 Silvia Lopez-Lastra1,2,3*
Silvia Lopez-Lastra1,2,3* James P. Di Santo1,2
James P. Di Santo1,2
- 1Innate Immunity Unit, Institut Pasteur, Paris, France
- 2Inserm U1223, Paris, France
- 3Université Paris-Sud (Paris-Saclay), Paris, France
Animal models have extensively contributed to our understanding of human immunobiology and to uncover the underlying pathological mechanisms occurring in the development of diseases. However, mouse models do not reproduce the genetic and molecular complexity inherent in human disease conditions. Human immune system (HIS) mouse models that are susceptible to human pathogens and can recapitulate human hematopoiesis and tumor immunobiology provide one means to bridge the interspecies gap. Natural killer cells are the founding member of the innate lymphoid cell family. They exert a rapid and strong immune response against tumor and pathogen-infected cells. Their antitumor features have long been exploited for therapeutic purposes in the context of cancer. In this review, we detail the development of highly immunodeficient mouse strains and the models currently used in cancer research. We summarize the latest improvements in adoptive natural killer (NK) cell therapies and the development of novel NK cell sources. Finally, we discuss the advantages of HIS mice to study the interactions between human NK cells and human cancers and to develop new therapeutic strategies.
Introduction
Since the generation of the first inbred mouse strains in the early 20th century, mice have served as model organisms to study mammalian biology. This approach has given birth to some of the most important scientific breakthroughs and discoveries that, in many cases, led to the development of successful treatments for previously untreatable diseases (e.g., acute promyelocytic leukemia) (1). However, Mus musculus and Homo sapiens have been evolving divergently for 85 million years, adapting to very different environments and undergoing selection for many traits, from the circadian rhythm to our body size (2). Thanks to the genome decoding, we can now appreciate that the one fifth of the genetic divergence between mice and humans is enriched in regions implicated in the immune system, metabolic processes, and stress responses (3). It is, therefore, not surprising that only less than 8% of the cancer studies in animal models reach clinical trials and that more than 80% of these eventually fail when tested in humans (4). The increasing knowledge of the molecular differences between mice and humans should allow us to evaluate the degree in which animal models may be suitable for translational research and when this is not the case, to then search for better systems.
With this aim, mice have been “humanized” by introducing human genes or genomic regions and by transferring human tissues or cells to study various aspects of human biology. The engraftment of human blood cells or blood-forming cells and organs into immunodeficient mice has opened a new era for translational immunology and the improvement of immunotherapies against human cancer and infectious diseases caused by pathogens with exclusive human tropism, such as HIV, HBV, and HCV.
Developing Human Immune System (HIS) Mice
Since the discovery of the nude athymic mutations in the 1960s, our knowledge of the host immune system and its ability to reject xenografts have led to the development of several mouse strains that permit long-term “take” and function of the human tissue grafts (5). Experiments performed in the 1980s with severe combined immunodeficient (SCID) mice (that lacked functional mouse adaptive lymphocytes due to mutations in the DNA-dependent protein kinase Prkdc) showed that these mice could be reconstituted with human peripheral blood mononuclear cells (PBMCs) or hematopoietic stem cells (HSCs) (6, 7). However, some residual adaptive (leakiness) and an essentially intact innate immunity in SCID mice limited the complete reconstitution of all human immune subsets. Moreover, SCID mice failed to engraft human tumor xenografts, thereby limiting the development of preclinical cancer models. An alternative system with analogous immunodeficiency was obtained by mutating the recombinant activating genes (Rag1, Rag2) loci that avoided genetic “leakiness” and, in contrast to SCID mice, did not result in host radiosensitivity (8, 9). Additional genetic modifications followed to further the immunodeficiency of host mice in order to promote tolerance to human cells. Two breakthroughs have remarkably boosted the advancement of the field. First, Greiner and colleagues found that the NOD strain supported an enhanced tolerance compared to other strains and, several years later, Takenaka’s team revealed that the molecular basis for this lies in the signal regulatory protein alpha (Sirpa) allele polymorphism (10–13). Contrarily to other strains, SIRPα from NOD mice binds to human CD47 ligand triggering a negative signal in mouse macrophages that prevents their phagocytosis (13, 14). This finding prompted the generation of transgenic mice expressing the human or NOD strain Sirpa allele thus conferring enhanced human cell engraftment (15–17). The second turning point for achieving a successful xenotransplantation was the common cytokine receptor gamma chain (γc, encoded at Il2rg), which leads to complete impairment of natural killer (NK) cell development and dendritic cell (DC) dysfunction (18, 19). Mice carrying Il2rg mutations were developed in various genetic backgrounds [NSG or NOG (both NOD PrkdcSCIDIl2rg−/−) and BRG (Balb/c Rag2−/−Il2rg−/−)] allowing robust, long-lasting de novo multilineage development of the HIS, including human thymopoiesis, and are the basis for most of the currently used models (20–23). From that point forward, a number of model variants have been developed to address specific questions or improve particular aspects of immunity, either by genetic manipulation, engraftment of additional human tissues, or exogenous administration of human factors. This is the case of the recently described Balb/c Rag2−/−Il2rg−/−Flt3−/− (BRGF) model with specific boost of conventional and plasmacytoid DCs after exogenous Flt3 ligand treatment. This model offers a great platform for screening of immune adjuvants and DC targeting therapies (24).
Human Cancer Models in “Humanized” Mice
Immunodeficient mice allow great flexibility for the study of human tumor immunobiology. Human tumors can be generated in NSG, NOG, BRGS, and other strains using established tumor cell lines, after transplantation of human primary tumors or following de novo induction of hematological neoplasms (Figure 1). These different models provide systems that better reflect the complexity of the disease. In order to allow human tumor to engraft and grow in mice, the host immune system is generally compromised leading to tumor kinetics that may not reflect the true patient situation. As discussed earlier, human immune components can be generated in vivo from human HSCs or other progenitors and “supported or potentiated” later on or infused once the tumor is established. These approaches provide “mixed” systems in which human immune cells and human tumors can co-exist allowing the dissection of immune deviation as well as studying immunotherapy.
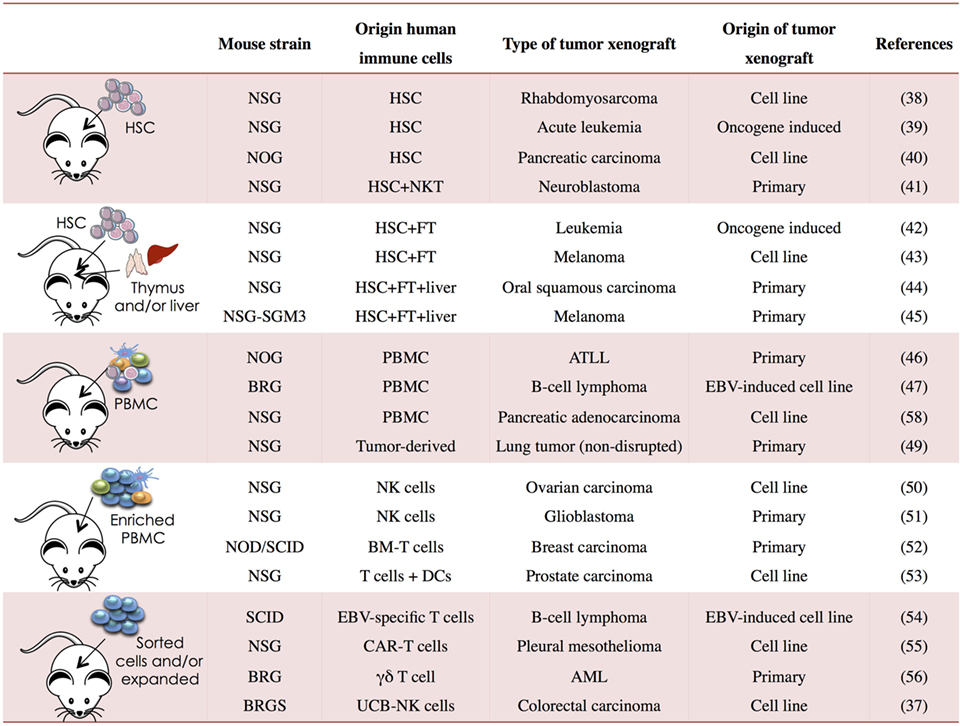
Figure 1. Human immune system (HIS) mouse models used in cancer research. PBMC, peripheral blood mononuclear cells; NSG, NOD PrkdcSCIDIl2rg−/−; BRG, Balb/c Rag2−/−Il2rg−/−; BRGS, Balb/c Rag2−/−Il2rg−/− SirpaNOD; HSC, hematopoietic stem cell; FT, fetal thymus; BM, bone marrow; CAR, chimeric antigen receptor; UCB, umbilical cord blood; ATLL, T-cell leukemia/lymphoma; AML, acute myeloid leukemia; EBV, Epstein–Barr virus (37–56).
A wide range of established tumor cell lines from different origins (brain, colon, breast, melanoma, ovarian, prostate, etc.) have been engrafted in immunocompromised mice and have greatly contributed to drug development and the preclinical assessment of potential therapies. However, the gradual accumulation of genetic and phenotypic aberrations in these cells due to their long-term culture impacts the surface markers and the tumorigenicity of the malignancy (25). These limitations have set aside these models to preliminary studies addressing specific questions like the ability of a potential therapy to target a certain molecule that has been overexpressed in the cell line. In recent years, the field has been, therefore, switching toward the engraftment of patient-derived primary tumors (PDX, patient-derived xenografts) that retain the phenotypic and genetic complexity observed in clinical samples thus better predicting drug efficacy and clinical translatability (26, 27). These include tumor stromal cells and tumor-associated lymphocytes that contribute greatly to tumor growth and metastasis and, therefore, to the therapeutic response. These PDX-HIS mouse models can engraft the tumor as efficiently as the non-humanized mice, they respond to standard chemotherapeutic drugs similarly to patients and they have proven to be responsive to newly derived immune modulators.
One of the better-characterized PDX models is the AML that has contributed to the identification of leukemia stem cells (LSC) by transplanting different stem-like cell fractions and analyzing the leukemia-initiating activity of each in SCID mice (28–31). The discovery of the concept of cancer stem cell (CSC) has been a breakthrough in cancer biology due to the clinical benefits for the long-term disease-free survival. CSC presence has been identified in numerous other malignancies through transfer into immunocompromised mice and, interestingly, markers associated to CSCs have been correlated to the tumorigenic potential (32, 33). Recent improvements in HIS mouse models by the transgenic expression of certain factors, like the NSG-SGM3 expressing human SCF, GM-SCF, and IL-3, have further ameliorate the engraftment and growth of human leukemia allowing the study of the tumor initiating cells (34). The demonstration that HIS mouse models reproduce the heterogeneity and behavior of human tumors creates great expectation on the better phenotyping of these tumor-initiating cells and the identification of drugs targeting this key population (35).
One of the advantages of modeling cancer in humanized mice is that we can study the systemic environment and the contribution of nearly all the immune cells to the pathogenesis. In this regard, a NSG model of treatment-refractory B-cell leukemia revealed that infiltration of leukemia cells into the bone marrow rewires the tumor microenvironment to inhibit engulfment of antibody-targeted tumor cells. This resistance could be overcome by combination regimens involving therapeutic antibodies and chemotherapy that lead to macrophage infiltration and phagocytic activity in the bone marrow improving the efficacy of targeted therapeutics (36).
Targeting NK Cells for Cancer Immunotherapy
To date, most immunomodulatory strategies have focused on agents or cell therapies targeting T cell immunity. In contrast, innate immune cells, such as NK cells, have been less exploited. Nevertheless, the fundamental role for these cells has been for long justified by the higher cancer incidence in individuals with defective NK function (57). Furthermore, a number of mouse models lacking or deficient for NK cell function have corroborated their importance in tumor immunosurveillance (58).
Natural killer cells exert an immediate cytotoxicity when encountering a malignant cell and they do so without a specific antigen priming but instead, by the integrated signal of an array of activating and inhibitory receptors. Among the first group, the C-type lectin-like receptors CD94/NKG2C and NKG2D and the natural cytotoxicity receptors NKp30, NKp44, and NKp46 as well as the Ig-like receptor DNAM-1 (CD266) mediate NK cell activation when they recognize tumor cells. On the other hand, polymorphic inhibitory killer cell immunoglobulin-like receptors (KIRs) with their cognate human–leukocyte–antigen (HLA) ligands as well as CD94/NKG2A with the non-classical class I molecule HLA-E as ligand provide inhibitory signaling. In addition to the contact mediated regulation of the activity, NK cells also respond to cytokines like IL-2, IL-12, IL-15, IL-18, and IL-21, as well as toll-like receptor ligands that shape their differentiation, proliferation, and activation status (59). Cytotoxicity activity is triggered through activation of the low-affinity activating receptor FcγRIIIa (CD16) that binds the Fc portion of immunoglobulin G1, which has been exploited in monoclonal antibody immunotherapies. NK cells kill virus-infected and tumor cells using a cargo of perforin and granzymes contained in cytotoxic granules and less efficiently by a mechanism dependent on FAS ligand, TNF, or TNF-related apoptosis-inducing ligand (60).
Given that NK cells in HSC-derived humanized mice express all the afore-mentioned receptors and respond similarly to the same cytokines (61, 62), these in vivo models represent a powerful platform to explore the pivotal role of NK cells in cancer immunosurveillance (63–65) (Lopez-Lastra et al., in revision). Additionally, environmental components such as inhibiting factors (TGF-β, IL-10, prostaglandin E2…) and immunosuppressive cells (Tregs, MDSCs) that influence the NK cell antitumor activity have also been described in HIS mice, enabling the evaluation of therapeutic strategies targeting the suppression of NK cells (66).
Although chemotherapy is still the core of the current clinical anticancer treatments, immunomodulators have now regained expectations after the revolutionary discovery of the CTLA-4 and PD-1 checkpoint inhibitors targeting T-cell activation (67). Humanized mice have proven to recapitulate the therapeutic effect of those antibodies as well as the side effects and have began to provide insights about the mechanism behind and possible strategies to improve them (68–70). The expression of these receptors on human NK cells suggests that they could also be targeted by checkpoint molecules and, therefore, contributes to the outcome of the therapy (71, 72). Indeed, mouse studies on a glioma model treated with activated NK cells preincubated with an anti-PD-1 blocking antibody showed an enhancement of the survival suggesting a role that must be explored in a human system (73).
Adoptive Transfer of NK Cells for Cancer Therapy
The potential of NK cells as innate effectors in cancer has been studied by the adoptive transfer of ex vivo expanded and/or activated NK cells in immunodeficient mice. Mice treated with adoptively transferred human NK cells show NK-mediated rejection of the engrafted human tumor and further administration of cytokines, such as IL-2 and IL-15 greatly improve the NK cell pool and their cytotoxic activity against transformed cell. These observations initially made in mice laid the foundation for the autologous NK cell infusion therapies started in the 1980s for metastatic cancers (74). Preclinical assessment of cytokine regimens in other cancer models, such as the low-dose IL-2 in the spontaneous EBV-associated B-cell lymphoma in PBL-SCID mice, demonstrated reduction of the tumor load and survival prolongation (75), and preceded a number of clinical trials for both hematological and solid tumors (76–79).
The discovery that inhibitory KIRs binding to MHC-I mediate inhibition of NK cells opened a new path on NK cell immunotherapies. NOD/SCID cancer models served as a platform to confirm the higher efficacy of alloreactive NK cells for the treatment of leukemia. Contrarily to T cells, NK cell do not provoke graft-versus-host disease (GVHD) in hematopoietic stem cell transplantation (HSCT) contexts but, instead, protect the patient against it and eliminate leukemia relapse and graft rejection (80). Later on, safety and efficacy of alloreactive NK cell infusion was confirmed in the clinic by Miller and colleagues in non-HSCT settings with patients suffering from metastatic melanoma, renal cell carcinoma, Hodgkin’s lymphoma, and refractory AML (81). For many years, allogeneic NK cell infusions have been tested in the clinic with positive results and rare cases of mild toxicity (82). Strikingly, a recent pediatric clinical study has reported some patients suffering from acute GVHD after infusion of ex vivo expanded donor NK cells in HLA-matched HSCT (83), rising the necessity to perform more robust preclinical testing in humanized models. One strategy to do so was illustrated in a recent study performed in NSG mice, in which an alloreactive NK cell subpopulation expressing KIR2DS2 but lacking inhibitory KIR-HLA mismatch had dominant functional activation advantage to kill patient-derived glioblastoma cells (84). The regulation of the activity on infused NK cells has been classically based on HLA-KIR matching; however, other inhibitory receptors are implicated on the inhibition of NK cell cytotoxicity. A recent study in NSG mice engrafted with human HSC has shown that anti-NKG2A antibodies can stimulate human NK cell killing in AML and ALL models bypassing the need to search for NK cell alloreactive donors (85). In vitro experiments have also pointed to an increased NK cell-mediated lysis of lymphoma and myeloma cells with allogeneic NK cell infusion in combination with monoclonal antibodies blocking inhibitory KIRs but this effect need to be confirmed in vivo (86, 87).
Another strategy to increase NK cell activity without aggravating the side-effects is the expression of chimeric antigen receptors (CARs) directed against tumor antigens. Preclinical evaluation of CD20 targeting primary NK cell infusion in humanized mice has led to a clinical trial on B-lineage acute lymphoblastic leukemia currently undergoing (88). Other preclinical trials using CAR-engineered primary human NK cells are now being performed in lymphoma, leukemia, carcinomas, and neuroblastoma mouse models.
Natural killer cells are often infused in combination with immunomodulators that boost their antitumor effects or regulate their activity. CD16 receptor is targeted by many of those modulators since it mediates antibody-dependent cellular cytotoxicity (ADCC) when it recognizes an antibody on a tumor cell, leading to target cell lysis. This mechanism has been exploited by using monoclonal antibodies targeted tumor antigens thus stimulating the endogenous or adoptive NK cells. Evidences of NK cell-mediated ADCC and mild to moderate toxicity were observed in preclinical models and then confirmed for some cases in the clinical setting. Malignancies such as non-Hodgkin lymphoma with rituximab (anti-CD20), metastatic breast cancer with trastuzumab (anti-HER2) or metastatic colorectal, and squamous cell carcinoma of the head and neck have been treated with monoclonal antibodies together with NK cell infusions or in combination regimes extending the disease-free survival and overall survival of thousands of patients (89–91).
As mentioned before, CSCs are emerging as necessary targets to achieve cancer cures since current treatments eliminate the bulk of the tumor cells but rare resistant CSCs persist and lead to later tumor relapse (92). The upregulation of stress-induced antigens together with the ability of NK cells to target non-proliferating cells suggest that NK cells could effectively eliminate CSCs. Indeed, recent studies in pancreatic carcinoma-bearing NSG mice demonstrated the capacity of activated transferred NK cells to reduce intratumoral CSCs and tumor burden (93–95).
Novel NK Cell Sources for Adoptive Therapy of Cancer
Two of the parameters to consider when evaluating the safety of NK cell products in clinical applications are the cell source and the culture conditions before the infusion. GM-CSF mobilized PBMCs, bone marrow, or umbilical cord blood (UCB) are the main sources of NK cells. With GM-CSF effects on NK function still to determine and BM being logistically difficult to obtain, UCB derived NK cells have been revealed as the best source of human material. Researchers are working on improving the expansion yield and purity as well as to enhance the activity of UCB derived NK cells before infusion in the patients. NSG mice demonstrated the capacity of these cells to migrate to BM, spleen, and liver and the inhibition of leukemia growth and prolongation of mice survival when combined with low-dose IL-15 (96). This preclinical result prompted a phase I clinical study in elderly AML patients that confirmed the safety and capacity of these cells to migrate and repopulate BM even in the absence of cytokine administration (97). This NK cell product aims at overcoming the major limitation of NK cell therapies in solid tumors, the delivery of high enough numbers of activated NK cells to the tumor site, and it is now under preclinical evaluation in the context of cervical and colorectal carcinomas (37, 98).
Alternative sources for NK cell therapy include embryonic stem cell (hESC)- or induced pluripotent stem cell (iPS)-derived NK cells, which are still under experimental development. Efficient generation of NK cells from hESC and iPS cells has been achieved, showing in vitro functional cytolytic activity against tumor cells, IFN-γ production, and expression of functional receptors (99). Very few reports are available regarding the in vivo activity of these products, with the most encouraging being in a NOD/SCID mouse model in which hESC-derived NK cells efficiently cleared a leukemia cell line tumor (100). Nevertheless, feeder-free conditions of NK cell generation need to be improved and the stability and safety of these NK cells products should be further proved in preclinical humanized models.
Finally, there is great prospect in NK cell lines as a potentially unlimited “pure” NK cell source. A clonal NK cell line NK-92 has shown the highest and most consistent cytotoxicity due to the combination of activating receptors it expresses and the absence of inhibitory KIRs (101). AML, myeloma, and melanoma are some of the numerous malignancies that have been partially eliminated from SCID mice after infusion of NK-92 (102–104). Clinical trials have further confirmed the safety and efficacy of this cell line in both solid and hematologic malignancies (105, 106). One further advantage of NK-92 is the ease of transfection with non-viral vectors allowing them to express IL-2 (required for their proliferation), thus representing a powerful “off-the-shelf” cell therapeutic (107). Additionally and inspired by the remarkable responses obtained by CAR-T cells and the early results in primary NK cells, NK-92 can be very easily transfected with a gene that expresses a tumor-CAR (108). The first preclinical tests in NSG mice have shed very optimistic results in leukemia models after CD19- or CD20-specific NK-92 infusions as well as in patient-derived glioblastoma with EGFR-specific NK-92 (109, 110). Still, these cellular therapies retain safety concerns including on-target/off-tumor effects and unregulated cytotoxicity. As such, suicide genes (including herpes-simplex-thymidine-kinase and inducible caspase-9) have been integrated into these cell products thus allowing their subsequent selective destruction (111, 112).
The latest of the NK cell therapeutic strategies was developed by Vallera and colleagues with the bi- or tri-specific killer cell engagers, BiKEs and TriKEs that are small molecules containing two or three single chain variable fragments from antibodies of different specificities (113). These are generated to bind CD16 on NK cells and one or two tumor antigens such as CD19 and CD20 (B-cell non-Hodgkin’s lymphoma) (114), CD33 or CD33 and CD123 (AML) (115), CD30 (Hodgkin’s lymphoma) (116), EGFR or EpCAM (EGFR/EpCAM overexpressing carcinomas) (117, 118), and many others. The initial preclinical evaluation in humanized mice proved very promising translational potential with results exceeding those of monoclonal antibodies, like in the case of CD16-CD19-CD20 TriKE versus rituximab, and also proved efficient for overpassing HLA-mediated inhibition in refractory AML blasts.
IL-15 is the master cytokine necessary for NK cell differentiation and survival and it is currently used in clinical trials alone or as an adjuvant for certain types of metastatic solid tumors to promote in vivo cell expansion and NK cell function (63, 119). Taking advantage of this, novel TriKE structures have been developed that use a human IL-15 as a modified cross-linker between the anti-CD16 and the antitumor antigen in order to promote in vivo NK cell proliferation. Assessment of the activity of a CD33 specific TriKE in an AML NSG model of NK cell adoptive transfer has shown in vivo persistence, high cytotoxic activity, and no toxicity to the construct (120). Clinical development is currently under progress and will probably obtain FDA approval in the upcoming months to be tested in patients.
Modeling Virally Induced Human Tumors Using HIS Mice
While NK cell first identification was based on its antitumor activities, it is also a critical innate effector against pathogen invasions particularly viral infections. Human NK cells have been proven essential for the immune response against members of the herpesvirus, poxvirus, and papillomavirus families, as demonstrated by the predisposition of NK deficient individuals to suffer from these virus infections (121, 122). Remarkably, in one fifth of human cancers viral infection and oncogenesis are intimately linked. Viruses act on carcinogenesis either by directly promoting the initiation of the disease or by interacting in the immune response and/or immune evasion (123). Particularly, Epstein–Barr virus (EBV), hepatitis B virus (HBV), hepatitis C virus (HCV), human papillomavirus, human T-cell lymphotropic virus, Kaposi’s sarcoma herpesvirus, and Merkel cell polyomavirus account for the majority of tumor cases linked to viral infection. Humanized mice offer a platform to access the molecular mechanisms behind that causal role and the receptor–ligand interactions occurring at the interface “NK cell-infected cell” that could eventually have therapeutic value. The engrafted human cells occupy relevant physiological sites, where they proliferate and function, and eventually interact with oncogenic viruses that spread and replicate to other cells or organs thus recapitulating the physiological human infection. Additionally, their unique susceptibility to infection by virus with exclusive human tropism and the possibility to manipulate the timing and dose of the infection render them indispensable for better understanding the virus–tumor interplay and disease progression as well as for developing therapeutic approaches.
Epstein–Barr virus is the most common human tumor virus worldwide (more than 200,000 associated malignancies every year) and is also the cause of infectious mononucleosis. It has been extensively studied in humanized mice modeling the different protein expression patterns of the virus that lead to latent infection 0, I, II, and III as well as low level lytic replication, although only latency III has been unequivocally demonstrated (124–129). Several studies have reported specific adaptive cellular and humoral immune response to EBV in humanized mice (128, 130, 131). Furthermore, transformation of B-cell in vivo has been also reported and this model has disclosed one of the viral genes (EBNA3B) responsible for tumor formation (125). Preclinical studies in HIS mice have been pivotal for the development of the therapeutic vaccines that are now undergoing clinical trials, including the EBV gp350 neutralizing antibody and infusion of EBV-specific T cells (132). The involvement of NK cells in EBV infection and disease progression was demonstrated by depletion of NK cells from EBV-infected NSG mice resulting in higher EBV DNA load in the spleen, exaggerated CD8+ T-cell responses to the virus and an increased risk of EBV-induced lymphoproliferation (65). Current investigations try to deepen our understanding of the NK cell-mediated control of primary EBV infection in HIS mice and will likely provide insight on the NK cell subset responsible for that viral control (133).
About 80% of hepatocellular carcinomas (HCC) are due to HBV or HCV infections. There are more than 250,000 new cases of HCC and an estimated half a million deaths due to this disease annually (123, 134). We and others have developed mouse models harboring both the immune system and human hepatocytes, allowing the natural course of acute infection and also chronic hepatitis, characterized by advanced liver disease and hepatocellular carcinoma genesis (17, 135, 136). In addition to the immunodeficiency, these mice have liver defects that allow engraftment and expansion of transplanted human hepatocytes. Several immune system–liver humanized models have been developed, including BRGS-uPA (BALB/c Rag2−/−Il2rg−/−SirpaNODuPAtg/tg) (17), uPA-NOG (uPA-NOD PrkdcSCIDIl2rg−/−) (135), and FRGN (Fah−/−Rag2−/−Il2rg−/− NOD) (136). These doubly humanized mice show high level of human liver chimerism and immune engraftment in primary and secondary lymphoid organs with reconstitution of myeloid and lymphoid populations at levels similar to the single HIS models. In BRGS-uPA mice, NK cells are present in spleen and liver in numbers even higher than BRGS mice and display the same NK receptor expression profile (unpublished data). Infection with HBV and HCV has been achieved in these mice and human immune responses have been detected as well as associated liver diseases that resemble the human pathology. Furthermore, both mimic the clinical response upon treatment with anti-HBs neutralizing antibodies and IFNα-2an, respectively, and prevented the leukocyte infiltration and liver fibrosis (137, 138). Any in depth analysis of the NK cell response against the virus or the role in tumorigenesis has been so far performed in these mice, other than the detection of CD56+ cells, to our knowledge.
Improving the NK Cell Compartment in HIS Mice
Given the central role of NK cells in immune responses in infection, malignancy and inflammation and the great therapeutic potential they hold, it is necessary to optimize the available models for understanding their biology and preclinically evaluate new therapies.
In previous sections, we discussed about two types of HIS mice for the study of NK cell biology, those in which the human immune cells develop in vivo from injected hematopoietic precursors and a second category that adoptively receive mature NK cells freshly isolated or derived from an ex vivo expansion or activation process, a cell line or an ES or iPS cell. The later have fewer requirements in terms of niche, cell–cell interactions, and soluble growth factors that are needed for NK cell development, and instead require cytokines for their survival and homeostatic proliferation. Common cytokine receptor γc cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21) play critical roles. In particular, IL-15 is responsible for NK cell maintenance and homeostatic proliferation through IL-15Rα presentation (139, 140), while IL-2 effect in vivo is oriented to the activation and induction of cytotoxicity through the regulation of the peripheral NK subsets. These humanized mice serve as platforms to understand the mechanisms underlying NK survival and function and provide preclinical information for the design of new therapeutics. Furthermore, they give valuable information about the cell migration capacity and synergistic effects with other cell types or immunomodulators.
As mentioned earlier, several immunodeficient hosts (NSG, NOG, BRG, BRGS) support multilineage development of human immune cells, including low levels of NK cells. In the BRGS model (16, 141), NK cells expressed CD56 and NKp46 as well as some level of CD16 and were able to degranulate moderately after stimulation with a cancer cell line. However, in both BRGS and NSG mice, NK cell displayed defects in maturation, functionality, and heterogeneity in comparison with the human counterparts due to a deficient cytokine signaling (142). The absence of human appropriate MHC class I expression on hematopoietic or stromal cells may result in the failure to “educate” or “license” developing NK cells in HIS mice. This could explain the abundance of immature NK cells (CD56brightCD16−KIR−) and their functional defects. In line with this idea, recent publications showed improved NK cell licensing in a HIS models expressing diverse educating HLA alleles (143, 144). This approach may allow better definition of the mechanisms underlying human NK cell education in vivo.
Based on their cytokine requirements, IL-15 has been exogenously administered either alone or as a complex with IL-15Rα resulting in an extensive NK cell proliferation and accumulation of CD16+KIR+ NK cells. Also, NK cell differentiation progressed from CD56+ to CD56lowCD16+, and finally to CD56lowCD16+KIR+ mimicking the human model (63). On the other hand, the constitutive high expression of the high-affinity heterotrimeric IL-2 receptor complex in CD56brightCD16± NK-cell subset and the effect of IL-2 in NK expansion and activation prompted the development of and IL-2 transgenic NOG mouse strain (145). When IL-2TgNOG mice were engrafted with human HSC, CD56+ massively developed with a highly active phenotype including IFN-γ production and cytotoxicity against tumor cells. Interestingly, treatment of these mice with a therapeutic humanized anti-CCR4 Ab (mogamulizumab) suppressed the growth of a CCR4+ lymphoma, suggesting that the human NK cells in the mice exerted active Ab-dependent cellular cytotoxicity in vivo. These cells expressed various NK receptors, including NKp30, NKp44, NKp46, NKG2D, and CD94, as well as a diverse set of killer cell Ig-like receptor molecules at levels comparable to normal human NK cells from the peripheral blood (62). Nevertheless, there are several limitations in this model due to the supra-physiological levels of IL-2 and, therefore, the high activation status of the NK cells.
It is well known that NK homeostasis and function are regulated by the interaction with other immune cells, particularly macrophages, DCs, and T cells. In addition, soluble factors released by those cells, like NKG2D ligands, IL-2, IL-12, or IL-15, signal on NK cells leading to proliferation and activation. Based on these crosstalk events, others and we have developed humanized mice that through the enhancement of the myeloid compartment, NK cell development results improved. As it happens for other lineages, human myelopoiesis is driven by soluble factors normally present in the BM niche and periphery, which are from murine origin in HIS mice. Some of these mouse cytokines cross-react to some extent with the human cells but others, the species-specific cytokines, do not. In order to circumvent this deficiency, human cytokines have been administered to HIS mice either as recombinant proteins (63) by cytokine-encoding plasmids (146) or by insertion of the cytokines either as transgenes in the mouse genome or by knocking in the human gene to replace the mouse counterpart (147, 148). As mentioned before, transgenic models provoke supra-physiological levels of the cytokine in the periphery and in the case of pro-myeloid factors, such as TPO, IL-3, GM-CSF, or M-CSF, also lead to the exacerbated mobilization and HSC exhaustion limiting the utility of the system. Swapping mouse coding exons for M-CSF, IL-3/GM-CSF, TPO, and SIRPα with their human counterparts allowed for the creation of the MISTRG strain (149). This host expresses these human cytokines under control of mouse regulatory elements and show superior human myeloid cell engraftment. Subsequently, MISTRG HIS mice showed an increased number of functional NK cells, including higher expression of KIR, CD94, and CD161 receptors (149). Nevertheless, cellular and humoral immune responses in MISTRG HIS mice are poor and these mice develop severe anemia.
The transpresentation of IL-15 occurs mainly through the IL-15Rα expressed by DCs, so efforts have been made to increase specifically this cell population in order to increase the NK pool avoiding the overdevelopment of other myeloid subsets. In our laboratory, Flt3-deficient BRG mice (BRGF) were created and after reconstitution with human HSC, human Flt3L was administered to the mice. The result was a specific increase of all the DC subsets and the promotion of NK cell hematopoiesis, with enhanced CD94, CD16, and KIR receptor expression. The combination of this system with the expression of the SirpaNOD protein in the BRGSF model has led to further augmentation of NK cell numbers and also an enhanced functional competency as demonstrated by their degranulation capacity and cytokine production activity (unpublished data). This HIS model provides a unique platform to study NK cell development, crosstalk mechanisms with other immune cells, and the preclinical assessment of new immunotherapies targeting innate cells.
The combination of the protocols detailed in the previous sections for modeling human cancer or infection with the abovementioned strategies to boost the NK cells in HIS mice will raise the potential to understand how NK cell interact with malignant or infected cells. Moreover, HSC-HIS mice offer the possibility to study the tissue specific interactions, the reservoirs, the migration patterns, and the crosstalks within the immune compartment that may be important to develop combinatorial therapies that avoid metastasis, tumor relapse, and “relocation” of the viruses.
Concluding Remarks and Future Perspectives
Therapies designed to induce or potentiate the immune response against tumors are an appealing strategy to control tumor growth and have been the object of intense research since their discovery in the 1970s. Despite representing the most promising cancer treatment since the emergence of chemotherapy, several cases of side effects or disappointing clinical results have downshifted the development of new immunotherapies. The better understanding of the tumor heterogeneity, the mechanisms of the immune response, and the interaction with the tumor microenvironment is a required step for the development of safe and effective therapies. Humanized mice have the potential to reproduce the HIS, the tumor growth and immune evasion, and the response to treatments targeting immune effector cells or immunomodulators. One of the most challenging aspects of tumor research has been to understand the variability within he same type of cancer among individuals and, therefore, the disparate responses and outcomes after therapy. Current efforts are being made to overcome these limitations by creating truly personalized HIS-PDX mouse models in which both the immune system and the tumor are derived form the same individual. These models will provide an invaluable bridge between immunotherapy discovery and the clinic, increasing the success rate of new therapies in human trials and improving the chances to beat cancer.
Natural killer cells have been for long time considered the only innate effectors of the lymphoid system but nowadays we appreciate that they belong to a larger family, the “innate lymphoid cells” (ILCs) (150). These recently described populations lack cytotoxic capacity but instead, they exert very potent cytokine production. In recent years, there has been a rapid advance in our understanding of their development, phenotypic, and functional diversity, which has been nicely reviewed elsewhere (151–155). ILCs come in three groups mirroring the cytokine and transcriptional profile of CD4+ helper T cells (Th1, Th2, and Th17/22). Given the myriad of cytokines they produce, ILCs have been involved in the early orchestration of immune responses against a number of pathogens, in tumor immunosurveillance and in inflammatory diseases. Recent works in mice have proven the antitumor effects of ILC1s whereas ILC3s have been found to exert both beneficial and tumor-promoting effects depending on the circumstances (156–158). The multifaceted functions of ILCs suggest new alternatives for immunotherapeutic approaches against tumors that need to be explored in in vivo humanized models. The improvement of human helper ILCs in HIS mice could open new avenues for harnessing innate immunity to treat cancer and inflammatory diseases.
Author Contributions
Both authors have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
JD is a stakeholder in AXENIS (founder, member of the executive board). The remaining author declares no conflict of interest.
Funding
This work received funding from the Institut Pasteur and INSERM. SL-L received support through the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7-PEOPLE-2012-ITN (NATURIMMUN project) under REA grant agreement no. 317013.
References
1. Bernardi R, Grisendi S, Pandolfi PP. Modelling haematopoietic malignancies in the mouse and therapeutical implications. Oncogene (2002) 21:3445–58. doi:10.1038/sj.onc.1205313
2. Springer MS, Murphy WJ. Mammalian evolution and biomedicine: new views from phylogeny. Biol Rev Camb Philos Soc (2007) 82:375–92. doi:10.1111/j.1469-185X.2007.00016.x
3. Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature (2014) 515:355–64. doi:10.1038/nature13992
4. Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res (2014) 6(2):114–8.
5. Rygaard J, Povsen CO. Heterotransplantation of a human malignant tumour to “nude” mice. APMIS (2007) 115:604–6. doi:10.1111/j.1600-0463.2007.apm_689a.x
6. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature (1988) 335:256–9. doi:10.1038/335256a0
7. Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science (1992) 255:1137–41. doi:10.1126/science.1372131
8. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell (1992) 68:869–77. doi:10.1016/0092-8674(92)90030-G
9. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell (1992) 68:855–67. doi:10.1016/0092-8674(92)90029-C
10. Greiner DL, Shultz LD, Yates J, Appel MC, Perdrizet G, Hesselton RM, et al. Improved engraftment of human spleen cells in NOD/LtSz-scid/scid mice as compared with C.B-17-scid/scid mice. Am J Pathol (1995) 146:888–902.
11. Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J Infect Dis (1995) 172:974–82. doi:10.1093/infdis/172.4.974
12. Pflumio F, Izac B, Katz A, Shultz LD, Vainchenker W, Coulombel L. Phenotype and function of human hematopoietic cells engrafting immune-deficient CB17-severe combined immunodeficiency mice and nonobese diabetic-severe combined immunodeficiency mice after transplantation of human cord blood mononuclear cells. Blood (1996) 88:3731–40.
13. Takenaka K, Prasolava TK, Wang JCY, Mortin-Toth SM, Khalouei S, Gan OI, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol (2007) 8:1313–23. doi:10.1038/ni1527
14. Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, et al. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood (2013) 121(8):1316–25. doi:10.1182/blood-2012-06-440354
15. Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Srick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRP α) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Natl Acad Sci U S A (2011) 108:13224–9. doi:10.1073/pnas.1101398108
16. Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C. Transgenic expression of human signal regulatory protein of human hematopoietic cells in humanized mice. Proc Natl Acad Sci U S A (2011) 108(32):13218–23. doi:10.1073/pnas.1109769108
17. Strick-Marchand H, Dusséaux M, Darche S, Huntington ND, Legrand N, Masse-Ranson G, et al. A novel mouse model for stable engraftment of a human immune system and human hepatocytes. PLoS One (2015) 10(3):e0119820. doi:10.1371/journal.pone.0119820
18. Di Santo JP, Vosshenrich CA. Bone marrow versus thymic pathways of natural killer cell development. Immunol Rev (2006) 214:35–46. doi:10.1111/j.1600-065X.2006.00461.x
19. Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity (1995) 2:223–38. doi:10.1016/1074-7613(95)90047-0
20. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood (2002) 100:3175–82. doi:10.1182/blood-2001-12-0207
21. Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti J-C, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science (2004) 304:104–7. doi:10.1126/science.1093933
22. González-Clemente JM, Mauricio D, Richart C, Broch M, Caixàs A, Megia A, et al. Diabetic neuropathy is associated with activation of the TNF-alpha system in subjects with type 1 diabetes mellitus. Clin Endocrinol (Oxf) (2005) 63:525–9. doi:10.1111/j.1365-2265.2005.02376.x
23. Gimeno R, Weijer K, Voordouw A, Uittenbogaart CH, Legrand N, Alves NL, et al. Monitoring the effect of gene silencing by RNA interference in human CD34+ cells injected into newborn RAG2-/- γc-/- mice: functional inactivation of p53 in developing T cells. Blood (2004) 104:3886–93. doi:10.1182/blood-2004-02-0656
24. Li Y, Mention J-J, Court N, Masse-Ranson G, Toubert A, Spits H, et al. A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur J Immunol (2016) 46:1291–9. doi:10.1002/eji.201546132
25. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell (2007) 131:1109–23. doi:10.1016/j.cell.2007.10.054
26. Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell (2016) 29:574–86. doi:10.1016/j.ccell.2016.03.008
27. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med (2015) 21:1318–25. doi:10.1038/nm.3954
28. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med (2011) 17:1086–93. doi:10.1038/nm.2415
29. Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia (2006) 20:2147–54. doi:10.1038/sj.leu.2404401
30. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med (1997) 3:730–7. doi:10.1038/nm0797-730
31. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367:645–8. doi:10.1038/367645a0
32. Lohberger B, Rinner B, Stuendl N, Absenger M, Liegl-Atzwanger B, Walzer SM, et al. Aldehyde dehydrogenase 1, a potential marker for cancer stem cells in human sarcoma. PLoS One (2012) 7:e43664. doi:10.1371/journal.pone.0043664
33. Rasheed Z, Wang Q, Matsui W. Isolation of stem cells from human pancreatic cancer xenografts. J Vis Exp (2010) 43:2169. doi:10.3791/2169
34. Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood (2011) 117:3076–86. doi:10.1182/blood-2010-08-301507
35. Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood (2015) 125(17):2630–40. doi:10.1182/blood-2014-11-570218
36. Pallasch CP, Leskov I, Braun CJ, Vorholt D, Drake A, Soto-Feliciano YM, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell (2014) 156:590–602. doi:10.1016/j.cell.2013.12.041
37. Veluchamy JP, Lopez-Lastra S, Spanholtz J, Bohme F, Kok N, Heideman DA, et al. In vivo efficacy of umbilical cord blood stem cell derived NK cells in the treatment of metastatic colorectal cancer. Front Immunol (2017) 8:87. doi:10.3389/fimmu.2017.00087
38. Seitz G, Pfeiffer M, Fuchs J, Warmann SW, Leuschner I, Vokuhl C, et al. Establishment of a rhabdomyosarcoma xenograft model in human-adapted mice. Oncol Rep (2010) 24:1067–72. doi:10.3892/or.2010.1067
39. Barabé F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science (2007) 316(5824):600–4. doi:10.1126/science.1139851
40. Suemizu H, Monnai M, Ohnishi Y, Ito M, Tamaoki N, Nakamura M. Identification of a key molecular regulator of liver metastasis in human pancreatic carcinoma using a novel quantitative model of metastasis in NOD/SCID/gammacnull (NOG) mice. Int J Oncol (2007) 31(4):741–51. doi:10.3892/ijo.31.4.741
41. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest (2012) 122:2221–33. doi:10.1172/JCI59535
42. Xia J, Hu Z, Yoshihara S, Li Y, Jin C-H, Tan S, et al. Modeling human leukemia immunotherapy in humanized mice. EBioMedicine (2016) 10:101–8. doi:10.1016/j.ebiom.2016.06.028
43. Hu Z, Xia J, Fan W, Wargo J, Yang Y-G, Hu Z, et al. Human melanoma immunotherapy using tumor antigen-specific T cells generated in humanized mice. Oncotarget (2016) 7:6448–59. doi:10.18632/oncotarget.7044
44. Kaur K, Cook J, Park S-H, Topchyan P, Kozlowska A, Ohanian N, et al. Novel strategy to expand super-charged NK cells with significant potential to lyse and differentiate cancer stem cells; differences in NK expansion and function between healthy and cancer patients. Front Oncol (2017) 7:9. doi:10.3389/fimmu.2017.00297
45. Aryee K-E, Brehm MA, Shultz LD, Jurczyk A. Modeling immune system-tumor interactions using humanized mice. J Immunol (2016) 196:212.12.
46. Ito A, Ishida T, Utsunomiya A, Sato F, Mori F, Yano H, et al. Defucosylated anti-CCR4 monoclonal antibody exerts potent ADCC against primary ATLL cells mediated by autologous human immune cells in NOD/Shi-scid, IL-2Rγnull mice in vivo. J Immunol (2009) 183(7):4782–91. doi:10.4049/jimmunol.0900699
47. Xiang Z, Liu Y, Zheng J, Liu M, Lv A, Gao Y, et al. Targeted activation of human Vγ9Vd2-T cells controls Epstein–Barr virus-induced B cell lymphoproliferative disease. Cancer Cell (2014) 26:565–76. doi:10.1016/j.ccr.2014.07.026
48. Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther (2014) 25:1003–12. doi:10.1089/hum.2013.209
49. Simpson-Abelson MR, Sonnenberg GF, Takita H, Yokota SJ, Conway TF, Kelleher RJ, et al. Long-term engraftment and expansion of tumor-derived memory T cells following the implantation of non-disrupted pieces of human lung tumor into NOD-scid IL2Rgamma(null) mice. J Immunol (2008) 180:7009–18. doi:10.4049/jimmunol.180.10.7009
50. Geller MA, Knorr DA, Hermanson DA, Pribyl L, Bendzick L, McCullar V, et al. Intraperitoneal delivery of human natural killer cells for treatment of ovarian cancer in a mouse xenograft model. Cytotherapy (2013) 15:1297–306. doi:10.1016/j.jcyt.2013.05.022
51. Lee SJ, Kang WY, Yoon Y, Jin JY, Song HJ, Her JH, et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer (2015) 15:1011. doi:10.1186/s12885-015-2034-y
52. Feuerer M, Beckhove P, Bai L, Solomayer E-F, Bastert G, Diel IJ, et al. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med (2001) 7:452–8. doi:10.1038/86523
53. Roth MD, Harui A. Human tumor infiltrating lymphocytes cooperatively regulate prostate tumor growth in a humanized mouse model. J Immunother cancer (2015) 3:12. doi:10.1186/s40425-015-0056-2
54. Koehne G, Doubrovin M, Doubrovina E, Zanzonico P, Gallardo HF, Ivanova A, et al. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nat Biotechnol (2003) 21:405–13. doi:10.1038/nbt805
55. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126:3130–44. doi:10.1172/JCI83092
56. Gründer C, van Dorp S, Hol S, Drent E, Straetemans T, Heijhuurs S, et al. γ9 and d2CDR3 domains regulate functional avidity of T cells harboring γ9d2TCRs. Blood (2012) 120(26):5153–62. doi:10.1182/blood-2012-05-432427
57. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132:515–25; quiz 526. doi:10.1016/j.jaci.2013.07.020
58. Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci U S A (2000) 97:2731–6. doi:10.1073/pnas.050588297
59. Colucci F, Caligiuri MA, Di Santo JP. What does it take to make a natural killer? Nat Rev Immunol (2003) 3:413–25. doi:10.1038/nri1088
60. Briercheck EL, Freud AG, Caligiuri MA. Natural Killer Cells: Basic Science and Clinical Application. Amsterdam, Boston, London: Elsevier/Academic Press (2010). p. 113–22.
61. Huntington ND, Alves NL, Legrand N, Lim A, Strick-Marchand H, Mention J-J, et al. IL-15 transpresentation promotes both human T-cell reconstitution and T-cell-dependent antibody responses in vivo. Proc Natl Acad Sci U S A (2011) 108:6217–22. doi:10.1073/pnas.1019167108
62. Katano I, Takahashi T, Ito R, Kamisako T, Mizusawa T, Ka Y, et al. Predominant development of mature and functional human NK cells in a novel human IL-2-producing transgenic NOG mouse. J Immunol (2015) 194:3513–25. doi:10.4049/jimmunol.1401323
63. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med (2009) 206:25–34. doi:10.1084/jem.20082013
64. Jost S, Chang J, Rands K, Tomezsko P, Doyle E, Daillere R, et al. Impact of NK cells on control of HIV infection in humanized BLT mice (P6207). J Immunol (2013) 190:118.27–118.27.
65. Chijioke O, Müller A, Feederle R, Barros MHM, Krieg C, Emmel V, et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein–Barr virus infection. Cell Rep (2013) 5:1489–98. doi:10.1016/j.celrep.2013.11.041
66. Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human tumour immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS One (2011) 6:e22842. doi:10.1371/journal.pone.0022842
67. Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol (2013) 94:25–39. doi:10.1189/jlb.1212621
68. Ma S-D, Xu X, Jones R, Delecluse H-J, Zumwalde NA, Sharma A, et al. PD-1/CTLA-4 blockade inhibits Epstein–Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog (2016) 12:e1005642. doi:10.1371/journal.ppat.1005642
69. Vudattu NK, Waldron-Lynch F, Truman LA, Deng S, Preston-Hurlburt P, Torres R, et al. Humanized mice as a model for aberrant responses in human T cell immunotherapy. J Immunol (2014) 193:587–96. doi:10.4049/jimmunol.1302455
70. Singh M, Singh P, Vaira D, Amand M, Rahmouni S, Moutschen M. Minocycline attenuates HIV-1 infection and suppresses chronic immune activation in humanized NOD/LtsZ-scidIL-2Rγ(null) mice. Immunology (2014) 142:562–72. doi:10.1111/imm.12246
71. Stojanovic A, Fiegler N, Brunner-Weinzierl M, Cerwenka A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK cell IFN-γ production in response to mature dendritic cells. J Immunol (2014) 192:4184–91. doi:10.4049/jimmunol.1302091
72. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol (2016) 34:539–73. doi:10.1146/annurev-immunol-032414-112049
73. Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One (2015) 10:e0134715. doi:10.1371/journal.pone.0134715
74. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313:1485–92. doi:10.1056/NEJM198512053132327
75. Baiocchi RA, Caligiuri MA. Low-dose interleukin 2 prevents the development of Epstein–Barr virus (EBV)-associated lymphoproliferative disease in scid/scid mice reconstituted i.p. with EBV-seropositive human peripheral blood lymphocytes. Proc Natl Acad Sci U S A (1994) 91:5577–81. doi:10.1073/pnas.91.12.5577
76. Boiardi A, Silvani A, Ruffini PA, Rivoltini L, Parmiani G, Broggi G, et al. Loco-regional immunotherapy with recombinant interleukin-2 and adherent lymphokine-activated killer cells (A-LAK) in recurrent glioblastoma patients. Cancer Immunol Immunother (1994) 39:193–7. doi:10.1007/BF01533386
77. Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Moore EJ, Pierz DM, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer (1995) 76:840–52. doi:10.1002/1097-0142(19950901)76:5<840::AID-CNCR2820760519>3.0.CO;2-R
78. Keilholz U, Scheibenbogen C, Brado M, Georgi P, Maclachlan D, Brado B, et al. Regional adoptive immunotherapy with interleukin-2 and lymphokine-activated killer (LAK) cells for liver metastases. Eur J Cancer (1994) 30A:103–5. doi:10.1016/S0959-8049(05)80028-0
79. Sutlu T, Alici E. Natural killer cell-based immunotherapy in cancer: current insights and future prospects. J Intern Med (2009) 266:154–81. doi:10.1111/j.1365-2796.2009.02121.x
80. Ruggeri L, Mancusi A, Burchielli E, Aversa F, Martelli MF, Velardi A. Natural killer cell alloreactivity in allogeneic hematopoietic transplantation. Curr Opin Oncol (2007) 19:142–7. doi:10.1097/CCO.0b013e3280148a1a
81. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi:10.1182/blood-2004-07-2974
82. Curti A, Ruggeri L, Parisi S, Bontadini A, Dan E, Motta MR, et al. Larger size of donor alloreactive NK cell repertoire correlates with better response to NK cell immunotherapy in elderly acute myeloid leukemia patients. Clin Cancer Res (2016) 22:1914–21. doi:10.1158/1078-0432.CCR-15-1604
83. Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood (2015) 125:784–92. doi:10.1182/blood-2014-07-592881
84. Gras Navarro A, Kmiecik J, Leiss L, Zelkowski M, Engelsen A, Bruserud Ø, et al. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J Immunol (2014) 193:6192–206. doi:10.4049/jimmunol.1400859
85. Ruggeri L, Urbani E, Andre P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica (2016) 101:626–33. doi:10.3324/haematol.2015.135301
86. Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol (2008) 180:6392–401. doi:10.4049/jimmunol.180.9.6392
87. Nijhof IS, van Bueren JJL, van Kessel B, Andre P, Morel Y, Lokhorst HM, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica (2015) 100(2):263–8. doi:10.3324/haematol.2014.117531
88. Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol (2015) 6:21. doi:10.3389/fphar.2015.00021
89. Du J, Lopez-Verges S, Pitcher BN, Johnson J, Jung S-H, Zhou L, et al. CALGB 150905 (alliance): rituximab broadens the antilymphoma response by activating unlicensed NK cells. Cancer Immunol Res (2014) 2:878–89. doi:10.1158/2326-6066.CIR-13-0158
90. Daldrup-Link HE, Meier R, Rudelius M, Piontek G, Piert M, Metz S, et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur Radiol (2005) 15:4–13. doi:10.1007/s00330-004-2526-7
91. Monteverde M, Milano G, Strola G, Maffi M, Lattanzio L, Vivenza D, et al. The relevance of ADCC for EGFR targeting: a review of the literature and a clinically-applicable method of assessment in patients. Crit Rev Oncol Hematol (2015) 95:179–90. doi:10.1016/j.critrevonc.2015.02.014
92. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer (2012) 12:767–75. doi:10.1038/nrc3368
93. Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, et al. NK cells preferentially target tumor cells with a cancer stem cell phenotype. J Immunol (2015) 195:4010–9. doi:10.4049/jimmunol.1500447
94. Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R, et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol (2013) 190:2381–90. doi:10.4049/jimmunol.1201542
95. Dionne LK, Driver ER, Wang XJ. Head and neck cancer stem cells: from identification to tumor immune network. J Dent Res (2015) 94:1524–31. doi:10.1177/0022034515599766
96. Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, de Vries J, et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rgnull mice. PLoS One (2013) 8(6):e64384. doi:10.1371/journal.pone.0064384
97. Dolstra H, Roeven MWH, Spanholtz J, Hangalapura B, Tordoir M, Maas F, et al. A phase I study of allogeneic natural Killer cell therapy generated from cord blood hematopoietic stem and progenitor cells in elderly acute myeloid leukemia patients. Blood (2015) 126:1357.
98. Veluchamy JP, Heeren AM, Spanholtz J, van Eendenburg JDH, Heideman DAM, Kenter GG, et al. High-efficiency lysis of cervical cancer by allogeneic NK cells derived from umbilical cord progenitors is independent of HLA status. Cancer Immunol Immunother (2016) 66(1):51–61. doi:10.1007/s00262-016-1919-1
99. Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol (2005) 175:5095–103. doi:10.4049/jimmunol.175.8.5095
100. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood (2009) 113(24):6094–101. doi:10.1182/blood-2008-06-165225
101. Gong J, Maki G, Klingemann H. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia (1994) 8:652–8.
102. Tam YK, Miyagawa B, Ho VC, Klingemann H-G. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother (1999) 8:281–90. doi:10.1089/106161299320316
103. Yan Y, Steinherz P, Klingemann HG, Dennig D, Childs BH, McGuirk J, et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res (1998) 4:2859–68.
104. Swift BE, Williams BA, Kosaka Y, Wang X-H, Medin JA, Viswanathan S, et al. Natural killer cell lines preferentially kill clonogenic multiple myeloma cells and decrease myeloma engraftment in a bioluminescent xenograft mouse model. Haematologica (2012) 97:1020–8. doi:10.3324/haematol.2011.054254
105. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10:625–32. doi:10.1080/14653240802301872
106. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15:1563–70. doi:10.1016/j.jcyt.2013.06.017
107. Boissel L, Campbell K, Toneguzzo F, Nichols K, Klingemann H. NK-92: an “off the shelf” target-specific cytotoxic cell therapeutic. Cytotherapy (2015) 17(6):S19. doi:10.1016/j.jcyt.2015.03.367
108. Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology (2014) 3:e28147. doi:10.4161/onci.28147
109. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep (2015) 5:11483. doi:10.1038/srep11483
110. Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology (2013) 2:e26527. doi:10.4161/onci.26527
111. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol (2014) 5:254. doi:10.3389/fphar.2014.00254
112. Zhou X, Di Stasi A, Brenner MK. iCaspase 9 suicide gene system. Methods Mol Biol (2015) 1317:87–105. doi:10.1007/978-1-4939-2727-2_6
113. Miller JS. Therapeutic applications: natural killer cells in the clinic. Hematol Am Soc Hematol Educ Program (2013) 2013:247–53. doi:10.1182/asheducation-2013.1.247
114. Kipriyanov SM, Cochlovius B, Schäfer HJ, Moldenhauer G, Bähre A, Le Gall F, et al. Synergistic antitumor effect of bispecific CD19 x CD3 and CD19 x CD16 diabodies in a preclinical model of non-Hodgkin’s lymphoma. J Immunol (2002) 169:137–44. doi:10.4049/jimmunol.169.1.137
115. Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia (2014) 28:1–10. doi:10.1038/leu.2014.62
116. Hartmann F, Renner C, Jung W, Deisting C, Juwana M, Eichentopf B, et al. Treatment of refractory Hodgkin’s disease with an anti-CD16/CD30 bispecific antibody. Blood (1997) 89:2042–7.
117. Elsässer D, Stadick H, Stark S, Van de Winkel JG, Gramatzki M, Schrott KM, et al. Preclinical studies combining bispecific antibodies with cytokine-stimulated effector cells for immunotherapy of renal cell carcinoma. Anticancer Res (1999) 19:1525–8.
118. Vallera DA, Zhang B, Gleason MK, Oh S, Weiner LM, Kaufman DS, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharm (2013) 28:274–82. doi:10.1089/cbr.2012.1329
119. Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, et al. Interleukin (IL)-15 is a novel cytokine that activates human natural-killer cells via component of the IL-2 receptor. J Exp Med (1994) 180:1395–403. doi:10.1084/jem.180.4.1395
120. Miller JS, Felice M, McElmurry R, McCullar V, Zhou X, Tolar J, et al. Trispecific killer engagers (TriKEs) that contain IL-15 to make NK cells antigen specific and to sustain their persistence and expansion. Blood (2015) 126:232.
121. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med (1989) 320:1731–5. doi:10.1056/NEJM198906293202605
122. Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect (2002) 4:1545–58. doi:10.1016/S1286-4579(02)00038-2
123. Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist (2010) 15(Suppl 4):5–13. doi:10.1634/theoncologist.2010-S4-05
124. Islas-Ohlmayer M, Padgett-Thomas A, Domiati-Saad R, Melkus MW, Cravens PD, Martin Mdel P, et al. Experimental infection of NOD/SCID mice reconstituted with human CD34+ cells with Epstein–Barr virus. J Virol (2004) 78:13891–900. doi:10.1128/JVI.78.24.13891-13900.2004
125. White RE, Rämer PC, Naresh KN, Meixlsperger S, Pinaud L, Rooney C, et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J Clin Invest (2012) 122(4):1487–502. doi:10.1172/JCI58092
126. Ma S-D, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A new model of Epstein–Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol (2011) 85:165–77. doi:10.1128/JVI.01512-10
127. Ma S-D, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, et al. An Epstein–Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol (2012) 86:7976–87. doi:10.1128/JVI.00770-12
128. Strowig T, Gurer C, Ploss A, Liu Y-F, Arrey F, Sashihara J, et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med (2009) 206:1423–34. doi:10.1084/jem.20081720
129. Kutok JL, Wang F. Spectrum of Epstein–Barr virus-associated diseases. Annu Rev Pathol (2006) 1:375–404. doi:10.1146/annurev.pathol.1.110304.100209
130. Yajima M, Imadome K-I, Nakagawa A, Watanabe S, Terashima K, Nakamura H, et al. A new humanized mouse model of Epstein–Barr virus infection that reproduces persistent infection, lymphoproliferative disorder, and cell-mediated and humoral immune responses. J Infect Dis (2008) 198:673–82. doi:10.1086/590502
131. Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, Tomizawa M, et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r null humanized mice. Proc Natl Acad Sci U S A (2010) 107:13022–7. doi:10.1073/pnas.1000475107
132. Cohen JI. Epstein–barr virus vaccines. Clin Transl Immunol (2015) 4:e32. doi:10.1038/cti.2015.4
133. Fujiwara S, Imadome K-I, Takei M. Modeling EBV infection and pathogenesis in new-generation humanized mice. Exp Mol Med (2015) 47:e135. doi:10.1038/emm.2014.88
134. El–Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology (2007) 132:2557–76. doi:10.1053/j.gastro.2007.04.061
135. Gutti TL, Knibbe JS, Makarov E, Zhang J, Yannam GR, Gorantla S, et al. Human hepatocytes and hematolymphoid dual reconstitution in treosulfan-conditioned uPA-NOG mice. Am J Pathol (2014) 184:101–9. doi:10.1016/j.ajpath.2013.09.008
136. Wilson EM, Bial J, Tarlow B, Bial G, Jensen B, Greiner DL, et al. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res (2014) 13:404–12. doi:10.1016/j.scr.2014.08.006
137. Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog (2014) 10:e1004032. doi:10.1371/journal.ppat.1004032
138. Keng CT, Sze CW, Zheng D, Zheng Z, Yong KSM, Tan SQ, et al. Characterisation of liver pathogenesis, human immune responses and drug testing in a humanised mouse model of HCV infection. Gut (2016) 65:1744–53. doi:10.1136/gutjnl-2014-307856
139. Prlic M, Blazar BR, Farrar MA, Jameson SC. In vivo survival and homeostatic proliferation of natural killer cells. J Exp Med (2003) 197:967–76. doi:10.1084/jem.20021847
140. Marçais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:450. doi:10.3389/fimmu.2013.00450
141. Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Natl Acad Sci U S A (2011) 108:13224–9. doi:10.1073/pnas.1101398108
142. Strowig T, Chijioke O, Carrega P, Arrey F, Meixlsperger S, Rämer PC, et al. Human NK cells of mice with reconstituted human immune system components require preactivation to acquire functional competence Human NK cells of mice with reconstituted human immune system components require preactivation to acquire functional competence. Blood (2010) 116:4158–67. doi:10.1182/blood-2010-02-270678
143. Landtwing V, Raykova A, Pezzino G, Béziat V, Marcenaro E, Graf C, et al. Cognate HLA absence in trans diminishes human NK cell education. J Clin Invest (2016) 126(10):3772–82. doi:10.1172/JCI86923
144. Boudreau JE, Liu XR, Zhao Z, Zhang A, Shultz LD, Greiner DL, et al. Cell-extrinsic MHC Class I molecule engagement augments human NK cell education programmed by cell-intrinsic MHC Class I. Immunity (2016) 45:280–91. doi:10.1016/j.immuni.2016.07.005
145. Caligiuri MA. Human natural killer cells. Blood (2008) 112:461–9. doi:10.1182/blood-2007-09-077438
146. Li Y, Chen Q, Zheng D, Yin L, Chionh YH, Wong LH, et al. Induction of functional human macrophages from bone marrow promonocytes by M-CSF in humanized mice. J Immunol (2013) 191:3192–9. doi:10.4049/jimmunol.1300742
147. Willinger T, Rongvaux A, Strowig T, Manz MG, Flavell RA. Improving human hemato-lymphoid-system mice by cytokine knock-in gene replacement. Trends Immunol (2011) 32:321–7. doi:10.1016/j.it.2011.04.005
148. Ito R, Takahashi T, Katano I, Kawai K, Kamisako T, Ogura T, et al. Establishment of a human allergy model using human IL-3/GM-CSF-transgenic NOG mice. J Immunol (2013) 191:2890–9. doi:10.4049/jimmunol.1203543
149. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol (2014) 32:364–72. doi:10.1038/nbt.2858
150. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi:10.1038/nri3365
151. Tait Wojno ED, Artis D. Emerging concepts and future challenges in innate lymphoid cell biology. J Exp Med (2016) 213:2229–48. doi:10.1084/jem.20160525
152. Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol (2015) 15(7):415–28. doi:10.1038/nri3855
153. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517:293–301. doi:10.1038/nature14189
154. Eberl G, Di Santo JP, Vivier E. The brave new world of innate lymphoid cells. Nat Immunol (2015) 16:1–5. doi:10.1038/ni.3059
155. Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med (2015) 21:698–708. doi:10.1038/nm.3892
156. Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17:765–74. doi:10.1038/ni.3489
157. Cording S, Medvedovic J, Cherrier M, Eberl G. Development and regulation of RORγt+ innate lymphoid cells. FEBS Lett (2014) 588:4176–81. doi:10.1016/j.febslet.2014.03.034
Keywords: humanized mouse models, innate lymphoid cell, natural killer cells, cancer immunotherapy, natural killer cell immunotherapy
Citation: Lopez-Lastra S and Di Santo JP (2017) Modeling Natural Killer Cell Targeted Immunotherapies. Front. Immunol. 8:370. doi: 10.3389/fimmu.2017.00370
Received: 01 February 2017; Accepted: 14 March 2017;
Published: 29 March 2017
Edited by:
Ulrike Koehl, Hannover Medical School, GermanyReviewed by:
Lutz Walter, Leibniz-Institute for Primate Research, GermanyPhilippe Saas, Etablissement Français du Sang BFC, France
Copyright: © 2017 Lopez-Lastra and Di Santo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Lopez-Lastra, c2lsbG9sYXNAcGFzdGV1ci5mcg==