 Rihab Gam
Rihab Gam Pranali Shah
Pranali Shah Rachel E. Crossland
Rachel E. Crossland Jean Norden
Jean Norden Anne M. Dickinson
Anne M. Dickinson Ralf Dressel
Ralf Dressel- 1Hematological Sciences, Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, UK
- 2Institute of Cellular and Molecular Immunology, University Medical Centre Göttingen, Göttingen, Germany
The outcome of hematopoietic stem cell transplantation (HSCT) is controlled by genetic factors among which the leukocyte antigen human leukocyte antigen (HLA) matching is most important. In addition, minor histocompatibility antigens and non-HLA gene polymorphisms in genes controlling immune responses are known to contribute to the risks associated with HSCT. Besides single-nucleotide polymorphisms (SNPs) in protein coding genes, SNPs in regulatory elements such as microRNAs (miRNAs) contribute to these genetic risks. However, genetic risks require for their realization the expression of the respective gene or miRNA. Thus, gene and miRNA expression studies may help to identify genes and SNPs that indeed affect the outcome of HSCT. In this review, we summarize gene expression profiling studies that were performed in recent years in both patients and animal models to identify genes regulated during HSCT. We discuss SNP–mRNA–miRNA regulatory networks and their contribution to the risks associated with HSCT in specific examples, including forkheadbox protein 3 and regulatory T cells, the role of the miR-155 and miR-146a regulatory network for graft-versus-host disease, and the function of MICA and its receptor NKG2D for the outcome of HSCT. These examples demonstrate how SNPs affect expression or function of proteins that modulate the alloimmune response and influence the outcome of HSCT. Specific miRNAs targeting these genes and directly affecting expression of mRNAs are identified. It might be valuable in the future to determine SNPs and to analyze miRNA and mRNA expression in parallel in cohorts of HSCT patients to further elucidate genetic risks of HSCT.
Introduction
A considerable proportion of the risk of adverse outcome after hematopoietic stem cell transplantation (HSCT) is genetically determined and can be attributed to various factors including human leukocyte antigen (HLA) matching, killer-immunoglobulin-like receptor matching, minor histocompatibility antigens (miHAg), and non-HLA gene polymorphisms. Outcomes such as acute and chronic graft-versus-host disease (aGvHD and cGvHD), relapse, and survival have been shown to be modified by functionally relevant polymorphisms in non-HLA genes that are involved in immune responses (1, 2). Such functional polymorphisms are complicated to pinpoint among other polymorphisms localized near these genes that have no direct effects on gene function. Reliable identification of polymorphisms that result in differences in gene expression or protein function and affect the outcome of HSCT is challenging in view of the complexity of the human genome (3).
The most frequent genetic variations of the human genome are single-nucleotide polymorphisms (SNPs), which occur on average in 1 out of 300 bp throughout the genome (4–6). The majority of the SNPs arise in non-coding regions including intronic, intergenic, and untranslated regions (UTRs) (7). Those which are within genes, including genes affecting the immune response, may alter the expression of the gene or the structure of the encoded proteins (8). In microRNAs (miRNAs), SNPs can alter regulatory properties, but elucidation of the functions of these SNPs is not straight forward (9). Understanding the biogenesis of miRNAs is key to comprehending the impact of SNPs on these molecules (Figure 1). The miRNAs are a class of small endogenous non-coding RNAs of 21–25 nucleotides in length that originate as primary transcripts (pri-miRNAs) from miRNA genes. After transcription of pri-miRNAs by RNA polymerase II, they are processed by DROSHA, a RNA specific ribonuclease enzyme complex, producing short precursor-miRNAs (pre-miRNAs) of approximately 70 nucleotides length (10). The pre-miRNAs are then transported from the nucleus to the cytoplasm by exportin 5 (10). In the cytoplasm, they undergo further cleavage by an endonuclease enzyme (DICER), resulting in the generation of mature miRNA (11, 12). Accordingly, functionally relevant SNPs can be present in miRNA biogenesis-related genes, in specific miRNA-encoding genomic loci or in the seed match sequence of target mRNA 3′ UTRs. SNPs may lead to either an alteration in miRNA expression level, a decreased or increased miRNA-target interaction, or a new miRNA-target interaction (13). Atarod and Dickinson (14) described the driving gears of GvHD as miRNA’s regulating gene expression, chemokine and cytokine secretion, while their expression in turn is affected by SNPs in mRNA genes (Figure 2).
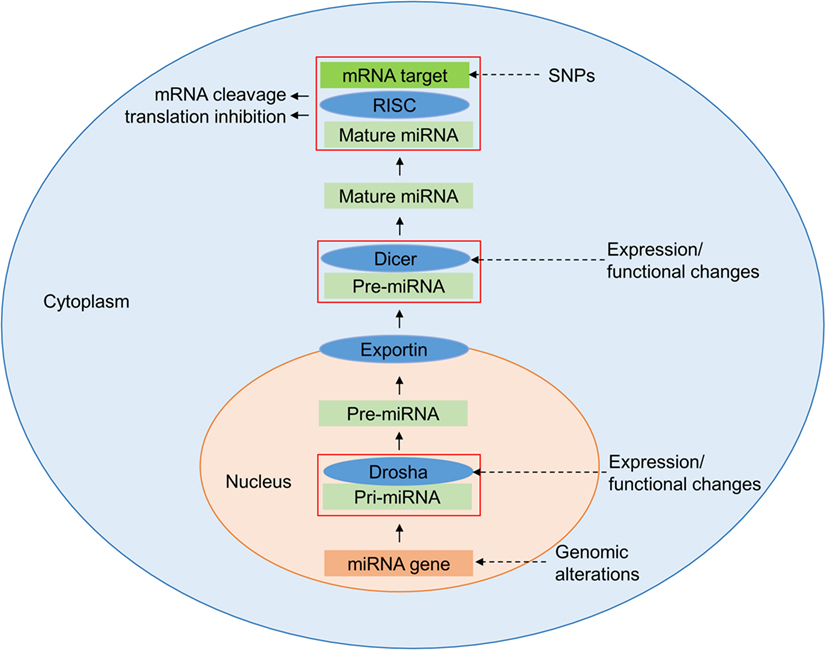
Figure 1. Regulation of microRNAs (miRNAs). Expression of miRNAs can be altered at various stages of its biogenesis by genomic [single-nucleotide polymorphisms (SNPs) and mutations] and epigenetic alterations. Changes in the expression and function of Drosha and Dicer, part of the miRNA processing machinery, lead to the deregulation of mature miRNAs. The figure has been adapted from Ref. (14, 15).
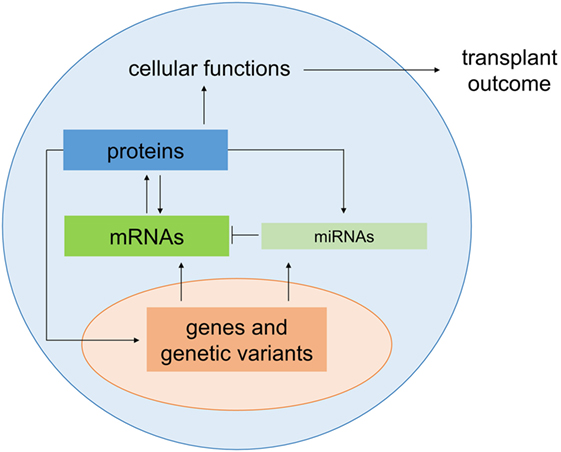
Figure 2. Genetic regulation of transplant outcome. Genetic variants in protein encoding genes and microRNAs (miRNAs) alter gene expression as well as protein and cellular functions which in turn contribute to the regulating the outcome of transplantation. The color code introduced here is used for genes, miRNAs, mRNAs, and proteins in Figures 3–5.
In this review, we will pinpoint SNP–mRNA–miRNA regulatory network alterations and their contribution to the risks associated with HSCT in specific examples, elucidating the consequences of the interaction between these three genetic elements. Moreover, we will summarize mRNA and miRNA profiling studies aiming to decipher genetic risks of HSCT.
Examples of SNP–mRNA–miRNA Regulatory Networks Controlling Outcome of HSCT
Forkheadbox Protein 3 (FOXP3) Polymorphisms, mRNA Expression, and FOXP3-Regulating miRNAs
Regulatory T cells (Tregs) have been the focus of several HSCT studies due to their ability to suppress alloreactivity during GvHD (16). Tregs, defined as CD4+CD25+ FOXP3+ T cells, are involved in the maintenance of immunological tolerance (17). They reduce the invasion of CD8+ effector T cells (Teff) in target tissue and ameliorate GvH tissue damage (18). Cuzzola and colleagues found an increased expression of FOXP3 mRNA in patients who were responsive to anti-GvHD therapies (19). These results are in concordance with previous data reporting an inverse correlation between the amount of Tregs and progression of aGvHD (20). Other studies showed correlation between a lower incidence of aGvHD and improved survival in HSCT recipients with an increased number of donor Tregs (21, 22). Low numbers of Tregs have also been associated with higher cGvHD incidence (23). Similarly, the severity of aGvHD and extent of cGvHD in patients were found to be associated with Treg numbers (24). Furthermore, inducing selective expansion of Tregs by the daily administration of low doses of interleukin (IL)-2 showed an improvement in clinical cGvHD symptoms in patients (25). Notably, not only CD4+ Tregs can mitigate GvHD but also CD8+FOXP3+ Tregs become induced during GvHD and can suppress the disease in mouse models (26, 27). CD8+ Tregs might have even advantageous over CD4+ Tregs since they have been reported not to abrogate graft-versus-leukemia (GvL) effects (28). The potency of CD8+ Tregs cells is further emphasized by their ability to prevent the rejection of heart allografts in rats (29).
Currently, 90 SNPs have been identified in the FOXP3 gene region, and several have been identified as risk factors for a number of malignant and autoimmune diseases (30). An SNP (rs3761548) in the promoter region of FOXP3 (Figure 3) resulting in an A/C base exchange causes loss of binding to the E47 and c-Myb factors and leads to defective transcription of the FOXP3 gene (31). In patients undergoing HSCT, this SNP has been associated with a higher incidence of hepatic veno-occlusive disease and cytomegalovirus (CMV) infection but a lower treatment-related mortality, resulting in a difference in the overall survival of patients with the CC genotype (32). However, the authors found no difference in the incidence of GvHD, relapse, or blood stream infection to be associated with this polymorphism (32).
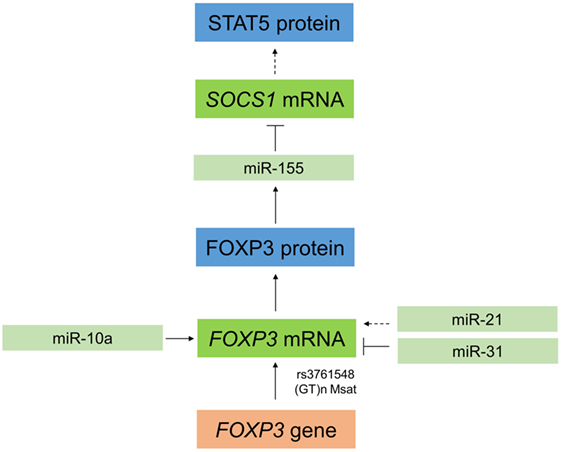
Figure 3. Interaction between microRNAs and forkheadbox protein 3 (FOXP3) in regulatory T cells (Tregs). In Tregs, miR-10a stabilizes FOXP3, and FOXP3 can positively regulate expression of miR-155. This leads to a downregulation of the target SOCS1 (⊤), which in turn results indirectly in increased expression of STAT5. FOXP3 can also be regulated by miR-21, which indirectly positively regulates FOXP3 in a process that is not yet completely understood. Moreover, FOXP3 is downregulated by miR-31 by direct targeting of the 3′ untranslated region. To further complicate this regulatory network, single-nucleotide polymorphisms (SNPs) in FOXP3 also affect its expression in Tregs, such as the SNP rs3761548 or a GT(n) microsatellites (Msat).
Posttransplant chimerism analysis in clinical HSCT largely uses polymorphisms of short tandem repeats of <10 nucleotides, or microsatellites (Msat). They have a higher degree of allelic polymorphism compared to SNPs, and therefore, a larger degree of information (33). An Msat studied in FOXP3 is the (GT)n polymorphism in the promoter/enhancer region of the FOXP3 gene (34). This polymorphism was shown to be associated with the development of auto- or alloimmune conditions, including type I diabetes, and graft rejection in renal transplant recipients (35). Moreover, this polymorphism has been associated with a lower incidence of grade III–IV GvHD in patients transplanted from donors carrying short alleles [≤(GT)15] (35). However, this polymorphism did not affect relapse, event free survival or overall survival in patients with aGvHD and cGvHD (35).
Recently, several miRNAs including miR-155 and miR-10a have been identified that impact T cell differentiation and function (36, 37). MiR-155 is required for Treg development (38) and for maintaining Treg homeostasis and survival by targeting SOCS1 (39). MiR-155 is an important positive regulator of natural Treg (nTreg) development and miR-155 gene transcription is driven by FOXP3 (Figure 3) (38). In mice, miR-155 is upregulated in mature Tregs (CD4+CD25+FOXP3+) relative to conventional T cells (CD4+CD25−FOXP3−) as well as in FOXP3+ double positive and single positive thymocytes (37, 40). MiR-155 knockout mice have reduced Treg numbers in both the thymus and periphery, and miR-155-deficient Tregs have a reduced proliferative potential and impaired IL-2 signaling (39). In this context, miR-155 promotes Treg survival and proliferation in the thymus and periphery by enhancing their sensitivity to IL-2 (39). As shown in Figure 3, miR-155 achieves this by targeting and downregulating SOCS1, an inhibitor of IL-2 signaling, thus increasing levels of activated STAT5 and enhancing IL-2 signaling (40). MiR-10a is functionally linked to stabilization of FOXP3 in Tregs (41) (Figure 3) and interestingly, although miR-10a has not been specifically investigated in relation to HSCT, an inverse correlation between miR-10a and susceptibility to autoimmune disease has been identified (41). With regard to miR-10a, it is uniquely expressed in Tregs, but not other T cells, where it is crucial for long-term maintenance of their stability and function (41). FOXP3 itself can be regulated by other miRNAs, including miR-21 and miR-31, which positively and negatively regulate FOXP3, respectively, thus having opposing effects on its expression (36) (Figure 3). MiR-31 can directly target FOXP3 by binding to a specific recognition site in the 3′ UTR region, while miR-21 regulation is believed to be indirect as no potential target sequence in FOXP3 was identified (36). The specific function of miR-21 and miR-31 in Tregs in the setting of HSCT is still to be explored.
The miR-155 and miR-146a Regulatory Network
Notably, miR-155 is involved in a larger regulatory network that affects the outcome of HSCT (Figure 4). Although miR-155 promotes the development of Tregs as explained above, it may also have pro-inflammatory functions. MiR-155 and miR-146a were found to be upregulated in the skin of rats suffering from aGVHD (42). Atarod and colleagues showed that low expression levels of both miR-155 and miR-146a were associated with higher incidence of aGVHD at day 28 post-allo-HSCT in patients and that both regulate expression of the transcription factor SPI1 (43). Pontoppidan and colleagues showed that miR-155 was increased in patient plasma at the time of maximal toxicity of preconditioning at day 7 posttransplantation and remained increased until day 21. This was inversely mirrored by miR-146a, which was significantly reduced from day 7 to day 21 after transplantation (44). Together, this suggests that miR-155 and miR-146a play opposite roles having pro-inflammatory and anti-inflammatory properties, respectively, and thus play a role in regulating the systemic inflammatory response during maximum toxicity of preconditioning in HSCT patients (44). Relevantly, miR-155 expression was upregulated in donor T cells in mice during aGvHD and mice receiving miR-155-deficient splenocytes developed less severe aGvHD and had increased survival rates compared to mice receiving wild type splenocytes (45). Specific targeting of miR-155 using antagomirs effectively mitigated aGvHD in mice and increased survival rates (45). MiR-155-deficiency in the dendritic cell (DC) compartment also protected mice from aGvHD since miR-155 appears to promote the migration of DC toward sites of tissue damage (46). MiR-155 expression was increased in mouse T cells as well as in intestinal patient biopsies during aGvHD (45). Expression of miR-155 can be stimulated by tumor necrosis factor (TNF) α (47), and similarly, miR-155 can promote TNF-α production in a positive feedback loop (48), thus exacerbating the inflammatory cascade (Figure 4). Altogether, these data indicate a role for miR-155 in the modulation of aGvHD.
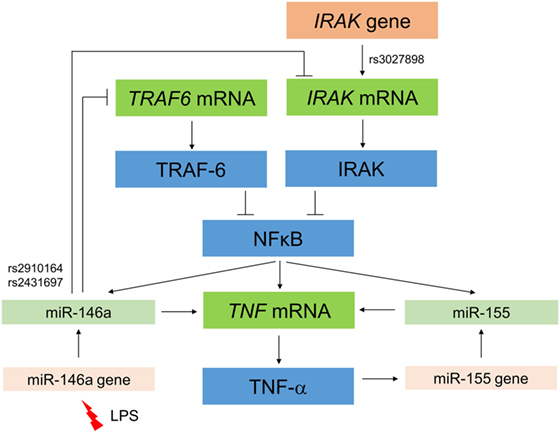
Figure 4. Interaction between miR-146 and miR-155, their effects on the nuclear factor (NF)-kB pathway, and the expression of IRAK1 and tumor necrosis factor (TNF)-α. Activation of the NF-kB pathway represents a hallmark of the pathophysiology of GvHD. NF-kB activation induces expression of miR-146a and in turn, miR-146a inhibits these pathways through targeting key adapter proteins, IRAK1(⊥) and TRAF6 (⊥). The presence of single-nucleotide polymorphisms in coding regions of these genes, such as rs3027898 in IRAK1, further influences expression within the network. MiR-146a expression can also be stimulated by lipopolysaccharide (LPS) release during GvHD conditioning. The miR-146a and miR-155 mediate an increase in TNF-α, which in turn can positively regulate miR-155 in a feedback loop.
MiR-146a is distinctly increased in response to lipopolysaccharide (LPS), a component of the cell wall of gram-negative bacteria. LPS is released in response to GvHD conditioning regimens (Figure 4) and acts as a potent enhancer of cytokine secretion (49). Using a genetically engineered mouse model, it was demonstrated that a deletion of miR-146a results in several immune pathologies (50). Specifically, lack of miR-146a expression increased responsiveness of macrophages to LPS and exacerbated the inflammatory response in LPS-challenged mice. TNF receptor-associated factor 6 (Traf6) and IL-1 receptor-associated kinase 1 (Irak1) genes have also been identified as targets of miR-146a (Figure 4), contributing to the phenotype of miR-146a-deficient mice (50). Both TRAF6 and IRAK1 act as adapter proteins in the nuclear factor (NF)-κB activation pathway and in addition to innate immune cells, miR-146a has also been shown to target these genes in T cells resulting in their downregulation (Figure 4). T cells that are lacking miR-146a are hyperactive in both acute antigenic responses and chronic inflammatory autoimmune responses (51). However, the presence of SNPs in these genes that may affect miR-146a binding, such as rs3027898 in the IRAK1 3′UTR may further complicate this already complex network of interactions. Furthermore, activation of NF-κB has been described to upregulate miR-146a expression, which in turn downregulated NF-κB via TRAF6 and IRAK1 repression in a negative feedback loop (52, 53) (Figure 4). More recently, miR-146a regulation of TRAF6 and IRAK1 was specifically associated with aGvHD, since upregulation of miR-146a expression was observed in T cells of mice developing aGvHD compared to untreated mice (52). When transplanted with miR-146a-deficient T cells, recipient mice developed GvHD of increased severity resulting in reduced survival, and they also had elevated TNF-α serum levels (52). The protein levels of TRAF6 were upregulated in miR-146a-deficient T cells following alloantigen stimulation, and this translated into increased NF-κB activity. Conversely, miR-146a overexpression reduced aGvHD severity. In an autoimmune setting, miR-146a expression can be induced by TNF-α (47), which in turn is stimulated by NF-κB, thus further confounding this complex regulatory network (Figure 4). Interestingly, another member of the miR-146 family, i.e., miR-146b, is highly expressed in CD4+CD25+FOXP3+ thymic-derived Tregs and has been reported to promote survival, proliferation, and suppressor function of these cells by targeting TRAF6 and subsequently increasing NF-κB activity (54).
Single-nucleotide polymorphisms within miRNA coding regions as well as those within their target mRNA seed regions can directly influence miRNA–mRNA interactions. Indeed, with regard to miR-146a, two SNPs rs2431697 and rs2910164 have been reported that cause single base changes and altered expression of the mature microRNA (Figure 4). The SNP rs2910164 specifically results in a change from a G:U pair to a C:U mismatch in the stem structure of the miR-146a precursor. This results in processing variation and lower expression of the mature miRNA, which has been associated with the development of a range of cancers (55). Stickel and colleagues reported that the minor CC genotype caused a decrease in miR-146a production (52). The same team also provided evidence that miR-146a acts as an important negative regulator in murine and human GVHD, consistent with an anti-inflammatory role for miR-146a, and suggested the exogenous increase of miR-146a as a potential novel strategy for therapeutic intervention in this disease (52). Further interactions that have been described between miRNAs and the induction of GvHD have been recently reviewed by Atarod and Dickinson (14).
MICA Polymorphisms, mRNA Expression, and MICA-Regulating miRNAs
The major histocompatibility complex (MHC) class I chain-related molecule A (MICA) is a highly polymorphic ligand for the activating natural killer (NK) cell receptor NKG2D (Figure 5). An SNP within this gene, rs1051792, which leads to an amino acid exchange from valine to methionine at position 129 (56), was investigated for its association with the outcome of HSCT. We found that the MICA-129Met variant was associated with an increased overall survival and a reduced risk to die from aGvHD, despite homozygous carriers of the MICA-129Val allele having an increased risk of developing aGvHD (57). The NKG2D pathway was expected to be directly related to the outcome of HSCT, since it is an activating receptor on NK cells (58) and a costimulatory receptor on CD8+ T cells (59). On the functional level, we found the MICA-129Met isoform triggered more cytotoxicity and interferon (IFN)-γ release by NK cells and it activated alloreactive cytotoxic T cells faster. This variant also induced more rapid and severe downregulation of NKG2D on NK and cytotoxic T cells (57). Normally, most cell types do not express MICA, but it becomes induced by cellular and genotoxic stress, including virus infection and malignant transformation. Therefore, it renders stressed cells susceptible to killing by NK cells and allows them, despite being non-professional antigen presenting cells (APCs), to directly activate cytotoxic T cells specific for antigens presented by these cells. Notably, MICA expression was found to be increased in GvHD-affected tissue samples from patients (60). The MICA-129Met variant can therefore initially confer a higher risk of aGvHD due to a faster activation of alloreactive cytotoxic T cells (57). However, in the longer perspective, the strong-counter regulation of NKG2D by this variant appears to be associated with a decreased risk of cGvHD and an increased risk of relapse due to lesser GvL effects by cytotoxic T cells and NK cells (61).
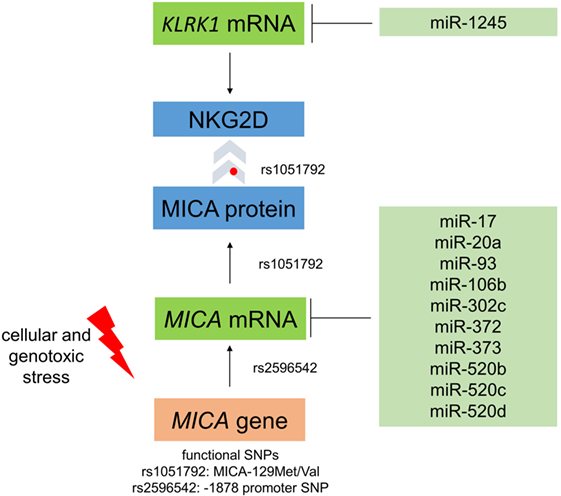
Figure 5. Regulation of MICA expression and interaction with NKG2D. The single-nucleotide polymorphism (SNP) rs1051792 results in a valine to a methionine exchange at position 129 of MICA and distinguishes MICA variants into those binding the receptor NKG2D with high (MICA-129Met) or low (MICA-129Val) avidity. This polymorphism also affects the cell surface expression of MICA protein. The SNP rs2596542 in the promoter of MICA affects mRNA expression. Moreover, several microRNAs target MICA and downregulate its expression. Moreover, cellular and genotoxic stress induces the expression of MICA. The MICA receptor NKG2D is encoded by the KLRK1 gene and can be targeted by miR-1245.
Interestingly, the biological effects of the MICA-129 variants were strongly influenced by MICA expression intensity (57). The MICA-129Met variant triggered increased NKG2D signals at low expression intensities, whereas the MICA-129Val variant elicited more NKG2D-mediated effects at high expression intensities. At high expression intensity, the functional effects of the MICA-129Met variant were impaired due to a rapid downregulation of NKG2D (57). Thus, MICA expression intensity could change the biological effect of this SNP, giving an interesting example of the complex functional interactions between SNPs and gene expression (Figure 5). Moreover, the SNP might interact with other SNPs in the NKG2D signaling pathway (62) including KLRK1, encoding NKG2D. Polymorphisms in the KLRK1 locus have been described also to affect the outcome of HSCT (63).
Notably, MICA expression intensities can vary for certain MICA alleles (64). The SNP at −1878 (rs2596542) in the promoter region of the MICA gene was described to affect the transcriptional activity (65). A polymorphic microsatellite in exon 5 encoding the transmembrane region of MICA modifies its plasma membrane expression (66). We have recently shown that the MICA-129Met/Val dimorphism also affects plasma membrane expression. Increased levels of the MICA-129Met variant were retained intracellularly and if expressed at the cell surface, the MICA-129Met variant was more prone to shedding than the MICA-129Val isoform (67).
Matching of donor and recipient for MICA alleles (68–71) and specifically for the MICA-129, polymorphism (72) has been found to be beneficial in HSCT, although not in all studies (73, 74). The effect of MICA matching appears hardly explainable solely by the avoidance of potential miHAg and further points toward an important biological function of MICA after HSCT.
Several stress pathways regulate the transcription of the MICA gene (75), and several miRNAs have been implicated in controlling MICA expression via posttranscriptional mechanisms (Figure 5). Stern-Ginossar and colleagues described that the expression of MICA was decreased by miR-17, miR-20a, miR-93, miR-106b, miR-372, miR-373, and miR-520d (76). Effects of miR-17 (77), miR-20a (77–80), miR-93 (77, 80–82), and miR-106b (80, 81) on MICA expression have also been confirmed in subsequent studies. Moreover, the IFN-γ-induced miR-520b can lead to a reduction in MICA plasma membrane expression intensity (83). Interestingly, miR-520d acts on both the MICA 3′-UTR and the promoter region to decrease MICA transcript levels (83). MiR-302c and miR-520c are two further miRNAs that can target MICA (84). Human CMV can also target MICA by US18 and US20, which promote the degradation of MICA in lysosomes (85). The regulation of NKG2D ligands by miRNAs has been recently reviewed in more detail (86, 87). Notably, also the expression of NKG2D has been found to be attenuated by miRNAs, specifically, miR-1245 (88).
miRNA and mRNA Expression Profiling in HSCT
A number of large-scale gene expression profiling studies have been performed in both patients and animal models to identify genes regulated during HSCT. In animals, both acute and chronic GvHD models were investigated. Studies performed on human samples mostly used blood or in cases of cGvHD, conjunctiva from patients. The advantage of using animal models for gene expression is the broad availability of specific target tissues of GvHD, such as liver and skin (89). A selection of the most relevant gene expression profiling studies is listed in Table 1. MiRNAs have also been studied in relation to HSCT, and there is increasing evidence to show that miRNAs are present in plasma, serum, saliva, urine, and other body fluids in a remarkably stable form that is protected from endogenous RNase activity (90). Circulating miRNAs have the potential to serve as novel and non-invasive biomarkers for various diseases such as cancer, cardiovascular disease, and organ transplant rejection and infection (91).
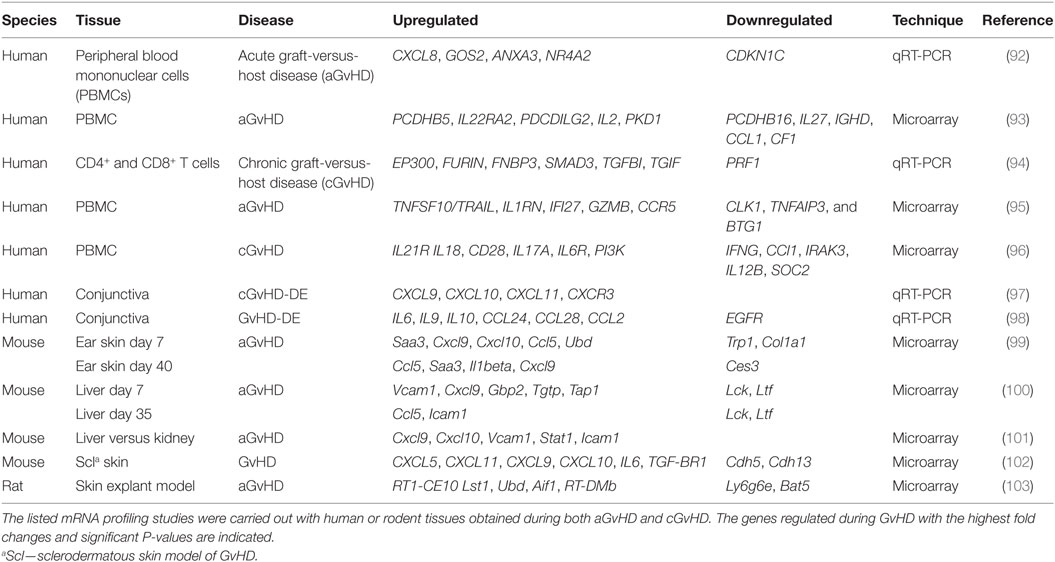
Table 1. Summary of large-scale mRNA expression profiling studies during GvHD.
miRNAs Targeting Immune-Related Genes and Their Application on Prediction of HSCT Outcome
In a recent clinical study, a micro-RNA-based model was developed to predict the probability of aGvHD comprising miR-423, miR-199a-3p, miR-93 and miR-377. Elevated levels of these miRNAs were detected in plasma before the onset of aGvHD (average 16 days before diagnosis), and their expression was associated with the severity of aGvHD as well as with a lower overall survival (91). In another profiling study of 48 miRNAs in the plasma of aGvHD patients, miR-586 expression was decreased upon the occurrence of aGvHD (104).
Interestingly, a recent publication by Wu and colleagues (105), which has been reviewed by Serody (106), has been shown that the miR-17–92 cluster, which is conserved among vertebrates, is important for the development of aGvHD. The miR-17–92 cluster is critical for the proliferation, survival and function of Th1 and Th17 effector T cells and inhibits Th2 and Treg differentiation (107). It has now also been shown that this cluster of miRNAs promotes the migration of CD8+ T cells to GvHD target organs and confers GvL effects (105). Donor T cells lacking the miR-17–92 complex gave rise to diminished GvHD in a mouse model of aGvHD. Blocking of miR-17 and miR-19b, which are included in the miR-17–92 complex, by systemic administration of antagomiRs (locked nucleic acid-modified oligonucleotides) significantly reduced aGvHD severity (105).
To better understand the regulation of neovascularization during GvHD, Leonhardt and colleagues focused on the role of miR-100 and showed that in intestinal tissue biopsies from patients undergoing allo-HSCT, miR-100 was downregulated when aGvHD evolved suggesting that this miRNA has a role as a negative regulator of aGvHD (108). MiR-100 was also downregulated in the inflamed intestinal tissue of mice developing aGvHD. In the mouse model, functional inactivation of miR-100 with antagomiRs enhanced aGvHD severity, indicating a protective role for miR-100 by blocking inflammatory neovascularization during aGvHD (108).
Other studies focusing on miR-34 showed that the miR-34 family mimics p53 effects by inducing cell cycle arrest and apoptosis in response to DNA damage (109). In the case of Fanconi anemia (FA), an inherited disorder characterized by developmental defects, genomic instability and progressive bone marrow failure (110), miR-34a expression in patient gut biopsies after HSCT was significantly higher in aGvHD grades II–IV compared to grade 0–I or with non-transplanted FA patient gut biopsies (111).
Association between Polymorphisms and Gene Expression Levels in Cytokine and Chemokine-Coding Genes with the Outcome of HSCT
Cytokine Gene Expression Variation and Its Impact on the Outcome of HSCT
Cytokines, such as INF-γ, IL-2, and TNF-α produced by Th1 cells, contribute to the induction phase of aGvHD (112). A number of polymorphisms in genes encoding IL-10, TNF-α, and IL-6 have been linked to an increased risk of GvHD. IL-2 has a role as a T cell growth factor and treatment, and prophylaxis of aGvHD involves frequently inhibition of IL-2 production by cyclosporine A (113). Moreover, in both animal and human studies, administration of monoclonal antibodies against the IL-2 receptor after HSCT have prevented aGvHD occurrence (114, 115). On the other hand, emerging data show that IL-2 is also necessary for the generation and the maintenance of CD4+CD25+Foxp3+ Tregs, so that inhibiting IL-2 may have a negative effect on the development of long-term tolerance after allo-HSCT (116, 117). Another cytokine of critical importance during aGvHD is IFN-γ, a pro-inflammatory cytokine, produced by several cell types such as activated T cells, NK, and NKT cells. IFN-γ and IL-2 both play a role in T cell proliferation, stimulation of cytotoxic T lymphocyte (CTL) and NK cell responses and production of IL-1 and TNF-α (118). A number of studies have reported a correlation between the expression of IFN-γ and severity of aGvHD (119–121). IFN-γ production occurs early in the cytokine cascade of GvHD. Acute GvHD is augmented by IFN-γ, which leads to the maturation of DC and stimulation of macrophages to generate cytokines and NO (118).
IFNG (IFN-γ) mRNA expression in the conjunctiva of patients was associated with dry-eye cGvHD (98) and in CD8+ T cells with cGvHD (122). Ichiba and colleagues studied the regulation of 7,329 genes in the hepatic tissue of mice on days 7 and 35, after allogeneic and syngeneic bone marrow transplantation (100). On day 7, 456 genes and on day 35, 554 genes were regulated. Interestingly, Ifng mRNA expression was not upregulated during hepatic GvHD on day 7 and no expression of Ifng was observed in the liver on day 35. However, the expression of many genes that are inducible by IFN-γ, such as interferon regulatory factor-1 (Irf1) and Irf7 were increased (100). Both IFNG and IL2 mRNA were increased in the peripheral blood mononuclear cells (PBMCs) of GvHD patients that received a donor lymphocyte infusion for the treatment of relapsed leukemia after allogeneic HSCT and IL2 mRNA expression correlated with the progression of GvHD (121). In another study, genes that contribute to control of inflammation, such as the IL-1 decoy receptor IL1R2, as well as pro-fibrotic genes have been found to be overexpressed in PBMCs of patients with cGvHD (123).
Gene expression of TNF, encoding TNF-α, a critical pro-inflammatory cytokine, was also elevated during aGvHD in PBMCs of patients (93). TNF-α is one of the most important factors involved in the pathogenesis of aGvHD, and it is important at various stages during the progression of the disease. The importance of this molecule in aGvHD was firstly described in a mouse model (124). Since then, several studies have shown that the neutralization of TNF-α can reduce symptoms of aGvHD (125). Tumor necrosis factor superfamily, member 10 (TNFSF10) mRNA expression was also elevated during aGvHD in human PBMCs (95).
Other cytokines regulated in GvHD include IL-15. In a murine aGvHD model, donor IL-15 was crucial for the development for aGvHD (126). Investigations on the role of IL-15 suggested that IL-15 could induce aGVHD by activating T cells and NK cells (127). In conjunctiva of patients with GvHD, IL15 mRNA was significantly increased (98). IL27 mRNA was strongly downregulated during aGvHD in PBMCs from patients (93). Previous reports indicate that IL-27 exhibits a pro-inflammatory response, is involved in activating Th1 cells, and enhances the immunological response to tumor cells (128). IL-35 is an inhibitory cytokine secreted by Tregs. The exact role of IL-35 in aGvHD is not known, although overexpression of IL-35 during murine aGvHD reduced its severity by suppressing activation of effector CD4+ T cells and expansion of CD4+Foxp3+ Tregs in target organs of aGvHD, while preserving a GvL effect (129). IL-35 could be a potential therapeutic target for prevention of aGvHD (129, 130).
SNPs in Cytokine Coding Genes and Association with HSCT Outcome
Given the dysregulation of many cytokines during acute and chronic GvHD, it is not surprising that SNPs in these genes have been associated with the outcome of HSCT. SNP association studies in HSCT have been recently reviewed by Dickinson and Norden (8). Since the original work by Middleton and colleagues (131), large cohort candidate gene association studies have been reported on SNPs in more than 20 genes that code for cytokines and other molecules involved in the biology of HSCT (132–135). Moreover, SNPs originally identified in NOD2 for their association with Crohn’s disease have since been associated with HSCT outcomes (136, 137). Individuals carrying just one variant of rs2066844 (SNP8), rs2066845 (SNP12), or rs41450053 (SNP13) have a twofold to fourfold increased risk of developing Crohn’s disease, which increases to approximately 20-fold in individuals who are homozygotes or compound heterozygotes (138, 139). NOD2 is mainly involved in defense against infection; it recognizes pathogen-associated patterns and induces a cytokine response, and is itself regulated by pro-inflammatory cytokines (140).
Goussetis and colleagues retrospectively analyzed specific polymorphisms in genes for IL-10, IL-6, TNF-α, and IFN-γ in a pediatric cohort of 57 HLA-identical sibling myeloablative transplants and found a significant association between the IL10 promoter haplotype polymorphisms at positions −1082, −819, and −592 with the occurrence of severe aGVHD (grades III–IV). Recipients with the haplotype GCC had a decreased risk of severe aGVHD in comparison with patients with other IL10 haplotypes (141). Chien and colleagues identified two SNPs in IL10, such as rs1800871 and rs1800872, which were associated with a 30% decrease of the risk for grade III–IV aGVHD (139). Moreover, the donor allele C for rs1800795 in IL6 was associated with a 20–50% increase in the risk for grade II–IV aGVHD, and the IL2 polymorphism rs2069762 in the donor genotype was associated with a 1.3-fold increase in risk of grade III–IV aGVHD (139).
Chemokine Gene Expression Variation and Its Impact on the Outcome of HSCT
Many genes encoding chemokines are regulated during GvHD. CXCR3 is an important chemokine receptor involved in lymphocyte recruitment and is expressed on T cells. CXCL9, CXCL10, and CXCL11, the ligands for CXCR3, are induced by the Th1 cytokines IFN-γ and TNF-α (142). CXCL9 is expressed by effector CD4+ Th1 cells and CD8+ CTL and has been shown to affect the migration of Teff to inflamed tissue during progression of GvHD (142). CXCL10 and CXCL11 mRNA expression were increased in patient skin biopsies with aGvHD (grades II–III) when compared to patients without or grade I GvHD (143). The mRNA expression of CXCL9 and CXCL10, along with their receptor CXCR3, was increased in cGvHD in conjunctival biopsies of 10 patients when compared to 10 healthy controls (97). Elevated mRNA expression of the CXCR3 ligands, CXCL9 and CXCL10 in target organs of GvHD, shows that CXCR3 could have a role in GvHD (144). CXCL8, encoding IL-8, was upregulated in PBMC from patients who developed aGVHD (92). Moreover, Cxcl9 (101) and Cxcl10 were also elevated during murine aGvHD (100). Interestingly, the use of CXCR3-transfected Tregs, as a novel therapeutic strategy, resulted in decreased severity of GvHD due to attraction of Tregs to the target tissues of GvHD (145).
Other chemokines involved in the stimulation and activation of T cells in lymphoid tissue (Cxcl1, Cxcl2, Cxcl9, and Cxcl20, Ccl2, Ccl5, Ccl6, Ccl7, Ccl8, Ccl9, Ccl11, and Ccl29) and chemokine receptors (Ccr1 and Ccr5) were elevated in the skin of mice during acute GvHD (99). The chemokines CCL2, CCL3, CCL4, and CCL5 are involved in the migration of donor cells to the target organs during GvHD (146). CCR5 mRNA was also increased during in aGvHD human PBMCs (95) and in murine skin during GvHD (102). In addition, Ccl2, Ccl5, Ccl17, Cxcl9 (Mig), Cxcl10 (IP-10), and Cxcl11 (1-TAC) mRNAs were also significantly regulated in mouse skin during GvHD (102). In conjunctival biopsies of patients with GvHD, the gene expression of CCL24, CCL18, and CCL2 was highly increased (98). Another chemokine mRNA, Ccl5 (RANTES), was elevated in the skin of mice during aGvHD (99) and profoundly upregulated during hepatic aGvHD (100).
Differential Expression of Genes Involved in Antigen Processing and Presentation during GvHD
The role of MHC molecules is of critical importance to the development of GvHD. Both class I (HLA-A, B, and C in human) and class II (HLA-DR, DQ, and DP in human) determine not only the histocompatibility but are also responsible for controlling T cell recognition (147). Expression of class II HLA molecules by professional APCs, mainly in the gastrointestinal tract epithelium and skin, allows CD4+ T cells to recognize foreign antigens, possibly contributing to the specific organ sites of aGvHD (148). During the afferent phase of the pathogenesis of aGvHD, the release of cytokines such as IFN-γ leads to an increased expression of MHC molecules. On day 7 of hepatic aGvHD in mice, MHC class II genes, including I-Aα, I-Aβ, I-Eα, and I-Eβ, were overexpressed and remained upregulated at day 35 of aGvHD. On the other hand, the expression of the MHC class I genes was not regulated in this study. However, the genes that encode alternative proteasome subunits and that alternate peptide production associated with MHC class I molecules proteasome subunit beta 9 (Psbm9) and Psbm8 (also known as lower molecular mass peptides LMP2 and LMP7) were increased in mouse liver during aGvHD (100). Moreover, Tap1 and Tap2 mRNAs were increased in mouse skin (99) as well as in liver during aGvHD (100). TAP1 and TAP2 are transporters associated with antigen processing 1 and 2, responsible for translocating peptides into the endoplasmic reticulum before loading on MHC class I molecules. In a rat skin explant model, an increase in expression of Tap1 as well as Psbm8 and Ubd mRNA during graft-versus-host reaction was observed (103). UBD, also known as FAT10, is involved in the proteasomal degradation of cytosolic proteins by providing a ubiquitin-independent signal (149). The differential expression of the MHC I and II genes in addition to genes involved in antigen processing that have been observed during GvHD is in agreement with the important role of MHC genes for HSCT outcomes.
Involvement of the miHAg in Immune Responses during HSCT
Mismatches of polymorphic peptides between donor and recipients cause miHag that can also elicit an alloimmune response (150). The extent of the desired GvL versus the unwanted GvHD responses is dependent on the expression profiles of these genes. In humans, miHag are mostly restricted by HLA class I molecules. Previously, mismatches for HA-1, HA-2, and HA-5 between donor and recipient have been described to be associated with an increased risk of GvHD (151). In a gene expression profiling study to identify the risk of GvHD and relapse posttransplant, the mRNA expression of miHag was assessed in 311 HLA-matched siblings from a single center (152). The HA-8 gene was expressed in almost all tissues, whereas ACC-1 gene had a restricted profile. Nonetheless, both HA-8 and ACC-1 miHag mismatches were found to be associated with occurrence of cGvHD (152).
Notably, whole exome sequencing studies have been performed recently to estimate the alloreactive potential between donors and recipients in HSCT. It has been found that non-synonymous and non-conservative SNPs were twice as frequent in HLA-matched unrelated compared to related donor–recipient pairs (153). The information on SNPs between donor and recipient can be used to predict candidate miHags by algorithms taking peptide binding to HLA class I molecules and the tissue distribution of the respective proteins into account (154). Modeling of T cell responses to these miHags potentially can help to identify more favorable donors or to adapt the immunosuppressive treatment after HSCT (155, 156).
Gene Expression Patterns in T Cells Associated with the Outcome of HSCT
Baron and colleagues compared the gene expression profiles of 50 allo-HSCT donors in CD4+ and CD8+ T cells to identify donors that are stronger allo-responders and could elicit a stronger GvHD response than others could. They suggested genes that regulate the transforming growth factor-β signaling and cell proliferation, in donor T cells, as the dangerous donor trait responsible for the occurrence of cGvHD in the corresponding recipients (94). Low levels of SMAD3 mRNA, which encodes a transcription factor that is activated in response to TFG-β in CD4+ T cells, was associated with the absence of GvHD, while high levels of SMAD3 were necessary but not sufficient for GvHD occurrence (94).
CD8+ T cells are important effectors in aGvHD (157), and perforin is a cytotoxic effector protease produced by CTL and NK cells. High expression of perforin 1 (PRF1) mRNA in CD8+ T cells was found to be associated with the incidence of GVHD in patients (94). In the skin of mice during aGvHD, granzyme B (Gzmb) was significantly elevated, in addition to the downstream effector caspase 7 (Casp7) (99). Other genes upregulated included the pro-apoptotic members of the BCL2 family, BCL2-antagonist/killer 1 (Bak1), BLC2-like 11 (Bcl2l11), and BCL2-associated X protein (Bax) (99). Thus, expression profiling can indicate ongoing pathophysiological processes contributing to GvHD, such as cellular cytotoxicity.
Notably, Sadeghi and colleagues observed an increase in gene expression of the adhesion molecules intracellular adhesion molecule 1 (Icam1) and vascular cell adhesion molecule 1 (Vcam1) during murine hepatic aGvHD. Increased expression of Vcam1 mRNA was also observed in the liver and kidney compared to the muscle during murine aGvHD, whereas Icam1 mRNA was upregulated only in the liver (101). Both adhesion molecules are expressed on endothelial cells and are critical for the migration of leukocytes to tissues during inflammation (158). Furthermore, Icam1 and Vcam1 genes were also upregulated in mouse skin during aGvHD, along with other adhesion molecules Cd18 or integrin subunit beta 2 (Itgb2), Ly69 or integrin beta 7 (Itgb7), and Psgl1 or selectin platelet ligand (Selplg) (99).
The expression of costimulatory molecules that have a role in regulating T cell activation, differentiation, and proliferation has been studied to determine their role in GvHD. CD28 and CD28/cytotoxic T lymphocyte antigen 4 (CTLA4) are the most well characterized costimulatory and inhibitory molecules, respectively (159). Both are present on T cells, while their ligands CD80 (B7-1) and CD86 (B7-2) are expressed primarily on APCs (160). CD28 mRNA was increased during cGvHD in PBMCs of patients (96). Interestingly, SNPs in CTLA4 could have an impact on its function. In patients, the presence of the A allele in both rs231775 and rs3087243 was associated with a reduced risk of aGvHD after HSCT (161). Another study showed that recipients with the +49A/G allele had a significantly lower disease-free survival and overall survival in comparison to recipients with the A/A genotype (162). In addition to the +49A/G polymorphism, −1722, −1661, −318 polymorphisms in CTLA4 were also evaluated after allo-HSCT, and a significant association between GA genotype (CTLA4 −1661) and GvHD was shown in males with GvHD compared to males without GvHD (163). In addition, inducible T cell costimulator (ICOS), a member of the CTLA4 family that is expressed on activated T cells, was shown to be associated with the occurrence of aGvHD (19). The exact role of ICOS in GvHD is not clear; however, ICOS mRNA was downregulated in aGvHD patients. In contrast, ICOS mRNA was elevated during cGvHD in activated T cells in canines (164). Furthermore, blockade of ICOS in vitro during mixed lymphocyte reaction (MLR) resulted in immunosuppression, suggesting that ICOS plays a role in graft rejection and blockade of ICOS could be a potential therapeutic strategy (164). Cuzzola and colleagues also observed an increase in mRNA expression of ICOS in patients responding to anti-GvHD therapies as well as other genes that are regulated by ICOS, including Th2 cytokines (IL4, STAT6, and IL18) (19).
In addition to gene and miRNA expression studies in T cells, the characterization of the T cell receptor (TCR) repertoire in patients who underwent HSCT might be informative to assess risks of GvHD or relapse. It has been reported recently that these complications were associated with a lower TCR repertoire and the expansion of certain T cell clones (165).
Gene Expression Profiles in B Cells Associated with Outcome of HSCT
B cells have been found to be important in contributing to cGvHD; however, the mechanisms involved in maintaining their activation are not known (166). Depletion of B cells reduced the incidence of cGvHD in mice (167). Elevated B cell-activating factor (BAFF), also known as tumor necrosis factor superfamily member 13b (TNFSF13B), mRNA levels were observed in patients with cGvHD and correlated to B cell activation (168). BAFF mRNA expression was also significantly upregulated in clinical GvHD patient biopsies in comparison to those with no GvHD (143). A differential pattern for gene expression for several genes was observed in the purified B cells from cGvHD patients on comparison with the B cells from healthy counterparts. Four of the genes, IL12A, interferon regulatory factor 4 (IRF4), CD40, and interferon gamma receptor 2 (IFNGR2), were downregulated whereas B cell linker (BLNK) mRNA was upregulated in B cells in patients with cGvHD (168). BLNK has been found to be important in proliferation and survival of B lymphocytes (169).
Genome-Wide Association Studies (GWAS) for HSCT Outcome
As a result of the Human Genome and the International HapMap Projects in the early 2000s (4, 5), GWAS became possible, expanding dramatically our capacity to understand genetic variability. A GWAS study of non-HLA SNPs in allogeneic HSCT was reported, which identified a number of SNP genotypes associated with severe aGvHD using a cohort of 1,298 patient donor pairs (139). The IL6 donor genotype for rs1800795 was confirmed to be associated an increased risk of severe aGvHD. In addition other genes associated with aGvHD, IL2, methylene tetrahydrofolate reductase (MTHFR), Heparanase (HPSE), and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) were identified in this GWAS cohort and illustrate (Figure 6) the fact that genomic control of immunoregulatory cytokines could alter the function of cells which in turn aid or reduce successful transplant outcome (170).
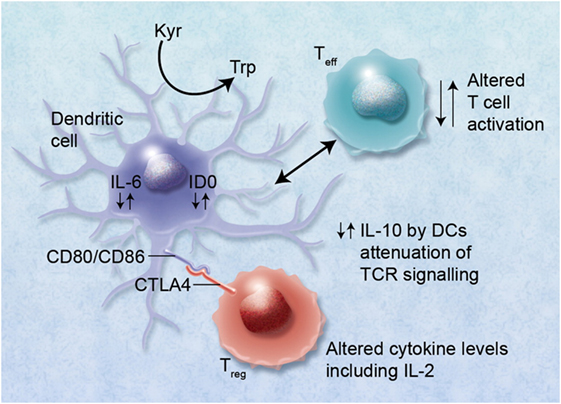
Figure 6. Alterations in cytokine levels, such as interleukin (IL)-10, IL-6, and IL-2, via immunoregulatory single-nucleotide polymorphisms (SNPs) can lead to altered immunoregulatory function of regulatory T cells (Tregs) and effector T cells (Teff). Binding of cytotoxic T lymphocyte antigen 4 (CTLA4) with its receptor (possibly also via functional single-nucleotide polymorphisms) with CD80/CD86 proteins on dendritic cells (DCs) can lead to induction of indoleamine 2,3 dioxygenase (IDO) and the catabolism of tryptophan into proapoptotic metabolites causing immunosuppression of Teff. Altered binding of CTLA4 may also lead to reduced immunosuppression via Tregs and GvHD. High IL-6 levels induced in DCs by Treg interaction can also cause alteration of Tregs to Th17 cells and may lead to exacerbation of GvHD. The figure has been taken from Ref. (170). Professional illustration by Alice Y. Chen.
The Ogawa group has performed GWAS study in large cohort involving 1,589 patients and donors to identify miHAg associated with aGvHD (171). They identified three new loci that were significantly associated with severe (grade II–IV) aGvHD including the SNP rs17473423 within the KRAS locus. In a further GWAS study on a smaller identification cohort of 68 patients and a validation cohort of 100 patients, two GvHD susceptibility loci (rs17114803 and rs17114808) within the “suppressor of fused homolog” (SUFU) gene have been found (172). The incidence of aGvHD was significantly higher in patients that were homozygous for CC at SUFU rs17114808, than in heterozygous patients. Functional studies showed that ectopic expression of SUFU in DCs reduced expression of HLA-DR and suppression of MLR, whereas an increased HLA-DR expression and enhanced MLR was observed on silencing of SUFU (172). In the future, GWAS studies in HSCT will require larger multicenter cohorts but are expected to reveal new genetic associations for HSCT outcomes (8, 147).
Problems with Gene Association Studies in HSCT
Genetic association studies have inherent difficulties when it comes to validation of results. Only robust genetic markers are able to be validated across HSCT cohorts. This is due to heterogeneity of transplants with regards to diagnosis, conditioning regimens, type of stem cells (e.g., peripheral blood, cord blood, or bone marrow), sibling or matched unrelated transplants and risk factors for outcome (CMV positivity; female to male donors, age of the transplant cohort) all of which alter the biology of the transplant itself. Problems associated with non-HLA genomics in HSCT have also been recently reviewed (8).
One example of this is within our own studies on SNP polymorphisms as risk factors for survival in chronic myeloid leukemia (CML). In the first study, presence of interleukin 1 receptor antagonist (IL1RN) allele 2 genotype in the donor (indicating downregulation of IL-1), absence of donor IL10 ATA/ACC genotype (indicating more downregulation of IL-10) and absence of tumor necrosis factor superfamily member 1B (TNFSF1B) 196R in the patient (indicating increased levels of soluble TNF-RII and decreased levels of TNF-α), all were associated with decreased survival and increased transplant relate mortality (173). In a validation cohort of matched unrelated transplants, none of the SNPs could be validated, and a comparison of the cohorts demonstrated differences in survival and clinical characteristics (174).
In addition, in a larger heterogeneous cohort, including CML and lymphoma (2), we developed a clinical and genetic score, which included the European Bone Marrow Transplantation (EBMT) Group score. This score incorporates clinical risk factors such as age of the patient and donor; time to transplant and type of transplant (175–178). Using a statistical analysis that included a bootstrap estimate of prediction error (179, 180), three further SNPs were associated with survival. A protective effect for the IL10 genotype ACC/ACC in the donors was observed, while estrogen receptor 1 (ESR1) rs9340799 in the patient, IL6 rs18000795 in the donors, and TIRAP (or MAL) rs177374 in the patient were associated with poorer survival (2). The subsequent clinical and genetic score assigned to each patient was shown to have a better predictive value than the EBMT score alone. In a more recent cohort studying acute leukemia transplant patients (181), three polymorphisms, presence of toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) (alternatively named MAL) allele T (rs8177374) in the patient, absence of the glucocorticoid receptor (GCR) haplotype (consisting of rs6198, rs33389, and rs33388) ACT in the patient and absence of HSPA1L (or HSP70-hom) +2437 (rs2227956) allele C in the patient were associated with decreased survival. The subsequent clinical and genetic score assigned to each patient was shown to have a better predictive value than the EBMT score alone.
Interestingly, in all cohorts, the SNPs associated with reduced survival were all involved in downregulating the immune response, suggesting that this downregulation may be linked to reduced GvL responses and therefore lower overall survival.
These studies indicate that although replication of the exact genomic profiles may be difficult to achieve, the overall influence of genomics on the biology of the transplant is comparable and leads to a similar outcome, demonstrating that genomic studies are important for understanding the overall biology of the transplant.
Conclusion
In this review, we have shown the differential expression patterns of a variety of mRNA, different miRNAs and SNPs in specific genes that have a significant impact on transplant outcome and development of GvHD. In addition, several SNP–mRNA–miRNA regulatory networks have been found to contribute to post-HSCT outcomes. Taken together, these findings demonstrate how expression of specific miRNAs can target the genes of key immune response modulators, directly affecting expression of mRNAs that influence the aGvHD response. However, mRNA and miRNA expression studies have not yet revealed a set of genes or miRNAs that can be used as reliable biomarkers for predicting the outcome of HSCT across different transplantation centers. Further, preferably multicentre, studies are required to determine SNPs and to analyze miRNA and mRNA expression in parallel in cohorts of HSCT patients to further elucidate genetic risks of HSCT. Such combined approaches have the potential to improve clinical practise of HSCT and eventually to benefit patients.
Author Contributions
RG and PS drafted the manuscript; RC and JN commented and edited the draft; AD and RD supervised and revised the manuscript; and all authors approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RC and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors would like to thank Dasaradha Jalapothu (Department of Molecular Medicine, Institute of Basic Medical Sciences, University of Oslo) for his comments during preparation of the manuscript.
Funding
The work of the authors was supported by the European Union grant FP7-PEOPLE-2012-ITN-315963 (CELLEUROPE). Furthermore, the authors acknowledge support by the Open Access Publication Funds of the Göttingen University.
References
1. Harkensee C, Oka A, Onizuka M, Middleton PG, Inoko H, Hirayasu K, et al. Single nucleotide polymorphisms and outcome risk in unrelated mismatched hematopoietic stem cell transplantation: an exploration study. Blood (2012) 119:6365–72. doi: 10.1182/blood-2012-01-406785
2. Balavarca Y, Pearce K, Norden J, Collin M, Jackson G, Holler E, et al. Predicting survival using clinical risk scores and non-HLA immunogenetics. Bone Marrow Transplant (2015) 50:1445–52. doi:10.1038/bmt.2015.173
3. Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature (2001) 409:928–33. doi:10.1038/35057149
4. International Hapmap Consortium. The international HapMap project. Nature (2003) 426:789–96. doi:10.1038/nature02168
5. International Hapmap Consortium. A haplotype map of the human genome. Nature (2005) 437:1299–320. doi:10.1038/nature04226
6. International Hapmap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature (2007) 449:851–61. doi:10.1038/nature06258
7. Engle LJ, Simpson CL, Landers JE. Using high-throughput SNP technologies to study cancer. Oncogene (2006) 25:1594–601. doi:10.1038/sj.onc.1209368
8. Dickinson AM, Norden J. Non-HLA genomics: does it have a role in predicting haematopoietic stem cell transplantation outcome? Int J Immunogenet (2015) 42:229–38. doi:10.1111/iji.12202
10. Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics (2010) 11:537–61. doi:10.2174/138920210793175895
11. Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annu Rev Med (2009) 60:167–79. doi:10.1146/annurev.med.59.053006.104707
12. Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer (2009) 8:102. doi:10.1186/1476-4598-8-102
13. Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer (2010) 10:389–402. doi:10.1038/nrc2867
14. Atarod S, Dickinson AM. MicroRNAs: the missing link in the biology of graft-versus-host disease? Front Immunol (2013) 4:420. doi:10.3389/fimmu.2013.00420
15. Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol (2009) 27:5848–56. doi:10.1200/JCO.2009.24.0317
16. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med (2002) 196:389–99. doi:10.1084/jem.20020399
17. Beres AJ, Drobyski WR. The role of regulatory T cells in the biology of graft versus host disease. Front Immunol (2013) 4:163. doi:10.3389/fimmu.2013.00163
18. Mavin E, Ahmed SS, O’boyle G, Turner B, Douglass S, Norden J, et al. Regulatory T cells inhibit CD8(+) T-cell tissue invasion in human skin graft-versus-host reactions. Transplantation (2012) 94:456–64. doi:10.1097/TP.0b013e31826205d6
19. Cuzzola M, Fiasche M, Iacopino P, Messina G, Martino M, Console G, et al. A molecular and computational diagnostic approach identifies FOXP3, ICOS, CD52 and CASP1 as the most informative biomarkers in acute graft-versus-host disease. Haematologica (2012) 97:1532–8. doi:10.3324/haematol.2011.059980
20. Magenau JM, Qin X, Tawara I, Rogers CE, Kitko C, Schlough M, et al. Frequency of CD4(+)CD25(hi)FOXP3(+) regulatory T cells has diagnostic and prognostic value as a biomarker for acute graft-versus-host-disease. Biol Blood Marrow Transplant (2010) 16:907–14. doi:10.1016/j.bbmt.2010.02.026
21. Rezvani K, Mielke S, Ahmadzadeh M, Kilical Y, Savani BN, Zeilah J, et al. High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood (2006) 108:1291–7. doi:10.1182/blood-2006-02-003996
22. Wolf D, Wolf AM, Fong D, Rumpold H, Strasak A, Clausen J, et al. Regulatory T-cells in the graft and the risk of acute graft-versus-host disease after allogeneic stem cell transplantation. Transplantation (2007) 83:1107–13. doi:10.1097/01.tp.0000260140.04815.77
23. Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood (2005) 106:2903–11. doi:10.1182/blood-2005-03-1257
24. Miura Y, Thoburn CJ, Bright EC, Phelps ML, Shin T, Matsui EC, et al. Association of Foxp3 regulatory gene expression with graft-versus-host disease. Blood (2004) 104:2187–93. doi:10.1182/blood-2004-03-1040
25. Koreth J, Matsuoka K, Kim HT, Mcdonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365:2055–66. doi:10.1056/NEJMoa1108188
26. Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, Drobyski WR. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol (2012) 189:464–74. doi:10.4049/jimmunol.1200886
27. Sawamukai N, Satake A, Schmidt AM, Lamborn IT, Ojha P, Tanaka Y, et al. Cell-autonomous role of TGFbeta and IL-2 receptors in CD4+ and CD8+ inducible regulatory T-cell generation during GVHD. Blood (2012) 119:5575–83. doi:10.1182/blood-2011-07-367987
28. Heinrichs J, Li J, Nguyen H, Wu Y, Bastian D, Daethanasanmak A, et al. CD8(+) Tregs promote GVHD prevention and overcome the impaired GVL effect mediated by CD4(+) Tregs in mice. Oncoimmunology (2016) 5:e1146842. doi:10.1080/2162402X.2016.1146842
29. Picarda E, Bezie S, Venturi V, Echasserieau K, Merieau E, Delhumeau A, et al. MHC-derived allopeptide activates TCR-biased CD8+ Tregs and suppresses organ rejection. J Clin Invest (2014) 124:2497–512. doi:10.1172/JCI71533
30. Eastell T, Group BS, Hinks A, Thomson W. SNPs in the FOXP3 gene region show no association with juvenile idiopathic arthritis in a UK Caucasian population. Rheumatology (Oxford) (2007) 46:1263–5. doi:10.1093/rheumatology/kem129
31. Shen Z, Chen L, Hao F, Wang G, Liu Y. Intron-1 rs3761548 is related to the defective transcription of Foxp3 in psoriasis through abrogating E47/c-Myb binding. J Cell Mol Med (2010) 14:226–41. doi:10.1111/j.1582-4934.2008.00370.x
32. Piao Z, Kim HJ, Choi JY, Hong CR, Lee JW, Kang HJ, et al. Effect of FOXP3 polymorphism on the clinical outcomes after allogeneic hematopoietic stem cell transplantation in pediatric acute leukemia patients. Int Immunopharmacol (2016) 31:132–9. doi:10.1016/j.intimp.2015.12.022
33. Tiercy JM. Immunogenetics of hematopoietic stem cell transplantation: the contribution of microsatellite polymorphism studies. Int J Immunogenet (2011) 38:365–72. doi:10.1111/j.1744-313X.2011.01026.x
34. Bassuny WM, Ihara K, Sasaki Y, Kuromaru R, Kohno H, Matsuura N, et al. A functional polymorphism in the promoter/enhancer region of the FOXP3/Scurfin gene associated with type 1 diabetes. Immunogenetics (2003) 55:149–56. doi:10.1007/s00251-003-0559-8
35. Noriega V, Martinez-Laperche C, Buces E, Pion M, Sanchez-Hernandez N, Martin-Antonio B, et al. The genotype of the donor for the (GT)n polymorphism in the promoter/enhancer of FOXP3 is associated with the development of severe acute GVHD but does not affect the GVL effect after myeloablative HLA-identical allogeneic stem cell transplantation. PLoS One (2015) 10:e0140454. doi:10.1371/journal.pone.0140454
36. Rouas R, Fayyad-Kazan H, El Zein N, Lewalle P, Rothe F, Simion A, et al. Human natural Treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur J Immunol (2009) 39:1608–18. doi:10.1002/eji.200838509
37. Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol (2013) 13:666–78. doi:10.1038/nri3494
38. Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol (2009) 182:2578–82. doi:10.4049/jimmunol.0803162
39. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity (2009) 30:80–91. doi:10.1016/j.immuni.2008.11.010
40. Cobb BS, Hertweck A, Smith J, O’connor E, Graf D, Cook T, et al. A role for Dicer in immune regulation. J Exp Med (2006) 203:2519–27. doi:10.1084/jem.20061692
41. Jeker LT, Zhou X, Gershberg K, De Kouchkovsky D, Morar MM, Stadthagen G, et al. MicroRNA 10a marks regulatory T cells. PLoS One (2012) 7:e36684. doi:10.1371/journal.pone.0036684
42. Jalapothu D, Boieri M, Crossland RE, Shah P, Butt IA, Norden J, et al. Tissue-specific expression patterns of microRNA during acute graft-versus-host disease in the rat. Front Immunol (2016) 7:361. doi:10.3389/fimmu.2016.00361
43. Atarod S, Ahmed MM, Lendrem C, Pearce KF, Cope W, Norden J, et al. miR-146a and miR-155 expression levels in acute graft-versus-host disease incidence. Front Immunol (2016) 7:56. doi:10.3389/fimmu.2016.00056
44. Pontoppidan PL, Jordan K, Carlsen AL, Uhlving HH, Kielsen K, Christensen M, et al. Associations between gastrointestinal toxicity, micro RNA and cytokine production in patients undergoing myeloablative allogeneic stem cell transplantation. Int Immunopharmacol (2015) 25:180–8. doi:10.1016/j.intimp.2014.12.038
45. Ranganathan P, Heaphy CE, Costinean S, Stauffer N, Na C, Hamadani M, et al. Regulation of acute graft-versus-host disease by microRNA-155. Blood (2012) 119:4786–97. doi:10.1182/blood-2011-10-387522
46. Chen S, Smith BA, Iype J, Prestipino A, Pfeifer D, Grundmann S, et al. MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood (2015) 126:103–12. doi:10.1182/blood-2014-12-617258
47. Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum (2008) 58:1001–9. doi:10.1002/art.23386
48. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science (2007) 316:604–8. doi:10.1126/science.1141229
49. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103:12481–6. doi:10.1073/pnas.0605298103
50. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med (2011) 208:1189–201. doi:10.1084/jem.20101823
51. Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med (2012) 209:1655–70. doi:10.1084/jem.20112218
52. Stickel N, Prinz G, Pfeifer D, Hasselblatt P, Schmitt-Graeff A, Follo M, et al. MiR-146a regulates the TRAF6/TNF-axis in donor T cells during GVHD. Blood (2014) 124:2586–95. doi:10.1182/blood-2014-04-569046
53. Zhao JL, Starczynowski DT. Role of microRNA-146a in normal and malignant hematopoietic stem cell function. Front Genet (2014) 5:219. doi:10.3389/fgene.2014.00219
54. Lu Y, Hippen KL, Lemire AL, Gu J, Wang W, Ni X, et al. miR-146b antagomir-treated human Tregs acquire increased GVHD inhibitory potency. Blood (2016) 128:1424–35. doi:10.1182/blood-2016-05-714535
55. Xu W, Xu J, Liu S, Chen B, Wang X, Li Y, et al. Effects of common polymorphisms rs11614913 in miR-196a2 and rs2910164 in miR-146a on cancer susceptibility: a meta-analysis. PLoS One (2011) 6:e20471. doi:10.1371/journal.pone.0020471
56. Isernhagen A, Malzahn D, Bickeboller H, Dressel R. Impact of the MICA-129Met/Val dimorphism on NKG2D-mediated biological functions and disease risks. Front Immunol (2016) 7:588. doi:10.3389/fimmu.2016.00588
57. Isernhagen A, Malzahn D, Viktorova E, Elsner L, Monecke S, Von Bonin F, et al. The MICA-129 dimorphism affects NKG2D signaling and outcome of hematopoietic stem cell transplantation. EMBO Mol Med (2015) 7:1480–502. doi:10.15252/emmm.201505246
58. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi:10.1126/science.285.5428.727
59. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol (2001) 2:255–60. doi:10.1038/85321
60. Gannage M, Buzyn A, Bogiatzi SI, Lambert M, Soumelis V, Dal Cortivo L, et al. Induction of NKG2D ligands by gamma radiation and tumor necrosis factor-alpha may participate in the tissue damage during acute graft-versus-host disease. Transplantation (2008) 85:911–5. doi:10.1097/TP.0b013e31816691ef
61. Boukouaci W, Busson M, Peffault De Latour R, Rocha V, Suberbielle C, Bengoufa D, et al. MICA-129 genotype, soluble MICA, and anti-MICA antibodies as biomarkers of chronic graft-versus-host disease. Blood (2009) 114:5216–24. doi:10.1182/blood-2009-04-217430
62. Isernhagen A, Malzahn D, Monecke S, Schilling D, Shah P, Multhoff G, et al. Functional consequences of genetic polymorphisms in the NKG2D receptor signaling pathway and putative gene interactions. Receptors Clin Investig (2016) 3:e1269. doi:10.14800/rci.1269
63. Espinoza JL, Takami A, Onizuka M, Sao H, Akiyama H, Miyamura K, et al. NKG2D gene polymorphism has a significant impact on transplant outcomes after HLA-fully-matched unrelated bone marrow transplantation for standard risk hematologic malignancies. Haematologica (2009) 94:1427–34. doi:10.3324/haematol.2009.008318
64. Shafi S, Vantourout P, Wallace G, Antoun A, Vaughan R, Stanford M, et al. An NKG2D-mediated human lymphoid stress surveillance response with high interindividual variation. Sci Transl Med (2011) 3:113ra124. doi:10.1126/scitranslmed.3002922
65. Lo PH, Urabe Y, Kumar V, Tanikawa C, Koike K, Kato N, et al. Identification of a functional variant in the MICA promoter which regulates MICA expression and increases HCV-related hepatocellular carcinoma risk. PLoS One (2013) 8:e61279. doi:10.1371/journal.pone.0061279
66. Ashiru O, Lopez-Cobo S, Fernandez-Messina L, Pontes-Quero S, Pandolfi R, Reyburn HT, et al. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem J (2013) 454:295–302. doi:10.1042/BJ20130194
67. Isernhagen A, Schilling D, Monecke S, Shah P, Elsner L, Walter L, et al. The MICA-129Met/Val dimorphism affects plasma membrane expression and shedding of the NKG2D ligand MICA. Immunogenetics (2016) 68:109–23. doi:10.1007/s00251-015-0884-8
68. Kitcharoen K, Witt CS, Romphruk AV, Christiansen FT, Leelayuwat C. MICA, MICB, and MHC beta block matching in bone marrow transplantation: relevance to transplantation outcome. Hum Immunol (2006) 67:238–46. doi:10.1016/j.humimm.2006.02.012
69. Parmar S, Del Lima M, Zou Y, Patah PA, Liu P, Cano P, et al. Donor-recipient mismatches in MHC class I chain-related gene A in unrelated donor transplantation lead to increased incidence of acute graft-versus-host disease. Blood (2009) 114:2884–7. doi:10.1182/blood-2009-05-223172
70. Askar M, Sun Y, Rybicki L, Zhang A, Thomas D, Kalaycio M, et al. Synergistic effect of major histocompatibility complex class I-related chain a and human leukocyte antigen-DPB1 mismatches in association with acute graft-versus-host disease after unrelated donor hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20:1835–40. doi:10.1016/j.bbmt.2014.07.019
71. Carapito R, Jung N, Kwemou M, Untrau M, Michel S, Pichot A, et al. Matching for the nonconventional MHC-I MICA gene significantly reduces the incidence of acute and chronic GVHD. Blood (2016) 128:1979–86. doi:10.1182/blood-2016-05-719070
72. Fuerst D, Neuchel C, Niederwieser D, Bunjes D, Gramatzki M, Wagner E, et al. Matching for the MICA-129 polymorphism is beneficial in unrelated hematopoietic stem cell transplantation. Blood (2016) 128:3169–76. doi:10.1182/blood-2016-05-716357
73. Anderson E, Grzywacz B, Wang H, Wang T, Haagenson M, Spellman S, et al. Limited role of MHC class I chain-related gene A (MICA) typing in assessing graft-versus-host disease risk after fully human leukocyte antigen-matched unrelated donor transplantation. Blood (2009) 114:authorrely4754–5. doi:10.1182/blood-2009-08-239301
74. Askar M, Sobecks R, Wang T, Haagenson M, Majhail N, Madbouly A, et al. MHC class I chain-related gene A (MICA) donor-recipient mismatches and MICA-129 polymorphism in unrelated donor hematopoietic cell transplantations has no impact on outcomes in acute lymphoblastic leukemia, acute myeloid leukemia, or myelodysplastic syndrome: a center for International Blood and Marrow Transplant Research Study. Biol Blood Marrow Transplant (2017) 23:436–44. doi:10.1016/j.bbmt.2016.11.021
75. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
76. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol (2008) 9:1065–73. doi:10.1038/ni.1642
77. Eissmann P, Evans JH, Mehrabi M, Rose EL, Nedvetzki S, Davis DM. Multiple mechanisms downstream of TLR-4 stimulation allow expression of NKG2D ligands to facilitate macrophage/NK cell crosstalk. J Immunol (2010) 184:6901–9. doi:10.4049/jimmunol.0903985
78. Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res (2014) 74:5746–57. doi:10.1158/0008-5472.CAN-13-2563
79. Xie J, Liu M, Li Y, Nie Y, Mi Q, Zhao S. Ovarian tumor-associated microRNA-20a decreases natural killer cell cytotoxicity by downregulating MICA/B expression. Cell Mol Immunol (2014) 11:495–502. doi:10.1038/cmi.2014.30
80. Yang H, Lan P, Hou Z, Guan Y, Zhang J, Xu W, et al. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer (2015) 112:112–21. doi:10.1038/bjc.2014.547
81. Kishikawa T, Otsuka M, Yoshikawa T, Ohno M, Takata A, Shibata C, et al. Regulation of the expression of the liver cancer susceptibility gene MICA by microRNAs. Sci Rep (2013) 3:2739. doi:10.1038/srep02739
82. Ohno M, Otsuka M, Kishikawa T, Shibata C, Yoshikawa T, Takata A, et al. Specific delivery of microRNA93 into HBV-replicating hepatocytes downregulates protein expression of liver cancer susceptible gene MICA. Oncotarget (2014) 5:5581–90. doi:10.18632/oncotarget.2143
83. Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. J Immunol (2009) 182:39–43. doi:10.4049/jimmunol.182.1.39
84. Min D, Lv XB, Wang X, Zhang B, Meng W, Yu F, et al. Downregulation of miR-302c and miR-520c by 1,25(OH)2D3 treatment enhances the susceptibility of tumour cells to natural killer cell-mediated cytotoxicity. Br J Cancer (2013) 109:723–30. doi:10.1038/bjc.2013.337
85. Fielding CA, Aicheler R, Stanton RJ, Wang EC, Han S, Seirafian S, et al. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog (2014) 10:e1004058. doi:10.1371/journal.ppat.1004058
86. Elias S, Mandelboim O. Battle of the midgets: innate microRNA networking. RNA Biol (2012) 9:792–8. doi:10.4161/rna.19717
87. Jasinski-Bergner S, Mandelboim O, Seliger B. The role of microRNAs in the control of innate immune response in cancer. J Natl Cancer Inst (2014) 106:dju257. doi:10.1093/jnci/dju257
88. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica (2012) 97:1295–303. doi:10.3324/haematol.2011.058529
89. Boieri M, Shah P, Dressel R, Inngjerdingen M. The role of animal models in the study of hematopoietic stem cell transplantation and GvHD: a historical overview. Front Immunol (2016) 7:333. doi:10.3389/fimmu.2016.00333
90. Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, et al. The microRNA spectrum in 12 body fluids. Clin Chem (2010) 56:1733–41. doi:10.1373/clinchem.2010.147405
91. Xiao B, Wang Y, Li W, Baker M, Guo J, Corbet K, et al. Plasma microRNA signature as a noninvasive biomarker for acute graft-versus-host disease. Blood (2013) 122:3365–75. doi:10.1182/blood-2013-06-510586
92. Verner J, Kabathova J, Tomancova A, Pavlova S, Tichy B, Mraz M, et al. Gene expression profiling of acute graft-vs-host disease after hematopoietic stem cell transplantation. Exp Hematol (2012) 40(899–905):e895. doi:10.1016/j.exphem.2012.06.011
93. Buzzeo MP, Yang J, Casella G, Reddy V. A preliminary gene expression profile of acute graft-versus-host disease. Cell Transplant (2008) 17:489–94. doi:10.3727/096368908785096042
94. Baron C, Somogyi R, Greller LD, Rineau V, Wilkinson P, Cho CR, et al. Prediction of graft-versus-host disease in humans by donor gene-expression profiling. PLoS Med (2007) 4:e23. doi:10.1371/journal.pmed.0040023
95. Takahashi N, Sato N, Takahashi S, Tojo A. Gene-expression profiles of peripheral blood mononuclear cell subpopulations in acute graft-vs-host disease following cord blood transplantation. Exp Hematol (2008) 36:1760–70. doi:10.1016/j.exphem.2008.07.007
96. Lai P, Weng J, Lu Z, Guo R, Luo C, Wu S, et al. Gene expression profiling-based identification of CD28 and PI3K as new biomarkers for chronic graft-versus-host disease. DNA Cell Biol (2011) 30:1019–25. doi:10.1089/dna.2011.1284
97. Westekemper H, Meller S, Citak S, Schulte C, Steuhl KP, Homey B, et al. Differential chemokine expression in chronic GVHD of the conjunctiva. Bone Marrow Transplant (2010) 45:1340–6. doi:10.1038/bmt.2009.346
98. Cocho L, Fernandez I, Calonge M, Martinez V, Gonzalez-Garcia MJ, Caballero D, et al. Gene expression-based predictive models of graft versus host disease-associated dry eye. Invest Ophthalmol Vis Sci (2015) 56:4570–81. doi:10.1167/iovs.15-16736
99. Sugerman PB, Faber SB, Willis LM, Petrovic A, Murphy GF, Pappo J, et al. Kinetics of gene expression in murine cutaneous graft-versus-host disease. Am J Pathol (2004) 164:2189–202. doi:10.1016/S0002-9440(10)63776-5
100. Ichiba T, Teshima T, Kuick R, Misek DE, Liu C, Takada Y, et al. Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood (2003) 102:763–71. doi:10.1182/blood-2002-09-2748
101. Sadeghi B, Al-Chaqmaqchi H, Al-Hashmi S, Brodin D, Hassan Z, Abedi-Valugerdi M, et al. Early-phase GVHD gene expression profile in target versus non-target tissues: kidney, a possible target? Bone Marrow Transplant (2013) 48:284–93. doi:10.1038/bmt.2012.120
102. Zhou L, Askew D, Wu C, Gilliam AC. Cutaneous gene expression by DNA microarray in murine sclerodermatous graft-versus-host disease, a model for human scleroderma. J Invest Dermatol (2007) 127:281–92. doi:10.1038/sj.jid.5700517
103. Novota P, Zinöcker S, Norden J, Wang XN, Sviland L, Opitz L, et al. Expression profiling of major histocompatibility and natural killer complex genes reveals candidates for controlling risk of graft versus host disease. PLoS One (2011) 6:e16582. doi:10.1371/journal.pone.0016582
104. Wang Y, Zhao X, Ye X, Luo H, Zhao T, Diao Y, et al. Plasma microRNA-586 is a new biomarker for acute graft-versus-host disease. Ann Hematol (2015) 94:1505–14. doi:10.1007/s00277-015-2414-z
105. Wu Y, Heinrichs J, Bastian D, Fu J, Nguyen H, Schutt S, et al. MicroRNA-17-92 controls T-cell responses in graft-versus-host disease and leukemia relapse in mice. Blood (2015) 126:1314–23. doi:10.1182/blood-2015-02-627356
106. Serody J. GVHD and miR: good things in small packages. Blood (2015) 126:1265–7. doi:10.1182/blood-2015-07-657114
107. Jiang S, Li C, Olive V, Lykken E, Feng F, Sevilla J, et al. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood (2011) 118:5487–97. doi:10.1182/blood-2011-05-355644
108. Leonhardt F, Grundmann S, Behe M, Bluhm F, Dumont RA, Braun F, et al. Inflammatory neovascularization during graft-versus-host disease is regulated by alphav integrin and miR-100. Blood (2013) 121:3307–18. doi:10.1182/blood-2012-07-442665
109. He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network – another piece in the tumour-suppression puzzle. Nat Rev Cancer (2007) 7:819–22. doi:10.1038/nrc2232
110. Kee Y, D’andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest (2012) 122:3799–806. doi:10.1172/JCI58321
111. Wang L, Romero M, Ratajczak P, Leboeuf C, Belhadj S, Peffault De Latour R, et al. Increased apoptosis is linked to severe acute GVHD in patients with Fanconi anemia. Bone Marrow Transplant (2013) 48:849–53. doi:10.1038/bmt.2012.237
112. Reddy P. Pathophysiology of acute graft-versus-host disease. Hematol Oncol (2003) 21:149–61. doi:10.1002/hon.716
113. Qian L, Wu Z, Shen J. Advances in the treatment of acute graft-versus-host disease. J Cell Mol Med (2013) 17:966–75. doi:10.1111/jcmm.12093
114. Ferrara JL, Marion A, Mcintyre JF, Murphy GF, Burakoff SJ. Amelioration of acute graft vs host disease due to minor histocompatibility antigens by in vivo administration of anti-interleukin 2 receptor antibody. J Immunol (1986) 137:1874–7.
115. Blaise D, Olive D, Hirn M, Viens P, Lafage M, Attal M, et al. Prevention of acute GVHD by in vivo use of anti-interleukin-2 receptor monoclonal antibody (33B3.1): a feasibility trial in 15 patients. Bone Marrow Transplant (1991) 8:105–11.
116. Zeiser R, Beilhack A, Negrin RS. Acute graft-versus-host disease-challenge for a broader application of allogeneic hematopoietic cell transplantation. Curr Stem Cell Res Ther (2006) 1:203–12. doi:10.2174/157488806776956896
117. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445:771–5. doi:10.1038/nature05543
118. Jaksch M, Mattsson J. The pathophysiology of acute graft-versus-host disease. Scand J Immunol (2005) 61:398–409. doi:10.1111/j.1365-3083.2005.01595.x
119. Dickinson AM, Sviland L, Hamilton PJ, Usher P, Taylor P, Jackson G, et al. Cytokine involvement in predicting clinical graft-versus-host disease in allogeneic bone marrow transplant recipients. Bone Marrow Transplant (1994) 13:65–70.
120. Tanaka J, Imamura M, Kasai M, Sakurada K, Miyazaki T. Cytokine gene expression after allogeneic bone marrow transplantation. Leuk Lymphoma (1995) 16:413–8. doi:10.3109/10428199509054427
121. Das H, Imoto S, Murayama T, Mizuno I, Sugimoto T, Taniguchi R, et al. Kinetic analysis of cytokine gene expression in patients with GVHD after donor lymphocyte infusion. Bone Marrow Transplant (2001) 27:373–80. doi:10.1038/sj.bmt.1702799
122. Poloni A, Sartini D, Emanuelli M, Trappolini S, Mancini S, Pozzi V, et al. Gene expression profile of cytokines in patients with chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation with reduced conditioning. Cytokine (2011) 53:376–83. doi:10.1016/j.cyto.2010.12.008
123. Kohrt HE, Tian L, Li L, Alizadeh AA, Hsieh S, Tibshirani RJ, et al. Identification of gene microarray expression profiles in patients with chronic graft-versus-host disease following allogeneic hematopoietic cell transplantation. Clin Immunol (2013) 148:124–35. doi:10.1016/j.clim.2013.04.013
124. Piguet PF, Grau GE, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs.-host disease. J Exp Med (1987) 166:1280–9. doi:10.1084/jem.166.5.1280
125. Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J Jr, et al. Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest (1998) 102:1882–91. doi:10.1172/JCI4285
126. Blaser BW, Roychowdhury S, Kim DJ, Schwind NR, Bhatt D, Yuan W, et al. Donor-derived IL-15 is critical for acute allogeneic graft-versus-host disease. Blood (2005) 105:894–901. doi:10.1182/blood-2004-05-1687
127. Kumaki S, Minegishi M, Fujie H, Sasahara Y, Ohashi Y, Tsuchiya S, et al. Prolonged secretion of IL-15 in patients with severe forms of acute graft-versus-host disease after allogeneic bone marrow transplantation in children. Int J Hematol (1998) 67:307–12. doi:10.1016/S0925-5710(97)00117-5
128. Shimizu M, Shimamura M, Owaki T, Asakawa M, Fujita K, Kudo M, et al. Antiangiogenic and antitumor activities of IL-27. J Immunol (2006) 176:7317–24. doi:10.4049/jimmunol.176.12.7317
129. Liu Y, Wu Y, Wang Y, Cai Y, Hu B, Bao G, et al. IL-35 mitigates murine acute graft-versus-host disease with retention of graft-versus-leukemia effects. Leukemia (2015) 29:939–46. doi:10.1038/leu.2014.310
130. Zhang XH, Zhou Y, Zhang JM, Zhou SY, Wang M, Feng R, et al. IL-35 inhibits acute graft-versus-host disease in a mouse model. Int Immunopharmacol (2015) 29:383–92. doi:10.1016/j.intimp.2015.10.025
131. Middleton PG, Taylor PR, Jackson G, Proctor SJ, Dickinson AM. Cytokine gene polymorphisms associating with severe acute graft-versus-host disease in HLA-identical sibling transplants. Blood (1998) 92:3943–8.
132. Cavet J, Middleton PG, Segall M, Noreen H, Davies SM, Dickinson AM. Recipient tumor necrosis factor-alpha and interleukin-10 gene polymorphisms associate with early mortality and acute graft-versus-host disease severity in HLA-matched sibling bone marrow transplants. Blood (1999) 94:3941–6.
133. Socie G, Loiseau P, Tamouza R, Janin A, Busson M, Gluckman E, et al. Both genetic and clinical factors predict the development of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Transplantation (2001) 72:699–706. doi:10.1097/00007890-200108270-00024
134. Mullighan CG, Bardy PG. New directions in the genomics of allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2007) 13:127–44. doi:10.1016/j.bbmt.2006.10.018
135. Espinoza JL, Takami A, Nakata K, Onizuka M, Kawase T, Akiyama H, et al. A genetic variant in the IL-17 promoter is functionally associated with acute graft-versus-host disease after unrelated bone marrow transplantation. PLoS One (2011) 6:e26229. doi:10.1371/journal.pone.0026229
136. Holler E, Rogler G, Brenmoehl J, Hahn J, Herfarth H, Greinix H, et al. Prognostic significance of NOD2/CARD15 variants in HLA-identical sibling hematopoietic stem cell transplantation: effect on long-term outcome is confirmed in 2 independent cohorts and may be modulated by the type of gastrointestinal decontamination. Blood (2006) 107:4189–93. doi:10.1182/blood-2005-09-3741
137. Grube M, Brenmoehl J, Rogler G, Hahn J, Herr W, Holler E. Donor nucleotide-binding oligomerization-containing protein 2 (NOD2) single nucleotide polymorphism 13 is associated with septic shock after allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2015) 21:1399–404. doi:10.1016/j.bbmt.2015.05.011
138. Economou M, Pappas G. New global map of Crohn’s disease: genetic, environmental, and socioeconomic correlations. Inflamm Bowel Dis (2008) 14:709–20. doi:10.1002/ibd.20352
139. Chien JW, Zhang XC, Fan W, Wang H, Zhao LP, Martin PJ, et al. Evaluation of published single nucleotide polymorphisms associated with acute GVHD. Blood (2012) 119:5311–9. doi:10.1182/blood-2011-09-371153
140. Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology (2003) 124:1001–9. doi:10.1053/gast.2003.50157
141. Goussetis E, Varela I, Peristeri I, Kitra V, Spanou K, Moraloglou O, et al. Cytokine gene polymorphisms and graft-versus-host disease in children after matched sibling hematopoietic stem cell transplantation: a single-center experience. Cell Mol Immunol (2011) 8:276–80. doi:10.1038/cmi.2011.4
142. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol (2011) 89:207–15. doi:10.1038/icb.2010.158
143. Ahmed SS, Wang XN, Norden J, Pearce K, El-Gezawy E, Atarod S, et al. Identification and validation of biomarkers associated with acute and chronic graft versus host disease. Bone Marrow Transplant (2015) 50:1563–71. doi:10.1038/bmt.2015.191
144. Bouazzaoui A, Spacenko E, Mueller G, Miklos S, Huber E, Holler E, et al. Chemokine and chemokine receptor expression analysis in target organs of acute graft-versus-host disease. Genes Immun (2009) 10:687–701. doi:10.1038/gene.2009.49
145. Hasegawa H, Inoue A, Kohno M, Lei J, Miyazaki T, Yoshie O, et al. Therapeutic effect of CXCR3-expressing regulatory T cells on liver, lung and intestinal damages in a murine acute GVHD model. Gene Ther (2008) 15:171–82. doi:10.1038/sj.gt.3303051
146. Castor MG, Pinho V, Teixeira MM. The role of chemokines in mediating graft versus host disease: opportunities for novel therapeutics. Front Pharmacol (2012) 3:23. doi:10.3389/fphar.2012.00023
147. Hansen JA, Chien JW, Warren EH, Zhao LP, Martin PJ. Defining genetic risk for graft-versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol (2010) 17:483–92. doi:10.1097/MOH.0b013e32833eb770
148. Goker H, Haznedaroglu IC, Chao NJ. Acute graft-vs-host disease: pathobiology and management. Exp Hematol (2001) 29:259–77. doi:10.1016/S0301-472X(00)00677-9
149. Hipp MS, Kalveram B, Raasi S, Groettrup M, Schmidtke G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol Cell Biol (2005) 25:3483–91. doi:10.1128/MCB.25.9.3483-3491.2005
150. Warren EH, Zhang XC, Li S, Fan W, Storer BE, Chien JW, et al. Effect of MHC and non-MHC donor/recipient genetic disparity on the outcome of allogeneic HCT. Blood (2012) 120:2796–806. doi:10.1182/blood-2012-04-347286
151. Dickinson AM, Charron D. Non-HLA immunogenetics in hematopoietic stem cell transplantation. Curr Opin Immunol (2005) 17:517–25. doi:10.1016/j.coi.2005.07.017
152. Turpeinen H, Ojala PJ, Ojala K, Miettinen M, Volin L, Partanen J. Minor histocompatibility antigens as determinants for graft-versus-host disease after allogeneic haematopoietic stem cell transplantation. Int J Immunogenet (2013) 40:495–501. doi:10.1111/iji.12051
153. Sampson JK, Sheth NU, Koparde VN, Scalora AF, Serrano MG, Lee V, et al. Whole exome sequencing to estimate alloreactivity potential between donors and recipients in stem cell transplantation. Br J Haematol (2014) 166:566–70. doi:10.1111/bjh.12898
154. Jameson-Lee M, Koparde V, Griffith P, Scalora AF, Sampson JK, Khalid H, et al. In silico derivation of HLA-specific alloreactivity potential from whole exome sequencing of stem-cell transplant donors and recipients: understanding the quantitative immunobiology of allogeneic transplantation. Front Immunol (2014) 5:529. doi:10.3389/fimmu.2014.00529
155. Toor AA, Kobulnicky JD, Salman S, Roberts CH, Jameson-Lee M, Meier J, et al. Stem cell transplantation as a dynamical system: are clinical outcomes deterministic? Front Immunol (2014) 5:613. doi:10.3389/fimmu.2014.00613
156. Abdul Razzaq B, Scalora A, Koparde VN, Meier J, Mahmood M, Salman S, et al. Dynamical system modeling to simulate donor T cell response to whole exome sequencing-derived recipient peptides demonstrates different alloreactivity potential in HLA-matched and -mismatched donor-recipient pairs. Biol Blood Marrow Transplant (2016) 22:850–61. doi:10.1016/j.bbmt.2015.11.1103
157. Theiss-Suennemann J, Jörss K, Messmann JJ, Reichardt SD, Montes-Cobos E, Lühder F, et al. Glucocorticoids attenuate acute graft-versus-host disease by suppressing the cytotoxic capacity of CD8(+) T cells. J Pathol (2015) 235:646–55. doi:10.1002/path.4475
158. Ren G, Zhao X, Zhang L, Zhang J, L’huillier A, Ling W, et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol (2010) 184:2321–8. doi:10.4049/jimmunol.0902023
159. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1:220–8. doi:10.1038/35105024
160. Manickasingham SP, Anderton SM, Burkhart C, Wraith DC. Qualitative and quantitative effects of CD28/B7-mediated costimulation on naive T cells in vitro. J Immunol (1998) 161:3827–35.
161. Karabon L, Markiewicz M, Partyka A, Pawlak-Adamska E, Tomkiewicz A, Dzierzak-Mietla M, et al. A CT60G>A polymorphism in the CTLA-4 gene of the recipient may confer susceptibility to acute graft versus host disease after allogeneic hematopoietic stem cell transplantation. Immunogenetics (2015) 67:295–304. doi:10.1007/s00251-015-0840-7
162. Mossallam GI, Samra MA. CTLA-4 polymorphism and clinical outcome post allogeneic hematopoietic stem cell transplantation. Hum Immunol (2013) 74:1643–8. doi:10.1016/j.humimm.2013.08.002
163. Iravani-Saadi M, Karimi MH, Yaghobi R, Geramizadeh B, Ramzi M, Niknam A, et al. Polymorphism of costimulatory molecules (CTLA4, ICOS, PD.1 and CD28) and allogeneic hematopoietic stem cell transplantation in Iranian patients. Immunol Invest (2014) 43:391–404. doi:10.3109/08820139.2013.879594
164. Sato M, Storb R, Loretz C, Stone D, Mielcarek M, Sale GE, et al. Inducible costimulator (ICOS) up-regulation on activated T cells in chronic graft-versus-host disease after dog leukocyte antigen-nonidentical hematopoietic cell transplantation: a potential therapeutic target. Transplantation (2013) 96:34–41. doi:10.1097/TP.0b013e318295c025
165. Yew PY, Alachkar H, Yamaguchi R, Kiyotani K, Fang H, Yap KL, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant (2015) 50:1227–34. doi:10.1038/bmt.2015.133
166. Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, Von Bergwelt-Baildon MS. The role of B cells in the pathogenesis of graft-versus-host disease. Blood (2009) 114:4919–27. doi:10.1182/blood-2008-10-161638
167. Schultz KR, Paquet J, Bader S, Hayglass KT. Requirement for B cells in T cell priming to minor histocompatibility antigens and development of graft-versus-host disease. Bone Marrow Transplant (1995) 16:289–95.
168. Allen JL, Tata PV, Fore MS, Wooten J, Rudra S, Deal AM, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood (2014) 123:2108–15. doi:10.1182/blood-2013-10-533562
169. Xu S, Tan JE, Wong EP, Manickam A, Ponniah S, Lam KP. B cell development and activation defects resulting in xid-like immunodeficiency in BLNK/SLP-65-deficient mice. Int Immunol (2000) 12:397–404. doi:10.1093/intimm/12.3.397
170. Dickinson AM. SNPs and GVHD prediction: where to next? Blood (2012) 119:5066–8. doi:10.1182/blood-2012-03-409078
171. Sato-Otsubo A, Nannya Y, Kashiwase K, Onizuka M, Azuma F, Akatsuka Y, et al. Genome-wide surveillance of mismatched alleles for graft-versus-host disease in stem cell transplantation. Blood (2015) 126:2752–63. doi:10.1182/blood-2015-03-630707
172. Bari R, Hartford C, Chan WK, Vong Q, Li Y, Gan K, et al. Genome-wide single-nucleotide polymorphism analysis revealed SUFU suppression of acute graft-versus-host disease through downregulation of HLA-DR expression in recipient dendritic cells. Sci Rep (2015) 5:11098. doi:10.1038/srep11098
173. Dickinson AM, Pearce KF, Norden J, O’brien SG, Holler E, Bickeboller H, et al. Impact of genomic risk factors on outcome after hematopoietic stem cell transplantation for patients with chronic myeloid leukemia. Haematologica (2010) 95:922–7. doi:10.3324/haematol.2009.016220
174. Pearce KF, Lee SJ, Haagenson M, Petersdorf EW, Norden J, Collin MP, et al. Analysis of non-HLA genomic risk factors in HLA-matched unrelated donor hematopoietic cell transplantation for chronic myeloid leukemia. Haematologica (2012) 97:1014–9. doi:10.3324/haematol.2011.053611
175. Gratwohl A, Hermans J, Goldman JM, Arcese W, Carreras E, Devergie A, et al. Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Chronic Leukemia Working Party of the European Group for blood and marrow transplantation. Lancet (1998) 352:1087–92. doi:10.1016/S0140-6736(98)03030-X
176. Gratwohl A, Brand R, Apperley J, Crawley C, Ruutu T, Corradini P, et al. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia in Europe 2006: transplant activity, long-term data and current results. An analysis by the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Haematologica (2006) 91:513–21.
177. Gratwohl A, Stern M, Brand R, Apperley J, Baldomero H, De Witte T, et al. Risk score for outcome after allogeneic hematopoietic stem cell transplantation: a retrospective analysis. Cancer (2009) 115:4715–26. doi:10.1002/cncr.24531
178. Gratwohl A. The EBMT risk score. Bone Marrow Transplant (2012) 47:749–56. doi:10.1038/bmt.2011.110
179. Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrika (1982) 69:239–41. doi:10.1093/biomet/69.1.239
180. Efron B. Estimating the error rate of a prediction rule: improvement on cross-validation. J Am Stat Assoc (1983) 78:316–31. doi:10.1080/01621459.1983.10477973
Keywords: gene expression profiling, regulatory networks, non-human leukocyte antigen single-nucleotide polymorphisms, microRNAs, forkheadbox protein 3, miR-155, miR-146a, MICA
Citation: Gam R, Shah P, Crossland RE, Norden J, Dickinson AM and Dressel R (2017) Genetic Association of Hematopoietic Stem Cell Transplantation Outcome beyond Histocompatibility Genes. Front. Immunol. 8:380. doi: 10.3389/fimmu.2017.00380
Received: 03 August 2016; Accepted: 16 March 2017;
Published: 03 April 2017
Edited by:
Seiamak Bahram, University of Strasbourg, FranceReviewed by:
Raphael Carapito, University of Strasbourg, FranceIgnacio Anegon, University of Nantes, France
Copyright: © 2017 Gam, Shah, Crossland, Norden, Dickinson and Dressel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf Dressel, cmRyZXNzZUBnd2RnLmRl
†These authors share first authorship.
‡These authors share senior authorship.