 Priya D. A. Issuree
Priya D. A. Issuree Charles P. Ng1†
Charles P. Ng1† Dan R. Littman
Dan R. Littman- 1The Kimmel Center for Biology and Medicine of the Skirball Institute, New York University School of Medicine, New York, NY, USA
- 2Howard Hughes Medical Institute, New York University School of Medicine, New York, NY, USA
The adaptive immune system is dependent on functionally distinct lineages of T cell antigen receptor αβ-expressing T cells that differentiate from a common progenitor in the thymus. CD4+CD8+ progenitor thymocytes undergo selection following interaction with MHC class I and class II molecules bearing peptide self-antigens, giving rise to CD8+ cytotoxic and CD4+ helper or regulatory T cell lineages, respectively. The strict correspondence of CD4 and CD8 expression with distinct cellular phenotypes has made their genes useful surrogates for investigating molecular mechanisms of lineage commitment. Studies of Cd4 and Cd8 transcriptional regulation have uncovered cis-regulatory elements that are critical for mediating epigenetic modifications at distinct stages of development to establish heritable transcriptional programs. In this review, we examine the epigenetic mechanisms involved in Cd4 and Cd8 gene regulation during T cell lineage specification and highlight the features that make this an attractive system for uncovering molecular mechanisms of heritability.
Introduction
Conrad Waddington first coined the term “epigenetics” to refer to the study of the causal mechanisms connecting genotype with phenotype (1). It is well established that distinct cellular phenotypes in a multicellular organism arise from differences in gene regulation and not heterogeneity in DNA sequence. Gene expression patterns are preserved through cell division by heritable modifications of DNA and chromatin that expand the Watson–Crick base pairing information content of the genome. These adaptations comprise the “epigenetic landscape” crucial to cell lineage specification, depicted by Waddington as a marble rolling downhill into one of several furrows representing differentiated cell types (2). Early cytological studies distinguished heterochromatin that remained condensed throughout the cell cycle from euchromatin that had a diffuse appearance during interphase (3). Heterochromatin is generally tightly packed and transcriptionally quiescent. It can be further classified as constitutive heterochromatin composed of repetitive sequence elements such as telomeres and centromeres and facultative heterochromatin composed of genes that become silenced through developmental cues (4). In contrast, actively transcribed genes are typically located in accessible euchromatin. We are now beginning to understand the epigenetic processes that underlie heritable gene expression programs and characterize the physical properties of heterochromatin and euchromatin. Epigenetic mechanisms act on DNA and histones, both of which can be modified to regulate gene expression, such as by covalent histone linkages or methylation of cytosines in DNA [5-methyl cytosine (5mC)]. The combinatorial output of these marks provides a rich and diverse template for the development of distinct tissues and cell lineages, and in the era of genomics and computational biology, we are beginning to characterize the “epigenetic landscape” driving gene expression. In contrast to epigenetic changes that can occur during plastic stages of differentiation, the epigenetic programs controlling irreversible cell fate choices are key to understand the mechanisms of heritability. As we discuss below, in vertebrates, development of T cells expressing αβ T cell antigen receptors (TCRs) is a tractable system to study the epigenetic mechanisms of bi-potential cell fate decisions and the maintenance of gene expression states in differentiated cells. With ordered stages of maturation and defined molecular checkpoints at each stage, αβ T cell development provides an opportunity to study how chromatin changes drive gene expression during differentiation of a somatic tissue in adult animals. Furthermore, the study of CD4 and CD8, surface receptors whose expression corresponds to distinct T cell fates, has led to new insights into epigenetic inheritance.
T Cell Development as a Model for Epigenetic Gene Regulation
T cell development begins when common lymphoid precursors from the bone marrow or fetal liver migrate through the blood to seed the thymus. Expression of the surface glycoproteins CD4 and CD8 distinguishes developmental stages of αβ T cells, with the most immature thymocytes being double negative (DN) for CD4 and CD8. Productive (in frame) VDJ recombination at the locus encoding the TCRβ chain is followed by assembly and signaling of the pre-TCR, which is composed of the beta chain paired with a germline-encoded pre-TCRα polypeptide. The signal induces robust proliferation and marks the passage of cells with productive rearrangements of their TCRβ chain genes, a process known as β-selection, to the next stage of development. This stage is characterized by upregulation of both CD4 and CD8, yielding double-positive (DP) thymocytes, and by rearrangement of the locus encoding the TCRα chain through VJ recombination. DP thymocytes that signal through sufficiently strong avidity interactions of their rearranged TCR with self-peptide/MHC molecules are positively selected, with CD4 and CD8 facilitating TCR signaling through their roles as co-receptors for MHC class II and MHC class I, respectively. Positive selection of cells with TCRs specific for MHC class II leads to the development of CD4+ single-positive (SP) thymocytes, composed of not only largely T helper cells but also some regulatory T cells with relatively high affinity TCRs. Conversely, thymocytes with TCRs that interact with MHC class I differentiate into CD8+ cytotoxic T cells. Meanwhile, cells with excessive TCR-MHC affinity are eliminated by negative selection to limit the release of autoreactive lymphocytes into the periphery.
There is a striking correspondence of co-receptor expression and commitment to functionally distinct lineages, indicating that regulation of CD4 and CD8 expression is linked to the functional programs of the developing T cells. This observation forms the basis of most models for lineage commitment, including the current kinetic signaling model (5, 6). The kinetic signaling model posits that as CD8 is downregulated following positive selection, leading to a CD4+CD8lo phenotype, continued CD4 co-receptor expression allows for prolonged or stronger MHC class II-TCR signaling, inducing the helper T cell fate (7). Meanwhile, common gamma chain cytokines such as IL-7 rescue cells that have received an interrupted MHC class I signal and induce CD8 co-receptor reversal and the cytotoxic phenotype. “Top-down” studies of proximal TCR signaling have yielded little insight into how recognition of different types of MHC molecules results in distinct transcriptional programs. For this reason, and because of the intimate link between co-receptor expression and lineage commitment, “bottom-up” studies of Cd4 and Cd8 locus regulation have been undertaken as a way toward identifying signaling differences between the lineages. These studies have characterized an extensive transcriptional network that includes T helper inducing POZ/Krueppel-like factor (Thpok), Runx3, Mazr, Tcf1, and lymphoid enhancer factor 1 (Lef1) (8–10). Thpok and Runx3 are required for thymocytes to commit to the CD4 and CD8 lineages, respectively. Antagonistic cross-regulation between Thpok and Runx3 is essential to drive helper versus cytotoxic lineage choice, whereby Runx complexes limit the Thpok expression to MHC class II selected cells and Thpok represses Runx3 expression during differentiation toward CD4+ T cells. However, these transcription factors differ in their abilities to “redirect” cells so that they adopt the wrong fate following TCR–MHC interaction (11). Additional transcription factors also have important roles in lineage specification or the activation of lineage-specific genes, even if they do not directly control lineage commitment or repress genes of the wrong lineage (12). For example, GATA3 is required for the specification of thymocytes to the CD4 lineage, controlling expression of Thpok, and also participates with Thpok in “locking in” the lineage-specific program of gene expression.
Despite growing knowledge of the key transcription factors involved in lineage commitment, the mechanisms by which they direct cell fate decisions through epigenetic mechanisms to establish heritable programs of gene expression remain largely unknown. The study of the transcriptional regulation of the Cd4 and Cd8 loci, with their exquisite use of regulatory elements and key transcription factors to dictate temporal aspects of gene transcription, is slowly unraveling the orchestration of key epigenetic processes that subsequently allow for heritable gene expression patterns. As we discuss in this review, stage-specific cis elements at the Cd4 locus have critical roles in establishing the epigenetic marks that allow for heritable transmission of gene states. This allows for a clear dissection of how these marks are deposited via transcription complexes and what epigenetic marks encode heritable information that is transmitted independently of these cis elements and transcription factors thereafter. In addition to being a tractable system whereby developmental stages can be easily followed, the Cd4 and Cd8 system also offers the potential to understand extracellular signaling cues that lead to the choreography of intricate epigenetic processes.
Epigenetic Mechanisms of Heritable Gene Expression
DNA Methylation
One of the best-studied epigenetic mechanisms of heritability is the covalent modification of cytosine to 5mC, a mark deposited by the DNA methyltransferase (DNMT) enzymes. DNA methylation occurs predominantly at cytosine residues that are followed by guanine (CpG) in mammalian genomes, and about 60–80% of CpGs are methylated in somatic tissues (13). The classic model of DNA methylation holds that de novo DNA methylation is deposited in the genome by Dnmt3a and Dnmt3b along with their non-enzymatic co-regulator Dnmt3L (14, 15). Maintenance DNA methylation is carried out by Dnmt1, which associates with the replication fork through PCNA and with hemimethylated CpGs through the E3 ubiquitin ligase Uhrf1 during DNA replication (16–18). However, these distinctions are not absolute as Dnmt1 has been shown to exhibit de novo methyltransferase function, and Dnmt3 can participate in the maintenance of methylation marks (19). Also, as discussed later, the model of DNA methylation was further revised with the discovery of an active enzymatic process of demethylation.
In the 1970s, two laboratories hypothesized that DNA methylation could act as a cellular mechanism of transcriptional memory through cell division due to the symmetrical nature of the CpG dinucleotide (20, 21). Since then, DNA methylation has been shown to be critical for genomic imprinting, X chromosome inactivation, and long-term repression of mobile genetic elements (22). Mechanistically, DNA methylation can lead to gene silencing by inhibiting the binding of factors that activate transcription through the addition of methyl groups in the major groove of the double helix or through the recruitment of repressive complexes (13). For example, the binding of CTCF, an insulator protein involved in the formation of chromosomal domains, is inhibited by DNA methylation, allowing enhancer-mediated activation of the paternal allele at the imprinted H19/Igf2 locus (23). DNA methylation can also mediate gene repression through methyl-CpG-binding domain proteins that bind to 5mC. Some of these proteins such as Mbd2 and Mbd3 have been found to associate with the nucleosome remodeling and deacetylase (NuRD) complex (nucleosome remodeling and histone deacetylation), thus linking DNA methylation with other epigenetic mechanisms such as histone deacetylation and nucleosome positioning (24, 25). Functions beyond gene silencing have also been reported, such as genomic DNA methylation defining introns and exons during splicing (26, 27).
Although DNA methylation is a stable epigenetic mark that can be propagated across cell divisions, DNA demethylation had been observed in different biological contexts (28).
In contrast to passive demethylation, which results in the loss of 5mC during successive rounds of replication in the absence of DNA methylation maintenance machinery, replication-independent demethylation processes had also been observed invoking the possibility of active enzymatic removal of 5mC marks (29). The discovery of the ten eleven translocation (TET) family of enzymes that can modify 5mC through iterative oxidation converting 5mC to 5-hydroxymethyl cytosine (5hmC), and subsequently to 5-formylcytosine and 5-carboxylcytosine (5caC), and the detection of these intermediates in vivo in mammalian DNA, were critical in elucidating the mechanism of active DNA demethylation (30–32). There are three TET family members in mammals (Tet1/2/3), each possessing a core catalytic domain at the carboxyl terminus, a double-stranded β-helix fold containing crucial metal-binding residues, and a CpG DNA-binding CXXC domain toward the amino terminus of the protein (33). Although Tet1 and Tet3 contain an internal CXXC domain, Tet2 partners with IDAX, an independent CXXC-containing protein (34). Following iterative oxidation of 5mC to 5caC by the TET enzymes, unmodified cytosines can be generated via passive dilution of the oxidized base or enzymatic removal via thymine DNA glycosylase and the base excision repair pathway (35–38).
Histone Modifications
In eukaryotes, DNA is wrapped around an octamer of histones composed of two copies of H2A, H2B, H3, and H4. The linker histone H1 is present where the DNA enters and exits the core nucleosome, and this nucleosome filament can be further compacted, facilitating the packing of large eukaryotic genomes within the limited space of the nucleus (39). Covalent changes in histones through posttranslational modifications (PTMs) are critical to epigenetic processes as they can alter chromatin structure or allow recruitment of activators or repressors of transcription (40). Examples of PTMs include acetylation, methylation, phosphorylation, ribosylation, and ubiquitination although it is unclear whether all of these are self-propagating through cell division, a typical criterion of epigenetic processes. One well-studied modification is histone acetylation at lysine residues mediated by histone acetyltransferases (HATs). Acetylation is typically associated with gene activation, whereas histone deacetylase enzymes (HDACs) that catalyze the hydrolysis of acetyl-lysine residues generally result in silencing and chromatin compaction (41–43). Histone methylation can also occur on lysine and arginine residues, and the methylation of distinct lysines on the histone H3 tail is associated with either gene silencing or activation. H3K9me2/3 is a conserved hallmark of heterochromatin among eukaryotes and serves as a binding site for heterochromatin protein 1 (HP1) family members to facilitate heterochromatin integrity (44). Furthermore, H3K9me3 also interacts with the DNA methylation machinery, and cooperation between these silencing marks may be vital for heritable silencing. For instance, Uhrf1 was found to bind H3K9me3 marks and facilitates H3 ubiquitination via its RING domain for recruitment of Dnmt1 at the replication fork (45–47). Deacetylation of histones can also provide H3 lysine substrates for the histone methyltransferase G9a, which also facilitates de novo DNA methylation at gene promoters through the recruitment of Dnmt3a and Dnmt3b in ES cells (48, 49). Interestingly, the histone methyltransferase activity of G9a is dispensable for de novo methylation at G9a-target gene promoters in ES cells (50). A more direct connection between DNA methylation and H3K9 methylation exists in the fungus Neurospora crassa in which the H3 histone methyltransferase dim-5 is required for DNA methylation through trimethylation of H3K9 (51). Other repressive histone marks include H3K27me2/3, mediated by the polycomb group 2 complex (PRC2), modifications associated with the silencing of genes during developmental decisions (52, 53). In contrast to K9/K27 methylation, H3K4 trimethylation (H3K4me3) is associated with active promoters, whereas H3K4 monomethylation (H3K4me1) is often a mark of enhancer function (54). H3K4me3 was also suggested to inhibit DNA methylation, as the de novo DNA methylation co-factor Dnmt3L was blocked from binding methylated H3K4 (55). Several histone modifications have also been found to be reversible by enzymes such as the histone demethylase Lsd1 that removes monoethyl or dimethyl marks from H3K4 (H3K4me1/2) (44).
Non-covalent histone modifications can also impact the state of chromatin through changes in nucleosome positioning or altered exposure of DNA along the nucleosome through ATP-dependent nucleosome remodelers. Indeed, nucleosome remodelers are critical for chromatin assembly and enable dynamic changes in genomic architecture in response to signaling or differentiation (56). Many of these complexes can both activate and repress transcription, possibly through combinatorial assembly of different subunits (57). Their specificity is thought to stem from recruitment by histone modifications, transcription factors, or DNA methylation, and therefore they have an intimate connection to epigenetic processes. Thus, both covalent and non-covalent histone modifications extend the capacity of chromatin to influence gene expression.
Histone Variants
Variants of the core histones H2A, H2B, and H3 and the linker histone H1 exist and differ from their canonical histone counterparts in primary sequence and expression pattern. In contrast to S phase-coupled expression of the canonical histones, histone variants are expressed throughout the cell cycle (39). Substitution of canonical histones with these variants is thought to modulate gene expression by altering the physical properties of nucleosomes through changes in PTMs, chromatin structure, or recruitment of additional co-activators or repressors (58). Some examples of histone variants that have a putative role in transcription include the H2A variant H2A.Z, which is enriched at the transcription start site (TSS) of active genes, and the H3.1/H3.2 variant H3.3, which has been suggested to have roles in both gene activation and heterochromatin integrity (59, 60).
Other Modes of Epigenetic Regulation
Although epigenetic marks are required for proper regulation of gene expression, an area of active interest is the contribution of small and long non-coding RNAs (ncRNAs) in directing chromatin modifications. In yeast, plants, and Drosophila, a role for small ncRNAs has been implicated in heterochromatin formation, which is disrupted upon interference with the RNAi machinery (61–63). Whether a similar pathway operates in mammals is still unclear as studies using genetic knockout of the endoribonuclease Dicer in ES cells, which is required for the biogenesis of siRNAs and microRNAs, resulted in different conclusions, with one study showing no effect on histone modification or DNA methylation status (64, 65). The PIWI-interacting RNA (piRNA) pathway has also been demonstrated to regulate heterochromatin in Drosophila by directing H3K9 methylation to transcriptionally silence transposons and interacting with HP1 (66, 67). In mice, the piRNA pathway seems to be required for sequence specific de novo methylation of the imprinted Rasgrf1 locus in the male germline (68). The epigenetic landscape can also be shaped via site-specific chromatin modifications mediated by long ncRNAs (lncRNAs). To date, X chromosome inactivation is the canonical model for the epigenetic regulation by lncRNAs. The X-inactive-specific transcript (Xist) is transcribed from the inactive X chromosome in female mammalian cells and has been shown to mediate silencing of the X chromosome in cis through the direct recruitment of PRC2 and YY1 (69, 70). Since the discovery of Xist, additional lncRNAs have been implicated in the modulation of epigenetic processes at other loci through interaction with chromatin modifiers (71). However, the in vivo functions of many lncRNAs have not yet been evaluated.
Taken together, epigenetic changes are dictated by numerous mechanisms that work in a combinatorial manner (Figure 1). An understanding of how these processes work in combination and how they are deployed for heritable transmission of gene expression has proven challenging. As we review below, studies of Cd4 and Cd8 regulation during lineage commitment may help to shape our basic understanding of these processes in a system that is highly tractable.
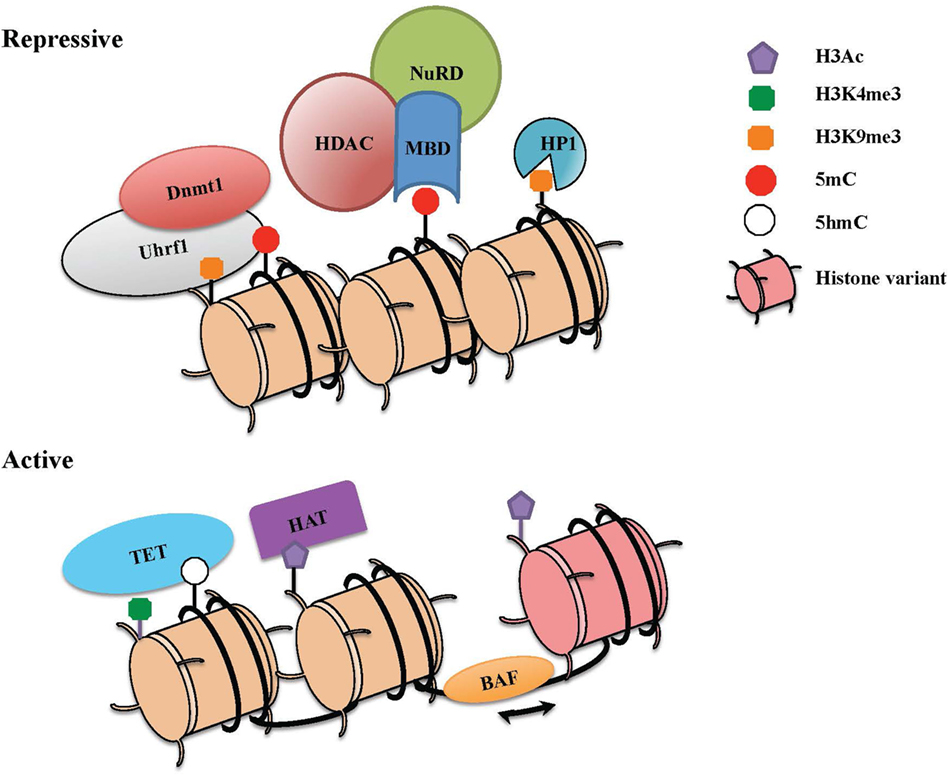
Figure 1. The combinatorial readout of chromatin dictates repressive versus active states of gene expression. Chromatin is a dynamic structure that enables gene regulation and packaging of the eukaryotic genome into the nucleus. The combinatorial output of chromatin modifications, including covalent histone linkages, histone variants, nucleosome occupancy, and DNA methylation patterns, dictate gene expression patterns in distinct tissues and cell lineages. (Top) Heterochromatin depicts a condensed and transcriptionally silent conformation of DNA. Silent genes in heterochromatin contain histone marks such as H3K9Me2/3 that can be bound by heterochromatin protein 1 (HP1), associates with DNA methylation machinery, and also recruits repressive complexes. High levels of DNA methylation are maintained through the action of Dnmt1 and its partner Uhrf1, which also interacts with H3K9me. Methyl-CpG-binding proteins that specifically recognize methylated DNA can recruit the nucleosome remodeling and deacetylase (NuRD) complex, which partakes in chromatin remodeling and catalyzes histone deacetylation though histone deacetylase enzymes (HDACs). (Bottom) To activate previously silent genes, activating transcription factors can recruit histone acetyltransferases (HATs) and chromatin remodeling complexes (BAF) to promoters and enhancers. Nucleosome remodeling can allow for transcription through nucleosome sliding and increased accessibility, exchange of a standard histone for a histone variant such as H2A.Z or eviction of the nucleosome. The DNA demethylase enzymes [ten eleven translocations (TETs)] also catalyze the conversion of methylcytosine to 5-hydroxymethylcytosine (5hmC) and other oxidation products, which in turn can inhibit the binding of repressive complexes and promote gene expression.
cis Elements and Epigenetic Regulation of the Cd4 Locus
The dynamic regulation of the CD4 co-receptor during αβT cell development is finely controlled via multiple cis-regulatory elements that were initially revealed by functionally testing DNase I hypersensitivity sites (DHSs). Four cis elements were reported to direct Cd4 expression in transgenic assays: a silencer (S4) situated 1 kb downstream of the Cd4 TSS, a proximal enhancer (PE) (E4P), a distal enhancer (E4D) situated 13 and 24 kb upstream of the Cd4 TSS, and a thymocyte enhancer (E4T) mapped 36 kb downstream. The in vivo roles of only three have thus far been assessed by targeted deletion of the endogenous locus. E4P and S4 have been found to direct expression of Cd4 in αβ T cells, and the thymocyte enhancer E4T was found to direct Cd4 expression in lymphoid tissu inducer (LTi) cells in the intestine (72, 73). Although E4D was reported to drive reporter expression in mature T cell lines, its function still remains to be assessed in vivo (74). Recently, an intronic enhancer adjacent to S4, ~2 kb downstream of the Cd4 TSS, which we refer to as E4M, was found to drive CD4 expression in αβ T cells and is described further below (Figure 2) (75).
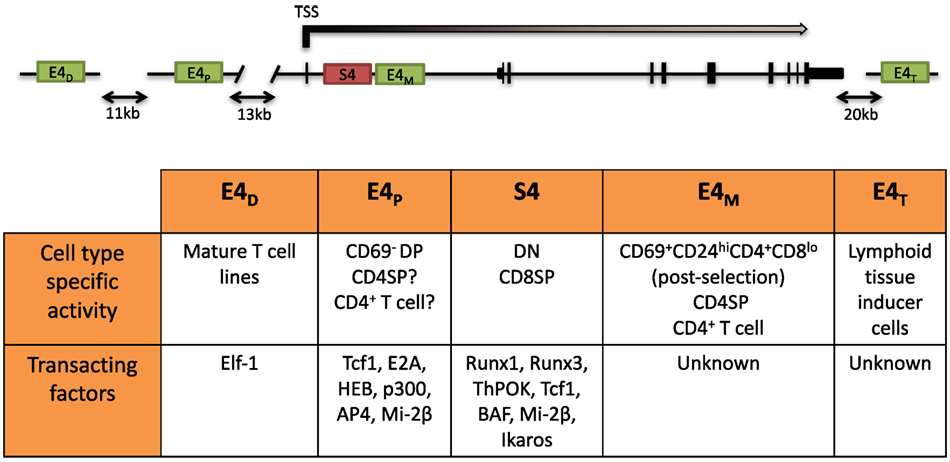
Figure 2. Schematic of the Cd4 locus and its cis-regulatory elements. (Top) Two positive cis elements, E4P and E4M, and a silencer element, S4, regulate CD4 expression in αβ T cells. E4P, consisting of a 339 bp DNA element, lies ~13 kb upstream from the Cd4 transcription start site (TSS) and is required for Cd4 expression in double-positive (DP) thymocytes and establishment of stable CD4 expression following positive selection of CD4 lineage cells. The 434 bp S4 element lies ~1.5 kb downstream of the Cd4 TSS in the first intron and represses Cd4 expression in double-negative (DN) and cytotoxic CD8+ T cells. E4M is immediately downstream of S4 and promotes Cd4 expression in postselected CD4 thymocytes and mature CD4+ T cells. The distal enhancer E4D was shown to promote CD4 reporter gene expression in T cell lines but was not assessed in vivo. The thymocyte enhancer E4T was shown to direct CD4 expression in lymphoid tissue inducer cells by mutation of the endogenous locus. (Bottom) Table of the cell type-specific activity of the different cis elements and known transacting factors reported to bind and modulate expression of Cd4 (description in text).
E4P
The E4P element was mapped to a 339 bp region of DNA that directed reporter activity in T cell lines and in vivo in T cells of transgenic reporter mice (76, 77). Germline targeting of E4P prevented CD4 upregulation during the DN–DP transition in the thymus, demonstrating its requirement for CD4 expression in preselection DP cells (73). After positive selection, CD4 was reexpressed on MHC class II selected cells, although at reduced levels compared to wild-type cells. Furthermore, CD4 expression was unstable during cell division, as a large proportion of CD4+ T cells in the periphery lost CD4 expression after activation. In these cells, H3 acetylation and H3K4Me3 within the Cd4 locus were also reduced. The loss of H3Ac and H3K4Me3 was observed in CD4− DP stage thymocytes, consistent with the loss of CD4 expression. To investigate whether E4P was required for regulating CD4 expression beyond the DP stage, Cre-mediated excision of a conditional allele was performed. In contrast to germline deletion, conditional deletion of E4P in mature CD4+ T cells did not impair CD4 expression during activation, and the levels of H3Ac and H3K4Me3 were similar to wild-type cells. This suggested that, during thymic development, E4P modulates a stable epigenetic state of the Cd4 locus that can be maintained in its absence thereafter.
Sequence analysis and nuclear extract binding studies suggested that the HMG-box transcription factors T cell factor 1 (Tcf1)/Lef1 and the basic helix-loop-helix (bHLH) proteins E2A/HEB bind to motifs within E4P (76, 78). Accordingly, gene targeted inactivation of the genes encoding E2A and HEB impaired CD4 expression during the DN–DP transition (79, 80). Similarly, mutation of Tcf7 (which encodes Tcf1) also reduced CD4 expression in DP thymocytes (81). ChIP analyses confirmed that these factors bound to E4P, suggesting that they directly promote expression of CD4 in DP thymocytes (81, 82). However none of these factors is individually required for CD4 expression in DP thymocytes, as single deletions do not phenocopy E4P−/− mice. Interestingly, the NuRD complex, typically associated with gene repression, was also implicated in CD4 activation (83). Deficiency in mi-2β, an ATPase chromatin remodeling subunit of the NuRD complex, led to impaired CD4 upregulation during the DN–DP transition (83). mi-2β also bound E4P by ChIP assays and co-immunoprecipitated with HEB and the HAT p300, linking histone acetylation with factors recruited to E4P. The cooperative mechanisms by which these transcription factors drive transcription of Cd4 and their contribution to epigenetic programs are yet to be assessed.
E4M
The observation that CD4 was re-expressed following positive selection in E4P−/− thymocytes suggested the existence of another regulatory element, now referred to as the “maturation enhancer,” E4M. The existence of such an element was also supported by a previous study employing a Cd4 transgene containing the Cd4 first intron, promoter, and E4P element, whereby transgenic reporter activity was lost in activated mature T cells without changes in endogenous Cd4 expression (84). Indeed, our recent ATAC-seq analysis of the Cd4 locus in CD4SP thymocytes revealed a previously uncharacterized chromatin-accessible region situated in the first intron of the gene, 3′ to the silencer (Priya D. A. Issuree and Dan R. Littman, unpublished results). The potential role of this region was highlighted by Egawa and colleagues, who showed that CD4 expression was unstable in T cells from mice with targeted deletion of 1.5 kb encompassing the silencer and the region 3′ to it, but was stable when only the core silencer was deleted (75). Our studies using mice lacking E4M revealed that this element controls Cd4 expression in postselected CD4SP thymocytes, and in the absence of both E4M and E4P, there is a complete loss of CD4 in T cells (Priya D. A. Issuree and Dan R. Littman, unpublished results). As observed in the absence of E4P, deletion of E4M resulted in the gradual loss of CD4 expression by activated CD4+ T cells following multiple rounds of cell division [(75) Priya D. A. Issuree and Dan R. Littman, unpublished results]. Analogous to E4P, retroviral Cre-mediated excision of this region in mature T cells did not diminish CD4 expression, suggesting that E4M may be required for establishing an epigenetically active state during development. However, it remains possible that both enhancers function cooperatively in the establishment of stable CD4 expression and are insufficient to do so individually. Importantly, these studies demonstrate that the main enhancers dictating CD4 expression in αβ T cells are E4P and E4M, and no additional elements are capable of compensating for their loss. The minimal region encompassing E4M activity and the transacting factors recruited there to drive expression are currently under investigation. It also remains to be determined whether the lack of E4M without a larger intronic deletion results in redirection of MHC class II selected cells into the CD8+ lineage, as was observed in mice with the 1.5-kb intronic deletion (75).
Cd4 Silencer (S4)
The silencer (S4) is a 434 bp core element in the first intron of Cd4 that was identified by its ability to inhibit expression from reporter constructs in cell lines and in both DN thymocytes and CD8+ T cells of transgenic mice (77, 85). Deletion of this element in the germline of mice caused CD4 derepression in DN thymocytes and CD8+ cytotoxic T cells, confirming its physiological function (86, 87). Importantly, Cre-mediated excision of a conditional LoxP-flanked S4 allele in mature CD8+ T cells did not reverse CD4 repression (87). The results demonstrated that S4 activity is reversible during the DN–DP transition, but becomes irreversible following positive selection, in mature cytotoxic T cells. Therefore, CD4 silencing in peripheral cytotoxic T cells is independent of S4 and mediated by a heritable silencing mechanism after CD8+ lineage commitment. To determine when S4-independent repression occurred, stage-specific S4 deletion with different Cre transgenic mice was carried out. By using CD4-Cre, which drives Cre expression in the late DN and early DP stages of development, DN thymocyte repression was intact but mature CD8+ T cells derepressed CD4 similarly to the germline deletion (87). S4 deletion with CD8-E8I Cre, which is active only in MHC class I-selected T cells, also caused CD4 derepression, suggesting that heritable Cd4 silencing occurs after positive selection. Deletion of S4 at additional stages after MHC class I-positive selection may provide more insight into the initiation of heritable CD4 silencing.
In contrast to uniform CD4 derepression in S4−/− mice, variegated CD4 derepression occurred with smaller deletions in S4 (88). This phenotype was reminiscent of position-effect variegation (PEV) studied extensively in Drosophila. PEV occurs when a gene is stochastically silenced in a population of cells due to spreading of heterochromatin from an adjacent locus (89). In agreement with the spreading of heterochromatin, CD4 repression was stable in cells that achieved silencing and the frequency of repression could be increased by overexpressing HP1β in vivo (88). There was no variegation observed in DN cells in which CD4 derepression occurred following mutations in S4. Instead, there was a partial but uniform derepression, consistent with active repression and the contribution of multiple transcription factor binding sites toward this process (88). Motif analysis revealed two sites in S4 that contained consensus binding motifs for members of the evolutionarily conserved family of Runx transcription factors (see Runx) (90). Deletion of both sites caused CD4 derepression in CD8+ T cells, and interaction between S4 and the Runt domain of Runx1 was found in a yeast-one-hybrid screen (91). Runx1 is expressed at the highest level in DN thymocytes, whereas Runx3 is predominantly expressed in CD8SP cells. Consistent with their expression pattern, loss of function studies demonstrated that Runx1 is indispensable for active repression of Cd4 in DN thymocytes, whereas Runx3 is responsible for establishing Cd4 silencing in CD8+ T cells (91–94). Interestingly, deficiency in the nucleosome remodeling Brg/Brahma-associated factor (BAF) complex was also reported to cause CD4 derepression in DN thymocytes (82). Combining BAF deficiency with point mutants in S4 significantly enhanced CD4 derepression, and BAF subunits were found to bind S4 (82). The BAF complex was subsequently implicated in regulating S4 accessibility for Runx1 binding in DN thymocytes (95). Finally, the zinc finger transcription factor Ikaros, which associates with BAF and other chromatin remodeling complexes, was also found to be required for CD4 repression in DN thymocytes (96). Active repression of CD4 hence appears to require remodeling of the locus and binding of Runx1, which is discussed further below (Figure 3).
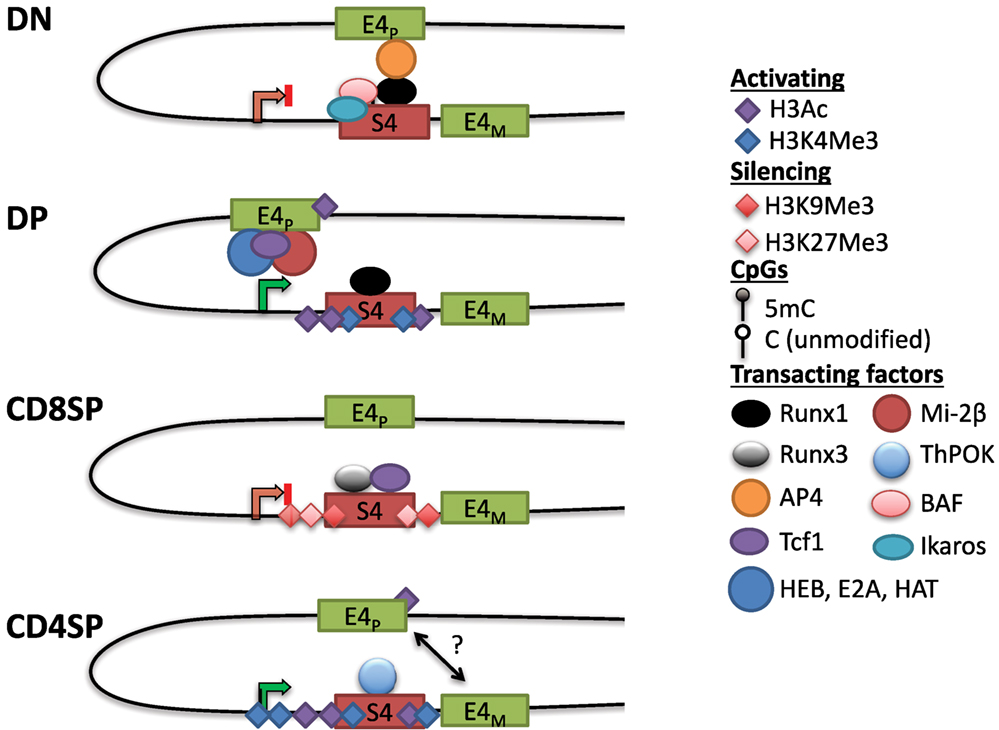
Figure 3. Dynamic Cd4 regulation during T cell development in the thymus. In double-negative (DN) thymocytes, Cd4 expression is repressed through S4, which may interact with the E4p enhancer, potentially preventing it from activating the Cd4 promoter. This interaction could be mediated via Runx1, which binds to S4 and associates with AP4 at E4P, possibly through chromatin looping (see text). Nucleosome remodeling complexes such as the SWI/SNF-like Brg/Brahma-associated factor (BAF) complex are thought to regulate accessibility to S4 to promote silencing. The Cd4 locus in DN thymocytes contains high levels of DNA methylation and lacks high levels of histone acetylation. In double-positive (DP) thymocytes, Cd4 expression is upregulated through E4P, potentially due to changes in conformation of the locus (see text). E4P serves as the recruitment site for numerous transcription factors including HEB, E2A, Tcf1, and lymphoid enhancer factor 1. The ATPase subunit of the nucleosome remodeling and deacetylase complex, mi-2β, is a positive regulator of Cd4 expression, possibly through the recruitment of histone acetyltransferases (HATs) to either E4P or S4. The locus acquires H3Ac and H3K4 trimethylation (H3K4Me3) at S4, but DNA methylation remains high (see Figure 4). In CD8SP cells, Cd4 expression is repressed through the transcription factor Runx3 and Tcf1 binding to S4. The locus acquires some additional DNA methylation and silencing histone marks H3K9Me3 and H3K27Me3. In CD4SP cells, E4M controls Cd4 potentially in a cooperative manner with E4P. S4 is bound by the CD4+ lineage commitment factor T helper inducing POZ/Krueppel-like factor (Thpok), which antagonizes activity of Runx complexes. The locus undergoes active demethylation and is thus hypomethylated in CD4SP cells relative to all other stages of T cell development (see Figure 4).
S4 deletion increased recruitment of HAT p300 at E4P and increased H3K9Ac at the promoter in DN thymocytes (93). Although E4P is 13 kb upstream of the Cd4 promoter, these elements have been suggested to physically communicate through looping (97). Chromatin conformation capture assays suggested that in DN thymocytes, Runx1 mediates a loop between S4 and E4P, which sequesters the positive transcription elongation factor (P-TEFb) from activating RNAPol-II at the promoter (97). Another factor that cooperates with Runx1 in Cd4 silencing at the DN stage is the bHLH transcription factor adaptor-related protein complex 4 (AP4), which binds to E4P (98). This raises the possibility that a loop forming between E4P and S4 via Runx1 and AP4 prevents transcription in DN thymocytes and, upon Runx1 downregulation during the DN–DP transition, P-TEFb associates with RNAPol-II at the promoter to drive expression. However, Runx1 has also been reported to bind to S4 in DP thymocytes, suggesting that its repressive function is overridden by an unknown mechanism at this stage. Another study using three-dimensional fluorescent in situ hybridization showed that the Cd4 and Cd8 loci are in close proximity when CD8 is expressed (in DP thymocytes and CD8+ T cells), but in the absence of S4, this association was lost, suggesting that S4 has a role in locus architecture (99). Strikingly, association of the co-receptor loci was conserved in human T cells even though Cd4 and Cd8 are on different chromosomes, suggesting that their proximity may be functionally important. Thus, S4 together with E4P and E4M are able to direct helper lineage-specific expression of CD4 in vivo.
Taken together, the study of cis elements controlling Cd4 expression during T cell development revealed a key role for these elements in the establishment of key epigenetic marks that can be transmitted thereafter in a heritable manner. Thus, a thorough analysis of the stage-specific activity of these elements provides a unique opportunity to delve into molecular requirements for heritable transmission.
DNA Methylation as a Key Epigenetic Mark for the Heritable Expression of Cd4
The nature of the epigenetic process employed to enable heritable silencing of Cd4 in mature CD8 T cells in the absence of S4 was a missing piece in our understanding until recently. An shRNA screen, designed to identify genes repressing Cd4 in mature CD8+ T cells independently of S4, led to the identification of Dnmt1 and the DNA methylation pathway as a central requirement to this process (100). Although previous experiments had concluded that DNA methylation was not involved in CD4 silencing, these results were obtained with 5-azacytidine, a potent inhibitor of DNA methylation, that likely obscured CD4 derepression due to toxicity in primary CD8+ T cells (87). Indeed, a subsequent study with reduced dosing of the inhibitor showed CD4 derepression (75). Additional genetic evidence demonstrating the role of DNA methylation in CD4 silencing was shown through the use of mice with mutations in Dnmt1 and the de novo DNMTs, Dnmt3a and Dnmt3b. Transfer of DNMT-deficient naïve CD8+ T cells into lymphopenic mice resulted in robust CD4 derepression following homeostatic proliferation. Interestingly CD4 expression in naïve T cells from Dnmt-deficient mice is normal, suggesting that CD4 derepression in the absence of DNA methylation machinery occurs during robust T cell proliferation. Whether the conditional inactivation of the Dnmt3 enzymes using CD4-Cre influences CD4 derepression due to effects on de novo rather than maintenance methylation remains to be determined.
The changes in DNA methylation at the Cd4 locus during thymic development were assessed using bisulfite CATCH-seq (clone-adapted template-capture-hybridization sequencing). A region displaying a high level of differential methylation [differentially methylated region (DMR)] between CD4+ and CD8+ T cells was identified in the first intron, extending from −0.7 kb to +3.2 kb relative to the Cd4 transcriptional start site. The levels of DNA methylation during development correlated with CD4 expression, such that the locus was hypermethylated in DN and CD8SP thymocytes compared to CD4SP thymocytes. Surprisingly, CD4 expression in DP thymocytes was uncoupled from DNA methylation, as the Cd4 locus was hypermethylated and resembled methylation patterns in CD8SP cells. However, a few additional methylated CpGs were found in CD8SP thymocytes compared to DP thymocytes, presumably due to the activity of Dnmt3 enzymes. It is possible that these methylated CpGs are sufficient to repress Cd4 transcription, for example, by influencing recruitment of activating or repressive transcription factors to the locus or decreasing chromatin accessibility through nucleosome remodeling. As bisulfite sequencing does not discriminate between 5mC and 5hmC, another possibility is that some of the methylated CpGs at the Cd4 locus are 5hmCs, whose presence would promote CD4 expression (101). It is also possible that additional chromatin modifications induced by S4 through lineage-specific transcription factors such as Runx3 may be required to initiate CD4 repression during commitment to the cytotoxic lineage. Analysis of the Cd4 locus revealed that the hypermethylation status of the DMR in CD8+ T cells was critically dependent on S4. In the absence of S4, the Cd4 DMR methylation levels in CD4+CD8+ cytotoxic T cells resembled those in WT CD4+ T cells. Thus, S4 may be required to prevent demethylation after commitment to the CD8+ lineage and to participate in the establishment of heritable methylation marks for stable repression of CD4 in CD8+ T cells. Future investigation will allow testing of these hypotheses and address the contribution of factors involved in Cd4 silencing such as Runx3 and Tcf1. Taken together, the results indicate that DNA methylation mediated by S4 is required, at least in part, for heritable silencing in the cytotoxic lineage.
A Role for TET-Mediated DNA Demethylation in Heritable Expression of CD4
The loss of methylation initially present in DN and DP cells during CD4+ T cell differentiation suggested that DNA demethylation is crucial for maintaining CD4 expression (100). As there is a lack of cell division during the DP to SP transition following positive selection, the decreased methylation in CD4SP cells suggested an active demethylation process rather than the lack of methylation maintenance during replication. Indeed, locus-specific oxidative bisulfite amplicon sequencing detected the presence of 5hmC at the Cd4 locus in CD4+CD8lo thymocytes as they differentiated into CD4SP cells. A subsequent study also found that 5hmC levels were correlated with gene activation in the thymus, and 5hmC was enriched in genomic regions harboring H3K4Me1 and H3K27Ac, marks associated with enhancers (102). The precise functions of the three TET enzymes in lineage commitment, and the mechanism of their recruitment to the Cd4 locus are not known. Remarkably, although S4 was associated with increased methylation in CD8SP cells, the PE E4P was critical for demethylation in CD4SP cells (Figure 4). E4P−/− naïve CD4+ T cells exhibited hypermethylation of the Cd4 DMR similar to WT CD8+ T cells. Furthermore, the loss of CD4 expression in activated E4P−/− helper T cells correlated with increased DNA methylation of the locus. Thus, E4P is required during development for the establishment of a heritable hypomethylated state at the Cd4 locus in mature CD4+ T cells. It will be interesting to determine how E4P coordinates DNA demethylation of the locus. In addition, in the absence of Thpok, the Cd4 locus was hypomethylated in MHC class II selected cells redirected to the CD8+ lineage compared to WT CD8+ lineage cells, suggesting that Thpok is partially dispensable for DNA demethylation (100). Taken together, these results reveal a critical role for cis-regulatory elements in coordinating the proper epigenetic machinery during development that is essential for heritable Cd4 expression (Figure 4). Elucidation of how transcription factors binding to these elements choreograph the functions of Dnmts and TET enzymes promises to reveal important aspects of differential signaling in positive selection of CD4 and CD8 SP cells.
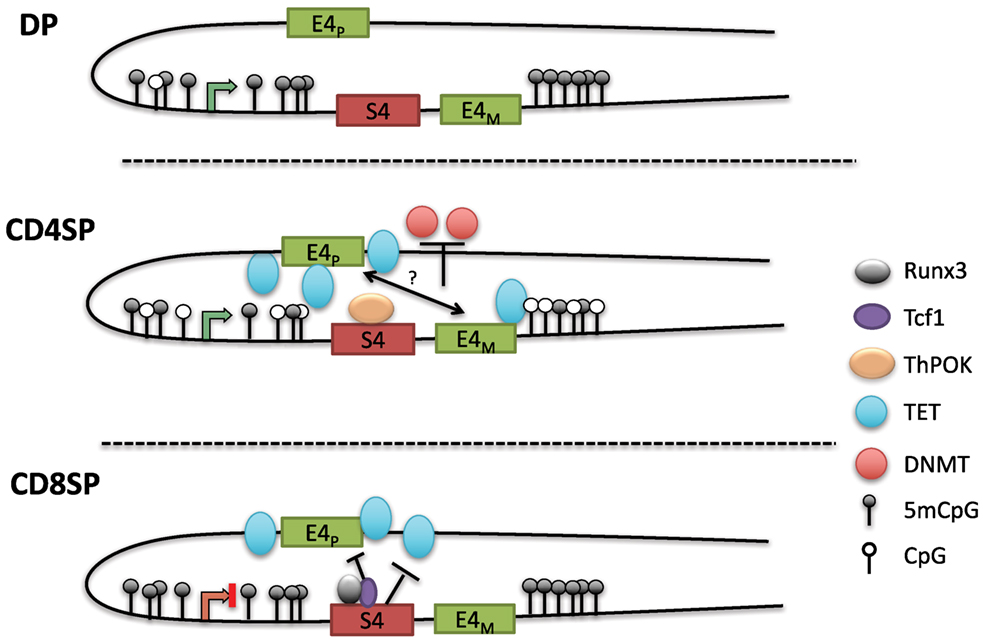
Figure 4. Model of methylation dynamics at the Cd4 locus during lineage commitment. The Cd4 locus is hypermethylated in double-positive (DP) thymocytes; as MHC class II selected cells downregulate CD8 to become CD4SP cells, the locus undergoes active demethylation presumably through iterative oxidation via ten eleven translocation (TET) enzymes. T helper inducing POZ/Krueppel-like factor (Thpok), which binds to S4 late during positive selection, partially contributes to the demethylation process. In the absence of E4P, the locus remains hypermethylated in CD4SP cells and resembles the methylation profile of CD8SP cells. Thus, E4P mediates demethylation of the locus, perhaps in cooperation with E4M following the selection by MHC class II molecules. In CD8SP cells, which undergo selection for MHC class I interaction, the Cd4 locus remains hypermethylated (with addition of a few novel methyl-CpGs). However, in the absence of S4, the locus undergoes demethylation similarly to CD4SP thymocytes, indicating that S4 potentially inhibits Cd4 demethylation by antagonizing ten eleven translocation (TET)-mediated demethylation.
cis Elements and Epigenetic Regulation of the Cd8 Locus
CD8 Enhancers
The Cd8 locus is composed of two linked genes, Cd8a and Cd8b1. In mice, they are found on chromosome 6, separated by 36 kb and aligned in the same transcriptional orientation. DP thymocytes and most cytotoxic TCRαβ T cells express CD8 as a CD8αβ heterodimer, whereas on intraepithelial lymphocytes (IELs) and a subset of dendritic cells, CD8 can be expressed as a homodimer of CD8αα. This suggests that both genes can be coordinately and independently regulated in different cell types. An 80-kb genomic fragment encompassing the Cd8 locus was found to drive developmental stage and lineage-specific expression of a reporter transgene (103, 104). Four DHS clusters within this fragment (CI-IV) contain at least six enhancers. E8I enhancer (CIII-1,2) was found to drive CD8 expression in CD8SP thymocytes, mature CD8+ T cells, and IELs (104–106). Interestingly, the onset of E8I activity was observed in positively selected thymocytes, suggesting a link between this element and cytotoxic T cell commitment. E8II (CIV-4,5) directed expression in both DP thymocytes and CD8+ T cells, while E8III (CIV-3) drove CD8 expression in immature DP thymocytes. The E8IV (CIV-1,2) element was active in DP and CD8+ T cells. In transgenic studies, E8V (CII) exhibited no enhancer function by itself, but a combination of E8V and E8I directed expression in both CD8+ T cells and DP thymocytes, unlike E8I alone, suggesting that stage-specific activity could arise through combinations of CD8 enhancers (107). Recently, another cis element termed E8VI (CIV-6) was described, which directed transgenic reporter expression in mature CD8+ T cells, memory-phenotype-like CD44hiCD62L+CD8+ T cells, and CD8αα+ dendritic cells (108). In summary, at least six enhancers direct CD8 expression in cytotoxic T cells (Figure 5).
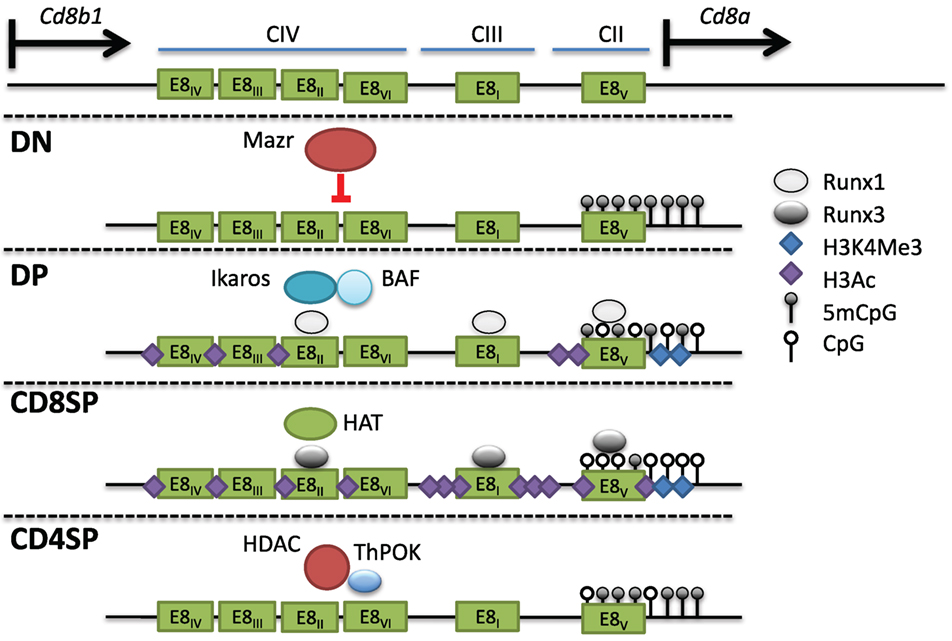
Figure 5. Multiple cis elements direct Cd8 expression during thymocyte development. (Top) The Cd8 locus consists of two linked genes, Cd8a and Cd8b1, on mouse chromosome 6. Cd8 expression is regulated by at least six enhancers (E8I-VI) found in three clusters of DNAse I hypersensitivity sites. In double-negative (DN) thymocytes, Mazr represses expression of Cd8, and the locus is hypermethylated and lacks activating histone marks. Following pre-T cell antigen receptor signaling and maturation to become double-positive (DP) thymocytes, Cd8 is upregulated via the control of multiple enhancers (see text). The chromatin remodeling Brg/Brahma-associated factor (BAF) complex and the transcription factor Ikaros promote Cd8 expression in DP thymocytes. Runx1 also binds to multiple Cd8 enhancers and was shown to promote Cd8 expression as well. The locus in DP thymocytes partially loses DNA methylation and gains H3Ac and H3K4Me3 at the Cd8a promoter. In CD8SP cells, Runx3 binds to numerous enhancers leading to a corresponding increase in H3Ac, possibly due to direct recruitment of histone acetyltransferases (HATs), as well as a decrease in DNA methylation. In CD4SP cells, T helper inducing POZ/Krueppel-like factor recruits histone deacetylase enzyme to actively repress expression.
Studies examining the regulation of the endogenous Cd8 locus through genetic deletion revealed both specific and cooperative modes of enhancer regulation. In E8I−/− mice, both TCRγδ and TCRαβ IELs showed a significant decrease in CD8α, while CD8 expression in DP thymocytes, CD8SP thymocytes, and peripheral CD8+ T cells was largely intact (106, 107). E8I was also found to be crucial for the maintenance of CD8α expression in activated CD8+ T cells (109). In some cases, combinatorial enhancer activity occurred as shown by combined deletion of E8I/E8II, resulting in variegated expression in DP thymocytes that did not occur with single deletion of either enhancer (110). In contrast, combined deletion of E8I/E8II had no additional effect on the maintenance of CD8 expression in activated CD8+ T cells than deletion of E8I alone (109). These results suggested that E8I might have a non-redundant role in maintaining transcription of CD8α during activation, as IELs are also thought to exist in a partially activated state (111). Similar to combined E8I/E8II deletion, variegated CD8 expression was observed during the DN–DP transition with deletion of E8V or combined deletion of E8II/E8III (112, 113). Taken together, these data indicate that in vivo the CD8 enhancers have both non-redundant and stage-specific functions.
Search for a CD8 Silencer Element
It is unclear whether there is a silencer in the Cd8 locus, analogous to that in Cd4, that restricts its expression to the cytotoxic lineage. Experiments in a hybridoma cell line suggested the possibility that DNA elements in cis to the Cd8 locus were able to silence transcription (114). Referred to as L2a, this region within E8V was identified as a matrix-associated region (MAR), an AT-rich sequence that mediates attachment to the nuclear matrix (115). In transgenic experiments, L2a was found to have negative transcriptional activity using an E8I/E8V hCD2 reporter construct that was reversed by the MAR binding protein, Satb1 (special AT-rich sequence binding protein 1) (116). Deletion at the endogenous locus as well as assays for position and orientation independence will help clarify whether this is a bona fide silencer. Thus, in contrast to the Cd4 locus, there is currently no known negative element that confers helper versus cytotoxic lineage specificity of CD8 expression. One ontological explanation is that there may be a need for greater plasticity in CD8 expression compared to CD4. For example, positive selection of DP thymocytes induces the downregulation of CD8, which is reversed during cytotoxic lineage commitment (7). The overexpression of Thpok in CD8+ cytotoxic T cells silences CD8 but does not induce CD4 expression (8, 117). In vivo, some CD4+ helper T cells in the intestine undergo transcriptional reprogramming toward a cytotoxic phenotype including upregulation of CD8αα following the downregulation of Thpok (118, 119). Thus, dynamic and flexible CD8 expression in T cells may require regulation mediated directly through a transcriptional circuit.
Epigenetic Regulation of CD8 Expression
The variegated CD8 expression in DP thymocytes due to deletion of E8I/II or E8V suggested that these enhancers were required to protect the Cd8 locus from repressive chromatin modifications. Interestingly, mutations in the SWI/SNF-like nucleosome remodeling BAF complex caused similar CD8 variegation in DP thymocytes as the enhancer-deficient mice (82, 110, 112). BAF likely regulates Cd8 directly, as it was found to bind to the locus by ChIP analysis (82). The DNA-binding transcriptional regulator Ikaros, which was reported to associate with the BAF complex, was also required for efficient CD8 upregulation during the DN–DP transition when on an Aiolos-deficient background (another Ikaros family member) (120, 121). In T cells, Ikaros was also found to associate with the repressive chromatin remodeling complex NuRD (122). The NuRD ATPase chromatin remodeling subunit, mi-2β, was found to bind the Cd8 locus until pre-TCR signaling, upon which its eviction coincided with increased DNAse I hypersensitivity and expression of CD8 (123). Thus, different chromatin remodeling complexes, presumably recruited by transcription factors such as Ikaros to the CD8 enhancers, can activate or repress CD8 expression at different stages of development.
Double-positive thymocytes that failed to upregulate CD8 in E8I/II-deficient mice showed decreased histone acetylation at the Cd8 locus concomitantly with a decrease in H3K4Me3, both consistent with the loss of expression (124). H3Ac was also decreased along with an increase in H3K27Me3 in activated cytotoxic T cells from E8I−/− mice that had lost CD8 expression (109). Treatment of E8I−/− CD8+ T cells with the HDAC inhibitor TSA partially rescued CD8 expression during activation although there was no restoration of CD8 in T cells that had already lost expression (109). Interestingly, the unstable CD8 expression during proliferation was reminiscent of CD4 expression in the absence of either E4M or E4P (73, 75).
Histone acetylation appears to play a particularly prominent role in CD8 regulation, as the deletion of HDAC1/2 with CD4-Cre caused CD8 derepression in CD4+ T cells (125). In contrast, Dnmt1 deletion did not affect CD8 expression in CD4+ T cells but caused CD8 derepression in TCRγδ cells, suggesting differential dependence on DNA methylation and histone modifications for CD8 silencing in different cell types (126). DNA methylation was also analyzed in thymocytes that had failed to upregulate CD8 during the DN–DP transition in E8I/II-deficient mice (124). Previous studies showed that DNA methylation patterns correlate with CD8 expression during thymocyte development and in peripheral T cells (126–129). However, in the absence of E8I/II, there was a gain in methylation of several CpGs at the Cd8 locus at E8V, and the expression of CD8 could be partially rescued by crossing the mice onto a Dnmt1-deficient background (124). These results suggest that factors binding to E8I/II promote Cd8 expression through inhibition of the DNA methylation machinery.
Key Regulators of the Cd4 and Cd8 Epigenetic Landscape
Mazr
The BTB-ZF family member Mazr was identified in a yeast-one-hybrid screen to bind to E8II (124). By ChIP-PCR, it was shown that Mazr also binds at numerous locations in the Cd8 locus at the DN stage, and this binding is reduced in DP thymocytes following CD8 upregulation. Ectopic expression of Mazr-induced variegated CD8 expression in DP thymocytes, while deletion of Mazr on an E8I/E8II-deficient background reduced CD8 variegation (124, 130). However, in WT cells, Mazr deficiency was insufficient to derepress CD8 expression in DN thymocytes, suggesting redundancy with other repressive factors or lack of activating factors to initiate expression. Mechanistically, Mazr was found to bind to the co-repressor N-CoR, a component of large repressor complexes that include HDACs, and presumably Mazr can recruit these complexes to silence CD8 in DN thymocytes. Mazr was also connected to the network of transcription factors that regulate lineage commitment through the discovery of its role in the regulation of the T helper commitment factor Thpok (130). In the absence of Mazr, Thpok was derepressed in DP thymocytes, leading to lineage redirection of MHC class I selected thymocytes into the helper lineage. The derepression of Thpok was enhanced when combined with mutations in Runx1 or Runx3 (131). CD4 derepression was also enhanced in DN thymocytes doubly deficient for Runx proteins and Mazr, supporting a cooperative role between these factors (131). Mazr likely regulates Thpok expression directly as it was found to bind the Thpok silencer element (130). Mazr-mediated silencing of Thpok is also important in the periphery as Mazr deficiency inhibited the induction of cytotoxic CD4+ T cells in the intestine through the inability to fully downregulate Thpok (118).
Thpok
The key transcription factor that directs commitment to the CD4+ lineage is Thpok, also known as Zbtb7b or cKrox. A role for Thpok in thymocyte development was discovered after the identification of a spontaneous mutant mouse strain deficient in T helper cells, referred to as helper-deficient (HD) mice (132, 133). The mutation was mapped to an amino acid substitution in the second zinc finger domain of Thpok, and the causal role of the mutation in the HD phenotype was confirmed by transgenic rescue of CD4+ lineage development with WT Thpok (133, 134). Furthermore, the HD mouse strain could not be rescued by an MHC class II restricted TCR transgene, and there was no evidence of impaired positive selection in the thymus, indicating that Thpok was downstream of TCR signaling. Another study investigated Thpok based on its significant upregulation following MHC class II selection in the thymus (134). Ectopic expression of Thpok redirected MHC class I selected thymocytes to the helper lineage, indicating it was both required and (in the context of expression of other factors, such as GATA3) sufficient for CD4+ lineage commitment (133, 134).
Mechanistically, Thpok plays a critical role in lineage commitment through its antagonism of the CD8+ lineage transcription factor Runx3. Thpok was found by ChIP to bind to the Cd4 silencer in CD4SP cells, and ectopic Thpok expression in DN thymocytes caused CD4 derepression, suggesting that Thpok directly antagonizes Runx activity at the silencer (92, 135). Thpok may continue to antagonize CD4 silencing in mature T cells, as peripheral deletion of Thpok led to aberrant populations of MHC class II restricted T cells that were CD4loCD8− and CD4+CD8+. These populations were reduced by the additional absence of Runx complexes, suggesting that postthymic Thpok is required for both the proper expression of CD4 and the repression of CD8 in helper T cells (119, 136). However, inducible inactivation of Thpok in peripheral T cells as well as the physiological downregulation of Thpok in the intestine did not produce a population of CD4−/CD4lo helper T cells (118, 137). This may be due to the leakiness of the transgenes used to drive Thpok excision in the different studies or technical limitations of inducible systems, and further studies are warranted to determine whether Thpok is needed for CD4 expression postthymically. Finally, analysis of mice with GFP inserted into the Thpok locus and YFP inserted into the Runx3 locus demonstrated that Thpok was required to repress Runx3 expression in MHC class II selected cells (138). Conversely, helper T cell differentiation in the absence of Thpok could be rescued by Runx deficiency (138). Taken together, these data suggest a mutual antagonism between these critical transcription factors for CD4+ and CD8+ T cell differentiation.
The above studies also suggested that, in contrast to the epigenetic silencing of CD4, CD8 silencing in mature CD4 lineage cells depends on the continuous activity of Thpok. As has been reported for other BTB-ZF family members, Thpok function has been linked to repression through HDAC activity (135, 139). At the Cd8 locus, Thpok has been found to occupy the CD8 enhancers and promoter in CD4SP thymocytes (139). The HDAC inhibitor TSA blocked the repressive activity of Thpok in luciferase assays, and numerous HDACs were found to associate with Thpok by Co-IP (139). Interestingly, a mutant form of Thpok that prevented its interaction with Hdac4 was unable to inhibit CD8 expression when overexpressed in CD8+ T cells (117, 139). Although the mutation did not affect Thpok binding to the Cd8 locus, it abrogated recruitment of Hdac4. In addition, in a CD4+ thymoma cell line, Thpok was unable to inhibit Runx-mediated repression of an E4p-S4 GFP reporter construct after TSA treatment (135). Thus, HDACs seem to be essential components for Thpok function, but additional in vivo studies of their complementary roles are needed.
Interestingly, initiation of demethylation of the Cd4 locus coincides with the upregulation of Thpok, which is first detected in postselection CD4+CD8lo thymocytes (100, 138). CD8+ T cells from ThpokGFP/− mice, which are MHC class II restricted cells redirected from the CD4SP lineage, express GFP, suggesting that MHC class II restricted positive selection is required and sufficient to turn on high Thpok expression (138). However, the Cd4 locus was still hypomethylated in MHC class II-selected cells redirected to the CD8+ lineage in the absence of Thpok compared to WT CD8+ lineage cells, suggesting that Thpok is partially dispensable for DNA demethylation. Therefore, additional unknown factors triggered during MHC class II-mediated TCR signaling may be responsible for coordinating TET-mediated demethylation of the locus (Figure 6).
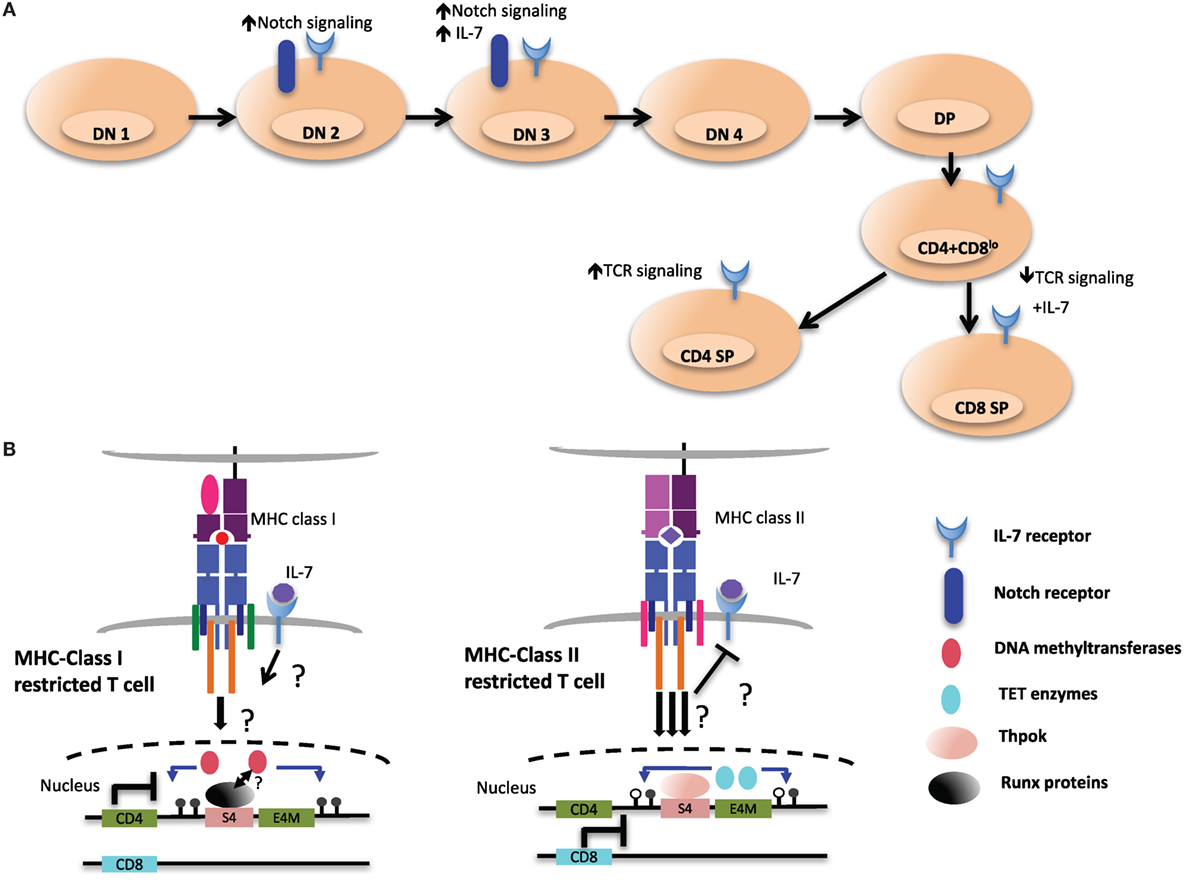
Figure 6. Environmental cues during T cell development may shape the epigenetic landscape of CD4 and CD8 T cells. (A) Extracellular signaling from the epithelial stroma is a key to guide the multiple stages of αβ T cell development in the thymus. Notch signaling during the early double-negative (DN) stages of T cell development inhibits potential for other lineages, such as B cell and myeloid cells. After thymocytes successfully pass β-selection, Notch signaling is turned off as a consequence of pre-T cell receptor signaling, and subsequent stages of T cell development exhibit very low levels of Notch signaling. Meanwhile IL-7 signaling at this particular stage is critical for the survival and the proliferation of β-selected cells. IL-7 signaling is terminated in immature CD4+CD8+ double-positive (DP) thymocytes through downregulation of receptor, which is re-expressed in postpositive selection CD4+CD8lo intermediate cells, which not only provides prosurvival signals but also cues for distinct T cell lineage differentiation. (B) Persistent T cell antigen receptor (TCR) signaling, which has been proposed to desensitize IL-7 signaling and to favor CD4 lineage differentiation, could be a critical environmental cue to instigate the expression and activity of ten eleven translocation (TET) enzymes at the Cd4 locus (right). In contrast, cessation of TCR signaling would allow IL-7R signaling in intermediate cells. This may contribute to CD8 lineage specification and may be a key signal to maintain the activity of DNA methyltransferases at the Cd4 locus and ensure methylation of the locus (left).
Given the sufficiency and requirement of Thpok in T helper cell differentiation, a series of studies were undertaken to characterize the cis elements regulating its expression (92, 140). A genomic region upstream of the Thpok distal promoter, the distal regulatory element (DRE), was found to drive expression in the CD4+CD8lo stage and in MHC class II selected cells. The DRE was subsequently dissected into two distinct elements, the Thpok silencer, which determined helper lineage specificity, and the thymic enhancer (TE), which drove expression early after positive selection (141). Interestingly, Thpok silencer deletion in mature CD8+ T cells did not cause derepression, indicating that Thpok was heritably silenced in cytotoxic T cells after thymic development, similar to the Cd4 locus (87, 142). DHS studies also revealed the presence of another region in the Thpok locus that lies 1.8 kb downstream of the proximal promoter of Thpok. Termed the proximal regulatory element (PRE), this region was more accessible in CD4 lineage cells. An enhancer within the PRE, referred to as the PE, was essential for efficient Thpok induction after MHC class II selection (92). Deletion of either the PE or the TE within the Thpok locus led to redirection of MHC class II-selected cells toward the cytotoxic lineage (92, 141). The general T lymphoid element (GTE), a region between the DRE and PRE, was found to direct reporter expression in both helper and cytotoxic T cell lineages in transgenic mice (140). Thus, Thpok expression is regulated by at least three enhancers and one silencer element. Several Runx motifs were found within the Thpok silencer, and their deletion in the germline revealed critical roles for Runx complexes in silencing Thpok (140, 143). Furthermore, mice with mutations in Runx1 and Runx3, or in the obligate Runx binding partner Cbfb, showed Thpok derepression in preselection DP thymocytes and loss of the cytotoxic lineage. Interestingly, Runx complexes were bound to the Thpok silencer in both CD4+ and CD8+ T cells, suggesting that Runx localization to the silencer was insufficient to repress Thpok (143). Indeed, Thpok was also bound to its own silencer, where it may antagonize Runx-mediated repression in helper T cells, thereby acting in a positive feedback loop.
Runx
The Runx family is composed of three evolutionally conserved transcription factors, Runx1, Runx2, and Runx3, which have important developmental roles including hematopoiesis, osteogenesis, and neurogenesis (90). Mammalian Runx genes make use of two promoters, a distal promoter (P1) and a proximal promoter (P2), to generate different transcripts. Expression of Runx1 and Runx3 in T cells is driven by distal P1-promoter activity (138, 144). Structurally, Runx proteins contain two conserved domains, an N-terminal DNA-binding Runt domain and the C-terminal VWRPY penta-peptide motif (145). The Runt domain binds to core-binding subunit-β (Cbfb) that is required for stabilizing the interaction of Runx proteins with DNA. The VWRPY motif is thought to act as a docking module for the Groucho/TLE co-repressor protein family, which is required for Runx-mediated repression of multiple genes. As discussed above, Runx1 and Runx3 are critical for CD4 silencing at the DN and CD8SP stages, respectively. Interestingly, silencing activity of Runx3 at the Cd4 locus depends on the VWRPY motif, and mice expressing mutant Runx1 and Runx3 lacking this motif showed complete CD4 derepression in CD8+ T cells (146, 147). In contrast, the Thpok silencer remained partly functional in the absence of both Runx VWRPY motifs, suggesting that these factors employ different modes of transcriptional regulation of Cd4 at different stages of development (147). Consistent with this observation, VWRPY-mediated recruitment of the co-repressor TLE2 was enriched at S4 relative to the Thpok silencer (147). VWRPY-independent silencing might involve Runx association with other repressive complexes. For instance, Runx proteins have been shown to associate with HDACs and Sin3A and thymic deletion of Sin3a in mice impairs the development of CD8+ T cells, reminiscent of Runx-deficient mice (143, 148–150). In addition to the Groucho and TLE co-repressor complexes, Runx1 was shown to interact with HDACs and SUV39H1 to repress transcription by way of a domain distinct from the VWRPY penta-peptide motif (151). However, whether this association plays a role in the epigenetic silencing of Cd4 or lineage commitment is unknown.
Although S4 is required for the proper methylation of the Cd4 locus in CD8+ T cells, it is presently unclear whether Runx3 plays a role in mediating methylation changes in CD8+ T cells. As the selection of CD8+ T cells has been proposed to be promoted by downregulation of CD8 expression at the CD4+CD8lo stage, due to transient reduction of avidity of TCR interaction with MHC class I complexes (7), it is tempting to speculate that TET enzymes are not recruited to the Cd4 locus in CD8+ T cells as a result of reduced TCR signaling. Alternatively, IL-7R signaling in MHC class I selected T cells may inhibit TET enzyme recruitment/activity by way of yet to be determined factors (Figure 6).
Runx complexes are also important for promoting CD8 expression during T cell development. ChIP experiments have revealed binding of Runx complexes to E8I, E8II, and E8IV in CD8SP and DP thymocytes (94). Runx1 deficiency reduced CD8 expression in DP thymocytes, while Runx3 deficiency reduced expression in CD8SP thymocytes (91, 152). In the absence of Runx3 or Cbfb, CD8 expression was also unstable in peripheral CD8+ T cells activated ex vivo (109). However, inactivation of Runx complexes in mature CD8+ T cells did not lead to loss of CD8 expression, suggesting that Runx complexes prime the Cd8 locus through enhancer activity during development, enabling heritable activation in peripheral T cells. The mechanisms by which Runx proteins mediate the heritable activation of CD8 expression in mature cytotoxic T cells and coordinate deposition of active chromatin marks is not known. In addition to Cd4 silencing and Cd8 activation, Runx proteins have multiple other important roles in T cell development. Deletion of Runx1 impaired β-selection, positive selection, and the survival of the helper lineage, while Runx3 has been shown to be essential for efficient CD8+ T cell differentiation by repressing CD4+ lineage genes such as Thpok and activating cytotoxic lineage genes such as those encoding perforin, granzyme B, and CD103 (143, 146, 152, 153). However, ectopic expression of Runx3 was insufficient to induce CD8+ lineage redirection, suggesting that it may be epistatic to other factors induced by MHC class II selection (11, 143).
Tcf1 and Lef1
T cell factor 1 and Lef1 (encoded by Tcf7 and Lef1 genes, respectively) are HMG transcription factors of the Tcf/Lef family that control key steps in development during T cell maturation (154). Notch signaling in early thymic progenitors induces expression of Tcf1, which then employs various mechanisms to ensure T cell lineage commitment, including promoting β-selection at the CD4−CD8− double-negative 3 stage and preventing malignant transformation (Figure 6, top) (155, 156). It was recently shown that Tcf1 and Lef1 are required for CD4+ lineage commitment, and a deficiency of Tcf1 and Lef1 results in lineage redirection of MHC class II selected cells into CD8+ T cells (157). This is in part mediated by direct regulation of Thpok as Tcf1 binds the GTE in the Thpok locus (140). In agreement with Tcf1 acting upstream of Thpok, ectopic expression of Thpok rescued the CD4+ T cell defect in Tcf7−/−Lef1−/− mice. Notably, the expression of additional transcription factors important for CD4+ T cell differentiation, GATA3, Myb, and Tox, was unchanged in the absence of Tcf1 and Lef1 (157).
Tcf7−/−Lef1−/− mice also showed derepression of CD4 in CD8SP cells, without changes in Runx3 expression. Tcf1 co-immunoprecipitated with Runx3, and ChIP analysis showed binding to S4. Mechanistically, Tcf1 likely cooperates with Runx complexes to silence CD4 as loss of Runx3 together with Tcf1 and Lef1 led to increased derepression of Cd4. Although Lef/Tcf members are known to interact with Groucho/TLE, physical interaction between Tcf1 and Runx3 was shown to occur independently of Groucho-TLE (158–160). Interestingly, an intrinsic HDAC domain in Tcf1 and Lef1 was required for the repression of genes associated with the CD4+ lineage, including Cd4, Cd40lg, FoxP3, and Rorc in CD8SP cells (158). Purified Tcf1 caused deacetylation of both H3K9Ac and H3K27Ac protein substrates in vitro, and this activity was abrogated in mutants lacking the putative 30 amino acid HDAC domain. In vivo, about 80% of Tcf1 target genes in CD8SP cells had elevated H3K27Ac and H3K9Ac in the absence of Tcf1 and Lef1. However, such marks were also found in a proportion of upregulated genes that did not harbor Tcf1-binding sites, suggesting indirect changes in acetylation as well. Furthermore, association with canonical HDACs could be responsible for deacetylation of certain genes, as physical interaction of Lef1 with HDAC1 was previously demonstrated (161). Tcf1-HDAC activity may also modify non-histone substrates. As Runx3 was found acetylated in some cancers, Tcf1 may act directly on Runx3 to modulate CD4 silencing (162, 163). It will be interesting to determine how Tcf1 and Lef1 HDAC activity is regulated, particularly with regards to target genes it activates, as deacetylation is typically associated with silencing.
Perspectives and Future Directions
In this review, we have emphasized how the studies of Cd4 and Cd8 regulation have revealed key insights into epigenetic mechanisms that mediate lineage commitment and maintain gene expression patterns that determine cell identity. The dynamic expression pattern of the co-receptor genes, from transient expression in DP thymocytes to stable expression or repression in CD4SP and CD8SP cells, provides a tractable system to understand mechanisms of heritability. Although cis elements are required for transcriptional control of Cd4, they are also required to direct epigenetic marks essential for heritable transmission in a stage-dependent manner. Recent discoveries have highlighted a key role for these elements in modulating DNA methylation changes that are key for heritable CD4 expression and histone acetylation in controlling dynamic CD8 expression. Excitingly, these studies have opened doors to understanding how antagonistic epigenetic processes can be co-regulated. For instance, it remains unclear how S4 inhibits DNA demethylation of the Cd4 locus while entraining the Dnmt enzymes in CD8+ T cells and how the Cd4 cis elements direct demethylation through the TET enzymes. The mechanisms regulating HDAC activity at the Cd8 locus for silencing expression in the CD4+ lineage also warrants additional study. Moreover, other branches shaping the epigenetic landscape have yet to be explored. Although the lncRNA transcriptome across bone marrow and thymic progenitors has been sequenced in humans, the functional contribution of these and other ncRNAs in T cell lineage commitment remains largely unexplored (164). Thus, it will be interesting to examine how lncRNA, enhancer RNA, microRNA, and other ncRNA species participate in lineage commitment and modulate chromatin at lineage-specific gene loci such as Cd4, Cd8, and Thpok. Furthermore, DNA demethylation at the Cd8 locus and its relationship with the CD8 cis elements is yet to be determined. The contribution of histone variants and their chaperones to gene expression is also likely to advance our understanding of lineage commitment, as is more detailed mapping of chromatin interactions between the different cis elements at the Cd4 and Cd8 loci. With advances in technology such as CRISPR/Cas9 genome editing, single-cell sequencing and locus-specific manipulation of chromatin, the pieces of the puzzle of heritability of gene expression can now begin to be assembled.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
DRL is an Investigator of the Howard Hughes Medical Institute. PDAI is the recipient of a fellowship from the Cancer Research Institute.
Abbreviations
E4M, maturity enhancer; E4P, proximal enhancer; S4, silencer; E4D, distal enhancer; E4T, thymocyte enhancer; Thpok, T helper inducing POZ/Krueppel-like factor; DNMT, DNA methyltransferase; MDB, methyl-CpG-binding domain; TET, ten eleven translocation; 5mC, 5-methyl cytosine; 5hC, 5-hydroxymethyl cytosine; HAT, histone acetyltransferase; HDAC, histone deacetylase enzyme; HP1, heterochromatin protein 1; DP, double positive; DN, double negative; CD4 SP, CD4 single positive; CD8 SP, CD8 single positive; Tcf1, T cell factor 1; Lef1, lymphoid enhancer factor 1; BAF, Brg/Brahma-associated factors; Uhrf1, ubiquitin like with PHD and ring finger domains 1; NuRD, nucleosome remodeling and histone deacetylase; AP4, adaptor-related protein complex 4.
References
3. Schulze SR, Wallrath LL. Gene regulation by chromatin structure: paradigms established in Drosophila melanogaster. Annu Rev Entomol (2007) 52:171–92. doi:10.1146/annurev.ento.51.110104.151007
4. Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell (2007) 28:1–13. doi:10.1016/j.molcel.2007.09.011
5. Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol (2008) 8(10):788–801. doi:10.1038/nri2416
6. Littman DR. How thymocytes achieve their fate. J Immunol (2016) 196:1983–4. doi:10.4049/jimmunol.1600032
7. Brugnera E, Bhandoola A, Cibotti R, Yu Q, Guinter TI, Yamashita Y, et al. Coreceptor reversal in the thymus: signaled CD4+8+ thymocytes initially terminate CD8 transcription even when differentiating into CD8+ T cells. Immunity (2000) 13(1):59–71. doi:10.1016/S1074-7613(00)00008-X
8. Egawa T. Regulation of CD4 and CD8 coreceptor expression and CD4 versus CD8 lineage decisions. Adv Immunol (2015) 125:1–40. doi:10.1016/bs.ai.2014.09.001
9. Taniuchi I. Views on helper/cytotoxic lineage choice from a bottom-up approach. Immunol Rev (2016) 271(1):98–113. doi:10.1111/imr.12401
10. Vacchio MS, Bosselut R. What happens in the thymus does not stay in the thymus: how T cells recycle the CD4+-CD8+ lineage commitment transcriptional circuitry to control their function. J Immunol (2016) 196(12):4848–56. doi:10.4049/jimmunol.1600415
11. Grueter B, Petter M, Egawa T, Laule-Kilian K, Aldrian CJ, Wuerch A, et al. Runx3 regulates integrin E/CD103 and CD4 expression during development of CD4-/CD8+ T cells. J Immunol (2005) 175(3):1694–705. doi:10.4049/jimmunol.175.9.6238
12. Wang L, Wildt KF, Zhu J, Zhang X, Feigenbaum L, Tessarollo L, et al. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4(+) T cells. Nat Immunol (2008) 9(10):1122–30. doi:10.1038/ni.1647
13. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol (2014) 6:a019133. doi:10.1101/cshperspect.a019133
14. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell (1999) 99:247–57. doi:10.1016/S0092-8674(00)81656-6
15. Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development (2002) 129:1983–93.
16. Bostick M, Kim JK, Estève PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science (2007) 317(5845):1760–4. doi:10.1126/science.1147939
17. Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature (2007) 450:908–12. doi:10.1038/nature06397
18. Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci U S A (1983) 80:5559–63. doi:10.1073/pnas.80.18.5559
19. Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci (2014) 39:310–8. doi:10.1016/j.tibs.2014.05.002
20. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Developmental clocks may depend on the enzymic modification of specific bases in repeated DNA sequences. Science (1975) 187:226–32. doi:10.1126/science.1111098
21. Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Genome Res (1975) 14:9–25. doi:10.1159/000130315
22. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet (2003) 33(Suppl):245–54. doi:10.1038/ng1089
23. Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature (2000) 405:482–5. doi:10.1038/35013100
24. Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet (1999) 23:58–61. doi:10.1038/12659
25. Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev (2001) 15:827–32. doi:10.1101/gad.876201
26. Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature (2010) 466:388–92. doi:10.1038/nature09147
27. Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res (2013) 23:1256–69. doi:10.1038/cr.2013.110
28. Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature (2000) 403(6769):501–2. doi:10.1038/35000656
29. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol (2010) 11(9):607–20. doi:10.1038/nrm2950
30. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324(5929):930–5. doi:10.1126/science.1170116
31. Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science (2009) 324(5929):929–30. doi:10.1126/science.1169786
32. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (2011) 333(6047):1300–3. doi:10.1126/science.1210597
33. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature (2013) 502(7472):472–9. doi:10.1038/nature12750
34. Ko M, An J, Bandukwala HS, Chavez L, Aijö T, Pastor WA, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature (2013) 497(7447):122–6. doi:10.1038/nature12052
35. Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science (2011) 334(6053):194. doi:10.1126/science.1212483
36. Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res (2011) 21(12):1670–6. doi:10.1038/cr.2011.189
37. Gehring M, Reik W, Henikoff S. DNA demethylation by DNA repair. Trends Genet (2009) 25(2):82–90. doi:10.1016/j.tig.2008.12.001
38. Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet (2009) 43:143–66. doi:10.1146/annurev-genet-102108-134205
39. Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev (2014) 28(7):672–82. doi:10.1101/gad.238873.114
40. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol (2015) 16:178–89. doi:10.1038/nrm3941
41. Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell (1996) 84:843–51. doi:10.1016/S0092-8674(00)81063-6
42. Kleff S, Andrulis ED, Anderson CW, Sternglanz R. Identification of a gene encoding a yeast histone H4 acetyltransferase. J Biol Chem (1995) 270:24674–7. doi:10.1074/jbc.270.42.24674
43. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science (1996) 272:408–11. doi:10.1126/science.272.5260.408
44. Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet (2010) 11:285–96. doi:10.1038/nrg2752
45. Nady N, Lemak A, Walker JR, Avvakumov GV, Kareta MS, Achour M, et al. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J Biol Chem (2011) 286:24300–11. doi:10.1074/jbc.M111.234104
46. Fang J, Cheng J, Wang J, Zhang Q, Liu M, Gong R, et al. Hemi-methylated DNA opens a closed conformation of UHRF1 to facilitate its histone recognition. Nat Commun (2016) 7:11197. doi:10.1038/ncomms11197
47. Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature (2013) 502(7470):249–53. doi:10.1038/nature12488
48. Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol (2006) 8(2):188–94. doi:10.1038/ncb1353
49. Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol (2008) 15(11):1176–83. doi:10.1038/nsmb.1476
50. Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J (2008) 27(20):2691–701. doi:10.1038/emboj.2008.193
51. Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature (2001) 414:277–83. doi:10.1038/35104508
52. Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature (2011) 469:343–9. doi:10.1038/nature09784
53. Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet (2011) 12(2):123–35. doi:10.1038/nrg2932
54. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet (2012) 13:343–57. doi:10.1038/nrg3173
55. Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature (2007) 448:714–7. doi:10.1038/nature05987
56. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem (2009) 78:273–304. doi:10.1146/annurev.biochem.77.062706.153223
57. Ho L, Crabtree GR. Chromatin remodelling during development. Nature (2010) 463:474–84. doi:10.1038/nature08911
58. Maze I, Noh KM, Soshnev AA, Allis CD. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat Rev Genet (2014) 15:259–71. doi:10.1038/nrg3673
59. Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell (2010) 140:678–91. doi:10.1016/j.cell.2010.01.003
60. Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell (2002) 9:1191–200. doi:10.1016/S1097-2765(02)00542-7
61. Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science (2002) 297(5588):1833–7. doi:10.1126/science.1074973
62. Pal-Bhadra M, Leibovitch BA, Gandhi SG, Chikka MR, Bhadra U, Birchler JA, et al. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science (2004) 303(5658):669–72. doi:10.1126/science.1092653
63. Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature (2007) 447(7143):418–24. doi:10.1038/nature05917
64. Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ. Characterization of dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci U S A (2005) 102(34):12135–40. doi:10.1073/pnas.0505479102
65. Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev (2005) 19(4):489–501. doi:10.1101/gad.1248505
66. Wang SH, Elgin SC. Drosophila Piwi functions downstream of piRNA production mediating a chromatin-based transposon silencing mechanism in female germ line. Proc Natl Acad Sci U S A (2011) 108(52):21164–9. doi:10.1073/pnas.1107892109
67. Brower-Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, et al. Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes Dev (2007) 21(18):2300–11. doi:10.1101/gad.1564307
68. Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science (2011) 332(6031):848–52. doi:10.1126/science.1203919
69. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science (2008) 322(5902):750–6. doi:10.1126/science.1163045
70. Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell (2011) 146(1):119–33. doi:10.1016/j.cell.2011.06.026
71. Lee JT. Epigenetic regulation by long noncoding RNAs. Science (2012) 338(6113):1435–9. doi:10.1126/science.1231776
72. Adlam M, Siu G. Hierarchical interactions control CD4 gene expression during thymocyte development. Immunity (2003) 18(2):173–84. doi:10.1016/S1074-7613(03)00021-9
73. Chong MM, Simpson N, Ciofani M, Chen G, Collins A, Littman DR. Epigenetic propagation of CD4 expression is established by the Cd4 proximal enhancer in helper T cells. Genes Dev (2010) 24(7):659–69. doi:10.1101/gad.1901610
74. Wurster AL, Siu G, Leiden JM, Hedrick SM. Elf-1 binds to a critical element in a second CD4 enhancer. Mol Cell Biol (1994) 14(10):6452–63. doi:10.1128/MCB.14.10.6452
75. Henson DM, Chou C, Sakurai N, Egawa T. A silencer-proximal intronic region is required for sustained CD4 expression in postselection thymocytes. J Immunol (2014) 192(10):4620–7. doi:10.4049/jimmunol.1302374
76. Sawada S, Littman DR. Identification and characterization of a T-cell-specific enhancer adjacent to the murine CD4 gene. Mol Cell Biol (1991) 11(11):5506–15. doi:10.1128/MCB.11.11.5506
77. Sawada S, Scarborough JD, Killeen N, Littman DR. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell (1994) 77(6):917–29. doi:10.1016/0092-8674(94)90140-6
78. Sawada S, Littman DR. A heterodimer of HEB and an E12-related protein interacts with the CD4 enhancer and regulates its activity in T-cell lines. Mol Cell Biol (1993) 13(9):5620–8. doi:10.1128/MCB.13.9.5620
79. Zhuang Y, Cheng P, Weintraub H. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2-2, and HEB. Mol Cell Biol (1996) 16(6):2898–905. doi:10.1128/MCB.16.6.2898
80. Barndt R, Dai MF, Zhuang Y. A novel role for HEB downstream or parallel to the pre-TCR signaling pathway during alpha beta thymopoiesis. J Immunol (1999) 163(6):3331–43.
81. Huang Z, Xie H, Ioannidis V, Held W, Clevers H, Sadim MS, et al. Transcriptional regulation of CD4 gene expression by T cell factor-1/beta-catenin pathway. J Immunol (2006) 176:4880–7. doi:10.4049/jimmunol.176.8.4880
82. Chi TH, Wan M, Zhao K, Taniuchi I, Chen L, Littman DR, et al. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature (2002) 418(6894):195–9. doi:10.1038/nature00876
83. Williams CJ, Naito T, Arco PG, Seavitt JR, Cashman SM, De Souza B, et al. The chromatin remodeler Mi-2beta is required for CD4 expression and T cell development. Immunity (2004) 20:719–33. doi:10.1016/j.immuni.2004.05.005
84. Manjunath N, Shankar P, Stockton B, Dubey PD, Lieberman J, von Andrian UH. A transgenic mouse model to analyze CD8(+) effector T cell differentiation in vivo. Proc Natl Acad Sci U S A (1999) 96(24):13932–7. doi:10.1073/pnas.96.24.13932
85. Siu G, Wurster AL, Duncan DD, Soliman TM, Hedrick SM. A transcriptional silencer controls the developmental expression of the CD4 gene. EMBO J (1994) 13(15):3570–9.
86. Leung RK, Thomson K, Gallimore A, Jones E, Van den Broek M, Sierro S, et al. Deletion of the CD4 silencer element supports a stochastic mechanism of thymocyte lineage commitment. Nat Immunol (2001) 2(12):1167–73. doi:10.1038/ni733
87. Zou YR, Sunshine MJ, Taniuchi I, Hatam F, Killeen N, Littman DR. Epigenetic silencing of CD4 in T cells committed to the cytotoxic lineage. Nat Genet (2001) 29(3):332–6. doi:10.1038/ng750
88. Taniuchi I, Sunshine MJ, Festenstein R, Littman DR. Evidence for distinct CD4 silencer functions at different stages of thymocyte differentiation. Mol Cell (2002) 10:1083–96. doi:10.1016/S1097-2765(02)00735-9
89. Elgin SC, Reuter G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb Perspect Biol (2013) 5(8):a017780. doi:10.1101/cshperspect.a017780
90. Levanon D, Groner Y. Structure and regulated expression of mammalian RUNX genes. Oncogene (2004) 23(24):4211–9. doi:10.1038/sj.onc.1207670
91. Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell (2002) 111(5):621–33. doi:10.1016/S0092-8674(02)01111-X
92. Muroi S, Naoe Y, Miyamoto C, Akiyama K, Ikawa T, Masuda K, et al. Cascading suppression of transcriptional silencers by ThPOK seals helper T cell fate. Nat Immunol (2008) 9(10):1113–21. doi:10.1038/ni.1650
93. Yu M, Wan M, Zhang J, Wu J, Khatri R, Chi T. Nucleoprotein structure of the CD4 locus: implications for the mechanisms underlying CD4 regulation during T cell development. Proc Natl Acad Sci U S A (2008) 105:3873–8. doi:10.1073/pnas.0800810105
94. Sato T, Ohno S, Hayashi T, Sato C, Kohu K, Satake M, et al. Dual functions of Runx proteins for reactivating CD8 and silencing CD4 at the commitment process into CD8 thymocytes. Immunity (2005) 22(3):317–28. doi:10.1016/j.immuni.2005.01.012
95. Wan M, Zhang J, Lai D, Jani A, Prestone-Hurlburt P, Zhao L, et al. Molecular basis of CD4 repression by the Swi/Snf-like BAF chromatin remodeling complex. Eur J Immunol (2009) 39:580–8. doi:10.1002/eji.200838909
96. Naito T, Gómez-Del Arco P, Williams CJ, Georgopoulos K. Antagonistic interactions between Ikaros and the chromatin remodeler Mi-2beta determine silencer activity and Cd4 gene expression. Immunity (2007) 27(5):723–34. doi:10.1016/j.immuni.2007.09.008
97. Jiang H, Peterlin BM. Differential chromatin looping regulates CD4 expression in immature thymocytes. Mol Cell Biol (2008) 28(3):907–12. doi:10.1128/MCB.00909-07
98. Egawa T, Littman DR. Transcription factor AP4 modulates reversible and epigenetic silencing of the Cd4 gene. Proc Natl Acad Sci U S A (2011) 108(36):14873–8. doi:10.1073/pnas.1112293108
99. Collins A, Hewitt SL, Chaumeil J, Sellars M, Micsinai M, Allinne J, et al. RUNX transcription factor-mediated association of Cd4 and Cd8 enables coordinate gene regulation. Immunity (2011) 34(3):303–14. doi:10.1016/j.immuni.2011.03.004
100. Sellars M, Huh JR, Day K, Issuree PD, Galan C, Gobeil S, et al. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat Immunol (2015) 16(7):746–54. doi:10.1038/ni.3198
101. Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One (2010) 5:e8888. doi:10.1371/journal.pone.0008888
102. Tsagaratou A, Äijö T, Lio CW, Yue X, Huang Y, Jacobsen SE, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A (2014) 111(32):E3306–15. doi:10.1073/pnas.1412327111
103. Hostert A, Tolaini M, Festenstein R, McNeill L, Malissen B, Williams O, et al. A CD8 genomic fragment that directs subset-specific expression of CD8 in transgenic mice. J Immunol (1997) 158(9):4270–81.
104. Hostert A, Tolaini M, Roderick K, Harker N, Norton T, Kioussis D. A region in the CD8 gene locus that directs expression to the mature CD8 T cell subset in transgenic mice. Immunity (1997) 7(4):525–36. doi:10.1016/S1074-7613(00)80374-X
105. Ellmeier W, Sunshine MJ, Losos K, Hatam F, Littman DR. An enhancer that directs lineage-specific expression of CD8 in positively selected thymocytes and mature T cells. Immunity (1997) 7(4):537–47. doi:10.1016/S1074-7613(00)80375-1
106. Ellmeier W, Sunshine MJ, Losos K, Littman DR. Multiple developmental stage-specific enhancers regulate CD8 expression in developing thymocytes and in thymus-independent T cells. Immunity (1998) 9(4):485–96. doi:10.1016/S1074-7613(00)80632-9
107. Hostert A, Garefalaki A, Mavria G, Tolaini M, Roderick K, Norton T, et al. Hierarchical interactions of control elements determine CD8alpha gene expression in subsets of thymocytes and peripheral T cells. Immunity (1998) 9(4):497–508. doi:10.1016/S1074-7613(00)80633-0
108. Sakaguchi S, Hombauer M, Hassan H, Tanaka H, Yasmin N, Naoe Y, et al. A novel Cd8-cis-regulatory element preferentially directs expression in CD44hiCD62L+ CD8+ T cells and in CD8alphaalpha+ dendritic cells. J Leukoc Biol (2015) 97(4):635–44. doi:10.1189/jlb.1HI1113-597RR
109. Hassan H, Sakaguchi S, Tenno M, Kopf A, Boucheron N, Carpenter AC, et al. Cd8 enhancer E8I and Runx factors regulate CD8 expression in activated CD8+ T cells. Proc Natl Acad Sci U S A (2011) 108:18330–5. doi:10.1073/pnas.1105835108
110. Ellmeier W, Sunshine MJ, Maschek R, Littman DR. Combined deletion of CD8 locus cis-regulatory elements affects initiation but not maintenance of CD8 expression. Immunity (2002) 16(5):623–34. doi:10.1016/S1074-7613(02)00309-6
111. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nature reviews. Immunology (2011) 11:445–56. doi:10.1038/nri3007
112. Garefalaki A, Coles M, Hirschberg S, Mavria G, Norton T, Hostert A, et al. Variegated expression of CD8 alpha resulting from in situ deletion of regulatory sequences. Immunity (2002) 16(5):635–47. doi:10.1016/S1074-7613(02)00308-4
113. Feik N, Bilic I, Tinhofer J, Unger B, Littman DR, Ellmeier W. Functional and molecular analysis of the double-positive stage-specific CD8 enhancer E8III during thymocyte development. J Immunol (2005) 174(3):1513–24. doi:10.4049/jimmunol.174.3.1513
114. Lee WH, Banan M, Harriss JV, Hwang I, Woodward E, Youn HJ, et al. cis-acting DNA elements and cell type-specific nuclear proteins which may play a role in regulation of mouse CD8α (Lyt-2) gene transcription. Int Immunol (1994) 6:1307–21. doi:10.1093/intimm/6.9.1307
115. Banan M, Rojas IC, Lee W-H, King HL, Harriss JV, Kobayashi R, et al. Interaction of the nuclear matrix-associated region (MAR)-binding proteins, SATB1 and CDP/Cux, with a MAR element (L2a) in an upstream regulatory region of the mouse CD8a gene. J Biol Chem (1997) 272:18440–52. doi:10.1074/jbc.272.29.18440
116. Yao X, Nie H, Rojas IC, Harriss JV, Maika SD, Gottlieb PD, et al. The L2a element is a mouse CD8 silencer that interacts with MAR-binding proteins SATB1 and CDP. Mol Immunol (2010) 48:153–63. doi:10.1016/j.molimm.2010.08.014
117. Jenkinson SR, Intlekofer AM, Sun G, Feigenbaum L, Reiner SL, Bosselut R. Expression of the transcription factor cKrox in peripheral CD8 T cells reveals substantial postthymic plasticity in CD4-CD8 lineage differentiation. J Exp Med (2007) 204(2):267–72. doi:10.1084/jem.20061982
118. Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol (2013) 14(3):281–9. doi:10.1038/ni.2523
119. Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat Immunol (2013) 14(3):271–80. doi:10.1038/ni.2518
120. O’Neill DW, Schoetz SS, Lopez RA, Castle M, Rabinowitz L, Shor E, et al. An ikaros-containing chromatin-remodeling complex in adult-type erythroid cells. Mol Cell Biol (2000) 20:7572–82. doi:10.1128/MCB.20.20.7572-7582.2000
121. Harker N, Naito T, Cortes M, Hostert A, Hirschberg S, Tolaini M, et al. The CD8alpha gene locus is regulated by the Ikaros family of proteins. Mol Cell (2002) 10(6):1403–15. doi:10.1016/S1097-2765(02)00711-6
122. Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity (1999) 10(3):345–55. doi:10.1016/S1074-7613(00)80034-5
123. Harker N, Garefalaki A, Menzel U, Ktistaki E, Naito T, Georgopoulos K, et al. Pre-TCR signaling and CD8 gene bivalent chromatin resolution during thymocyte development. J Immunol (2011) 186(11):6368–77. doi:10.4049/jimmunol.1003567
124. Bilic I, Koesters C, Unger B, Sekimata M, Hertweck A, Maschek R, et al. Negative regulation of CD8 expression via Cd8 enhancer-mediated recruitment of the zinc finger protein MAZR. Nat Immunol (2006) 7(4):392–400. doi:10.1038/ni1311
125. Boucheron N, Tschismarov R, Göschl L, Moser MA, Lagger S, Sakaguchi S, et al. CD4(+) T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat Immunol (2014) 15(5):439–48. doi:10.1038/ni.2864
126. Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity (2001) 15:763–74. doi:10.1016/S1074-7613(01)00227-8
127. Carbone AM, Marrack P, Kappler JW. Demethylated CD8 gene in CD4+ T cells suggests that CD4+ cells develop from CD8+ precursors. Science (1988) 242:1174–6. doi:10.1126/science.2460926
128. Hamerman A, Page ST, Pullen AM. Distinct methylation states of the C8beta gene in peripheral T cells and intraepithelial lymphocytes. J Immunol (1997) 159:1240–6.
129. Harland KL, Day EB, Apte SH, Russ BE, Doherty PC, Turner SJ, et al. Epigenetic plasticity of Cd8a locus during CD8(+) T-cell development and effector differentiation and reprogramming. Nat Commun (2014) 5:3547. doi:10.1038/ncomms4547
130. Sakaguchi S, Hombauer M, Bilic I, Naoe Y, Schebesta A, Taniuchi I, et al. The zinc-finger protein MAZR is part of the transcription factor network that controls the CD4 versus CD8 lineage fate of double-positive thymocytes. Nat Immunol (2010) 11(5):442–8. doi:10.1038/ni.1860
131. Sakaguchi S, Hainberger D, Tizian C, Tanaka H, Okuda T, Taniuchi I, et al. MAZR and Runx factors synergistically repress ThPOK during CD8+ T cell lineage development. J Immunol (2015) 195(6):2879–87. doi:10.4049/jimmunol.1500387
132. Dave VP, Allman D, Keefe R, Hardy RR, Kappes DJ. HD mice: a novel mouse mutant with a specific defect in the generation of CD4(+) T cells. Proc Natl Acad Sci U S A (1998) 95(14):8187–92. doi:10.1073/pnas.95.14.8187
133. He X, He X, Dave VP, Zhang Y, Hua X, Nicolas E, et al. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature (2005) 433(7028):826–33. doi:10.1038/nature03338
134. Sun G, Liu X, Mercado P, Jenkinson SR, Kypriotou M, Feigenbaum L, et al. The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nat Immunol (2005) 6(4):373–81. doi:10.1038/ni1183
135. Wildt KF, Sun G, Grueter B, Fischer M, Zamisch M, Ehlers M, et al. The transcription factor Zbtb7b promotes CD4 Expression by antagonizing Runx-mediated activation of the CD4 silencer. J Immunol (2007) 179(7):4405–14. doi:10.4049/jimmunol.179.7.4405
136. Vacchio MS, Wang L, Bouladoux N, Carpenter AC, Xiong Y, Williams LC, et al. A ThPOK-LRF transcriptional node maintains the integrity and effector potential of post-thymic CD4+ T cells. Nat Immunol (2014) 15(10):947–56. doi:10.1038/ni.2960
137. Wang L, Wildt KF, Castro E, Xiong Y, Feigenbaum L, Tessarollo L, et al. The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity (2008) 29(6):876–87. doi:10.1016/j.immuni.2008.09.019
138. Egawa T, Littman DR. ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nat Immunol (2008) 9(10):1131–9. doi:10.1038/ni.1652
139. Rui J, Liu H, Zhu X, Cui Y, Liu X. Epigenetic silencing of CD8 genes by ThPOK-mediated deacetylation during CD4 T cell differentiation. J Immunol (2012) 189(3):1380–90. doi:10.4049/jimmunol.1201077
140. He X, Park K, Wang H, He X, Zhang Y, Hua X, et al. CD4-CD8 lineage commitment is regulated by a silencer element at the ThPOK transcription-factor locus. Immunity (2008) 28(3):346–58. doi:10.1016/j.immuni.2008.02.006
141. Muroi S, Tanaka H, Miyamoto C, Taniuchi I. Cutting edge: fine-tuning of Thpok gene activation by an enhancer in close proximity to its own silencer. J Immunol (2013) 190(4):1397–401. doi:10.4049/jimmunol.1203006
142. Tanaka H, Naito T, Muroi S, Seo W, Chihara R, Miyamoto C, et al. Epigenetic Thpok silencing limits the time window to choose CD4(+) helper-lineage fate in the thymus. EMBO J (2013) 32(8):1183–94. doi:10.1038/emboj.2013.47
143. Setoguchi R, Tachibana M, Naoe Y, Muroi S, Akiyama K, Tezuka C, et al. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science (2008) 319:822–5. doi:10.1126/science.1151844
144. Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F, et al. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med (2007) 204:1749–55. doi:10.1084/jem.20062456
145. Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer (2015) 15(2):81–95. doi:10.1038/nrc3877
146. Yarmus M, Woolf E, Bernstein Y, Fainaru O, Negreanu V, Levanon D, et al. Groucho/transducin-like enhancer-of-split (TLE)-dependent and -independent transcriptional regulation by Runx3. Proc Natl Acad Sci U S A (2006) 103(19):7384–9. doi:10.1073/pnas.0602470103
147. Seo W, Tanaka H, Miyamoto C, Levanon D, Groner Y, Taniuchi I. Roles of VWRPY motif-mediated gene repression by Runx proteins during T-cell development. Immunol Cell Biol (2012) 90(8):827–30. doi:10.1038/icb.2012.6
148. Cowley SM, Iritani BM, Mendrysa SM, Xu T, Cheng PF, Yada J, et al. The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol Cell Biol (2005) 25(16):6990–7004. doi:10.1128/MCB.25.16.6990-7004.2005
149. Fenrick R, Amann JM, Lutterbach B, Wang L, Westendorf JJ, Downing JR, et al. Both TEL and AML-1 contribute repression domains to the t(12;21) fusion protein. Mol Cell Biol (1999) 19(10):6566–74. doi:10.1128/MCB.19.10.6566
150. Durst KL, Lutterbach B, Kummalue T, Friedman AD, Hiebert SW. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol (2003) 23(2):607–19. doi:10.1128/MCB.23.2.607-619.2003
151. Reed-Inderbitzin E, Moreno-Miralles I, Vanden-Eynden SK, Xie J, Lutterbach B, Durst-Goodwin KL, et al. RUNX1 associates with histone deacetylases and SUV39H1 to repress transcription. Oncogene (2006) 25(42):5777–86. doi:10.1038/sj.onc.1209591
152. Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med (2007) 204(8):1945–57. doi:10.1084/jem.20070133
153. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med (2009) 206:51–9. doi:10.1084/jem.20081242
154. Steinke FC, Xue HH. From inception to output, Tcf1 and Lef1 safeguard development of T cells and innate immune cells. Immunol Res (2014) 59:45–55. doi:10.1007/s12026-014-8545-9
155. Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to notch signaling. Proc Natl Acad Sci U S A (2011) 108(50):20060–5. doi:10.1073/pnas.1110230108
156. Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity (2012) 37(5):813–26. doi:10.1016/j.immuni.2012.08.009
157. Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, et al. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat Immunol (2014) 15(7):646–56. doi:10.1038/ni.2897
158. Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol (2016) 17(6):695–703. doi:10.1038/ni.3456
159. Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol (2005) 12(4):364–71. doi:10.1038/nsmb912
160. Brantjes H, Roose J, van De Wetering M, Clevers H. All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Res (2001) 29(7):1410–9. doi:10.1093/nar/29.7.1410
161. Billin AN, Thirlwell H, Ayer DE. Beta-catenin-histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol Cell Biol (2000) 20(18):6882–90. doi:10.1128/MCB.20.18.6882-6890.2000
162. Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science (2011) 333(6043):765–9. doi:10.1126/science.1201662
163. Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, et al. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J Biol Chem (2004) 279(28):29409–17. doi:10.1074/jbc.M313120200
Keywords: DNA methyltransferase, ten eleven translocation enzymes, T cell development, gene silencing, RUNX3, Runx1, T helper inducing POZ/Krueppel-like factor, TCF transcription factors
Citation: Issuree PDA, Ng CP and Littman DR (2017) Heritable Gene Regulation in the CD4:CD8 T Cell Lineage Choice. Front. Immunol. 8:291. doi: 10.3389/fimmu.2017.00291
Received: 21 December 2016; Accepted: 28 February 2017;
Published: 22 March 2017
Edited by:
Benjamin Youngblood, St. Jude Children’s Research Hospital, USAReviewed by:
Stephen Nutt, Walter and Eliza Hall Institute of Medical Research, AustraliaKimberly Sue Schluns, University of Texas MD Anderson Cancer Center, USA
Copyright: © 2017 Issuree, Ng and Littman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan R. Littman, ZGFuLmxpdHRtYW5AbWVkLm55dS5lZHU=
†These authors have contributed equally to this work.