 Kuan-Yu I1
Kuan-Yu I1 Yi-Shu Huang1†
Yi-Shu Huang1† Ching-Hsun Hu1
Ching-Hsun Hu1 Wen-Yi Tseng2†Chia-Hsin Cheng1
Wen-Yi Tseng2†Chia-Hsin Cheng1 Martin Stacey3
Martin Stacey3 Siamon Gordon1,4Gin-Wen Chang1
Siamon Gordon1,4Gin-Wen Chang1 Hsi-Hsien Lin1,5,6*
Hsi-Hsien Lin1,5,6*
- 1Department of Microbiology and Immunology, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- 2Division of Rheumatology, Allergy and Immunology, Chang Gung Memorial Hospital-Keelung, Keelung, Taiwan
- 3Faculty of Biological Sciences, School of Molecular and Cellular Biology, University of Leeds, Leeds, UK
- 4Sir William Dunn School of Pathology, University of Oxford, Oxford, UK
- 5Department of Anatomic Pathology, Chang Gung Memorial Hospital-Linkou, Taoyuan, Taiwan
- 6Chang Gung Immunology Consortium, Chang Gung Memorial Hospital, Chang Gung University, Taoyuan, Taiwan
EMR2/ADGRE2 is a human myeloid-restricted adhesion G protein-coupled receptor critically implicated in vibratory urticaria, a rare type of allergy caused by vibration-induced mast cell activation. In addition, EMR2 is also highly expressed by monocyte/macrophages and has been linked to neutrophil migration and activation. Despite these findings, little is known of EMR2-mediated signaling and its role in myeloid biology. In this report, we show that activation of EMR2 via a receptor-specific monoclonal antibody promotes the differentiation of human THP-1 monocytic cell line and induces the expression of pro-inflammatory mediators, including IL-8, TNF-α, and MMP-9. Using specific signaling inhibitors and siRNA knockdowns, biochemical and functional analyses reveal that the EMR2-mediated signaling is initiated by Gα16, followed by the subsequent activation of Akt, extracellular signal-regulated kinase, c-Jun N-terminal kinase, and nuclear factor kappa-light-chain-enhancer of activated B cells. Our results demonstrate a functional role for EMR2 in the differentiation and inflammatory activation of human monocytic cells and provide potential targets for myeloid cell-mediated inflammatory disorders.
Introduction
Professional phagocytes such as macrophages (Mφ), neutrophils (Nφ), and dendritic cells (DCs) are critical for the recognition and elimination of invading pathogens (1, 2). The processes of phagocytosis as well as the subsequent microbial killing and immune activation/resolution by these innate immune effector cells are largely mediated via a diverse array of receptors and their signaling reactions (1, 3). In this regard, one receptor of interest is EMR2/ADGRE2, a human myeloid-restricted adhesion G protein-coupled receptor (aGPCR) highly homologous to F4/80, the widely acclaimed surface marker that defines murine tissue Mφ (4–6).
As a human ortholog of F4/80, EMR2 similarly contains multiple epidermal growth factor-like modules in its extracellular domain (ECD), which binds to its endogenous ligand dermatan sulfate (4, 7, 8). Initially identified as a myeloid-restricted transcript expressed in monocytes (Mos)/Mφ, Nφ, and myeloid DC (4), EMR2 protein expression was later shown to be upregulated during the in vitro differentiation of Mφ but downregulated following DC maturation (9). On the other hand, the strongest in vivo EMR2 protein signal was detected in CD16+ blood Mos and BDCA-3+ myeloid DC (10). Foamy Mφ in atherosclerotic vessels and splenic Gaucher cells are highly EMR2-positive, whereas multiple sclerosis brain foam cells express little if any EMR2 (11). The differential expression patterns of EMR2 in distinct myeloid populations strongly suggest a regulatory role of EMR2 in myeloid cell function (12, 13).
Indeed, binding and activation of EMR2 by a ECD-specific 2A1 monoclonal antibody (mAb) strongly enhanced the inflammatory responses of Nφ to a panel of stimuli, while 2A1 treatment alone (without inflammatory stimuli) did not seem effective (14). In addition, 2A1-induced EMR2 activation was shown to modulate the production of multiple cytokines and survival of lipopolysaccharide-stimulated Nφ (15). Hence, EMR2 activation seems to have a priming effect on Nφ activation. Furthermore, upregulated EMR2 expression was identified in Nφ of patients suffering from systemic inflammatory response syndrome (SIRS), and a significant association was noted between the percentage of EMR2-expressing Nφ and the extent of organ failure in SIRS patients. As a result, EMR2 was proposed recently as a novel Nφ biomarker for SIRS (14, 16). A more recent study demonstrated that Nφ of liver cirrhosis patients with infection have higher EMR2 expression levels, which showed strong correlation with disease severity and predicted overall mortality (17). Likewise, we previously showed that Mφ activated by 2A1-induced EMR2 ligation promoted secretion of several pro-inflammatory cytokines (18). More recently, a missense EMR2-C492Y variant was identified as the disease protein responsible for the autosomal dominant vibratory urticaria, a dermal vibration-induced hives. It was shown that the disease-associated EMR2 variant was less stable and prone to sensitize mast cells for aberrant histamine release upon vibratory stimulation in the presence of dermatan sulfate or 2A1 (19).
Adhesion G protein-coupled receptors represent a selective group of seven transmembrane (7TM) receptors with a large ECD that usually contains multiple tandem repeats of cell adhesion-like protein motifs and a GPCR autoproteolysis-inducing (GAIN) domain (20–22). During receptor biosynthesis, aGPCRs are normally bisected at a consensus GPCR proteolysis site via the GAIN domain-mediated autoproteolytic reaction into a N-terminal ECD-fragment (NTF) and a C-terminal 7TM-fragment (CTF), which remain conjugated as a dual-subunit receptor (13, 21). Recent advances indicate that aGPCR activation is likely mediated by ligand-induced NTF displacement, followed by the unfolding and binding of an internal agonist peptide to the 7TM core of CTF (23, 24). The mechanistic insights of the “tethered agonism” of aGPCRs are increasingly being unraveled, including the coupling of unique G proteins to distinct aGPCR members (21, 25–27). However, an orderly depiction of aGPCR-mediated signaling pathways is currently lacking. In the present report, we investigated and identified the involvement of Gα16/Akt/mitogen-activated protein kinase (MAPK)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in EMR2 receptor-mediated signaling. Our results indicate that EMR2 activation/signaling plays a functional role in the differentiation and inflammatory activation of human monocytic cells. The EMR2-induced signaling cascades reported here may help identify potential targets for the therapeutic management of inflammatory disorders, such as SIRS and vibratory urticaria.
Materials and Methods
Reagents and Antibodies
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. Anti-mAbs used for Western blotting against extracellular signal-regulated kinase (ERK)1/2, p-ERK1/2, p38, p-p38 (Thr180/Tyr182), c-Jun N-terminal kinase (JNK), p-JNK (Thr183/Tyr185), IκB-α, p-IκB-α (Ser32), p-Ikkα/β (Ser176/180), and p-Akt (Ser473) were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-Gα16 mAb was from Abcam (Cambridge, UK). Anti-F(ab′)2 fragment goat anti-mouse (GAM) IgG (H + L) was from Jackson ImmunoResearch (West Grove, PA, USA). Anti-CD11b-PE, anti-CD62L-PE, anti-CD81-PE, anti-CD9-APC, and anti-CD4-FITC for flow cytometry and anti-phosphotyrosine, anti-β-actin mAb for Western blotting were purchased from BD Biosciences. The mAbs used for cell stimulation were 2A1 (EMR2-specific mAb) (AbD Serotec) and mouse monoclonal IgG1 (Clone 11711) (R&D System) as described previously (18).
Cell Culture
THP-1 (ATCC®TIB-202™), HL-60 (ATCC®CCL-240™), and U937 (ATCC®CRL-1593.2™) human monocytic cell lines were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Thermo HyClone), 1% l-glutamate, 1% penicillin, and 1% streptomycin. All cells were cultured at 37°C in a 5% CO2 incubator. For the induction of Mφ-like differentiation, THP-1 cells were treated with 10 nM PMA for up to 4 days. Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors’ blood by Ficoll-Plague PLUS gradient centrifugation (Amersham Bioscience, Ltd.) as described previously (18). All procedures were approved by the Chang Gung Memorial Hospital Ethics Committee (CGMH IRB No: 97-2288B) and performed according to the guideline set by the Committee. Mos were subsequently isolated from PBMCs by immunomagnetic separation using human CD14 MicroBeads MACS cell separation kit (Miltenyi Biotec, Inc.) and cultured in complete RPMI 1640 medium. When required, cells were serum starved for 16–20 h before experiment. When indicated, cell culture plates were pre-coated with appropriate mAbs (usually 10 µg/ml) in 1 × PBS at 4°C for 16 h.
For treatment with inhibitors, cells were pre-incubated with the indicated reagents at 37°C: PD98059 (20 μM/90 min) (Cayman chemical), U0126 (10 μM/60 min) (Promega), Wortmannin (10 μM/60 min) (Cayman chemical), SB203580 (5 μM/15 min) (Cayman chemical), SP600125 (10 μM/60 min) (Sigma-Aldrich), LY294002 (20 μM/30 min) (Cayman chemical), and BAY11-7082 (10 μM/30 min) (InvivoGen). For siRNA-mediated gene silencing, 200 nM of EMR2- and Gα16-specific siRNAs (Invitrogen) were delivered into THP-1 cells using DharmaFECT-2 Transfection reagent (GE Dharmacon) as suggested by the manufacturer and incubated for 24–48 h prior to the subsequent experiments. The sequence information of the siRNAs used is listed below: #1 EMR2-siRNA: 5′GCU CGA CUG GAA UCA GGC ACA GAA A 3′, #2 EMR2-siRNA: 5′ CAG UGA UCC CGA GAC AGA AGG UGC U 3′, #3 EMR2-siRNA: 5′ GAA CAC AAG GAU GCU GGC AUU UAA A 3′, #1 GNA15-siRNA: 5′GGC CAG AAG UCA GAG CGU AAG AAA U 3′, #2 GNA15-siRNA: 5′CCA AGA GGU UCA UCC UGG ACA UGU A 3′, #3 GNA15-siRNA: 5′ GGA CUA UCC UGG AAC UAC CCU GGU U3′.
Cell Adhesion Assay
THP-1 cells were serum starved in RPMI medium for 16 h, harvested, and re-suspended in complete RPMI medium at 2 × 105 cells/100 μl/well in a 96-well plate pre-coated with mAbs. After 1 h at 37°C, cells were washed carefully at least six times with HBSS before being fixed with 2% glutaraldehyde for 20 min at room temperature. Cells were stained with 1% methylene blue for 30 min and lysed with 100 µl of 75% ethanol after excess dye was washed off with water. Eighty microliters of lysate samples were transferred into a new ELISA plate, and absorbance was measured at OD595 nm.
Flow Cytometry Assay
Cells were harvested and fixed with fresh 2% paraformaldehyde solution at 4°C for 20 min. Cells (1 × 106 cells/ml) were suspended in blocking buffer (1× PBS containing 1% BSA and 5% normal goat serum) at 4°C for 1 h. Cells were subsequently incubated with appropriate concentration of first Ab in blocking buffer at 4°C for 1 h, washed thoroughly with blocking buffer, and incubated with appropriate fluorophore-conjugated second Ab (1:200 in PBS) at 4°C for 1 h. Following extensive washes with cold PBS buffer, cells were analyzed by FACScan flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Flowjo).
Phenotypic Analysis of Neutrophil Activation
Peripheral blood Nφ were isolated from fresh venous blood donated by healthy volunteers using the Ficoll Hypaque gradient centrifugation method as described previously (15). For the morphological analysis, Nφ (5 × 105 cells/ml) were incubated at 37°C for 10 min with f-MLF (1 × 10−7 M) or conditioned medium (CM) of THP-1 cells under various stimulated conditions as indicated. Cell images were recorded under a light microscope at a magnification of 400×. For flow cytometry analysis of the expression of cell adhesion molecules, Nφ (2 × 106 cells/ml) were incubated at 37°C with the CM of THP-1 cells in the absence or presence of f-MLF (1 × 10−7 M) for 15 min. Cells were fixed with 2% paraformaldehyde/PBS at 4°C for immunostaining with anti-CD11b-PE or anti-CD62L-PE as described elsewhere and analyzed using CellQuest software (BD Biosciences). For the detection of reactive oxygen species (ROS) generation, Nφ (2 × 106 cells/ml) were incubated with 2 µM dihydrorhodamine-123 (DHR123; Molecular Probes) or 4 mM CM-H2DCFDA (Molecular Probes) for 25 min at room temperature. Cells were then incubated for 20–30 min with the CM of THP-1 cells stimulated as indicated. The accumulation of H2O2 was immediately analyzed by flow cytometer as described (14). For chemotactic cell migration assay, Boyden chamber-type transwells (Millipore) with a polycarbonate filter of 5.0 µm pore size were employed. Nφ (5 × 105 cells/ml) were seeded into the upper chambers in a total volume of 100 µl RPMI1640 containing 0.5% BSA. Relevant CM samples (400 µl) were added to the lower chambers. When necessary, f-MLF (1 × 10−7 M) was added in CM and used as a positive control. The transwell chamber was incubated at 37°C for 50 min, and cells migrated to the lower chambers were harvested and analyzed.
Gelatin Zymography
Gelatin zymography assay was performed as described previously (28). In brief, serum-starved THP-1 cells (2 × 106 cells/200 μl/well) were cultured in serum-free medium in 12-well plates pre-coated with or without mAbs at 37°C for 16 h. Culture medium was then collected by centrifugation at 1,500 rpm for 5 min at 4°C. Supernatant was collected and diluted 1:1 (v/v) with 2× sample buffer, heated for 30 min at 37°C before being subjected to gel electrophoresis in 8% SDS-PAGE gels containing 1 mg/ml gelatin. Following electrophoresis, gel was washed with 2.5% Triton X-100 for 10 min at RT twice, transferred into developing buffer (50 mM Tris–HCl, pH 7.4, 0.2 M NaCl, 5 mM CaCl2) with constant shaking at 25 rpm for 15 min at RT. Fresh developing buffer was replenished to allow for a further 48 h incubation at 37°C. Gel was subsequently transferred into fixing buffer (5% methanol, 10% acetic acid) with constant shaking at 25 rpm for 15 min at RT. Finally, gel was stained with 0.1% Coomassie brilliant blue in fixing buffer at RT, followed by destaining with fixing buffer with constant changing of the fixing buffer every 15 min until digested bands are clear.
Cytokine Elisa Assay
Cells were seeded at 2 × 106 cells/well into 12-well plates pre-coated with or without mAbs and incubated at 37°C for 16 h. After incubation, medium was collected by centrifugation at 1,500 rpm for 5 min at 4°C. Supernatant was transferred into new 1.5-ml eppendorf tubes. The levels of human IL-8 and TNF-α were measured by DuoSet® ELISA Development Systems (R&D System) according to the protocol suggested by the manufacturer.
Western Blotting Analysis
Cell lysate proteins for Western blot analysis were collected at indicated time points. In brief, cells were harvested by centrifugation at 1,500 rpm for 5 min at 4°C, washed once with ice-cold 1× HBSS, and lysed in 100-µl ice-cold modified cell lysis buffer as described previously (29). Proteins were quantified using Bicinchoninic acid protein assay kit (PIERCE, Rockford, IL, USA) by reading absorbance at 550 nm. Protein samples were separated in SDS-polyacrylamide gels by electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, MA, USA). Blotted PVDF membranes were blocked for 1 h in blocking buffer (5% of BSA in washing buffer) with agitation, then incubated for 1 h with the indicated first Ab (2–4 µg/ml in blocking buffer). Following extensive washes, membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated second Ab (1:2,000–1:5,000 in blocking buffer). Finally, membranes were extensively washed before detection of bound HRP by chemilluminescence (ECL, Amersham Life Science Ltd. or SuperSignal West Pico Plus, Pierce) for 5 min.
Statistical Analysis
Quantitative analysis was performed based on results of six independent experiments unless indicated otherwise. Differences between groups were determined by Student’s t-test using the Prism 5 software and shown as mean ± SD. In all cases, a probability value of p value < 0.05 was accepted to reject the null hypothesis. The statistical significance of p was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Ligation and Activation of EMR2 Receptor, a Novel Surface Marker of Mφ Differentiation, Promotes Mφ-Like Differentiation in THP-1 Cell
Consistent with our previous findings (9), EMR2 expression was indeed persistently upregulated during the in vitro differentiation of PMA-treated THP-1 cells, which gradually displayed characteristic Mφ-like phenotypes such as increased cell adherence, enlarged cell size and granularity, and expressional changes of specific differentiation/maturation markers including CD4, CD9, CD11b, and CD81 (Figure S1 in Supplementary Material) (30). Notably, the upregulated EMR2 expression levels correlated very closely with those of Mφ phenotypic markers. Therefore, EMR2 might be considered a novel surface marker of human Mφ differentiation.
Interestingly, a similar Mφ-like phenotype akin to that of PMA-treated cells was detected when THP-1 cells were cultured on plates pre-coated with the EMR2-specific 2A1 mAb, which bound and ligated surface EMR2 receptor (Figure 1; Figure S2 in Supplementary Material). Importantly, no such phenotypic changes were noted in cells cultured on plates coated with control mIgG1 or in the presence of soluble 2A1 mAb (Figure 1; Figure S2 in Supplementary Material and data not shown). Finally, enhanced cell adherence induced by the immobilized 2A1 was greatly diminished in cells transfected with siRNAs that silenced EMR2 expression, confirming the specific effect of EMR2 ligation on inducing THP-1 differentiation (Figure 1E). These results indicate clearly a functional role of EMR2 activation and signaling in promoting the Mφ-like differentiation of THP-1 cells.

Figure 1. Ligation of EMR2 receptor induces macrophage (Mφ)-like differentiation in THP-1 cell. (A,B) Microscopic observation (A) and flow cytometry analysis (B) of morphological changes of THP-1 cells incubated with immobilized 2A1 monoclonal antibody (mAb) (10 µg/ml) at day 4. mIgG1 was a negative control and PMA-treated cells were a positive control. Scale bar: 10 µm. SSC: side scatter for cell granularity; FSC: forward scatter for cell size. (C,D) Flow cytometry analysis of specific surface marker expression of 2A1-ligated THP-1 cells shows a macrophage-like phenotype. Cells were treated with or without immobilized 2A1 mAb (10 µg/ml) for 4 days. mIgG1 was included as a negative control and PMA-treated cells were a positive control. CD4, CD9, CD11b, and CD81 were used as cell surface markers of Mφ-like differentiation of THP-1 cells. Data are one representative of three independent experiments with similar results. (E) The effect of EMR2 ligation and activation on THP-1 cell adhesion was evaluated by cell adhesion assay in cells transfected without or with EMR2-specific siRNAs and incubated with immobilized 2A1 mAb (n = 6, mean ± SD; *p < 0.05).
EMR2 Receptor Activation in Mos Induces Inflammatory Responses
Monocyte/macrophage activation usually results in dynamic changes in the surrounding milieu, further recruiting and modulating the activities of other immune cells. To explore the functional significance of EMR2 activation, CM of 2A1-ligated THP-1 cells (2A1-CM) was collected and tested for its ability to activate peripheral blood Nφ, one of the first-line innate immune effector cells. Interestingly, chemotactic cell migration assay showed that 2A1-CM was able to induce Nφ chemotaxis as well as f-MLF (Figure 2A). Cell adhesion and spreading was also observed in Nφ cultured in the presence of 2A1-CM, similar to cells treated with f-MLF (Figure 2B). By contrast, CM of THP-1 cells cultured alone or with the control mIgG1 did not induce such morphological changes in Nφ. These results strongly suggested that 2A1-CM specifically activated Nφ, which was subsequently ascertained by the identification of phenotypic changes including the upregulation of CD11b and shedding of CD62L on the cell surface (Figure 2C). Intriguingly, these phenotypic changes were more profound in Nφ under combined treatment of 2A1-CM and f-MLF than those treated singly with 2A1-CM or f-MLF, suggesting a possible synergetic effect. Moreover, it was found that while 2A1-CM treatment by itself did not activate ROS production in Nφ, combined treatment of 2A1-CM and f-MLF generated much more ROS than did cells treated with f-MLF alone (Figure 2D). Taken together, we conclude that 2A1-mediated EMR2 ligation activates THP-1 cells, which in turn induces inflammatory responses on Nφ, most likely due to the production of pro-inflammatory mediators.
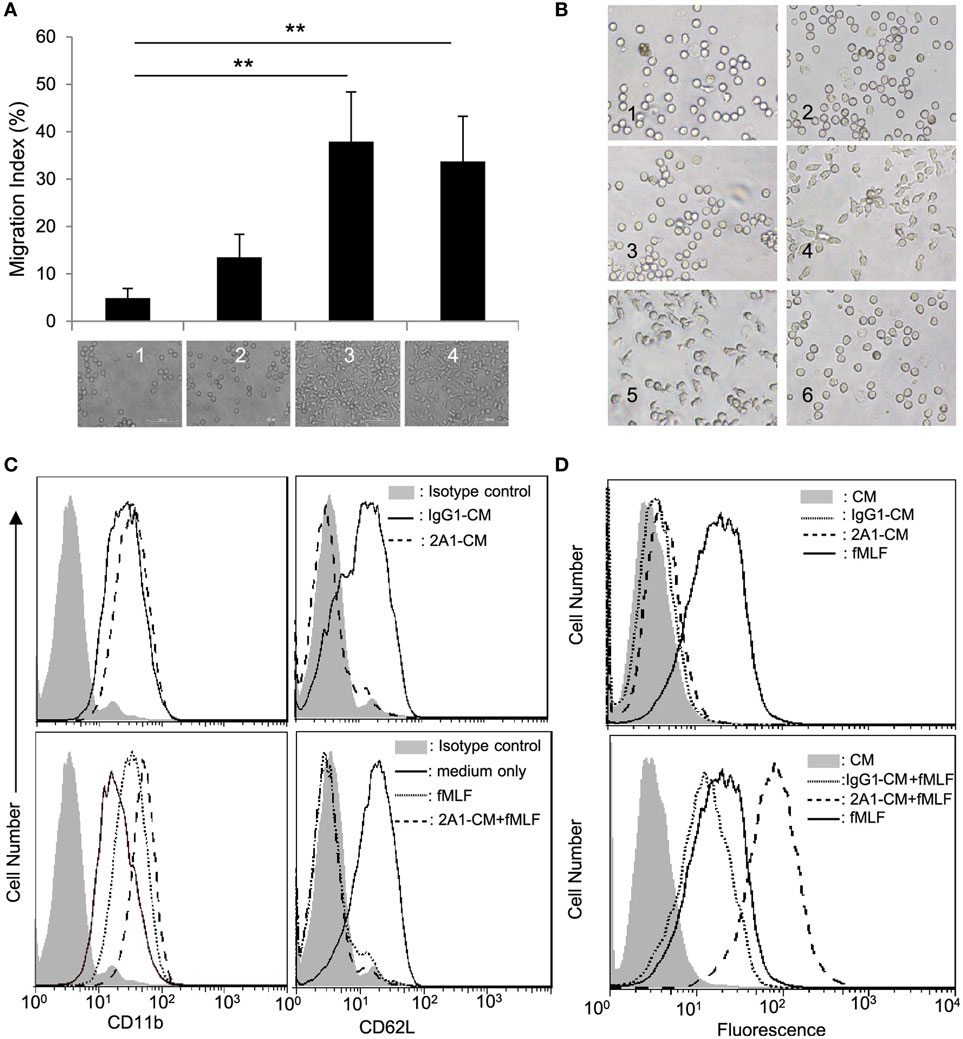
Figure 2. EMR2 ligation and activation in THP-1 cells induces inflammatory responses. (A) Chemotactic cell migration analysis of neutrophils (Nφ) was performed using Boyden chamber-based transwells in which the bottom wells were loaded with conditioned medium (CM) of THP-1 cells stimulated with none (THP-1 CM) (lane 1), with immobilized mIgG1 control (10 µg/ml) (IgG1-CM) (lane 2), with immobilized 2A1 (10 µg/ml) (2A1-CM) (lane 3), or THP-1 CM plus f-MLF (100 nM) (lane 4) as chemoattractants. The bottom pictures showed Nφ that have migrated across the membrane (n = 3, mean ± SD; **p < 0.01). (B) CM of 2A1-stimulated THP-1 cells induces morphological changes of Nφ. Nφ were incubated with 10 µg/ml soluble 2A1 (panel 2), 100 nM f-MLF (panel 4), or THP-1 CM (panel 1), IgG1-CM (panel 3), 2A1-CM (panel 5), or soluble 2A1 (10 µg/ml) (panel 6). Microscopic images are of 400× magnification. (C,D) Flow cytometry analysis of Nφ activation by detecting the expression levels of CD11b and CD62L (C) and ROS generation (D). Nφ were treated without (top panels) or with (bottom panels) 100 nM f-MLF in the absence or presence of IgG1-CM or 2A1-CM.
EMR2 Receptor Activation in Mos Promotes IL-8, TNF-α, and MMP-9 Production
In good agreement with our previous results (18), enhanced levels of TNF-α, IL-8, and MMP-9 were detected in 2A1-CM from THP-1 cells, as well as that of two other EMR2-expressing HL-60 and U937 cell lines (Figure 3; Figure S3 in Supplementary Material). More importantly, EMR2 ligation-induced pro-inflammatory mediator production was shown to be 2A1 dose-dependent, cell number-dependent, and time-dependent (Figures 3A,B; Figure S3B in Supplementary Material). Finally, the specificity of 2A1-induced functional effect was confirmed by two independent experiments. The first is to incubate cells first with soluble 2A1, followed by the addition of the F(ab′)2 fragment of a GAM Ab to cross-link receptor-bound 2A1. Interestingly, cells under this Ab-cross-linking condition also displayed similar inflammatory phenotypes as observed in those cultured with plate-bound immobilized 2A1, while cells treated with soluble 2A1 alone or GAM F(ab′)2 only did not (Figure 3C; Figure S3C in Supplementary Material and data not shown). The second strategy is to use THP-1 cells whose EMR2 expression was knocked down (KD) by siRNA-mediated gene silencing (Figure 3D). As shown in Figures 3E,F, EMR2-KD THP-1 cells displayed reduced MMP-9 and IL-8 production, as well as lower CD9 and CD81 levels in comparison to those transfected with a scrambled control siRNA. These results indicate that EMR2 activation cannot be achieved simply by the binding of 2A1, but requires receptor ligation (cross-linking) brought about by immobilized 2A1, which eventually leads to THP-1 cell differentiation and production of pro-inflammatory mediators.
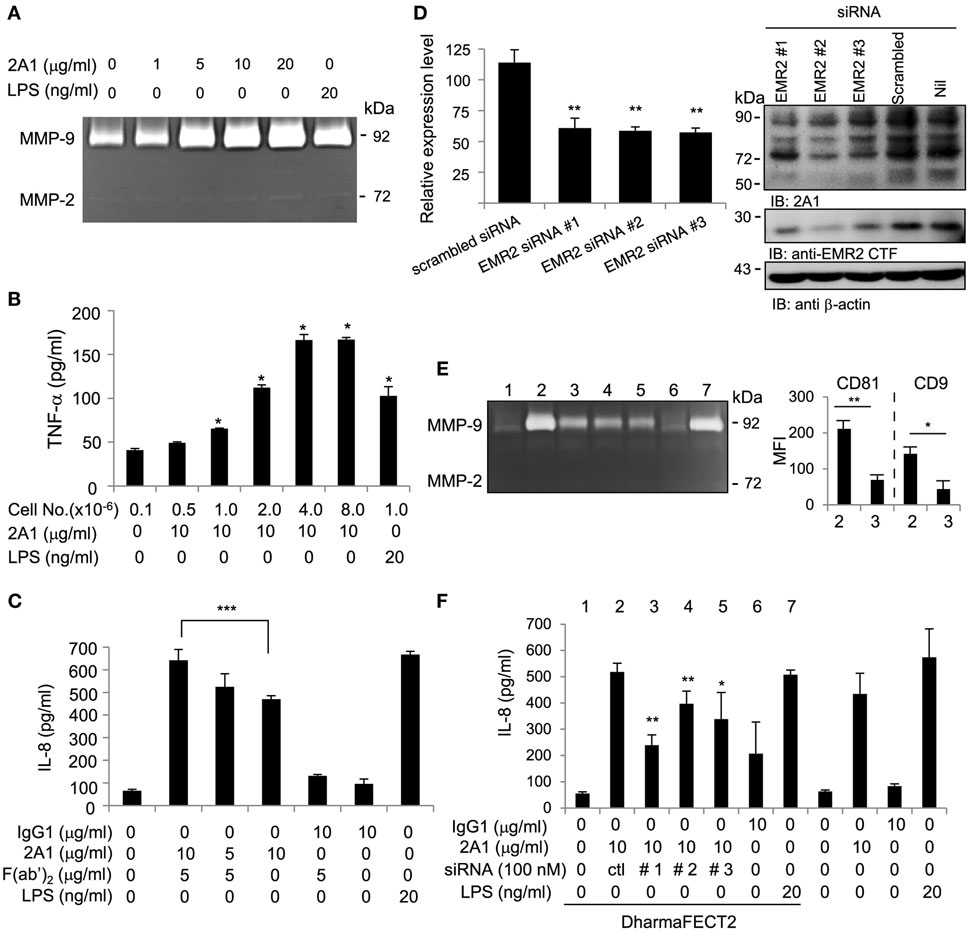
Figure 3. EMR2 ligation and activation in THP-1 cells promotes IL-8, TNF-α, and MMP-9 secretion. (A,B) Ligation of EMR2 on THP-1 by immobilized 2A1 mAb promoted MMP-9 and TNF-α production in a dose-dependent and cell number-dependent manner (n = 3, mean ± SD; *p < 0.05, **p < 0.01). (C–F) The specific effect of 2A1-activated EMR2 on THP-1 cells was confirmed alternatively by incubating cells at 4°C with 2A1 for 30 min followed by cross-linking with or without the F(ab′)2 fragment of a Goat anti-mouse Ab (5 µg/ml) as indicated. (C), or by EMR2-knock down in THP-1 cells that were transfected with EMR2-specific siRNAs (EMR2-siRNA#1, #2, or #3) (D). Reduced EMR2 expression was demonstrated by flow cytometry analysis of surface EMR2 levels (left panel) and Western blot analysis of lysates (right panel) of transfectant cells. The specific effect of EMR2 knockdown on THP-1 cell phenotype was shown by reduced MMP-9 production [(E) left panel], decreased expression of macrophage-like differentiation markers (CD9 and CD81) [(E) right panel], and reduced IL-8 secretion (F) in cells cultured on immobilized 2A1 for 4 days. Nos. 1–7 in panel (E) represented cell treatment conditions as listed in panel (F). In all experiments, mIgG1 and lipopolysaccharide (LPS) were used as a negative and a positive control, respectively (n = 6, mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001).
EMR2 Activation in Mos Induced Specific MAPK Phosphorylation
As an aGPCR, EMR2 activation is expected to turn on specific signaling pathways, including in principle G protein(s). To explore the EMR2-induced signaling cascades, we first examined the status of global tyrosine phosphorylation of THP-1 cell lysate following EMR2 engagement (Figure S4 in Supplementary Material). Indeed, EMR2 ligation induced fast and transient tyrosine phosphorylation of cell lysate proteins, reaching the peak at ~5 min and returned to the basal levels after ~30 min. This result clearly suggests the activation of unique signaling molecules and prompts us to survey the possible candidates using a wide range of selective signaling inhibitors. As shown in Figures 4A,B, EMR2 activation-induced IL-8 and MMP-9 production was mitigated agreeably by many inhibitors tested, including PI3K inhibitors (Wortmannin and LY294002), MAPK/ERK kinase inhibitors (PD98059, U0126), and JNK inhibitor (SP600125). By contrast, the p38 inhibitor (SB203580) in fact further enhanced IL-8 production induced by EMR2 activation. These results strongly suggest that 2A1-mediated EMR2 ligation activates multiple signaling pathways, some of which might be involved in a negative feedback loop.
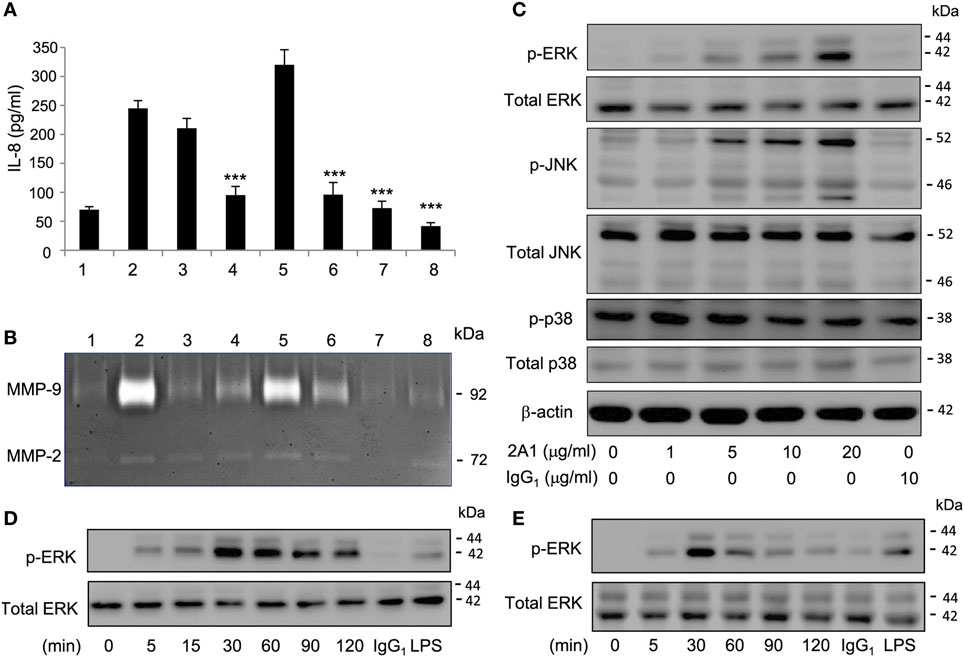
Figure 4. EMR2 activation in THP-1 and primary monocytes (Mos) induced specific phosphorylation of Mitogen-activated protein kinases molecules. (A,B) THP-1 cells were pretreated with different signaling inhibitors as indicated, followed by incubation with immobilized 2A1 (10 µg/ml) for 16 h. Culture supernatant was collected for the detection of IL-8 (A) and MMP-9 (B). Lane #1, control with no 2A1 coating; #2, DMSO; #3, Wortmannin (40 µM); #4, LY294002 (50 µM); #5, SB203580 (20 µM); #6, PD98059 (20 µM); #7, SP600125 (40 µM); and #8, U0126 (10 µM) (n = 6, mean ± SD; ***p < 0.001). (C) Western blot analysis of indicated signaling molecules in THP-1 cells incubated with or without 2A1 as indicated. β-actin is a loading control for the immunoblotting. (D,E) Western blot analysis of EMR2 activation-induced extracellular signal-regulated kinase (ERK) phosphorylation in THP-1 cell (D) and primary Mos (E) incubated with immobilized 2A1 at the indicated time points. In all experiments, mIgG1 and lipopolysaccharide (LPS) treatment was a negative and positive control, respectively.
As the MAPK signaling cascade is a well-known inflammation-associated signaling pathway and down stream targets of many GPCRs (31–34), we next confirmed EMR2-induced activation of specific MAPKs by Western blotting. Consistent with earlier results with the use of selective inhibitors, increased phosphorylation of ERK and JNK, but not p38, was detected following 2A1-mediated EMR2 ligation in a Ab dose-dependent and time-dependent fashion (Figures 4C–E). Importantly, EMR2 activation-induced phosphorylation of ERK and JNK was comparably detected in THP-1 cells as well as in primary blood Mos. As expected, phosphorylation of the two MAPK molecules was efficiently inhibited by specific inhibitors in a dose-dependent manner, again both in THP-1 cell and primary Mos (Figures 4D,E; Figures S5 and S6 in Supplementary Material). Simultaneously, EMR2 activation-induced pro-inflammatory mediator production was inhibited by these inhibitors (Figures S5 and S6 in Supplementary Material). We hence conclude that 2A1-mediated EMR2 ligation in Mos induces phosphorylation and activation of specific MAPK molecules, including ERK and JNK.
EMR2-Induced Signaling Is Mediated in Part via NF-κB Activation
In addition to the MAPK signaling pathway, NF-κB is another major signaling molecule critical for the induction of inflammatory reactions (35, 36). In essence, NF-κB activation involves the phosphorylation and subsequent degradation of IκB, which is initiated in turn through phosphorylation of the IκB kinases (IKK) α/β (37). As shown, increased phosphorylation of IKK α/β and IκB proteins and the concurrent loss of IκB were readily detected following 2A1-mediated EMR2 ligation in THP-1 cells, again in a Ab dose-dependent and time-dependent fashion (Figure 5; Figure S7 in Supplementary Material). Furthermore, EMR2 ligation-induced NF-κB activation was specifically mitigated in THP-1 cells pretreated with two independent IKK inhibitors, namely BAY 11-7082 and TPCA-1. Coincidentally, production of IL-8, TNF-α, and MMP-9 in 2A1-stimulated THP-1 cells was dose-dependently inhibited by the treatment of BAY 11-7082 and TPCA-1 (Figure 5; Figure S7 in Supplementary Material). These data reveal clearly the induction of NF-κB signaling pathway by EMR2 ligation and activation in THP-1 cells.
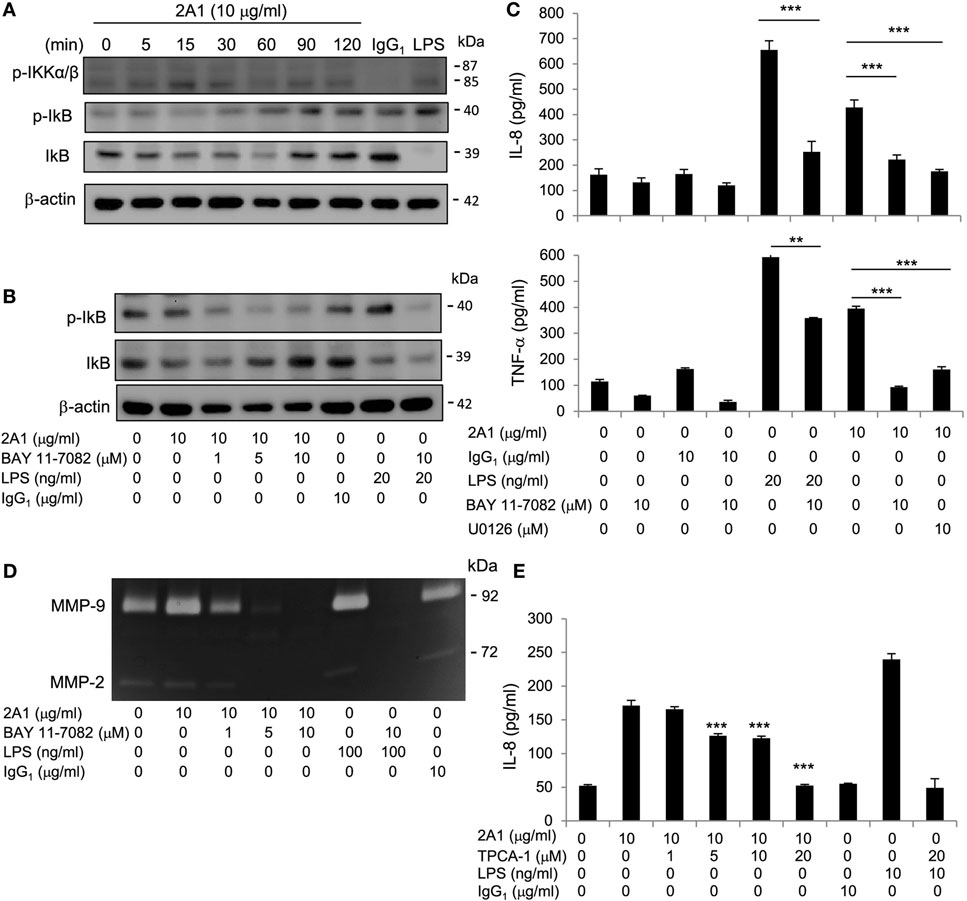
Figure 5. EMR2-induced signaling in THP-1 cells is mediated in part via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation. (A) Western blot analysis of NF-κB activation of THP-1 cells incubated with or without 2A1 (10 µg/ml) at different time points as indicated. Blots were probed to detect phospho-IKKα/β, phospho-IκB, IκB, and β-actin level. (B) Western blot analysis of NF-κB activation of THP-1 cells pretreated with different concentrations of Bay11-7082 for 1 h, followed by incubation with 2A1 (10 µg/ml) for 1 h. Blots were probed to detect phospho-IκB, IκB, and β-actin level. (C,D) Culture supernatants of THP-1 cells treated with indicated conditions for 16 h were collected for the detection of TNF-α and IL-8 by ELISA (C) and MMP-9 activity by gelatin zymography (D). (E) THP-1 cells were pretreated with different concentrations of TPCA-1 for 1h, followed by incubation with 2A1 (10 µg/ml) for 16 h. Culture supernatants were collected for the detection of IL-8 by ELISA. In all experiments, mIgG1 and lipopolysaccharide (LPS) treatment was a negative and positive control, respectively (n = 6, mean ± SD; **p < 0.01, ***p < 0.001).
EMR2 Activation in Monocytic Cells Signals via the Gα16/PLC/Akt Pathways
Several aGPCRs have recently been shown to signal through specific G proteins (21, 38); however, very little is known for EMR2. CD97/ADGRE5, a close homolog of EMR2, was reported to heterodimerize with the lysophosphatidic acid (LPA) receptor 1 and signal via Gα12/13 to induce LPA-dependent Rho and ERK activation in prostate cancer cells (39). On the other hand, the CD97-LPAR1 heterodimer was shown to mediate the lysophosphatidylethanolamine-induced intracellular Ca2+ increase in MDA-MB-231 breast cancer cells by the pertussis toxin (PTX)-sensitive Gαi/o protein and phospholipase C (PLC) (40). In a transient over-expression system in heterologous HEK293 cells, Gupte et al. have shown that co-transfection of EMR2 and Gα15 resulted in constitutive signaling (41).
To delineate the G protein(s) involved in EMR2-mediated signaling in monocytic cells, we first showed that PTX is ineffective in inhibiting EMR2 activation, thus excluding the involvement of Gαi/o proteins (Figure S8A in Supplementary Material). Next, the role of Gα16 (human ortholog of murine Gα15) was examined using Gα16-specific siRNAs that effectively dampened its expression (Figure S8B in Supplementary Material). Interestingly, EMR2 ligation-induced IL-8 production was diminished when Gα16 expression was KD (Figure 6A), suggesting that Gα16 is indeed involved in EMR2-mediated signaling. Gα15/16 proteins are known to couple GPCRs to PLC-β, which hydrolyzes phosphatidylinositol biphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (42, 43). We, therefore, incubated THP-1 cells in the presence of U73122, a potent inhibitor of G protein–PLC coupling and activation. As expected, production of IL-8 and MMP-9 was inhibited dose-dependently in cells treated with U73122 (Figure 6B). In addition, diminished ERK and NF-κB activation was observed in U73122-treated cells, which is consistent with the idea that PLC-β activation is upstream of MAPK and NF-κB signaling (Figure 6B). These results show for the first time the coupling of EMR2 to PLC-β activation via Gα16.
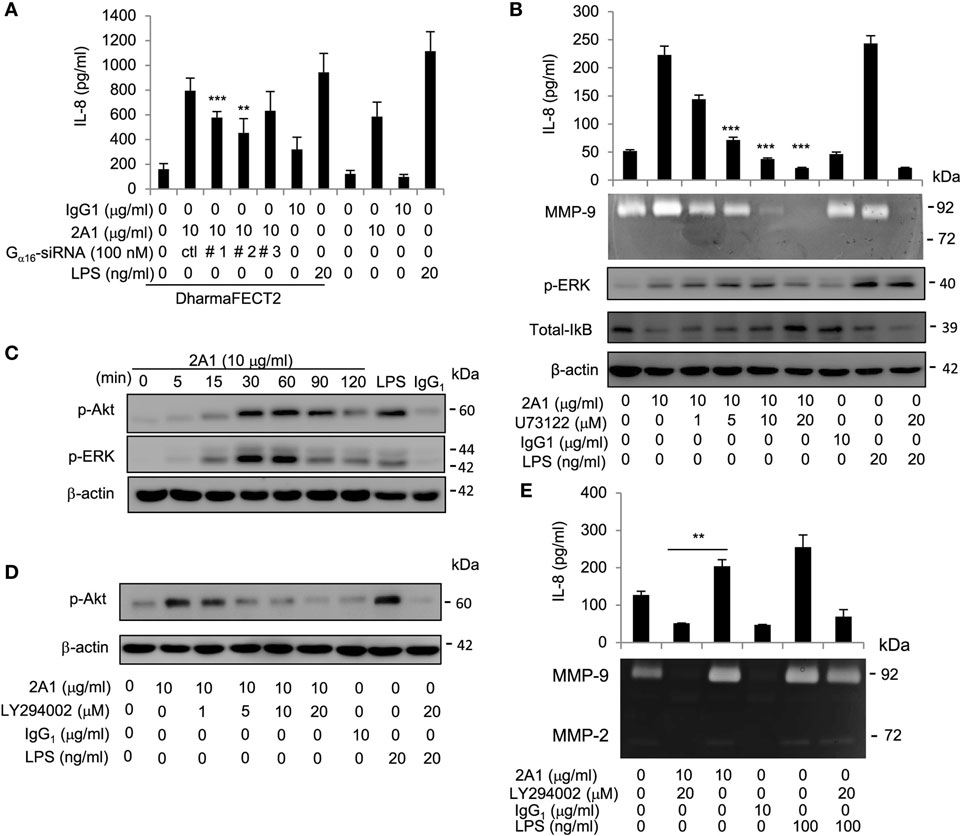
Figure 6. EMR2 activation in monocytic cells signals via the Gα16/PLC/Akt pathways. (A) The role of Gα16 in EMR2-mediated signaling was evaluated in THP-1 cells that were transfected with Gα16-specific siRNAs (Gα16-siRNA#1, #2, or #3). IL-8 levels resulted from EMR2 ligation were compared as described. (B) The involvement of PLC-β in EMR2-mediated signaling was evaluated in THP-1 cells that were pretreated with different concentrations of U73122 for 1 h, followed by incubation with 2A1 (10 µg/ml). Conditioned medium (CM) was analyzed for IL-8 by ELISA and MMP-9 activity by gelatin zymography, respectively (top panels). Western blot analyses were performed to detect phospho-extracellular signal-regulated kinase (ERK), IκB, and β-actin levels (lower panels). (C,D) Western blot analysis of EMR2 activation-induced Akt phosphorylation in THP-1 cells. Cells were pretreated without (C) or with different concentrations of LY294002 (D) for 1 h, followed by incubation with 2A1 (10 µg/ml) as shown. Blots were probed to detect phospho-Akt, phospho-ERK, and β-actin levels. (E) The effect of the PI3K inhibitor (LY294002) on EMR2 activation in THP-1 cells was analyzed by comparing IL-8 levels and MMP-9 activities of CM from cells pretreated without or with LY294002 as described. In all experiments, mIgG1 and lipopolysaccharide (LPS) treatment was a negative and positive control, respectively (n = 6, mean ± SD; **p < 0.01, ***p < 0.001).
Finally, the role of PIP2 metabolism by PLC-β and the fact that two PI3K inhibitors (Wortmannin and LY294002) were effective in modulating EMR2 activation (Figure 4) prompted us to investigate the involvement of Akt in EMR2-mediated signaling cascade. The PI3K-Akt pathway is initiated by activated PI3K that phosphorylated PIP2 to phosphatidylinositol 3,4,5 trisphosphate (PIP3), which then recruited and activated Akt (44, 45). Importantly, PI3K can be activated by many cell surface receptors, including GPCRs, primarily via dissociated Gβγ subunits (46, 47). As shown in Figure 6C, activated/phosphorylated Akt was indeed identified in THP-1 cells cultured with immobilized 2A1 in a time-dependent manner very similar to ERK activation. Moreover, Akt activation and production of IL-8 and MMP-9 were dose-dependently reduced by LY294002 in 2A1-treated THP-1 cells (Figures 6D,E), suggesting that the PI3K-Akt pathway is also involved in EMR2-mediated signaling. Taken together, we conclude that the Gα16/PLC/Akt pathways are coupled and activated by the ligated EMR2 receptor in monocytic cells.
Discussion
The identification of EMR2/ADGRE2 as the vibratory urticaria-inducing molecule attests to the functional importance of this myeloid-restricted aGPCR (19). While accumulating evidence is emerging for a signaling role of EMR2 in myeloid cells, the actual pathways and significance of EMR2-mediated signaling are not understood. Herein, we show that EMR2 receptor is a novel surface marker of Mφ differentiation and EMR2 activation is mainly coupled to the Gα16 protein, which subsequently activates the PLC and Akt/PI3K pathways, eventually leading to the MAPK and NF-κB signaling (Figures 4–6; Figures S1–S8 in Supplementary Material). EMR2 activation promotes Mφ-like differentiation of THP-1 cells and evokes inflammatory responses by stimulating the production of pro-inflammatory mediators (Figures 1–3). In summary, EMR2 is a previously uncharacterized surface protein regulator of the differentiation/maturation process and inflammatory activation of Mφ subpopulations.
The coupling of Gα16 protein to EMR2 is one of a limited number of specific aGPCR–G protein partnership identified to date, but it is in line with previous results. First, it was shown recently that co-transfection of EMR2 and murine Gα15 in heterologous cells specifically induced cellular activation (41). Second, human Gα16 protein (and murine Gα15) are known to be restricted to cells of certain hematopoietic lineages, including myeloid cells (48). Finally, Gα16/Gα15 proteins belong to the PTX-insensitive Gαq subfamily and are known to couple many GPCRs to activate PLC-β (42, 43). Due to the fact that EMR2 and Gα16 are co-expressing in human Mos and that EMR2 activation is impaired by siRNA-induced Gα16 downregulation and a pharmacological inhibitor of PLC but is unresponsive to PTX, the coupling of EMR2 activation to Gα16 in Mos is the most likely signaling pathway (Figure 6; Figure S8 in Supplementary Material). Nevertheless, the possibility of the involvement of other G proteins in EMR2-mediated signaling is not completely excluded.
Interestingly, the homologous CD97 receptor was shown to couple separately to Gα12/13 or Gαi/o proteins in response to different stimuli in distinct cancer cell types (39, 40). Hence, it is possible that other Gα proteins might also be involved in EMR2-mediated signaling depending on the nature of EMR2 stimulation. Likewise, it remains to be determined whether the similar Gα16-mediated signaling cascades is employed in EMR2 activation in different myeloid cell types such as Nφ, mast cell, and DC.
G protein-coupled receptor-induced Akt activation is known to be mediated by all four major Gα protein subfamilies via distinct mechanisms (47, 49). The involvement of PI3K/Akt pathway in EMR2-mediated signaling suggested the possible participation of the dissociated Gβγ subunits and/or other means such as increased intracellular Ca2+ brought about by Gα16-induced PLC-β activity (47). Regardless of the exact mechanism, the inclusion of PLC-β/Akt-signaling pathways described here clearly indicates that phosphinositide-dependent metabolism is a significant component of EMR2-induced signaling events. This idea is further ascertained by the identification of EMR2-activated MAPKs (ERK and JNK) and NF-κB, which are validated downstream targets of the PLC-β and Akt-signaling pathways and many GPCRs (50–53).
Both MAPKs and NF-κB are well-established signaling molecules involved in the induction of inflammatory responses (31, 32, 35, 54). Thus, the activation of ERK/JNK and NF-κB by the Gα16/PLC/Akt pathways is consistent with the role of EMR2 in promoting Mφ-like differentiation and production of pro-inflammatory mediators. Hence, it seems that EMR2-activated signaling pathways are rather similar to the ones occurred in Mφ under inflammatory conditions. However, due to the fact that multiple signaling pathways normally cross-talk and that phagocytes usually encounter and require numerous stimuli to achieve optimal cellular activation (55), in the future it will be interesting to investigate how the EMR2-mediated signaling cross-interact and contribute to the overall Mφ activity in the presence of other stimuli.
The recent revelation of the tethered agonistic activation mechanism of aGPCRs strongly hints at the notion that some aGPCRs might function as a mechanical sensor because a physical/mechanical signal is likely needed for the dissociation of aGPCR-NTF from its CTF (38, 56, 57). Indeed, this idea was substantiated by several lines of evidences recently. These include the findings of the enhanced vibration-induced NTF-CTF dissociation of the less stable EMR2-C492Y variant in vibratory urticaria patients and the shear stress-induced shedding of CD97-NTF from leukocytes that interacted with its CD55 ligand (19, 58). While we did not directly address this issue in the present study where all experiments were performed in a static condition, it is interesting to note that the 2A1-induced EMR2 activation only occurs when the mAb is immobilized, presumably to provide enough contact/interaction to cross-link the receptor. In the future studies, it will be interesting to examine whether EMR2-NTF is dissociated from cell surface under this static condition or alternatively to test whether vibration can further enhance 2A1-induced EMR2 signaling in Mos. It will be also of interest to know whether the EMR2-C492Y variant induces stronger and/or faster signaling in Mos, apart from enhancing mast cell degranulation. Taken together, our systematic analysis of EMR2-mediated signaling not only provides a direct evidence for its role in the differentiation and inflammatory reaction of Mos but also a starting point to investigate the activation mechanism and possible manipulation of aGPCR function in diseases, such as SIRS and vibratory urticaria.
Author Contributions
K-YI, Y-SH, C-HH, W-YT, C-HC, and G-WC performed experiments and analyzed data. MS, SG, and H-HL helped in analysis and interpretation of the data. K-YI, Y-SH, and H-HL wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank other members of Lin laboratory for insightful discussion and suggestion.
Funding
This study was supported in part by grants from the Ministry of Science and Technology, Taiwan (MOST-104-2320-B-182-035-MY3) and the Chang Gung Memorial Hospital (CMRPD1C0631-3, CORPD1F0061 and BMRP860).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00373/full#supplementary-material.
Abbreviations
aGPCR, adhesion GPCR; CM, conditioned medium; CTF, C-terminal fragment; DAG, diacylglycerol; DC, dendritic cell; ECD, extracellular domain; EMR2, EGF-like module-containing mucin-like hormone receptor-like 2; ERK, extracellular signal-regulated kinase; f-MLF, N-formyl-methionyl-leucyl-phenylalanine; GAIN, GPCR autoproteolysis-inducing; GPCR, G protein-coupled receptor; GPS, GPCR proteolysis site; IP3, inositol triphosphate; JNK, c-Jun N-terminal kinase; LPA, lysophosphatidic acid; LPE, lysophosphatidylethanolamine; LPS, lipopolysaccharide; Mφ, macrophage; mAb, monoclonal antibody; MAPK, Mitogen-activated protein kinases; MMP, matrix metalloproteinases; Mo, monocyte; Nφ, neutrophil; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NTF, N-terminal fragment; PLC, phospholipase C; PMA, phorbol 12-myristate 13-acetate; PI3K, Phosphoinositide 3-kinase; PIP2, phosphatidylinositol biphosphate; PIP3, phosphatidylinositol 3,4,5 trisphosphate; PTX, pertussis toxin; ROS, reactive oxygen species; SIRS, systemic inflammatory response syndrome; TNF, tumor necrosis factor; 7TM, seven transmembrane.
References
1. Gordon S. Phagocytosis: an immunobiologic process. Immunity (2016) 44(3):463–75. doi:10.1016/j.immuni.2016.02.026
2. Yona S, Gordon S. From the reticuloendothelial to mononuclear phagocyte system – the unaccounted years. Front Immunol (2015) 6:328. doi:10.3389/fimmu.2015.00328
3. Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol (2005) 23:901–44. doi:10.1146/annurev.immunol.23.021704.115816
4. Lin HH, Stacey M, Hamann J, Gordon S, McKnight AJ. Human EMR2, a novel EGF-TM7 molecule on chromosome 19p13.1, is closely related to CD97. Genomics (2000) 67(2):188–200. doi:10.1006/geno.2000.6238
5. Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol (1981) 11(10):805–15. doi:10.1002/eji.1830111013
6. Gordon S, Hamann J, Lin HH, Stacey M. F4/80 and the related adhesion-GPCRs. Eur J Immunol (2011) 41(9):2472–6. doi:10.1002/eji.201141715
7. Stacey M, Chang GW, Davies JQ, Kwakkenbos MJ, Sanderson RD, Hamann J, et al. The epidermal growth factor-like domains of the human EMR2 receptor mediate cell attachment through chondroitin sulfate glycosaminoglycans. Blood (2003) 102(8):2916–24. doi:10.1182/blood-2002-11-3540
8. Kwakkenbos MJ, Kop EN, Stacey M, Matmati M, Gordon S, Lin HH, et al. The EGF-TM7 family: a postgenomic view. Immunogenetics (2004) 55(10):655–66. doi:10.1007/s00251-003-0625-2
9. Chang GW, Davies JQ, Stacey M, Yona S, Bowdish DM, Hamann J, et al. CD312, the human adhesion-GPCR EMR2, is differentially expressed during differentiation, maturation, and activation of myeloid cells. Biochem Biophys Res Commun (2007) 353(1):133–8. doi:10.1016/j.bbrc.2006.11.148
10. Kwakkenbos MJ, Chang GW, Lin HH, Pouwels W, de Jong EC, van Lier RA, et al. The human EGF-TM7 family member EMR2 is a heterodimeric receptor expressed on myeloid cells. J Leukoc Biol (2002) 71(5):854–62.
11. van Eijk M, Aust G, Brouwer MS, van Meurs M, Voerman JS, Dijke IE, et al. Differential expression of the EGF-TM7 family members CD97 and EMR2 in lipid-laden macrophages in atherosclerosis, multiple sclerosis and Gaucher disease. Immunol Lett (2010) 129(2):64–71. doi:10.1016/j.imlet.2010.02.004
12. Kwakkenbos MJ, Matmati M, Madsen O, Pouwels W, Wang Y, Bontrop RE, et al. An unusual mode of concerted evolution of the EGF-TM7 receptor chimera EMR2. FASEB J (2006) 20(14):2582–4. doi:10.1096/fj.06-6500fje
13. Yona S, Lin HH, Siu WO, Gordon S, Stacey M. Adhesion-GPCRs: emerging roles for novel receptors. Trends Biochem Sci (2008) 33(10):491–500. doi:10.1016/j.tibs.2008.07.005
14. Yona S, Lin HH, Dri P, Davies JQ, Hayhoe RP, Lewis SM, et al. Ligation of the adhesion-GPCR EMR2 regulates human neutrophil function. FASEB J (2008) 22(3):741–51. doi:10.1096/fj.07-9435com
15. Chen TY, Hwang TL, Lin CY, Lin TN, Lai HY, Tsai WP, et al. EMR2 receptor ligation modulates cytokine secretion profiles and cell survival of lipopolysaccharide-treated neutrophils. Chang Gung Med J (2011) 34(5):468–77.
16. Lewis SM, Treacher DF, Edgeworth J, Mahalingam G, Brown CS, Mare TA, et al. Expression of CD11c and EMR2 on neutrophils: potential diagnostic biomarkers for sepsis and systemic inflammation. Clin Exp Immunol (2015) 182(2):184–94. doi:10.1111/cei.12679
17. Huang C-H, Jeng W-J, Ho Y-P, Wei-Teng, Hsieh Y-C, Chen W-T, et al. Increased EMR2 expression on neutrophils correlates with disease severity and predicts overall mortality in cirrhotic patients. Sci Rep (2016) 6:38250. doi:10.1038/srep38250
18. Huang YS, Chiang NY, Hu CH, Hsiao CC, Cheng KF, Tsai WP, et al. Activation of myeloid cell-specific adhesion class G protein-coupled receptor EMR2 via ligation-induced translocation and interaction of receptor subunits in lipid raft microdomains. Mol Cell Biol (2012) 32(8):1408–20. doi:10.1128/MCB.06557-11
19. Boyden SE, Desai A, Cruse G, Young ML, Bolan HC, Scott LM, et al. Vibratory urticaria associated with a missense variant in ADGRE2. N Engl J Med (2016) 374(7):656–63. doi:10.1056/NEJMoa1500611
20. Arac D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Sudhof TC, et al. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J (2012) 31(6):1364–78. doi:10.1038/emboj.2012.26
21. Hamann J, Aust G, Arac D, Engel FB, Formstone C, Fredriksson R, et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev (2015) 67(2):338–67. doi:10.1124/pr.114.009647
22. Lin HH. Adhesion family of G protein-coupled receptors and cancer. Chang Gung Med J (2012) 35(1):15–27. doi:10.4103/2319-4170.106170
23. Liebscher I, Schon J, Petersen SC, Fischer L, Auerbach N, Demberg LM, et al. A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep (2014) 9(6):2018–26. doi:10.1016/j.celrep.2014.11.036
24. Stoveken HM, Hajduczok AG, Xu L, Tall GG. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc Natl Acad Sci U S A (2015) 112(19):6194–9. doi:10.1073/pnas.1421785112
25. Langenhan T, Aust G, Hamann J. Sticky signaling – adhesion class G protein-coupled receptors take the stage. Sci Signal (2013) 6(276):re3. doi:10.1126/scisignal.2003825
26. Monk KR, Hamann J, Langenhan T, Nijmeijer S, Schoneberg T, Liebscher I. Adhesion G protein-coupled receptors: from in vitro pharmacology to in vivo mechanisms. Mol Pharmacol (2015) 88(3):617–23. doi:10.1124/mol.115.098749
27. Chiang NY, Peng YM, Juang HH, Chen TC, Pan HL, Chang GW, et al. GPR56/ADGRG1 activation promotes melanoma cell migration via NTF dissociation and CTF-mediated Galpha12/13/RhoA signaling. J Invest Dermatol (2017) 137(3):727–36. doi:10.1016/j.jid.2016.10.031
28. Hsiao CC, Wang WC, Kuo WL, Chen HY, Chen TC, Hamann J, et al. CD97 inhibits cell migration in human fibrosarcoma cells by modulating TIMP-2/MT1-MMP/MMP-2 activity – role of GPS autoproteolysis and functional cooperation between the N- and C-terminal fragments. FEBS J (2014) 281(21):4878–91. doi:10.1111/febs.13027
29. Chang GW, Hsiao CC, Peng YM, Vieira Braga FA, Kragten NA, Remmerswaal EB, et al. The adhesion G protein-coupled receptor GPR56/ADGRG1 is an inhibitory receptor on human NK cells. Cell Rep (2016) 15(8):1757–70. doi:10.1016/j.celrep.2016.04.053
30. Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One (2010) 5(1):e8668. doi:10.1371/journal.pone.0008668
31. Hommes DW, Peppelenbosch MP, van Deventer SJ. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut (2003) 52(1):144–51. doi:10.1136/gut.52.1.144
32. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy – from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta (2005) 1754(1–2):253–62. doi:10.1016/j.bbapap.2005.08.017
33. Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene (2007) 26(22):3122–42. doi:10.1038/sj.onc.1210407
34. Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci (2001) 22(7):368–76. doi:10.1016/S0165-6147(00)01678-3
35. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest (2001) 107(1):7–11. doi:10.1172/JCI11830
36. Colquhoun MC, Daly M, Stewart P, Beeley L. Interaction between warfarin and miconazole oral gel. Lancet (1987) 1(8534):695–6. doi:10.1016/S0140-6736(87)90470-3
37. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi:10.1186/1476-4598-12-86
38. Schoneberg T, Liebscher I, Luo R, Monk KR, Piao X. Tethered agonists: a new mechanism underlying adhesion G protein-coupled receptor activation. J Recept Signal Transduct Res (2015) 35(3):220–3. doi:10.3109/10799893.2015.1072978
39. Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, Goldsmith PK, et al. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res (2011) 71(23):7301–11. doi:10.1158/0008-5472.CAN-11-2381
40. Park SJ, Lee KP, Kang S, Chung HY, Bae YS, Okajima F, et al. Lysophosphatidylethanolamine utilizes LPA(1) and CD97 in MDA-MB-231 breast cancer cells. Cell Signal (2013) 25(11):2147–54. doi:10.1016/j.cellsig.2013.07.001
41. Gupte J, Swaminath G, Danao J, Tian H, Li Y, Wu X. Signaling property study of adhesion G-protein-coupled receptors. FEBS Lett (2012) 586(8):1214–9. doi:10.1016/j.febslet.2012.03.014
42. Offermanns S, Simon MI. G alpha 15 and G alpha 16 couple a wide variety of receptors to phospholipase C. J Biol Chem (1995) 270(25):15175–80. doi:10.1074/jbc.270.25.15175
43. Zhu X, Birnbaumer L. G protein subunits and the stimulation of phospholipase C by Gs-and Gi-coupled receptors: lack of receptor selectivity of Galpha(16) and evidence for a synergic interaction between Gbeta gamma and the alpha subunit of a receptor activated G protein. Proc Natl Acad Sci U S A (1996) 93(7):2827–31. doi:10.1073/pnas.93.7.2827
44. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med (2005) 9(1):59–71. doi:10.1111/j.1582-4934.2005.tb00337.x
45. Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal (2002) 14(5):381–95. doi:10.1016/S0898-6568(01)00271-6
46. Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J Biol Chem (1998) 273(30):19080–5. doi:10.1074/jbc.273.30.19080
47. New DC, Wu K, Kwok AW, Wong YH. G protein-coupled receptor-induced Akt activity in cellular proliferation and apoptosis. FEBS J (2007) 274(23):6025–36. doi:10.1111/j.1742-4658.2007.06116.x
48. Amatruda TT III, Steele DA, Slepak VZ, Simon MI. G alpha 16, a G protein alpha subunit specifically expressed in hematopoietic cells. Proc Natl Acad Sci U S A (1991) 88(13):5587–91. doi:10.1073/pnas.88.13.5587
49. New DC, Wong YH. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J Mol Signal (2007) 2:2. doi:10.1186/1750-2187-2-2
50. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci (2011) 36(6):320–8. doi:10.1016/j.tibs.2011.03.006
51. Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta (2007) 1773(8):1161–76. doi:10.1016/j.bbamcr.2007.01.002
52. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and raptor in association with IKK. Genes Dev (2008) 22(11):1490–500. doi:10.1101/gad.1662308
53. Gutkind JS. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE (2000) 2000(40):re1. doi:10.1126/stke.2000.40.re1
54. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol (2009) 1(6):a001651. doi:10.1101/cshperspect.a001651
55. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6(3):173–82. doi:10.1038/nri1785
56. Liebscher I, Monk KR, Schoneberg T. How to wake a giant. Oncotarget (2015) 6(27):23038–9. doi:10.18632/oncotarget.5112
57. Scholz N, Gehring J, Guan C, Ljaschenko D, Fischer R, Lakshmanan V, et al. The adhesion GPCR latrophilin/CIRL shapes mechanosensation. Cell Rep (2015) 11(6):866–74. doi:10.1016/j.celrep.2015.04.008
Keywords: cytokine, EMR2, GPCR, macrophage, inflammation, signaling
Citation: I K-Y, Huang Y-S, Hu C-H, Tseng W-Y, Cheng C-H, Stacey M, Gordon S, Chang G-W and Lin H-H (2017) Activation of Adhesion GPCR EMR2/ADGRE2 Induces Macrophage Differentiation and Inflammatory Responses via Gα16/Akt/MAPK/NF-κB Signaling Pathways. Front. Immunol. 8:373. doi: 10.3389/fimmu.2017.00373
Received: 23 December 2016; Accepted: 15 March 2017;
Published: 03 April 2017
Edited by:
Céline Cougoule, Centre national de la recherche scientifique (CNRS), FranceReviewed by:
Frédéric Velard, Université de Reims Champagne-Ardenne, FranceJunjie Zhang, University of Southern California, USA
Copyright: © 2017 I, Huang, Hu, Tseng, Cheng, Stacey, Gordon, Chang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hsi-Hsien Lin, aGhsaW5AbWFpbC5jZ3UuZWR1LnR3
†Present address: Yi-Shu Huang and Wen-Yi Tseng, Kennedy Institute of Rheumatology, University of Oxford, Oxford, UK