 Fangli Lu1,2*
Fangli Lu1,2* Shiguang Huang3*
Shiguang Huang3*
- 1Department of Parasitology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China
- 2Key Laboratory of Tropical Disease Control Sun Yat-sen University, Ministry of Education, Guangzhou, China
- 3School of Stomatology, Jinan University, Guangzhou, China
Protozoan parasites such as Plasmodium spp., Leishmania spp., Trypanosoma spp., and Toxoplasma gondii are major causes of parasitic diseases in both humans and animals. The immune system plays a critical role against protozoa, but their immune mechanism remains poorly understood. This highlights the need to investigate the function of immune cells involved in the process of parasite infections and the responses of host immune system to parasite infections. Mast cells (MCs) are known to be central players in allergy and anaphylaxis, and it has been demonstrated that MCs have crucial roles in host defense against a number of different pathogens, including parasites. To date, there are many studies that have examined the interaction of helminth-derived antigens and MCs. As one of the major effector cells, MCs also play an important role in the immune response against some parasitic protozoa, but their role in protozoan infections is, however, less well characterized. Herein, we review the current knowledge about the roles of MCs and their mediators during infections involving highly pathogenic protozoa including Plasmodium spp., Leishmania spp., Trypanosoma spp., and T. gondii. We offer a general review of the data from patients and experimental animal models infected with the aforementioned protozoa, which correlate MCs and MC-derived mediators with exacerbated inflammation and disease progression as well as protection against the parasitic infections in different circumstances. This review updates our current understanding of the roles of MCs during parasitic protozoan infections, and the participation of MCs in parasitic protozoan infections could be of a potential therapeutic target.
Introduction
Plasmodium spp., Leishmania spp., Trypanosoma spp., and Toxoplasma gondii are some of the most important medical protozoan parasites that cause diseases in humans. Plasmodium spp. is a group of mosquito-borne parasitic protozoa. After being bitten by an Anopheles mosquito, sporozoites penetrate the liver cells of the host and produce thousands of free merozoites, which invade erythrocytes and then burst the cells to release the merozoites to invade other erythrocytes and cause clinical symptoms (1). Leishmania spp. comprises several species and causes leishmaniasis, which affects more than 300 million people worldwide (2). This parasite has a complex life cycle composed of two distinct stages: the promastigote form found in the female sandfly vector and the amastigote form replicated in the mammalian host (3). Trypanosoma brucei causes the fatal illness human African trypanosomiasis (4), which is adapted to parasitize the mammalian bloodstream after inoculation by the tsetse fly (Glossina spp.). Trypanosoma cruzi causes American trypanosomiasis or Chagas disease. This parasite chronically infects millions of people, and up to 30% of the infected individuals ultimately develop chronic cardiomyopathy or gastrointestinal disease. Transmission of this parasite occurs when trypomastigotes in vector (triatomine bug) feces enter bite wounds, mucous membranes of the nose, oral cavity, or conjunctiva of the new host. In addition, transmission can also occur through an oral route by ingestion of food contaminated with triatomine bugs or their feces (5, 6). T. gondii is spreading all over the world, which can infect a vast number of intermediate hosts and causes toxoplasmosis in both humans and animals (7). Toxoplasmic encephalitis is a subsequent risk for all severely immunocompromised patients (8). Moreover, infection during pregnancy may cause serious lesions to the fetus through congenital infection (9).
Mast cells (MCs) are tissue-resident, granule-containing cells, which participate in the regulation of innate and adaptive immune responses (10). In healthy individuals, MCs are involved in tissue homeostasis, tissue repair, and host defense via the release of different kind of pro-inflammatory mediators, proteases, and cytokines (11). Degranulation of MCs is essential for host defense against parasitic infections (12). It is well known that MCs play an important role in parasitic helminth infections (13). Accumulating evidences have demonstrated that MCs have pivotal roles in parasitic protozoan diseases (14, 15); this has led us to focus on the role of MCs in the immune responses against parasitic infections including Plasmodium spp., Leishmania spp., Trypanosoma spp., and T. gondii. The main function of MCs in the aforementioned protozoa is summarized in Table 1.
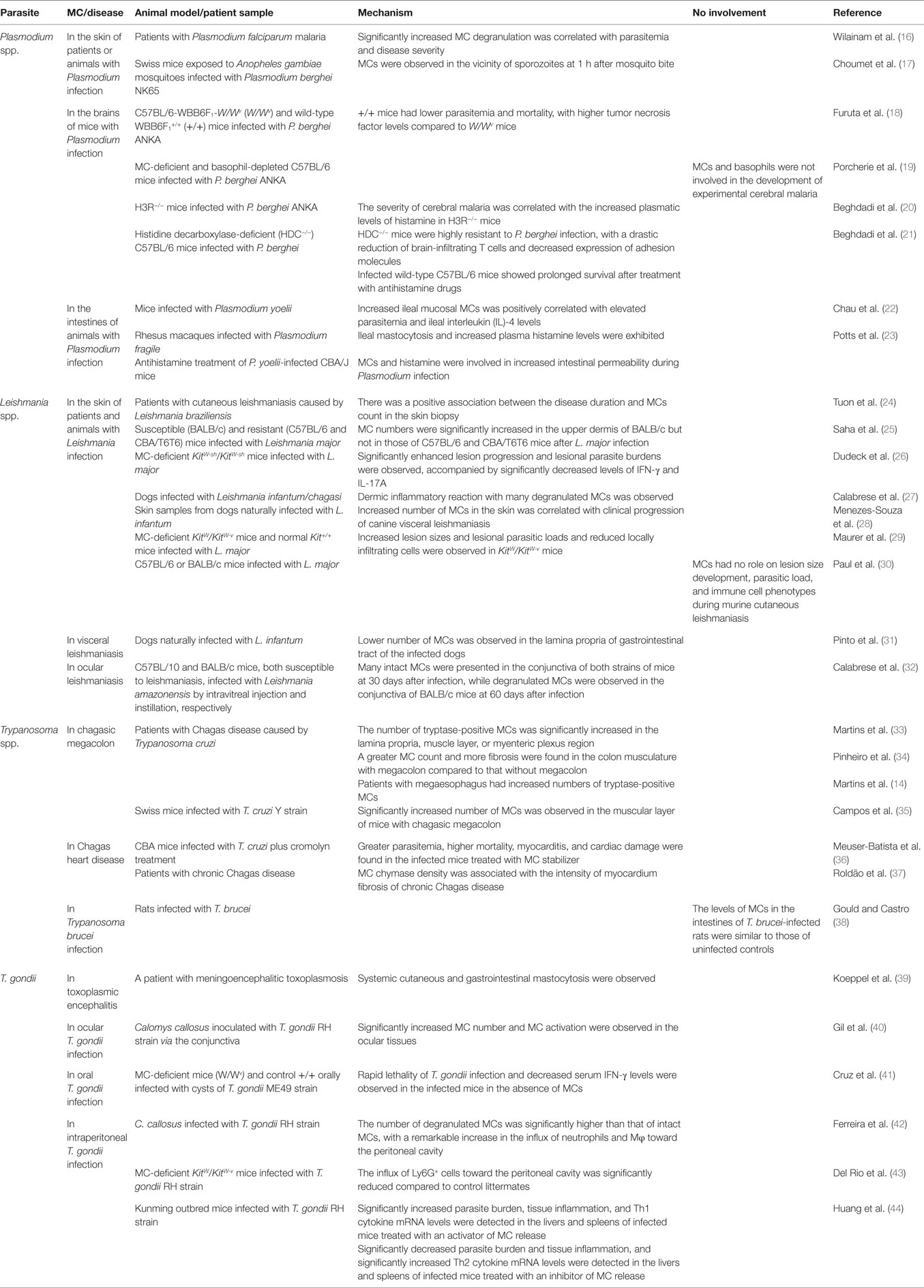
Table 1. The function of mast cells (MCs) in Plasmodium spp., Leishmania spp., Trypanosoma spp., and Toxoplasma gondii infections.
MCs in Plasmodium spp. Infection
MCs in the Skin of Patients and Animals with Plasmodium Infection
Mast cells are abundant in tissues exposed to the external environment, including the skin (45). Malaria parasites may promote malaria pathogenesis by triggering MCs. Plasmodium berghei-infected Anopheles gambiae mosquito saliva can trigger mouse dermal MC degranulation as little as 5 min after the mosquito bite. One hour after the bite, MCs were observed in the vicinity of sporozoites on skin sections from mice bitten by P. berghei-infected An. gambiae mosquitoes (17). Saliva-induced activation of dermal MCs causes lymph node swelling via the recruitment of T cells, B cells, dendritic cells (DC), and monocytes/macrophages (Mφ) as well as neutrophils (46, 47). Importantly, it was shown that there was an increase in MC activation and degranulation in the skin dermis of severe Plasmodium falciparum malaria patients, compared to controls. The percentage of MC degranulation was significantly correlated with parasitemia and disease severity, which are relevant to MC mediators (16).
MCs in Cerebral Malaria (CM)
Mast cells are found in the central nervous system, especially along the blood vessels and leptomeninges (48). Kenyan children with mild and severe malaria were shown to have increased plasma levels of Flt3 ligand (Flt3L) (15). Elevated serum Flt3L levels and DC expansion were found in patients infected by P. falciparum and mice with Plasmodium infection. MCs are an important source of Flt3L, a soluble cytokine that influences DC function, with the subsequent activation of pathogenic CD8+ T cells, a critical effector of the disease (49). After infection with P. berghei ANKA, compared to MC-deficient WBB6F1-W/Wv mice, the control littermate WBB6F1+/+ C57BL/6 mice had lower parasitemia and mortality with higher tumor necrosis factor (TNF) levels. Malarial antigens from P. berghei ANKA are able to stimulate Mφ and MCs to secrete TNF in vitro. An in vivo study further demonstrated that MCs are a critical source of TNF in addition to Mφ and T cells in murine malaria. Therefore, MCs and MC-derived TNF play an important role in protection against experimental cerebral malaria (ECM) (18). Furthermore, P. berghei ANKA peroxiredoxin induces a significant amount of MC-derived TNF secretion from IgE-mediated protection through FcεRI on MCs and innate immunity by means of toll-like receptor (TLR) 4 with myeloid differentiation primary response gene 88 and MD-2 and plays a role in innate and acquired immune responses in malaria (50). Conversely, one study reported that malaria developed in MC-deficient and basophil-depleted C57BL/6 mice infected with P. berghei ANKA was similar to that developed in wild-type mice, suggesting that MCs and basophils were not involved in malaria pathogenesis in this model (19).
Histamine has four different receptors, namely H1R, H2R, H3R, and H4R, which mediate numerous different effects (51). Histamine is the major MC mediator in malaria, and its signaling has been associated with the severity of P. falciparum malaria (21). In addition, the significant elevation of the blood concentrations of IgE and IgE-antimalarial antibodies has also been linked to the disease severity in falciparum malaria patients (52). As a DC modulator, especially during early phases of the immune response, histamine causes increased vascular permeability and subsequent extensive vascular damage to endothelial cells during malaria infection (47). Experiments have demonstrated that histamine binding to H1R and H2R increases the susceptibility of infection with P. berghei in H1R−/− and H2R−/− mice (21). H3R−/− mice infected with P. berghei ANKA have an accelerated onset of CM and mortality, accompanied by an earlier loss of blood–brain barrier integrity, earlier formation of hemorrhagic lesions, higher sequestration of CD4+ and CD8+ T cells in the brain, and higher serum histamine levels compared to C57BL/6 wild-type mice. The severity of CM is related to the increased plasmatic levels of histamine in H3R−/− mice during the infection (20). Mice genetically deficient in the histidine decarboxylase (HDC−/−) gene, thus lacking histamine, were highly resistant to lethal infection by P. berghei ANKA and P. berghei NK65, associated with decreased brain-infiltrating T cells and expression of adhesion molecules. After treatment with antihistamine drugs, mice infected with P. berghei had prolonged survival compared to infected mice without antihistamine treatment (21).
Vascular endothelial growth factor (VEGF) is both neuroprotective and pro-inflammatory in the brain. VEGF was shown to accumulate intracellularly in P. falciparum-infected red blood cells in vitro, and inhibition of VEGF receptor (VEGFR)-2 signaling reduced intraerythrocytic growth of P. falciparum (53). VEGF and soluble VEGFR (sVEGFR)-2 are increased in CM patients compared to healthy adults (54). Plasma VEGF concentrations in Kenyan children with CM are associated with an increased risk of neurological sequelae (55). sVEGFR-1 may play a pathological role during chronic placenta malaria and hypertension in first-time mothers (56). Both P. falciparum and Plasmodium vivax crude antigens induce VEGF release from the human MC line HMC-1 or the human basophilic cell line KU812 in vitro. Increased parasitemia of P. berghei ANKA was observed in anti-VEGF Ab-treated mice compared to non-treated mice (54). Furthermore, VEGF was shown to promote malaria-associated acute lung injury induced by P. berghei ANKA in mice (57). The pro-inflammatory cytokine interleukin (IL)-33 is strongly enhanced in infants (<5 years) with severe malaria from P. falciparum infection (58). IL-33 contributes to the stimulation and release of VEGF in human MCs (hMCs) (59). Conversely, IL-33 prevents the development of ECM in C57BL/6 mice infected with P. berghei ANKA and reduces the production of inflammatory mediators IFN-γ, IL-12, and TNF-α (60).
MCs in the Intestines of Animals with Plasmodium Infection
In the gastrointestinal tract, MCs regulate vascular and epithelial permeability, ion secretion, angiogenesis, peristalsis, fibrosis, tissue repair, and innate and adaptive immunity (61). Increased numbers of mucosal MCs (MMCs) in the ileal villi and crypts and increased histamine levels in the ileum were detected in Plasmodium yoelii-infected mice. The increase in ileal MMCs was positively correlated with elevated parasitemia and IL-4 mRNA levels in the same tissue. An additional study found that P. yoelii nigeriensis-infected mice develop an l-arginine deficiency, which is associated with intestinal mastocytosis, elevated levels of plasma histamine, and enhanced intestinal permeability (22). Plasmodium fragile-infected rhesus macaques have been shown to exhibit ileal mastocytosis and increased plasma histamine levels. Antihistamine treatment during P. yoelii infection results in decreased intestinal permeability in CBA/J mice, suggesting that MCs and histamine are involved in increased intestinal permeability during Plasmodium infection (23).
MCs in Leishmania spp. Infection
Leishmaniasis is caused by several different species of Leishmania, and each infection has a different clinical outcome; human leishmaniasis is usually classified as cutaneous, mucocutaneous, or visceral. Leishmania parasites primarily live in Mφ (25), but Leishmania major and Leishmania infantum promastigotes can also bind to MC membranes and infect MCs (62).
MCs in the Skin of Patients and Animals with Leishmania Infection
Cutaneous leishmaniasis is an important public health concern in many parts of the world, especially in Africa (63). MCs degranulate and release inflammatory mediators such as TNF-α after infection and recruit polymorphonuclear leukocytes (PMNs) to the site of infection (64). MCs can be important in cutaneous leishmaniasis and are involved in healing lesions. The MC count was higher in the skin biopsy of patients with cutaneous leishmaniasis caused by Leishmania braziliensis with earlier healing after treatment, and there was a positive association between the disease duration and MC count (24).
After infection of Leishmania amazonensis in susceptible (C57BL/10 and CBA), relatively resistant (DBA/2), and resistant (C3H.He) mice, the primary lesions in footpads and draining lymph nodes showed a predominance of eosinophils and MCs in the initial phase of infection in all the infected mice (65). MC numbers increased significantly in the upper dermis in susceptible (BALB/c) but not in resistant (C57BL/6 and CBA/T6T6) mice after L. major infection. However, the number of degranulating MCs was higher in CBA/T6T6 mice during early L. major infection. In addition, MC-derived cytokines, such as TNF-α, play a proparasitic role in a susceptible strain (BALB/c) of mice, but an antiparasitic role in resistant strains (C57BL/6 and CBA/T6T6) of mice, suggesting that the susceptible and resistant mouse strains may have different modes of producing the antileishmanial immune response by differential regulation of MC function (25). After L. major inoculation, MC activation and parasite uptake by skin-resident Mφ occurred, followed by neutrophil and monocyte immigration and DC activation. Therefore, MC-dependent recruitment of Mφ, PMN, and DC to the skin is involved in controlling leishmaniasis (66). Furthermore, using MC-deficient KitW-sh/KitW-sh mice for infection with L. major promastigotes results in a worse disease outcome, e.g., significantly enhanced lesion progression and lesional parasite burdens, accompanied by significantly decreased levels of IFN-γ and IL-17A, but significantly increased IL-4 and IL-10, compared to wild-type mice, indicating that MCs play a crucial role against Leishmania parasites by promoting Th1 and Th17 responses in vivo (26). L. infantum/chagasi-infected skins, from dogs of two different leishmaniasis endemic areas of Brazil, showed different skin infection patterns; however, dogs from both areas showed dermic inflammatory infiltrates composed of numerous degranulated MCs compared to normal skin, indicating that MCs modulate the immune response and participate in the host defense against Leishmania infection (27). Dogs naturally infected with L. infantum showed increased inflammatory infiltrates in the skin of animals with severe forms of canine visceral leishmaniasis and a high parasite density. The increased number of Mφ and decreased number of lymphocytes, eosinophils, and MCs in the skin correlate with clinical progression of canine visceral leishmaniasis (28).
After susceptible BALB/c and resistant C57BL/6 mice were infected with L. major and pretreated with compound 48/80 (a MC activator), both BALB/c and C57BL/6 mice displayed smaller lesions in footpads compared to controls, indicating that MC degranulation contributes to susceptibility to L. major infection, and in the absence of granulated MCs, BALB/c and C57BL/6 mice had increased resistance to L. major infection. Although IL-4 and MC degranulation are important to Leishmania-associated pathogenesis, the MC-mediated susceptibility seems to be independent of IL-4 (67). MC-deficient C57BL/6-KitW/KitW-v mice and congenic wild-type Kit+/+ mice were infected with metacyclic promastigotes of L. major by intradermal injections, resulted in significantly increased lesion sizes and lesional parasitic loads, and significantly reduced locally infiltrating cells in KitW/KitW-v mice. In addition, pronounced MC degranulation was observed in infected skin sites after intradermal injection of L. major in C57BL/6 mice, indicating that MCs provide protection against L. major infection (29). An in vitro study showed that stimulation with Leishmania mexicana lipophosphoglycan led to a significant increase in degranulation in bone marrow-derived MCs (BMMCs) from BALB/c mice compared to BMMCs from C57BL/6 mice. Moreover, an in vivo study showed that after infection with L. mexicana, the number of MCs increased more rapidly and to a greater level, with significantly higher levels of parasites in the lesions of BALB/c mice compared to C57BL/6 mice. This indicates that MCs regulate the outcome of leishmaniasis and is dependent on the genetic background of the host (68). Conversely, a recent study showed that MC has no impact on the severity of cutaneous leishmaniasis in mice infected with L. major. By using Kit mutant mice with different genetic backgrounds, it was shown that MC deficiency did not affect lesion size development after L. major infection, suggesting that MCs are not involved in murine cutaneous Leishmania infections (30).
MCs in Visceral Leishmaniasis
Visceral leishmaniasis is a serious public health problem that causes high morbidity and mortality (69). Analysis of Th1, Th2, and Th17 cytokine responses by cultured peripheral blood mononuclear cells from patients who had developed kala-azar caused by Leishmania donovani, or who were protected against kala-azar, showed that IL-17 and IL-22 are the cytokines most strongly associated with protection against kala-azar (70). It has been reported that higher numbers of plasma cells, lymphocytes, and Mφ but lower number of MCs are present in the lamina propria of gastrointestinal tract of dogs naturally infected with L. infantum compared to non-infected controls, in all gastrointestinal tract segments (31). Thus, MCs may play different roles in visceral leishmaniasis and cutaneous leishmaniasis.
MCs in Mucocutaneous Leishmaniasis
Mucosal leishmaniasis is a chronic infection that affects the upper respiratory tract and/or the oral mucosa (71). Some patients diagnosed with mucosal leishmaniasis have oral lesions. Scraping cytology examination in patients with oral leishmaniasis presented free Leishman-Donovan bodies or Mφ loaded with Leishman-Donovan bodies, acute and chronic inflammatory cells, histiocytes, multinucleated giant cells, MCs, and plasma cells (72).
MCs in Ocular Leishmaniasis
Ocular involvement is an unusual presentation of leishmaniasis and commonly limited to the eyelid skin (73). Both C57BL/10 and BALB/c mice are susceptible to leishmaniasis, infected with L. amazonensis by intravitreal injection and instillation, respectively. Independent of the infective routes, C57BL/10 mice infected intravitreally presented an intense inflammatory reaction in the epithelium of the eyelids, as well as the presence of many intact MCs in the conjunctiva of the eyes from 30 days postinfection (p.i.). On the other hand, BALB/c mice infected via the instillation route presented no lesions but an enhancement of intact MCs in the conjunctival region at 30 days p.i., and a discreet inflammatory infiltrate and degranulated MCs were observed in the conjunctival region at 60 days p.i. (32).
MC–TLRs Interaction during Leishmania Infection
To date, the regulatory effect of MCs on the pathogenesis of leishmaniasis is still unclear. The clearance of L. major strongly depends on TLRs (24). TLR9-deficient Mφ had reduced expressions of CD40, IL-12, and TNF-α (74). MCs respond to TLR ligands by secreting cytokines, chemokines, and lipid mediators, and some studies have found that TLR ligands can also cause MC degranulation (75). The HMC-1 stimulated by promastigotes of L. braziliensis has a significantly greater release of histamine and IL-4 compared to control cells treated with medium (76). MCs release IL-3 and IL-4 to render Mφ susceptible to Leishmania infection in vitro (25).
MCs in Trypanosoma spp. Infection
MCs in T. cruzi Infection: Chagasic Megacolon
Megacolon is frequently observed in patients with Chagas disease caused by T. cruzi infection, which has been considered a consequence of an inflammatory process, and inflammatory infiltrates are composed of lymphocytes, Mφ, natural killer cells, MCs, and eosinophils. Morphometric analyses in the lamina propria, muscle layer, or myenteric plexus region revealed that the numbers of both tryptase-immunoreactive MCs and eosinophils are significantly increased in patients with megacolon compared to uninfected individuals. MC and eosinophil activation, as well as their physical interaction, were observed by electron microscopy (33). T. cruzi-induced injury resulted in intramuscular fibrosis and increased thickness of the colon wall in patients with chagasic megacolon, and there was a greater MC count and more fibrosis in the circular colon musculature of chronic Chagas patients with megacolon compared to Chagas cases without megacolon (34). The density of MCs was significantly higher in the esophagus and large intestine in patients with AIDS plus Chagas disease reactivation compared to chronic chagasic patients without AIDS. This suggests that MCs may play a major role in esophageal and intestinal inflammation during Chagas disease reactivation in HIV-coinfected patients (77). MC-specific proteases (tryptase and chymase) influence the activation of inflammatory cells. Increased numbers of tryptase-immunoreactive MCs were found in the esophagus sections of T. cruzi-infected individuals with or without megaesophagus. However, increased numbers of chymase-immunoreactive MCs were only found in the esophagus sections of infected individuals without megaesophagus compared to the control groups. Therefore, patients with megaesophagus had increased levels of tryptase-immunoreactive MCs (14).
One animal study showed that, after infection with the Y strain of T. cruzi, there were no significant differences in MC counts in the acute phase in Swiss mice. However, there was a significant increase in the number of MCs in the muscular layer of chronically infected Swiss mice with chagasic megacolon compared to non-infected control mice, accompanied by increased thickness of the colon wall, diffuse muscle cell hypertrophy, and increased collagen deposition (35), which may be associated with MC functions.
MCs in T. cruzi Infection: Chagas Heart Disease
The density of MCs in the myocardium was shown to be significantly higher in the chronic chagasic patients compared to control groups (77). The autopsied chagasic patient group showed higher MC chymase and MC tryptase densities and a higher percentage of collagen in the lingual muscles and myocardium compared to the non-chagasic patient group, and MC chymase level was associated with the intensity of myocardium fibrosis of chronic Chagas disease (37). Infiltrated T cells, Mφ, B cells, and MCs were all observed in the myocardium of patients with Chagas cardiopathy, who died at an early mean age or at older ages. However, the numbers of T-lymphocytes and MCs were significantly higher in the cases who suffered early cardiac death (78).
One animal study showed that T. cruzi-infected CBA mice treated with cromolyn (a MC stabilizer) presented much greater parasitemia and IFN-γ levels, higher mortality, myocarditis, and cardiac damage, indicating that MCs control blood and tissue parasitemia, IFN-γ production, cardiac inflammation, and susceptibility to infection, suggesting that MCs are involved in resistance to this infection (36).
MCs in T. brucei Infection
Trypanosoma brucei is a protozoan parasite that causes human African trypanosomiasis. Rats initially infected with T. brucei, followed by infection with Trichinella spiralis, showed that T. brucei infection does not significantly alter the number of MCs generated by T. spiralis infection, while the intestinal MC numbers in rats infected with only T. brucei were similar to those in uninfected rats (38).
MCs in T. gondii Infection
MCs in Toxoplasmic Encephalitis
Toxoplasmic encephalitis in patients with AIDS is a life-threatening disease, mostly due to the reactivation of T. gondii cysts in the brain (79). It has been reported that a patient with meningoencephalitic toxoplasmosis was associated with systemic cutaneous and gastrointestinal mastocytosis, suggesting a possible relationship between MC proliferation and the parasitic infection (39).
MCs in Ocular T. gondii Infection
Calomys callosus (Rodentia: Cricetidae) animals were inoculated intraperitoneally or via the conjunctiva with tachyzoites of the RH strain of T. gondii, resulting in the presence of the parasites and inflammatory cells and a significant increase in the number of MCs. Furthermore, MC activation in the ocular tissues was observed after infection, suggesting that MCs play an important role in the acute inflammatory response against T. gondii (40).
MCs in Oral T. gondii Infection
Oral infection is the natural toxoplasmosis route, and the MC population is highly abundant in intestinal mucosa. In MC-deficient mice (W/Wv) and their control +/+ counterparts orally infected with cysts of the ME49 strain of T. gondii, rapid lethality and decreased IFN-γ levels were observed in the serum of infected mice in the absence of MCs. This demonstrated that MCs play a primordial role in resistance to oral infection with T. gondii, and MCs are required for survival of mice after oral infection with T. gondii (41). RBL-2H3 MCs infected with T. gondii type I (RH), II (PTG), or III (CTG or VEG) tachyzoites showed that acute T. gondii infection inhibits antigen-mediated MC degranulation, irrespective of the genotype of parasite used, and that tachyzoite attachment but not invasion is necessary for inhibiting degranulation. Ca2+ mobilization is a central and well-studied aspect of IgE/FcεRI-mediated signaling in MCs, and T. gondii infection has been shown to inhibit MC degranulation by suppressing antigen-mediated Ca2+ responses (80).
MCs in Intraperitoneal T. gondii Infection
After intraperitoneal infection with the RH strain of T. gondii, the number of non-degranulated MCs was significantly lower than that of degranulated cells in the peritoneal cavity, submandibular and dorsal lymph nodes, and ileum in infected C. callosus compared to uninfected animals. After the MC degranulation, a remarkable increase in the influx of neutrophils and Mφ but a decrease in lymphocyte influx toward the peritoneal cavity of the infected animals were observed. MCs were observed interacting with other parasitized cells including Mφ, and extracellular parasites were destroyed during the interaction with MCs exhibiting degranulation. This suggests that MC is an important cell type during the inflammatory response against T. gondii (42). The use of MC-deficient KitW/KitW-v mice demonstrated that the influx of Ly6G+ cells toward the peritoneal cavity was significantly reduced compared to control littermates, indicating that MCs are an important chemokine source driving PMN recruitment to the peritoneal cavity during T. gondii infection (43). In both wild-type and serglycin-deficient mice, intraperitoneal infection with T. gondii resulted in highly increased extracellular levels of glycosaminoglycans, including hyaluronan and chondroitin sulfate A, suggesting that serglycin proteoglycan is dispensable for normal secretion and activity of MC proteases in response to T. gondii infection (81). A murine model showed that infection of T. gondii increased not only the number of MCs at the site of infection but also a noticeable degree of MC degranulation. Kunming outbred mice were infected intraperitoneally with the RH strain of T. gondii and treated by compound 48/80 or disodium cromoglycate (a MC stabilizer). The MC activator aggravated the pathology and increased the parasitic load, accompanied by upregulated mRNA levels of Th1 cytokines (IFN-γ, IL-12p40, or TNF-α) in the livers and spleens of T. gondii-infected mice. Conversely, the MC stabilizer improved the pathology and decreased the parasitic load, accompanied by increased mRNA levels of Th2 cytokines (IL-4 and IL-10) in the livers and spleens of mice infected with this parasite. In addition, significantly increased inflammatory foci of neutrophil infiltrates in different tissues occurred as a result of MC degranulation after the parasite infection (44). Thus, the activation or inhibition of MCs is a key factor determining the fate of the infection and associated immunopathology.
MC–T. gondii Interaction In Vitro
When T. gondii tachyzoites and MCs were incubated together, the tachyzoites adhered to the surface of the MCs, followed by MC degranulation. MC histamine and LTB4 release was significantly increased after incubation with the tachyzoites, which resulted in damage to the tachyzoites. MC-treated tachyzoites were found to be incapable of infection and replication in murine peritoneal Mφ. Therefore, LTB4 released by MCs and other inflammatory cells may be a key factor in the host defense against T. gondii (82). hMCs co-cultured with T. gondii RH tachyzoites that were opsonized with IgG directed against the surface antigen 1 exhibited a polarized degranulation toward the invading parasites and resulted in the death of more than 70% of the parasites during the process. On the other hand, non-opsonized T. gondii rapidly infected MCs without triggering any detectable degranulation process, and only 20% of the parasites died during the process. hMC treated with a chymase inhibitor did not affect parasite mortality, whereas hMC treated with a tryptase inhibitor significantly decreased the number of dead parasites in contact with degranulated MCs. Thus, IgG-opsonized T. gondii resulted in tryptase-dependent parasite death (83). TS-4 strain T. gondii infection significantly increased the expression of metalloproteinases (MMP)-2 and MMP-9 in P815 murine mastocytoma cells, and the invading parasites can elicit Erk1/2 phosphorylation, leading to NF-κB activation in the cytoplasm. This pathway for generating MMP-2 and MMP-9 is important in host defense mechanisms against T. gondii (84). The mediators of activated MCs play an important role in modulating acute inflammatory pathogenesis and parasite clearance in T. gondii infection (85).
Concluding Remarks
In this review, we have highlighted that MCs influence the outcome and immune response to Plasmodium spp., Leishmania spp., Trypanosoma spp., and T. gondii infections, and these protozoan parasites can all trigger MC activation, exhibiting an increase in the number of MCs and the degree of their degranulation, and have fundamentally diverse impacts on protozoan infections in different settings, i.e., protozoan parasite infections can be controlled or may deteriorate through the release of different MC mediators, proteases, and cytokines, etc. In P. berghei-infected mice, MCs and MC-derived TNF play protective roles in murine malaria. In Leishmania infection, the MC count is positively associated with the disease duration of cutaneous leishmaniasis. During T. gondii infection, an increased MC number and greater MC activation are observed in infected animals. Furthermore, MCs are required for mouse survival after oral infection with T. gondii. However, MCs can also worsen the outcome of a protozoan infection under certain circumstances. For example, MC degranulation is significantly correlated with parasitemia and disease severity in human malaria; histamine-mediated signaling contributes to malaria pathogenesis. In T. cruzi infection, greater MC counts with more fibrosis are found in the colon musculature of chronic Chagas patients with megacolon or the myocardium of patients with Chagas cardiopathy. MC activation by MC stimulators can deteriorate the pathology and increase the parasitic load in acute T. gondii-infected mice. Moreover, some studies have shown that MCs have no impact on malaria pathogenesis caused by P. berghei ANKA and no effect on lesion size development by L. major infection in mouse models. In addition, the numbers of MCs in the intestines of T. brucei-infected rats are not significantly different compared to uninfected controls. Mediators from MCs play a key role in inflammation and in the pathogenesis of the protozoan parasitic diseases. Understanding the mechanisms by which MCs regulate pathogenesis during different protozoan parasite infections may potentially lead to the development of a new and unique therapeutic target for protozoan-related diseases. Therefore, further studies that evaluate the clinical importance of MC-protozoan interactions may lead to new therapeutic approaches for these protozoan parasitic diseases.
Author Contributions
FL conceived and wrote the manuscript, and SH participated in the writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This publication was supported in part by the Natural Science Foundation of China (no. 81471973) and the Science and Technology Planning Project of Guangdong Province, China (nos. 2014A020212108, 2014A020212212, and 2013B021800043).
References
1. Haldar K, Mohandas N. Erythrocyte remodeling by malaria parasites. Curr Opin Hematol (2007) 14:203–9. doi:10.1097/MOH.0b013e3280f31b2d
2. Gurung P, Kanneganti TD. Innate immunity against Leishmania infections. Cell Microbiol (2015) 17:1286–94. doi:10.1111/cmi.12484
3. Séguin O, Descoteaux A. Leishmania, the phagosome, and host responses: the journey of a parasite. Cell Immunol (2016) 309:1–6. doi:10.1016/j.cellimm.2016.08.004
4. Thomas SM, Purmal A, Pollastri M, Mensa-Wilmot K. Discovery of a carbazole-derived lead drug for human African trypanosomiasis. Sci Rep (2016) 6:32083. doi:10.1038/srep32083
5. Rassi A Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet (2010) 375:1388–402. doi:10.1016/S0140-6736(10)60061-X
6. de Noya BA, Gonzalez ON. An ecological overview on the factors that drives to Trypanosoma cruzi oral transmission. Acta Trop (2015) 151:94–102. doi:10.1016/j.actatropica.2015.06.004
7. Dubey JP. History of the discovery of the life cycle of Toxoplasma gondii. Int J Parasitol (2009) 39:877–82. doi:10.1016/j.ijpara.2009.01.005
8. Landrith TA, Harris TH, Wilson EH. Characteristics and critical function of CD8+ T cells in the Toxoplasma-infected brain. Semin Immunopathol (2015) 37:261–70. doi:10.1007/s00281-015-0487-3
9. Maldonado YA, Read JS; Committee on Infectious Diseases. Diagnosis, treatment, and prevention of congenital toxoplasmosis in the United States. Pediatrics (2017) 139:e20163860. doi:10.1542/peds.2016-3860
10. Bulfone-Paus S, Bahri R. Mast cells as regulators of T cell responses. Front Immunol (2015) 6:394. doi:10.3389/fimmu.2015.00394
11. Arthur G, Bradding P. New developments in mast cell biology: clinical implications. Chest (2016) 150:680–93. doi:10.1016/j.chest.2016.06.009
12. Halova I, Draberova L, Draber P. Mast cell chemotaxis-chemoattractants and signaling pathways. Front Immunol (2012) 3:119. doi:10.3389/fimmu.2012.00119
13. Vukman KV, Lalor R, Aldridge A, O’Neill SM. Mast cells: new therapeutic target in helminth immune modulation. Parasite Immunol (2016) 38:45–52. doi:10.1111/pim.12295
14. Martins PR, Nascimento RD, de Souza Lisboa A, Martinelli PM, d’Ávila Reis D. Neuroimmunopathology of Trypanosoma cruzi-induced megaoesophagus: is there a role for mast cell proteases? Hum Immunol (2014) 75:302–5. doi:10.1016/j.humimm.2014.02.003
15. Theoharides TC. Mast cells promote malaria infection? Clin Ther (2015) 37:1374–7. doi:10.1016/j.clinthera.2015.03.014
16. Wilainam P, Nintasen R, Viriyavejakul P. Mast cell activation in the skin of Plasmodium falciparum malaria patients. Malar J (2015) 14:67. doi:10.1186/s12936-015-0568-8
17. Choumet V, Attout T, Chartier L, Khun H, Sautereau J, Robbe-Vincent A, et al. Visualizing non-infectious and infectious Anopheles gambiae blood feedings in naive and saliva-immunized mice. PLoS One (2012) 7:e50464. doi:10.1371/journal.pone.0050464
18. Furuta T, Kikuchi T, Iwakura Y, Watanabe N. Protective roles of mast cells and mast cell-derived TNF in murine malaria. J Immunol (2006) 177:3294–302. doi:10.4049/jimmunol.177.5.3294
19. Porcherie A, Mathieu C, Peronet R, Schneider E, Claver J, Commere PH, et al. Critical role of the neutrophil-associated high-affinity receptor for IgE in the pathogenesis of experimental cerebral malaria. J Exp Med (2011) 208:2225–36. doi:10.1084/jem.20110845
20. Beghdadi W, Porcherie A, Schneider BS, Morisset S, Dubayle D, Peronet R, et al. Histamine H-3 receptor-mediated signaling protects mice from cerebral malaria. PLoS One (2009) 4:e6004. doi:10.1371/journal.pone.0006004
21. Beghdadi W, Porcherie A, Schneider BS, Dubayle D, Peronet R, Huerre M, et al. Inhibition of histamine-mediated signaling confers significant protection against severe malaria in mouse models of disease. J Exp Med (2008) 205:395–408. doi:10.1084/jem.20071548
22. Chau JY, Tiffany CM, Nimishakavi S, Lawrence JA, Pakpour N, Mooney JP, et al. Malaria-associated l-arginine deficiency induces mast cell-associated disruption to intestinal barrier defenses against nontyphoidal Salmonella bacteremia. Infect Immun (2013) 81:3515–26. doi:10.1128/IAI.00380-13
23. Potts RA, Tiffany CM, Pakpour N, Lokken KL, Tiffany CR, Cheung K, et al. Mast cells and histamine alter intestinal permeability during malaria parasite infection. Immunobiology (2016) 221:468–74. doi:10.1016/j.imbio.2015.11.003
24. Tuon FF, Amato VS, Bacha HA, Almusawi T, Duarte MI, Amato Neto V. Toll-like receptors and leishmaniasis. Infect Immun (2008) 76:866–72. doi:10.1128/IAI.01090-07
25. Saha B, Tonkal AM, Croft S, Roy S. Mast cells at the host-pathogen interface: host-protection versus immune evasion in leishmaniasis. Clin Exp Immunol (2004) 137:19–23. doi:10.1111/j.1365-2249.2004.02505.x
26. Dudeck A, Suender CA, Kostka SL, von Stebut E, Maurer M. Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur J Immunol (2011) 41:1883–93. doi:10.1002/eji.201040994
27. Calabrese KS, Cortada VM, Dorval ME, Souza Lima MA, Oshiro ET, Souza CS, et al. Leishmania (Leishmania) infantum/chagasi: histopathological aspects of the skin in naturally infected dogs in two endemic areas. Exp Parasitol (2010) 124:253–7. doi:10.1016/j.exppara.2009.10.005
28. Menezes-Souza D, Guerra-Sá R, Carneiro CM, Vitoriano-Souza J, Giunchetti RC, Teixeira-Carvalho A, et al. Higher expression of CCL2, CCL4, CCL5, CCL21, and CXCL8 chemokines in the skin associated with parasite density in canine visceral leishmaniasis. PLoS Negl Trop Dis (2012) 6:e1566. doi:10.1371/journal.pntd.0001566
29. Maurer M, Lopez Kostka S, Siebenhaar F, Moelle K, Metz M, Knop J, et al. Skin mast cells control T cell-dependent host defense in Leishmania major infections. FASEB J (2006) 20:2460–7. doi:10.1096/fj.06-5860com
30. Paul C, Wolff S, Zapf T, Raifer H, Feyerabend TB, Bollig N, et al. Mast cells have no impact on cutaneous leishmaniasis severity and related Th2 differentiation in resistant and susceptible mice. Eur J Immunol (2016) 46:114–21. doi:10.1002/eji.201545613
31. Pinto AJ, de Amorim IF, Pinheiro LJ, Madeira IM, Souza CC, Chiarini-Garcia H, et al. Glycol methacrylate embedding for the histochemical study of the gastrointestinal tract of dogs naturally infected with Leishmania infantum. Eur J Histochem (2015) 59:2546. doi:10.4081/ejh.2015.2546
32. Calabrese KS, Silva LS, Hardoim DJ, Souza CS, Abreu-Silva AL. Ocular experimental leishmaniasis in C57BL/10 and BALB/c mice induced by Leishmania amazonensis infection. Exp Parasitol (2013) 133:156–61. doi:10.1016/j.exppara.2012.11.008
33. Martins PR, Nascimento RD, Lopes JG, Santos MM, de Oliveira CA, de Oliveira EC, et al. Mast cells in the colon of Trypanosoma cruzi-infected patients: are they involved in the recruitment, survival and/or activation of eosinophils? Parasitol Res (2015) 114:1847–56. doi:10.1007/s00436-015-4371-9
34. Pinheiro SW, Rua AM, Etchebehere RM, Cançado CG, Chica JE, Lopes ER, et al. Morphometric study of the fibrosis and mast cell count in the circular colon musculature of chronic Chagas patients with and without megacolon. Rev Soc Bras Med Trop (2003) 36:461–6. doi:10.1590/S0037-86822003000400005
35. Campos CF, Cangussú SD, Duz AL, Cartelle CT, Noviello Mde L, Veloso VM, et al. Enteric neuronal damage, intramuscular denervation and smooth muscle phenotype changes as mechanisms of chagasic megacolon: evidence from a long-term murine model of Tripanosoma cruzi infection. PLoS One (2016) 11:e0153038. doi:10.1371/journal.pone.0153038
36. Meuser-Batista M, Corrêa JR, Carvalho VF, de Carvalho Britto CF, Moreira OC, Batista MM, et al. Mast cell function and death in Trypanosoma cruzi infection. Am J Pathol (2011) 179:1894–904. doi:10.1016/j.ajpath.2011.06.014
37. Roldão JA, Beghini M, Ramalho LS, Porto CS, Rodrigues DB, Teixeira VP, et al. Comparison between the collagen intensity and mast cell density in the lingual muscles and myocardium of autopsied chronic chagasic and non-chagasic patients. Parasitol Res (2012) 111:647–54. doi:10.1007/s00436-012-2882-1
38. Gould SS, Castro GA. Suppression by Trypanosoma brucei of anaphylaxis-mediated ion transport in the small intestine of rats. Immunology (1994) 81:468–74.
39. Koeppel MC, Abitan R, Angeli C, Lafon J, Pelletier J, Sayag J. Cutaneous and gastrointestinal mastocytosis associated with cerebral toxoplasmosis. Br J Dermatol (1998) 139:881–4. doi:10.1046/j.1365-2133.1998.02518.x
40. Gil CD, Mineo JR, Smith RL, Oliani SM. Mast cells in the eyes of Calomys callosus (Rodentia: Cricetidae) infected by Toxoplasma gondii. Parasitol Res (2002) 88:557–62. doi:10.1007/s00436-002-0593-8
41. Cruz A, Mendes ÉA, de Andrade MV, do Nascimento VC, Cartelle CT, Arantes RM, et al. Mast cells are crucial in the resistance against Toxoplasma gondii oral infection. Eur J Immunol (2014) 44:2949–54. doi:10.1002/eji.201344185
42. Ferreira GLS, Mineo JR, Oliveira JG, Ferro EAV, Souza MA, Santos AAD. Toxoplasma gondii and mast cell interactions in vivo and in vitro: experimental infection approaches in Calomys callosus (Rodentia, Cricetidae). Microbes Infect (2004) 6:172–81. doi:10.1016/j.micinf.2003.11.007
43. Del Rio L, Bennouna S, Salinas J, Denkers EY. CXCR2 deficiency confers impaired neutrophil recruitment and increased susceptibility during Toxoplasma gondii infection. J Immunol (2001) 167:6503–9. doi:10.4049/jimmunol.167.11.6503
44. Huang B, Huang S, Chen Y, Zheng H, Shen J, Lun ZR, et al. Mast cells modulate acute toxoplasmosis in murine models. PLoS One (2013) 8:e77327. doi:10.1371/journal.pone.0077327
45. Cohen PR. Solitary mastocytoma presenting in an adult: report and literature review of adult-onset solitary cutaneous mastocytoma with recommendations for evaluation and treatment. Dermatol Pract Concept (2016) 6:31–8. doi:10.5826/dpc.0603a07
46. Owhashi M, Harada M, Suguri S, Ohmae H, Ishii A. The role of saliva of Anopheles stephensi in inflammatory response: identification of a high molecular weight neutrophil chemotactic factor. Parasitol Res (2001) 87:376–82. doi:10.1007/s004360000355
47. Mecheri S. Contribution of allergic inflammatory response to the pathogenesis of malaria disease. Biochim Biophys Acta (2012) 1822:49–56. doi:10.1016/j.bbadis.2011.02.005
48. Khalil M, Ronda J, Weintraub M, Jain K, Silver R, Silverman AJ. Brain mast cell relationship to neurovasculature during development. Brain Res (2007) 1171:18–29. doi:10.1016/j.brainres.2007.07.034
49. Guermonprez P, Helft J, Claser C, Deroubaix S, Karanje H, Gazumyan A, et al. Inflammatory Flt3l is essential to mobilize dendritic cells and for T cell responses during Plasmodium infection. Nat Med (2013) 19:730–8. doi:10.1038/nm.3197
50. Furuta T, Imajo-Ohmi S, Fukuda H, Kano S, Miyake K, Watanabe N. Mast cell-mediated immune responses through IgE antibody and toll-like receptor 4 by malarial peroxiredoxin. Eur J Immunol (2008) 38:1341–50. doi:10.1002/eji.200738059
51. Walter M, Stark H. Histamine receptor subtypes: a century of rational drug design. Front Biosci (Schol Ed) (2012) 4:461–88. doi:10.2741/s279
52. Enwonwu CO, Afolabi BM, Salako LO, Idigbe EO, Bashirelah N. Increased plasma levels of histidine and histamine in falciparum malaria: relevance to severity of infection. J Neural Transm (Vienna) (2000) 107:1273–87. doi:10.1007/s007020070017
53. Hempel C, Hoyer N, Staalsø T, Kurtzhals JA. Effects of the vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor SU5416 on in vitro cultures of Plasmodium falciparum. Malar J (2014) 13:201. doi:10.1186/1475-2875-13-201
54. Furuta T, Kimura M, Watanabe N. Elevated levels of vascular endothelial growth factor (VEGF) and soluble vascular endothelial growth factor receptor (VEGFR)-2 in human malaria. Am J Trop Med Hyg (2010) 82:136–9. doi:10.4269/ajtmh.2010.09-0203
55. Casals-Pascual C, Idro R, Gicheru N, Gwer S, Kitsao B, Gitau E, et al. High levels of erythropoietin are associated with protection against neurological sequelae in African children with cerebral malaria. Proc Natl Acad Sci U S A (2008) 105:2634–9. doi:10.1073/pnas.0709715105
56. Muehlenbachs A, Mutabingwa TK, Edmonds S, Fried M, Duffy PE. Hypertension and maternal-fetal conflict during placental malaria. PLoS Med (2006) 3:e446. doi:10.1371/journal.pmed.0030446
57. Epiphanio S, Campos MG, Pamplona A, Carapau D, Pena AC, Ataíde R, et al. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog (2010) 6:e1000916. doi:10.1371/journal.ppat.1000916
58. Ayimba E, Hegewald J, Ségbéna AY, Gantin RG, Lechner CJ, Agosssou A, et al. Proinflammatory and regulatory cytokines and chemokines in infants with uncomplicated and severe Plasmodium falciparum malaria. Clin Exp Immunol (2011) 166:218–26. doi:10.1111/j.1365-2249.2011.04474.x
59. Shaik-Dasthagirisaheb YB, Varvara G, Murmura G, Saggini A, Potalivo G, Caraffa A, et al. Vascular endothelial growth factor (VEGF), mast cells and inflammation. Int J Immunopathol Pharmacol (2013) 26:327–35. doi:10.1177/039463201302600206
60. Besnard AG, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, et al. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog (2015) 11:e1004607. doi:10.1371/journal.ppat.1004607
61. Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol (2007) 7:93–104. doi:10.1038/nri2018
62. Bidri M, Vouldoukis I, Mossalayi MD, Debré P, Guillosson JJ, Mazier D, et al. Evidence for direct interaction between mast cells and Leishmania parasites. Parasite Immunol (1997) 19:475–83. doi:10.1046/j.1365-3024.1997.d01-153.x
63. Aflatoonian MR, Sharifi I, Aflatoonian B, Shirzadi MR, Gouya MM, Kermanizadeh A. A review of impact of bam earthquake on cutaneous leishmaniasis and status: epidemic of old foci, emergence of new foci and changes in features of the disease. J Arthropod Borne Dis (2016) 10:272–81.
64. von Stebut E. Cutaneous Leishmania infection: progress in pathogenesis research and experimental therapy. Exp Dermatol (2007) 16:340–6. doi:10.1111/j.1600-0625.2007.00554.x
65. de Oliveira Cardoso F, de Souza Cda S, Mendes VG, Abreu-Silva AL, Gonçalves da Costa SC, Calabrese KS. Immunopathological studies of Leishmania amazonensis infection in resistant and in susceptible mice. J Infect Dis (2010) 201:1933–40. doi:10.1086/652870
66. Kautz-Neu K, Schwonberg K, Fischer MR, Schermann AI, von Stebut E. Dendritic cells in Leishmania major infections: mechanisms of parasite uptake, cell activation and evidence for physiological relevance. Med Microbiol Immunol (2012) 201:581–92. doi:10.1007/s00430-012-0261-2
67. Romão PR, Da Costa Santiago H, Ramos CD, De Oliveira CF, Monteiro MC, De Queiroz Cunha F, et al. Mast cell degranulation contributes to susceptibility to Leishmania major. Parasite Immunol (2009) 31:140–6. doi:10.1111/j.1365-3024.2008.01084.x
68. Villaseñor-Cardoso MI, Salaiza N, Delgado J, Gutiérrez-Kobeh L, Pérez-Torres A, Becker I. Mast cells are activated by Leishmania mexicana LPG and regulate the disease outcome depending on the genetic background of the host. Parasite Immunol (2008) 30:425–34. doi:10.1111/j.1365-3024.2008.01042.x
69. Roy S, Mandal C. Leishmania donovani utilize sialic acids for binding and phagocytosis in the macrophages through selective utilization of siglecs and impair the innate immune arm. PLoS Negl Trop Dis (2016) 10:e0004904. doi:10.1371/journal.pntd.0004904
70. Pitta MG, Romano A, Cabantous S, Henri S, Hammad A, Kouriba B, et al. IL-17 and IL-22 are associated with protection against human kala azar caused by Leishmania donovani. J Clin Invest (2009) 119:2379–87. doi:10.1172/JCI38813
71. Cruz AF, Resende RG, Albuquerque DR, de Lacerda JC, Leite CF, Ferreira Aguiar MC. Mucosal leishmaniasis in Brazilian patients: two case reports with similar clinical presentation and different approaches. Oral Surg Oral Med Oral Pathol Oral Radiol (2016) 122:e199–203. doi:10.1016/j.oooo.2016.02.017
72. Daneshbod Y, Oryan A, Davarmanesh M, Shirian S, Negahban S, Aledavood A, et al. Clinical, histopathologic, and cytologic diagnosis of mucosal leishmaniasis and literature review. Arch Pathol Lab Med (2011) 135:478–82. doi:10.1043/2010-0069-OA.1
73. Nikandish M, Goyonlo VM, Taheri AR, Kiafar B. Ocular leishmaniasis treated by intralesional amphotericin B. Middle East Afr J Ophthalmol (2016) 23:153–5. doi:10.4103/0974-9233.171801
74. Pandey SP, Doyen N, Mishra GC, Saha B, Chandel HS. TLR9-deficiency reduces TLR1, TLR2 and TLR3 expressions in Leishmania major-infected macrophages. Exp Parasitol (2015) 154:82–6. doi:10.1016/j.exppara.2015.04.005
75. Sandig H, Bulfone-Paus S. TLR signaling in mast cells: common and unique features. Front Immunol (2012) 3:185. doi:10.3389/fimmu.2012.00185
76. de Oliveira MP, Lima MC, Calheiros AS, Martins MA, Antas PR, De Luca PM, et al. Leishmania (Viannia) braziliensis: human mast cell line activation induced by logarithmic and stationary promastigote derived-lysates. Exp Parasitol (2005) 109:72–9.
77. Gattoni CM, Aleixo IF, de Araujo MF, Teixeira Vde P, Rodrigues DB, Pereira SA. Chagas disease reactivation in HIV-coinfected patients: histopathological aspects. Immunobiology (2015) 220:656–62. doi:10.1016/j.imbio.2014.11.013
78. Cabral HR, Novak IT, Glocker TM, Castro Viera GA. Chagas cardiopathy: identification and quantification of infiltrating cells in the hearts of cardiac death patients of different ages. Rev Fac Cien Med Univ Nac Cordoba (2002) 59:83–9. [Article in Spanish].
79. Ajzenberg D, Lamaury I, Demar M, Vautrin C, Cabié A, Simon S, et al. Performance testing of PCR assay in blood samples for the diagnosis of toxoplasmic encephalitis in AIDS patients from the French departments of America and genetic diversity of Toxoplasma gondii: a prospective and multicentric study. PLoS Negl Trop Dis (2016) 10:e0004790. doi:10.1371/journal.pntd.0004790
80. Smith NL, Abi Abdallah DS, Butcher BA, Denkers EY, Baird B, Holowka D. Toxoplasma gondii inhibits mast cell degranulation by suppressing phospholipase Cγ-mediated Ca2+ mobilization. Front Microbiol (2013) 4:179. doi:10.3389/fmicb.2013.00179
81. Sawesi O, Spillmann D, Lundén A, Wernersson S, Åbrink M. Serglycin-independent release of active mast cell proteases in response to Toxoplasma gondii infection. J Biol Chem (2010) 285:38005–13. doi:10.1074/jbc.M110.118471
82. Henderson WR Jr, Chi EY. The importance of leukotrienes in mast cell-mediated Toxoplasma gondii cytotoxicity. J Infect Dis (1998) 177:1437–43. doi:10.1086/517833
83. Joulia R, Gaudenzio N, Rodrigues M, Lopez J, Blanchard N, Valitutti S, et al. Mast cells form antibody-dependent degranulatory synapse for dedicated secretion and defence. Nat Commun (2015) 6:6174. doi:10.1038/ncomms7174
84. Wang MF, Lu CY, Lai SC. Up-regulation of matrix metalloproteinases-2 and -9 via an Erk1/2/NF-kappaB pathway in murine mast cells infected with Toxoplasma gondii. J Comp Pathol (2013) 149:146–55. doi:10.1016/j.jcpa.2013.03.002
Keywords: mast cell, Plasmodium spp., Leishmania spp., Trypanosoma spp., Toxoplasma gondii
Citation: Lu F and Huang S (2017) The Roles of Mast Cells in Parasitic Protozoan Infections. Front. Immunol. 8:363. doi: 10.3389/fimmu.2017.00363
Received: 15 August 2016; Accepted: 14 March 2017;
Published: 06 April 2017
Edited by:
Heinrich Korner, University of Tasmania, AustraliaReviewed by:
Julia Walochnik, Medical University of Vienna, AustriaMarisa Mariel Fernandez, University of Buenos Aires, Argentina
Dirk Schlüter, Otto-Von-Gutricke University Magdeburg, Germany
Copyright: © 2017 Lu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fangli Lu, ZmFuZ2xpbHVAeWFob28uY29t;
Shiguang Huang, dGhzaGdAMTI2LmNvbQ==