 Rani Soni
Rani Soni Drista Sharma
Drista Sharma Praveen Rai
Praveen Rai Bhaskar Sharma
Bhaskar Sharma Tarun K. Bhatt
Tarun K. Bhatt- Department of Biotechnology, School of Life sciences, Central University of Rajasthan, Ajmer, India
Irrespective of various efforts, malaria persist the most debilitating effect in terms of morbidity and mortality. Moreover, the existing drugs are also vulnerable to the emergence of drug resistance. To explore the potential targets for designing the most effective antimalarial therapies, it is required to focus on the facts of biochemical mechanism underlying the process of parasite survival and disease pathogenesis. This review is intended to bring out the existing knowledge about the functions and components of the major signaling pathways such as kinase signaling, calcium signaling, and cyclic nucleotide-based signaling, serving the various aspects of the parasitic asexual stage and highlighted the Toll-like receptors, glycosylphosphatidylinositol-mediated signaling, and molecular events in cytoadhesion, which elicit the host immune response. This discussion will facilitate a look over essential components for parasite survival and disease progression to be implemented in discovery of novel antimalarial drugs and vaccines.
Introduction
Plasmodium falciparum is one of the major afflictions to the human health. The annual report of malaria speculated around 2.1 million cases of disease and more than 0.4 million mortality cases in 2015 (1). The emergence of drug resistance species appended the severity of the problem. The intracellular inhabitation of Plasmodium makes the substantial modification in the host cell environment. After exoerythrocytic schizogony, the merozoites are released from hepatocytes into the blood stream and targeted to host erythrocytes, which marks the beginning of erythrocytic phase. Inside the erythrocyte, they multiply asexually and released following the rupture of RBC. These newly released merozoites make the recurrence of the same process for the fresh erythrocytes (2). Infection of fresh erythrocyte requires the egress from infected erythrocyte and reinvasion to fresh one. During egress and internalization process, multiple molecular interactions between the surface proteins of merozoites and receptors on the host erythrocytes come into play (3). Moreover, the parasitic entry into the host cell modulates the host environment to suit its own needs and to stay clear from the host defense. The modulation processes are coupled with a well-defined signaling mechanism, which can be described at cellular and molecular levels (4, 5). A few of the large repertoire of parasite proteins involved in modulating the host signaling pathways are summarized in the Table 1. Despite of unraveling of the functions and involvement of molecules in signaling pathways during the parasite life cycle, certain proteins remained uncharacterized. The analysis of different signaling mechanism during the asexual erythrocytic stage (6, 7) will be advantageous in understanding the strategies used by parasite to thrive successfully in the host, which would give novel input in planning an effective antimalarial therapeutic approach.

Table 1. Plasmodium protein triggering signals associated with modulation in host response.
Cyclic Nucleotide-Based Signaling During Malaria
Signals from the extracellular environment are transmitted inside the cell through the secondary messenger molecules like cyclic adenylyl monophosphate (cAMP) and cyclic guanylyl monophosphate (cGMP). The homologous genes for enzymatic components involved in cyclic nucleotide-based signaling like adenylyl cyclase (AC), guanylyl cyclase (GC), cGMP-dependent protein kinase [protein kinase G (PKG)], and a regulatory and catalytic subunit of cAMP-dependent protein kinase, and nucleotide phosphodiesterase (PDE) have been identified (18, 19) in malaria parasite.
Plasmodium falciparum Protein Kinase A (PfPKA) and cAMP
The first evidence of the cAMP signaling in malaria parasite arose through an experimental study in which the addition of external cAMP to Plasmodium culture was shown to positively affect the exflagellation or gametocyte formation during the ring stage of the parasite (20). However, the parasitic AC differs biochemically from that of host counterpart. Forskolin and Alf4, the activators of mammalian AC, and GTPγs, the activator of G protein, are unable to cause stimulation in parasitic AC (21). Moreover, expression of G stimulatory α have been demonstrated in early asexual stage and mature sexual stage. So, it was assumed that G protein might be implicated in the signaling during gametogenesis. However, this finding leads to a dilemma because the Plasmodium genome is devoid of gene corresponding to the G protein (22).
Adenylyl cyclase and cAMP signaling were demonstrated to play an important role during the infection of hepatocytes by sporozoites. The migration of sporozoites across the host hepatocytes results in their activation and triggering of apical regulated exocytosis. The sporozoites can be activated externally by calcium ionophore, which are then able to infect liver cell without migration (23). The gene knockout experiment of ACα in Plasmodium berghei explained the prevention of the exocytosis along with the reduced infectivity. However, the results were reciprocated after the reintroduction of the ACα gene into the mutant. Thus, the involvement of cAMP-mediated signaling in the initial phase of infection was confirmed. Besides this, the ACα also share homology with K+ channels, which are required for exocytosis in sporozoites (24). Not only sporozoites but also merozoites invasion process also involves cAMP-dependent signaling. During invasion, there occurs the formation of tight junctions with host cells, which leads to the secretion of apical organelle containing several proteins like gliding-associated protein 45 and apical membrane antigen 1 (AMA-1) (25, 26). The whole event of invasion is regulated by the cAMP-dependent phosphorylation of protein AMA-1 mediated by PfPKA (Figure 1). The mutational analysis of AMA-1 showed the hampering of the invasion process due to a change in phosphorylation site (serine 610) (27). It was evidenced from another study that merozoite proteins, particularly localized to the microneme and rhoptry organelle, get secreted and interact with receptors on fresh erythrocytes. The secretion of proteins involves a stepwise signaling cascade initiated due to the exposure of the low K+ extracellular environment (28–32). Low K+ triggers activation of PfACβ followed by an increase in cAMP. The cAMP activates Epac (exchange protein activated by cAMP) pathway, which subsequently cause an increase in PfPKA coupled with elevation of Ca2+ level (33). In the Epac pathway, Ras-proximate-1 (Rap1) converted to Rap1-GTP and further activates phospholipase C (PLC). The activated PLC induces calcium-dependent protein kinase 1 and calcineurin, which eventually leads to the secretion of microneme and rhoptry proteins (33–35) (Figure 1). Intense analysis of the pathway and its regulating components rendered a better understanding of overall mechanism, which can be implemented for inhibiting parasitic growth, invasion, and malaria prevention.
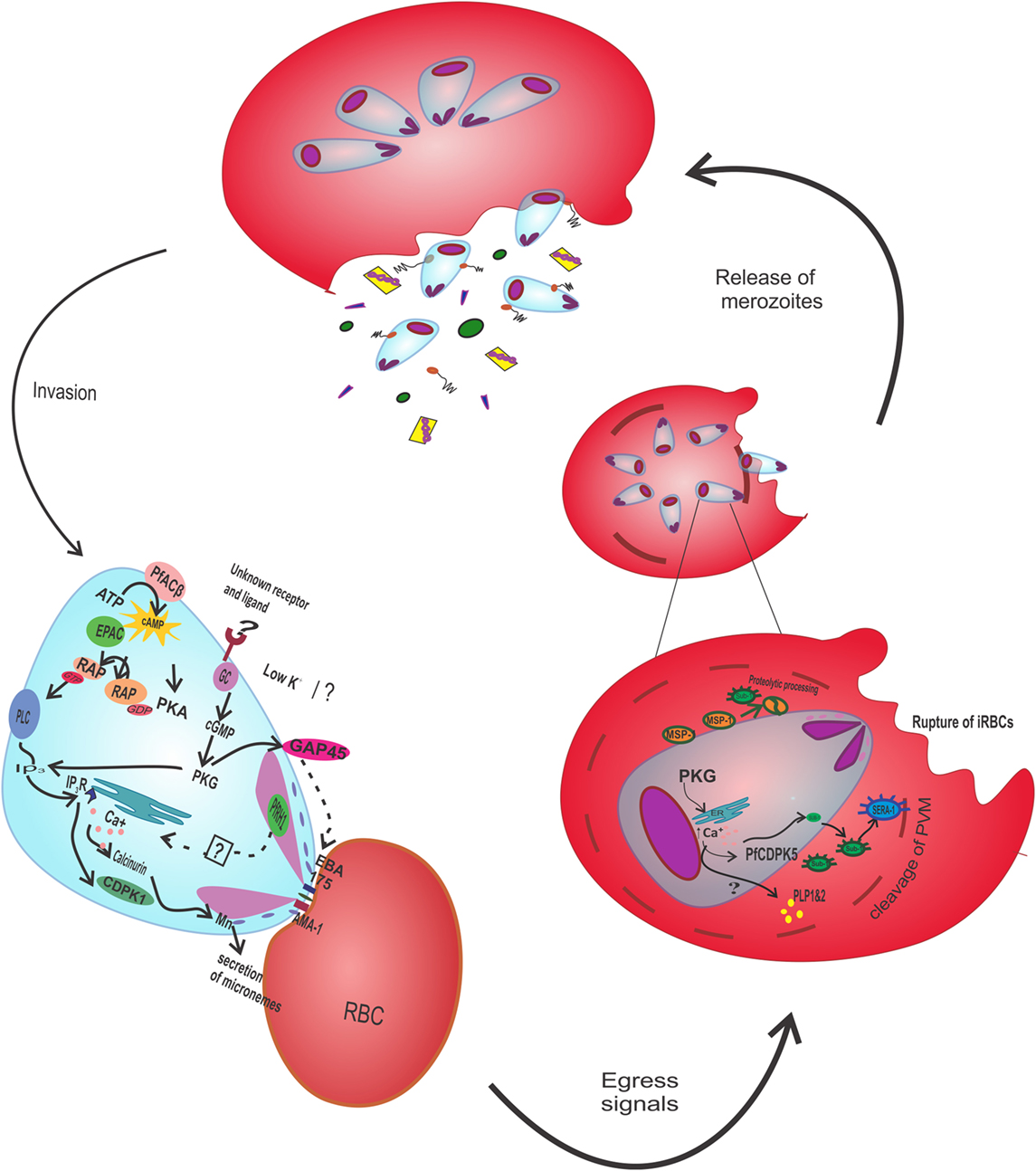
Figure 1. Signaling during egress and invasion of merozoites: invasion is mediated through the secretion of apical organelle, microneme and rhoptry organelle containing AMA-1 and GAP45, and EBA175. PfACβ triggers the cAMP level, boosting Epac pathway thereby phosphorylating RAP-GDP into RAP-GTP. RAP-GTP activates PLC to produce IP3 that binds to IP3R on endoplasmic reticulum, releasing calcium ions. Calcium ions bind to calcineurin and also activate CDPK1, required for the discharge of microneme and rhoptry. During egress, the cleavage of parasitophorous vacuole and rupture of infected erythrocyte require the proteolytic processing of PfSERA and PfMSP-1. The activation of PfSERA and PfMSP-1 occurs through the proteolytic activity of PfSUB-1, caused by increment of intracellular calcium ions in response to PKG. Calcium ions also regulates the release of PLP1 and PLP2 and activates PfCDPK5. AMA-1, apical membrane antigen 1; GAP45, glideosome-associated protein 45; EBA175, erythrocyte-binding antigen 175; PfACβ, Plasmodium falciparum adenylyl cyclase-β; cAMP, cyclic adenylyl monophosphate; Epac, exchange protein activated by cAMP; Rap-GDP, Ras proximate guanylyl diphosphate; PLC, phospholipase C; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; CDPK1, calcium dependent protein kinase 1; PfSERA, Plasmodium falciparum serine-like repeat antigen; PfMSP-1, Plasmodium falciparum merozoite surface protein; PfSUB-1, Plasmodium falciparum subtilisin like protease; PKG, protein kinase G; PLP, perforin-like protein.
The parasitic infection remodels the membrane of host erythrocytes for the nutrient acquisition and maintaining the balance of electrolytes. The electrophysiology-based studies indicated a peak of conductance of anionic channels during Plasmodium infection (36). The conductance of anions across the infected host cell membrane is mainly regulated by cAMP signaling. The experimental addition of PKA and ATP to the uninfected erythrocyte caused the upregulation of anion conductance (26), while the process overturned on the addition of alkaline phosphatase (ALP) or by dephosphorylation (37, 38). In this contest, the dependency of anion channel regulation on cAMP was further confirmed through an experiment with PfPKI (H89 cAMP-dependent protein kinase inhibitor) or by developing transgenic parasite with higher expression of PfPKA-R (phosphokinase A-regulatory subunit), which binds to cAMP, thereby downregulating the process and directly affecting the parasitic growth (39, 40). The signaling cascade either involves parasitic components or host is still controversial. Also, little is known about the actual substrates of PKA. The above discussion presents the essentiality of cAMP. Cyclic nucleotides are produced by AC and GC on hydrolysis, which requires PDE (19, 41). Therefore, PDE regulates the production and functioning of cAMP and cGMP. In P. falciparum, PfPDE1 was the first-reported PDE, which is specific for cGMP (42). Among various PDE types, PDE4 is predominant in the immune cells. Implementation of PDE4 inhibitors was found to enhance chemokines production and elicit inflammatory response. To delineate the regulatory mechanism of cyclic nucleotide, parasite-specific PDE inhibitors were developed. Zaprinast, a PDE5 inhibitor, and 5-benzyl-3-isopropyl-1H-pyrazolo[4, 3-d] pyrimidin-7(6H)-one, inhibitor of PfPDEα, were potentially most effective inhibitors blocking the parasite proliferation (43). PDE inhibitors can be explored in developing antimalarial therapy (44).
PfPKG and cGMP
The protein sequence analysis of PfPKG with respect to vertebrate counterpart revealed certain differences between their sequence features. For instance, the presence of three cAMP/cGMP binding motif, a degenerated cGMP binding motif and lack of leucine zipper motif required for dimerization and insensitivity towards cGMP analogue (45, 46). Along with its role in gametogenesis, cGMP has been demonstrated to express its functionality during the ring and schizogony stage (45). A known PKG inhibitor, 4-[2-(4-fluorophenyl)-5-(1-methylepiperidine-4-yl)-1H pyrrol-3-yl] pyridine (compound 1), has a retarding effect on parasitemia level (47, 48). In contrast, the mutant strain with genetically modified PfPKG showed normal development in the presence of inhibitor compound 1. These findings established the key role of cGMP signaling during the asexual phase of parasite development.
Conditional knockout study carried on P. berghei PKG (PfPKG) at late liver sporozoites (LS) stage depicted that parasite can infect hepatic HePG2 cell lines, but failed to get released from hepatocytes as merosome. Sporozoites at this stage elicited protective immune response in the host (49). Thus, it reflects an essentiality of cGMP signaling during the formation and release of merosome. So far, the precise role of PKG and trigger for its stimulation is not clear. A deep insight into the signaling involved in LS would be opportune to control disease pathologies at the erythrocytic stage and can be advantageous in search of prospective medication of malaria and preerythrocytic vaccine development (49).
During the egress cascade, PKG is required in proteolytic processing of proteins such as proteolytic processing of P. falciparum serine-like repeat antigen (PfSERAs) and PfMSP-1 by P. falciparum subtilisin-like protease (PfSUB1) (Figure 1), which in turn are involved in merozoite egress and secretion of apical organelle (50, 51). The proteolytic processing of PfMSP-1 was prevented with the inhibitor of PfPKG, compound 1 without affecting the activity of SUB1. Noticeable, in an experiment involving schizonts deficient in PfCDPK5 showed normal processing of PfMSP-1 in the presence of compound 1. Therefore, it indicates the role of PfCDPK5 downstream to PKG (52). The phosphoproteomics and chemical genetic approach involving the use of PKG inhibitor, compound 2 4-[7-[(dimethylamino) methyl]-2-(4-fluorophenyl)imidazo[1,2-α]pyridin-3-yl]pyrimidin-2-amine along with strain containing PKG mutant allele (PfPKGT618Q), resistant to compound 2 further demonstrated the role of PKG in the signaling event. Comparative analysis of the effect of treatment with compound 2 in wild-type and PfPKG mutant brought out various cellular targets of PfPKG involved in egress and invasion (53). The significance of PfPKG upstream to calcium signaling was indicated by a study in which phosphorylation of PfCDPK1 occurred in a PKG-dependent manner (50, 53). Moreover, PKG plays a significant role in regulating the cytosolic calcium signaling both during egress and at the asexual stage (54). The key importance of PfPKG in phosphorylation of the substrates gives a reflection that both PfPKG and its substrates are the therapeutic candidates in malaria treatment and in transmission blocking (55).
ATP as Signaling Molecule in Infected Erythrocytes
During malaria, extensive modification in erythrocytes is accompanied by changes in membrane permeability (56, 57). It has been well documented that hypoxic signals or surface deformation of RBC accounts for the release of ATP (58, 59). An elevated level of ATP has been recorded both in the extracellular environment and in the cytoplasm of host and parasite (60, 61). Parasitemia is proved to be directly linked to level of ATP (62). Any diminution of ATP in the medium rendered the Plasmodium ineffective to infect the fresh erythrocytes (60, 63, 64). The essentiality of ATP in the invasion process of parasite was confirmed by thwarting the entry of the parasite through inhibitor-mediated blocking of purinergic receptor and the addition of apyrase (65). The underlying mechanism of this inhibition involved the boosting up of the cytoplasmic calcium levels by extracellular ATP (65). The effect of purinergic signaling is mediated through phosphorylation of skeleton protein like spectrin present in erythrocyte (64, 65). Purinoceptor signaling is linked to induction of the new permeation pathway. Therefore, purinoceptors antagonist, suramin, reduces the membrane permeability and leads to deterioration in parasitic growth, both in vivo and in vitro (66). Purinoceptor blocker, suramin, and pyridoxal phosphate-6-azophenyle-2′,4′-disulphonic acid alter the proteolytic processing of protein like MSP-1, which is involved in the invasion, while the addition of ATP was found to trigger the intracellular proteolytic event in P. berghie and Plasmodium yoelii (67). It suggests the involvement of purinergic receptors in ATP signaling. The purinergic receptors that are expressed on the parasite surface are phylogenetically distinct from human counterparts. Thus, these receptors can be exploited as targets so as to design inhibitors of parasite invasion process (68, 69). ATP is also thought to be involved in the host-induced inflammation following the malaria infection (70).
P-21 Activated Protein Kinase (PAK)-MEK Signaling Pathway in Infected Erythrocytes
In eukaryotes, the regulation of cell cycle is dependent on mitogen-activated protein kinases (MAPKs). In this pathway, signals are transmitted successively to its respective components, MAPKKKs (MEKKs), MAPKKs [MEK/extracellular signal regulated kinase (ERK)], and finally to MAPK (71, 72). The Plasmodium kinome study revealed the presence of two homologs of human MAPK, namely, Pfmap-1 (73, 74) and Pfmap-2 (74, 75). Pfmap-1 is expressed in both asexual and gametocyte stage, while the expression of Pfmap-2 is found in gametocytes only. The reverse genetic-based approach demonstrated the essentiality of Pfmap-2 in the asexual development of P. falciparum (74). The sequence alignment depicted that the homology of gene PfPK7 with human MAPKK3 at its C-terminal region and the N-terminal region was aligned with fungi PKA. The assay conducted to analyze the inhibition via phosphokinase inhibitors, namely, PKI and H89 showed that PfPK7 activity was not affected. Similarly, MEK inhibitor, U0126, had no inhibitory effect on PfPK7 activity. It was suggested that PfPK7 is not an ortholog of MAPKK due to the absence of MAPKK activation site (76). The data provide an evidence of the absence of a regular MAPK pathway in Plasmodium. Due to the absence in parasite, the role of MEK of host erythrocytes in parasite development was hypothesized and confirmed. The immunological experiment evidenced the modulation of the host erythrocyte MAPK pathway. When compared, the level of phosphorylated MEK in infected erythrocytes was found much higher than uninfected ones. Moreover, MEK inhibitors, U0126 and PD184352, had parasiticidal effect on trophozoite, while the invasion process remained ineffective (13). It was found that activation of MEK is relying on MEKK-independent mechanism, which involves PAK (77). The involvement of PAK-1 was evident by observing the inhibition of parasitic growth due to reduction in the phosphorylation of MEK-1 by the use PAK-1 inhibitor, IPA-3 (78). The activation of PAK-1 occurs as a consequence of erythrocyte remodeling during parasite infection (79, 80). Furthermore, the importance and requirement of MEK-1 and substrate of MEK-1-PAK-1 pathway for parasite are yet obscured (13). Targeting the human kinase would provide a discrete strategy for the development of antimalarial therapy. It could be advantageous as many kinase inhibitors are known to pass the phases of clinical trials as anticancerous agents. If such inhibitors have “cidal” effect on parasite, then they can be used as an antimalarial in a cost-effective manner. The overall lengthy process of drug development can also be reduced. Second, targeting host protein will aid in circumventing the problem of drug resistance (13). Most common examples are the pyridinyl imidazoles SB203580 and SB202190 known to inhibit the activation of human p38MAPK. These p38MAPK inhibitors were found to be inhibitory for protozoan parasite as well. Similarly MAPK p38 inhibitors like pyridinyl imidazole RWJ67657 and pyrrolobenzimidazole RWJ68198 impede the growth of P. falciparum.
Ca2+ Signaling
Ca2+ is one of the important secondary messenger molecule involved in the signal transduction. It plays multiple roles in different aspects of parasite lifecycle, such as egress, invasion, growth, development, motility, and secretion (64, 81, 82). Therefore, maintenance of Ca2+ homeostasis is crucial for the parasite survival. Ca2+ ions regulate varied cellular events by binding to the effector molecules. But it is difficult to characterize the components of Ca2+-dependent signaling due to the lack of homology of effector molecules between Plasmodium and higher eukaryotes (83). Some effectors like Ca2+ transporters, PfCHA (PF3D7_0603500), similar to Ca2+/H+ exchanger (84), P. falciparum sarco-endoplasmic reticulum calcium ATPase (SERCA-type ATPase), PfATP6 (PF3D7_0106300), and PfATP4 (85) have been identified, but their functions need to be investigated.
Ca2+ Signaling during Egress
Live cell fluorescent video microscopy involving the use of fluo-4 Ca2+ probe, Ca2+ chelators, and inhibitors of Ca2+ ATPase revealed the differential role of various effector molecules in egress and the invasion of the merozoites. It has been shown that before egress of merozoites there is a constant increase in Ca2+ level in the cytoplasm of both parasite and infected RBC (iRBC) independent of extracellular Ca2+ ions.
Inhibition of Ca2+ ATPase from ER inhibitors and addition of Ca2+ ionophore enhanced the process of egress, while chelating agents like bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra acetic acid inhibited the process of egress (64, 86). The mechanism of inhibition involved the prevention of permeabilization of host membrane. The Ca2+ is found to regulate the release of perforin-like protein 1 (PLP1) from microneme to the membrane where it begins its lytic activity. Likewise, the expression of PLP2 also occurs during the asexual blood stage, but its role is not clear. The reverse genetic studies would be helpful in disclosure of the details of PLP1 and PLP2(86). P. falciparum subtilisin-like protease (PfSUB1) is another effector of calcium signaling, which is found to be required for the PfSERAs. The inhibition of PfSUB1 by the use of Ca2+ chelators prevented the rupture of the parasitophorous vacuole and consequently the egress of merozoites (86).
Ca2+ Signaling during Invasion
The invasion process requires the discharge of rhoptry and microneme, which is dependent on the elevated levels of Ca2+. On exposure to low K+ level, cytosolic Ca2+ level increases through PLC and it triggers the export of erythrocyte-binding antigen 175 (EBA175) and AMA-1 from microneme to merozoite surface (Figure 1). The interaction with the receptors of RBC brings the increased level of calcium to the basal level, which induces the release of rhoptry protein (29). P. falciparum reticulocyte-binding protein homologs-1 (PfRH1), present in trace amounts in rhoptry neck, was found to trigger the release of calcium ions, which further initiates the cascade. The use of PfRH1 antibody inhibited the invasion process by blocking the calcium signaling and halting the interaction of EBA175 with host receptors. Therefore, it is assumed that an alternative pathway, namely, K+ ion-dependent pathway for the release of protein from apical organs, might exist. In spite of the above findings, the mechanism of triggering of signals to release calcium ions is yet to be probed (87). A study indicated the role of PfCDPK1 in the discharge of microneme protein for invasion. The mutational analysis further analyzed the inhibition of invasion process upon mutating its active site residues (88, 89). Microneme secretion and thereby invasion was also found to be impaired on deletion of another calcium lipid-binding Doc 2 protein, PfDoc2. However, the actual mechanism of action is unclear (47, 90).
Calcium-Dependent Cell Cycle Regulation
The cell cycle of parasite is known to be regulated by the coordinated release of calcium ions. The efflux of calcium ions from ER occurs through inositol 1,4,5-trisphosphate (IP3) channels. It was demonstrated that exogenous addition of IP3 also causes the release of ions. Melatonin hormone was found to induce the production of IP3 through PLC, which further opens the Ca2+ channels in the ER (91–93). An increase in calcium level on incubation with tumor necrosis factor (TNF)-α caused the downregulation of P. falciparum’s proliferating cell nuclear antigen-1 and ultimately the retardation of parasite growth (94). Calcium mediates its activity through PfCDPK7. The role of PfCDPK7 was confirmed by knockout studies in which PfCDPK7 mutant showed drastic retardation in growth. It can be assumed that the role of calcium might be correlated with protein kinase, PfPK7 and cdc2-related protein kinase, because the inhibition of these genes also demonstrated to retard parasitic growth, but this hypothesis needs further validation (95). Expression of PfCDK2 was also indicated to be at peak during the ring and trophozoite stage. However, the function of PfCDK2 has not been deciphered yet (96). A different group of Ca2+-binding orthologs of cytoskeletal-binding protein centrin (PfCEN), expressed during asexual and the gametocyte stage of parasite, was found to be colocalized with the centrosome. The role of centrin in cell division of Plasmodium yet concealed, while the knockout studies in Leishmania donovani revealed their involvement in the growth and cytokinesis (97).
There is an extensive involvement of calcium signaling in various important pathways of parasite. Any interruption would be deleterious for invasion, egress, and ultimately the growth of parasite. On these grounds, components of calcium signaling are considered for therapeutic interventions.
Toll-Like Receptor (TLR)-Mediated Signals During Malaria
Pathological symptoms during the severity of malaria are corresponding to the elevated level of pro-inflammatory cytokine. With the release of liver schizonts, a relative increase in the production of pro-inflammatory cytokine, such as interleukin (IL)-12, IL-8, and interferon (IFN)-γ, has been noticed in infected individuals (98–103). Any flaw in the inflammatory response will be responsible for the severity of disease (101, 104, 105). Antigen recognition by TLRs is one of the common mechanisms for the activation of the innate immune response. The mechanism involves the triggering of the signals from TLRs, which will ultimately cause the activation of the pro-inflammatory response (106–108) (Figure 2). In a case study of severe malaria, higher levels of TLR2, TLR4, and TLR8 were found (109). Owing to their role in severe malaria pathologies, TLRs are considered to be good candidate for in-depth research to elucidate the mechanism of innate immune response during parasite infection. Also, TLR ligands can be implicated for therapeutic intervention (110). The TLR role was investigated by correlating liver inflammation with parasite infection. In this study, cytotoxic activity of hepatic lymphocytes was induced by IL-12, produced in response to TLR-myeloid differentiation factor 88 (MyD88) mediated signaling pathway. On the contrary, the normal IL-12 level was found in MyD88-deficient mice. The cascade initiates with the interaction of a ligand with TLR extracellular domain, which is followed by the transfer of signal to intracellular Toll/interleukin-1 receptor (TIR) domain (111). Afterward, the signals are transferred to downstream targets and followed by the activation of transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1. The signals from the TIR domain are transduced via intracellular adaptors such as MyD88, TLR2, TLR4, TLR9, and TLR7 (112).
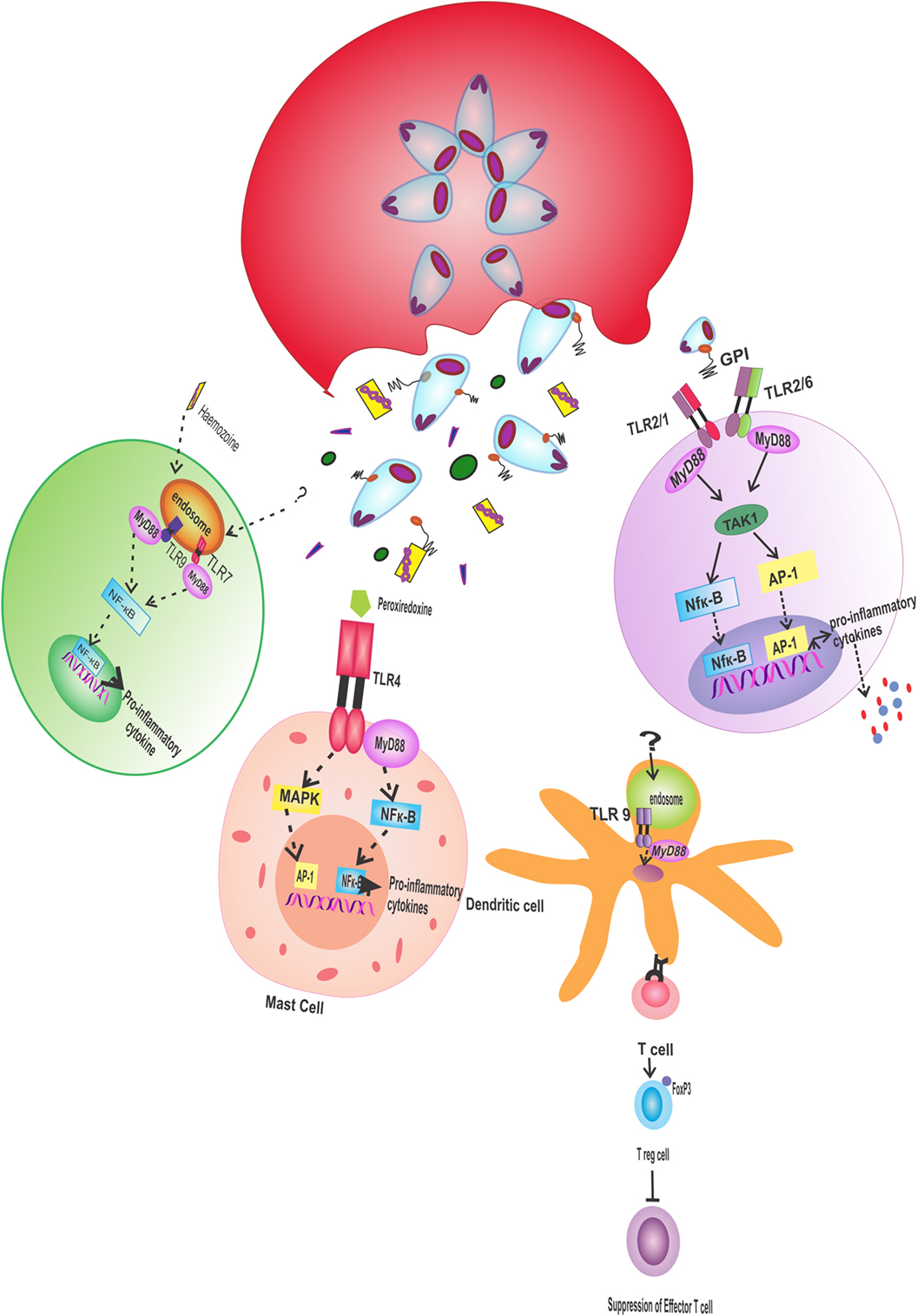
Figure 2. Pro-inflammatory response through TLR: various antigenic molecules are recognized differentially by TLRs, which produce pro-inflammatory responses. Hemozoin, released on rupture of infected erythrocyte, triggers MyD88-mediated signals through TLR9 receptor in host immune cells, and TLR7 is activated by unknown ligand to produce the same response. In dendritic cells, TLR9 activated by unknown ligand causes the differentiation of T cells to regulatory T cell, which suppress the activity of effector T cells. Antigenic protein peroxiredoxin stimulates TLR4 on mast cells, which activate MAPK and NF-κB pathway in MyD88-dependent manner, thereby ultimately releasing pro-inflammatory cytokines. TLR2 coupled with TLR 1 or TLR6 in host immune cells recognizes GPIs on parasitic surface to induce the production of pro-inflammatory cytokine through the activation of NF-κB and AP-1. TLR, toll-like receptor; MyD 88, myeloid differentiated factor 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; AP-1, activator protein 1; MyD88, myeloid differentiation factor 88; GPI, glycosylphosphatidylinositol.
Role of TLR9
During malaria, TLR9 is involved in providing protective immune response in a MyD88-dependent manner (113). Hemozoin (HZ) was indicated to stimulate this response (114–116). In a study, the ligand property of purified HZ against TLR was studied. The studies performed in TLR9-deficient mice showed the inadequate functioning of HZ in the production of chemokine, cytokine, and costimulatory signals (117). Parroche et al. (118) identified that natural HZ but not purified hemin can induce TLR9. Experimentally, it was explored that the stimulation was forbidden due to the lack of binding to TLR9 in the presence of nuclease. Later, it was confirmed that DNA present on the surface of HZ interacts with TLR9 (118). Another piece of information supporting the role of TLR signaling during malaria was derived from the finding, which has demonstrated the activation of regulatory T (Treg) cells in a TLR9-dependent manner. During the P. yoelii infection, an unknown protein of parasite stimulates the TLR9 present on dendritic cells. These signals ultimately trigger off the Treg cells, which in turn suppress the effector T cells. Evidence suggests that mice deficient in TLR9 are more resistant to malaria infection due to the activation of the effective response of T cells (15).
Role of TLR4
The pro-inflammatory response is not restricted to be mediated through TLR9, but in recent past, natural HZ has been shown to bind with host fibrinogen, which interacts with fibrinogen receptor TLR4 on monocytes, and this interaction leads to downstream activation of NF-κB and MAPK, thereby arousing the oxidative burst and elevated expression of TNF and other pro-inflammatory cytokines (119). Similarly in case of murine malaria, 2-Cys peroxiredoxin antigenic protein of Plasmodium stimulates TLR4 on mast cell and macrophages to produce TNF-α (120) (Figure 2).
Role of TLR7
A study carried out in Plasmodium chabaudi elucidated the role of TLR7 in the production of IFN-1, IL-12, and IFN-γ. Earlier, TLR9 was reported as the key sensor of infection. But the experiments conducted in the absence of TLR7 and MyD88 showed the remarkable reduction in the pro-inflammatory cytokine-like IFN-1. Contrary to it, no influence on IFN production was observed in mice deficient in TLR2, TLR4, TLR9, interleukin-1 receptor, or IL18R. Rational for this disparity is that the activation of TLR9 or TLR 7 depending on the time or stage of infection and alteration in the available ligand (16). Although the parasitic ligand triggering TLR7 has not yet been proven, however, based on finding that single-stranded RNA is required for TLR7-dependent production of IFN-I during viral infection (121), it was hypothesized that the RNA of parasite might work as a ligand against the receptor (16). Despite this speculation, the actual receptor–ligand interaction needs to be elucidated.
Role of TLR2
TLR2 recognize glycosylphosphatidylinositol (GPI) with TLR1 and TLR6 in a heterodimeric form (110) and induces the inflammatory cytokine production (122) (Figure 2). Severity of malaria was found to be correlated to allelic variation in TLR1 (123). TLR2 signaling in liver stage initiates the production of pro-inflammatory response, which hampers the parasite’s development (124).
GPI-Based Signaling and Immune Response
Several factors are involved in immune modulation leading to malaria pathologies. For instance, GPI is involved in the elicitation of innate immune response. GPI, ubiquitously found in eukaryotes, but are more prominent on parasitic surface. GPIs of different species exhibit structural diversity. GPIs are considered as toxic due to their deleterious effect of inducing of pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, IFN-γ, and nitric oxide (NO) in macrophage. The induced cytokines lead to the development of symptoms like hypoglycemia, pyrexia, fever, illness, and lethal cachexia (125–129). Conversely, the anti-GPI antibody significantly diminishes the pro-inflammatory response. Consistent with this, data show that surviving individuals after sever malaria have higher level of anti-GPI (130, 131). The GPI manifest its effect through the activation of protein tyrosine kinase and phosphokinase C consecutively activating NF-κB (125, 129, 132). Studies with knockout mice demonstrated that TLR-2 (110) and to a lesser extent TLR-4 (133) recognize GPIs, on the surface of merozoites (Figure 2). Mice deficient in MyD88 and CD36 showed reduced TNFα secretion in the presence of GPI. This indicates that GPI pass down its signals through TLR2 and CD36 (134). Elucidating the signaling cascade activated by GPI in murine peritoneal and bone marrow-derived macrophages, it was found that GPIs from Plasmodium can differentially stimulate the MAPK pathway like ERK, P38, and c jun N-terminal kinase (JNK) (128, 134). Of the above three MAPK pathway, ERK is not involved in GPI-induced secretion of TNF-α and NO (128). Reflecting on JNK its two isoforms, JNK1 and JNK 2 participate differentially in GPI-mediated cytokine production. On the induction of macrophage with the GPI, unaffected production of IL-6 and NO was observed in both JNK1−/− and JNK2−/−. But IL-12 and TNF-α levels were reduced in JNK2−/−, thus indicating the essential requirement of JNK2 to produce TNF-α and IL-12 (134). The crucial role of GPI in the activation of pro-inflammatory response and highly conserved nature suggest the synthetic GPIs as potential vaccine candidates (135). Targeting GPIs for designing the antimalarial therapy would be beneficial and secured from emergence of drug resistance (136). GPIs exerts their effect by imitating host GPIs, thereby modulating the normal host signaling pathways, thus candidature of GPI as a vaccine target was further verified (136, 137).
In an effort to find inhibitory molecules against GPIs a molecule, human C1 inhibitor (C1INH) with an anti-inflammatory effect, secreted by liver cell, was brought to attention (138). C1INH directly interact with GPIs of P. falciparum, and this binding inhibits the invasion of parasite to fresh erythrocyte and also it obstructs the interaction of iRBC to CD36 and chondroitin sulfate A, thereby halting parasite sequestration and consequently suppress the production of the pro-inflammatory cytokine as well (139). The effectiveness of C1INH was questioned due to the insufficiency of the endogenously produced molecule in controlling the disease pathogenesis. The presence of a mechanism like elastase, responsible for the weakening of the effect of C1INH in Pseudomonas aeruginosa, was hypothesized to be present in P. falciparum (139, 140) but has no experimental clues.
Cytoadhesion and Related Signaling During Malaria Infection
Most of the malaria pathologies are associated with the cellular interaction between host and parasitic proteins (7, 10, 141) (Table 1). Therefore, the focus on the response produced by cytoadhesion will help in overviewing of mechanisms of pathogenesis. During trophozoite stage, a large repertoire of proteins is exported on iRBC surface. Of hitherto of proteins, interaction of PfEMP1 with endothelial cell surface receptors or receptors on immune cells is most widely explored (142–145). Most of the exported proteins form a knob-like structure with adhesive property (4). Binding of these proteins to the EC, other iRBC or fresh RBCs cause the sequestration of iRBC in different tissue. As a consequence, it reduces the blood flow and bypasses the main flow to stay off from the splenic clearance (146). On the EC, several different types of receptors such as CD36, interstitial cell adhesion molecule (ICAM-1), endothelial protein C receptor (EPCR), P-selectin, and E-selectin are expressed in a tissue-specific manner (10, 141, 147, 148).
Recently, it was suggested that shedding of protective glycocalyx from endothelium is responsible for increased permeability and procoagulation state (149, 150). It makes direct access of the iRBC on the endothelial receptors. This finding provides a new line for the development of adjunct therapies, which can prevent the damage to the glycocalyx (151).
CD36 scavenger receptor is an important signaling molecule for various ligands and responsible for producing a pro-inflammatory response during inflammatory disease. The importance of CD36-mediated sequestration in parasitic growth was demonstrated through mutant of P. berghie with deficient CD36-binding ligand (152). The adherence of the iRBC to CD36 on vascular endothelium activates the intracellular signaling cascade, which in turn intensifies the affinity of the interaction of receptor for its ligand (11, 153). A downstream signaling cascade of CD36-iRBC interaction was explained by employing cross linking anti-CD36 or recombinant domain CD36-binding domain of PfEMP. This interaction stimulates src-dependent kinase, which activates ecto-ALP present on the surface of the EC (Table 1). The ALP subsequently potentiates the affinity of the receptor by dephosphorylation (153). It has been shown that recruitment of α5β1 integrin due to src signaling is responsible for an increase in affinity. This also leads to the rearrangement of cytoskeleton protein through phosphorylation of (P130Cas) Crk-associated substrate (P130Cas) adaptor protein (61, 154). The src kinase also activates Erk1/2, but it is not involved in receptor interaction. In monocytes, it avails in phagocytosis of iRBC but not concerned with TNF, responsible for sever pathologies (155, 156). During acute lung injury in P. berghei infection, splenic monocytes recruited to the lung tissue cause CD36-mediated phagocytosis of iRBC (157). Future research is expected for unrevealing the prospects of phagocytosis and the involvement of this pathway in activation of adaptive immunity.
In childhood malaria, PfEMP1 containing domain cassettes 8 and 13 domain binds to specific EPCR. EPCR-bound activated protein C (APC) activates protease activation receptor-1, which leads to induction of protective signals. APC on getting released binds to membrane phospholipids on platelets and causes the inactivation of the coagulation factor (14). However, during malaria, the interaction of PfEMP1 with EPCR inhibits the activation of protein C then pro-inflammatory cytokines from EC cause the shedding of EPCR (158). But it is matter to ponder upon that whether to implicate EPCR-mediated interaction for restricting iRBC sequestration? Would providing APC exogenously restore the cytoprotective and anticoagulant state? It would then provide novel concept to deal with the severity of malaria (158).
Host immune response causes the subtle changes in the functioning of blood–brain barrier, which affects the cellular trafficking of lymphocytes (159). The infiltration of lymphocytes from endothelium is mediated through the interactions between ICAM-1 and cell surface integrin (160). In P. falciparum infection, interaction of iRBCs with ICAM-1 is one of the reasons for the development of cerebral pathologies (161, 162). During the inflammatory response, the expression level of ICAM-1 increases. The rosetting of iRBC and their adherence to ICAM-1 was suggested to be responsible for bacterial enteric infection in children with severe malaria (163). Severity of disease involves damage in the microvasculature and organ. So, implementation of anti-adhesion agents coupled with anti-malarials are needed to combat the disease (164). The application of antiadhesion therapies would provide a new perspective in the reduction of the interactions related to severe pathologies. In the line of this effort, a truncated ICAM-1 biophore peptide (IBT213) was designed through in silico approach, which can specifically block the binding of PfEMP1 to ICAM-1(165).
Conclusion
The impression of the involvement of signaling pathways during the asexual stage of parasite is clear. The different signaling strategies are involved to gain access or to modulate the host environment. Several techniques such as pharmacological inhibition and reverse genetic approach and techniques for single-cell live imaging revealed various indispensable components responsible for the invasion, egress, parasite growth, and survival. The non-homologous nature of most of the signaling molecules such as PfPKG, PfPKA, and PfCDK gives an opportunity to exploit them as targets. The uniqueness of these molecules to the Apicomplexan provides an additional benefit. Despite this, several host receptors such as TLRs and immune cells are also awaiting (112) for a ligand search. Along with the ligand screening, a few parasitic molecules involved in the regulation of parasite signals are remained to be explored. More emphasized and detailed studies focused on the characterization of such signaling molecules will provide a new insight for inviting valuable treatment therapy to combat malaria.
Author Contributions
RS, DS, PR, and BS collected the data. RS wrote the manuscript. Overall monitoring was done by TB.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors are thankful to Department of Science and Technology, SERB, Government of India for providing financial assistance to the lab (SB/FT/LS-191/2012). RS is CSIR-UGC Senior Research Fellow, and PR is DBT Junior Research Fellow.
References
2. Bousema T, Okell L, Felger I, Drakeley C. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol (2014) 12:833–40. doi:10.1038/nrmicro3364
3. Cowman AF, Berry D, Baum J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J Cell Biol (2012) 198:961–71. doi:10.1083/jcb.201206112
4. Deitsch KW, Wellems TE. Membrane modifications in erythrocytes parasitized by Plasmodium falciparum. Mol Biochem Parasitol (1996) 76:1–10. doi:10.1016/0166-6851(95)02575-8
5. Parker PD, Tilley L, Klonis N. Plasmodium falciparum induces reorganization of host membrane proteins during intraerythrocytic growth. Blood (2004) 103:2404–6. doi:10.1182/blood-2003-08-2692
6. Miller LH, Good MF, Milon G. Malaria pathogenesis. Science (1994) 264:1878–83. doi:10.1126/science.8009217
7. Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature (2002) 415:673–9. doi:10.1038/415673a
8. Singh M, Mukherjee P, Narayanasamy K, Arora R, Sen SD, Gupta S, et al. Proteome analysis of Plasmodium falciparum extracellular secretory antigens at asexual blood stages reveals a cohort of proteins with possible roles in immune modulation and signaling. Mol Cell Proteomics (2009) 8:2102–18. doi:10.1074/mcp.M900029-MCP200
9. Spillman NJ, Dalmia VK, Goldberg DE. Exported epoxide hydrolases modulate erythrocyte vasoactive lipids during Plasmodium falciparum infection. MBio (2016) 7:e1538–1516. doi:10.1128/mBio.01538-16
10. Ho M, White NJ. Molecular mechanisms of cytoadherence in malaria. Am J Physiol (1999) 276:C1231–42.
11. Yipp BG, Robbins SM, Resek ME, Baruch DI, Looareesuwan S, Ho M. Src-family kinase signaling modulates the adhesion of Plasmodium falciparum on human microvascular endothelium under flow. Blood (2003) 101:2850–7. doi:10.1182/blood-2002-09-2841
12. Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun (2006) 74:3262–70. doi:10.1128/iai.01625-05
13. Sicard A, Semblat J-P, Doerig C, Hamelin R, Moniatte M, Dorin-Semblat D, et al. Activation of a PAK-MEK signalling pathway in malaria parasite-infected erythrocytes. Cell Microbiol (2011) 13:836–45. doi:10.1111/j.1462-5822.2011.01582.x
14. Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature (2013) 498:502–5. doi:10.1038/nature12216
15. Hisaeda H, Tetsutani K, Imai T, Moriya C, Tu L, Hamano S, et al. Malaria parasites require TLR9 signaling for immune evasion by activating regulatory T cells. J Immunol (2008) 180:2496–503. doi:10.4049/jimmunol.180.4.2496
16. Baccarella A, Fontana MF, Chen EC, Kim CC. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun (2013) 81:4431–42. doi:10.1128/IAI.00923-13
17. Bhatt TK, Khan S, Dwivedi VP, Banday MM, Sharma A, Chandele A, et al. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat Commun (2011) 2:530. doi:10.1038/ncomms1522
18. Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature (2002) 419:498–511. doi:10.1038/nature01097
19. Baker DA. Cyclic nucleotide signalling in malaria parasites. Cell Microbiol (2011) 13:331–9. doi:10.1111/j.1462-5822.2010.01561.x
20. Kaushal DC, Carter R, Miller LH, Krishna G. Gametocytogenesis by malaria parasites in continuous culture. Nature (1980) 286:490–2. doi:10.1038/286490a0
21. Read LK, Mikkelsen RB. Plasmodium falciparum-infected erythrocytes contain an adenylate cyclase with properties which differ from those of the host enzyme. Mol Biochem Parasitol (1991) 45:109–19. doi:10.1016/0166-6851(91)90032-2
22. Dyer M, Day K. Expression of Plasmodium falciparum trimeric G proteins and their involvement in switching to sexual development. Mol Biochem Parasitol (2000) 108:67–78. doi:10.1016/S0166-6851(00)00205-X
23. Mota MM, Hafalla JC, Rodriguez A. Migration through host cells activates Plasmodium sporozoites for infection. Nat Med (2002) 8:1318–22. doi:10.1038/nm785
24. Ono T, Cabrita-Santos L, Leitao R, Bettiol E, Purcell LA, Diaz-Pulido O, et al. Adenylyl cyclase alpha and cAMP signaling mediate Plasmodium sporozoite apical regulated exocytosis and hepatocyte infection. PLoS Pathog (2008) 4:e1000008. doi:10.1371/journal.ppat.1000008
25. Alexander DL, Arastu-Kapur S, Dubremetz JF, Boothroyd JC. Plasmodium falciparum AMA1 binds a rhoptry neck protein homologous to TgRON4, a component of the moving junction in Toxoplasma gondii. Eukaryot Cell (2006) 5:1169–73. doi:10.1128/ec.00040-06
26. Treeck M, Zacherl S, Herrmann S, Cabrera A, Kono M, Struck NS, et al. Functional analysis of the leading malaria vaccine candidate AMA-1 reveals an essential role for the cytoplasmic domain in the invasion process. PLoS Pathog (2009) 5:e1000322. doi:10.1371/journal.ppat.1000322
27. Leykauf K, Treeck M, Gilson PR, Nebl T, Braulke T, Cowman AF, et al. Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathog (2010) 6:e1000941. doi:10.1371/journal.ppat.1000941
28. Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell (2006) 124:755–66. doi:10.1016/j.cell.2006.02.006
29. Singh S, Alam MM, Pal-Bhowmick I, Brzostowski JA, Chitnis CE. Distinct external signals trigger sequential release of apical organelles during erythrocyte invasion by malaria parasites. PLoS Pathog (2010) 6:e1000746. doi:10.1371/journal.ppat.1000746
30. Gaur D, Chitnis CE. Molecular interactions and signaling mechanisms during erythrocyte invasion by malaria parasites. Curr Opin Microbiol (2011) 14:422–8. doi:10.1016/j.mib.2011.07.018
31. Baum J. A complete molecular understanding of malaria parasite invasion of the human erythrocyte: are we there yet? Pathog Glob Health (2013) 107:107. doi:10.1179/2047772413z.000000000121
32. Sharma P, Chitnis CE. Key molecular events during host cell invasion by Apicomplexan pathogens. Curr Opin Microbiol (2013) 16:432–7. doi:10.1016/j.mib.2013.07.004
33. Dawn A, Singh S, More KR, Siddiqui FA, Pachikara N, Ramdani G, et al. The central role of cAMP in regulating Plasmodium falciparum merozoite invasion of human erythrocytes. PLoS Pathog (2014) 10:e1004520. doi:10.1371/journal.ppat.1004520
34. Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol (2002) 4:901–6. doi:10.1038/ncb874
35. Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol (2010) 50:355–75. doi:10.1146/annurev.pharmtox.010909.105714
36. Desai SA, Bezrukov SM, Zimmerberg J. A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature (2000) 406:1001–5. doi:10.1038/35023000
37. Decherf G, Egee S, Staines HM, Ellory JC, Thomas SL. Anionic channels in malaria-infected human red blood cells. Blood Cells Mol Dis (2004) 32:366–71. doi:10.1016/j.bcmd.2004.01.008
38. Verloo P, Kocken CH, Van der Wel A, Tilly BC, Hogema BM, Sinaasappel M, et al. Plasmodium falciparum-activated chloride channels are defective in erythrocytes from cystic fibrosis patients. J Biol Chem (2004) 279:10316–22. doi:10.1074/jbc.M311540200
39. Syin C, Parzy D, Traincard F, Boccaccio I, Joshi MB, Lin DT, et al. The H89 cAMP-dependent protein kinase inhibitor blocks Plasmodium falciparum development in infected erythrocytes. Eur J Biochem (2001) 268:4842–9. doi:10.1046/j.1432-1327.2001.02403.x
40. Merckx A, Nivez M-P, Bouyer G, Alano P, Langsley G, Deitsch K, et al. Plasmodium falciparum regulatory subunit of cAMP-dependent PKA and anion channel conductance. PLoS Pathog (2008) 4:e19. doi:10.1371/journal.ppat.0040019
41. Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr Opin Cell Biol (2000) 12:174–9. doi:10.1016/S0955-0674(99)00073-3
42. Yuasa K, Mi-Ichi F, Kobayashi T, Yamanouchi M, Kotera J, Kita K, et al. PfPDE1, a novel cGMP-specific phosphodiesterase from the human malaria parasite Plasmodium falciparum. Biochem J (2005) 392:221–9. doi:10.1042/bj20050425
43. Howard BL, Harvey KL, Stewart RJ, Azevedo MF, Crabb BS, Jennings IG, et al. Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem Biol (2015) 10:1145–54. doi:10.1021/cb501004q
44. Seebeck T, Sterk GJ, Ke H. Phosphodiesterase inhibitors as a new generation of antiprotozoan drugs: exploiting the benefit of enzymes that are highly conserved between host and parasite. Future Med Chem (2011) 3:1289–306. doi:10.4155/fmc.11.77
45. Deng W, Baker DA. A novel cyclic GMP-dependent protein kinase is expressed in the ring stage of the Plasmodium falciparum life cycle. Mol Microbiol (2002) 44:1141–51. doi:10.1046/j.1365-2958.2002.02948.x
46. Deng W, Parbhu-Patel A, Meyer DJ, Baker DA. The role of two novel regulatory sites in the activation of the cGMP-dependent protein kinase from Plasmodium falciparum. Biochem J (2003) 374:559–65. doi:10.1042/BJ20030474
47. Gurnett AM, Liberator PA, Dulski PM, Salowe SP, Donald RG, Anderson JW, et al. Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites. A novel chemotherapeutic target. J Biol Chem (2002) 277:15913–22. doi:10.1074/jbc.M108393200
48. Diaz CA, Allocco J, Powles MA, Yeung L, Donald RG, Anderson JW, et al. Characterization of Plasmodium falciparum cGMP-dependent protein kinase (PfPKG): antiparasitic activity of a PKG inhibitor. Mol Biochem Parasitol (2006) 146:78–88. doi:10.1016/j.molbiopara.2005.10.020
49. Falae A, Combe A, Amaladoss A, Carvalho T, Menard R, Bhanot P. Role of Plasmodium berghei cGMP-dependent protein kinase in late liver stage development. J Biol Chem (2010) 285:3282–8. doi:10.1074/jbc.M109.070367
50. Yeoh S, O’Donnell RA, Koussis K, Dluzewski AR, Ansell KH, Osborne SA, et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell (2007) 131:1072–83. doi:10.1016/j.cell.2007.10.049
51. Koussis K, Withers-Martinez C, Yeoh S, Child M, Hackett F, Knuepfer E, et al. A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J (2009) 28:725–35. doi:10.1038/emboj.2009.22
52. Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, Hopp CS, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science (2010) 328:910–2. doi:10.1126/science.1188191
53. Alam MM, Solyakov L, Bottrill AR, Flueck C, Siddiqui FA, Singh S, et al. Phosphoproteomics reveals malaria parasite protein kinase G as a signalling hub regulating egress and invasion. Nat Commun (2015) 6:7285. doi:10.1038/ncomms8285
54. Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, et al. Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca(2)(+) signals at key decision points in the life cycle of malaria parasites. PLoS Biol (2014) 12:e1001806. doi:10.1371/journal.pbio.1001806
55. Taylor HM, McRobert L, Grainger M, Sicard A, Dluzewski AR, Hopp CS, et al. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot Cell (2010) 9:37–45. doi:10.1128/EC.00186-09
56. Chen Q, Schlichtherle M, Wahlgren M. Molecular aspects of severe malaria. Clin Microbiol Rev (2000) 13:439–50. doi:10.1128/CMR.13.3.439-450.2000
57. Sherman IW, Eda S, Winograd E. Erythrocyte aging and malaria. Cell Mol Biol (Noisy-le-grand) (2004) 50:159–69.
58. Moehlenbrock MJ, Price AK, Martin RS. Use of microchip-based hydrodynamic focusing to measure the deformation-induced release of ATP from erythrocytes. Analyst (2006) 131:930–7. doi:10.1039/b605136g
59. Wan J, Ristenpart WD, Stone HA. Dynamics of shear-induced ATP release from red blood cells. Proc Natl Acad Sci U S A (2008) 105:16432–7. doi:10.1073/pnas.0805779105
60. Levano-Garcia J, Dluzewski AR, Markus RP, Garcia CRS. Purinergic signalling is involved in the malaria parasite Plasmodium falciparum invasion to red blood cells. Purinergic Signal (2010) 6:365–72. doi:10.1007/s11302-010-9202-y
61. Davis SP, Amrein M, Gillrie MR, Lee K, Muruve DA, Ho M. Plasmodium falciparum-induced CD36 clustering rapidly strengthens cytoadherence via p130CAS-mediated actin cytoskeletal rearrangement. FASEB J (2012) 26:1119–30. doi:10.1096/fj.11-196923
62. Eaton JW, Brewer GJ. Red cell ATP and malaria infection. Nature (1969) 222:389–90. doi:10.1038/222389a0
63. Ayi K, Liles WC, Gros P, Kain KC. Adenosine triphosphate depletion of erythrocytes simulates the phenotype associated with pyruvate kinase deficiency and confers protection against Plasmodium falciparum in vitro. J Infect Dis (2009) 200:1289–99. doi:10.1086/605843
64. Glushakova S, Lizunov V, Blank PS, Melikov K, Humphrey G, Zimmerberg J. Cytoplasmic free Ca 2+ is essential for multiple steps in malaria parasite egress from infected erythrocytes. Malar J (2013) 12:1. doi:10.1186/1475-2875-12-41
65. Rangachari K, Dluzewski A, Wilson RJ, Gratzer WB. Control of malarial invasion by phosphorylation of the host cell membrane cytoskeleton. Nature (1986) 324:364–5. doi:10.1038/324364a0
66. Tanneur V, Duranton C, Brand VB, Sandu CD, Akkaya C, Kasinathan RS, et al. Purinoceptors are involved in the induction of an osmolyte permeability in malaria-infected and oxidized human erythrocytes. FASEB J (2006) 20:133–5. doi:10.1096/fj.04-3371fje
67. Fleck SL, Birdsall B, Babon J, Dluzewski AR, Martin SR, Morgan WD, et al. Suramin and suramin analogues inhibit merozoite surface protein-1 secondary processing and erythrocyte invasion by the malaria parasite Plasmodium falciparum. J Biol Chem (2003) 278:47670–7. doi:10.1074/jbc.M306603200
68. Akkaya C, Shumilina E, Bobballa D, Brand VB, Mahmud H, Lang F, et al. The Plasmodium falciparum-induced anion channel of human erythrocytes is an ATP-release pathway. Pflugers Arch (2009) 457:1035–47. doi:10.1007/s00424-008-0572-8
69. Burnstock G, Verkhratsky A. Evolutionary origins of the purinergic signalling system. Acta Physiol (Oxf) (2009) 195:415–47. doi:10.1111/j.1748-1716.2009.01957.x
70. Cruz LN, Wu Y, Craig AG, Garcia CR. Signal transduction in Plasmodium-red blood cells interactions and in cytoadherence. An Acad Bras Cienc (2012) 84:555–72. doi:10.1590/S0001-37652012005000036
71. Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol (1999) 11:211–8. doi:10.1016/S0955-0674(99)80028-3
72. Raman M, Cobb MH. MAP kinase modules: many roads home. Curr Biol (2003) 13:R886–8. doi:10.1016/j.cub.2003.10.053
73. Doerig CM, Parzy D, Langsley G, Horrocks P, Carter R, Doerig CD. A MAP kinase homologue from the human malaria parasite, Plasmodium falciparum. Gene (1996) 177:1–6. doi:10.1016/0378-1119(96)00281-8
74. Dorin-Semblat D, Quashie N, Halbert J, Sicard A, Doerig C, Peat E, et al. Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Mol Microbiol (2007) 65:1170–80. doi:10.1111/j.1365-2958.2007.05859.x
75. Dorin D, Alano P, Boccaccio I, Ciceron L, Doerig C, Sulpice R, et al. An atypical mitogen-activated protein kinase (MAPK) homologue expressed in gametocytes of the human malaria parasite Plasmodium falciparum. Identification of a MAPK signature. J Biol Chem (1999) 274:29912–20. doi:10.1074/jbc.274.42.29912
76. Dorin D, Semblat JP, Poullet P, Alano P, Goldring JP, Whittle C, et al. PfPK7, an atypical MEK-related protein kinase, reflects the absence of classical three-component MAPK pathways in the human malaria parasite Plasmodium falciparum. Mol Microbiol (2005) 55:184–96. doi:10.1111/j.1365-2958.2004.04393.x
77. Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol (2003) 162:281–91. doi:10.1083/jcb.200212141
78. Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J, et al. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol (2008) 15:322–31. doi:10.1016/j.chembiol.2008.03.005
79. Parrini MC, Matsuda M, de Gunzburg J. Spatiotemporal regulation of the Pak1 kinase. Biochem Soc Trans (2005) 33:646–8. doi:10.1042/bst0330646
80. Park ER, Eblen ST, Catling AD. MEK1 activation by PAK: a novel mechanism. Cell Signal (2007) 19:1488–96. doi:10.1016/j.cellsig.2007.01.018
81. Varotti FP, Beraldo FH, Gazarini ML, Garcia CR. Plasmodium falciparum malaria parasites display a THG-sensitive Ca2+ pool. Cell Calcium (2003) 33:137–44. doi:10.1016/S0143-4160(02)00224-5
82. Brochet M, Billker O. Calcium signalling in malaria parasites. Mol Microbiol (2016) 100:397–408. doi:10.1111/mmi.13324
83. Prole DL, Taylor CW. Identification of intracellular and plasma membrane calcium channel homologues in pathogenic parasites. PLoS One (2011) 6:e26218. doi:10.1371/journal.pone.0026218
84. Rotmann A, Sanchez C, Guiguemde A, Rohrbach P, Dave A, Bakouh N, et al. PfCHA is a mitochondrial divalent cation/H+ antiporter in Plasmodium falciparum. Mol Microbiol (2010) 76:1591–606. doi:10.1111/j.1365-2958.2010.07187.x
85. Krishna S, Woodrow C, Webb R, Penny J, Takeyasu K, Kimura M, et al. Expression and functional characterization of a Plasmodium falciparum Ca2+-ATPase (PfATP4) belonging to a subclass unique to Apicomplexan organisms. J Biol Chem (2001) 276:10782–7. doi:10.1074/jbc.M010554200
86. Garg S, Agarwal S, Kumar S, Yazdani SS, Chitnis CE, Singh S. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat Commun (2013) 4:1736. doi:10.1038/ncomms2725
87. Gao X, Gunalan K, Yap SSL, Preiser PR. Triggers of key calcium signals during erythrocyte invasion by Plasmodium falciparum. Nat Commun (2013) 4:2862–73. doi:10.1038/ncomms3862
88. Azevedo MF, Sanders PR, Krejany E, Nie CQ, Fu P, Bach LA, et al. Inhibition of Plasmodium falciparum CDPK1 by conditional expression of its J-domain demonstrates a key role in schizont development. Biochem J (2013) 452:433–41. doi:10.1042/bj20130124
89. Bansal A, Singh S, More KR, Hans D, Nangalia K, Yogavel M, et al. Characterization of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1) and its role in microneme secretion during erythrocyte invasion. J Biol Chem (2013) 288:1590–602. doi:10.1074/jbc.M112.411934
90. Jean S, Zapata-Jenks MA, Farley JM, Tracy E, Mayer DC. Plasmodium falciparum double C2 domain protein, PfDOC2, binds to calcium when associated with membranes. Exp Parasitol (2014) 144:91–5. doi:10.1016/j.exppara.2014.06.015
91. Hotta CT, Gazarini ML, Beraldo FH, Varotti FP, Lopes C, Markus RP, et al. Calcium-dependent modulation by melatonin of the circadian rhythm in malarial parasites. Nat Cell Biol (2000) 2:466–8. doi:10.1038/35017112
92. Beraldo FH, Almeida FM, da Silva AM, Garcia CR. Cyclic AMP and calcium interplay as second messengers in melatonin-dependent regulation of Plasmodium falciparum cell cycle. J Cell Biol (2005) 170:551–7. doi:10.1083/jcb.200505117
93. Alves E, Bartlett PJ, Garcia CR, Thomas AP. Melatonin and IP3-induced Ca2+ release from intracellular stores in the malaria parasite Plasmodium falciparum within infected red blood cells. J Biol Chem (2011) 286:5905–12. doi:10.1074/jbc.M110.188474
94. Cruz LN, Wu Y, Ulrich H, Craig AG, Garcia CRS. Tumor necrosis factor reduces Plasmodium falciparum growth and activates calcium signaling in human malaria parasites. Biochim Biophys Acta (2016) 1860:1489–97. doi:10.1016/j.bbagen.2016.04.003
95. Kumar P, Tripathi A, Ranjan R, Halbert J, Gilberger T, Doerig C, et al. Regulation of Plasmodium falciparum development by calcium-dependent protein kinase 7 (PfCDPK7). J Biol Chem (2014) 289:20386–95. doi:10.1074/jbc.M114.561670
96. Solyakov L, Halbert J, Alam MM, Semblat J-P, Dorin-Semblat D, Reininger L, et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat Commun (2011) 2:565. doi:10.1038/ncomms1558
97. Mahajan B, Selvapandiyan A, Gerald NJ, Majam V, Zheng H, Wickramarachchi T, et al. Centrins, cell cycle regulation proteins in human malaria parasite Plasmodium falciparum. J Biol Chem (2008) 283:31871–83. doi:10.1074/jbc.M800028200
98. Malaguarnera L, Musumeci S. The immune response to Plasmodium falciparum malaria. Lancet Infect Dis (2002) 2:472–8. doi:10.1016/S1473-3099(02)00344-4
99. Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun (2004) 72:5630–7. doi:10.1128/iai.72.10.5630-5637.2004
100. Stevenson MM, Riley EM. Innate immunity to malaria. Nat Rev Immunol (2004) 4:169–80. doi:10.1038/nri1311
101. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol (2005) 5:722–35. doi:10.1038/nri1686
102. Clark IA, Budd AC, Alleva LM, Cowden WB. Human malarial disease: a consequence of inflammatory cytokine release. Malar J (2006) 5:85. doi:10.1186/1475-2875-5-85
103. Riley EM, Wahl S, Perkins DJ, Schofield L. Regulating immunity to malaria. Parasite Immunol (2006) 28:35–49. doi:10.1111/j.1365-3024.2006.00775.x
104. Clark IA, Al Yaman FM, Jacobson LS. The biological basis of malarial disease. Int J Parasitol (1997) 27:1237–49. doi:10.1016/S0020-7519(97)00121-5
105. Mackintosh CL, Beeson JG, Marsh K. Clinical features and pathogenesis of severe malaria. Trends Parasitol (2004) 20:597–603. doi:10.1016/j.pt.2004.09.006
106. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4:499–511. doi:10.1038/nri1391
107. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
108. Medvedev AE. Toll-like receptor polymorphisms, inflammatory and infectious diseases, allergies, and cancer. J Interferon Cytokine Res (2013) 33:467–84. doi:10.1089/jir.2012.0140
109. Sobota RS, Dara A, Manning JE, Niangaly A, Bailey JA, Kone AK, et al. Expression of complement and toll-like receptor pathway genes is associated with malaria severity in Mali: a pilot case control study. Malar J (2016) 15:1–12. doi:10.1186/s12936-016-1189-6
110. Gowda DC. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol (2007) 23:596–604. doi:10.1016/j.pt.2007.09.003
111. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi:10.1016/j.cell.2006.02.015
112. Anderson KV. Toll signaling pathways in the innate immune response. Curr Opin Immunol (2000) 12:13–9. doi:10.1016/S0952-7915(99)00045-X
113. Gowda NM, Wu X, Gowda DC. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol (2012) 188:5073–85. doi:10.4049/jimmunol.1102143
114. Sherry BA, Alava G, Tracey KJ, Martiney J, Cerami A, Slater AF. Malaria-specific metabolite hemozoin mediates the release of several potent endogenous pyrogens (TNF, MIP-1 alpha, and MIP-1 beta) in vitro, and altered thermoregulation in vivo. J Inflamm (1995) 45:85–96.
115. Coban C, Ishii KJ, Sullivan DJ, Kumar N. Purified malaria pigment (hemozoin) enhances dendritic cell maturation and modulates the isotype of antibodies induced by a DNA vaccine. Infect Immun (2002) 70:3939–43. doi:10.1128/IAI.70.7.3939-3943.2002
116. Olivier M, Van Den Ham K, Shio MT, Kassa FA, Fougeray S. Malarial pigment hemozoin and the innate inflammatory response. Front Immunol (2014) 5:25. doi:10.3389/fimmu.2014.00025
117. Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med (2005) 201:19–25. doi:10.1084/jem.20041836
118. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A (2007) 104:1919–24. doi:10.1073/pnas.0608745104
119. Barrera V, Skorokhod OA, Baci D, Gremo G, Arese P, Schwarzer E. Host fibrinogen stably bound to hemozoin rapidly activates monocytes via TLR-4 and CD11b/CD18-integrin: a new paradigm of hemozoin action. Blood (2011) 117:5674–82. doi:10.1182/blood-2010-10-312413
120. Furuta T, Imajo-Ohmi S, Fukuda H, Kano S, Miyake K, Watanabe N. Mast cell-mediated immune responses through IgE antibody and toll-like receptor 4 by malarial peroxiredoxin. Eur J Immunol (2008) 38:1341–50. doi:10.1002/eji.200738059
121. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303:1529–31. doi:10.1126/science.1093616
122. Zhu J, Krishnegowda G, Li G, Gowda DC. Proinflammatory responses by glycosylphosphatidylinositols (GPIs) of Plasmodium falciparum are mainly mediated through the recognition of TLR2/TLR1. Exp Parasitol (2011) 128:205–11. doi:10.1016/j.exppara.2011.03.010
123. Hahn WO, Harju-Baker S, Erdman LK, Krudsood S, Kain KC, Wurfel MM, et al. A common TLR1 polymorphism is associated with higher parasitaemia in a Southeast Asian population with Plasmodium falciparum malaria. Malar J (2016) 15:1–7. doi:10.1186/s12936-015-1071-y
124. Zheng H, Tan Z, Zhou T, Zhu F, Ding Y, Liu T, et al. The TLR2 is activated by sporozoites and suppresses intrahepatic rodent malaria parasite development. Sci Rep (2015) 5:18239. doi:10.1038/srep18239
125. Schofield L, Hackett F. Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J Exp Med (1993) 177:145–53. doi:10.1084/jem.177.1.145
126. Gerold P, Dieckmann-Schuppert A, Schwarz RT. Glycosylphosphatidylinositols synthesized by asexual erythrocytic stages of the malarial parasite, Plasmodium falciparum. Candidates for plasmodial glycosylphosphatidylinositol membrane anchor precursors and pathogenicity factors. J Biol Chem (1994) 269:2597–606.
127. Tachado SD, Gerold P, McConville MJ, Baldwin T, Quilici D, Schwarz RT, et al. Glycosylphosphatidylinositol toxin of Plasmodium induces nitric oxide synthase expression in macrophages and vascular endothelial cells by a protein tyrosine kinase-dependent and protein kinase C-dependent signaling pathway. J Immunol (1996) 156:1897–907.
128. Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem (2005) 280:8606–16. doi:10.1074/jbc.M413541200
129. Nebl T, De Veer M, Schofield L. Stimulation of innate immune responses by malarial glycosylphosphatidylinositol via pattern recognition receptors. Parasitology (2005) 130:S45–62. doi:10.1017/S0031182005008152
130. Naik RS, Branch OH, Woods AS, Vijaykumar M, Perkins DJ, Nahlen BL, et al. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J Exp Med (2000) 192:1563–76. doi:10.1084/jem.192.11.1563
131. Mbengue B, Niang B, Niang MS, Varela ML, Fall B, Fall MM, et al. Inflammatory cytokine and humoral responses to Plasmodium falciparum glycosylphosphatidylinositols correlates with malaria immunity and pathogenesis. Immun Inflamm Dis (2016) 4:24–34. doi:10.1002/iid3.89
132. Tachado SD, Gerold P, Schwarz R, Novakovic S, McConville M, Schofield L. Signal transduction in macrophages by glycosylphosphatidylinositols of Plasmodium, Trypanosoma, and Leishmania: activation of protein tyrosine kinases and protein kinase C by inositolglycan and diacylglycerol moieties. Proc Natl Acad Sci U S A (1997) 94:4022–7. doi:10.1073/pnas.94.8.4022
133. Coban C, Ishii KJ, Horii T, Akira S. Manipulation of host innate immune responses by the malaria parasite. Trends Microbiol (2007) 15:271–8. doi:10.1016/j.tim.2007.04.003
134. Lu Z, Serghides L, Patel SN, Degousee N, Rubin BB, Krishnegowda G, et al. Disruption of JNK2 decreases the cytokine response to Plasmodium falciparum glycosylphosphatidylinositol in vitro and confers protection in a cerebral malaria model. J Immunol (2006) 177:6344–52. doi:10.4049/jimmunol.177.9.6344
135. Berhe S, Schofield L, Schwarz RT, Gerold P. Conservation of structure among glycosylphosphatidylinositol toxins from different geographic isolates of Plasmodium falciparum. Mol Biochem Parasitol (1999) 103:273–8. doi:10.1016/S0166-6851(99)00125-5
136. Schofield L, Hewitt MC, Evans K, Siomos M-A, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature (2002) 418:785–9. doi:10.1038/nature00937
137. Boutlis CS, Riley EM, Anstey NM, de Souza JB. Glycosylphosphatidylinositols in malaria pathogenesis and immunity: potential for therapeutic inhibition and vaccination. Curr Top Microbiol Immunol (2005) 297:145–85. doi:10.1007/3-540-29967-X_5
138. Davis AE III, Cai S, Liu D. The biological role of the C1 inhibitor in regulation of vascular permeability and modulation of inflammation. Adv Immunol (2004) 82:331–63. doi:10.1016/s0065-2776(04)82008-x
139. Mejia P, Diez-Silva M, Kamena F, Lu F, Fernandes SM, Seeberger PH, et al. Human C1-Inhibitor suppresses malaria parasite invasion and cytoadhesion via binding to parasite glycosylphosphatidylinositol and host cell receptors. J Infect Dis (2016) 213:80–9. doi:10.1093/infdis/jiv439
140. Catanese J, Kress LF. Enzymatic inactivation of human plasma C1-inhibitor and alpha 1-antichymotrypsin by Pseudomonas aeruginosa proteinase and elastase. Biochim Biophys Acta (1984) 789:37–43. doi:10.1016/0167-4838(84)90057-8
141. Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol (1999) 29:927–37. doi:10.1016/S0020-7519(99)00049-1
142. Flick K, Chen Q. var genes, PfEMP1 and the human host. Mol Biochem Parasitol (2004) 134:3–9. doi:10.1016/j.molbiopara.2003.09.010
143. Kraemer SM, Smith JD. A family affair: var genes, PfEMP1 binding, and malaria disease. Curr Opin Microbiol (2006) 9:374–80. doi:10.1016/j.mib.2006.06.006
144. Pasternak ND, Dzikowski R. PfEMP1: an antigen that plays a key role in the pathogenicity and immune evasion of the malaria parasite Plasmodium falciparum. Int J Biochem Cell Biol (2009) 41:1463–6. doi:10.1016/j.biocel.2008.12.012
145. Avril M, Tripathi AK, Brazier AJ, Andisi C, Janes JH, Soma VL, et al. A restricted subset of var genes mediates adherence of Plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proc Natl Acad Sci U S A (2012) 109:E1782–90. doi:10.1073/pnas.1120534109
146. Dondorp AM, Kager PA, Vreeken J, White NJ. Abnormal blood flow and red blood cell deformability in severe malaria. Parasitol Today (2000) 16:228–32. doi:10.1016/S0169-4758(00)01666-5
147. Newbold C, Warn P, Black G, Berendt A, Craig A, Snow B, et al. Receptor-specific adhesion and clinical disease in Plasmodium falciparum. Am J Trop Med Hyg (1997) 57:389–98. doi:10.4269/ajtmh.1997.57.389
148. Yipp BG, Anand S, Schollaardt T, Patel KD, Looareesuwan S, Ho M. Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum-infected erythrocytes to microvascular endothelial cells under flow. Blood (2000) 96:2292–8.
149. Hempel C, Hyttel P, Kurtzhals JA. Endothelial glycocalyx on brain endothelial cells is lost in experimental cerebral malaria. J Cereb Blood Flow Metab (2014) 34:1107–10. doi:10.1038/jcbfm.2014.79
150. Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, et al. Brain swelling and death in children with cerebral malaria. N Engl J Med (2015) 372:1126–37. doi:10.1056/NEJMoa1400116
151. Hempel C, Pasini EM, Kurtzhals JAL. Endothelial glycocalyx: shedding light on malaria pathogenesis. Trends Mol Med (2016) 22:453–7. doi:10.1016/j.molmed.2016.04.004
152. Fonager J, Pasini EM, Braks JAM, Klop O, Ramesar J, Remarque EJ, et al. Reduced CD36-dependent tissue sequestration of Plasmodium-infected erythrocytes is detrimental to malaria parasite growth in vivo. J Exp Med (2012) 209:93–107. doi:10.1084/jem.20110762
153. Ho M, Hoang HL, Lee KM, Liu N, MacRae T, Montes L, et al. Ectophosphorylation of CD36 Regulates cytoadherence of Plasmodium falciparum to microvascular endothelium under flow conditions. Infect Immun (2005) 73:8179–87. doi:10.1128/IAI.73.12.8179-8187.2005
154. Davis SP, Lee K, Gillrie MR, Roa L, Amrein M, Ho M. CD36 Recruits α(5)β(1) integrin to promote cytoadherence of P. falciparum-infected erythrocytes. PLoS Pathog (2013) 9:e1003590. doi:10.1371/journal.ppat.1003590
155. McGilvray ID, Serghides L, Kapus A, Rotstein OD, Kain KC. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum–parasitized erythrocytes: a role for CD36 in malarial clearance. Blood (2000) 96:3231–40.
156. Patel SN, Serghides L, Smith TG, Febbraio M, Silverstein RL, Kurtz TW, et al. CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis (2004) 189:204–13. doi:10.1086/380764
157. Lagassé HAD, Anidi IU, Craig JM, Limjunyawong N, Poupore AK, Mitzner W, et al. Recruited monocytes modulate malaria-induced lung injury through CD36-mediated clearance of sequestered infected erythrocytes. J Leukoc Biol (2016) 99:659–71. doi:10.1189/jlb.4HI0315-130RRR
158. Aird WC, Mosnier LO, Fairhurst RM. Plasmodium falciparum picks (on) EPCR. Blood (2014) 123:163–7. doi:10.1182/blood-2013-09-521005
159. McEver RP. Leukocyte – endothelial cell interactions. Curr Opin Cell Biol (1992) 4:840–9. doi:10.1016/0955-0674(92)90109-P
160. Petruzzelli L, Takami M, Herrera R. Adhesion through the interaction of lymphocyte function-associated antigen-1 with intracellular adhesion molecule-1 induces tyrosine phosphorylation of p130 and its association with c-CrkII. J Biol Chem (1996) 271:7796–801. doi:10.1074/jbc.271.13.7796
161. Turner G. Cerebral malaria. Brain Pathol (1997) 7:569–82. doi:10.1111/j.1750-3639.1997.tb01075.x
162. Gray C, McCormick C, Turner G, Craig A. ICAM-1 can play a major role in mediating P. falciparum adhesion to endothelium under flow. Mol Biochem Parasitol (2003) 128:187–93. doi:10.1016/S0166-6851(03)00075-6
163. Church JA, Nyamako L, Olupot-Olupot P, Maitland K, Urban BC. Increased adhesion of Plasmodium falciparum infected erythrocytes to ICAM-1 in children with acute intestinal injury. Malar J (2016) 15:1–6. doi:10.1186/s12936-016-1110-3
164. Bell A, Boehm D. Anti-disease therapy for malaria – ‘resistance proof’? Curr Pharm Des (2013) 19:300–6. doi:10.2174/138161213804070366
Keywords: malaria, Plasmodium, cyclic nucleotide signaling, toll-like receptor, calcium signaling, glycosylphoshatidylinositol, cytoadhesion
Citation: Soni R, Sharma D, Rai P, Sharma B and Bhatt TK (2017) Signaling Strategies of Malaria Parasite for Its Survival, Proliferation, and Infection during Erythrocytic Stage. Front. Immunol. 8:349. doi: 10.3389/fimmu.2017.00349
Received: 12 December 2016; Accepted: 10 March 2017;
Published: 28 March 2017
Edited by:
José Roberto Mineo, Federal University of Uberlandia, BrazilReviewed by:
Hridayesh Prakash, University of Hyderabad, IndiaLaura Noelia Cariddi, Universidad Nacional de Río Cuarto, Argentina
Copyright: © 2017 Soni, Sharma, Rai, Sharma and Bhatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tarun K. Bhatt, dGFydW5AY3VyYWouYWMuaW4=