 Jungmin Yang1
Jungmin Yang1 Kang-Mu Lee1Sangjun Park1Yoeseph Cho1,2Eunju Lee1Jong-Hwan Park3
Kang-Mu Lee1Sangjun Park1Yoeseph Cho1,2Eunju Lee1Jong-Hwan Park3 Ok Sarah Shin4Junghyun Son2
Ok Sarah Shin4Junghyun Son2 Sang Sun Yoon1
Sang Sun Yoon1 Je-Wook Yu1*
Je-Wook Yu1*
- 1Department of Microbiology and Immunology, Institute for Immunology and Immunological Diseases, Brain Korea 21 PLUS Project for Medical Science, Yonsei University College of Medicine, Seoul, South Korea
- 2Doping Control Center, Korea Institute of Science and Technology, Seoul, South Korea
- 3Laboratory Animal Medicine, College of Veterinary Medicine and BK 21 PLUS Project Team, Chonnam National University, Gwangju, South Korea
- 4Department of Biomedical Sciences, College of Medicine, Korea University Guro Hospital, Seoul, South Korea
Inflammasome signaling can contribute to host innate immune defense against bacterial pathogens such as Pseudomonas aeruginosa. However, bacterial evasion of host inflammasome activation is still poorly elucidated. Quorum sensing (QS) is a bacterial communication mechanism that promotes coordinated adaptation by triggering expression of a wide range of genes. QS is thought to strongly contribute to the virulence of P. aeruginosa, but the molecular impact of bacterial QS on host inflammasome defense is completely unknown. Here, we present evidence that QS-related factors of the bacterial secretant (BS) from P. aeruginosa can dampen host inflammasome signaling in mouse bone marrow-derived macrophages. We found that BS from QS-defective ΔlasR/rhlR mutant, but not from wild-type (WT) P. aeruginosa, induces robust activation of the NLRC4 inflammasome. P. aeruginosa-released flagellin mediates this inflammasome activation by ΔlasR/rhlR secretant, but QS-regulated bacterial proteases in the WT BS impair extracellular flagellin to attenuate NLRC4 inflammasome activation. P. aeruginosa-secreted proteases also degrade inflammasome components in the extracellular space to inhibit the propagation of inflammasome-mediated responses. Furthermore, QS-regulated virulence factor pyocyanin and QS autoinducer 3-oxo-C12-homoserine lactone directly suppressed NLRC4- and even NLRP3-mediated inflammasome assembly and activation. Taken together, our data indicate that QS system of P. aeruginosa facilitates bacteria to evade host inflammasome-dependent sensing machinery.
Introduction
The inflammasome, a cytoplasmic caspase-1-activating molecular complex, provides host with an innate immune defense against invading bacterial pathogens through induction of pyroptosis and production of pro-inflammatory cytokines such as interleukin-1 beta (IL-1β) and IL-18 (1). Multiple inflammasome sensor proteins are activated upon direct or indirect sensing of bacterial components in the cytoplasm (2). The cytosolic presence of bacterial flagellin or type 3 secretion system (T3SS)-associated proteins can promote the activation of the nucleotide-binding oligomerization domain-like receptor containing a caspase recruitment domain 4 (NLRC4) inflammasome with the engagement of neuronal apoptosis inhibitory protein, a direct sensor of bacterial proteins (3–5). In addition, bacterial double-stranded DNA (dsDNA) from phagocytosed bacteria is recognized in the cytoplasm by binding directly to absent in melanoma 2 (AIM2), which then assembles into the AIM2 inflammasome complex using dsDNA as a platform (6, 7). Furthermore, diverse bacterial toxins including pore-forming toxins (e.g., hemolysin, pneumolysin) and Clostridium difficile toxin B (TcdB) stimulate activation of NLRP3 and pyrin inflammasomes, respectively, by inducing intracellular alterations such as an efflux of potassium or the inactivation of Rho GTPases (8–10). Upon bacterial infections, these inflammasome sensor components assemble the inflammasome complex with ASC and procaspase-1 leading to caspase-1-dependent pyroptotic cell death and IL-1β or IL-18 secretion for efficient clearance of invading bacteria.
Pseudomonas aeruginosa is a Gram-negative, flagellated bacterium and an opportunistic pathogen (11). Immunocompromised hosts, such as cystic fibrosis patients, are likely to succumb to P. aeruginosa infection and suffer from chronic respiratory symptoms such as pneumonia (12, 13). This opportunistic bacterial pathogen produces multiple virulence factors involved in pathogenesis. Similar to other Gram-negative bacteria, P. aeruginosa uses T3SS to inject effector molecules such as exotoxins (e.g., ExoS, ExoU, ExoT, and ExoY) into the cytoplasm of host target cells (14). In this regard, the T3SS is required for P. aeruginosa virulence. Among the four previously mentioned exotoxins, ExoU is considered the most potent cytotoxin causing target cell death through its phospholipase activity (14). In addition to exotoxins, the flagella also contribute to successful P. aeruginosa pathogenesis through flagella-mediated bacterial motility and adhesion (15). Quorum sensing (QS), a form of bacterial cell-to-cell communication, also plays an important role in the pathogenicity of P. aeruginosa (16, 17). In the QS system of P. aeruginosa, N-acyl homoserine lactones (AHLs) such as N-3-oxo-dodecanoyl-l-homoserine lactone (3-oxo-C12-HSL) or N-butyryl-l-homoserine lactone (C4-HSL) are the most important communication signaling molecules and are referred to as autoinducer (18). Once the level of AHLs reaches a threshold because of increased bacterial communications, AHL binds to LasR or RhlR proteins. The resulting complex then acts as a transcription factor to induce the expression of many genes involved in the antibiotic production, biofilm formation, and virulence factors (18). However, the molecular impact of bacterial QS on the host innate immune response remains poorly described.
Innate immune defense against P. aeruginosa largely depends on the immediate recognition of essential bacterial virulence factors. In particular, monomeric flagellin, a basic component of flagella, is sensed by toll-like receptor 5 (TLR5) on the host cell surface leading to the production of pro-inflammatory cytokines (19). Furthermore, both flagellin and T3SS components such as PscC are required for P. aeruginosa-triggered activation of the NLRC4 inflammasome in a TLR5-independent manner (20, 21). Despite the critical role of bacterial QS in the virulence of P. aeruginosa, it remains poorly understood whether this bacterial communication signaling affects host inflammasome-mediated immune responses. Here, we explored a potential effect of QS in P. aeruginosa on inflammasome signaling and present molecular evidence that P. aeruginosa QS-related products could suppress the activation of inflammasome pathways.
Materials and Methods
Mice
C57BL/6 mice were purchased from Orient Bio. Nlrp3−/− mice were obtained from The Jackson Laboratory. Nlrc4−/− mice were kindly provided by Dr. G. Nunez (University of Michigan, Ann Arbor, MI, USA). All mice were on C57BL/6 background and 8–10 weeks male mice were used for the experiments. All mice were maintained under specific pathogen-free conditions.
Ethics Statement
Protocols for the animal experiments were approved by the Institutional Ethical Committee, Yonsei University College of Medicine (2013-0164). All experiments were performed in accordance with the approved guidelines of the Institutional Ethical Committee, adhered to Guide for the Care and Use of Laboratory Animals of National Research Council (USA).
Cell Cultures
Mouse primary bone marrow-derived macrophages (BMDMs) were differentiated from bone marrow cells isolated from mouse femurs. Immortalized wild-type (WT), Asc−/−, NLRP3-GFP-expressing BMDMs and ASC-GFP-expressing THP-1 cells were kindly provided by Dr. E. S. Alnemri (Thomas Jefferson University, Philadelphia, PA, USA). All BMDMs were maintained in L929 supplements-conditioned Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. THP-1-ASC-GFP cells were grown in RPMI-1640 supplemented with 10% FBS, 2 mM glutamine, 10 mM HEPES, 1 mM sodium pyruvate, and 0.05 mM 2-mercaptoethanol and antibiotics. THP-1-ASC-GFP cells were differentiated into macrophage-like cells using a phorbol 12-myristate 13-acetate treatment and then used for experiments. A549 cells were cultured in DMEM supplemented with 10% FBS and antibiotics. Cell death was determined by the extracellular release of lactate dehydrogenase using a CytoTox96 non-radioactive cytotoxicity assay kit (Promega).
Bacterial Cultures
Pseudomonas aeruginosa strain PAO1 was used. PAO1 deletion mutants (ΔlasR/rhlR, ΔlasR, ΔrhlR, ΔfliC, ΔlasR/rhlR/fliC, ΔlasR/fliC, and ΔlasB) were created by allelic replacement as previously described (22). For bacterial infection, all PAO1 strains were grown overnight in Luria-Bertani (LB) medium at 37°C with aeration, then diluted 1:50 with fresh LB medium, and grown for an additional 2 h. Bacteria were then pelleted by centrifugation and resuspended in Opti-MEM medium. The bacterial suspension was then added to the culture medium of BMDMs at the indicated multiplicity of infection. To prepare bacterial lysates, cultured PAO1 were lysed, sonicated, and centrifuged to remove insoluble pellets. The supernatants were normalized and used for immunoblotting.
Bacterial Secretants (BSs)
For preparation of BS, PAO1 strains were grown overnight in LB medium and then diluted with fresh LB medium and grown for an additional 6 h. The bacterial culture was then normalized by the number of bacteria. The normalized bacterial suspension was centrifuged and filtered to remove bacteria, and the final filtrate was used as BS. For heat treatment, BSs were brought to heat at 98°C for 30 min to inactivate bacterial proteases activity. For protease treatment, BSs were incubated with proteinase K (100 μg/ml) at 37°C for 1 h and subsequently boiled at 98°C for 30 min to eliminate the proteinase K activity.
Reagents and Antibodies
Flagellin purified from P. aeruginosa was purchased from InvivoGen. DOTAP liposomal transfection reagent was purchased from Sigma-Aldrich. Lipopolysaccharide (LPS), ATP, nigericin, pyocyanin, and proteinase K were purchased from Sigma-Aldrich. N-3-oxo-dodecanoyl-l-homoserine lactone (3-oxo-C12-HSL) and N-butyryl-l-homoserine lactone (C4-HSL) were obtained from Cayman. Antibodies were acquired to detect mouse caspase-1 (Adipogen, 20B-0042), mouse IL-1β (R&D, AF-401-NA), mouse IL-6 (Cell Signaling, 12912), mouse NLRP3 (Adipogen, 20B-0014), mouse ASC (Santa Cruz, SC-22514-R), and flagellin (InvivoGen, mabg-flapa).
Immunoblot Analysis
Cells were harvested and then lysed in 20mM HEPES (pH 7.5) buffer containing 0.5% Non-idet P-40, 50mM KCl, 150mM NaCl, 1.5mM MgCl2, 1mM EGTA, and protease inhibitors. After centrifugation to remove cell debris, soluble lysates were fractionated by SDS-polyacrylamide gel electrophoresis, transferred to PVDF membranes (Bio-Rad), and then immunoblotted by western blotting. For some experiments, BMDM culture supernatants were collected after inflammasome stimulations, and proteins were precipitated by the addition of a methanol/chloroform mixture as reported previously (23), and then immunoblotted. All blots shown are representative images of at least three independent experiments. Images have been cropped for presentation.
Determination of Inflammasome Activation
To trigger classical NLRP3 inflammasome activation, BMDMs were primed with LPS (0.25 μg/ml, 3 h) and then treated with ATP (2 mM, 45 min) or nigericin (5 μM, 45 min). To induce AIM2 inflammasome activation, BMDMs were transfected with poly dA:dT using lipofectamine according to the manufacturer’s protocol. Inflammasome activation was then determined by the detection of active caspase-1 p20 and active IL-1β in culture supernatants using immunoblots, and by quantification of extracellular IL-1β using the mouse IL-1β Quantikine ELISA kit (R&D systems). The levels of IL-6 in culture supernatants were quantified by mouse IL-6 Quantikine ELISA kit (R&D systems). For some experiments, BMDM supernatants were incubated with BS, subjected to protein precipitation by methanol/chloroform, and then immunoblotted. To deliver flagellin into the cytosol of BMDMs, flagellin (0.35 μg) was incubated with DOTAP liposomal transfection reagent (3.5 μl) in serum-free Opti-MEM medium (50 μl) at 25°C for 30 min. Then, 300 μl Opti-MEM medium was added, and the final mixture (250 μl total) was used to transfect BMDMs in a six-well plate.
Determination of ASC Oligomerization
To detect ASC oligomerization, a chemical cross-linking assay was performed as described previously (6). Briefly, cells were lysed and centrifuged at 6,000 rpm for 10 min. The pellets were washed and resuspended in phosphate-buffered saline. The resuspended pellets were cross-linked with 1 mM discuccinimidyl suberate [disuccinimidyl suberate (DSS); Pierce, Rockford, IL, USA] for 30 min. The cross-linked pellets and soluble lysates were fractionated by SDS-PAGE and immunoblotted with anti-ASC antibody. To visualize ASC speck-like aggregates, THP-1 cells stably expressing ASC-GFP were observed by confocal microscopy.
Quantification of mRNA
To measure mRNA levels, quantitative real-time PCR was performed. Briefly, total cellular RNA was isolated using the TRIzol reagent (Invitrogen) and reverse transcribed using PrimeScript RT Master Mix (Takara) according to the manufacturer’s instructions. Template DNA was amplified by quantitative real-time PCR using SYBR Premix Ex TaqII (Takara). Primers were as follows: 5′-GCC CAT CCT CTG TGA CTC AT-3′ and 5′-AGG CCA CAG GTA TTT TGT CG-3′ (Il-1b); 5′-AGT TGC CTT CTT GGG ACT GA-3′ and 5′-TCC ACG ATT TCC CAG AGA AC-3′ (Il-6); and 5′-CGC GGT TCT ATT TTG TTG GT-3′ and 5′-AGT CGG CAT CGT TTA TGG TC-3′ (Rn18s).
Sample Preparation for Metabolomic Analysis
Selected solvents with different polarities (hexane, ethyl acetate, and acetonitrile) were used to extract metabolites from the samples. Each solvent was added to the same volume of sample, and the mixture was vortexed for 1 min. Samples were centrifuged (2,500 rpm, 5 min), the supernatant was removed and centrifuged again (15,000 rpm, 5 min), and 200 μl of the final supernatant was pipetted into sample vials with inserts.
Ultra-Fast Liquid Chromatography (UFLC) Condition
The UFLC system was equipped with a DGU-20A5R degasser, LC-20AD XR binary pump system, SIL-20AC XR autosampler at 15°C, and CTO-20AC column oven at 35°C (Shimadzu). Chromatographic separation was conducted on a reversed-phase column (Imtakt Scherzo S-C18 column, 100 mm × 2.0 mm, i.d., 3 μm) at flow rate of 0.3 ml min−1. The mobile phases used were (A) water and (B) methanol, both containing 0.1% formic acid. The gradient program was set as follows: 98% (A) and 2% (B), held for 1 min; linear increase from 2 to 95% (B) over 14 min, held for 3 min; return to initial conditions in 0.01 min and equilibration for 1.99 min.
High-Resolution Mass Spectrometry (HRMS)
High-resolution mass spectrometry detection was performed on both Exactive Plus Orbitrap and Q-Exactive Plus Quadrupole-Orbitrap mass spectrometers (Thermo) equipped with an electrospray ionization source and operated in positive mode. Screening and confirmation of the substances were performed using full scan mode in Exactive Plus and parallel reaction monitoring (PRM) mode in Q-Exactive Plus. For identification of selected molecules, the scan range and collision energy were applied to each substance at a resolution of 35,000 in PRM mode.
UFLC–HRMS Data Processing
The raw data acquired by Xcalibur software (Thermo) were processed using MZmine2 software (24). After peak picking, mass detection, chromatogram building, and deconvolution, peaks were grouped depending on m/z and retention time value by RANSAC alignment and peak finder. The expected structure of candidates based on isotope rate and MS/MS (PRM mode) were identified by entering the formula and fragment data into mzCloud,1 MetFrag,2 and PubChem.3 The results for the most suitable candidates were then compared to standards by MS and MS/MS spectra.
Statistical Analysis
All values were expressed as the mean and SE of three independent experiments unless otherwise indicated. Data were analyzed using one-way analysis of variance followed by Bonferroni post hoc test after checking the assumptions of normal distribution by Shapiro test and equal variance by Bartlett or Brown–Forsythe test. Otherwise, Welch’s t-tests were used for unequal variances. The p values ≤0.05 were considered significant. Statistical analyses were carried out using GraphPad Prism and R software.
Results
Prolonged P. aeruginosa Infection Impairs Inflammasome-Associated Molecules
To examine a potential role of P. aeruginosa QS in host inflammasome activation, we prepared a QS-defective ΔlasR/rhlR double mutant strain of P. aeruginosa PAO1 bacteria lacking LasR and RhlR, two principal transcriptional regulators of P. aeruginosa QS system. At 12 h postinfection, ΔlasR/rhlR mutant showed a reduced cytotoxicity to human lung epithelial A549 cells compared with WT P. aeruginosa (Figure 1A), suggesting that QS-deficient ΔlasR/rhlR mutant is less virulent. Then, we assessed inflammasome activation in BMDMs upon infection with WT and QS mutant P. aeruginosa. At 4 h postinfection, both WT and ΔlasR/rhlR mutant strain promoted robust inflammasome activation as determined by the secretion of active caspase-1 (p20) and IL-1β into BMDMs culture supernatant (Figure 1B). Of notice, prolonged infection with WT P. aeruginosa (6 or 8 h), but not with ΔlasR/rhlR, caused a manifest degradation of caspase-1 and IL-1β in the culture supernatants (Figure 1B). Consistently, levels of secreted pro-inflammatory cytokines IL-1β and IL-6 at 8 h postinfection were significantly lower in BMDMs infected with WT P. aeruginosa than in those with the ΔlasR/rhlR mutant (Figures 1C,D). This apparent QS-mediated impairment of inflammasome signaling is not likely a consequence of reduced growth of the WT strain, because the growth kinetics of WT and QS-defective strains were similar (Figure 1E). These observations indicate that prolonged infection with P. aeruginosa could impair host inflammasome-mediated responses in a QS-dependent manner.
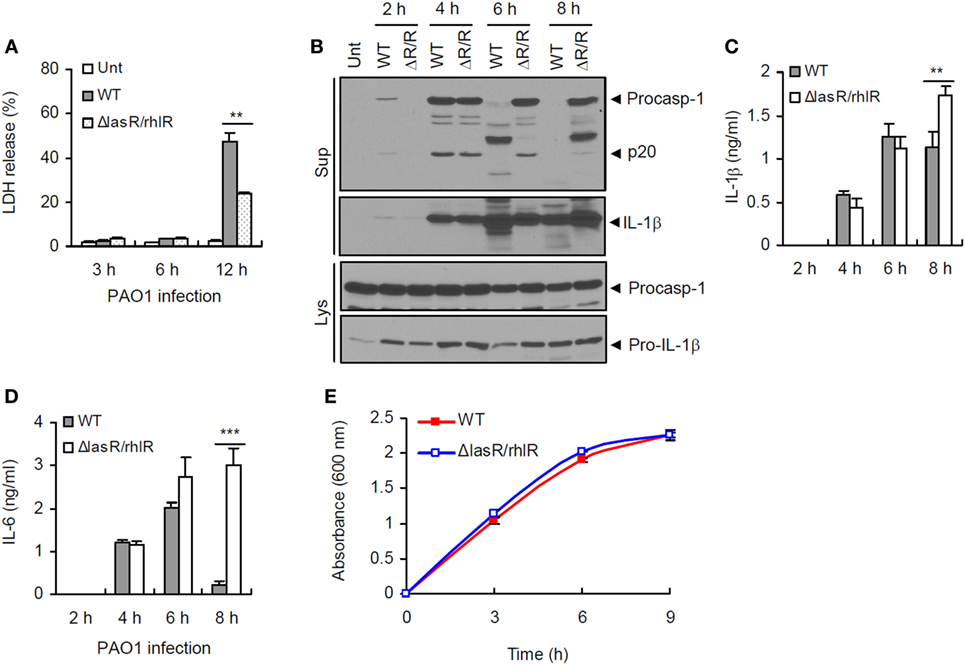
Figure 1. Quorum sensing-dependent impairment of inflammasome signaling by prolonged Pseudomonas aeruginosa infection. (A) Human lung epithelial A549 cells were infected with wild-type (WT) or ΔlasR/rhlR P. aeruginosa PAO1 at a multiplicity of infection (MOI) 3. At 3–12 h postinfection, A549 cell death was determined by lactate dehydrogenase (LDH) assay. Asterisk indicates significant difference between WT and ΔlasR/rhlR (n = 3). (B) Mouse bone marrow-derived macrophages (BMDMs) were untreated (Unt) or infected with WT or ΔlasR/rhlR (ΔR/R) P. aeruginosa PAO1 at MOI 3. At 2–8 h postinfection, culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. (C,D) Mouse BMDMs were infected with WT or ΔlasR/rhlR PAO1 at MOI 2. Extracellular levels of interleukin-1 beta (IL-1β) (C) or IL-6 (D) were quantified by ELISA in the culture supernatants of BMDMs at the indicated time postinfection. Asterisks indicate significant difference between WT and ΔlasR/rhlR (n = 6). (E) Bacterial growth curve of WT or ΔlasR/rhlR P. aeruginosa. Statistical significance was determined by one-way analysis of variance with a Bonferroni post-test or Welch’s t-test (**P < 0.01; ***P < 0.001).
P. aeruginosa Secretant Degrades Host Inflammasome Components in a QS-Dependent Manner
To test whether bacterial secreted factors are responsible for the impairment of host inflammasome-associated molecules shown above, we collected bacteria-free supernatant (referred to as bacterial secretant (BS)) from P. aeruginosa cultures (Figure 2A). Simultaneously, we collected culture supernatant from BMDMs upon stimulation with NLRP3 inflammasome-activating LPS/ATP, and this LPS/ATP-stimulated BMDM supernatant was then incubated with BS from the WT P. aeruginosa culture (Figure 2A). Both procaspase-1 and active caspase-1 p20 in the BMDMs supernatant were clearly processed into fragments by BS, as was the IL-1β secreted from BMDMs (Figure 2B).
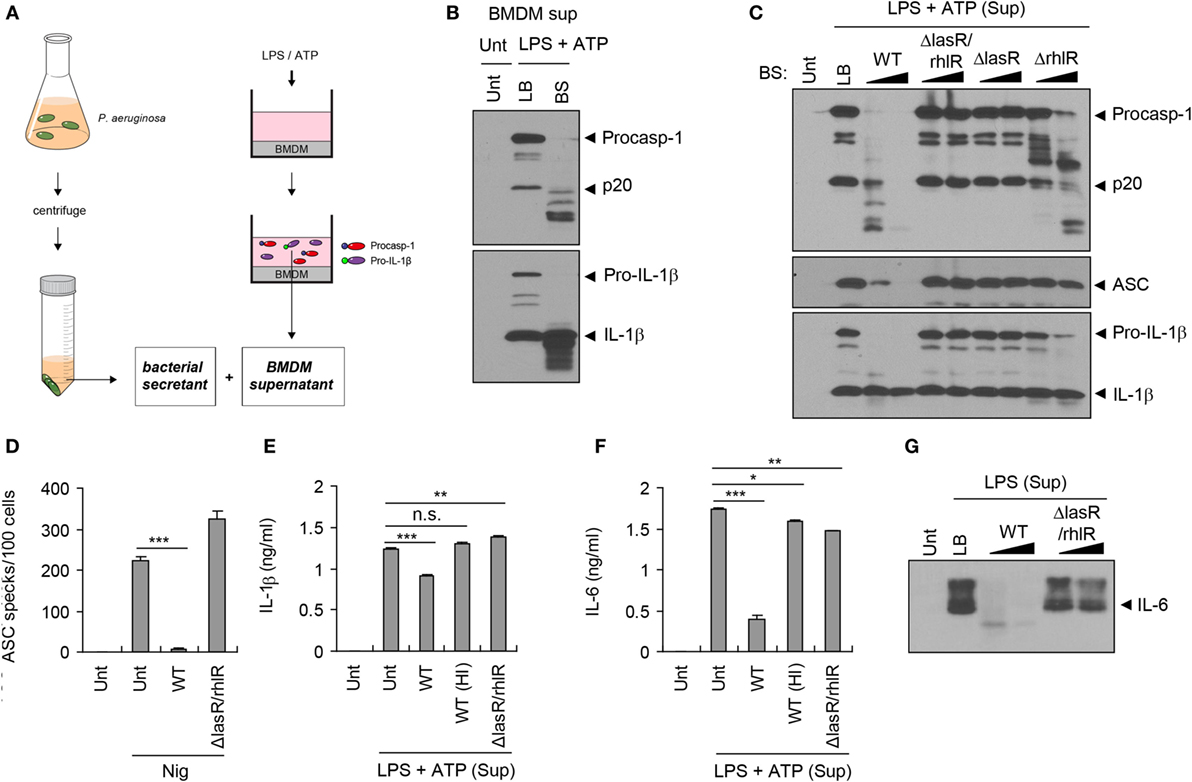
Figure 2. Quorum sensing-dependent degradation of inflammasome components by Pseudomonas aeruginosa secretant. (A) Illustration of experimental scheme showing a collection of bacterial secretant (BS) and bone marrow-derived macrophages (BMDMs) supernatant containing inflammasome components. BMDM culture supernatant was incubated with P. aeruginosa BS (1/10 volume of BMDM supernatant) at 37°C for 10–120 min. (B) Untreated or lipopolysaccharide (LPS)/ATP-stimulated BMDM culture supernatants were incubated with Luria-Bertani (LB) medium or PAO1 BS for 45 min. The mixtures were then precipitated and immunoblotted with the indicated antibodies. (C) LPS/ATP-stimulated BMDM culture supernatants were incubated with wild-type (WT), ΔlasR/rhlR, ΔlasR, or ΔrhlR secretant for 10 or 60 min, and immunoblotted with the indicated antibodies. (D) THP-1-ASC-GFP cells were treated with nigericin (5 μM, 3 h) in the presence or absence of WT or ΔlasR/rhlR secretant. The number of ASC specks was counted and displayed as specks per 100 cells. Asterisk indicates significant difference (n = 4). (E,F) LPS/ATP-stimulated BMDM culture supernatants were incubated with intact or heat-inactivated WT or ΔlasR/rhlR secretant at 37°C for 30 min. The levels of interleukin-1 beta (IL-1β) (E) or IL-6 (F) in the mixture were quantified by ELISA. Asterisks indicate significant differences (n = 3). HI, heat inactivation. (G) Untreated or LPS-treated (0.5 μg/ml, 5 h) BMDM culture supernatants were incubated with WT or ΔlasR/rhlR secretant for 10, 60 min, and then immunoblotted with anti-IL-6 antibody. Statistical significance was determined by one-way analysis of variance with a Bonferroni post-test (n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001).
Pseudomonas aeruginosa produces QS-regulated proteases such as elastase (LasB) and alkaline protease (AprA) to degrade host defense proteins such as extracellular matrix (ECM) components and cytokines (11, 25). To examine whether P. aeruginosa proteases are involved in the degradation of inflammasome-associated molecules, BS was heated to eliminate protease activity. Consequently, heat inactivation of BS markedly abolished the degradation of inflammasome components such as caspase-1 and ASC that was observed after incubation with intact P. aeruginosa secretant (Figure S1A in Supplementary Material). Furthermore, the secretant of the ΔlasB strain, lacking the major P. aeruginosa QS-dependent protease LasB, failed to efficiently degrade inflammasome components (Figure S1B in Supplementary Material), indicating that LasB plays a role in this degradation.
LasR is an essential transcriptional regulator in P. aeruginosa that induces the expression of many QS-related genes including proteases (18). Unlike WT P. aeruginosa secretant, neither ΔlasR/rhlR nor ΔlasR secretant was able to process inflammasome components (Figure 2C), confirming that the QS system is necessary for the degradation of host inflammasome components by BS. Although both LasR and RhlR are the principal transcriptional regulators of P. aeruginosa QS, LasR is a prerequisite regulator for the expression of RhlR (26). Partial processing of caspase-1 was detected in ΔrhlR mutant secretant (Figure 2C), suggesting that the secretant contains a minimal base level of proteases. Indeed, previous studies showed that ΔrhlR mutant produces a kind of proteases including AprA (27, 28).
Caspase-1 and IL-1β secreted from P. aeruginosa-infected BMDMs were also degraded by WT P. aeruginosa secretant, but not by heat-inactivated or ΔlasR/rhlR secretant (Figures S1C,D in Supplementary Material). Along with caspase-1 and IL-1β, aggregates of inflammasome adaptor protein ASC are secreted from BMDMs upon inflammasome activation, which could propagate the inflammatory responses (29, 30). Monomeric ASC in the LPS/ATP-stimulated supernatant was also degraded by WT BS, but not by ΔlasR/rhlR secretant (Figure 2C). In ASC-GFP-expressing THP-1 cells, WT P. aeruginosa secretant clearly reduced nigericin-triggered formation of ASC aggregates, which appear as specks by confocal microscopy, while ΔlasR/rhlR secretant failed to decrease the number of ASC specks (Figure 2D; Figure S1E in Supplementary Material). Consistently, the level of secreted IL-1β from LPS/ATP-treated BMDMs was significantly impaired by the incubation with WT secretant, but not with heat-inactivated nor ΔlasR/rhlR secretant (Figure 2E). Pro-inflammatory cytokine IL-6 secreted by BMDMs was also degraded by incubation with WT secretant, but less with QS mutant secretant (Figures 2F,G). All of these findings clearly demonstrate that BS from P. aeruginosa can degrade host inflammasome components and cytokines, enabling QS-dependent proteases to disrupt innate immune defenses.
BS from QS Mutant, but Not from WT P. aeruginosa, Promotes Inflammasome Activation
Given the host protein-processing effect of the P. aeruginosa QS system, we further examined a potential role of BS, which contains diverse QS-dependent virulence factors, in host inflammasome signaling pathways. Thus, we treated BMDMs with bacteria-free WT or QS-defective P. aeruginosa culture secretants (Figure 3A). To our surprise, BS from ΔlasR/rhlR or ΔlasR mutant strains promoted robust caspase-1 activation and IL-1β secretion, while WT secretant failed to trigger caspase-1 activation (Figure 3B). Meanwhile, ΔrhlR secretant isolated from 6- or 9-h preculture did not cause a robust caspase-1 processing. Because ΔrhlR mutant seems to still secrete some proteases, the deficiency of caspase-1 and IL-1β in the supernatant of BMDMs treated with ΔrhlR secretant could be explained by bacterial protease-mediated degradation.
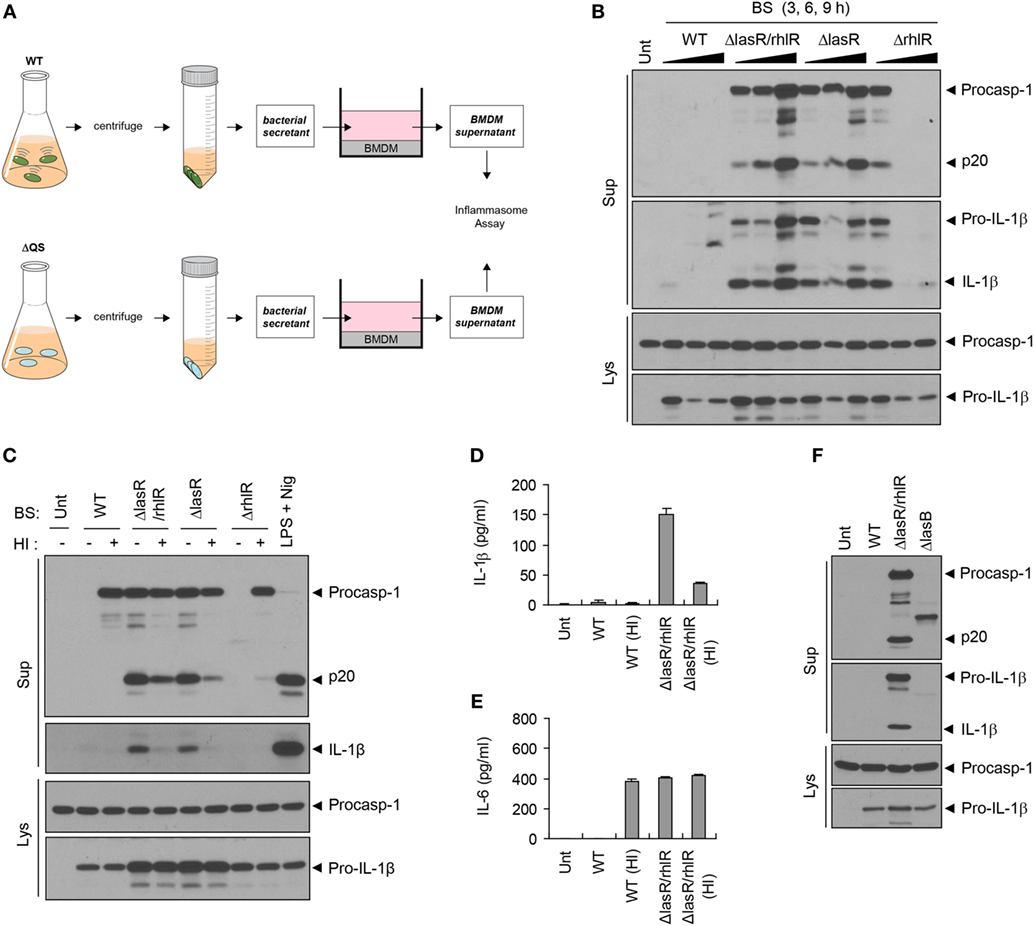
Figure 3. Quorum sensing (QS)-defective Pseudomonas aeruginosa secretant triggers inflammasome activation. (A) Illustration of experimental scheme to determine a potent role of bacterial secretant (BS) in inflammasome activation. BSs were prepared from wild-type (WT) or QS-defective mutant (ΔQS) P. aeruginosa cultures. BSs were then added to bone marrow-derived macrophage (BMDM) culture (at a 1:10 ratio) for 6 h, and BMDM culture supernatants were assayed for the inflammasome activity. (B) Mouse BMDMs were left untreated (Unt) or treated with BSs from WT, ΔlasR/rhlR, ΔlasR, or ΔrhlR P. aeruginosa 3-, 6-, or 9-h precultures. (C) Mouse BMDMs were treated with intact or heat-inactivated WT, ΔlasR/rhlR, ΔlasR, or ΔrhlR secretants for 6 h, or primed with lipopolysaccharide (0.25 μg/ml, 3 h), followed by treatment with nigericin (Nig, 5 μM, 45 min). (D,E) Mouse BMDMs were treated with intact or heat-inactivated WT or ΔlasR/rhlR secretants for 6 h. Cytokine levels of interleukin-1 beta (IL-1β) (D) or IL-6 (E) in the culture supernatants were quantified by ELISA (n = 3). HI, heat inactivation. (F) Mouse BMDMs were treated with WT, ΔlasR/rhlR, and ΔlasB secretants for 6 h. (B,C,F) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies.
Consistent with caspase-1 activation, ΔlasR/rhlR mutant secretant, but not that of WT bacteria, caused a robust production of IL-1β and IL-6 by BMDMs (Figures S2A,B in Supplementary Material), while secretants from both WT and ΔlasR/rhlR P. aeruginosa induced upregulation of Il-1β and Il-6 mRNA (Figures S2C,D in Supplementary Material). As for IL-6, it is likely that WT P. aeruginosa secretant could degrade IL-6 secreted from BMDMs via its proteases, because WT P. aeruginosa secretant triggered mRNA production of IL-6, subsequently leading to its secretion. On the other hand, the absence of caspase-1 and IL-1β in the supernatant of BMDMs treated with WT secretant could be explained by two possible scenarios. First, it is plausible that WT secretant may be unable to trigger inflammasome activation. The second hypothesis is that WT secretant may promote inflammasome activation, leading to the secretion of inflammasome proteins, but bacterial secreted proteases could then degrade caspase-1 and IL-1β in the extracellular supernatant. To test these possibilities, we performed similar experiments with heat-inactivated BS to avoid the involvement of proteases. As a result, WT secretant failed to induce caspase-1 activation and IL-1β secretion even after heat inactivation (Figures 3C,D). Instead, heat inactivation resulted in the appearance of procaspase-1 by WT secretant (Figure 3C), suggesting that secreted procaspase-1 may be degraded by bacterial proteases. These data demonstrate that WT secretant is unable to promote inflammasome activation regardless of protease activity. However, heat-treated WT secretant caused robust secretion of IL-6 (Figure 3E), suggesting that proteases from the WT secretant had been responsible for processing IL-6 in the extracellular space. To further clarify the involvement of proteases in the failure of inflammasome activation by WT secretant, BS from ΔlasB mutant, which is devoid of elastase expression, was examined. Treatment with ΔlasB secretant partially restored procaspase-1 in the supernatant but caused no caspase-1 processing (Figure 3F). This finding further supports that WT secretant is unable to promote inflammasome activation.
BS from QS Mutant P. aeruginosa Triggers NLRC4 Inflammasome Activation and Assembly
Next, we examined which sensor molecule is implicated in inflammasome activation by ΔlasR/rhlR secretant. In accordance with previous studies, P. aeruginosa infection in BMDMs resulted in NLRC4-dependent inflammasome activation (Figure 4A). Likewise, inflammasome activation by ΔlasR/rhlR secretant was abrogated in Nlrc4−/− BMDMs (Figure 4B), indicating that ΔlasR/rhlR secretant triggered NLRC4-dependent inflammasome activation. A deficiency of ASC also diminished the caspase-1 activation and IL-1β secretion by ΔlasR/rhlR secretant (Figure 4C). By contrast, robust inflammasome activation was observed in Nlrp3-deficient BMDMs in response to ΔlasR/rhlR secretant (Figure 4D). Therefore, our data demonstrated that ΔlasR/rhlR secretant promotes NLRC4-mediated, but not NLRP3-mediated inflammasome activation.
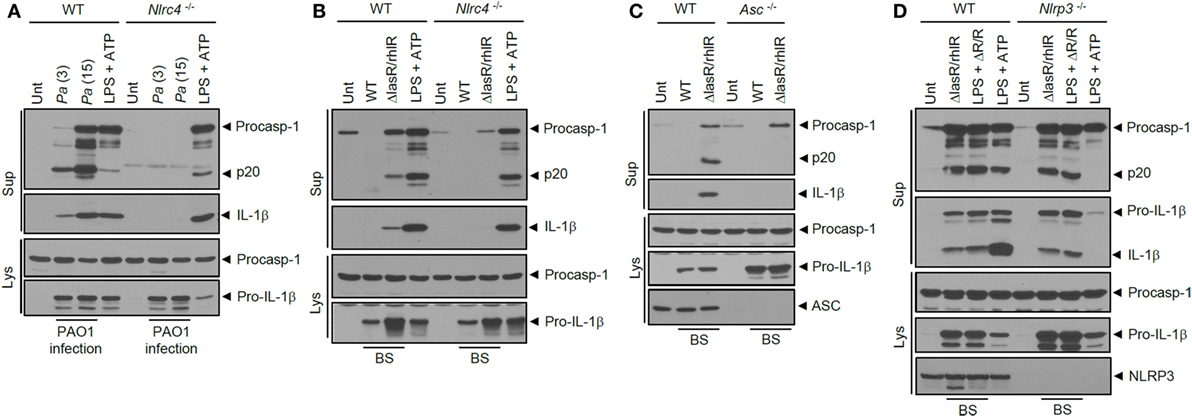
Figure 4. NLRC4 inflammasome activation by quorum sensing-defective Pseudomonas aeruginosa (Pa) secretant. (A) Wild-type (WT) or Nlrc4−/− mouse bone marrow-derived macrophages (BMDMs) were untreated or infected for 3 h with WT Pa at the indicated multiplicity of infection, or treated with lipopolysaccharide (LPS) (0.25 μg/ml, 3 h), followed by treatment with ATP (2.5 mM, 45 min). (B) WT or Nlrc4−/− mouse BMDMs were untreated (Unt) or treated with WT or ΔlasR/rhlR P. aeruginosa secretants for 6 h, or primed with LPS (0.25 μg/ml, 3 h), followed by treatment with ATP (2 mM, 40 min). (C) WT or Asc−/− mouse immortalized BMDMs were treated with WT or ΔlasR/rhlR secretants for 6 h. (D) WT or Nlrp3−/− mouse BMDMs were treated with ΔlasR/rhlR (ΔR/R) secretants in the presence or absence of LPS (0.25 μg/ml) for 6 h, or primed with LPS (3 h), followed by treatment with ATP (2.5 mM, 40 min). (A–D) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. BS, bacterial secretant.
Previously, it was shown that Salmonella infection triggered ASC oligomerization, considered as an essential event of inflammasome assembly (31, 32). Similarly, P. aeruginosa infection caused a robust oligomerization of ASC as determined by a DSS-mediated cross-linking assay (Figure S3 in Supplementary Material). The secretant of ΔlasR/rhlR P. aeruginosa, but not of WT, also triggered the formation of ASC dimeric and oligomeric structures only in the presence of NLRC4 (Figures 5A,B). The failure of WT secretant to induce ASC oligomerization further supports the hypothesis that it could not activate inflammasome signaling regardless of protease-mediated protein degradation. Moreover, even heat-inactivated WT secretant failed to promote the oligomerization of ASC inside cells (Figure 5C). Consistent with these data, BS from ΔlasR/rhlR mutant P. aeruginosa, but not from WT, triggered the formation of speck-like aggregates of ASC in human monocytic THP-1 cells expressing ASC-GFP (Figure 5D). These findings collectively demonstrate that the ΔlasR/rhlR mutant secretant, but not WT secretant, promotes the assembly and activation of NLRC4 inflammasome.
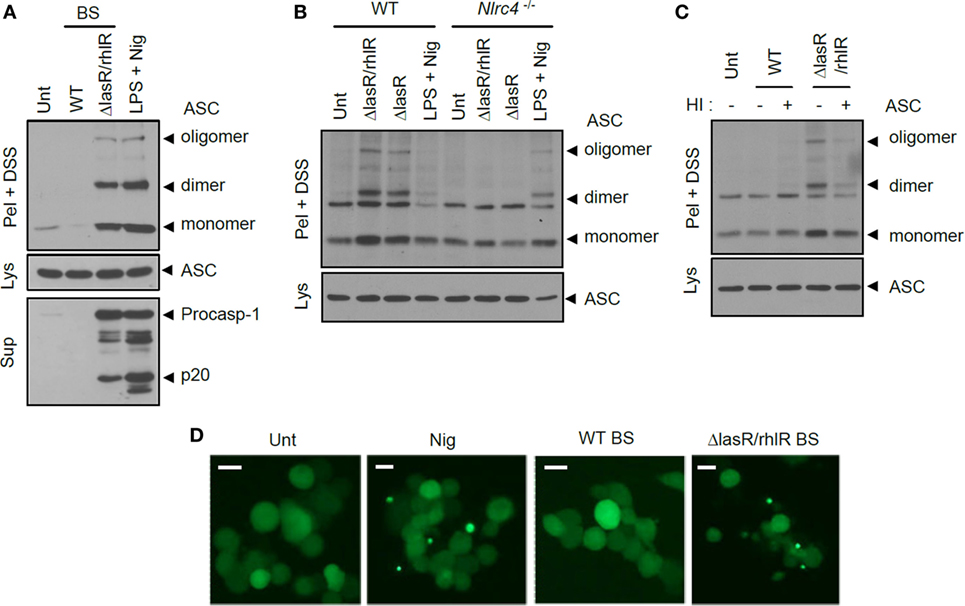
Figure 5. Quorum sensing-defective Pseudomonas aeruginosa secretant triggers NLRC4-dependent oligomerization of ASC. (A) Mouse bone marrow-derived macrophages (BMDMs) were untreated or treated with wild-type (WT) or ΔlasR/rhlR secretants for 6 h, or primed with lipopolysaccharide (LPS) (3 h), followed by treatment with nigericin (5 μM, 45 min). (B) WT or Nlrc4−/− mouse BMDMs were treated with ΔlasR/rhlR or ΔlasR secretants for 6 h, or primed with LPS, followed by treatment with nigericin. (C) Mouse BMDMs were treated with intact or heat-inactivated WT or ΔlasR/rhlR secretants. HI, heat inactivation. (A–C) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Disuccinimidyl suberate (DSS)-cross-linked pellets (Pel + DSS) were immunoblotted with ASC antibody. (D) THP-1-ASC-GFP cells were untreated or treated with WT or ΔlasR/rhlR secretants (8 h), or with nigericin (Nig, 5 μM, 40 min). Cells were then observed by confocal microscopy. Scale bars, 10 μm. Data shown are a representative image from eight independent samples.
Extracellular Flagellin Mediates Inflammasome Activation by QS Mutant Secretant
To further provide molecular insight into the inflammasome-activating capability of ΔlasR/rhlR secretant, we first examined whether specific protein components of BS drive inflammasome activation. Heat inactivation of ΔlasR/rhlR secretant significantly reduced caspase-1 activation and IL-1β secretion (Figure 6A; Figure S4A in Supplementary Material). This is consistent with the above results in Figures 3C,D, and these findings demonstrate that heat treatment significantly impairs BS-promoted inflammasome activation. Moreover, proteinase K treatment of ΔlasR/rhlR secretant completely abolished the inflammasome activation (Figure 6A; Figure S4A in Supplementary Material). These data strongly indicate that unidentified proteins in the secretant could be responsible for the secretant-triggered inflammasome activation. According to previous report, cytosolic presence of bacterial flagellin is critical to induce NLRC4 inflammasome activation upon P. aeruginosa infection (21). Related to this finding, we found that BS from the ΔfliC mutant lacking FliC flagellin was unable to promote inflammasome activation (Figure S4B in Supplementary Material). Intriguingly, the ablation of FliC in ΔlasR/rhlR or ΔlasR P. aeruginosa completely abolished the inflammasome activation by ΔlasR/rhlR or ΔlasR BS (Figure 6B), suggesting that flagellin in ΔlasR/rhlR secretant might be the main stimulus for inflammasome activation.
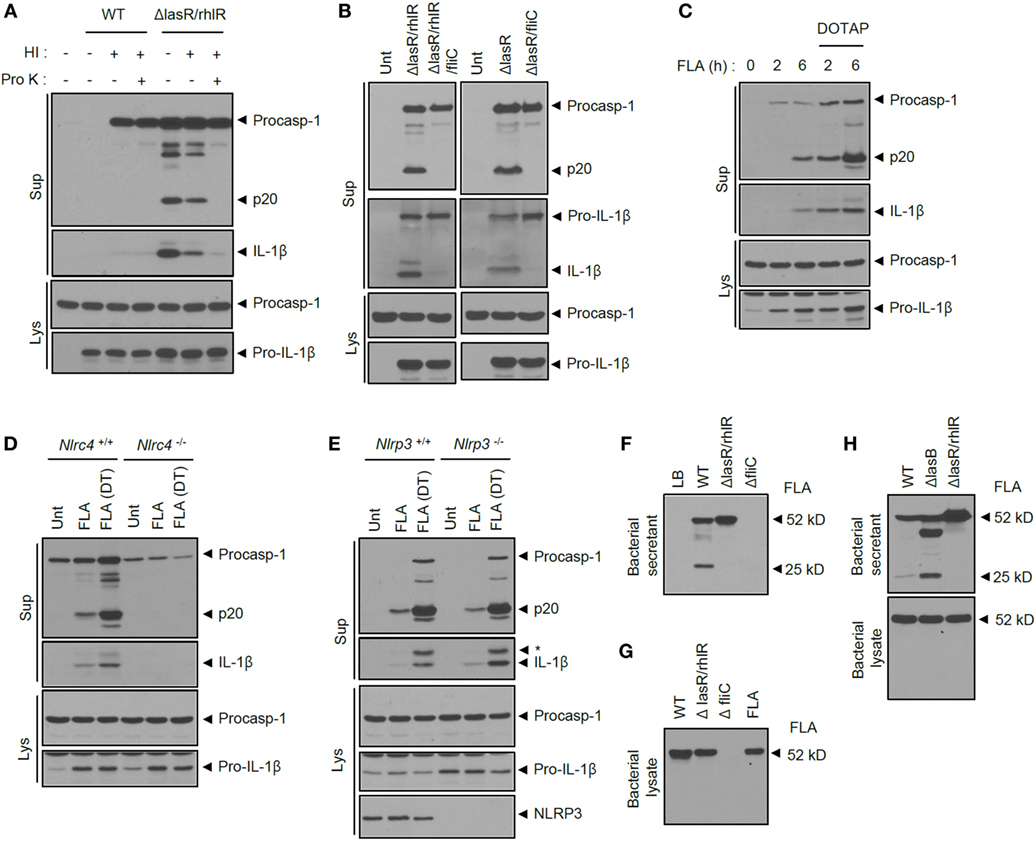
Figure 6. Extracellular flagellin is critical for inflammasome activation by quorum sensing-defective Pseudomonas aeruginosa secretant. (A) Mouse bone marrow-derived macrophages (BMDMs) were untreated or treated with wild-type (WT) or ΔlasR/rhlR P. aeruginosa secretants with or without heat inactivation (HI) and proteinase K (Pro K) treatment as indicated. (B) Mouse BMDMs were treated for 6 h with ΔlasR/rhlR, ΔlasR/rhlR/fliC, ΔlasR, or ΔlasR/fliC secretants as indicated. (C) Mouse BMDMs were treated with P. aeruginosa flagellin (FLA, 0.25 μg) for 2 or 6 h with or without pre-mixing with DOTAP liposomal transfection reagent. (D,E) WT, Nlrc4−/− (D) or Nlrp3−/− (E) mouse BMDMs were treated with flagellin (0.25 μg) with or without pre-mixing with DOTAP (DT) reagent for 6 h. (A–E) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. (F) Bacterial secretants (BSs) from WT, ΔlasR/rhlR or ΔfliC P. aeruginosa cultures were immunoblotted with anti-flagellin antibody. (G) WT, ΔlasR/rhlR, and ΔfliC P. aeruginosa bacterial lysates or purified P. aeruginosa flagellin (FLA) were immunoblotted with anti-flagellin antibody. (H) BSs or bacterial lysates from WT, ΔlasB, or ΔlasR/rhlR P. aeruginosa cultures were immunoblotted with anti-flagellin antibody.
To verify this possibility that extracellular flagellin released from P. aeruginosa could trigger inflammasome activation, we treated BMDMs with purified flagellin from P. aeruginosa. Previous reports showed that intracellular delivery of flagellin using a liposomal agent such as DOTAP is essential for activation of the NLRC4 inflammasome (3, 33). Treatment of flagellin for 1–3 h failed to activate caspase-1 and IL-1β secretion in BMDMs (3, 21). However, in our study, 6-h incubation of flagellin in the extracellular medium was able to promote caspase-1 activation and IL-1β secretion (Figure 6C). This extracellular flagellin-triggered inflammasome activation depended on NLRC4 but not NLRP3 (Figures 6D,E). To explain WT secretant’s inability to activate the inflammasome, the presence of flagellin was assayed in the secretant from P. aeruginosa cultures. Of interest, intact flagellin was observed in the BS of the ΔlasR/rhlR mutant, whereas flagellin was severely impaired in the WT secretant (Figure 6F). Indeed, flagellin levels were markedly reduced in WT secretant, but not in QS-defective mutants secretant (Figure S4C in Supplementary Material). By contrast, no degradation of flagellin was observed in the bacterial lysate of WT P. aeruginosa (Figure 6G), indicating that secreted flagellin could be degraded by bacterial proteases in the secretant rather than within the cells. The degradation of flagellin was also observed, but slightly reduced in ΔlasB mutant, suggesting that both elastase and other protease are responsible for the impairment of flagellin (Figure 6H). These data demonstrate that QS-dependent production of protease from WT P. aeruginosa culture led to degradation of flagellin and thereby eliminated its ability to activate the NLRC4 inflammasome.
QS-Regulated Virulence Factor Pyocyanin Attenuates Inflammasome Activation
To further delineate the potential contribution of QS to inflammasome activation, metabolomic analysis was performed with BSs from WT and ΔlasR/rhlR P. aeruginosa using liquid chromatography–mass spectrometry. Unfortunately, under our experimental conditions, we did not identify any candidate molecule that was only present in ΔlasR/rhlR secretant, but not in WT secretant. However, pyocyanin was identified as the sole candidate molecule present in WT secretant but not in ΔlasR/rhlR secretant (Figure 7A; Figures S5A,B in Supplementary Material). Pyocyanin is one of the well-known QS-dependent major virulence factors of P. aeruginosa (17, 18) and was reported to damage host cells via its oxidative stress-based cytotoxicity (34, 35). Thus, we tested a potential role of pyocyanin in the inflammasome activation. Of note, pyocyanin remarkably suppressed the inflammasome activation and IL-1β secretion triggered by ΔlasR/rhlR secretant (Figures 7B,C). Furthermore, pyocyanin treatment resulted in a strong suppression of NLRP3-mediated caspase-1 activation and IL-1β secretion upon LPS/ATP or LPS/nigericin stimulation (Figures 7D,E). Pyocyanin also significantly attenuated LPS/nigericin-triggered cell death of BMDMs (Figure 7F). This might be due to reduced caspase-1 activation by pyocyanin. However, pyocyanin failed to affect poly dA:dT-triggered AIM2 inflammasome activation (Figure 7G). Supporting these observations, pyocyanin clearly reduced NLRC4- or NLRP3-dependent ASC oligomerization in response to ΔlasR/rhlR secretant or LPS/nigericin treatment (Figures 7H,I). These findings demonstrate that the QS-dependent virulence factor pyocyanin could suppress the activation of both the bacterial infection-promoted NLRC4 inflammasome and the host endogenous danger signals-triggered NLRP3 inflammasome.
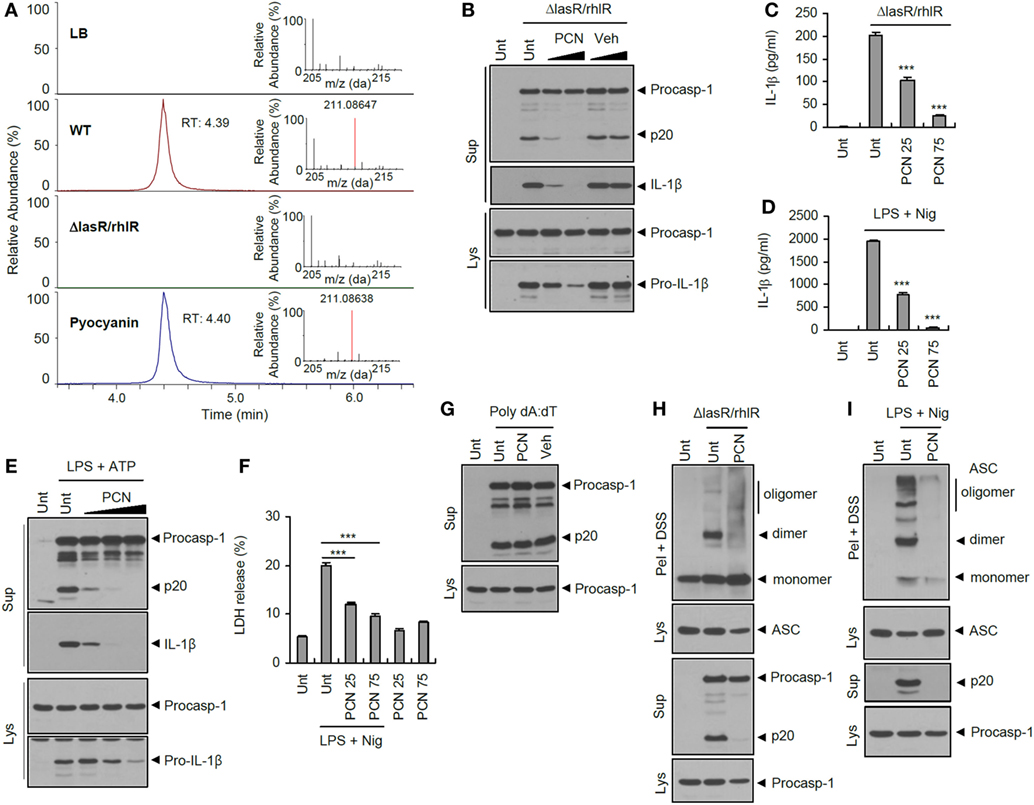
Figure 7. Suppression of inflammasome activation by quorum sensing-regulated product pyocyanin. (A) Extracted ion chromatogram for m/z 211.08647 (tolerance; 5 ppm) and mass spectra (RT 4.3–4.5) of each samples and standard pyocyanin in full scan mode. (B) Mouse bone marrow-derived macrophages (BMDMs) were treated with ΔlasR/rhlR Pseudomonas aeruginosa secretants in the presence of pyocyanin (PCN, 60 or 120 μM) or DMSO vehicle for 6 h. (C) Mouse BMDMs were treated with ΔlasR/rhlR secretant in the presence of pyocyanin (25 or 75 μM, 30 min before secretant) for 6 h. (D) Mouse BMDMs were primed with lipopolysaccharide (LPS) (0.25 μg/ml, 3 h) in the presence of pyocyanin (25 or 75 μM, 30 min before LPS), followed by treatment with nigericin (Nig, 5 μM, 45 min). (C,D) The levels of interleukin-1 beta (IL-1β) were quantified by ELISA in BMDM supernatants. Asterisks indicate significant differences compared with ΔlasR/rhlR-treated (C) or LPS/nig-treated (D) group (n = 3). (E) Mouse BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of pyocyanin (20, 50, and 100 μM) pretreatment (30 min before LPS), followed by treatment with ATP (2 mM, 45 min). (F) Mouse BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of pyocyanin pretreatment (25 or 75 μM, 30 min before LPS), followed by treatment with nigericin (5 μM, 30 min). Extracellular release of lactate dehydrogenase (LDH) was assayed. Asterisks indicate significant differences (n = 3). (G) Mouse BMDMs were untreated or transfected with poly dA:dT (1.5 μg, 5 h) in the presence of pyocyanin (50 μM, 30 min pretreatment). (H) Mouse BMDMs were treated with ΔlasR/rhlR secretants in the presence of pyocyanin (120 μM) for 6 h. (I) Mouse BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of pyocyanin (120 μM, 30 min before LPS), followed by treatment with nigericin (5 μM, 40 min). (B,E,G–I) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Disuccinimidyl suberate (DSS)-cross-linked pellets (Pel + DSS) were immunoblotted with ASC antibody. Statistical significance was determined by one-way analysis of variance with a Bonferroni post-test (***P < 0.001).
QS Autoinducer 3-oxo-C12-HSL Attenuates Inflammasome Activation
Bacterial QS systems use acylated homoserine lactones as a QS signaling regulator, called an autoinducer. In the case of P. aeruginosa, two signaling molecules—3-oxo-C12-HSL, produced by LasI, and C4-HSL, produced by RhlI—are employed for bacterial communication to control the expression of many virulence factors (18). So far, these QS autoinducers have never been examined for their potential effect on inflammasome signaling. Interestingly, 3-oxo-C12-HSL, but not C4-HSL, significantly dampened inflammasome activation triggered by ΔlasR/rhlR secretant (Figure 8A). Moreover, 3-oxo-C12-HSL completely blocked the NLRP3 inflammasome activation in response to LPS/ATP stimulation (Figure 8B). This P. aeruginosa autoinducer 3-oxo-C12-HSL also strongly attenuated IL-1β secretion upon ΔlasR/rhlR secretant or LPS/ATP treatment (Figures 8C,D). Similar to pyocyanin, two P. aeruginosa autoinducers showed no effect on the AIM2-dependent caspase-1 activation triggered by poly dA:dT transfection (Figure 8E), indicating that 3-oxo-C12-HSL could suppress NLRC4 and NLRP3 inflammasome signaling. Supporting these findings, 3-oxo-C12-HSL diminished the NLRC4- and NLRP3-mediated ASC oligomerization by cross-linking experiments (Figures 8F,G). However, NLRP3 inflammasome-dependent cell death was not abolished, but rather increased by 3-oxo-C12-HSL (Figure 8H). Indeed, 3-oxo-C12-HSL showed a basal cytotoxicity to BMDMs regardless of inflammasome activity. Of notice, QS-associated factors, pyocyanin and 3-oxo-C12-HSL, significantly inhibited the oligomerization of NLRP3 in response to classical NLRP3 stimulators (Figure 8I; Figure S6 in Supplementary Material). These data suggest that the suppression of inflammasome signaling by two QS-related factors might be the upstream event of oligomerization of inflammasome sensor molecule such as NLRP3. These findings collectively demonstrate that two QS-related products, pyocyanin and 3-oxo-C12-HSL, are bona fide inhibitors of inflammasome activation, and that bacterial QS could provide an evasion mechanism to avoid the host inflammasome defense system.
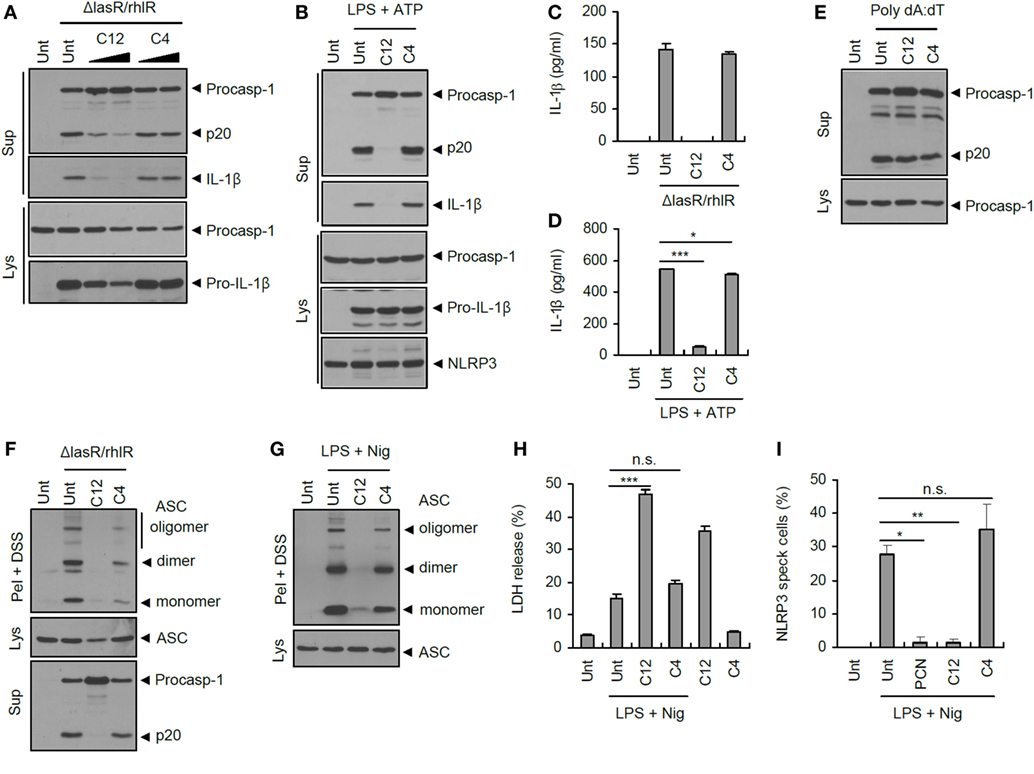
Figure 8. Suppression of inflammasome activation by quorum sensing autoinducer 3-oxo-C12-HSL. (A) Mouse bone marrow-derived macrophages (BMDMs) were treated with ΔlasR/rhlR secretants in the presence of 3-oxo-C12-HSL (C12) or C4-HSL (C4) (30, 80 μM, 30 min pretreatment) for 6 h. (B) Mouse BMDMs were primed with lipopolysaccharide (LPS) (0.25 μg/ml, 3 h) in the presence of C12-HSL or C4-HSL pretreatment (40 μM), followed by treatment with ATP (2 mM, 30 min). (C) Mouse BMDMs were treated with ΔlasR/rhlR secretants in the presence of 3-oxo-C12-HSL or C4-HSL pretreatment (80 μM) for 6 h. (D) Mouse BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of 3-oxo-C12-HSL or C4-HSL (40 μM, 30 min before LPS) for 3 h, followed by treatment with ATP (2 mM, 40 min). (C,D) The levels of interleukin-1 beta (IL-1β) were quantified by ELISA in BMDM supernatants. Asterisks indicate significant difference (n = 3). (E) Mouse BMDMs were untreated or transfected with poly dA:dT (1.5 μg, 5 h) in the presence of C12-HSL or C4-HSL (50 μM, 30 min pretreatment). (F) Mouse BMDMs were treated with ΔlasR/rhlR secretants in the presence of 3-oxo-C12-HSL or C4-HSL (80 μM, 30 min pretreatment) for 6 h. (G) Mouse BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of 3-oxo-C12-HSL or C4-HSL (40 μM, 30 min before LPS), followed by treatment with nigericin (5 μM, 30 min). (H) Mouse BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of 3-oxo-C12-HSL or C4-HSL (20 μM, 30 min before LPS), followed by treatment with nigericin (5 μM, 30 min). Extracellular release of lactate dehydrogenase (LDH) was assayed. Asterisk indicates significant differences (n = 3). (I) NLRP3-GFP-expressing BMDMs were primed with LPS (0.25 μg/ml, 3 h) in the presence of pyocyanin (PCN), 3-oxo-C12-HSL, or C4-HSL (50 μM, 30 min before LPS), followed by treatment with nigericin (5 μM, 40 min). Cells were observed by confocal microscope, and NLRP3 speck-containing cells were counted. Asterisks indicate significant difference (n = 3). (A,B,E–G) Culture supernatants (Sup) or cellular lysates (Lys) were immunoblotted with the indicated antibodies. Disuccinimidyl suberate (DSS)-cross-linked pellets (Pel + DSS) were immunoblotted with ASC antibody. Statistical significance was determined by one-way analysis of variance with a Bonferroni post-test (*P < 0.05; ***P < 0.001).
Discussion
Despite the recent advances in knowledge regarding bacterial recognition pathways leading to inflammasome activation, bacterial evasion of inflammasome activation remains still poorly understood. Moreover, most studies focus on the host cell’s response after uptake of bacteria. Thus, the potent role of extracellular bacteria-secreted factors during pathogenesis has not been fully investigated. In the present study, we attempted to dissect the role of P. aeruginosa factors that are secreted from that of internalized bacteria using bacteria-free secretant. Here, we present novel evidence demonstrating that P. aeruginosa makes use of bacterial QS-dependent secretant as an inflammasome evasion strategy.
Upon reaching a threshold density of bacterial growth, the bacterial QS system initiates the expression of an array of virulence determinants using AHLs as an autoinducer (36). Consequently, this regulated expression of virulence genes is thought to confer a selective advantage to bacteria over host defenses and to impair host protective functions in various ways (17, 36). For example, P. aeruginosa-secreted proteases, whose expression was greatly increased by the QS system, degrade many host defense proteins such as complement, surfactant, and ECM proteins (11). In addition, P. aeruginosa elastase is known to lead to degradation of pro-inflammatory cytokines such as IL-6 or IL-8 to avoid host immune defense (37). A recent report also showed that thrombin cleaved by P. aeruginosa elastase attenuated TLR-mediated pro-inflammatory responses (38). In correlation with these studies, our results clearly demonstrate that P. aeruginosa proteases degrade inflammasome components including caspase-1 and ASC, as well as pro-inflammatory cytokines in the extracellular space (Figure 9). Thus, QS-mediated production of proteases by P. aeruginosa provides the bacterial community an opportunity to debilitate the host’s innate immune protection.
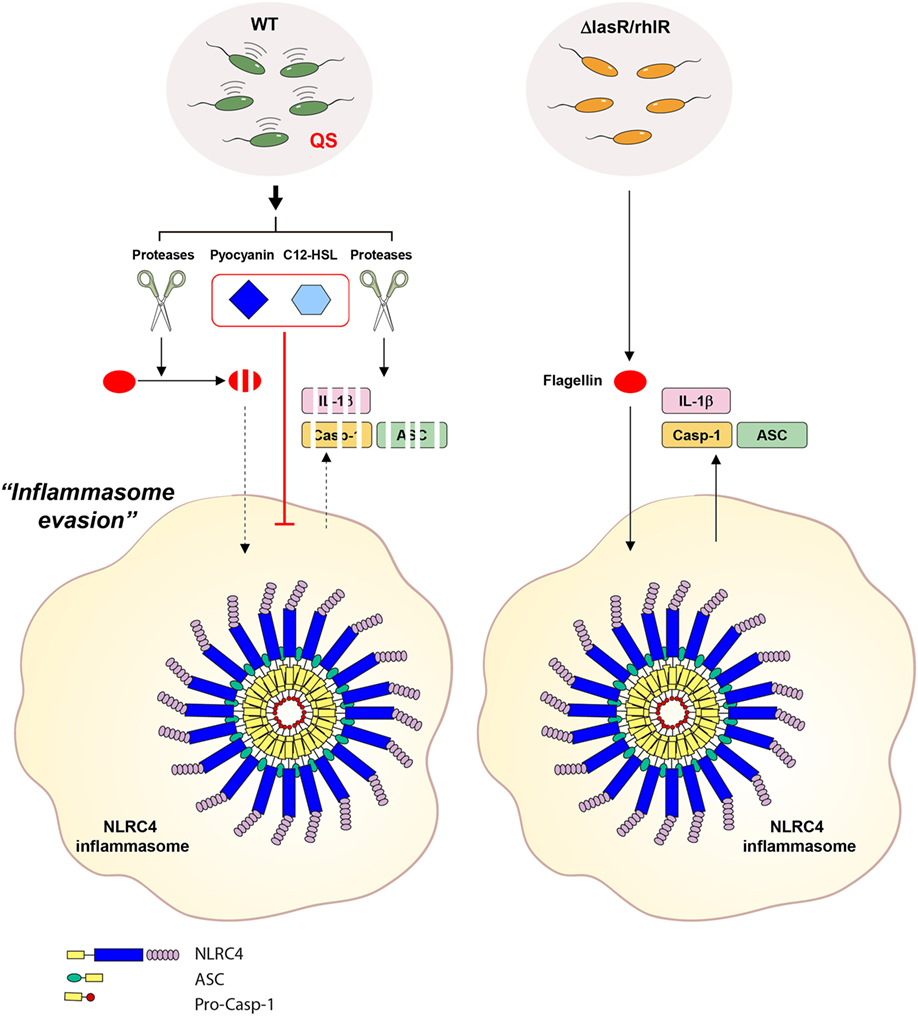
Figure 9. Proposed mechanism for the contribution of Pseudomonas aeruginosa secretant to NLRC4 inflammasome activation. Wild-type (WT) P. aeruginosa secrets quorum sensing (QS)-regulated factors such as proteases, pyocyanin and 3-oxo-C12-HSL in a QS-dependent manner. In the extracellular space, secreted proteases degrade inflammasome-associated molecules from host cells. Proteases also degrade extracellular flagellin to impair its potent NLRC4-stimulating ability. Of note, extracellular flagellin originating from P. aeruginosa can promote the activation of the NLRC4 inflammasome without bacterial invasion into host cells. Furthermore, two QS factors, pyocyanin and C12-HSL, suppress the activation of the NLRC4 inflammasome. QS-defective P. aeruginosa fail to produce the QS-regulated factors, mentioned above. Therefore, QS mutants could lead to deregulated inflammasome activation in certain contexts.
The P. aeruginosa QS system also controls the production of redox-active phenazine derivatives such as pyocyanin (17). We detected pyocyanin in WT, but not QS-defective mutant, P. aeruginosa secretant using a metabolomic analysis. Pyocyanin, one of the major toxins secreted by P. aeruginosa, was shown to damage or kill host target cells mainly by inducing oxidative stress (34, 39). Pyocyanin also plays an important role in the pathogenesis of cystic fibrosis upon P. aeruginosa infection (40). Of particular interest, the level of pyocyanin is elevated up to 100 μM in P. aeruginosa-infected patients with cystic fibrosis (41). Moreover, virulence was attenuated in pyocyanin-deficient P. aeruginosa-infected mice (42), indicating that pyocyanin is one of the major virulence factors of P. aeruginosa. In terms of its contribution to host inflammatory response, pyocyanin can exert a pro-inflammatory role based on its reactive oxygen species-producing ability (39). By contrast, pyocyanin-deficient P. aeruginosa infection caused increased inflammation in the bronchoalveolar lavage of mice (35), suggesting potent anti-inflammatory function of pyocyanin. In line with these data, our results present the first evidence that pyocyanin could suppress NLRC4- and even NLRP3-mediated inflammasome activation. Although the underlying mechanism by which pyocyanin inhibits the activation of inflammasome signaling remains to be elucidated, our data suggest the novel concept that P. aeruginosa could attenuate inflammasome activation through QS-dependent production of pyocyanin.
In addition to the contribution of proteases and pyocyanin to host inflammasome signaling, we also unveiled a novel function of P. aeruginosa QS autoinducer C12-HSL in suppression of inflammasome activation. An anti-inflammatory role of C12-HSL was previously reported. C12-HSL inhibits LPS-triggered production of pro-inflammatory cytokines, such as TNF-α and MCP-10, in peritoneal macrophages and BMDMs (43–45). Furthermore, C12-HSL, but not C4-HSL, causes apoptosis in BMDMs, which impairs the host immune defense against P. aeruginosa infection (46). These observations indicate that P. aeruginosa C12-HSL can not only trigger bacterial virulence gene expression as a QS autoinducer but also attenuate host innate immune defense. The present data provide additional insight into the anti-inflammatory role of C12-HSL in diminishing the activation of NLRP3 and NLRC4 inflammasome pathways. These findings suggest that the P. aeruginosa QS system could dampen host inflammasome-mediated protection via QS-associated products such as pyocyanin and C12-HSL (Figure 9).
The primary role of NLRC4 inflammasome signaling in response to P. aeruginosa infection is to control bacterial replication. Previous studies showed that inflammasome-deficient mice lacking NLRC4, caspase-1, IL-1β, or IL-1 receptor resulted in elevated bacterial burden and tissue damages upon P. aeruginosa infection (20, 47–49). Pattern-recognition host cells detect key virulence determinants of P. aeruginosa, such as flagellin or T3SS protein PscC, leading to NLRC4 inflammasome activation (21, 50, 51). In terms of bacterial evasion of host innate defense, P. aeruginosa major exotoxin ExoU was shown to attenuate NLRC4 inflammasome activation, likely through its phospholipase activity (47). Also, alkaline protease AprA, a QS-dependent secreted protease, degrades bacterial monomeric flagellin to evade TLR5-mediated immune responses (52). In close association with these previous findings, our data suggest that QS-dependent production of P. aeruginosa proteases degrade extracellular flagellin, leading to the impairment of flagellin-triggered inflammasome activation. Until now, it was generally accepted that cytosolic delivery of flagellin through bacterial secretion system or in vitro liposomal transfection reagent is prerequisite for inflammasome activation (3, 33). Intriguingly, our study shows that extracellular secreted flagellin alone can induce inflammasome activation. One possible explanation for this discrepancy could be the different treatment times. Previous studies examined the inflammasome activation in BMDMs only upon less than 3 h Salmonella flagellin treatment (3, 21); however, we obtained inflammasome activation after 6 h treatment of P. aeruginosa flagellin. Nevertheless, we still believe that the delivery of flagellin into cytosol is required to activate inflammasome. Further study will be needed to clarify how extracellular flagellin is able to penetrate host cell membrane.
In contrast to its protective role, inflammasome activation has also been proposed to exacerbate host tissue damages in response to P. aeruginosa infection (53, 54). It is possible that the initial activation of inflammasome signaling is preferable for host defense, especially upon acute P. aeruginosa infection. However, it is highly plausible that persistently dysregulated inflammasome activation triggered by P. aeruginosa may cause host tissue damage. Bacterial QS is thought to significantly enhance pathogenicity by promoting the acute expression of diverse virulence factors. Paradoxically, QS-defective strains such as LasR-lacking P. aeruginosa were highly represented in the clinical isolates from cystic fibrosis patients (55) and these patients are associated with exacerbated lung pathology (56). Although it is not completely understood why QS mutant P. aeruginosa, considered a strain with reduced virulence, were present in cystic fibrosis patients, our data provide a possible explanation. Specifically, QS-defective mutants could promote hyper-activation of inflammasome signaling by disarming inflammasome-suppressing machinery, which might lead to the exacerbated pathology of cystic fibrosis patients. Of note, QS products pyocyanin and C12-HSL effectively suppressed NLRP3 inflammasome activation triggered by classical NLRP3 stimulators. Given that P. aeruginosa flagellin was also proposed to induce NLRP3 inflammasome activation (57), chronic infection with P. aeruginosa could lead to excessive NLRP3 inflammasome activation. In this regard, QS-defective mutants are implicated in the dysregulation of NLRP3 inflammasome activation that accounts for the severe pathology of cystic fibrosis. Further studies will be needed to reveal the molecular mechanisms linking P. aeruginosa QS to NLRP3 inflammasome signaling. On the basis of our data, P. aeruginosa QS can be used to evade host inflammasome activation in the early stage of infection, but the absence of QS can cause excessive inflammation in the context of chronic infections. Our study also highlights the importance of extracellular bacterial secreted factors in modulating host innate immune responses.
Author Contributions
JY designed and performed most of experiments. K-ML, SP, YC, EL, and JS performed experiments. J-HP, OS, and SY provided materials and scientific advices. J-WY supervised the entire project and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dong-Su Jang, MFA (Medical Illustrator, Medical Support Section, Yonsei University College of Medicine) for his help with the illustrations. They also thank Dr. Hyun Seok Kim (Yonsei University College of Medicine) for assistance with statistical analysis.
Funding
This work was supported by National Research Foundation of Korea grants funded by the Korean Government (2014R1A4A1008625), and by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (2015M3A9B6073856).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00333/full#supplementary-material.
Footnotes
References
1. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13(4):325–32. doi: 10.1038/ni.2231
2. Storek KM, Monack DM. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev (2015) 265(1):112–29. doi:10.1111/imr.12289
3. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat Immunol (2006) 7(6):576–82. doi:10.1038/ni1346
4. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi:10.1038/nature10394
5. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477(7366):596–600. doi:10.1038/nature10510
6. Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol (2010) 11(5):385–93. doi:10.1038/ni.1859
7. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol (2015) 16(5):476–84. doi:10.1038/ni.3119
8. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One (2009) 4(10):e7446. doi:10.1371/journal.pone.0007446
9. McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog (2010) 6(11):e1001191. doi:10.1371/journal.ppat.1001191
10. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature (2014) 513(7517):237–41. doi:10.1038/nature13449
11. Gellatly SL, Hancock RE. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis (2013) 67(3):159–73. doi:10.1111/2049-632X.12033
12. Davies JC. Pseudomonas aeruginosa in cystic fibrosis: pathogenesis and persistence. Paediatr Respir Rev (2002) 3(2):128–34. doi:10.1016/S1526-0550(02)00003-3
13. Banerjee D, Stableforth D. The treatment of respiratory Pseudomonas infection in cystic fibrosis: what drug and which way? Drugs (2000) 60(5):1053–64. doi:10.2165/00003495-200060050-00006
14. Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol (2009) 7(9):654–65. doi:10.1038/nrmicro2199
15. Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, Tang H, et al. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun (1998) 66(1):43–51.
16. Smith RS, Iglewski BH. P. aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol (2003) 6(1):56–60. doi:10.1016/S1369-5274(03)00008-0
17. O’Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A (2013) 110(44):17981–6. doi:10.1073/pnas.1316981110
18. Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev (2012) 76(1):46–65. doi:10.1128/MMBR.05007-11
19. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature (2001) 410(6832):1099–103. doi:10.1038/35074106
20. Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol (2007) 37(11):3030–9. doi:10.1002/eji.200737532
21. Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci U S A (2008) 105(7):2562–7. doi:10.1073/pnas.0712183105
22. Lee KM, Park Y, Bari W, Yoon MY, Go J, Kim SC, et al. Activation of cholera toxin production by anaerobic respiration of trimethylamine N-oxide in Vibrio cholerae. J Biol Chem (2012) 287(47):39742–52. doi:10.1074/jbc.M112.394932
23. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9(8):847–56. doi:10.1038/ni.1631
24. Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics (2010) 11:395. doi:10.1186/1471-2105-11-395
25. Schmidtchen A, Holst E, Tapper H, Bjorck L. Elastase-producing Pseudomonas aeruginosa degrade plasma proteins and extracellular products of human skin and fibroblasts, and inhibit fibroblast growth. Microb Pathog (2003) 34(1):47–55. doi:10.1016/S0882-4010(02)00197-3
26. Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol Microbiol (1996) 21(6):1137–46. doi:10.1046/j.1365-2958.1996.00063.x
27. Wilder CN, Diggle SP, Schuster M. Cooperation and cheating in Pseudomonas aeruginosa: the roles of the las, rhl and pqs quorum-sensing systems. ISME J (2011) 5(8):1332–43. doi:10.1038/ismej.2011.13
28. Nouwens AS, Beatson SA, Whitchurch CB, Walsh BJ, Schweizer HP, Mattick JS, et al. Proteome analysis of extracellular proteins regulated by the las and rhl quorum sensing systems in Pseudomonas aeruginosa PAO1. Microbiology (2003) 149(Pt 5):1311–22. doi:10.1099/mic.0.25967-0
29. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ’prionoid’ activities that propagate inflammation. Nat Immunol (2014) 15(8):727–37. doi:10.1038/ni.2913
30. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi:10.1038/ni.2919
31. Hwang I, Park S, Hong S, Kim EH, Yu JW. Salmonella promotes ASC oligomerization-dependent caspase-1 activation. Immune Netw (2012) 12(6):284–90. doi:10.4110/in.2012.12.6.284
32. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A (2014) 111(20):7403–8. doi:10.1073/pnas.1402911111
33. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7(6):569–75. doi:10.1038/ni1344
34. Ran H, Hassett DJ, Lau GW. Human targets of Pseudomonas aeruginosa pyocyanin. Proc Natl Acad Sci U S A (2003) 100(24):14315–20. doi:10.1073/pnas.2332354100
35. Allen L, Dockrell DH, Pattery T, Lee DG, Cornelis P, Hellewell PG, et al. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J Immunol (2005) 174(6):3643–9. doi:10.4049/jimmunol.174.6.3643
36. Smith RS, Iglewski BH. Pseudomonas aeruginosa quorum sensing as a potential antimicrobial target. J Clin Invest (2003) 112(10):1460–5. doi:10.1172/JCI20364
37. LaFayette SL, Houle D, Beaudoin T, Wojewodka G, Radzioch D, Hoffman LR, et al. Cystic fibrosis-adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci Adv (2015) 1(6):e1500199. doi:10.1126/sciadv.1500199
38. van der Plas MJ, Bhongir RK, Kjellstrom S, Siller H, Kasetty G, Morgelin M, et al. Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat Commun (2016) 7:11567. doi:10.1038/ncomms11567
39. Look DC, Stoll LL, Romig SA, Humlicek A, Britigan BE, Denning GM. Pyocyanin and its precursor phenazine-1-carboxylic acid increase IL-8 and intercellular adhesion molecule-1 expression in human airway epithelial cells by oxidant-dependent mechanisms. J Immunol (2005) 175(6):4017–23. doi:10.4049/jimmunol.175.6.4017
40. Caldwell CC, Chen Y, Goetzmann HS, Hao Y, Borchers MT, Hassett DJ, et al. Pseudomonas aeruginosa exotoxin pyocyanin causes cystic fibrosis airway pathogenesis. Am J Pathol (2009) 175(6):2473–88. doi:10.2353/ajpath.2009.090166
41. Wilson R, Sykes DA, Watson D, Rutman A, Taylor GW, Cole PJ. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect Immun (1988) 56(9):2515–7.
42. Lau GW, Ran H, Kong F, Hassett DJ, Mavrodi D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect Immun (2004) 72(7):4275–8. doi:10.1128/IAI.72.7.4275-4278.2004
43. Telford G, Wheeler D, Williams P, Tomkins PT, Appleby P, Sewell H, et al. The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-l-homoserine lactone has immunomodulatory activity. Infect Immun (1998) 66(1):36–42.
44. Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, et al. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science (2008) 321(5886):259–63. doi:10.1126/science.1156499
45. Grabiner MA, Fu Z, Wu T, Barry KC, Schwarzer C, Machen TE. Pseudomonas aeruginosa quorum-sensing molecule homoserine lactone modulates inflammatory signaling through PERK and eI-F2alpha. J Immunol (2014) 193(3):1459–67. doi:10.4049/jimmunol.1303437
46. Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, et al. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun (2003) 71(10):5785–93. doi:10.1128/IAI.71.10.5785-5793.2003
47. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med (2007) 204(13):3235–45. doi:10.1084/jem.20071239
48. Reiniger N, Lee MM, Coleman FT, Ray C, Golan DE, Pier GB. Resistance to Pseudomonas aeruginosa chronic lung infection requires cystic fibrosis transmembrane conductance regulator-modulated interleukin-1 (IL-1) release and signaling through the IL-1 receptor. Infect Immun (2007) 75(4):1598–608. doi:10.1128/IAI.01980-06
49. Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J Immunol (2012) 189(9):4231–5. doi:10.4049/jimmunol.1201447
50. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107(7):3076–80. doi:10.1073/pnas.0913087107
51. Arlehamn CS, Petrilli V, Gross O, Tschopp J, Evans TJ. The role of potassium in inflammasome activation by bacteria. J Biol Chem (2010) 285(14):10508–18. doi:10.1074/jbc.M109.067298
52. Bardoel BW, van der Ent S, Pel MJ, Tommassen J, Pieterse CM, van Kessel KP, et al. Pseudomonas evades immune recognition of flagellin in both mammals and plants. PLoS Pathog (2011) 7(8):e1002206. doi:10.1371/journal.ppat.1002206
53. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest (2013) 123(4):1630–7. doi:10.1172/JCI66142
54. Faure E, Mear JB, Faure K, Normand S, Couturier-Maillard A, Grandjean T, et al. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. Am J Respir Crit Care Med (2014) 189(7):799–811. doi:10.1164/rccm.201307-1358OC
55. Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A (2006) 103(22):8487–92. doi:10.1073/pnas.0602138103
56. Hoffman LR, Kulasekara HD, Emerson J, Houston LS, Burns JL, Ramsey BW, et al. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J Cyst Fibros (2009) 8(1):66–70. doi:10.1016/j.jcf.2008.09.006
Keywords: bacterial secretant, flagellin, inflammasome, Pseudomonas aeruginosa, quorum sensing
Citation: Yang J, Lee K-M, Park S, Cho Y, Lee E, Park J-H, Shin OS, Son J, Yoon SS and Yu J-W (2017) Bacterial Secretant from Pseudomonas aeruginosa Dampens Inflammasome Activation in a Quorum Sensing-Dependent Manner. Front. Immunol. 8:333. doi: 10.3389/fimmu.2017.00333
Received: 16 January 2017; Accepted: 08 March 2017;
Published: 27 March 2017
Edited by:
Pablo Pelegrin, Biomedical Research Institute of Murcia – Hospital Universitario Virgen Arrixaca (IMIB-Arrixaca), SpainCopyright: © 2017 Yang, Lee, Park, Cho, Lee, Park, Shin, Son, Yoon and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Je-Wook Yu, amV3b29reXVAeXVocy5hYw==