT1 after TCR activation leads to increased glucose uptake and enhanced expression and activity of glycolytic enzymes. During glycolysis, glucose is not fully oxidized in the mitochondria but rather broken down into pyruvate that is converted into lactate even though in presence of sufficient oxygen (81). Glucose could be also derived toglucose-6phosphate and further directed into the PPP, providing precursors for the synthesis of nucleotides and aromatic amino acids (77).
It has also been reported that T-cell cytokine production is also relying on glucose. In fact, data showed enhanced T-cell cytokine production such as IL-2 and IFN-γ in transgenic model expressing GLUT1 specifically in T cells (78). In contrast, glucose deprivation has been shown to strongly inhibit cytokine production and to decrease cytolytic activity of CD8+ T cells, marked by reduced granzyme and perforin production. Thus, failure to properly upregulate glucose metabolism during T-cell activation can lead to impaired proliferation. As a consequence, T cells can enter to anergy if they survive this metabolic stress, or they die by apoptosis. Collectively, glucose is fundamental to support proliferation and effector functions that accompany clonal expansion of Teff. Besides, Treg cells do not depend on high rates of glucose as they express low levels of GLUT1 and rely on lipid oxidation for energy (39).
Glutamine
Glutamine is a nonessential amino acid and the most abundant nutrient in the blood. Glutamine constitutes also a critical substrate for T cells activation and growth process. Following T-cell activation through efficient TCR signaling, the uptake and biosynthesis of amino acids or amino acid transporter expression are dramatically increased (82, 83).
Glutamine catabolism is dramatically induced in active T cells providing intermediate molecules necessary for different pathways of biosynthesis and substrates for mitochondria (84, 85). During glutaminolysis, glutamine carbon backbone can be converted to α-ketoglutarate to maintain homeostasis of the TCA, or to lactate that generates NAD and NADPH (86). During T-cell activation glutamine can be used, providing pyruvates to overcome intense aerobic glycolysis levels (87). Further, activated T cells selectively increase glutamine uptake. This increase has been suggested to be concomitant with induced expression of glutamine transporters, recognized as members of the sodium-dependent neutral amino acid transporter (SNAT) family. In fact, the previous study demonstrates rapidly enhanced mRNA expression of SNAT1 and SNAT2 isoforms after in vivo stimulation of T cells (82). However, lack of glutamine can result in profound inhibition of cell growth, proliferation, and cytokine production (88). Since T-cell activation is strongly impacted by glutamine, thus different aspects of glutamine metabolism could serve as novel targets for immune modulation.
Tryptophan and Arginine
In addition to glutamine, other limiting amino acids such as tryptophan and arginine have been suggested to be crucial for T-cell activation and function. This concept has gained interest especially in cancer context, where tumor-induced extracellular depletion of these amino acids alters T-cell activity and causes their anergy. Tryptophan is an essential amino acid required for the production of several important molecules and its catabolism through the kynurenine pathway generate metabolites such as kynurenine, kynurenic acid, 3-hydroxy-kynurenine, and 3-hydroxy-anthranilic acid (89). Numerous studies showed that tryptophan plays a key role in T-cell survival and activation whereas its metabolites eliminate T-cell function and are able to induce T-cell apoptosis (90). Teff are affected by the decrease in tryptophan concentrations and high rates of toxic tryptophan-metabolites induced by mature antigen-presenting cells expressing enzymes that catabolize tryptophan (91). Tryptophan degradation is one of a resistance mechanisms adopted by tumors to avoid immune suppression (92, 93). Three enzymes were identified to control tryptophan degradation through the kynurenine pathway: tryptophan-2,3-dioxygenase, indoleamine 2,3-dioxygenase 1, and indoleamine 2,3-dioxygenase. Hence, T-cell cycle progression is prevented and Teff cells shift to anergy and apoptosis. In hostile tumor microenvironment context, such inhibition is resulting in suppression of antitumor immune responses (94).
In addition to tryptophan, arginine has gained much attention as an important amino acid in T-cell function. Arginine is a versatile amino acid engaged in protein synthesis and in generating many metabolites precursors including, polyamines, and nitric oxide involved in immunometabolism (95). Indeed, deficiency in extracellular arginine or in enzymes responsible of de novo synthesizing arginine [argininosuccinate 1 (ASS1)], has been found to critical during activation (96). Low levels of arginine impair T-cell proliferation, aerobic glycolysis and reduce cytokine production and expression of activation markers such as CD25 and CD28 (97, 98). Further, deletion of ASS1 blunt in vitro Th1 and Th17 cell polarization, even in the presence of extracellular arginine (99). Interestingly, recent study showed that increased arginine levels display improved survival capacity of T memory cells and antitumor activity (95). Taken together and according to the beneficial effect of arginine and tryptophan on T-cell metabolic adaptation and antitumor activity, both amino acids would be exploited as an attractive target for therapeutic intervention in antitumor response (96).
Warburg Effect or How Cancer Cell Rewire Metabolic Program
It is well established that cancer cells must reprogram cellular pathways to enable their growth and proliferation. Tumor cells reprogram their metabolic pathways and rely upon increased glucose uptake and high rate lactate production, principally through aerobic glycolysis (100), regardless of the level of oxygen (101). Metabolic switch of cancer cell supports biosynthesis of essential macromolecules (nucleic acids, lipids, and amino acids), through interconnected pathways. This metabolic program was recognized since 1920s by Otto Warburg as the “Warburg effect” (102, 103), a strategic metabolic adaptation enhancing rapid tumor growth, proliferation, and to dampen antitumor immunity, thus representing one additional hallmark of cancers. Since 1923, Otto Warburg has reported that cancer cells acquire irreversible switch of their energy-producing machinery from mitochondrial OXPHOS respiration, to aerobic glycolysis (104). Glycolysis is a predominant energy source for cancer cells, occurring either under aerobic or hypoxic conditions to produce large amounts of lactate, and much less efficient than OXPHOS for producing ATP (105, 106) (Figure 1B). This reprogramming of cancer cell metabolism has been acknowledged recently as a hallmark of cancer with many faces (107, 108). By analogy to immune cells, similar metabolic features with T cells during activation are observed. But, despite an apparent similarity, there is deep down a wide difference between glycolysis in activated T cells and cancer cells. Such metabolic transitions in T cells are part of a physiological adaptation process. However, intrinsic genetic mutations and external responses to the tumor microenvironment monitor the metabolic phenotype of tumor cells (109, 110). Cellular dysregulation of oncogenic signaling pathways are the result of the loss of tumor suppressors (such as p53) or the activation of oncoproteins (such as PI3K) (111). As a consequence, cancer cells thereby gain selective growth and survival (112).
Cancer cells use the Warburg effect as strategic metabolic adaptation to satisfy their urgent requirements for growth and proliferation under tumor microenvironmental limitations for oxygen and nutrients (113, 114). Under hypoxic conditions, cancer cells accelerate metabolism that lead to increased NADPH rate to cope with higher ROS levels (115, 116). Thus, the Warburg effect also supports tightly controlled redox balance for cancer cells, considered as important survival mechanism (117).
Glucose is considered as prominent player in the alterations of metabolism and energetic of cancer cells (118). Increased glucose uptake lead to upregulated glycolysis and thus more pyruvate is produced even in normoxia conditions. Under limited oxygen availability (hypoxia), more pyruvate avoids TCA cycle and generates excess of lactate secreted thereby in the tumor microenvironment (118, 119). In addition to its central role as a carbohydrate nutrient for ATP synthesis, new evidence revealed that high glucose uptake is also important for biomass synthesis needed for rapidly proliferating cancer cells. Upregulation of glycolysis increased several metabolic intermediates that may be shunted to interconnected pathways, as PPP (120, 121). The resulting glycolytic intermediates such fructose-6-phosphate, glyceraldehyde-3-phosphate, and 3-phosphoglycerate are critical for de novo synthesis of ribonucleotides, amino acids, and phospholipids, respectively (122).
Glutamine is the most abundant free amino acid and essential source of carbohydrate for proliferating cells. Cancer cells display increased glutamine demand and consumption. Interestingly, the glutamine dependence extends beyond protein synthesis to other important requirements (123). Rapidly proliferating, cancer cells use glutamine to fuel biosynthesis of nucleotides, to replenish TCA cycle intermediates through a process called anaplerosis, or to be taken from the mitochondria and then modified into lactate (glutaminolysis) (124, 125). Glutamine metabolism occurs in cancer cells, in general, with concomitant production of NADPH that not only maintains cellular redox but also reduces agent in varied biosynthetic pathways–underlying de novo fatty acid synthesis (126).
The molecular drivers that lead to the shift of cancer cell from oxidative to glycolytic metabolism are distinct and tend to happen simultaneously. Cancer metabolism adaptation to the anabolic program has been suggested to be under direct management by various transcription factors, such as Myc and hypoxia-inducible factor 1 (HIF-1) (127, 128).
Myc is a transcription factor upregulated in tumors and considered as master regulator of normoxic cancer cell reprogramming (129). Indeed, Myc contributes to cancer cells switch to aerobic metabolism by facilitating cellular glucose uptake and activating the expression of numerous genes essential for glycolysis. Furthermore, Myc plays important role to promote macromolecules synthesis and mitochondrial biogenesis, critical for fast developing cancer cells (130, 131).
Upon rapid proliferation, hypoxia becomes a key mediator of the Warburg effect and a common feature of human tumors. Extensive studies have provided evidence that cancer cells utilize hypoxia as physiological adaptation pathway that promotes metabolic changes in fast growing tumors (132). Indeed, under hypoxic tumor microenvironment, the uptake of glucose and the glycolytic flux are increased. This metabolic adaptation is mainly orchestrated through the upregulation of the transcription factor, HIF-1α. HIF-1α is induced by low oxygen conditions and recognized as independent marker of poor prognosis (133, 134). The activated tumor glycolytic flux involving HIF-1α implies upregulation and increased activity of several glycolytic protein including key glycolytic enzymes (HK2, PFK-L, PKM2, and LDH-A) and GLUT (GLUT1 and GLUT3) (135, 136). In contrast to Myc, HIF-1 strongly inhibits mitochondrial respiration and biogenesis (111).
Furthermore, PI3K/Akt/mTOR is one of the most frequently altered signaling pathway known to play an important role in glycolysis, cancer metabolism and cancer cell proliferation (137, 138) (Figure 1B). It is well known that this pathway is activated under the loss of function of the tumor suppressor gene phosphatqase and tensin homolog. The best studied driver of tumor glycolytic program in such pathway. The latter has been reported to induce GLUT expression and to stimulate phosphorylation of key glycolytic enzymes (139). In addition, AKT1 strongly activates mTOR signaling pathway. Hence, mTOR is constitutively activated during tumorigenesis (140) and constitutes a key metabolic issue, coupling cell growth to protein, and lipid biosynthesis (141).
Tumor Microenvironment Abrogates T-Cell Metabolic and Immune Checkpoints
Immuno-metabolism plays a key role of adaptive immunity and is particularly central to effective antitumor T-cell responses. T cells, following the metabolic strategies of growing tumors, have to start their effector programs. However, most of human tumors proliferate in spite of the presence of tumor associated antigen-specific T cells. In fact, tumor microenvironment may impose several limitations to dampen T-cell immunity (142) and deplete crucial nutrient availability and handling, such as glucose or amino acids (143). It can also stimulate conserved negative feedback mechanisms, such as through PD-1 (144). Besides, tumor cells must evade the checkpoint controls under such stressful metabolic conditions.
Tumor microenvironment is a forbidding environment that can pose significant metabolic challenges for infiltrating T cells to impair the effectiveness T-cell response. It is likely that T cells undergo immune suppressive networks that impair their specific functions and thereby enable tumor escape (145, 146). Many different molecular and cellular mechanisms have been proposed to contribute to the failure of T cells in tumor eradication. Recent studies have started to reveal that the feature and function of Teff in tumors are severely influenced by the tumor microenvironment context (147). Indeed, tumor microenvironment components form a very complex immunosuppressive network in cancer (148), lead to metabolic and immune checkpoints abrogation, which limits T-cell activation and induces T-cell dysfunction (149, 150). However, the exact mechanisms remain insufficiently understood.
Evidence is beginning to emerge suggesting that alterations of the T-cell metabolic pathways are critical to impair antitumor immunity, supporting immune escape (151). Cancer cells are recognized to be the most important players in tumor microenvironment mediating immune suppression. In fact, metabolic interplay and nutrient (glucose and glutamine) competition between cancer cells and T cells exist. Such competition is recognized as a key driver of cancer progression (152, 153). Due to high demand for energy and increased glucose addiction and glycolysis rate, fast growing cancer cells consumes most nutrients and specifically increases rate of glucose intake, from the surrounding environment (154). As a consequence, tumor-imposed metabolic restrictions can mediate T-cell hypo-responsiveness during cancer. T cells dramatically reduced glycolysis and become unable to produce cytokines and to develop into tumor-specific Teff cells, leading to a state of anergy (155) (Figure 1B). Thus, Treg cells differentiation is favored to inhibit antitumor immune response, instead of expansion of tumor-specific T cells (156, 157). As a contrast to Teff that suffer from a hostile tumor microenvironment, Treg cells, feel comfortable with a similar environment (158). This is possibly the result of to the flow in growth factors (such as transforming growth factor-β) and chemokines (such as CCL22) promoting Treg differentiation and recruitment (156, 159). One molecular explanation is that alteration of functional fate of T cells due to nutrient limitation could occur through modulation of metabolically sensitive signaling pathways. Under tumoral context, the balance between Teff and Treg may be directly disturbed when AMPK signaling pathway inhibits mTORC (56, 160). Opposing to mTORC, AMPK is activated in conditions where nutrients are limiting and promote oxidative metabolism (161) (Figure 1B). AMPK can be highly phosphorylated and activated in Treg. Consequently, Teff function is impaired while Treg cells are promoted. Furthermore, Treg cells have also been reported to be induced under hypoxic tumor microenvironment, through over activated HIF-1α (12, 162). The presence of Treg in solid tumors essentially correlates with poor prognosis (27).
Immunosuppressive tumor microenvironment is also characterized by elevated rates of ROS (115). Besides cancer cells, tumor-infiltrating leukocytes, including myeloid-derived suppressor cells, tumor-associated macrophages, and Treg, also generate excessive ROS (163). It has been demonstrated that high level of ROS in the tumor microenvironment downregulates T-cell activity and enhanced T-cell apoptosis, inhibiting subsequently antitumor immune response (164). However, although high levels of ROS impair T-cell metabolism and function, ROS at a low or moderate-concentration is indispensable for T-cell activation and effector function (165). Considering the paradoxal effect of ROS on T-cell function a tight balance between production and consumption of ROS should be accomplished to potentiate antitumor activity compromising Teff function.
Under immunosuppressive tumor microenvironment T cells acquire an “exhausted” phenotype highlighted by upregulation of inhibitory receptors. Interestingly, to eradicate effectiveness of antitumor immune response, tumor hostile environment act not only to impair metabolic checkpoints of Teff cells encountering tumor antigens, but also to abrogate immune checkpoints. Indeed, several negative feedback mechanisms are stimulated, such as PD-1 and CTLA4 pathways (166, 167), which can both promote T cells exhaustion (Figure 1B). Hence, further research is needed to identify new target to reverse exhaustion in addition to PD-1 and CTLA4.
Programmed death-1 is the major inhibitory receptor in T cells regulating T-cell exhaustion. Interaction of PD-1 with its ligand programmed death ligand-1 (PD-L1), allows the tumor to evade immune system by inhibiting T-cell function (168, 169). Recently, it has been reported that upon ligation, T cells receiving PD-1 signals can lower the capacity of T cells to express GLUT1, uptake glucose, and become unable to engage in glycolysis, glutaminolysis, or metabolism of branched-chain amino acids (144). Interestingly, PD-1 displayed an increased rate of FAO of endogenous lipids, and lipolysis is indicated by elevation of the lipase ATGL and by release of fatty acids (144). PD-1 signaling is associated with reduced cMyc expression and inhibition of activity of the PI3K/Akt/mTOR pathway, necessary for effector function (50, 170, 171). Besides, PD-L1 directly regulates tumor metabolism. Surface expressed PD-L1 is important for Akt/mTOR signaling to promote mTOR activity and glycolytic metabolism in tumor cells (172, 173).
Nonetheless, CTLA4 signaling also plays a key role in tumor immune escape since it inhibits CD28-mediated costimulation of Teff and favors Treg expansion (174, 175). Subsequently, CTLA4 may broadly impair Teff cell activation against antigenic stimulation in part by reducing the capability of Akt to enhance GLUT1 expression, glucose uptake, and aerobic glycolysis, but without enhanced FAO as for PD-1 pathway (151).
Immune Checkpoints Targeting for Enhancing T-Cell Function: Relationship with Metabolism
Metabolic reprogramming plays a pivotal role for appropriate T-cell activation that supports antitumor immunity. However, T-cell function is compromised by the immunosuppressive tumor microenvironment. Nevertheless, multiple mechanisms that instruct the development of immune suppression may exist to prevent effective antitumor response, but remain largely unclear. Metabolic and functional pathways in T cells may uncover new targets and challenges for cancer therapy (176). Therefore, manipulating metabolism may be a way to beneficially enhance or temper antitumor immunity. Current attractive therapeutic approaches which specially target T-cell metabolism are meant to use immunotherapy directed against several negative immunologic regulators CTLA-4 and PD-1/PD-L1 pathway (177–179).
In recent studies, it has been reported that mice exhibiting or transplanted with tumors were treated with checkpoint blockade therapy, such blockade increased the glucose concentrations in the extracellular tumor milieu and TILs from these mice had increased glucose uptake, glycolytic rates, activated mTORC1 pathway, and IFN-γ production (152). The same effects were reported after PD-L1 blockade or RNA interference directed against PD-L1 in cultured tumor cells (152). More importantly, T cells in allogeneic PD-L1−/− bone marrow transplant recipients had elevated levels of GLUT1 and lactate production, suggesting a normal in vivo role for PD-1 signaling to restrain T-cell glucose metabolism (180).
Although there is a promising efficacy of immunotherapy, the clinical benefit has been restricted by tumor-derived immunosuppression and its related coinhibitory signals. Indeed, to escape antitumor immune response, tumors develop different strategies including secretion of immunosuppressive cytokines and chemokines (TGF-β, IL-10, VEGF, CCL2, and CCL12) (181) or immunosuppressive converting tryptophan and arginine enzymes [indoleamine-2,3-dioxygenase (IDO) and arginase, respectively] (98, 182, 183). In light of this, it would be reasonable to combine immunotherapy with an immunosuppression-blocking protocol. In particular, IDO is an attractive area for exploitation to potentiate immunotherapy, since it is highly expressed in the microenvironments of various tumors (184). Recently, preclinical studies demonstrate the efficiency of two IDO inhibitors to attenuate tumor growth (185, 186). Currently, IDO inhibitors entered clinical trials (187). Interestingly, in vivo study has been conducted on mouse melanoma model where synergistic immunotherapy strategy that locally targets PD-1 and IDO for the treatment of melanoma has been developed. The preliminary results are quite encouraging and showed enhanced Teff cells and antitumor efficacy (188).
Therefore, the use of metabolism-targeting drugs working with checkpoint inhibitors might not only change the activation and differentiation program of tumor-specific T cells but also prohibit the generation of exhausted T cells. Currently, there is a lack of data taking into consideration the metabolic consequences occurring in T cells and/or tumor cells by targeting these immune checkpoint pathways. Nevertheless, combined immunotherapeutic strategies would be exciting and show promise to improve the anticancer efficacy of immunotherapy in the future.
Concluding Remarks
It is widely admitted that tumors are not autonomous masses of cells but function as organs composed of many interdependent cells supporting malignant cell survival, growth and progression. To ensure tumor growth and immune evasion, the tumor stromal components undergo numerous metabolic adaptations, reprogramming the mode of energy generation. T cells play key role in the orchestration of the immune response and T-cell metabolic adaptation acts as crucial checkpoint hijacked by tumors to dampen antitumor immunity as T cells are rendered dysfunctional, unable to carry out their effector functions. Accumulating evidence indicate that the diverse functions of the immune system require several bioenergetic processes and that T-cell metabolic reprograming relies upon the activation of distinct transcriptional and signaling pathways. In the context of tumor microenvironment, tumors impose several limitations to dampen T-cell immunity as T cells, experiencing the metabolic framework of growing tumors, fail to activate distinct pathways to accomplish their functional requirements. Tumor microenvironmental hypoxia is in this regard a relevant example demonstrating how the tumor microenvironment of a tumor can paralyze and neutralize T-cell functions. In fact, O2 is a master regulator of the CD8+ T-cell response and T lymphocytes face pathologically low O2 tensions within the tumor bed at which they will have to function. It has become clear that tumor-imposed metabolic restrictions may result in an impairment of T-cell function and that either some programmed changes or pathologic manifestations can inhibit the required energy essential for their several functions. Accordingly, attempts are made to identify approaches aiming at manipulating the reprogramming of T-cell metabolic pathways for therapeutic purposes, in particular, antitumor immunity.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by a grant “Equipe labellisée par la Ligue Contre le Cancer” (EL2015.LNCC/SaC).
Abbreviations
AKT, serine/threonine-specific protein kinase; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; CTLs, cytolytic T cells; CTLA-4, cytotoxic T-lymphocyte antigen; FAO, fatty acid oxidation; GLUT, glucose transporters; HIF, hypoxia-inducible factor; IFN-γ, interferon-γ; MHC, major-histocompatibility complex; mTOR, mammalian target of rapamycin; c-Myc, avian myelocytomatosis virus oncogene cellular homolog; OXPHOS, oxidative phosphorylation; PD-1, programmed death-1; PD-L1, programmed death ligand-1; PI3K, phosphatidylinositol-3 kinase; PPP, pentose phosphate pathway; PTEN, phosphatqase and tensin homolog; ROS, reactive oxygen species; TAA, tumor associated antigen; TCA, tricarboxylic acid; TCR, T-cell receptor; Teff, effector T cells; Th, helper T cells; TNF-α, tumor necrosis factor; Treg, regulatory T cells.
References
1. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
2. Ramsay G, Cantrell D. Environmental and metabolic sensors that control T cell biology. T Cell Biol (2015) 6:99. doi:10.3389/fimmu.2015.00099
3. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi:10.1016/j.cmet.2014.05.004
4. Cherayil BJ. Iron and immunity: immunological consequences of iron deficiency and overload. Arch Immunol Ther Exp (Warsz) (2010) 58(6):407–15. doi:10.1007/s00005-010-0095-9
5. Kemp JD, Thorson JA, Gomez F, Smith KM, Cowdery JS, Ballas ZK. Inhibition of lymphocyte activation with anti-transferrin receptor Mabs: a comparison of three reagents and further studies of their range of effects and mechanism of action. Cell Immunol (1989) 122(1):218–30. doi:10.1016/0008-8749(89)90162-7
6. Jason J, Archibald LK, Nwanyanwu OC, Bell M, Jensen RJ, Gunter E, et al. The effects of iron deficiency on lymphocyte cytokine production and activation: preservation of hepatic iron but not at all cost. Clin Exp Immunol (2001) 126(3):466–73. doi:10.1046/j.1365-2249.2001.01707.x
7. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
8. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
9. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
10. Dai C, Lin F, Geng R, Ge X, Tang W, Chang J, et al. Implication of combined PD-L1/PD-1 blockade with cytokine-induced killer cells as a synergistic immunotherapy for gastrointestinal cancer. Oncotarget (2016) 7(9):10332–44. doi:10.18632/oncotarget.7243
11. Noman MZ, Messai Y, Carré T, Akalay I, Méron M, Janji B, et al. Microenvironmental hypoxia orchestrating the cell stroma cross talk, tumor progression and antitumor response. Crit Rev Immunol (2011) 31(5):357–77. doi:10.1615/CritRevImmunol.v31.i5.10
12. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L-P, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475(7355):226–30. doi:10.1038/nature10169
13. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell (2007) 129(3):465–72. doi:10.1016/j.cell.2007.04.019
14. Chouaib S, Messai Y, Couve S, Escudier B, Hasmim M, Noman MZ. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front Immunol (2012) 3:21. doi:10.3389/fimmu.2012.00021
15. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev (2012) 249(1):14–26. doi:10.1111/j.1600-065X.2012.01155.x
16. Slack M, Wang T, Wang R. T cell metabolic reprogramming and plasticity. Mol Immunol (2015) 68(2 Pt C):507–12. doi:10.1016/j.molimm.2015.07.036
17. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43. doi:10.1016/j.immuni.2013.04.005
18. Sanchez-Lockhart M, Rojas AV, Fettis MM, Bauserman R, Higa TR, Miao H, et al. T cell receptor signaling can directly enhance the avidity of CD28 ligand binding. PLoS One (2014) 9(2):e89263. doi:10.1371/journal.pone.0089263
19. Beyersdorf N, Kerkau T, Hünig T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. Immunotargets Ther (2015) 4:111–22. doi:10.2147/ITT.S61647
20. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi:10.1172/JCI69589
21. Liu H, Yang H, Chen X, Lu Y, Zhang Z, Wang J, et al. Cellular metabolism modulation in T lymphocyte immunity. Immunology (2014). doi:10.1111/imm.12321
22. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science (2011) 334(6060):1278–83. doi:10.1126/science.1211485
23. Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi:10.1084/jem.20151159
24. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13(10):907–15. doi:10.1038/ni.2386
25. Marelli-Berg FM, Fu H, Mauro C. Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology (2012) 136(4):363–9. doi:10.1111/j.1365-2567.2012.03583.x
26. Nguyen HD, Chatterjee S, Haarberg KM, Wu Y, Bastian D, Heinrichs J, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest (2016) 126(4):1337–52. doi:10.1172/JCI82587
27. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33(4):168–73. doi:10.1016/j.it.2012.01.010
28. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi:10.1016/j.jaci.2009.12.980
29. Petrova G, Ferrante A, Gorski J. Cross-reactivity of T cells and its role in the immune system. Crit Rev Immunol (2012) 32(4):349–72. doi:10.1615/CritRevImmunol.v32.i4.50
30. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control Th17 responses in a Stat3-dependent manner. Science (2009) 326(5955):986–91. doi:10.1126/science.1172702
31. Obar JJ, Lefrançois L. Memory CD8+ T cell differentiation. Ann N Y Acad Sci (2010) 1183:251–66. doi:10.1111/j.1749-6632.2009.05126.x
32. Arens R, Schoenberger SP. Plasticity in programming of effector and memory CD8 T-cell formation. Immunol Rev (2010) 235(1):190–205. doi:10.1111/j.0105-2896.2010.00899.x
33. Yang K, Chi H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin Immunol (2012) 24(6):421–8. doi:10.1016/j.smim.2012.12.004
34. Chisolm DA, Weinmann AS. TCR-signaling events in cellular metabolism and specialization. Front Immunol (2015) 6:292. doi:10.3389/fimmu.2015.00292
35. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. mTOR differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi:10.1016/j.immuni.2009.04.014
36. Araujo L, Khim P, Mkhikian H, Mortales CL, Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife (2017) 6:e21330. doi:10.7554/eLife.21330
37. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi:10.1126/science.1241165
38. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
39. Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev (2013) 252(1):52–77. doi:10.1111/imr.12029
40. Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res (2015) 7:gkv047. doi:10.1093/nar/gkv047
41. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab (2016) 24(6):807–19. doi:10.1016/j.cmet.2016.10.008
42. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi:10.1084/jem.20110278
43. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi:10.1016/j.immuni.2012.10.020
44. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi:10.1038/nm.3704
45. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A (2013) 110(35):14336–41. doi:10.1073/pnas.1221740110
46. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi:10.1038/nature08097
47. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. doi:10.1016/j.immuni.2011.12.007
48. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi:10.1038/nri3405
49. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi:10.1016/j.immuni.2009.04.014
50. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi:10.1111/j.1600-065X.2012.01152.x
51. Pham K, Shimoni R, Charnley M, Ludford-Menting MJ, Hawkins ED, Ramsbottom K, et al. Asymmetric cell division during T cell development controls downstream fate. J Cell Biol (2015) 210(6):933–50. doi:10.1083/jcb.201502053
52. Verbist KC, Guy CS, Milasta S, Liedmann S, Kamiński MM, Wang R, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature (2016) 532(7599):389–93. doi:10.1038/nature17442
53. Pollizzi KN, Sun I-H, Patel CH, Lo Y-C, Oh M-H, Waickman AT, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8+ T cell differentiation. Nat Immunol (2016) 17(6):704–11. doi:10.1038/ni.3438
54. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A (2008) 105(22):7797–802. doi:10.1073/pnas.0800928105
55. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by carma1 and MALT1, but not Bcl10. Sci Signal (2014) 7(329):ra55–55. doi:10.1126/scisignal.2005169
56. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol (2012) 12(5):325–38. doi:10.1038/nri3198
57. Yang K, Shrestha S, Zeng H, Karmaus PWF, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic programming. Immunity (2013) 39(6):1043–56. doi:10.1016/j.immuni.2013.09.015
58. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med (2012) 209(13):2441–53. doi:10.1084/jem.20112607
59. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer (2008) 8(11):851–64. doi:10.1038/nrc2501
60. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi:10.1016/j.immuni.2011.09.021
61. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458(7239):762–5. doi:10.1038/nature07823
62. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem (1994) 269(38):23757–63.
63. Discher DJ, Bishopric NH, Wu X, Peterson CA, Webster KA. Hypoxia regulates beta-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. J Biol Chem (1998) 273(40):26087–93. doi:10.1074/jbc.273.40.26087
64. Roman J, Rangasamy T, Guo J, Sugunan S, Meednu N, Packirisamy G, et al. T-cell activation under hypoxic conditions enhances IFN-gamma secretion. Am J Respir Cell Mol Biol (2010) 42(1):123–8. doi:10.1165/rcmb.2008-0139OC
65. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
66. Shi LZ, Wang R, Green D, Chi H. Metabolic control of T cell fate decision: the HIF1α-glycolysis axis in the differentiation of TH17 and iTreg cells. J Immunol (2012) 188(1 Suppl):17–163.
67. Gualdoni GA, Mayer KA, Göschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J (2016) 30(11):3800–9. doi:10.1096/fj.201600522R
68. Mockler MB, Conroy MJ, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front Oncol (2014) 4:107. doi:10.3389/fonc.2014.00107
69. Loftus RM, Finlay DK. Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem (2016) 291(1):1–10. doi:10.1074/jbc.R115.693903
70. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol (2015) 36(2):81–91. doi:10.1016/j.it.2014.12.005
71. Araki K, Ahmed R. AMPK: a metabolic switch for CD8+ T-cell memory. Eur J Immunol (2013) 43(4):878–81. doi:10.1002/eji.201343483
72. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKα1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol (2013) 43(4):889–96. doi:10.1002/eji.201243008
73. Sun Y, Tian T, Gao J, Liu X, Hou H, Cao R, et al. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J Neuroimmunol (2016) 292:58–67. doi:10.1016/j.jneuroim.2016.01.014
74. Araki K, Youngblood B, Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev (2010) 235(1):234–43. doi:10.1111/j.0105-2896.2010.00898.x
75. Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. Adenosine-mono-phosphate-activated protein kinase-independent effects of metformin in T cells. PLoS One (2014) 9(9):e106710. doi:10.1371/journal.pone.0106710
76. Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev (2010) 236:190–202. doi:10.1111/j.1600-065X.2010.00911.x
77. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol (2008) 84(4):949–57. doi:10.1189/jlb.0108024
78. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi:10.4049/jimmunol.180.7.4476
79. Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem (1994) 269(50):31484–90.
80. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med (2013) 34(0):121–38. doi:10.1016/j.mam.2012.07.001
81. Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol (2009) 92(3):329–33. doi:10.1016/j.radonc.2009.06.025
82. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi:10.4049/jimmunol.0903586
83. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249(1):27–42. doi:10.1111/j.1600-065X.2012.01150.x
84. Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol (1985) 70(4):473–89. doi:10.1113/expphysiol.1985.sp002935
85. Newsholme P, Curi R, Curi TCP, Murphy CJ, Garcia C, de Melo MP. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: its importance in health and disease 1. J Nutr Biochem (1999) 10(6):316–24. doi:10.1016/S0955-2863(99)00022-4
86. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104(49):19345–50. doi:10.1073/pnas.0709747104
87. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi:10.1016/j.immuni.2014.12.030
88. Hammami I, Chen J, Bronte V, DeCrescenzo G, Jolicoeur M. l-glutamine is a key parameter in the immunosuppression phenomenon. Biochem Biophys Res Commun (2012) 425(4):724–9. doi:10.1016/j.bbrc.2012.07.139
89. Chen Y, Guillemin GJ. Kynurenine pathway metabolites in humans: disease and healthy states. Int J Tryptophan Res (2009) 2:1–19.
90. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189(9):1363–72. doi:10.1084/jem.189.9.1363
91. Mellor AL, Munn DH. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi:10.1038/nri1457
92. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol (2015) 6:34. doi:10.3389/fimmu.2015.00034
93. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi:10.1038/nm934
94. Muller AJ, Sharma MD, Chandler PR, DuHadaway JB, Everhart ME, Johnson BA, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A (2008) 105(44):17073–8. doi:10.1073/pnas.0806173105
95. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. l-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829.e–42.e. doi:10.1016/j.cell.2016.09.031
96. Qiu F, Huang J, Sui M. Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett (2015) 364(1):1–7. doi:10.1016/j.canlet.2015.04.020
97. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. l-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol (2003) 171(3):1232–9. doi:10.4049/jimmunol.171.3.1232
98. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
99. Tarasenko TN, Gomez-Rodriguez J, McGuire PJ. Impaired T cell function in argininosuccinate synthetase deficiency. J Leukoc Biol (2015) 97(2):273–8. doi:10.1189/jlb.1AB0714-365R
100. Li C, Zhang G, Zhao L, Ma Z, Chen H. Metabolic reprogramming in cancer cells: glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J Surg Oncol (2016) 14(1):15. doi:10.1186/s12957-016-0769-9
101. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (review). Oncol Lett (2012) 4(6):1151–7. doi:10.3892/ol.2012.928
102. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi:10.1038/nrc3038
103. Yi M, Xiang B, Li X, Li G. [Metabolic reprogramming in cancer: the art of balance]. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2013) 38(11):1177–87. doi:10.3969/j.issn.1672-7347.2013.11.016
104. Amoêdo ND, Valencia JP, Rodrigues MF, Galina A, Rumjanek FD. How does the metabolism of tumour cells differ from that of normal cells. Biosci Rep (2013) 33(6):e00080. doi:10.1042/BSR20130066
105. Sheng H, Tang W. Glycolysis inhibitors for anticancer therapy: a review of recent patents. Recent Pat Anticancer Drug Discov (2016) 11(3):297–308. doi:10.2174/1574892811666160415160104
106. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget (2016) 7(25):38908–26. doi:10.18632/oncotarget.7676
107. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov (2012) 2(10):881–98. doi:10.1158/2159-8290.CD-12-0345
108. Nakajima EC, Van Houten B. Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol Carcinog (2013) 52(5):329–37. doi:10.1002/mc.21863
109. Brock A, Krause S, Ingber DE. Control of cancer formation by intrinsic genetic noise and microenvironmental cues. Nat Rev Cancer (2015) 15(8):499–509. doi:10.1038/nrc3959
110. Scott JG, Hjelmeland AB, Chinnaiyan P, Anderson AR, Basanta D. Microenvironmental variables must influence intrinsic phenotypic parameters of cancer stem cells to affect tumourigenicity. PLoS Comput Biol (2014) 10(1):e1003433. doi:10.1371/journal.pcbi.1003433
111. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep (2015) 42(4):841–51. doi:10.1007/s11033-015-3858-x
112. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11(2):85–95. doi:10.1038/nrc2981
113. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
114. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211–8. doi:10.1016/j.tibs.2015.12.001
115. Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1α differentially in cancer and ischemia. Mol Cell Biol (2008) 28(16):5106–19. doi:10.1128/MCB.00060-08
116. Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol (2015) 89(8):1209–26. doi:10.1007/s00204-015-1520-y
117. Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P. The Warburg effect: insights from the past decade. Pharmacol Ther (2013) 137(3):318–30. doi:10.1016/j.pharmthera.2012.11.003
118. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer (2016) 16(10):635–49. doi:10.1038/nrc.2016.77
119. Granja S, Pinheiro C, Reis RM, Martinho O, Baltazar F. Glucose addiction in cancer therapy: advances and drawbacks. Curr Drug Metab (2015) 16(3):221–42. doi:10.2174/1389200216666150602145145
120. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5(8):592–602. doi:10.1007/s13238-014-0082-8
121. Lucarelli G, Galleggiante V, Rutigliano M, Sanguedolce F, Cagiano S, Bufo P, et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget (2015) 6(15):13371–86. doi:10.18632/oncotarget.3823
122. Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med (2015) 79:253–63. doi:10.1016/j.freeradbiomed.2014.08.027
123. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35(8):427–33. doi:10.1016/j.tibs.2010.05.003
124. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol (2015) 11(1):9–15. doi:10.1038/nchembio.1712
125. Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, et al. Mitochondrial localization and structure-based phosphate activation mechanism of glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A (2012) 109(4):1092–7. doi:10.1073/pnas.1112495109
126. Li Y, Erickson JW, Stalnecker CA, Katt WP, Huang Q, Cerione RA, et al. Mechanistic basis of glutaminase activation: a key enzyme that promotes glutamine metabolism in cancer cells. J Biol Chem (2016) 291(40):20900–10. doi:10.1074/jbc.M116.720268
127. Matijevic Glavan T, Cipak Gasparovic A, Vérillaud B, Busson P, Pavelic J. Toll-like receptor 3 stimulation triggers metabolic reprogramming in pharyngeal cancer cell line through Myc, MAPK, and HIF. Mol Carcinog (2016). doi:10.1002/mc.22584
128. Dang CV, Kim J, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer (2008) 8(1):51–6. doi:10.1038/nrc2274
129. Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res (2009) 15(21):6479–83. doi:10.1158/1078-0432.CCR-09-0889
130. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med (2013) 3(8):a014217. doi:10.1101/cshperspect.a014217
131. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med (2014) 4(6):a014241. doi:10.1101/cshperspect.a014241
132. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem (2002) 277(26):23111–5. doi:10.1074/jbc.M202487200
133. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol (2016) 18(4):356–65. doi:10.1038/ncb3330
134. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab (2014) 2:3. doi:10.1186/2049-3002-2-3
135. Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab (2013) 1:8. doi:10.1186/2049-3002-1-8
136. Lu H, Li X, Luo Z, Liu J, Fan Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol Cancer Ther (2013) 12(10):2187–99. doi:10.1158/1535-7163.MCT-12-1245
137. Zhao X, Jiang P, Deng X, Li Z, Tian F, Guo F, et al. Inhibition of mTORC1 signaling sensitizes hepatocellular carcinoma cells to glycolytic stress. Am J Cancer Res (2016) 6(10):2289–98.
138. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. Recent Results Cancer Res (2016) 207:39–72. doi:10.1007/978-3-319-42118-6_3
139. Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G, et al. Signaling through the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma. J Biol Chem (2015) 290(28):17495–504. doi:10.1074/jbc.M115.660498
140. Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature (2016) 534(7606):272–6. doi:10.1038/nature17963
141. Agani F, Jiang BH. Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets (2013) 13(3):245–51. doi:10.2174/1568009611313030003
143. Frey AB. Suppression of T cell responses in the tumor microenvironment. Vaccine (2015) 33(51):7393–400. doi:10.1016/j.vaccine.2015.08.096
144. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
145. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol (2015) 35(Suppl):S185–98. doi:10.1016/j.semcancer.2015.03.004
146. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi:10.1111/j.1365-2567.2007.02587.x
147. Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol (2011) 23(2):286–92. doi:10.1016/j.coi.2010.11.013
148. Bianchi G, Borgonovo G, Pistoia V, Raffaghello L. Immunosuppressive cells and tumour microenvironment: focus on mesenchymal stem cells and myeloid derived suppressor cells. Histol Histopathol (2011) 26(7):941–51. doi:10.14670/HH-26.941
149. Taylor ES, McCall JL, Girardin A, Munro FM, Black MA, Kemp RA. Functional impairment of infiltrating T cells in human colorectal cancer. Oncoimmunology (2016) 5(11):e1234573. doi:10.1080/2162402X.2016.1234573
150. Quail D, Joyce J. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi:10.1038/nm.3394
151. Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med (2016) 5:29. doi:10.1186/s40169-016-0110-9
152. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi:10.1016/j.cell.2015.08.016
153. Zhang Y, Ertl HCJ. Starved and asphyxiated: how can CD8(+) T cells within a tumor microenvironment prevent tumor progression. Front Immunol (2016) 7:32. doi:10.3389/fimmu.2016.00032
154. Nakaigawa N, Kondo K, Ueno D, Namura K, Makiyama K, Kobayashi K, et al. The acceleration of glucose accumulation in renal cell carcinoma assessed by FDG PET/CT demonstrated acquisition of resistance to tyrosine kinase inhibitor therapy. BMC Cancer (2017) 17(1):39. doi:10.1186/s12885-016-3044-0
155. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25(2):214–21. doi:10.1016/j.coi.2012.12.003
156. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel) (2016) 4(3):E28. doi:10.3390/vaccines4030028
157. Yaqub S, Henjum K, Mahic M, Jahnsen FL, Aandahl EM, Bjørnbeth BA, et al. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother (2008) 57(6):813–21. doi:10.1007/s00262-007-0417-x
158. Chaudhary B, Abd Al Samid M, al-Ramadi BK, Elkord E. Phenotypic alterations, clinical impact and therapeutic potential of regulatory T cells in cancer. Expert Opin Biol Ther (2014) 14(7):931–45. doi:10.1517/14712598.2014.900539
159. Ruter J, Barnett BG, Kryczek I, Brumlik MJ, Daniel BJ, Coukos G, et al. Altering regulatory T cell function in cancer immunotherapy: a novel means to boost the efficacy of cancer vaccines. Front Biosci (Landmark Ed) (2009) 14:1761–70. doi:10.2741/3338
160. Shaw RJ. LKB1 and AMPK control of mTOR signalling and growth. Acta Physiol (Oxf) (2009) 196(1):65–80. doi:10.1111/j.1748-1716.2009.01972.x
161. Chaube B, Bhat MK. AMPK, a key regulator of metabolic/energy homeostasis and mitochondrial biogenesis in cancer cells. Cell Death Dis (2016) 7(1):e2044. doi:10.1038/cddis.2015.404
162. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res (2001) 61(12):4766–72.
163. Wei J, Zhang M, Zhou J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res (2015) 228(3):695–701. doi:10.1016/j.psychres.2015.06.002
164. Chen X, Song M, Zhang B, Zhang Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev (2016) 2016:1580967. doi:10.1155/2016/1580967
165. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195(1):59–70. doi:10.1084/jem.20010659
166. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity (2016) 45(2):358–73. doi:10.1016/j.immuni.2016.07.008
167. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol (2015) 33(17):1974–82. doi:10.1200/JCO.2014.59.4358
168. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26(1):677–704. doi:10.1146/annurev.immunol.26.021607.090331
169. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med (2013) 5(200):200ra116. doi:10.1126/scitranslmed.3006504
170. Celada LJ, Rotsinger JE, Young A, Shaginurova G, Shelton D, Hawkins C, et al. Programmed death-1 inhibition of PI3K/AKT/mTOR signaling impairs sarcoidosis CD4+ T cell proliferation. Am J Respir Cell Mol Biol (2016) 56(1):74–82. doi:10.1165/rcmb.2016-0037OC
171. Waickman AT, Powell JD. Mammalian target of rapamycin integrates diverse inputs to guide the outcome of antigen recognition in T cells. J Immunol (2012) 188(10):4721–9. doi:10.4049/jimmunol.1103143
172. Lastwika KJ, Wilson W, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT–mTOR pathway in non-small cell lung cancer. Cancer Res (2016) 76(2):227–38. doi:10.1158/0008-5472.CAN-14-3362
173. Dong L, Lv H, Li W, Song Z, Li L, Zhou S, et al. Co-expression of PD-L1 and p-AKT is associated with poor prognosis in diffuse large B-cell lymphoma via PD-1/PD-L1 axis activating intracellular AKT/mTOR pathway in tumor cells. Oncotarget (2016) 7(22):33350–62. doi:10.18632/oncotarget.9061
174. McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol (1999) 77(1):1–10. doi:10.1046/j.1440-1711.1999.00795.x
175. Wells AD, Walsh MC, Bluestone JA, Turka LA. Signaling through CD28 and CTLA-4 controls two distinct forms of T cell anergy. J Clin Invest (2001) 108(6):895–903. doi:10.1172/JCI13220
176. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest (2015) 125(9):3384–91. doi:10.1172/JCI80011
177. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi:10.1038/nature13988
178. Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med (2012) 209(2):201–9. doi:10.1084/jem.20112275
179. Hamanishi J, Mandai M, Matsumura N, Abiko K, Baba T, Konishi I. PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int J Clin Oncol (2016) 21(3):462–73. doi:10.1007/s10147-016-0959-z
180. Saha A, Aoyama K, Taylor PA, Koehn BH, Veenstra RG, Panoskaltsis-Mortari A, et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood (2013) 122(17):3062–73. doi:10.1182/blood-2013-05-500801
181. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi:10.1146/annurev.immunol.25.022106.141609
182. Eleftheriadis T, Pissas G, Antoniadi G, Spanoulis A, Liakopoulos V, Stefanidis I. Indoleamine 2,3-dioxygenase increases p53 levels in alloreactive human T cells, and both indoleamine 2,3-dioxygenase and p53 suppress glucose uptake, glycolysis and proliferation. Int Immunol (2014) 26(12):673–84. doi:10.1093/intimm/dxu077
183. Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res (2001) 61(3):1100–6.
184. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72(21):5435–40. doi:10.1158/0008-5472.CAN-12-0569
185. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med (2005) 11(3):312–9. doi:10.1038/nm1196
186. Hou D-Y, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res (2007) 67(2):792–801. doi:10.1158/0008-5472.CAN-06-2925
187. Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology (2014) 3(10):e957994. doi:10.4161/21624011.2014.957994
Keywords: immune system, T-lymphocytes, tumor cell metabolism, cancer, hypoxia, tumor microenvironment
Citation: Kouidhi S, Elgaaied AB and Chouaib S (2017) Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk with Tumor Microenvironment. Front. Immunol. 8:270. doi: 10.3389/fimmu.2017.00270
Received: 15 November 2016; Accepted: 24 February 2017;
Published: 13 March 2017
Edited by:
Yongsheng Li, Third Military Medical University, ChinaReviewed by:
Bruno Silva-Santos, Universidade de Lisboa, PortugalKankana Bardhan, Massachusetts General Hospital, USA
Luca Gattinoni, National Cancer Institute, USA
Zong Sheng Guo, Harvard University, USA
Copyright: © 2017 Kouidhi, Elgaaied and Chouaib. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salem Chouaib, c2FsZW0uY2hvdWFpYkBndXN0YXZlcm91c3N5LmZy