 Sonia Carta
Sonia Carta Claudia Semino
Claudia Semino Roberto Sitia
Roberto Sitia Anna Rubartelli
Anna Rubartelli- 1Cell Biology Unit, IRCCS AOU San Martino-IST, Genova, Italy
- 2Unit of Protein Transport and Secretion, Division of Genetics and Cell Biology, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Vita-Salute San Raffaele University, Milan, Italy
Infectious and sterile inflammation is induced by activation of innate immune cells. Triggering of toll-like receptors by pathogen-associated molecular pattern or damage-associated molecular pattern (PAMP or DAMP) molecules generates reactive oxygen species that in turn induce production and activation of pro-inflammatory cytokines such as IL-1β. Recent evidence indicates that cell stress due to common events, like starvation, enhanced metabolic demand, cold or heat, not only potentiates inflammation but may also directly trigger it in the absence of PAMPs or DAMPs. Stress-mediated inflammation is also a common feature of many hereditary disorders, due to the proteotoxic effects of mutant proteins. We propose that harmful mutant proteins can induce dysregulated IL-1β production and inflammation through different pathways depending on the cell type involved. When expressed in professional inflammatory cells, stress induced by the mutant protein activates in a cell-autonomous way the onset of inflammation and mediates its aberrant development, resulting in the explosive responses that hallmark autoinflammatory diseases. When expressed in non-immune cells, the mutant protein may cause the release of transcellular stress signals that trigger and propagate inflammation.
Introduction
The term “autoinflammation” (1) groups syndromes with different etiologies characterized by systemic inflammation in the absence of detectable infections and/or autoimmunity. Autoinflammatory diseases are disorders of the innate immune system, sharing recurrent episodes of fever, rash, joint pain, neutrophilia, and increased inflammatory markers. Most of them are monogenic, and the causative gene relates to the innate immune system. Examples are MEFV/pyrin in familial Mediterranean fever (FMF), TNFRSF1A/TNF receptor type 1 in TNF receptor-associated periodic syndrome (TRAPS), and nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing 3 (NLRP3) in cryopyrin-associated periodic syndromes (CAPS) (2).
The reversal of clinical symptoms in CAPS patients upon treatment with recombinant IL-1 receptor antagonist (Anakinra) or with IL-1β blocking agents (e.g., Canakinumab, a neutralizing antibody) provided compelling ex adjuvantibus evidence for the key role of IL-1β (3). The efficacy of anti-IL-1 drugs suggested that “gain-of-function” mutations in NLRP3, a central component of the inflammasome, cause uncontrolled IL-1β production, in turn responsible for the severe inflammatory symptoms (4, 5). Less expectedly, the same drugs displayed strong therapeutic effects also in autoinflammatory diseases, where the causative gene is not directly involved in IL-1β production and regulation (2, 3). A representative case is TRAPS, a disease characterized by recurrent episodes of long-lasting fever, pain, and fasciitis. Despite TRAPS is caused by mutations in p55 TNF receptor type I, patients showed no or modest response to TNFα inhibition (6), whereas IL-1β-blocking agents have high efficacy (7). These observations suggest that the presence of a mutated protein in inflammatory cells, independently from its function, activates mechanisms converging on dysregulated IL-1β secretion.
In this perspective article, we propose a pro-inflammatory role for cell stress and the responses it elicits in some hereditary diseases, and suggest that stress is a central player in the pathophysiology of autoinflammatory disorders, due to its presence in innate immune cells.
Stress and Inflammation
Inflammation is traditionally defined as a reaction to infectious or sterile injuries, aimed at recruiting molecules and cells of the immune system to the tissue where the damage is taking place and restoring homeostasis. Inflammation is initiated by activation of pattern recognition receptors on inflammatory cells, by two subclasses of ligands responsible for infectious and sterile inflammation, respectively (8, 9): pathogen-associated molecular patterns and damage-associated molecular patterns (PAMPs and DAMPs). The former are part of pathogens, while the latter are components of cells or extracellular matrix released or degraded upon cell and tissue damage. Additional factors concur in determining the onset, duration, and intensity of inflammatory responses. Among these, particularly important is cell stress due to starvation, enhanced metabolic demand, cold or heat, altered proteostasis. The most common and well studied cell stresses are endoplasmic reticulum (ER) stress and oxidative stress that are counteracted by highly conserved responses. These responses share common traits, for example, eIF2α phosphorylation, with transient translational inhibition and transcriptional activation of chaperones and antioxidants (10). This integrated stress response prevents the toxicity caused by misfolded proteins [named “proteotoxicity” (10)] and limits reactive oxygen species (ROS)-based vicious circles. If excessive or prolonged, however, virtually all stress responses become maladaptive and induce inflammation due to activation of chemokine genes or, in case of cell damage, release of DAMPs that recruit inflammatory cells (11).
Oxidative stress is due to excessive production and/or deficient detoxification of ROS. These can be abundantly generated by mitochondria during oxidative phosphorylation (12) and by flavoenzymes like NADPH oxidases (NOX) (13). In cells of the innate immune system, phagocytosis and toll-like receptor (TLR) triggering activate NOX to produce abundant H2O2 (14). H2O2 is released into phagosomes to clear microorganisms and induces pro-inflammatory cytokines and inflammation: however, it may generate oxidative stress (13, 14). ROS are also produced in the ER as a by-product of oxidative protein folding, particularly in conditions of ER stress, which elicit the unfolded protein response (UPR) (15, 16). ER stress occurs when misfolded proteins accumulate in the secretory pathway, and also during infections, lipid unbalance, and other metabolic defects (15). UPR, a complex set of intracellular signaling pathways, has evolved to respond to protein misfolding and restore ER homeostasis. In addition, UPR signaling has a recognized role in immunity and inflammation (16). Oxidative and ER stresses are intimately linked: the former can induce misfolding of secretory proteins impacting disulfide bond formation. On the other hand, ER stress leads to ROS production (17). In concert with ROS, a prolonged UPR can induce NF-κB-mediated chemokine production and recruit inflammatory cells. In turn, PAMP or DAMP can potentiate the UPR (16).
These vicious circuits are evident in many chronic disorders such as type 2 diabetes (18), obesity (19), lung respiratory disease (20), inflammatory bowel disease (21), non-alcoholic fatty liver disease (22), and cancer (23).
Also in many hereditary diseases, the mutant protein may alter proteostasis: if this occurs, stress and inflammation are induced. For example, in cystic fibrosis, different mechanisms contribute to the inflammatory lung disease that is the major cause of morbidity and mortality in patients affected by this disease. Firstly, the mutated cystic fibrosis transmembrane conductance regulator (CFTR) protein cannot fold properly into the ER lumen, causing accumulation of misfolded CFTR aggregates, ER stress, and UPR. In turn, UPR activates NF-κB inducing production of chemokines, such as IL-8, that recruit polymorphonuclear leukocytes (PMN). PMN increase the oxidative burden in the lung, with generation of ROS that amplify the production of IL-8 thus locally increasing PMNs (24). Moreover, upregulation of ROS inhibits autophagy with consequent accumulation of protein aggregates and lung inflammation (25). Finally, the mutant CFTR transporter is unable to channel antioxidants into the airways: oxidative stress is worsened and concurs to the hyperinflammatory phenotype (24).
In Duchenne muscle dystrophy, due to the defect of dystrophin, oxidative stress and UPR-activated NF-κB interactively promote fiber necrosis. Recruited macrophages generate inflammatory cytokines and ROS, thereby triggering vicious inflammatory waves (26, 27).
Differently from autoinflammatory disorders, in these cases, the mutant protein, being synthesized by epithelia or muscle, determines the release of stress signals that recruit leukocytes ultimately causing inflammation. These signals include small molecules like ROS and antioxidants, and proteins such as thioredoxin (28) and chemokines (as described above for cystic fibrosis, 24), which induce inflammation transcellularly, i.e., by recruiting and activating other cells (Figure 1A). When instead it is a professional inflammatory cell that produces a proteotoxic mutant, inflammation is generated in a cell-autonomous way and the onset, development, and outcome of it will be much worse for the host (Figure 1B).
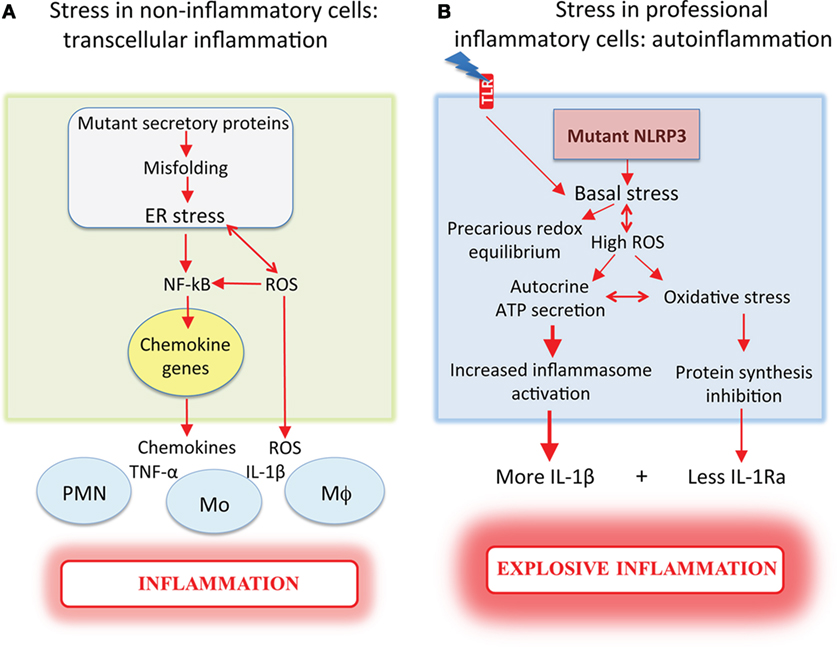
Figure 1. Mutant proteins induce more severe inflammation when expressed in professional inflammatory cells. (A) Non-inflammatory cells (e.g., epithelia or muscle), which express mutant proteins that undergo aberrant folding in the ER, exhibit ER stress and increased ROS, and promote NF-κB-mediated chemokine induction. The release of chemokines recruits inflammatory cells that secrete pro-inflammatory cytokines, ultimately causing inflammation. (B) Inflammatory cells from cryopyrin-associated periodic syndrome patients, which express mutated NLRP3, display cell stress with high reactive oxygen species (ROS) and antioxidant levels resulting in a precarious redox equilibrium that is deranged by toll-like receptor (TLR) stimulation. The high ROS levels facilitate autocrine ATP secretion, with increased and accelerated IL-1β secretion. When the antioxidant responses collapse, oxidative stress occurs with inhibition of protein synthesis responsible for the decrease of IL-1Ra secretion. Dysregulated cytokine production results in explosive inflammation. ROS are released in both conditions, triggering loops of amplification of stress and inflammation.
Stress in CAPS Monocytes
This hypothesis is supported by the observation that monocytes from CAPS patients, which express mutated NLRP3 molecules, display redox distress even before PAMP stimulation. Why mutant NLRP3 causes stress is unclear. A possible explanation is that it changes the affinity for the other components of the inflammasome complex (4), causing a disruption of the cytosolic homeostasis with induction of stress and integrated stress responses (10). Whatever the reason of NLRP3-induced stress, CAPS monocytes have higher basal ROS levels than monocytes from healthy donors but also higher expression of antioxidant systems (29–31) that allow them to maintain the redox homeostasis despite their stressed state. This equilibrium is, however, precarious, and CAPS monocytes can easily be induced to overreact, through pathways that largely depend on extracellular ATP, the most common inflammasome-activating signal (32). ATP is released by injured tissues, activated platelets, and other cells through pathways that are still ill defined (32). Unlike other pro-inflammatory cells, however, human monocytes do not need ATP from external sources. The accumulation of ROS upon TLR triggering (33) induces them to secrete ATP (34) that autocrinally or paracrinally stimulates cognate purinergic receptors (P2X7R) at the cell surface (32, 34). The ensuing lower intracellular [K+] induces inflammasome assembly and IL-1β secretion (35). The higher ROS levels in CAPS monocytes following TLR triggering facilitate ATP release that increases and accelerates IL-1β secretion (31) (Figure 1B).
Cell stress also decreases the threshold for IL-1β processing and secretion: minute amounts of TLR agonists, that in healthy monocytes are sufficient to trigger pro-IL-1β synthesis but not its processing and secretion, drive large amounts of IL-1β release in CAPS monocytes (31). Probably owing to their “pre-activated state,” small doses of TLR agonists increase ROS, inducing abundant ATP release, and IL-1β processing and secretion (31). This circuit explains why small traumas or infections that go undetected in healthy subjects can cause severe inflammatory manifestations in CAPS patients.
The effects described above occur soon after TLR stimulation. In later phases, the precarious redox equilibrium of CAPS monocytes is broken as antioxidant responses collapse. CAPS monocytes display damaged mitochondria (30), a further indication of the presence of oxidative stress [(12), Figure 2]. Interestingly, mitochondria are normal in CAPS lymphocytes, which do not express NLRP3, and in monocytes from healthy donors, which express wild-type NLRP3 (30), suggesting that mutant NLRP3 is indeed the causative agent of the oxidative stress. In this crucial phase, stress impacts also the production of IL-1Ra, normally secreted by activated monocytes a few hours after IL-1β to limit inflammation [(30), Figure 1]. Thus, deficient IL-1Ra production likely concurs in increasing the severity of the disease. Highlighting the dangerous stress-inflammation liaisons, insufficient IL-1Ra production may depend on eIF2α phosphorylation. Indeed, TLR-activated monocytes from CAPS patients, but not from healthy donors, display attenuated protein translation (30). Thus, IL-1ra mRNA is transcribed but stress prevents translation. Once more, oxidative and ER stress appear to be linked because IL-1Ra secretion is restored by antioxidants.
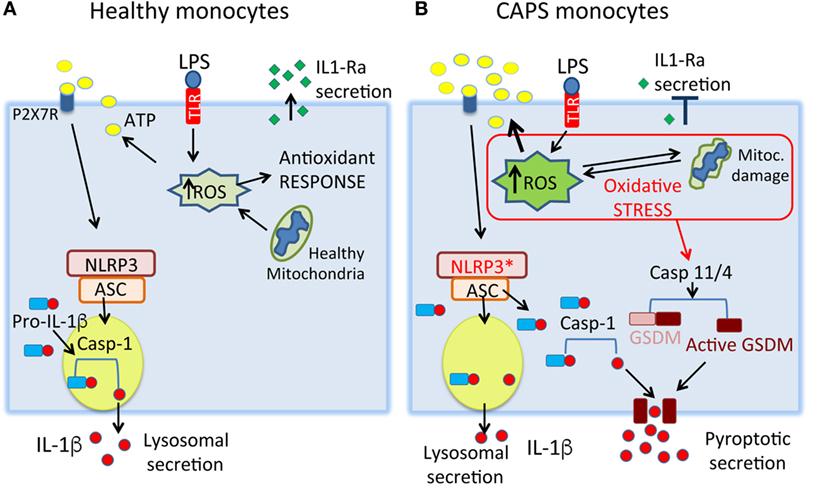
Figure 2. A model for stress-mediated cytokine secretion in cryopyrin-associated periodic syndromes (CAPS) monocytes. (A) In healthy monocytes, toll-like receptor (TLR) stimulation induces the production of low amounts of reactive oxygen species (ROS), rapidly neutralized by the antioxidant response. The ROS-induced ATP release is low, resulting in processing and secretion of little amounts of IL-1β through secretory lysosomes. The anti-inflammatory cytokine IL-1Ra is produced, contributing to switch off the inflammatory response. (B) In CAPS monocytes, small doses of TLR agonists induce a strong increase of ROS resulting in release of large amounts of endogenous ATP and IL-1β. The state of stress may trigger pyroptotic secretion of IL-1β through activation of caspase-11/4 that cleaves gasdermin D (GSDMD) generating a toxin-like N-terminal peptide that forms pores on the plasma membrane. Mature IL-1β, cleaved by the NLRP3 inflammasome, will be released through gasdermin D-formed pores. Later, failure of antioxidant response and mitochondria dysfunctions lead to severe oxidative stress, with impaired production of IL-1Ra. NLRP3*, mutated NLRP3.
Hyper-Stimulated Healthy Monocytes Recapitulate the Behavior of CAPS Monocytes
The above observations suggest that the increased IL-1β/IL-1Ra ratio in CAPS depends on the synergistic effects of NLRP3 mutations and stress. Combinations of PAMPs that stimulate surface and intracellular TLRs (LPS, R848, zymosan) were then used to induce a CAPS-like stress state in healthy monocytes (36). When given alone, each TLR agonist triggered the secretion of IL-1β and IL-1Ra by healthy monocytes. When provided simultaneously, however, they induced a superstimulation resulting in enhanced secretion of IL-1β but impaired release of IL-1Ra (36). The underlying molecular mechanisms are similar to those described in CAPS monocytes (29–31): super-stimulation induces ROS accumulation, responsible of the massive ATP release and IL-1β secretion, and of the consequent oxidative stress leading to inhibition of IL-1Ra production, despite normal IL-1Ra mRNA levels. Antioxidants restore IL-1Ra release by super-stimulated healthy monocytes, confirming the role of oxidative stress and recapitulating the phenotype of CAPS monocytes (36). However, the latter are constitutively stressed by the mutation (37) so that stimulation with low doses of a single TLR agonist strongly increases stress that drives prompt and abundant IL-1β secretion and, in a second phase, lowers IL-1Ra (30, 31). In healthy monocytes with balanced basal redox state (29, 36), instead, multiple TLR co-stimulation is needed to cause cell stress and derange the normal cytokine network (36). These observations may suggest that, in CAPS, mutations in NLRP3 are more important indirectly, triggering and enduring stress, than directly activating inflammasome.
Different Mechanisms for IL-1β Secretion: Does Stress Determine the Pathways of Secretion?
Since IL-1β is a potent and potentially dangerous mediator of inflammation, its production is tightly controlled virtually at all levels, including post-translationally (38, 39). IL-1β is synthesized as an inactive precursor, pro-IL-1β, and processed mainly by caspase-1, which in turn must be activated by the inflammasome. Only mature, 17 kDa IL-1β is then secreted. The underlying mechanisms are still poorly understood. Indeed, IL-1β secretion has been a problem for cell biologists, since it was shown that the cytokine lacks a secretory signal sequence (40). Initially, a popular view was that the cytokine was released by dying cells. However, further studies demonstrated that secretion of mature IL-1β is an active process, requires living cells, avoids the ER-Golgi route, and involves secretory lysosomes (41–44). In addition to this pathway, recent studies revealed another route for IL-1β release, involving pyroptosis. This is a highly inflammatory form of programmed cell death, which has been proposed to mediate IL-1β secretion under condition of strong stimulation such as infection with intracellular pathogens (45–48). According to this model of secretion, stressful stimuli (e.g., intracellular LPS) activate caspase-11 (the mouse homologous of human caspase-4). In turn, caspase-11 cleaves gasdermin D, generating toxin-like peptides that form pores on the plasma membrane, which allow secretion of mature IL-1β, but not of the 33 kDa precursor (48, 49). It remains to be determined how the pores guarantee transport selectivity.
The two pathways are not mutually exclusive, and the choice of lysosomal or pyroptotic secretion may depend on the strength of pro-inflammatory signals (Figure 2). Mild stimuli, such as low amounts of PAMPs triggering surface bound TLRs, would induce the less efficient but more regulated lysosomal pathway. Accordingly, low doses of LPS induce pro-IL-1β synthesis, but not ATP secretion (31): in the absence of a second trigger, therefore, pro-IL-1β is degraded by lysosomal proteases (31, 42) preventing unnecessary inflammation. Stronger stimuli, such as intracellular infections with gram-negative bacteria (45) could instead induce pyroptosis, causing massive release of IL-1β and possibly DAMPs, and dysregulated cytokine production (36).
Support to this hypothesis comes from our preliminary observations that human monocytes display more IL-1β-containing lysosomes when stimulated with LPS alone than with three agonists simultaneously triggering extra- and intracellular TLRs (unpublished results). Moreover, only in monocytes stimulated with extracellular LPS alone, do drugs interfering with lysosomal function modulate IL-1β secretion. Conversely, caspase-4 inhibitors block IL-1β release only in super-stimulated monocytes (unpublished results).
It is possible that the secretory lysosome-mediated mechanism is more active in low pathogen load or small trauma, as a way to restore the homeostasis. Differently, the pyroptosis-mediated secretion would intervene in severe inflammatory responses, characterized by strong or multiple stimuli such as it may occur in sepsis (50), diabetes (51) or cancer (52).
The ongoing stress could also determine the route of IL-1β secretion. Owing to the high ROS levels that favor ATP release, we predict that CAPS monocytes utilize preferentially the pyroptotic pathway. Accordingly, caspase-4 inhibition blocks IL-1β secretion by CAPS monocytes stimulated with a single TLR agonist, a condition that neither involves caspase-4 nor induces pyroptosis in healthy monocytes (unpublished) (Figure 2).
Cell-Autonomous Proteotoxic Stress in Monocytes Increases IL-1β Secretion in Autoinflammatory Diseases
Increased IL-1β secretion has been reported in vitro by monocytes from other autoinflammatory diseases, including FMF (53), TRAPS (54) hyperimmunoglobulinemia D syndrome (55), pyogenic sterile arthritis, pyoderma gangrenosum and acne (PAPA) (56), and also in the milder NLRP-12-associated periodic syndrome (57). As introduced above, anti-IL-1β therapies are the standard of care in these syndromes (58), suggesting that IL-1β is a key culprit. Nonetheless, the links between the mutated gene and IL-1β secretion are elusive. Remarkably, in these diseases, the mutant genes are expressed by monocytes that are under stress (53, 57, 59–61). It is tempting to speculate that stress and the ensuing responses converge to induce excessive IL-1β secretion, possibly switching from lysosomal to pyroptotic secretion (Figure 2). The consequences on disease severity are many, since pyroptosis-mediated secretion would alter the networks of pro- and anti-inflammatory cytokine production.
Stress-induced hyperinflammatory response may occur in other inherited diseases that are not (yet) classified as autoinflammatory diseases. This is the case of chronic granulomatous disease (CGD), a disorder linked to mutations in NOX2. Because of these mutations, phagocytes of CGD patients fail to produce ROS with consequent deficiency in bactericidal activity and increased susceptibility to infections (62). In addition, and consistent with the evidence that CGD is associated with increased inflammasome activation (63–65), patients often develop hyperinflammatory traits. Moreover, Anakinra induced significant clinical improvement in two cases with colitis (66). Thus, CGD was defined as a potentially lethal combination of immunodeficiency and excess inflammation (67), most likely due to cell-autonomous stress responses. Likewise, evidence is accumulating for a role of stress and inflammation in the pathogenesis of Gaucher disease, the inherited deficiency of lysosomal glucocerebrosidase (68). Monocyte/macrophages from these patients display increased secretion of IL-1β that depends on increased inflammasome activation, in turn due to the impaired autophagy secondary to the lysosomal enzyme deficiency (68). A further example is mucopolysaccharidosis type I, where, in innate immune cells, stress induced by lysosomal storage defects can upregulate immunity-related genes. In turn, these may be responsible for the severe inflammation-dependent pathologies observed in patients (69).
Conclusion and Perspectives
In essence, we propose that stress hallmarks monocytes from patients affected by autoinflammatory syndromes (and possibly other inherited diseases) that express mutant proteins not necessarily directly involved in IL-1β production. Stress induces inflammation and is, therefore, a key pathogenetic factor in these diseases. The stress levels contribute to determine the severity of the disease, and so do individual differences in resistance to oxidative stress, as proposed for chronic inflammation-mediated diseases such as diabetes (70). Accordingly, we showed clear correlations among basal stress, ongoing antioxidant responses, and disease severity in two CAPS patients sharing the same NLRP3 mutation (31). Extending these concepts, we suggest that a similar stress-related mechanism may be operative in other genetic diseases, where the mutant protein is present in monocytes and inflammation participates to disease progression. Considering that individual tolerance plays a major role (71), improving the responses to stress represents a promising therapeutic opportunity for these serious diseases.
Author Contributions
SC, CS, RS, and AR designed, wrote, and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Prof. M. E. Bianchi from Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Milan, Italy, for discussion and support and Dr. M. Gattorno from Pediatrics II Unit, G. Gaslini Institute, Genoa, Italy, for advice.
Funding
This study was supported in part by the Italian Ministry of Health (“Cinque per mille” and Ricerca Corrente to AR; PE-2011-02352286 to RS), by Telethon, Italy (grants no. GGP14144 to AR and GGP15059 to RS), by Associazione Italiana per la Ricerca sul Cancro (grants no. IG15434 to AR and IG18824 to RS), and Fondazione Cariplo (2015-0591 to RS).
Abbreviations
CAPS, cryopyrin-associated periodic syndromes; DAMPs, damage-associated molecular patterns; ER, endoplasmic reticulum; IL, interleukin; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing 3; NOX, NADPH oxidases; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TLRs, toll-like receptors; TRAPS, TNFRSF1A/TNF receptor type 1 in TNF receptor-associated periodic syndrome; UPR, unfolded protein response.
References
1. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Cen-tola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNFreceptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell (1999) 97:133–44. doi:10.1016/S0092-8674(00)80721-7
2. Rubartelli A. Autoinflammatory diseases. Immunol Lett (2014) 161:226–30. doi:10.1016/j.imlet.2013.12.013
3. Ter Haar N, Lachmann H, Ozen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis (2013) 72:678–85. doi:10.1136/annrheumdis-2011-201268
4. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity (2004) 20:319–25. doi:10.1016/S1074-7613(04)00046-9
5. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet (2001) 29:301–5. doi:10.1038/ng756
6. Jacobelli S, André M, Alexandra JF, Dodé C, Papo T. Failure of anti-TNF therapy inTNF receptor 1-associated periodic syndrome (TRAPS). Rheumatology (Oxford) (2007) 46:1211–2. doi:10.1093/rheumatology/kel298
7. Gattorno M, Pelagatti MA, Meini A, Obici L, Barcellona R, Federici S, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum (2008) 58:1516–20. doi:10.1002/art.23475
8. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
9. Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev (2007) 220:60–81. doi:10.1111/j.1600-065X.2007.00579.x
10. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep (2016) 17:1374–95. doi:10.15252/embr.201642195
11. Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell (2014) 54:281–8. doi:10.1016/j.molcel.2014.03.030
12. Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J (2011) 278:941–54. doi:10.1111/j.1742-4658.2011.08010.x
13. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol (2015) 12:5–23. doi:10.1038/cmi.2014.89
14. Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J Immunol (2004) 173:3589–93. doi:10.4049/jimmunol.173.6.3589
15. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest (2005) 115:2656–64. doi:10.1172/JCI26373
16. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol (2016) 16:469–84. doi:10.1038/nri.2016.62
17. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal (2014) 21:396–413. doi:10.1089/ars.2014.5851
18. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell (2010) 140:900–17. doi:10.1016/j.cell.2010.02.034
19. Pagliassotti MJ, Kim PY, Estrada AL, Stewart CM, Gentile CL. Endoplasmic reticulum stress in obesity and obesity-related disorders: an expanded view. Metabolism (2016) 65:1238–46. doi:10.1016/j.metabol.2016
20. Osorio F, Lambrecht B, Janssens S. The UPR and lung disease. Semin Immunopathol (2013) 35:293–306. doi:10.1007/s00281-013-0368-6
21. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134:743–56. doi:10.1016/j.cell.2008.07.021
22. Henkel A, Green RM. The unfolded protein response in fatty liver disease. Semin Liver Dis (2013) 33:321–9. doi:10.1055/s-0033-1358522
23. Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov (2015) 5:586–97. doi:10.1158/2159-8290.CD-14-1490
24. Blohmke CJ, Mayer ML, Tang AC, Hirschfeld AF, Fjell CD, Sze MA, et al. Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to p38 MAPK-mediated innate immune responses. J Immunol (2012) 189:5467–75. doi:10.4049/jimmunol.1103661
25. Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol (2010) 12:863–75. doi:10.1038/ncb2090
26. Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest (2007) 117:889–901. doi:10.1172/JCI30556
27. Hulmi JJ, Hentilä J, DeRuisseau KC, Oliveira BM, Papaioannou KG, Autio R, et al. Effects of muscular dystrophy, exercise and blocking activin receptor IIB ligands on the unfolded protein response and oxidative stress. Free Radic Biol Med (2016) 99:308–22. doi:10.1016/j.freeradbiomed.2016.08.017
28. Rubartelli A, Sitia R. Stress as an intercellular signal: the emergence of stress-associated molecular patterns (SAMP). Antioxid Redox Signal (2009) 11:2621–9. doi:10.1089/ars.2009.2377
29. Tassi S, Carta S, Delfino L, Caorsi R, Martini A, Gattorno M, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc Natl Acad Sci U S A (2010) 107:9789–94. doi:10.1073/pnas.1000779107
30. Carta S, Tassi S, Delfino L, Omenetti A, Raffa S, Torrisi MR, et al. Deficient production of IL-1 receptor antagonist and IL-6 coupled to oxidative stress in cryopyrin-associated periodic syndrome monocytes. Ann Rheum Dis (2012) 71:1577–81. doi:10.1136/annrheumdis-2012-201340
31. Carta S, Penco F, Lavieri R, Martini A, Dinarello CA, Gattorno M, et al. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci U S A (2015) 112:2835–40. doi:10.1073/pnas.1424741112
32. Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol (2013) 3:414. doi:10.3389/fimmu.2012.00414
33. Tassi S, Carta S, Vené R, Delfino L, Ciriolo MR, Rubartelli A. Pathogen-induced interleukin-1beta processing and secretion is regulated by a biphasic redox response. J Immunol (2009) 183:1456–62. doi:10.4049/jimmunol.0900578
34. Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligandsand induces IL-1β and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A (2008) 105:8067–72. doi:10.1073/pnas.0709684105
35. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi:10.1016/j.immuni.2013.05.016
36. Lavieri R, Piccioli P, Carta S, Delfino L, Castellani P, Rubartelli A. TLR costimulation causes oxidative stress with unbalance of proinflammatory and anti-inflammatory cytokine production. J Immunol (2014) 192:5373–81. doi:10.4049/jimmunol.1303480
37. Varga G, Gattorno M, Foell D, Rubartelli A. Redox distress and genetic defects conspire in systemic autoinflammatory diseases. Nat Rev Rheumatol (2015) 11:670–80. doi:10.1038/nrrheum.2015.105
38. Rubartelli A, Bajetto A, Allavena G, Cozzolino F, Sitia R. Post-translational regulation of interleukin 1 beta secretion. Cytokine (1993) 5:117–24. doi:10.1016/1043-4666(93)90050-F
39. Dinarello CA. Blocking IL-1 in systemic inflammation. J Exp Med (2005) 201:1355–9. doi:10.1084/jem.20050640
40. Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM, et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A (1984) 81:7907–11. doi:10.1073/pnas.81.24.7907
41. Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J (1990) 9:1503–10.
42. Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell (1999) 10:1463–75. doi:10.1091/mbc.10.5.1463
43. Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci U S A (2004) 101:9745–50. doi:10.1073/pnas.0308558101
44. Gardella S, Andrei C, Lotti LV, Poggi A, Torrisi MR, Zocchi MR, et al. CD8(+) T lymphocytes induce polarized exocytosis of secretory lysosomes by dendritic cells with release of interleukin-1beta and cathepsin D. Blood (2001) 98:2152–9. doi:10.1182/blood.V98.7.2152
45. Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A (2015) 112:6688–93. doi:10.1073/pnas.1421699112
46. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi:10.1038/nature15541
47. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi:10.1038/nature15514
48. Broz P. Immunology: caspase target drives pyroptosis. Nature (2015) 526:642–3. doi:10.1038/nature15632
49. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi:10.1038/nature18590
50. Aziz M, Jacob A, Wang P. Revisiting caspases in sepsis. Cell Death Dis (2014) 5:e1526. doi:10.1038/cddis.2014.488
51. Pillon NJ, Chan KL, Zhang S, Mejdani M, Jacobson MR, Ducos A, et al. Saturated fatty acids activate caspase-4/5 in human monocytes, triggering IL-1β and IL-18 release. Am J Physiol Endocrinol Metab (2016) 311:825–35. doi:10.1152/ajpendo.00296.2016
52. Chu Q, Jiang Y, Zhang W, Xu C, Du W, Tuguzbaeva G, et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget (2016) 7(51):84658–65. doi:10.18632/oncotarget.12384
53. Omenetti A, Carta S, Delfino L, Martini A, Gattorno M, Rubartelli A. Increased NLRP3-dependent interleukin 1β secretion in patients with familial Mediterranean fever: correlation with MEFV genotype. Ann Rheum Dis (2014) 73:462–9. doi:10.1136/annrheumdis-2012-202774
54. Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U S A (2010) 107:9801–6. doi:10.1073/pnas.0914118107
55. Drenth JP, van der Meer JW, Kushner I. Unstimulated peripheral blood mononuclear cells from patients with the hyper-IgD syndrome produce cytokines capable of potent induction of C-reactive protein and serum amyloid A in Hep3B cells. J Immunol (1996) 157:400–4.
56. Omenetti A, Carta S, Caorsi R, Finetti M, Marotto D, Lattanzi B, et al. Disease activity accounts for long-term efficacy of IL-1 blockers in pyogenic sterile arthritis pyoderma gangrenosum and severe acne syndrome. Rheumatology (Oxford) (2016) 55:1325–35. doi:10.1093/rheumatology/kew031
57. Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum (2011) 63:830–9. doi:10.1002/art.30170
58. Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med (2014) 65:223–44. doi:10.1146/annurev-med-061512-150641
59. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med (2011) 208:519–33. doi:10.1084/jem.20102049
60. Bachetti T, Chiesa S, Castagnola P, Bani D, Di Zanni E, Omenetti A, et al. Autophagy contributes to inflammation in patients with TNFR-associated periodic syndrome (TRAPS). Ann Rheum Dis (2013) 72:1044–52. doi:10.1136/annrheumdis-2012-201952
61. van der Burgh R, Nijhuis L, Pervolaraki K, Compeer EB, Jongeneel LH, van Gijn M, et al. Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J Biol Chem (2014) 289:5000–12. doi:10.1074/jbc.M113.536920
62. Goldblatt D. Recent advances in chronic granulomatous disease. J Infect (2014) 69(Suppl 1):S32–5. doi:10.1016/j.jinf.2014.07.013
63. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood (2010) 116:1570–3. doi:10.1182/blood-2010-01-264218
64. van Bruggen R, Köker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood (2010) 115:5398–400. doi:10.1182/blood-2009-10-250803
65. van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A (2010) 107:3030–3. doi:10.1073/pnas.0914795107
66. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A (2014) 111:3526–31. doi:10.1073/pnas.1322831111
67. Deresinski S. Chronic granulomatous disease: a potentially lethal combination of immunodeficiency and excess inflammation. Clin Infect Dis (2014) 59:III–IV. doi:10.1093/cid/ciu351
68. Aflaki E, Moaven N, Borger DK, Lopez G, Westbroek W, Chae JJ, et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell (2016) 15:77–88. doi:10.1111/acel.12409
69. Khalid O, Vera MU, Gordts PL, Ellinwood NM, Schwartz PH, Dickson PI, et al. Immune-mediated inflammation may contribute to the pathogenesis of cardiovascular disease in mucopolysaccharidosis type I. PLoS One (2016) 11:e0150850. doi:10.1371/journal.pone.0150850
70. Ramprasath T, Selvam GS. Potential impact of genetic variants in Nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Curr Med Chem (2013) 20:4680–93. doi:10.2174/09298673113209990154
Keywords: autoinflammatory syndromes, endoplasmic reticulum stress, IL-1β, inflammation, monocytes, NLRP3 inflammasome, oxidative stress, toll-like receptor
Citation: Carta S, Semino C, Sitia R and Rubartelli A (2017) Dysregulated IL-1β Secretion in Autoinflammatory Diseases: A Matter of Stress? Front. Immunol. 8:345. doi: 10.3389/fimmu.2017.00345
Received: 22 December 2016; Accepted: 10 March 2017;
Published: 04 April 2017
Edited by:
Alessandra Soriano, Università Campus Bio-Medico, ItalyReviewed by:
Anja Schwenzer, Oxford University, UKNicolas Riteau, National Institutes of Health, USA
Copyright: © 2017 Carta, Semino, Sitia and Rubartelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Rubartelli, YW5uYS5ydWJhcnRlbGxpQGhzYW5tYXJ0aW5vLml0
†These authors have contributed equally to this work.